
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom non-innocent to guilty: on the role of redox-active ligands in the electro-assisted reduction of CO2 mediated by a cobalt(II)-polypyridyl complex†
N.
Queyriaux‡
 *a,
K.
Abel
a,
J.
Fize
b,
J.
Pécaut
c,
M.
Orio
*d and
L.
Hammarström
*a
*a,
K.
Abel
a,
J.
Fize
b,
J.
Pécaut
c,
M.
Orio
*d and
L.
Hammarström
*a
aDepartment of Chemistry, Ångström Laboratories, Uppsala University, Box 523, 751 20 Uppsala, Sweden. E-mail: nicolas.queyriaux@lcc-toulouse.fr; leif.hammarstrom@kemi.uu.se
bUniv. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, 17 rue des Martyrs, F-38054, Grenoble Cedex, France
cUniv. Grenoble Alpes, CEA, CNRS, IRIG, SYMMES, 38000 Grenoble, France
dAix Marseille Univ., CNRS, Centrale Marseille, iSm2, 13397 Marseille, France. E-mail: maylis.orio@univ-amu.fr
First published on 13th May 2020
Abstract
The electrochemical behavior of [Co(bapbpy)Cl]+ [1-Cl]+, a pentacoordinated polypyridyl cobalt(II) complex containing a redox-active tetradentate ligand (bapbpy: 6,6′-bis-(2-aminopyridyl)-2,2′-bipyridine) has been investigated in DMF. Cyclic voltammograms (CV), recorded in the presence of increasing amounts of chloride anions, highlighted the existence of an equilibrium with the neutral hexacoordinated complex. Under a CO2 atmosphere, CVs of [Co(bapbpy)Cl]+ exhibit significant current enhancement assigned to CO2 catalytic reduction. Controlled-potential electrolysis experiments confirmed formation of CO and HCOOH as the only identifiable products. The addition of water or chloride ions was shown to affect the distribution of the products obtained, as well as the faradaic efficiency associated with their electrocatalytic generation. A combination of electrochemical techniques, chemical reductions, spectroscopic measurements (UV-vis and IR) and quantum chemical calculations suggests that the ability of the bapbpy ligand to be reduced at moderately negative potentials drastically limits the catalytic performances of [1-Cl]+, by stabilizing the formation of a catalytically-competent CO2-adduct that only slowly reacts with oxide acceptors to evolve towards the desired reduction products.
Introduction
Carbon dioxide (CO2) is a major side-product of most human activities, from the transport sector to home heating and industrial processes.1 The increasing concentration in the atmosphere of this thermodynamically stable greenhouse gas significantly and durably contributes to global warming.2 Molecular systems capable of photochemical and/or electrochemical reductive conversion of CO2 into carbon-based fuels, such as CO, HCOOH, CH3OH or CH4, have thus attracted much attention as they may offer an elegant way to finally close the anthropogenic carbon cycle.3–5 However, direct one-electron reduction of CO2 to its radical anion CO2˙− requires high overpotential (eqn (1)).6 Such uphill process can be circumvented by multielectron reduction processes generating a range of C1-products with lower energy requirements, at 25 °C in pH = 7 aqueous solution (eqn (2)–(6)).7 Due to the various chemicals that can be produced from CO2 under closely related experimental conditions and the competitive reduction of protons to H2 (eqn (7)), product selectivity remains a challenge:8 as far as molecular approaches are considered, mixtures of two-electron reduction products are generally obtained.| CO2 + e− → CO2˙− (Eo′ = −1.90 V vs. NHE) | (1) |
| CO2 + 2H+ + 2e− → CO + H2O (Eo′ = −0.53 V vs. NHE) | (2) |
| CO2 + 2H+ + 2e− → HCO2H (Eo′ = −0.61 V vs. NHE) | (3) |
| CO2 + 4H+ + 4e− → HCHO + H2O (Eo′ = −0.48 V vs. NHE) | (4) |
| CO2 + 6H+ + 6e− → CH3OH + H2O (Eo′ = −0.38 V vs. NHE) | (5) |
| CO2 + 8H+ + 8e− → CH4 + H2O (Eo′ = −0.24 V vs. NHE) | (6) |
| 2H+ + 2e− → H2 (Eo′ = −0.42 V vs. NHE) | (7) |
Transition metal catalysts usually favor the desired multielectron processes, owing to their ability to accommodate multiple reduction steps within the range of considered potentials. Their relatively versatile and tunable properties have allowed rapid development of a wide diversity of electro- and photocatalytic systems, some of the most efficient and selective catalysts being based on rhenium9–11 and ruthenium12–14 complexes. Although studies on these two families of compounds have clearly demonstrated the essential role played by the metal center during the catalytic cycle,15,16 it is interesting to note that the first reduction steps initiating the catalytic process are centered on the ligand. This mechanistic specificity, associated with the cost-prohibitive nature of the rare metals being used, has certainly contributed to the great attention and efforts directed towards the development of first-row transition metal complexes capable to efficiently reduce CO2.17 While several complexes based on Fe,18,19 Co,20–22 Ni23,24 or Mn25,26 have been reported, improvements in terms of both selectivity and stability are still demanded.
Cobalt polypyridyl complexes have recently emerged as interesting candidates for both CO2 reduction and hydrogen evolution.21,27–29 This type of coordination sphere has been shown to offer a wide range of advantages, from their ability to stabilize low-valent cobalt centers involved over the course of the catalytic process to their synthetic modularity and robustness. Generally based on polydentate ligands, these ligand frameworks are also proposed to contribute to the high stability of the resulting cobalt complexes by virtue of the chelate effect. The relevance of cobalt polypyridyl complexes for the electrocatalytic reduction of CO2 was first evidenced with the use of [Co(phen)3]2+ by Durand and co-worker in 1988.30 Further investigations have led to the development of a range of bis-triimine complexes capable to electro-assist the reduction of CO2 to generate mixtures of C1-products whose selectivity can be preferentially oriented towards HCOOH or CO.31–33 More recently, tetradentate platforms have been developed and allowed good selectivity towards formation of carbon monoxide to be reached.21,34–37 These encouraging results have stimulated the development of heterogenization strategies, either by their attachment onto transparent conducting oxide and carbonaceous materials (graphene and carbon nanotubes, typically) or by integrating them into polymer chains.38–42
The role played by the ligand set in catalytic mechanisms has recently attracted much attention,43–45 different functions susceptible to increase catalytic performance being envisaged: proton relays (amine, thiol or phenol groups),18,21,46–48 electron reservoirs (pyrazine, dithiolene or quinone moieties),49–51 effective catalytic sites52 or a combination of those functions.53,54 In the case of the electro-assisted H2 evolution, proton relay sites and redox-active units have clearly demonstrated their ability to improve catalytic kinetic features or to significantly reduce the overpotential required to drive the reaction.55 Examples are scarcer when it comes to CO2 reduction and mechanistic dead-ends have been established in several situations. While Savéant, Costentin and co-workers found that introduction of multiple proton relays dramatically enhanced the CO2-to-CO electrocatalytic properties of iron porphyrin,18 similar modifications performed on a ruthenium(II) complex by the group of Fujita resulted in a catalytically-inactive complex under CO2 atmosphere.56 Such unexpected behavior has been assigned to new deactivation pathways in which a carbonate species has been formed by the reaction between the reduced ligand and CO2. Similarly, Elgrishi et al. investigated the electro-assisted CO2-to-CO conversion mediated by [M(tpy)2]2+ complexes (M = Mn, Fe, Co, Ni, Cu, Zn) and suggested carboxylation of the reduced tpy ligand as a potential deactivation pathway (tpy = 2,2′:6′,2′′-terpyridine).32
Herein, we describe the electrocatalytic behavior of a cobalt(II) polypyridyl complex displaying an unusual combination of features, containing both a redox-active moiety and protic sites,55,57 potentially capable to set up a stabilizing hydrogen bond network around the bound CO2 molecule. Electrochemical measurements provide insights on the nature of the doubly reduced cobalt catalyst, the first reduction having a predominantly metal-based character and the second electron transfer being mostly localized on the ligand scaffold. According to theoretical calculations, the interaction of CO2 with this two-electron reduced complex results in a poorly reactive adduct. Nevertheless, this catalytically-competent intermediate is capable to slowly release CO and HCOOH, with a product distribution depending on the reaction conditions.
Results and discussions
Synthesis and structural characterizations
The bapbpy ligand (bapbpy: 6,6′-bis-(2-aminopyridyl)-2,2′-bipyridine, Fig. 1A) and its cobalt complex were synthesized according to published procedures.55,58 Interestingly, two sets of single crystals – varying slightly in their colors – suitable for X-ray analysis were obtained by layering of diisopropyl ether over a methanolic solution of the complex. The structure determined for the dark golden crystals of [CoII(bapbpy)Cl]+ [1-Cl]+ (Fig. 1B) has already been described elsewhere55 and differs from the one established with lighter yellow crystals of [CoII(bapbpy)Cl2] [2-Cl2] (Fig. 1C) in the number of chloride ions that are coordinated to the CoII centre. While [1-Cl]+ adopts a pentacoordinated structure displaying a highly distorted square pyramidal geometry (τ = 0.52),59 [2-Cl2] rather coordinates two chloride anions in a trans configuration featuring a distorted octahedral structure. Selected bond lengths and angles are provided in Table S1.† A significant Co–Cl bond elongation is observed in [2-Cl2] (2.5701 Å and 2.5499 Å) compared to [1-Cl]+ (2.3085 Å), reflecting a weakening of the metal-chloride bonds in the hexacoordinated complex. In contrast, Co–N bond lengths remain essentially unaffected in both complexes. In the pentacoordinated structure, the chloride ion is sitting close to an ancillary amine group, allowing the establishment of a hydrogen bond (N–H⋯Cl being described by a length of 2.331 Å and an angle of 157.57°).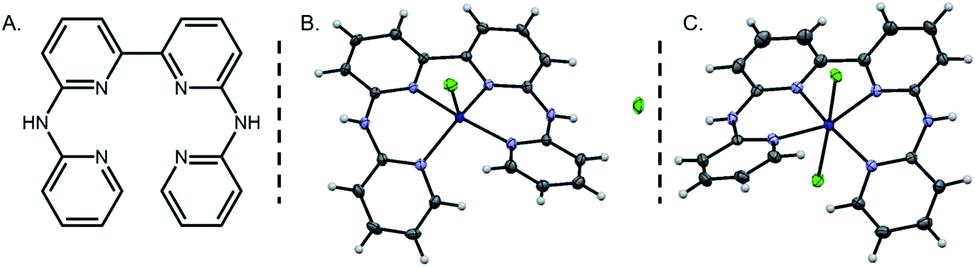 | ||
| Fig. 1 Structure of the bapbpy ligand (A) and crystal structures of pentacoordinated complex [1-Cl]+ (B) and hexacoordinated complex [2-Cl2] (C). Thermal ellipsoids are drawn at the 50% probability level. | ||
Due to the steric constraints originating from the two hydrogen atoms in ortho positions on the pyridine units, the bapbpy ligand adopts a saddle-shaped structure. To gain some insight on this type of twisted configuration, Bonnet and co-workers introduced the use of specific dihedral angles (Table S2†).60 Interestingly, the angles measuring terminal pyridines (β) and coordination plane torsions (γ and γ′) remain mostly unaffected by the change in the coordination number. On the contrary, the one describing the degree of rotation between the two rings forming the bipyridine motif (α) is dramatically modified, rising up from 6.53° in the pentacoordinated structure [1-Cl]+ to 16.66° in the hexacoordinated configuration [2-Cl2]. A combination of DFT calculations and electrochemical measurements (vide infra) have been undertaken in order to investigate the respective weight of each of these two structures in solution, highlighting the prominence of the pentacoordinated structure [1-Cl]+.
Electrochemical properties
The electrochemical behavior of [1-Cl]+ was assessed in anhydrous dimethylformamide (DMF) in the presence of 0.1 M of nBu4NPF6 as the supporting electrolyte. Fig. 2 shows the cyclic voltammogram (CV) obtained using a glassy carbon working electrode under inert atmosphere of argon. Complex [1-Cl]+ displays two quasi-reversible redox processes at −0.11 V and −1.44 V vs. Fc+/Fc, respectively assigned to the CoIII/CoII (Fig. 2, process I) and CoII/CoI (Fig. 2, process II) couples.49,61 At a more negative potential of −1.99 V vs. Fc+/Fc, a last irreversible process, attributed to ligand-based reduction centered on the bipyridine moiety, is observed (Fig. 2, process III).55 Plotting peak currents as a function of the square root of the scan rates (from 50 mV s−1 to 500 mV s−1) provides linear trends that confirm the diffusional nature of the electrochemical processes (Fig. S1 and S2†).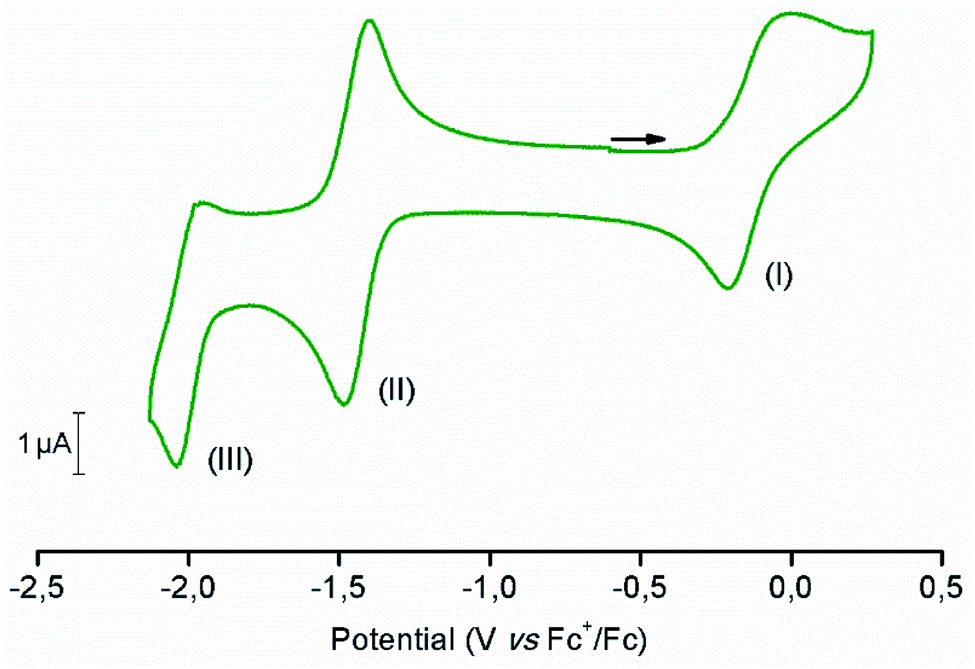 | ||
| Fig. 2 Cyclic voltammogram of [1-Cl]+ recorded in anhydrous DMF (containing 0.1 M nBu4NPF6 as supporting electrolyte), under argon atmosphere. Scan rate: 100 mV s−1. Working electrode: glassy carbon. Counter-electrode: Pt wire. Reference electrode: Ag/AgNO3. | ||
In an effort to further support the assignments of the different redox processes observed for complex [1-Cl]+, DFT calculations were conducted. The geometries of [1-Cl]+ and these one- and two-electron reduced species were thus optimized (Fig. S3†) and their electronic structures investigated in detail. When compared with the experimentally-obtained X-ray structure, the DFT-optimized structure of [1-Cl]+ appears in good agreement confirming that the pentacoordinated complex can be best described as a high-spin CoII complex with a quartet ground spin state (S = 3/2, Table S3†). One-electron reduction is metal-based, forming a species with a triplet ground spin state (S = 1, Tables S3 and S4†): the singly occupied molecular orbitals (SOMO) of [1-Cl]0 indeed have a dominant metal-based character (Fig. 3). The lability of the chloride ligand following mono-reduction has been evaluated, suggesting that chloride loss provides a slightly more stable adduct (Table S5†).
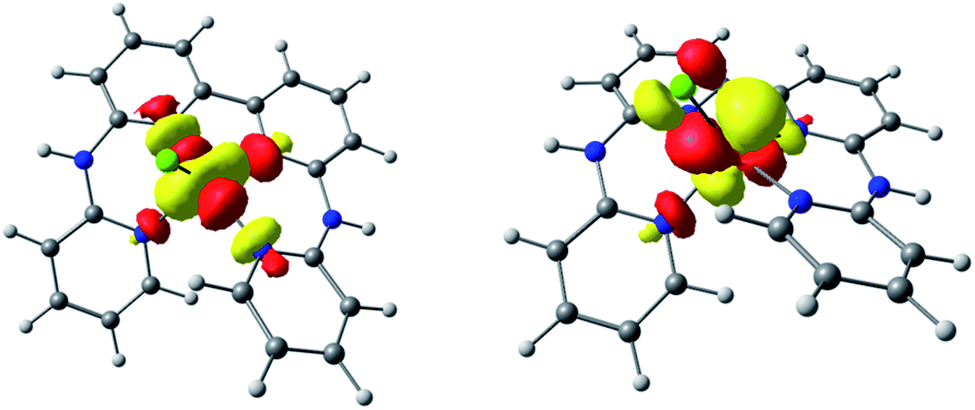 | ||
| Fig. 3 Localized SOMOs of the one-electron reduced complex [1-Cl]0. | ||
However, should the chloride ligand be released upon reduction to the CoI oxidation state, the reversibility of the CoII/CoI couple should be significantly affected. Indeed, [CoII(bapbpy)Cl]+ would be first reduced to [CoI(bapbpy)Cl]0, but only [CoI(bapbpy)]+ would be present for re-oxidation in the anodic sweep. As a consequence, the remarkable reversibility of the CoII/CoI electrochemical system is a strong evidence in favor of chloride retention on the timescale of the cyclic voltammetry measurements. This suggests slow kinetics associated with the chloride loss from the one-electron reduced species, [1-Cl]0. The second reduction process occurs on the bipyridine moiety of the bapbpy ligand and generates a delocalized ligand-based radical species having a quartet ground spin state (S = 3/2, Tables S3 and S6†), so that two-electron reduced species can be best described as a [CoI(bapbpy˙−)] complex, [1]0 (Fig. 4). Indeed, calculations strongly support chloride ligand expulsion upon reduction, providing a reasonable explanation to the poor reversibility of the more cathodic redox system (Table S7†).
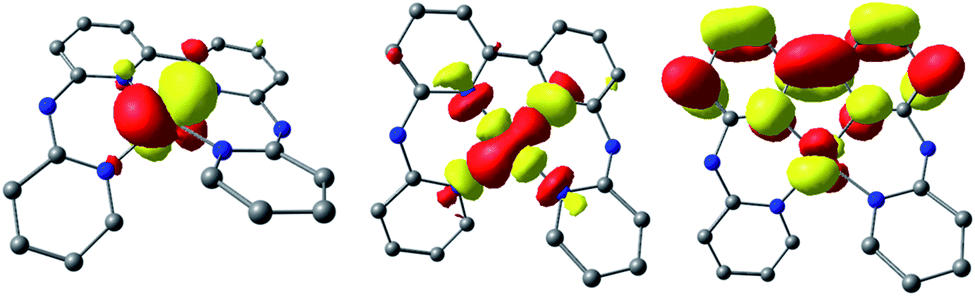 | ||
| Fig. 4 Localized SOMOs of the two-electron reduced complex [1]0. | ||
As previously described, two different forms of the CoII complex have been identified in the solid state: the positively charged five-coordinate complex [1-Cl]+ and the neutral six-coordinate complex [2-Cl2]. If these two species coexist in solution, some measurable metrics describing their electrochemical behavior are likely to be affected by the equilibrium described in eqn (8).62,63 From that perspective, it is thus interesting to note that: (i) increasing scan rate leads to an apparent decrease of the current function (Fig. S4†) and (ii) the ratio ip,a/ip,c describing the reversibility of the CoII/CoI couple is maintained over the unity and further increases with scan rate (Fig. S5†). Taken together, these results provide evidences in favor of the existence of an equilibrium preceding the reduction process, according to a CE-type mechanism (where E corresponds to an electron transfer step and C to a chemical reaction).
| [CoII(bapbpy)Cl]+ [1-Cl]+ + Cl− ↔ [CoII(bapbpy)Cl2] [2-Cl2], Ka | (8) |
To further support this assignment, we sought to change the balance between the pentacoordinated [1-Cl]+ and hexacoordinated [2-Cl2] complexes by modification of the free chloride anion concentration (Fig. S6†). Plotting the half-wave potential of the CoII/CoI couple as a function of the logarithm of the chloride concentration provides meaningful information related to the chloride coordination to the metal center (Fig. 5). From this data, two main regimes can be defined. At low concentration (≤1 mM), the half-wave potential of the CoII/CoI couple is only slightly modified suggesting the establishment of a plateau. At concentrations above 3 mM; a linear decrease of potential with log([Cl−]) is observed with a slope of −52 mV dec−1, the CoII/CoI potential shifting towards more negative potentials as the chloride concentration increases.
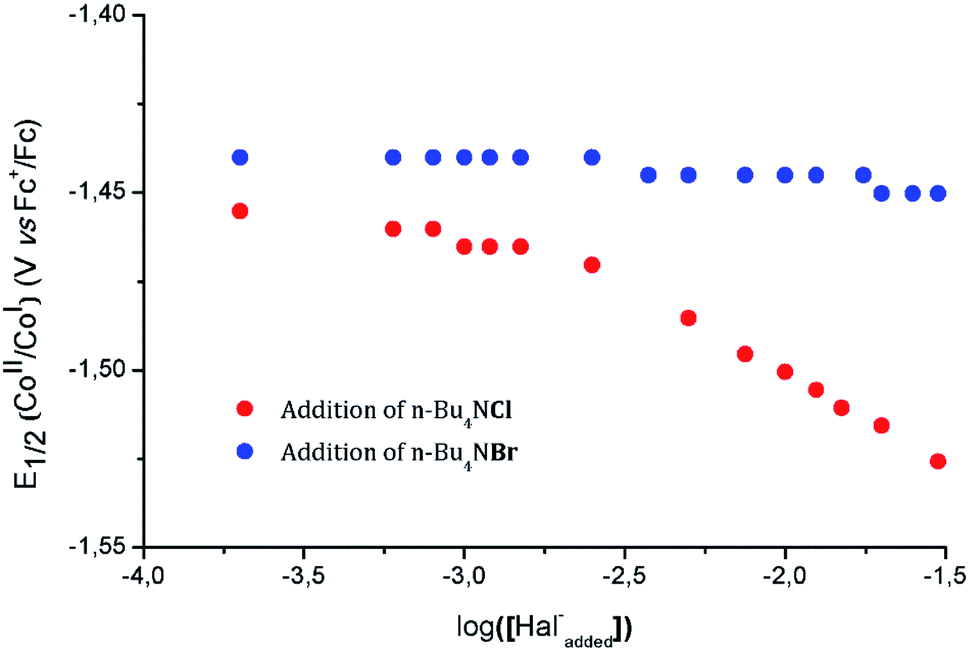 | ||
| Fig. 5 Plots of the variation of the CoII/CoI half-wave potential upon addition of an increasing concentration of tetrabutylammonium chloride (nBu4NCl, red dots) or tetrabutylammonium bromide (nBu4NBr, blue dots). | ||
Theoretical description of similar system has previously been proposed by Savéant and Xu.64 However, the exact concentration of free chloride ions in solution prior to addition being unknown, only a rough estimate of the equilibrium constant −3.5 < log(Ka) < −2.5 can be determined from the intersection of the plateau and the linearly decaying trace. This set of data is in complete agreement with the existence of an equilibrium preceding the redox events, [1-Cl]+ being the dominant species in solution in the absence of any added chloride. Using tetrabutylammonium bromide in the same concentration range did not significantly affect the potential of the CoII/CoI couple, indicating that hard ligands – such as chloride anions – are required to trigger the formation of hexacoordinated species. DFT calculations further support the slightly higher stability of the pentacoordinated complex [1-Cl]+, when compared to [2-Cl2] (Table S8†).
Electrocatalytic behavior
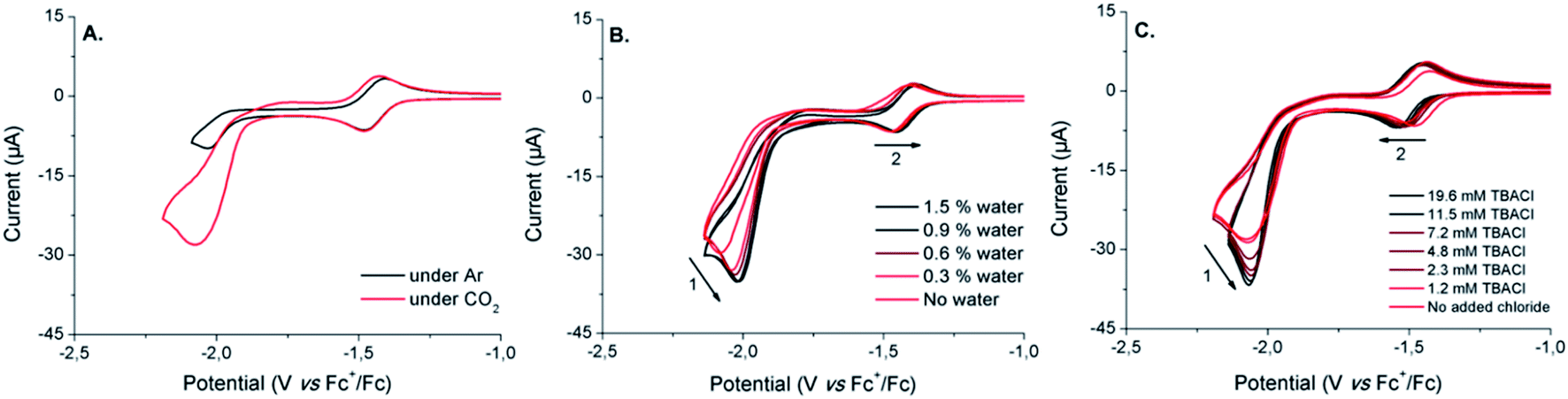 | ||
| Fig. 6 Cyclic voltammograms of DMF solutions (containing 0.1 M n-Bu4NPF6 as supporting electrolyte) of [1-Cl]+ (1 mM): (A) under Ar (black trace) or CO2 (red trace), (B) with increasing amounts of water under CO2 (from red to black traces) and (C) with increasing amounts of nBu4NCl under CO2 (from red to black traces). Scan rate: 25 mV s−1. Working electrode: glassy carbon. Counter-electrode: Pt wire. Reference electrode: Ag/AgNO3. | ||
Due to its ability to serve as an oxide acceptor and to trigger proton-coupled electron transfers, addition of water has been shown to usually dramatically facilitate electro-assisted CO2 reduction.20,65–67 Increasing the water content of the electrolyte up to 3.0 vol% induces some noticeable changes on the CVs shape (Fig. 6B). The electrocatalytic wave was indeed anodically shifted together with a significant increase of the catalytic current, reaching a plateau above about 1% of water in the medium (Fig. S8†). This apparent enhancement of the catalytic ability of [1-Cl]+ in the presence of water also appears to affect the CoII/CoI couple. While maintaining its remarkable reversibility, the CoII/CoI couple is indeed shifted towards anodic potentials when increasing amounts of water are added. This observation strongly suggests the existence of protonation steps that are coupled to the first reduction of [1-Cl]+, as previously demonstrated in the presence of weak acids.55 Plotting the evolution of the CoII/CoI process as a function of the concentration of water in the electrolyte, resulted in a saturation curve (Fig. S9†): the CoII/CoI reduction is facilitated in the presence of low contents of water (<1.5%), any further addition having no observable effect.
As previously shown, the speciation of the cobalt complex [1-Cl]+ can be manipulated by adding excess chloride anions. In order to determine if the number of chloride ligands initially coordinated to the Co(II) center is likely to affect the electrocatalytic properties of the system, we investigated the electrochemical behavior of [1-Cl]+ under CO2 and in the presence of increasing amounts of tetrabutylammonium chloride (TBACl). Under CO2, the progressive addition of TBACl to the electrolyte solution resulted in some modifications of the electrochemical response of [1-Cl]+ (Fig. 6C). Beyond the changes observed on the CoII/CoI couple and previously investigated (vide supra), it is interesting to note that the electrocatalytic wave is doubly affected by: (i) an increased magnitude of the catalytic peak current (Fig. S10†) and (ii) a slight shift of the catalytic wave onset towards the more negative potentials. Such dual consequences probably emerge from conflicting effects of chloride addition to the medium. The increased catalytic current may arise from chloride assistance to the catalytic process as a kinetically-efficient carrier of proton traces, as previously demonstrated by Darensbourg and co-workers68,69 In contrast, the negative shift of the onset potential could be attributed either to (i) chloride coordination to the metal centre disfavoring CO2 binding or (ii) bapbpy ligand flexibility loss due to effective hydrogen bonds. Alternatively, chloride binding to a catalytic intermediate may contribute to both shift of the potential at more negative values and increased catalytic rate.
| Entry | [1-Cl]+ (M) | Additives | Charge (C) | FECO (%) | FEHCOOH (%) | FEH2 (%) | FEtotal (%) |
|---|---|---|---|---|---|---|---|
| A | 1 × 10−3 | — | 5.93 | 7.4 | 7.9 | 0 | 15.3 |
| B | 1 × 10−3 | H2O 2% | 7.85 | 15.7 | 5.4 | 2.9 | 24.0 |
| C | 1 × 10−3 | H2O 10% | 11.59 | 11.3 | 7.7 | 7.9 | 26.9 |
| D | 1 × 10−3 | TBACl 50 mM | 5.18 | 8.9 | 7.4 | 1.3 | 17.6 |
| E | 0 | — | 0.503 | 0 | 0 | 0 | 0 |
When performed in DMF containing 2% of water (Table 1, entry B), CPE allows clear improvement of the product selectivity towards the generation of CO (FECO = 15.7% and FEHCOOH = 5.4%, at −1.95 V vs. Fc+/Fc). Increasing further the water content, however, has a negative impact on product distribution and faradaic efficiencies (Table 1, entry C). This behavior strongly contrasts with the situation found in the recent studies of McCrory65 and Fujita,28 where high faradaic efficiency towards CO or HCOOH formation was reached at high water contents only. It is interesting to note that, although still quite limited, hydrogen evolution starts to occur under such conditions. In the presence of a large excess of chloride anions (Table 1, entry D), only a slight increase of the faradaic efficiency associated with the formation of CO was observed.
Regardless the nature of the electrolyzed mixture, an initial current build-up was observed together with darkening of the initial yellow solution. Interestingly, this color change persists – even after the end of the CPE – as long as the solution is maintained under CO2 and kept free of oxygen. When opening the electrochemical cell to the air, the solution color turns back to the initial yellow color. This observation, associated with the modest faradaic efficiencies and UV-vis characterization of the chemically reduced catalyst (vide infra), suggest the existence of a reduced species that is able to trap two electrons and thus to hamper further catalytic processes. Such electron sink is expected to actually accounts for about 15 to 35% of the overall electrons passed through the system during CPE. In that prospect, three situations can be considered: (i) a poor interaction between the two-electron reduced cobalt complex [1]0 and CO2, (ii) a low reactivity of a [CoI(bapbpy˙−)(COO)] adduct towards the formation of the reduction products or (iii) a carbonyl-trapped cobalt complex.
Mechanistic considerations
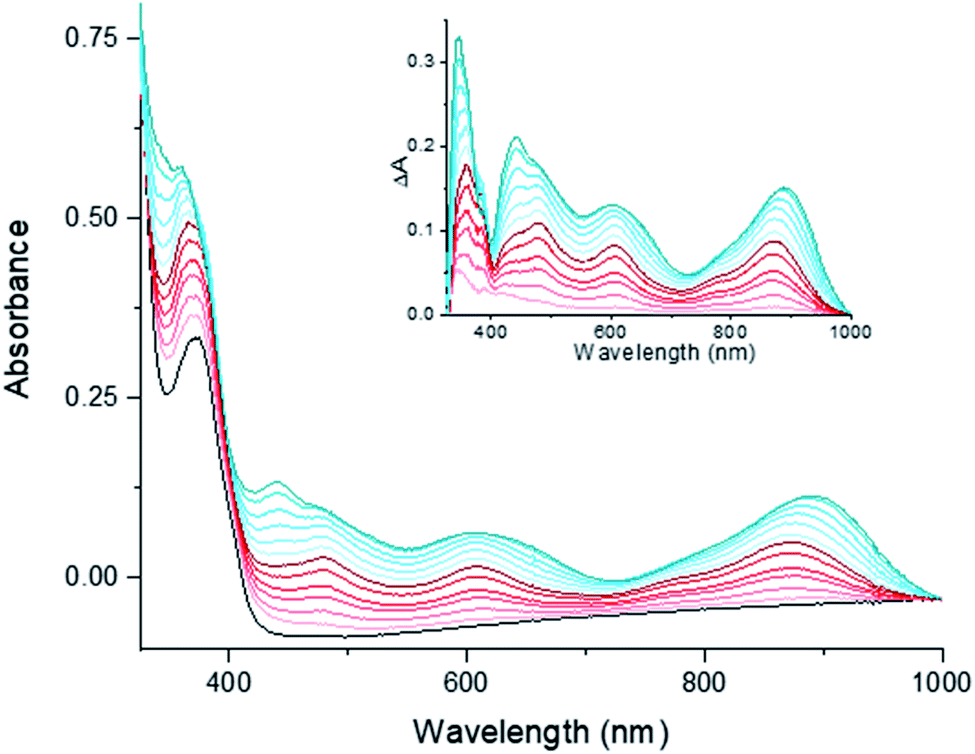 | ||
| Fig. 7 Evolution of the UV-vis spectrum of [1-Cl]+ in dry DMF upon titration by decamethylcobaltocene, Co(Cp*)2. From black to dark red trace, addition of the first equivalent. From dark red to dark blue trace, addition of a second equivalent of reductant. | ||
TD-DFT calculations were performed on the series of complexes and the predicted spectroscopic data provides calculated spectra that compare well with the experimental observations (Tables S9–S11 and Fig. S11†). Our computations support that the UV-vis spectrum of [1-Cl]+ is dominated by three main absorption bands of different intensity while progressive reduction of the complex with one and two electrons resulted in the appearance of new electronic transitions in the [400–900] nm range. [1-Cl]0 and [1]0 display similar UV-vis spectra but with an increased intensity for the low-energy bands of the latter. The spectral features characterizing [1-Cl]+ are all assigned to charge transfers from the ligand to metal (LMCT, Fig. S12†). For [1-Cl]0 and [1]0 the additional transitions observed in the visible region display mixed characters with contributions from the metal and the ligand in the donor states and only from the ligand in the acceptor states. The NIR absorptions for the reduced complexes correspond to charge transfers with similar contributions from the metal and the ligand in both donor and acceptor states, but where the donor state involves the reduced bpy fragment of the ligand while the acceptor state involves the pyridine fragments with less excess electron density due to reduction of the complex (Fig. S13 and S14†). Our TD-DFT calculations thus adequately reproduce the energy of the key features of the experimental spectra which further support the geometries and electronic properties of [1-Cl]+, its reduced one- and two-electron reduced states.
The reactivity of [1-Cl]0 and [1]0 towards carbon dioxide was investigated by subjecting DMF solutions of the reduced species to 1 bar of CO2 in Fisher–Porter tubes. To avoid potential side reactions related to the presence of different cobalt species in the mixture, KC8 was employed as the reductant for this series of measurements providing similar UV-vis spectra for the one- and two-electron reduced species (Fig. S15A†). Whereas a rapid color change from dark brown to dark yellow was observed in the case of [1]0, no significant modifications were observed for the singly-reduced compound [1-Cl]0 within the same timeframe, indicating that CO2 is quickly reacting with the doubly reduced species (Fig. S15B†). Two main situations are possible: (i) the formation of a low-reactive CO2-adduct or (ii) the generation of a carbonyl-bearing cobalt complex. To deeper investigate these possibilities, the IR spectrum of the reduced species under CO2 was recorded. Due to the DMF absorptions, the IR region under investigation was limited to 1800–2700 cm−1 where cobalt carbonyl complexes have previously been evidenced.28,70 In our case, no new CO stretching bands were observed supporting the CO2-adduct hypothesis.
Interestingly, the CO2-adduct is best described as a [CoI(bapbpy˙−)(COO)] complex having a doublet ground spin state due to an effective antiferromagnetic coupling between the metal center and the ligand-based radical (Fig. S20, S21 and Table S14†). In consequence, coordination of CO2 to the metal center does not result in significant charge redistribution within the newly-bound CO2 ligand-centered orbitals. The merely increased electron density in the bound CO2 molecule only results in a moderate bending of the O–C–O angle of 32°. Such electronic reorganization has been proposed as a crucial step of the catalytic process.14,15 As a result, the bound CO2 molecule is only weakly activated due to an effective electron trapping within the bapbpy ligand platform. Further reactivity with oxide acceptors, such as carbon dioxide or protons, is thus expected to be significantly altered limiting catalytic performances.
A plausible mechanism for the electro-assisted reduction of CO2 by [1-Cl]+ is displayed on Fig. 8. Complex [1-Cl]+ undergoes two one-electron transfers associated with two different loci: a first metal-based reduction that generates [CoI(bapbpy)Cl]0 and a second ligand-centered process yielding [CoI(bapbpy˙−)Cl]−. Chloride loss, followed by CO2 binding produced the two-electron reduced complex [CoI(bapbpy˙−)(CO2)]0. The limited electronic reorganization of the two-electron reduced species upon CO2 binding is characteristic of an electron trapping within the bapbpy scaffold. Such behavior impedes the efficient formation of a metallocarboxylate intermediates and thus hampers further reactivity towards the formation of the two-electron reduced products (CO and HCOOH).
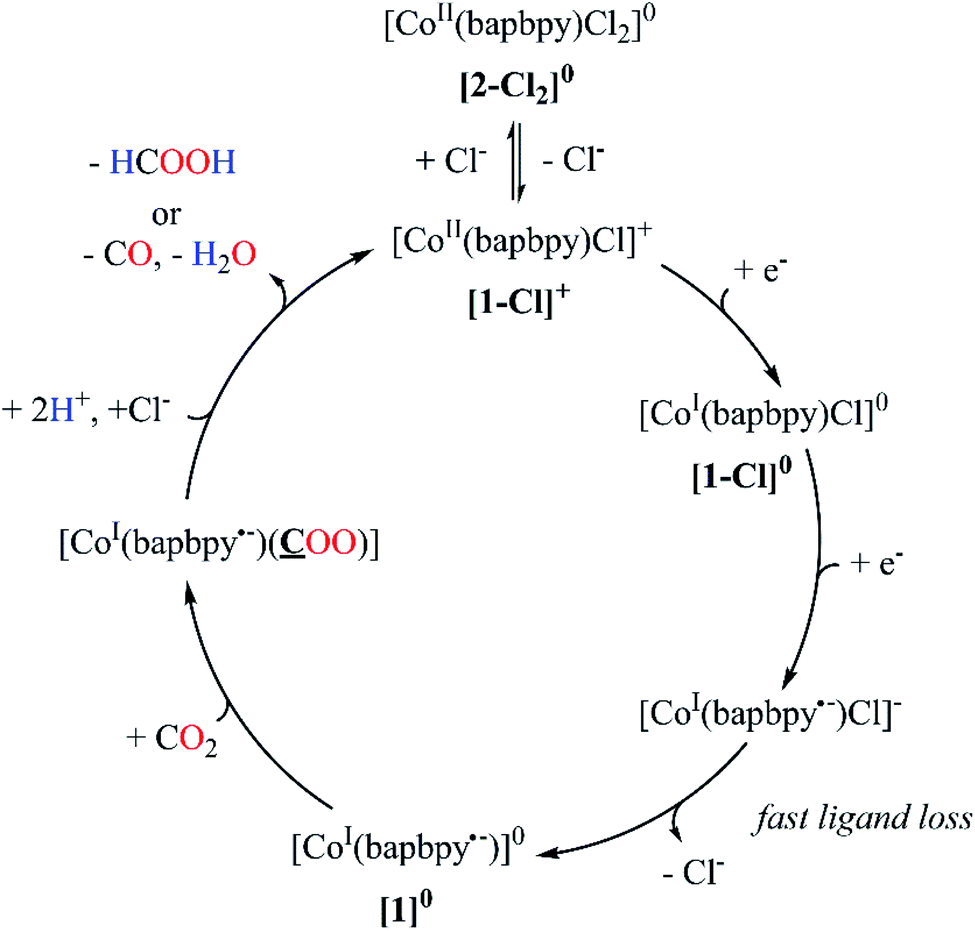 | ||
| Fig. 8 Proposed mechanism for the reduction of CO2 using [1-Cl]+ as an electrocatalyst. | ||
Conclusion
In summary, the cobalt(II) polypyridyl complex [Co(bapbpy)Cl]+, previously reported as an active catalyst for electro-assisted H2 evolution, also proved reactive for CO2 electroreduction. Using a combination of electrochemical tools, UV-vis spectroscopy and DFT calculations, we evidenced that the second electron required over the catalytic cycle is unambiguously located within the bapbpy ligand platform. This propensity to store electrons, however, appears detrimental to the catalytic activity by preventing effective charge delocalization into the bound CO2 substrate. Although activated, the CO2 molecule remains poorly reactive towards oxide acceptors, therefore limiting turnover frequency. While the potential of redox-active ligand in multi-electrons/multi-protons catalysis has often been highlighted, this study shows that the use of such ligand can be a double-edge sword as it can alter the metal-based reactivity.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Foundation Olle Engkvist Byggmästare (grant 2016/3), the Swedish Energy Agency (Grant no. 11674-8) and the NordForsk network NordCO2. MO gratefully acknowledges research support of this work by the ANR (ANR-19-CE05_0030_01). N. Q. also thanks Dr Vincent Artero & Dr Charlène Esmieu for fruitful discussions regarding this study. JF gratefully acknowledges support of the Agence Nationale de la Recherche (Labex ARCANE, CBH-EUR-GS, ANR-17-EURE-0003).References
- R. Quadrelli and S. Peterson, Energy Policy, 2007, 35, 5938–5952 CrossRef.
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- H. A. Schwarz and R. W. Dodson, J. Phys. Chem., 1989, 93, 409–414 CrossRef CAS.
- E. Fujita, Coord. Chem. Rev., 1999, 185–186, 373–384 CrossRef CAS.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- H. Hori, F. P. A. Johnson, K. Koike, K. Takeuchi, T. Ibusuki and O. Ishitani, J. Chem. Soc., Dalton Trans., 1997, 1019–1024, 10.1039/a607058b.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef PubMed.
- Y. Tamaki, K. Koike and O. Ishitani, Chem. Sci., 2015, 6, 7213–7221 RSC.
- K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211–218 CrossRef CAS.
- B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed., 2016, 55, 1825–1829 CrossRef CAS PubMed.
- C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2014, 136, 16285–16298 CrossRef CAS PubMed.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- L. Chen, Z. Guo, X. G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabehere-Mallart, K. C. Lau, T. C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918–10921 CrossRef CAS PubMed.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef CAS PubMed.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulie, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Isci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS PubMed.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS PubMed.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304–305, 3–19 CrossRef CAS PubMed.
- T. Shimoda, T. Morishima, K. Kodama, T. Hirose, D. E. Polyansky, G. F. Manbeck, J. T. Muckerman and E. Fujita, Inorg. Chem., 2018, 57, 5486–5498 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- T. C. Simpson and R. R. Durand, Electrochim. Acta, 1988, 33, 581–583 CrossRef CAS.
- C. Arana, S. Yan, M. Keshavarz-K, K. T. Potts and H. D. Abruna, Inorg. Chem., 1992, 31, 3680–3682 CrossRef CAS.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC.
- K.-M. Lam, K.-Y. Wong, S.-M. Yang and C.-M. Che, J. Chem. Soc., Dalton Trans., 1995, 1103–1107, 10.1039/dt9950001103.
- M. Isaacs, J. C. Canales, M. J. Aguirre, G. Estiú, F. Caruso, G. Ferraudi and J. Costamagna, Inorg. Chim. Acta, 2002, 339, 224–232 CrossRef CAS.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, ACS Catal., 2018, 8, 3411–3417 CrossRef CAS.
- C. Cometto, L. Chen, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, Organometallics, 2018, 38, 1280–1285 CrossRef.
- M. Wang, L. Chen, T.-C. Lau and M. Robert, Angew. Chem., Int. Ed., 2018, 57, 7769–7773 CrossRef CAS PubMed.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Nat. Commun., 2019, 4, 666–672 CAS.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y.-F. Han, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- J. J. Leung, J. A. Vigil, J. Warnan, E. Edwardes Moore and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 7697–7701 CrossRef CAS PubMed.
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354–365 CrossRef CAS.
- G.-G. Luo, H.-L. Zhang, Y.-W. Tao, Q.-Y. Wu, D. Tian and Q. Zhang, Inorg. Chem. Front., 2019, 6, 343–354 RSC.
- R. H. Crabtree, New J. Chem., 2011, 35, 18–23 RSC.
- C. Costentin, J.-M. Savéant and C. Tard, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9104–9109 CrossRef CAS PubMed.
- M. Rakowski Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef CAS PubMed.
- M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller and S. C. Marinescu, ACS Cent. Sci., 2018, 4, 397–404 CrossRef CAS PubMed.
- J. W. Jurss, R. S. Khnayzer, J. A. Panetier, K. A. El Roz, E. M. Nichols, M. Head-Gordon, J. R. Long, F. N. Castellano and C. J. Chang, Chem. Sci., 2015, 6, 4954–4972 RSC.
- A. Z. Haddad, D. Kumar, K. Ouch Sampson, A. M. Matzner, M. S. Mashuta and C. A. Grapperhaus, J. Am. Chem. Soc., 2015, 137, 9238–9241 CrossRef CAS PubMed.
- T. Straistari, J. Fize, S. Shova, M. Réglier, V. Artero and M. Orio, ChemCatChem, 2017, 9, 2262–2268 CrossRef CAS.
- B. H. Solis, A. G. Maher, D. K. Dogutan, D. G. Nocera and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 485–492 CrossRef CAS PubMed.
- A. Z. Haddad, B. D. Garabato, P. M. Kozlowski, R. M. Buchanan and C. A. Grapperhaus, J. Am. Chem. Soc., 2016, 138, 7844–7847 CrossRef CAS PubMed.
- A. Z. Haddad, S. P. Cronin, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2017, 56, 11254–11265 CrossRef CAS PubMed.
- N. Queyriaux, D. Sun, J. Fize, J. Pécaut, M. J. Field, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2020, 142, 274–282 CrossRef CAS PubMed.
- L. Duan, G. F. Manbeck, M. Kowalczyk, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2016, 55, 4582–4594 CrossRef CAS PubMed.
- N. Queyriaux, E. Giannoudis, C. D. Windle, S. Roy, J. Pécaut, A. G. Coutsolelos, V. Artero and M. Chavarot-Kerlidou, Sustainable Energy Fuels, 2018, 2, 553–557 RSC.
- E. Molenbroek, N. Straathof, S. Dück, Z. Rashid, J. H. van Lenthe, M. Lutz, A. Gandubert, R. J. M. Klein Gebbink, L. De Cola and S. Bonnet, Dalton Trans., 2013, 42, 2973–2984 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356, 10.1039/DT9840001349.
- I. Gamba, I. Mutikainen, E. Bouwman, J. Reedijk and S. Bonnet, Eur. J. Inorg. Chem., 2013, 2013, 115–123 CrossRef CAS.
- W. K. C. Lo, C. E. Castillo, R. Gueret, J. Fortage, M. Rebarz, M. Sliwa, F. Thomas, C. J. McAdam, G. B. Jameson, D. A. McMorran, J. D. Crowley, M.-N. Collomb and A. G. Blackman, Inorg. Chem., 2016, 55, 4564–4581 CrossRef CAS PubMed.
- J. Orsini and W. E. Geiger, Organometallics, 1999, 18, 1854–1861 CrossRef CAS.
- P. Zanello, C. Nervi, F. Fabrizi de Biani, In Inorganic Electrochemistry: Theory, Practice and Application, The Royal Society of Chemistry, 2003, pp. 49–136, 10.1039/9781847551146-00049.
- J. M. Savéant and F. Xu, J. Electroanal. Chem. Interfacial Electrochem., 1986, 208, 197–217 CrossRef.
- W. Nie and C. C. L. McCrory, Chem. Commun., 2018, 54, 1579–1582 RSC.
- H. Y. Ching, X. Wang, M. He, N. Perujo Holland, R. Guillot, C. Slim, S. Griveau, H. C. Bertrand, C. Policar, F. Bedioui and M. Fontecave, Inorg. Chem., 2017, 56, 2966–2976 CrossRef CAS PubMed.
- E. Portenkirchner, E. Kianfar, N. S. Sariciftci and G. Knor, ChemSusChem, 2014, 7, 1347–1351 CrossRef CAS PubMed.
- M. Y. Darensbourg and M. M. Ludvig, Inorg. Chem., 1986, 25, 2894–2898 CrossRef CAS.
- X. Zhao, Y.-M. Hsiao, C.-H. Lai, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2002, 41, 699–708 CrossRef CAS PubMed.
- S. Fernández, F. Franco, C. Casadevall, V. Martin-Diaconescu, J. M. Luis and J. Lloret-Fillol, J. Am. Chem. Soc., 2020, 142, 120–133 CrossRef PubMed.
- E. Fujita, C. Creutz, N. Sutin and B. S. Brunschwig, Inorg. Chem., 1993, 32, 2657–2662 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1970564 and 1970565. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0se00570c |
| ‡ Current address: CNRS, LCC (Laboratoire de Chimie de Coordination), 31077 Toulouse, France. |
| This journal is © The Royal Society of Chemistry 2020 |