
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamics of carbon formation during the catalytic hydrodeoxygenation of raw bio-oil†
Idoia
Hita
 *ab,
Tomás
Cordero-Lanzac
b,
Giuseppe
Bonura
c,
Francesco
Frusteri
c,
Javier
Bilbao
b and
Pedro
Castaño
ab
*ab,
Tomás
Cordero-Lanzac
b,
Giuseppe
Bonura
c,
Francesco
Frusteri
c,
Javier
Bilbao
b and
Pedro
Castaño
ab
aKing Abdullah University of Science and Technology, KAUST Catalysis Center (KCC), Multiscale Reactor Engineering, Thuwal 23955-6900, Saudi Arabia. E-mail: idoia.hitadelolmo@kaust.edu.sa
bDepartment of Chemical Engineering, University of the Basque Country (UPV/EHU), PO Box 644-48080, Bilbao, Spain
cCNR-ITAE, Istituto di Tecnologie Avanzate per l'Energia “Nicola Giordano”, Via S. Lucia sopra Contesse, 5-98126 Messina, Italy
First published on 27th May 2020
Abstract
The formation, growth and transformation of the carbon residue (coke) deposited on the catalyst during the raw bio-oil hydrodeoxygenation have been studied. These deposits have a great impact on the overall process performance, and they have been formed in accelerated deactivation conditions (450 °C, 65 bar, space time of 0.09 gcat h gbio-oil−1) using a continuous fixed bed reactor and a FeMoP/HZSM-5 catalyst. Coke deposition causes partial deactivation of the catalyst, which reaches a pseudosteady state of constant activity and also contant yields of interesting chemicals. The evolution of the coke in the transient state has been studied through temperature-programmed oxidation, Raman spectroscopy and elemental analysis. We have identified three different types of coke, whose composition evolves with time on stream towards condensed and stable structures. The assessment of the evolution of the reaction medium composition and the application of the principal component analysis (PCA) methodology have evidenced that the dynamics of coke have three stages: (1) it is controlled by the thermally-induced deposition of thermal lignin; (2) followed by the interconversion into intermediate coke through aging reactions; and (3) it ends up in a pseudosteady state dominated by the formation of catalytic coke species originating from both deoxygenated and carbonized intermediate coke as well as the condensation of aromatics in the reaction medium.
Introduction
Biomass-derived pyrolysis oil, commonly referred to as bio-oil, is steadily becoming a valid alternative for the production of renewable fuels and platform chemicals, aiding in the transition from a fossil oil-based economy to a more sustainable and circular one.1,2 Bio-oil is composed of a significant amount of water (15–30 wt%) and a plethora of oxygenated organic components (50–65 wt%), among which one can find acids, aldehydes, ketones, furans, phenolics, guaiacols, syringols and sugars.3,4 The very reactive nature of some of these renders bio-oil unstable upon storage, highly viscous and corrosive, showcasing the necessity of bio-oil pre-treatment for adjusting its composition for any further downstream transformation.5,6Among the different alternatives for bio-oil stabilization and upgrading, hydroprocessing is conceived as the most efficient route, providing highly stable products with a high H/C ratio.7,8 To date, traditional transition metal-based (Ni, Co, Mo, W) hydrotreatment catalysts have proven their efficiency in hydrodesulfurization (HDS) and hydrodenitrogenation (HDN) reactions of complex and heavy feedstock.9–11 On the other hand, noble metal-based hydrocracking (HC) catalysts have also been addressed when enhanced catalytic and cracking activity is required.12–15 Nonetheless, hydrodeoxygenation (HDO) becomes the imperative hydroprocessing goal when dealing with heavily oxygenated feedstock like bio-oil and, in this context, metal phosphides present interesting possibilities.16,17 The higher HDO activity of phosphided catalysts is related to the presence of P–OH groups which act as Brønsted sites, providing active hydrogen.18 Unsupported FeMoP catalysts were reported by Rensel et al.19,20 as highly selective materials towards aromatic compounds, also with a high catalyst stability and recyclability. In a complementary study, they correlated the Lewis acidity of FexMo2−xP catalyst with the lower activation energy required for the C–O bond cleavage in phenol HDO.21 A high selectivity towards benzene (up to 90%) from the HDO of phenol was also showcased by Jain et al.22 with a FeMoP catalyst, in contrast to NiMoP and RuMoP catalysts at the same conditions.
Zeolites are particularly interesting as HDO catalyst supports, providing high hydrothermal stability in a reaction medium with a high water content and also tunable properties, which is of crucial relevance for directing catalyst selectivity towards preferred products and limiting the deactivation due to the formation of solid carbonaceous deposits.23,24 In a previous work dealing with raw bio-oil HDO, we demonstrated that a HZSM-5 zeolite support was preferred over HY and Hβ zeolites for FeMoP catalysts due to a balance between (i) a moderate catalyst acidity, (ii) a suitable proportion of weak and moderately acidic sites and (iii) a pore structure (MFI framework) that allows the sweeping of coke precursors towards the reaction medium, partially preventing catalyst deactivation.16
Any scalable approach in bio-oil HDO should consider the massive production of carbon residues (coke) and the resulting catalyst deactivation.25,26 This step is primarily a consequence of fast repolymerization of the unstable and highly reactive oxygenates in bio-oil,27,28 and can lead to reactor and pipeline clogging, directly affecting process stability and feasibility.29 Hence, understanding catalyst deactivation mechanisms, with a special focus on the initial hours of reaction, is of paramount importance regarding the viability of raw bio-oil HDO. However, this task remains highly challenging, considering the complexity of the reaction medium and the great amount of reaction synergies taking place.30 In previous works on the HDO of raw bio-oil,16,31 we determined that the process goes through an initial period of fast coke deposition at low times on stream (1–4 h), which causes a significant activity decay, and after which a subsequent pseudosteady catalytic activity state was attained.
To date, the most detailed deactivation mechanism for the HDO of raw bio-oil has been described by Cordero-Lanzac et al.,30 by establishing two parallel deactivation pathways dominated by bio-oil oxygenates and aromatic hydrocarbon precursors, respectively, at the pseudosteady state. They also showcased the crucial relevance of achieving a stable pseudosteady activity state in order to successfully implement a continuous raw bio-oil HDO process. On top of that, these preliminary results indicate that the dynamics of coke strongly influence the overall catalytic performance and reaction network. The coke formed on the catalyst is not an inert. Much on the contrary, it is constituted by a set of species with complex composition that dynamically evolve over time and affect the reaction pathways and the surface of the catalyst.
This manuscript targets to analyze the evolution with time on stream (TOS) of the nature of the coke deposited over a FeMoP/HZSM-5 catalyst during the HDO of a raw bio-oil. The HDO runs have been conducted in a continuous fixed bed reactor at 450 °C, 65 bar and a space time of 0.09 gcat h gbio-oil−1. Then, we have correlated the evolution of the reaction medium composition with that of coke, in order to find the link between the reaction and deactivation pathways. The properties of the formed coke have been analyzed by thermogravimetric temperature-programmed oxidation (TG-TPO), elemental analysis and Raman spectroscopy. The reaction medium has been analyzed by individual analysis of the liquid (aqueous and organic) phases. A statistical correlation between the compositions of all the phases has been performed trough multivariate data analysis using principal component analysis (PCA). Finally, we have proposed a simplified reaction mechanism which explains the dynamics of the reactions involved in the transformation of coke over TOS.
Experimental
Catalyst synthesis and characterization
For the preparation of the FeMoP/HZSM-5 catalyst, FeNO3·9H2O (Sigma Aldrich, 99%), (NH4)6Mo7O24·4H2O (Panreac, 99%) and (NH4)2HPO4 (Baker Chemicals B.V., 98%) were added sequentially in a stirred round bottom flask to an aqueous citric acid solution (0.4 M), in order to obtain a Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo:P molar ratio of 1:1:1. The citric acid was present in a concentration such as to ensure a molar ratio of 2:1 with respect to all metals. A commercial ZSM-5 zeolite (Alfa Aesar; SiO2:Al2O3, 30:1) was used in its ammonium form. After the metal precursors were dissolved in citric acid, the zeolite sample was impregnated by incipient wetness (20 wt%), then dried overnight in an oven at 200 °C and calcined in a muffle at 700 °C for 5 h. Complementary XRD measurements conducted in a Philips X-Pert diffractometer, operating at 40 kV, 30 mA and a scan step of 0.05 s−1, with a Ni β-filtered Cu Kα radiation (λ=1.5406 Å), showed that no significant modification/destruction of the HZSM-5 zeolite framework occurred upon calcination at this temperature (Fig. S1†). In addition, this high-temperature calcination also favors the formation of Lewis acidic sites, which prevent secondary undesired reactions, typically enhanced by stronger Brønsted acidic sites.32 The activation of the catalyst was carried out by programmed temperature treatment at the following conditions: heating at 100 °C for 1 h under Ar atmosphere (30 STP cm3 min−1), followed by reduction under a H2 atmosphere (100 STP cm3 min−1) up to 650 °C for 1 h (heating rate of 5 °C min−1). After reduction, the FeMoP/HZSM-5 catalyst was cooled to room temperature under a H2 atmosphere and then passivated using a 1% O2/He mixture (30 STP cm3 min−1) for 1 h.
:Mo:P molar ratio of 1:1:1. The citric acid was present in a concentration such as to ensure a molar ratio of 2:1 with respect to all metals. A commercial ZSM-5 zeolite (Alfa Aesar; SiO2:Al2O3, 30:1) was used in its ammonium form. After the metal precursors were dissolved in citric acid, the zeolite sample was impregnated by incipient wetness (20 wt%), then dried overnight in an oven at 200 °C and calcined in a muffle at 700 °C for 5 h. Complementary XRD measurements conducted in a Philips X-Pert diffractometer, operating at 40 kV, 30 mA and a scan step of 0.05 s−1, with a Ni β-filtered Cu Kα radiation (λ=1.5406 Å), showed that no significant modification/destruction of the HZSM-5 zeolite framework occurred upon calcination at this temperature (Fig. S1†). In addition, this high-temperature calcination also favors the formation of Lewis acidic sites, which prevent secondary undesired reactions, typically enhanced by stronger Brønsted acidic sites.32 The activation of the catalyst was carried out by programmed temperature treatment at the following conditions: heating at 100 °C for 1 h under Ar atmosphere (30 STP cm3 min−1), followed by reduction under a H2 atmosphere (100 STP cm3 min−1) up to 650 °C for 1 h (heating rate of 5 °C min−1). After reduction, the FeMoP/HZSM-5 catalyst was cooled to room temperature under a H2 atmosphere and then passivated using a 1% O2/He mixture (30 STP cm3 min−1) for 1 h.
Inductively coupled plasma mass spectrometry (ICP-MS) was used to analyze the chemical composition of the sample in a Perkin-Elmer DRC-e apparatus. The textural properties of the fresh and used catalysts were determined through N2 adsorption–desorption at −196 °C in a Micromeritics ASAP 2020 apparatus. Prior to analysis samples were degassed at 150 °C for 8 h. Specific surface area (SBET) was calculated using the Brunauer–Emmett–Teller equation while the micropore volume (Vmicropore) was calculated using the t-plot method. The mesopore volume (Vmesopore) was estimated by difference between the total pore volume (at relative pressure of 0.995, Vpore) and the Vmicropore. The total acidity of the fresh and used catalysts was measured through temperature programmed desorption (TPD) of tert-butylamine (tBA) at 100 °C in a Setaram DSC-111 calorimeter. The sample was firstly pretreated at 550 °C under a continuous flow of He in order to remove impurities. After saturation of the sample with tBA, a temperature programmed desorption (TPD) was launched at a 5 °C min−1 rate up to 500 °C, while simultaneously the tBA cracking products were registered in a Balzers Quadstar 422 mass spectrometer (MS), with butene being the main cracking product (m/z = 56). The main physico-chemical properties of the fresh FeMoP/HZSM-5 catalyst are summarized in Table 1.
| Property | Value |
|---|---|
| Fe (wt%) | 5.4 |
| Mo (wt%) | 7.7 |
| P (wt%) | 4.7 |
| S BET (m2 g−1) | 312 |
| V pore (cm3 g−1) | 0.218 |
| V micropore (cm3 g−1) | 0.113 |
| V mesopore (cm3 g−1) | 0.105 |
| Total acidity (mmoltBA g−1) | 0.47 |
Bio-oil feedstock, HDO runs and product analysis
The raw bio-oil was obtained in a pilot plant (25 kg h−1) from the fast pyrolysis of black poplar sawdust,33 and used as received with no further treatment. The raw bio-oil stock was stored in a refrigerated environment (6 °C). Its elemental and chemical composition (both on a dry bio-oil basis) are provided in Table 2. Besides a water content of 49.0 wt%, bio-oil presents a noteworthy concentration of levoglucosan (24.0 wt%) and acetic acid (23.5 wt%) in its composition, together with a significant amount of phenolic compounds (13.0 wt%). EA analysis revealed an oxygen content of 40.7 wt% in bio-oil.| Elemental composition (wt%) | |
|---|---|
| C | 52.2 |
| H | 7.1 |
| O | 40.7 |
| Water content (wt%) | 49.0 |
| Chemical composition (wt%) | |
|---|---|
| Acids and esters | 9.5 |
| Acetic acid | 23.5 |
| Aldehydes | 4.8 |
| Ketones | 8.7 |
| 1-Hydroxy-2-propanone | 7.0 |
| Phenols | 13.0 |
| Alcohols | 6.5 |
| Ethers | 2.0 |
| Levoglucosan | 24.0 |
| Non-identified | 1.0 |
The raw bio-oil HDO runs were carried out in a continuous down-flow stainless steel fixed bed reactor by PID Eng & Tech (catalyst bed height: 10 cm; i.d.: 1 cm), and schematized in detail elsewhere,9 at 450 °C, 65 bar, 0.09 gcat h gbio-oil−1; 0.05 cm3 min−1 bio-oil; 90 STP cm3 min−1 H2; and varying times on stream up to 8 h. The catalyst mixed with SiC was loaded in the reactor following the specifications of Van Herk et al.35 including SiC layers 20 mm thick above and below the catalyst bed, in order to minimize heat losses and gas bypassing. Prior to every reaction, the catalyst was pre-treated at 400 °C for 12 h under a continuous gas flow mix of 30 STP cm3 min−1 H2 and 50 STP cm3 min−1 N2. After exiting the reactor, liquid products were sent to a Peltier cell (gas/liquid separator), then sampled and weighed every hour for mass balance calculation purposes. Liquid products consisted of an aqueous and an organic phase that were separated by decantation and analyzed individually. The composition of the organic product phase was assessed through two-dimensional Gas Chromatography (GCxGC) coupled with Mass Spectrometry (MS) in an Agilent 7890A apparatus connected on-line with an Agilent 5975C series MS. The GCxGC was provided with two columns of different polarities connected through a flow modulator, being the first column a non-polar DB-5MS (length = 30 m; internal diameter = 0.25 mm), while the second one was a polar TRB-50 HT (length = 5 m; internal diameter = 0.25 mm). On the other hand, the aqueous phase was analyzed by means of gas chromatography-mass spectrometry in a Shimadzu GC-MS QP2010 unit provided with a BPX5 column (length = 50 m; internal diameter = 0.22 mm). The water content in the aqueous product phase was quantified by Karl-Fischer titration using a Metrohm830 KF Titrino plus apparatus.
The yields of liquid carbon product (C-prod) and water are defined by eqn (1) on a total bio-oil basis (including water):
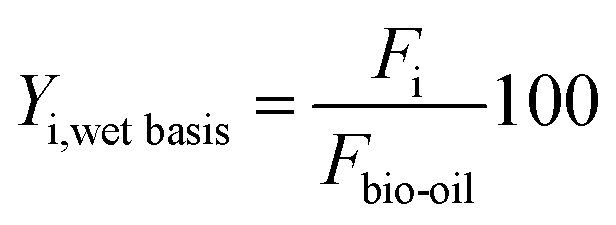 | (1) |
Liquid carbon products were classified in lumps of different chemical nature, as follows: oxygenates (comprising: methanol, MeOH; acetone, AC and; acetic acid, AA); alkanes, AL; ketones, KT; phenol, PH; alkylphenolics, APH; 1 ring aromatics (A1); 2+ ring aromatics (A2+) and; other oxygenates (acids and esters), OT. Their yields on a dry basis are defined by eqn (2) as:
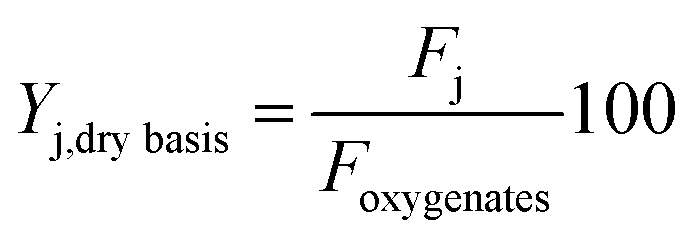 | (2) |
Coke characterization
The deactivated catalyst samples were analyzed for coke quantification through temperature-programmed oxidation (TPO) in a TA Instruments TGA Q5000 IR thermobalance (TG-TPO), without any previous coke separation. Prior to analysis, samples were subjected to a stripping pre-treatment under a N2 atmosphere (100 cm3 min−1) at 450 °C in order to remove organic adsorbed species. TPO analyses were conducted under a continuous air flow (100 cm3 min−1) applying a ramp up to 700 °C at a 5 °C min−1 rate, which ensures total combustion of the formed coke. The TG-TPO results allow for the quantification of the different coke fractions.Elemental analysis of the used catalyst samples was performed in a EuroVector EA3000 Elemental Analyzer. The oxygen content was determined by difference from the wt% contents of C, H and N.
Raman spectroscopy analyses were carried out in a Renishaw InVia confocal microscope, consisting of a spectrometer and two lasers coupled to a Leica optical microscope. An excitation wavelength of 514 nm was used, and the fluorescence caused by the material was subtracted.
Multivariate data analysis
A principal component analysis (PCA) routine developed in Matlab was applied for the multivariate analysis of the reaction data. A dataset of 15 variables from 21 uncorrelated runs was configurated and processed as previously detailed by Alvira et al.36 The Varimax rotation was applied in order to facilitate interpretation of the results. The assessed variables comprise reaction parameters (time on stream, TOS; space time, W/F), coke composition (contents of the fractions of thermal lignin, TL; intermediate coke, IC; and catalytic coke, CC), and reaction medium composition (on the basis of the lumps previously defined: MeOH, AC, AA, AL, KT, PH, APH, A1, A2, and OT).Results and discussion
Evolution of product yields and composition over TOS
The evolution with time on stream (TOS) of the yields of the main product fractions in a wet bio-oil basis (gas + coke, water, and carbon products in the organic (Cprod-org) and aqueous (Cprod-aq) phases) as defined by eqn (1) are displayed in Fig. 1a. At TOS > 2 h the main component at the reactor exit is water (>51.8 wt%), which originates from raw bio-oil itself and is also formed as end product of HDO mechanisms. The yield of total carbon products (Cprod-org + Cprod-aq) presents an increasing trend over time, with a parallel decrease in the gas + coke lump. Interestingly, while carbon products originate solely from an aqueous carbon product phase at TOS = 1–2 h (Cprod-aq = 2.3–9.8 wt%), an incipient formation of an organic product phase (Cprod-org) is detected at TOS > 3 h. A higher increase in the total carbon product yield is then attained up to TOS = 5 h, after which constant values of 27.3 wt%, 54.3 wt% water and 18.4 wt% gas + coke are obtained (average values at TOS = 5–8 h). The evolution over time on stream of the water content in the aqueous phase, displayed in Fig. S2,† shows a similar behavior. From an initial water content of 92.5 wt% at TOS = 1 h, a drop of about 9 wt% is measured once a constant water content of 84.2 wt% is achieved. These results suggest that HDO reactions deactivate more preferentially over time, being particularly affected during the initial 4 h on stream, where the water concentration in the aqueous phase steadily decreases. Accordingly, the formation of the organic carbon product lump is also promoted in a greater extent (Fig. 1a).16,31 In this case, the high water concentration of this raw bio-oil (49 wt%) is favoring the stability not only of the catalyst but of the overall process. It is known that, in the catalytic conversion of raw bio-oils,37 a higher water concentration in the reaction medium is beneficial and performs a coke precursor stripping effect, hence maintaining a higher remaining catalyst activity over time on stream, which ultimately will aid to the overall process stability.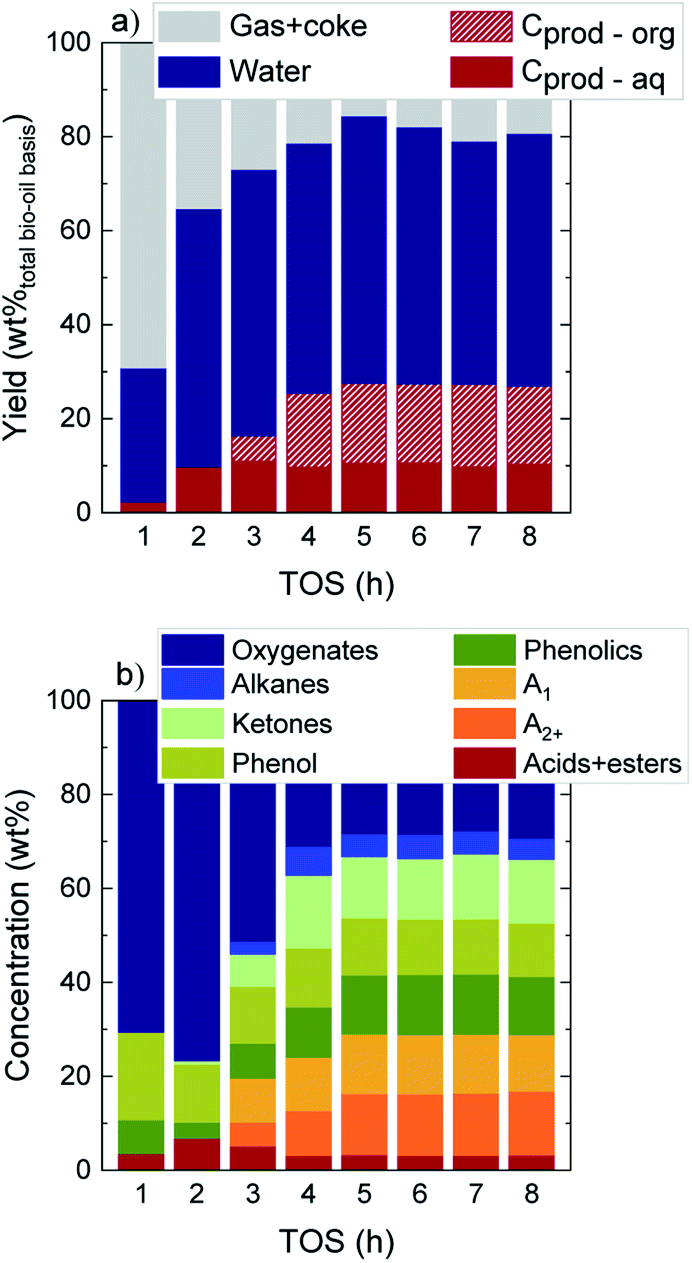 | ||
| Fig. 1 Evolution with TOS of (a) product yields on a wet bio-oil basis and (b) the concentration of the different lumps in liquid carbon products. | ||
These results are indicative of two different reaction stages taking place during bio-oil HDO: (i) an initial transient stage at TOS = 1–5 h where an important loss in catalyst activity due to coke deposition occurs, and (ii) a pseudosteady activity stage, where product yields are constant and carbon products are maximized.16,31 In agreement with other studies on the conversion of raw bio-oil with different catalysts, a fast catalyst deactivation primarily occurs during the transient state because of the condensation of the highly reactive alkyl(methoxy)phenols, forming the so-called thermal lignin (TL). This solid product from the thermal repolymerization of bio-oil oxygenates can later on evolve towards a heavier and more condensed coke.30,38
Fig. 1b establishes the focus on the concentration of the different lumps in C-products, which account for a total 53.5 wt% in a dry bio-oil basis at the pseudosteady state (see Fig. S3†). During the initial 2 h on stream, the lump of oxygenates is predominant (70.6–76.6 wt%), whereas a significant concentration of aromatics, ketones, phenol and phenolics is detected at TOS > 3 h. Eventually, values of 25.7 wt% total aromatics, 13.3 wt% ketones, 11.8 wt% phenol and 12.6 wt% phenolics are attained at the pseudosteady activity state. The appearance over time on stream of significant amounts of unsaturated and oxygenated compounds reconfirms the catalyst activity drop, mainly during the initial hours of reaction.
The concentration of the three compounds comprised in the oxygenated lump in Fig. S4† (methanol, acetic acid and acetone), with a majority of acetic acid through the whole reaction time (73–83 wt%) indicates that the HDO activity of the FeMoP/HZSM-5 catalyst is lower than that achieved with a noble metal-based catalyst at the same experimental conditions.31 This difference is explained by the higher capacity of noble metal-based catalysts for C–O bond cleavage and demethoxylation of alkyl(methoxy)phenols, hence leading to a heavily hydrogenated end liquid product.39,40 All in all, when bio-oil HDO is intended for the production of certain platform chemicals, the pore blockage and masking of acidic sites caused by coke deposits results beneficial in order to limit the hydrogenation capability of the catalyst, resulting in higher yields of unsaturated compounds. The acidic and structural properties of the used catalysts are reported in Table S1.† As plotted in Fig. 2, we observed that, after 1 h on stream, about 48% of the total catalyst acidity is lost in comparison to fresh catalyst conditions, as well as 62% of the specific surface, detecting a pronounced decrease in both parameters through the whole transient reaction stage (TOS = 1–4 h). Once the pseudosteady state is attained (TOS > 5 h), these values further decrease down to 93% and 97%, respectively, hence indicating that almost complete coverage and blockage of the acidic sites and pores occurs due to the severe coke formation in the process.
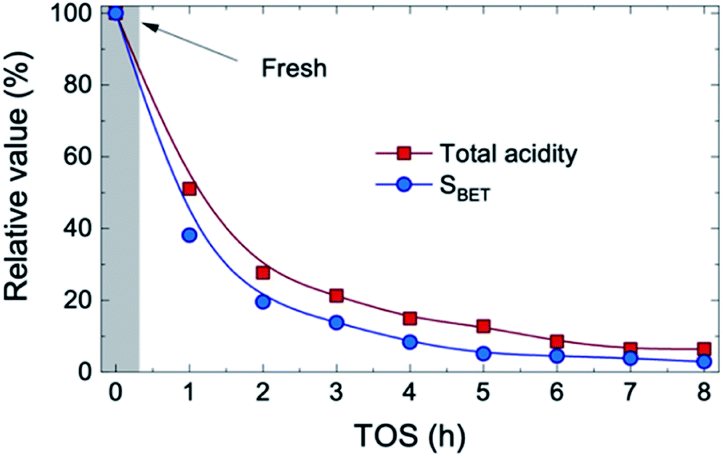 | ||
| Fig. 2 Evolution with TOS of the relative values for total acidity and specific surface of the catalyst. | ||
Coke characterization
Combined thermogravimetric and combustion techniques provide relevant information on coke content and nature.41 On the basis of their combustion behavior and the role they play in coke formation mechanisms, three different carbonaceous species are defined: (i) thermal lignin (TL, combustion peak maximum at 365–370 °C), intermediate coke (IC, peak maximum at 445–450 °C) and catalytic coke (CC, peak maximum at 515–520 °C). Detailed TPO profiles and deconvolutions at every TOS can be found in Fig. S5.† The thermal origin of this material was corroborated from a blank experiment with no catalyst. In this experiment, the reactor rapidly clogged due to the very fast formation of carbonaceous deposits within the reactor. The fraction of these deposits collected from the entrance of the reactor presented the same characteristics than those of the thermal lignin, hence confirming its formation from thermal mechanisms with no catalytic intervention.The evolution with time on stream of the total coke content as well as the different coke species is displayed in Fig. 3a. Regarding total coke content, a pronounced increase is observed from 21 wt% at TOS = 1 h up to 56 wt% at TOS = 3 h, where a maximum value is reached. After that, the total coke content shows a decreasing tendency until an almost constant value is attained at TOS = 5 h, with a value of 38 wt% (average TOS = 5–8 h value). In terms of the evolution of the different species, both TL and IC show similar tendencies, with an increasing formation up to TOS = 3 h, where the tendency reverses. Maximum values of 31 wt% and 16 wt% of TL and IC are attained at TOS = 3 h, decreasing down to 16 wt% and 9 wt%, respectively, at the pseudosteady activity state. On the other hand, CC presents a steadily increasing formation over time on stream, increasing from 4 wt% at TOS = 1 h and up to 13 wt% at the pseudosteady state (Fig. 2a). In terms of the concentration of the different carbon species (Fig. 3b), TL is dominant over the course of the whole reaction, with concentrations in the range of 77–56% in the transient stage, and 41% in the pseudosteady activity state. This decrease in the total amount of coke at TOS > 3 h can be explained by the relevance of the condensation mechanisms of the coke fractions, causing gas product and water formation, and also by the gasification of coke due to a high water content in the reaction media at TOS > 3 h. Note that the gasification of the solid product is possible at such a low temperature because of the high reaction pressure.
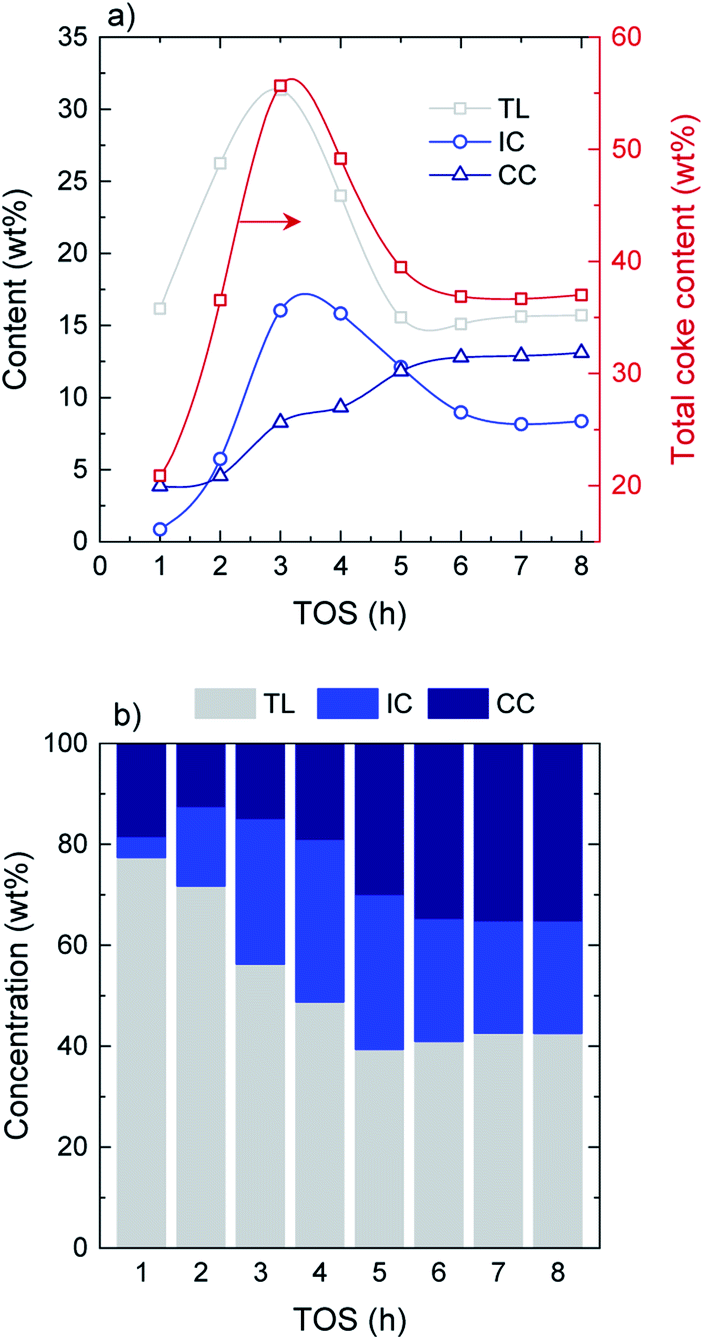 | ||
| Fig. 3 Evolution with TOS of the (a) amount of total coke and the different coke species and (b) concentration of the different deactivating species in coke. | ||
Reaching the pseudosteady state leads to the supression of the (additional) coke formation and stabilization of its composition as depicted in Fig. 4. The results (derived from EA analyses) indicate a highly oxygenated coke being formed at the initial hours of reaction (oxygen content of 77%), with an O/C ratio of 3.0 and H/C ratio ca. 1.4, which evolves into less oxygenated and more condensed structures over time on stream. At TOS > 4 h, upon progress of hydrodeoxygenation, condensation and dehydration reactions,27 the coke in the used catalyst samples present O/C values of 0.50–0.66 (corresponding to oxygen contents between 39–45%) and lower H/C ratios of 1.0–1.2. This is in agreement with the coke compositions reported in Fig. 3b, where TL conforms the majority of the formed carbon residues in the TOS = 1–3 h range. Later on, the coke evolves towards more dehydrogenated and deoxygenated structures, as a consequence of the dominance of oxygen-free unsaturated compounds in the reaction medium (Fig. 1b).
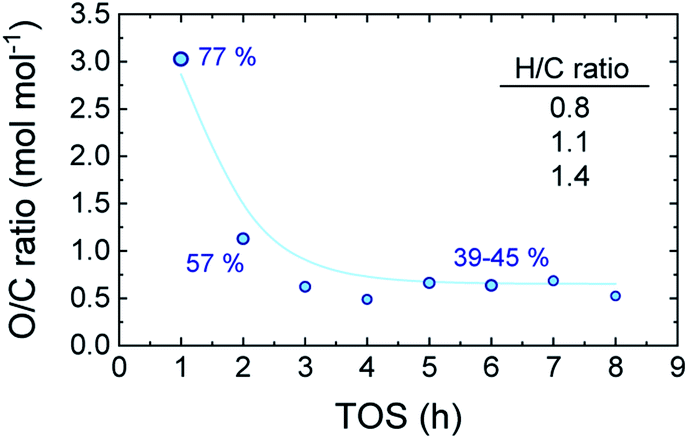 | ||
| Fig. 4 Evolution with TOS of the O/C molar ratio of the coke in the used catalysts. | ||
The normalized Raman spectra of the used catalyst samples in Fig. S6† presents two main bands characteristic of carbonaceous structures.42,43 The D band (1350 cm−1) is attributed to the breathing mode of poorly structured aromatic clusters, while the G band (1580–1610 cm−1) is related to in-plane stretching of ordered aromatic carbons.42 Both the D and G bands become narrower for used catalysts deactivated at longer times on stream (Fig. S6a†), indicating that coke develops a higher degree of structural order.44 In addition, the evolution of the D/G band height ratio with time on stream in Fig. S6b† proves that structural changes are notable at TOS < 4 h, with little variations at longer reaction times, reaching an almost constant composition.45
Multivariate data analysis
In complex processes where several variables are liable to conditioning the process and important reaction synergies occur, proposing accurate kinetic schemes that describe either the process or coke formation over time on stream becomes a challenging task. In this context, principal component analysis (PCA) is a valuable statistical tool that can be applied for categorizing the different process variables by replacing them with a set of new ones (principal components). This strategy aids for describing the main trends in the process and aid for detecting hidden correlations.36,46 From the experimental data obtained from 21 different HDO runs of uncorrelated experiments derived from the present and previous works on raw bio-oil HDO,16,31 we established a dataset including the variables defined in the Experimental section. Through PCA, data was simplified to three main components (PC1-3). In the Varimax loading plot for PC1 and PC2, shown in Fig. 5a, certain clustering concerning the reaction medium composition is observed, on the basis of (i) oxygen-free components (alkanes + aromatics) and (ii) ketones, phenol and alkylphenolics. The latter presents the highest loadings along PC1, while the ones of the rest of oxygenates (acetic acid, acetone, methanol, acids and esters) along PC1 are noticeably lower. Time on stream also presents a significant loading along PC1, directly correlated with the reaction medium composition. These variables are also directly correlated with the formation of CC, and inversely correlated with the other two coke species (TL and IC). All the aforementioned correlations are in agreement with the results plotted in Fig. 1b and 3a, which show that after 3 h on stream phenolic and aromatic compounds appear in a greater extent (as the organic product phase is formed). The amounts of both TL and IC suffer a decrease in favor of a greater CC formation from catalytic mechanisms. In line with the dual coke formation mechanism proposed by Cordero-Lanzac et al.30 for the HDO of raw bio-oil, dominated by alkyl(methoxy)phenolic and aromatic precursors respectively, PCA analysis points out the contribution of both of these component classes on the formation of the more condensed CC.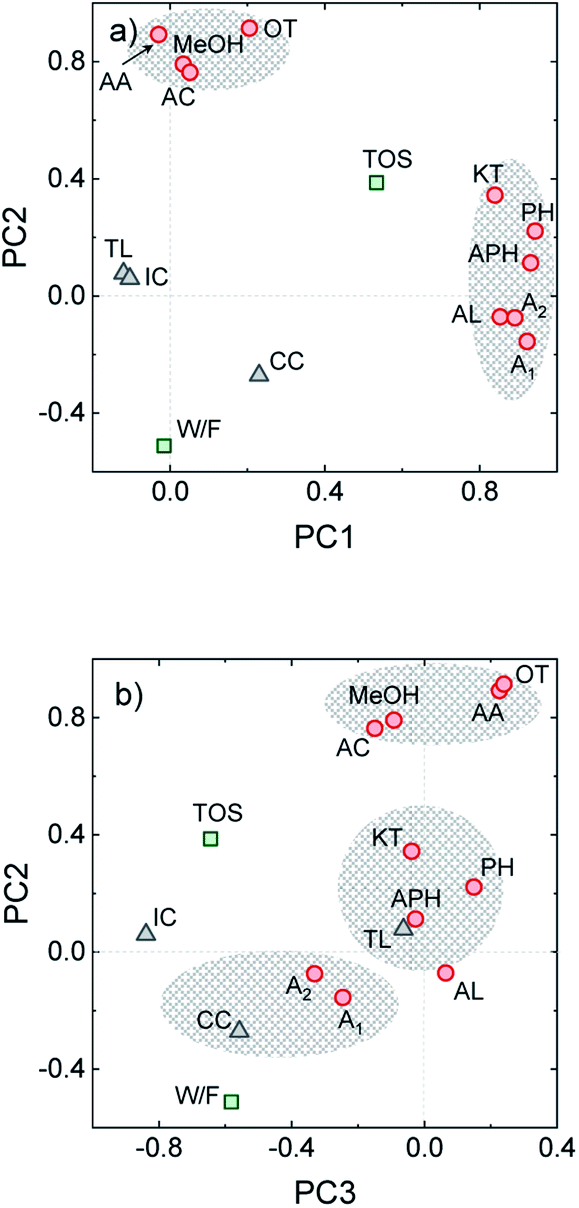 | ||
| Fig. 5 Varimax loading plots for (a) PC2 vs. PC1 and (b) PC2 vs. PC3. | ||
On the other hand, the highest loadings along PC2 are those of the components in the lumps of oxygenates (methanol, acetic acid, acetone) and other oxygenates (acids + esters), with lower loadings of the rest of the components. Oxygenated compounds (ketones, phenol and phenolics) present lower loadings, inversely correlated with the yields of alkanes and aromatics. The former leads to the latter through hydrodeoxygenation, and the oxygen-free compounds play the role of CC precursors, hence explaining the direct correlation between CC, A1 and A2 along PC2. These results indicate that the compounds in the lump of oxygenates play a secondary role on coke formation despite being present in relatively important amounts (ca. 30 wt% of the carbon product at the pseudosteady state, see Fig. 1b).
The distribution of variables along PC3 (Fig. 5b) indicates the impact that reaction conditions (namely time on stream and space time) have on the formation of CC, but not on that of TL. As previously discussed, TL is thermally induced upon entrance of the raw bio-oil feedstock in the reactor, even prior to its contact with the catalyst. The higher dependence of TL on thermal effects rather than on time on stream or reaction medium composition is confirmed by the very low loadings of TL along all PCs.
The statistical fitting obtained through PCA analysis provides a solid mathematical support for discussion on the coke formation mechanism presented herein and in a previous work.30 The conclusions extracted from the fit of a random set of experimental data are consistent with our hypotheses, highlighting the influence of the reaction medium composition on the solid deposition during the transient and pseudosteady states.
Coke formation mechanism and discussion
The results presented in the previous sections indicate the existence of different coke formation/transformation stages that directly control the corresponding two reaction stages (transient and pseudosteady) and impact the observed product distribution (Fig. 1). The coke formation/transformation stages can be tracked in terms of coke content (Fig. 3) and composition (Fig. 4 and S6†). From these results, a mechanism for describing the formation of coke over time on stream in the HDO of raw bio-oil is proposed in Fig. 6. Representative molecules of each one of the main chemical groups involved in coke formation (precursors) are included in the scheme, as well as proposed structures for the three different coke fractions.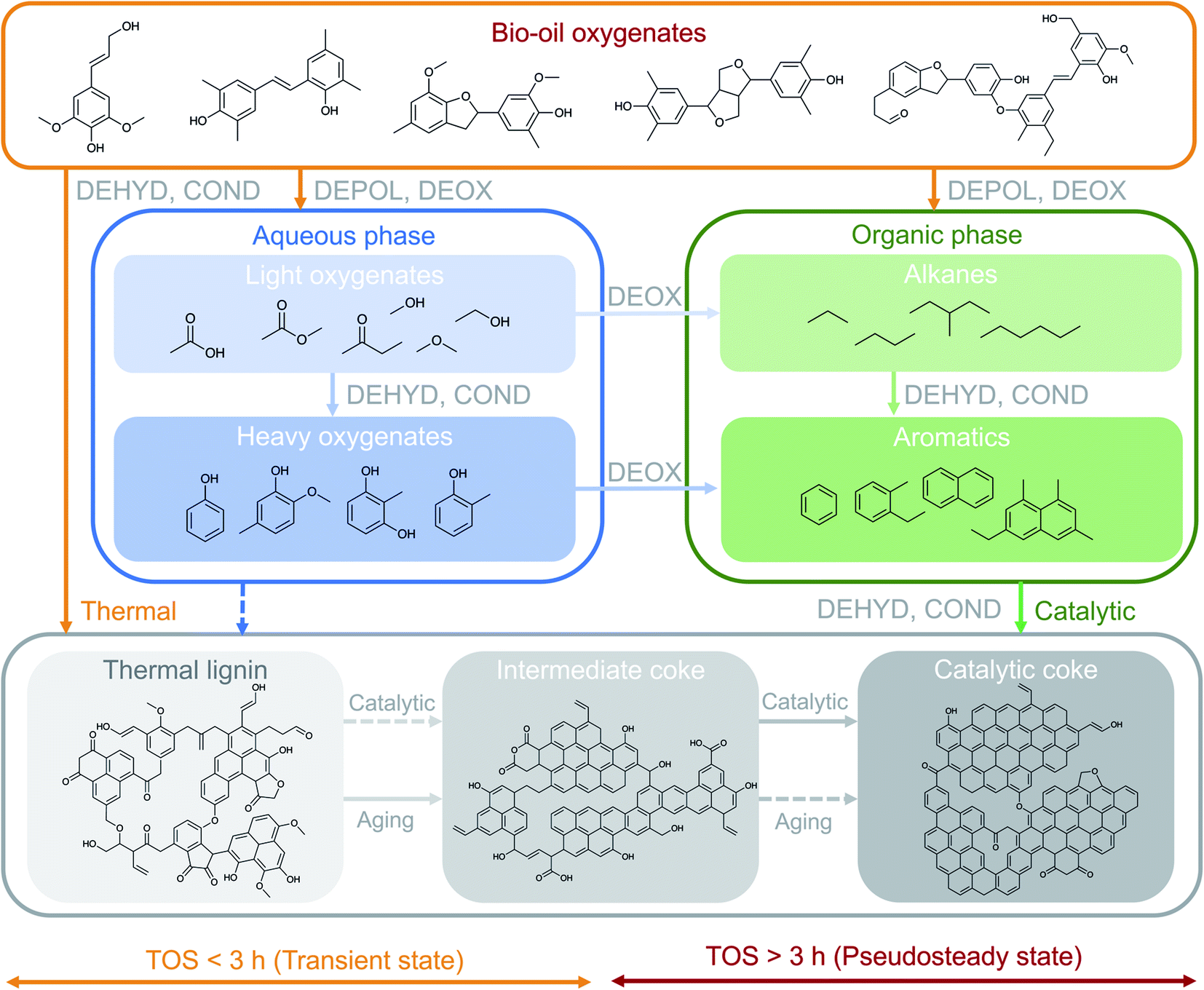 | ||
| Fig. 6 Proposed scheme for the parallel thermal/catalytic formation of the different coke fractions in the raw bio-oil HDO over time on stream. The main sets of reactions have been abbreviated as: depolymerization, DEPOL; dehydrogenation, DEHYD; deoxygenation, DEOX; condensation, COND. | ||
On the basis of the well-established HDO deactivation mechanisms,27,30 the scheme in Fig. 6 considers that TL originates from the thermally-induced repolymerization of unstable and highly reactive oxygenates present in bio-oil, and acts as a precursor for more evolved coke. The evolution (aging) of TL occurs through a series of catalyst-activated reactions: gasification and condensation-carbonization. Ibañez et al.47 and Valle et al.48 established a direct correlation between the concentration of the bio-oil components in the feed, particularly polyols and (alkyl)phenolics, with the amount of deposited coke on the catalyst in the catalytic conversion of raw bio-oil + methanol mixtures over a HZSM-5 based catalyst at 450 °C. The results in Fig. 3 evidence not only the nature of TL as precursor, but also the role of IC as intermediate in coke formation mechanisms, with increasing trends up to 3 h on stream, after which this trend inverts. The steady formation of CC over time on stream, on the other hand, denotes its nature of final product in the mechanism. This evolution of the content of the different species of coke indicates the occurrence of a thermal/catalytic parallel coke formation mechanism over time on stream. In line with the scheme in Fig. 6, TL is initially formed through thermally-induced repolymerization of bio-oil oxygenates, and its further transformation into IC can be associated with two possible pathways: (i) a gasification mechanism (which entails condensation of the remaining coke) due to the higher content of water in the reaction medium and the high reaction temperature and pressure,49 and; (ii) a catalytic mechanism which subjects coke precursors to dehydration, deoxygenation, condensation and hydrogen transfer reactions towards heavily condensed catalytic coke (CC). The formation of TL, to a much lesser extent, is also plausible from the repolymerization of the heavier oxygenates (phenolics) in the aqueous phase, which at the same time can be formed from dehydration and condensation reactions of lighter oxygenates. As a consequence of aging reactions, important amounts of CO, CO2 and water are produced.30 These reactions imply a mass loss in the total coke, as well as the TL and IC fractions, as shown in Fig. 3a.
This interpretation of the coke evolution mechanism is in agreement with the evolution of the HDO products over TOS. During the initial hours of reaction (TOS = 0–2 h), the composition of the reaction media consists of solely light oxygenates (i.e. acetic acid, ketones, alcohols, etc.) mixed with a significant amount of water originating from the raw bio-oil feedstock itself (49 wt%) and formed as an end product of HDO mechanisms (see Fig. 1b). In this reaction period, the concentration of acetic acid, an important coke precursor,50 is also significant in the aqueous product phase (Fig. 3b and S4†). At these conditions, the thermal mechanism of TL formation is promoted, leading to a fast catalyst deactivation and causing partial activity loss at TOS < 3 h. Afterwards, as a consequence of the aforementioned activity loss, the presence of unsaturated oxygen-free aromatics becomes significant (ca. 25 wt% in the pseudosteady state). These aromatics originate from depolymerization and deoxygenation of the bio-oil oxygenates, as well as dehydration and deoxygenation of the heavy oxygenates in the aqueous phase and give way to the catalytic mechanism of coke formation from aromatic precursors. This capacity of aromatic compounds for forming coke is justified by their condensation activity over the acidic sites of the catalyst.30,51
Consequently, the overall coke nature evolves towards a more condensed (Fig. S6†) and less oxygenated nature (Fig. 4) upon increasing the time on stream, indicating that this catalytic route from hydrocarbon precursors is dominant during the pseudosteady stage of the reaction, while the oxygenated precursor route (mainly thermally affected) controls the transient state. At the pseudosteady state, CC formation occurs from the catalytic conversions of both (i) the aromatic reaction products originated from the raw bio-oil HDO and (ii) condensation–carbonization IC species, as demonstrated in Fig. 3. Despite the co-existence of both routes at the pseudosteady activity state, the controlling role of the catalytic route from aromatic precursors at high reaction temperatures is suggested, while milder temperatures would favor the continuous formation of coke deposits from oxygenated precursors.30
Conclusions
This work exposes the complexity of coke formation mechanisms and the evolution of its composition and structure over time on stream in the HDO of raw bio-oil using a FeMoP/HZSM-5 catalyst. This evolution corresponds to a transient stage of 3 h, after which coke content and structure remains constant and, consequently, the catalyst reaches a pseudosteady state where product yields remain stable over time on stream. In these conditions, the production of interesting platform chemicals is maximized.A statistical principal component analysis (PCA) can correlate coke composition with the reaction variable and the reaction media composition. The concentration of oxygen-free and oxygenated unsaturated compounds, namely aromatics and (alkyl)phenolics, is confirmed to be the determinant factor conditioning the coke formation, followed by other oxygenates (acetic acid, methanol, acetone, acids and esters), and with a lower influence of reaction conditions (time on stream and space time).
The discernible coke dynamics of formation and transformation control the two reaction stages (transient and pseudosteady). In the transient state, the content of thermal lignin increases due to thermally-induced repolymerization of bio-oil oxygenates. This step coincides with a composition of products dominated by the oxygenated compounds (acetic acid, methanol, acetone, phenol and phenolics). Then, and also in the transient state, thermal lignin evolves and ages into intermediate coke through gasification–condensation reactions. In the pseudosteady state, a parallel catalytic coke formation route acquires relevance from both (i) the further aging of intermediate coke and (ii) condensation of the aromatics in the reaction media. Consequently, the overall coke composition evolves from highly oxygenated structures in the transient state to a more deoxygenated and condensed aromatic-type material at the pseudosteady state, with a higher structural order. This coke stability explains the constant activity of the catalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been carried out with the financial support of the Ministry of Economy and Competitiveness of the Spanish Government, some co-founded with ERDF funds (CTQ2015-67425-R, CTQ2016-79646-P), the Basque Government (IT1218-19), and the European Commission (Horizon H2020-MSCA RISE-2018, Contract No. 823745). Dr Idoia Hita is grateful for her postdoctoral grant awarded by the Department of Education, University and Research of the Basque Government (POS_2015_1_0035).References
- S. Pang, Biotechnol. Adv., 2019, 37, 589–597 CrossRef CAS PubMed.
- Z. Yang, Y. Wu, Z. Zhang, H. Li, X. Li, R. I. Egorov, P. A. Strizhak and X. Gao, Renewable Sustainable Energy Rev., 2019, 103, 384–398 CrossRef CAS.
- E. Lazzari, T. Schena, M. C. A. Marcelo, C. T. Primaz, A. N. Silva, M. F. Ferrão, T. Bjerk and E. B. Caramão, Ind. Crops Prod., 2018, 111, 856–864 CrossRef CAS.
- Z. Si, X. Zhang, C. Wang, L. Ma and R. Dong, Catalysts, 2017, 7, 169–191 CrossRef.
- X. Yuan, X. Ding, L. Leng, H. Li, J. Shao, Y. Qian, H. Huang, X. Chen and G. Zeng, Energy, 2018, 154, 110–118 CrossRef CAS.
- J. Xing, T. Li, Y. Yu, C. Chen and J. Chang, Int. J. Adhes. Adhes., 2018, 87, 91–97 CrossRef CAS.
- A. N. Kay Lup, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, J. Ind. Eng. Chem., 2017, 56, 1–34 CrossRef CAS.
- M. Patel and A. Kumar, Renewable Sustainable Energy Rev., 2016, 58, 1293–1307 CrossRef CAS.
- I. Hita, A. Gutiérrez, M. Olazar, J. Bilbao, J. M. Arandes and P. Castaño, Fuel, 2015, 145, 158–169 CrossRef CAS.
- Y. Villasana, F. J. Méndez, M. Luis-Luis and J. L. Brito, Fuel, 2019, 235, 577–588 CrossRef CAS.
- K. Guo, Y. Ding and Z. Yu, Appl. Catal., B, 2018, 239, 433–440 CrossRef CAS.
- X.-J. Liu, F. Wang, L.-L. Zhai, Y.-P. Xu, L.-F. Xie and P.-G. Duan, Fuel, 2019, 249, 418–426 CrossRef CAS.
- A.R. Ardiyanti, A. Gutierrez, M.L. Honkela, A.O.I. Krause and H.J. Heeres, App. Cat. A, 2011, 407, 56–66 CrossRef CAS.
- I. Hita, T. Cordero-Lanzac, A. Gallardo, J. M. Arandes, J. Rodríguez-Mirasol, J. Bilbao, T. Cordero and P. Castaño, Catal. Commun., 2016, 78, 48–51 CrossRef CAS.
- T. Cordero-Lanzac, I. Hita, F. J. García-Mateos, P. Castaño, J. Rodríguez-Mirasol, T. Cordero and J. Bilbao, Chem. Eng. J., 2020, 124679, DOI:10.1016/j.cej.2020.124679.
- I. Hita, T. Cordero-Lanzac, G. Bonura, C. Cannilla, J. M. Arandes, F. Frusteri and J. Bilbao, J. Ind. Eng. Chem., 2019, 80, 392–400 CrossRef CAS.
- R. K. Chowdari, S. Agarwal and H. J. Heeres, ACS Sustainable Chem. Eng., 2019, 7, 2044–2055 CrossRef CAS PubMed.
- K. Li, R. Wang and J. Chen, Energy and Fuels, 2011, 25, 854–863 CrossRef CAS.
- D. J. Rensel, J. Kim, Y. Bonita and J. C. Hicks, Appl. Catal., A, 2016, 524, 85–93 CrossRef CAS.
- D. J. Rensel, S. Rouvimov, M. E. Gin and J. C. Hicks, J. Catal., 2013, 305, 256–263 CrossRef CAS.
- D. J. Rensel, J. Kim, V. Jain, Y. Bonita, N. Rai and J. C. Hicks, Catal. Sci. Technol., 2017, 7, 1857–1867 RSC.
- V. Jain, Y. Bonita, A. Brown, A. Taconi, J. C. Hicks and N. Rai, Catal. Sci. Technol., 2018, 8, 4083–4096 RSC.
- Á. Ibarra, I. Hita, M. J. Azkoiti, J. M. Arandes and J. Bilbao, J. Ind. Eng. Chem., 2019, 78, 372–382 CrossRef.
- H. Shafaghat, P. S. Rezaei and W. M. A. W. Daud, J. Ind. Eng. Chem., 2016, 35, 268–276 CrossRef CAS.
- X. Lan, E. J. M. Hensen and T. Weber, Appl. Catal., A, 2018, 550, 57–66 CrossRef CAS.
- E. Furimsky and F. E. Massoth, Catal. Today, 1999, 52, 381–495 CrossRef CAS.
- I. Hita, P. J. Deuss, G. Bonura, F. Frusteri and H. J. Heeres, Fuel Process. Technol., 2018, 179, 143–153 CrossRef CAS.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- B. Valle, N. García-Gómez, A. Arandia, A. Remiro, J. Bilbao and A. G. Gayubo, Int. J. Hydrogen Energy, 2019, 44, 12593–12603 CrossRef CAS.
- T. Cordero-Lanzac, R. Palos, I. Hita, J. M. Arandes, J. Rodríguez-Mirasol, T. Cordero, J. Bilbao and P. Castaño, Appl. Catal., B, 2018, 239, 513–524 CrossRef CAS.
- I. Hita, T. Cordero-Lanzac, F. J. García-Mateos, M. J. Azkoiti, J. Rodríguez-Mirasol, T. Cordero and J. Bilbao, Appl. Catal., B, 2019, 118112, DOI:10.1016/j.apcatb.2019.118112.
- N. Kosinov, F. J. A. G. Coumans, G. Li, E. Uslamin, B. Mezari, A. S. G. Wijpkema, E. A. Pidko and E. J. M. Hensen, J. Catal., 2017, 346, 125–133 CrossRef CAS.
- A. R. Fernandez-Akarregi, J. Makibar, G. Lopez, M. Amutio and M. Olazar, Fuel Process. Technol., 2013, 112, 48–56 CrossRef CAS.
- T. Cordero-Lanzac, R. Palos, J. M. Arandes, P. Castaño, J. Rodríguez-Mirasol, T. Cordero and J. Bilbao, Appl. Catal., B, 2017, 203, 389–399 CrossRef CAS.
- D. van Herk, P. Castaño, M. Quaglia, M. T. Kreutzer, M. Makkee and J. A. Moulijn, Appl. Catal., A, 2009, 365, 110–121 CrossRef CAS.
- J. I. Alvira, I. Hita, E. Rodríguez, J. M. Arandes and P. Castaño, Processes, 2018, 6, 243–258 CrossRef CAS.
- Á. Ibarra, I. Hita, J. M. Arandes and J. Bilbao, Energy Fuels, 2019, 33, 7458–7465 CrossRef.
- I. Hita, H. J. Heeres and P. J. Deuss, Bioresour. Technol., 2018, 267, 93–101 CrossRef CAS PubMed.
- B. Lin, R. Li, R. Shu, C. Wang, Z. Yuan, Y. Liu and Y. Chen, J. Energy Inst., 2020, 93, 847–856 CrossRef CAS.
- A. S. Ouedraogo and P. R. Bhoi, J. Cleaner Prod., 2020, 253, 119957 CrossRef CAS.
- A. Ochoa, B. Valle, D. E. Resasco, J. Bilbao, A. G. Gayubo and P. Castaño, ChemCatChem, 2018, 10, 2311–2321 CrossRef CAS.
- A. Ochoa, B. Aramburu, M. Ibáñez, B. Valle, J. Bilbao, A. G. Gayubo and P. Castaño, ChemSusChem, 2014, 7, 2597–2608 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- K. Chen, H. Zhang, U.-K. Ibrahim, W. Xue, H. Liu and A. Guo, Fuel, 2019, 246, 60–68 CrossRef CAS.
- I. Hita, E. Rodríguez, M. Olazar, J. Bilbao, J. M. Arandes and P. Castaño, Energy Fuels, 2015, 29, 5458–5466 CrossRef CAS.
- T. Rato, M. Reis, E. Schmitt, M. Hubert and B. De Ketelaere, AIChE J., 2016, 62, 1478–1493 CrossRef CAS.
- M. Ibáñez, B. Valle, J. Bilbao, A. G. Gayubo and P. Castaño, Catal. Today, 2012, 195, 106–113 CrossRef.
- B. Valle, P. Castaño, M. Olazar, J. Bilbao and A. G. Gayubo, J. Catal., 2012, 285, 304–314 CrossRef CAS.
- P. Magnoux, H. S. Cerqueira and M. Guisnet, Appl. Catal., A, 2002, 235, 93–99 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2619–2626 CrossRef CAS.
- Á. Ibarra, A. Veloso, J. Bilbao, J. M. Arandes and P. Castaño, Appl. Catal., B, 2016, 182, 336–346 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Bio-oil composition, water content in aqueous products, evolution with TOS of total carbon product yield, evolution with TOS of the oxygenated lump composition, physico-chemical properties of used catalysts, TG-TPO deconvolution profiles of coked catalysts. See DOI: 10.1039/d0se00501k |
| This journal is © The Royal Society of Chemistry 2020 |