
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The success story of graphite as a lithium-ion anode material – fundamentals, remaining challenges, and recent developments including silicon (oxide) composites
Jakob
Asenbauer†
ab,
Tobias
Eisenmann†
ab,
Matthias
Kuenzel†
 ab,
Arefeh
Kazzazi
ab,
Zhen
Chen
*ab and
Dominic
Bresser
*ab
ab,
Arefeh
Kazzazi
ab,
Zhen
Chen
*ab and
Dominic
Bresser
*ab
aHelmholtz Institute Ulm (HIU), Helmholtzstrasse 11, 89081 Ulm, Germany. E-mail: dominic.bresser@kit.edu; chen.zhen@kit.edu
bKarlsruhe Institute of Technology (KIT), P.O. Box 3640, 76021 Karlsruhe, Germany
First published on 7th May 2020
Abstract
Lithium-ion batteries are nowadays playing a pivotal role in our everyday life thanks to their excellent rechargeability, suitable power density, and outstanding energy density. A key component that has paved the way for this success story in the past almost 30 years is graphite, which has served as a lithium-ion host structure for the negative electrode. And despite extensive research efforts to find suitable alternatives with enhanced power and/or energy density, while maintaining the excellent cycling stability, graphite is still used in the great majority of presently available commercial lithium-ion batteries. A comprehensive review article focusing on graphite as lithium-ion intercalation host, however, appeared to be missing so far. Thus, herein, we provide an overview on the relevant fundamental aspects for the de-/lithiation mechanism, the already overcome and remaining challenges (including, for instance, the potential fast charging and the recycling), as well as recent progress in the field such as the trade-off between relatively cheaper natural graphite and comparably purer synthetic graphite and the introduction of relevant amounts of silicon (oxide) to boost the energy and power density. The latter, in fact, comes with its own challenges and the different approaches to overcome these in graphite/silicon (oxide) composites are discussed herein as well.
1. Introduction and outline
Lithium-ion batteries (LIBs) have been on the market for almost thirty years now and have rapidly evolved from being the powering device of choice for relatively small applications like portable electronics to large-scale applications such as (hybrid) electric vehicles ((H)EVs) and even stationary energy storage systems.1–7 One key step during these years has been the development of graphite-based anodes, replacing the initially used soft and hard carbons, allowing for greatly enhanced full-cell energy densities due to its low de-/lithiation potential and high (theoretical) gravimetric capacity of 372 mA h g−1.1,8This development has been made possible by several important milestones, which are – together with more recent developments – summarized illustratively in Fig. 1. The possibility to form lithium intercalation compounds with graphite up to a maximum lithium content of LiC6 using molten lithium or compressed lithium powder has been known, in fact, since 1975.9–11 Initial attempts in the 1970s to reversibly intercalate lithium into graphite electrochemically, however, failed due to the continuous co-intercalation and decomposition of the commonly used liquid organic electrolytes based on, e.g., propylene carbonate (PC) and the resulting graphite exfoliation.12,13 The first successful attempt was reported in 1983 by Yazami and Touzain,14 employing a solid polymer electrolyte. In 1990, Dahn and co-workers15 eventually found that the reversible electrochemical lithium intercalation is possible also for liquid electrolyte when using ethylene carbonate (EC) as electrolyte co-solvent thanks to the formation of a stable solid electrolyte interphase (SEI) – a term that had earlier been proposed by Peled16 – on the graphite surface. Shortly after this, the first lithium-ion battery was commercialized by Sony in 1991; at that time, though, still incorporating PC as electrolyte solvent and a coke anode.1 The subsequent quest for suitable electrolyte compositions based on EC, which were compatible also with 4 V-cathodes, while providing suitable ionic conductivity and electrode wettability, led to the development of mixtures comprising EC and dimethyl carbonate (DMC), as reported by Guyomard and Tarascon in 1993.17 As a result, starting from 1994, almost all commercial LIBs were (and still are) based on graphite as active material for the negative electrode.18 Since then, the performance of graphite anodes – especially the first cycle coulombic efficiency, but also the reversible capacity and cycling stability – was continuously improved, for instance, via the identification of suitable film-forming electrolyte additives and coatings, as also discussed herein in the following. Further progress in the early 2000s included the development of aqueous and sustainable electrode processing technologies, replacing the toxic and harmful polyvinylidene fluoride (PVdF) and N-methyl-2-pyrrolidone (NMP) as binder and solvent, respectively. Latest research and development efforts focused especially on the increase in energy and power density by incorporating silicon (oxide) as secondary active material.
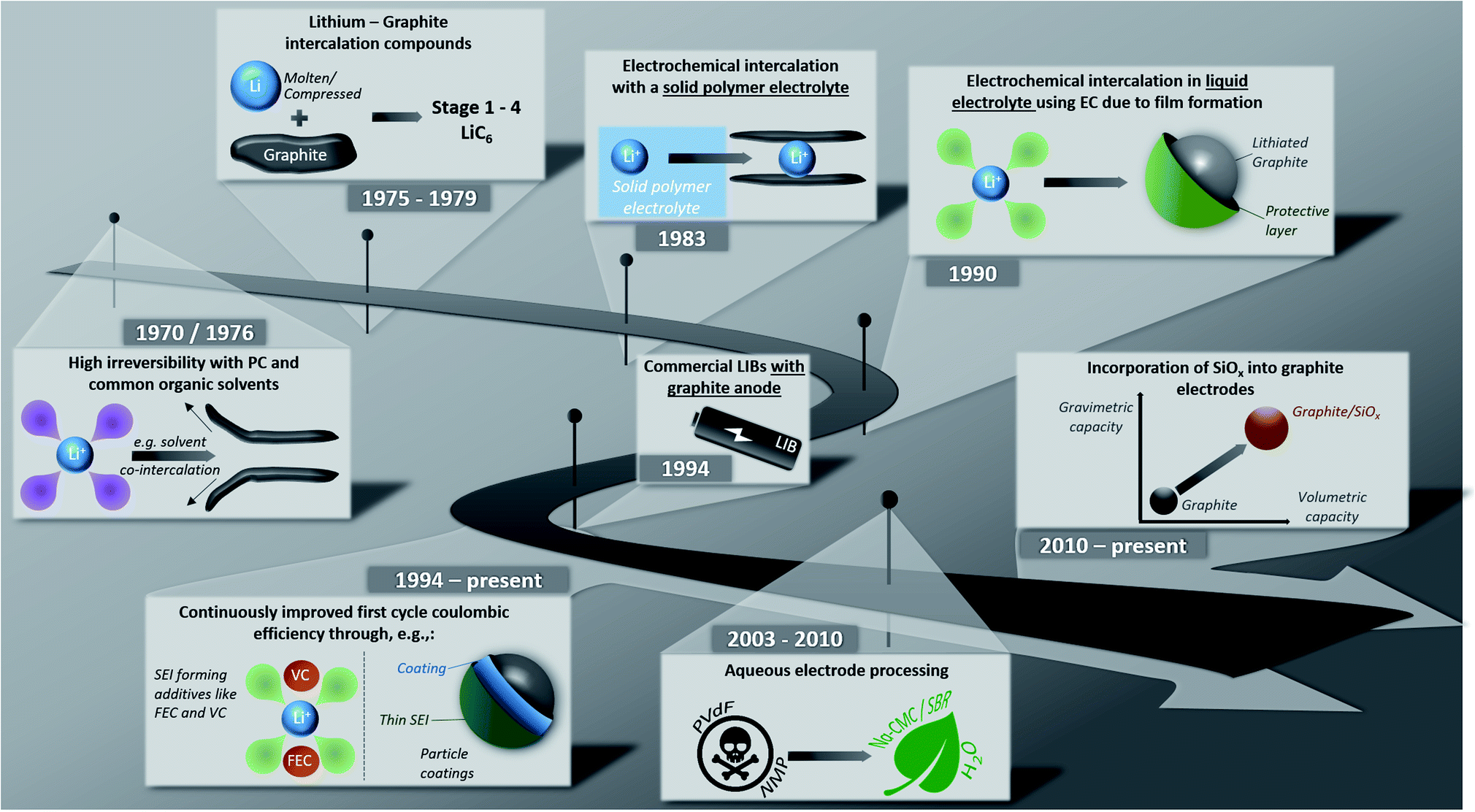 | ||
| Fig. 1 Illustrative summary of major milestones towards and upon the development of graphite negative electrodes for lithium-ion batteries. | ||
Remarkably, despite extensive research efforts on alternative anode materials,19–25 graphite is still the dominant anode material in commercial LIBs. Even more remarkably in this regard maybe is the fact that there have been several review articles published in recent years on these alternatives, including alloying-, conversion-, and conversion/alloying-type anodes,19–27 but – to the best of our knowledge – no comparable review on graphite anodes, apart from a few book chapters dealing with this subject.8,28–31
With this review article we are attempting to fill this gap and provide a comprehensive overview on the recent development and anticipated progress, starting from some rather fundamental considerations concerning the de-/lithiation mechanism. Subsequently, we briefly summarize the importance of the formation of a suitable SEI and its impact on the reversible lithium de-/intercalation – as far as it is important for the further reading – and highlight the great advantages of graphite as lithium-ion host structure, while pointing out also the remaining challenges – especially when it comes to the first cycle irreversibility and rate capability. After these rather fundamental issues, we discuss also very applied considerations, including the potential recycling and the benefits and drawbacks of using either synthetic or natural graphite – both aspects being highly relevant with respect to the potential criticality of natural graphite32 and the rapid increase in (H)EV sales.7 Finally, we review the most recent development for commercial LIB anodes, the incorporation of silicon oxide, silicon–metal alloys, or elemental silicon, with a particular focus on those systems, which are or will presumably be of practical relevance within the near- to mid-term future and various cost-efficient approaches to overcome the associated challenges. Covering all these aspects, we may anticipate that this review article will serve as introductory starting point for researchers newly entering the field of LIB research and development, while potentially providing some insightful information and serving as reference work also for experienced scientists and engineers.
2. Fundamentals – the lithium storage mechanism
2.1 General suitability as host material
Solid carbon materials essentially exist in two hybridization states, i.e., sp3 (diamond) or sp2 (e.g., graphite). In case of graphite, the sp2 hybridized graphene layers are linked by rather weak van der Waals forces and π–π interactions of the delocalized electron orbitals.33 These layers can be stacked either in the thermodynamically more stable ABAB sequence with hexagonal symmetry or in the less stable ABCABC sequence with rhombohedral symmetry.34–36 Bulk graphite commonly comprises both phases (Fig. 2a), although the rhombohedral phase accounts usually for less than 30% as a result of its inferior thermodynamic stability.37 As a consequence of this layered structure, graphite particles are usually characterized by a flake-like particle morphology with two different kinds of surfaces – basal planes and edge planes, the latter also referred to as prismatic planes, as schematically illustrated in Fig. 2b and indicated for an SEM (scanning electron microscopy) micrograph in Fig. 2c. The prismatic planes can be further divided into the so-called zig-zag surfaces and arm-chair surfaces (Fig. 2d), depending on the corresponding stacking sequence. Generally, the prismatic planes show a higher surface reactivity than the basal planes due to their higher surface energy.38 As a matter of fact, the 2D layered structure does not only lead to an anisotropy concerning surface energy, but moreover has a dramatic effect on its electronic, physicochemical, and mechanical properties in general. For instance, the electronic and thermal conductivity are very high (about 2.26 × 104 Ω−1 cm−1 (ref. 39) and 140 to 500 W mK−1 (ref. 40), respectively) along the basal planes (due to the metallic character of the in-plane bonding), but rather poor perpendicular to them (i.e., 5.9 Ω−1 cm−1 (ref. 39) and 3 to 10 W mK−1 (ref. 40), respectively).33 The rather weak van der Waals forces between the graphene layers, however, enable the intercalation (i.e., insertion into 2D layered structures) of ionic and molecular species across the prismatic surfaces, as they allow for an expansion of the interlayer distance, eventually accompanied by a re-stacking of the graphene layers.41 The general concept of such graphite intercalation compounds (GICs) has been established as early as 1840.42 In principle, there are two different kinds of GICs, namely covalent GICs (e.g., graphite oxide43 and graphite fluoride44) and ionic GICs such as alkali-metal GICs.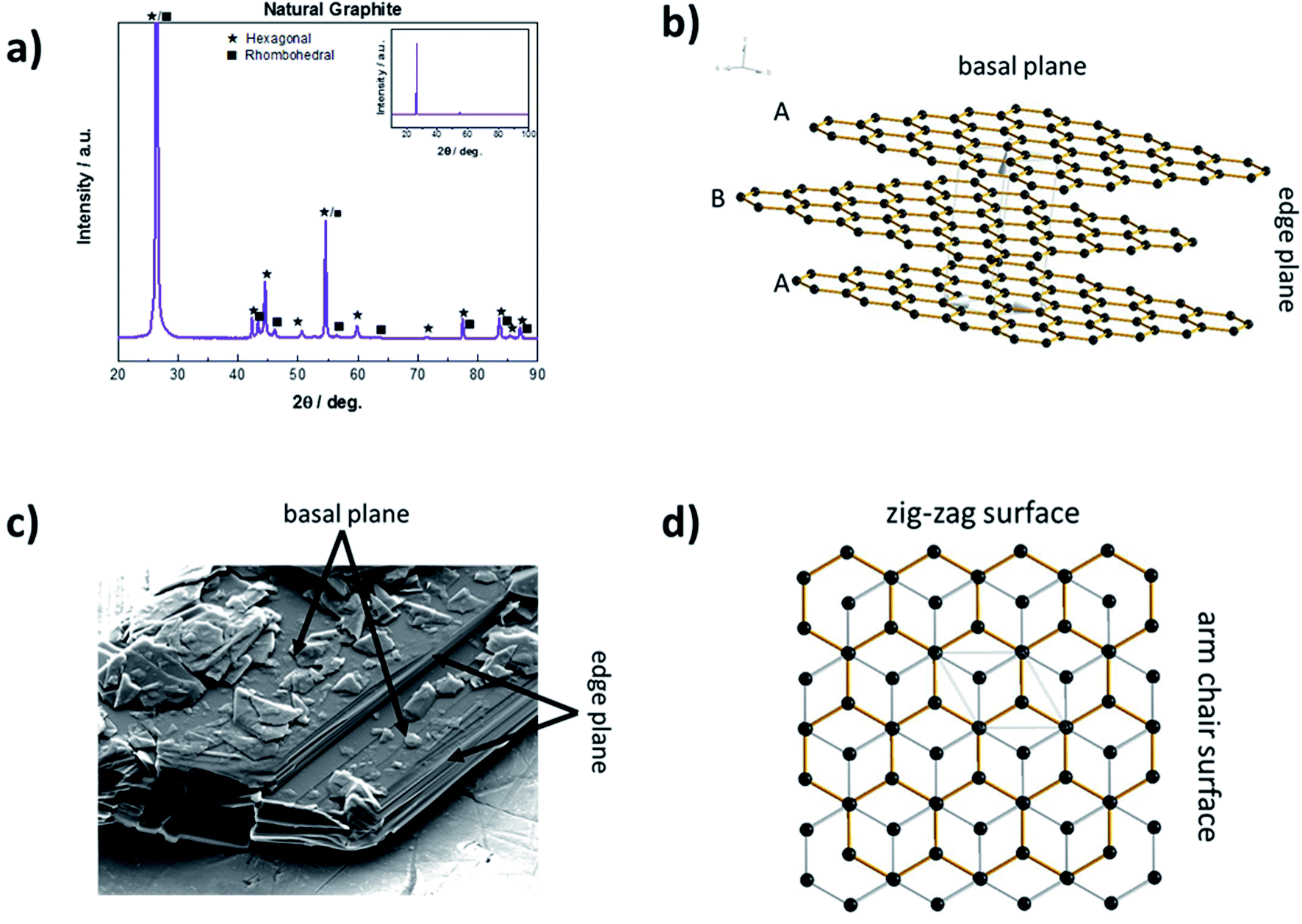 | ||
| Fig. 2 (a) Representative XRD (X-ray diffraction) pattern for natural graphite, indicating the presence of hexagonal and rhombohedral graphite structures, with a relatively lower fraction of the latter. (b) Schematic illustration of the layered structure and the resulting presence of basal and edge planes. (c) Exemplary SEM micrograph, indicating the basal and edge planes for a graphite particle. (d) Schematic illustration of the layered graphite structure in bird's eye view (i.e., along the c axis), indicating the difference between zig-zag and arm chair surfaces. | ||
2.2 The structural changes upon intercalation via ‘staging’
The intercalation of secondary species like ions or molecules (generally referred to as intercalants) into the graphite host structure reveals a characteristic concentration-dependent feature, the so-called staging mechanism, as developed for the first time by Rüdorff and Hofmann in 1938.45,46 According to this mechanism each ‘nth stage compound’ is characterized by a periodic sequence of intercalant layers, i.e., a sequence of layers incorporating the intercalated species, separated by layers not incorporating any intercalated species, for which ‘n’ defines the number of graphene layers separating two intercalate layers (Fig. 3, upper path). Consequently, as the concentration of the intercalant in the graphite host structure increases, the number of empty layers (i.e., ‘n-1’) decreases. Thermodynamically, this staging phenomenon is related to the interplay of (i) the energy required to expand the gap between two graphene layers and (ii) the repulsive interaction between intercalant layers.47 A substantial limitation of this model, however, remains to be the missing ability to explain the transition from Stage 3 to Stage 2, for instance, when assuming that the layers span the entire graphite crystal, as also discussed by Rüdorff and Hofmann in their seminal work.45,46 In fact, such a transition would require the deintercalation of a complete layer and the re-intercalation of the intercalants into a neighboring layer, as the diffusion across the basal planes has been discarded. In an attempt to overcome this limitation, Daumas and Herold postulated a modified model,48 for which for all stages with n > 1 the graphene layers are flexible and deforming around domains (or as initially referred to ‘islands’) of the intercalated species (Fig. 3, lower path). These domains are rather small compared to the crystallite size and stacked according to the initial model by Rüdorff and Hofmann. Thus, the ordering according to the earlier mentioned staging is maintained locally, but not in such rigid fashion. For the transition between, e.g., Stage 3 and Stage 2, the intercalated species would simply need to diffuse within the same layer (or via the “sliding motion” of the intercalant islands49), forming the next lower or higher stage upon intercalation or deintercalation, respectively. While several studies provided experimental evidence for such a “domain-dominated” structure for GICs according to the model of Daumas and Herold – based on, e.g., simulations,50 high-resolution electron microscopy,51 scanning ion microprobe,52 or a combination of optical microscopy and Raman spectroscopy53 – the staging mechanism, i.e., the transition from one stage to another remains to be fully elucidated and more complex than initially proposed.53 A very recent study, combining optical microscopy, scanning transmission electron microscopy (STEM), and voltammetry indicated that the de-/intercalation transport is substantially affected by defects in the crystal structure, while the expectations of both models may be valid only in average for the bulk sample.54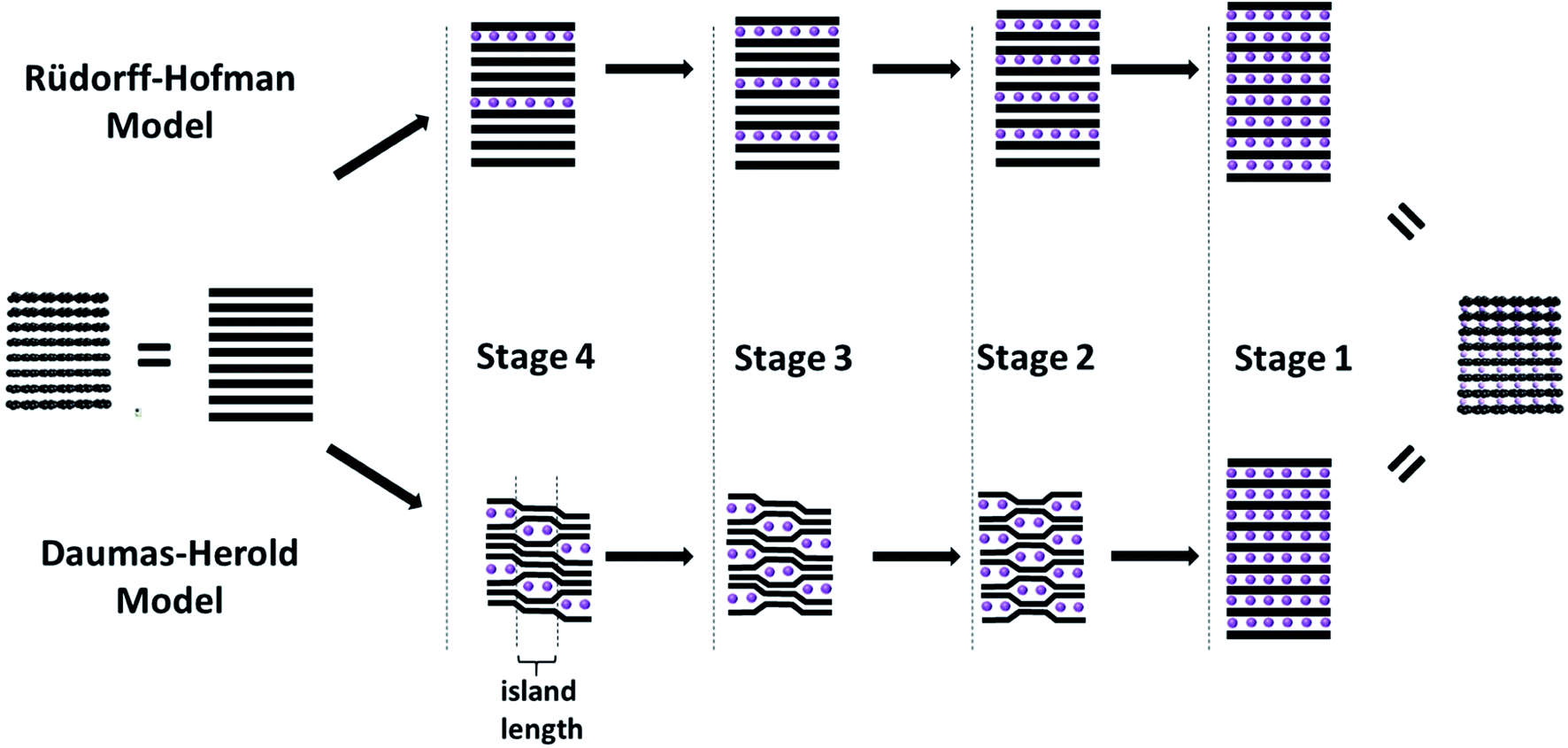 | ||
| Fig. 3 Schematic illustration of the intercalation mechanism of secondary species (i.e., the intercalant; purple spheres) into the layered graphite structure (illustrated by the black lines) according to the model of Rüdorff and Hofmann (upper path) and Daumas and Herold (lower path; figure partially redrawn from ref. 69). | ||
2.3 The intercalation of Li+ for secondary lithium-ion battery anodes
While the previous considerations are applicable to any potential intercalant, the greatest commercial attention has certainly been on the application of graphite as host structure for the reversible intercalation of lithium cations, i.e., its employment as active material for the negative electrode of lithium-ion batteries (LIBs), as introduced by Yazami and Touzain in 1983.14 The de-/intercalation process, however, essentially follows the general mechanism discussed above with Stage 1 as the final stage for the lithium ion intercalation, i.e., an overall stoichiometry of LiC6 as the highest possible Li content at ambient conditions, accounting for a theoretical specific and volumetric capacity of 372 mA h g−1 and 850 mA h cm−3, respectively.8 To accommodate such a high lithium content, the single graphene layers slightly slide with respect to each other, resulting in a symmetry change from an ABABA stacking in pristine graphite to an AIAIA stacking in the fully lithiated state (LiC6; with I being the intercalant layer).55 Simultaneously, the interlayer distance between the graphene layers is moderately increasing by around 10.4%, i.e., from 3.35 Å for lithium-free graphite to 3.70 Å for LiC6.56 The detailed de-/lithiation steps, however, slightly vary with respect to the general staging mechanism (Fig. 4). In fact, for increasing lithium contents, the consecutive occurrence of the Stages n = 1L, 4, 3, 2L, and 2 (before eventually reaching Stage 1) has been observed, for which the additional letter ‘L’ indicates that the lithium ions are not perfectly ordered within the layers, but organized in a rather liquid-like manner.57 The first stage, Stage 1L, represents a random, solid-solution-type lithium intercalation throughout the graphite particle, i.e., in every interslab of the graphite lattice. Subsequently, a first-order phase transition occurs from Stage 1L to Stage 4, followed by the transition to Stage 3 along a sloped decrease in potential. Despite this indication for the absence of a first-order transition, the detailed mechanism is still not fully clarified and might, indeed, be rather complex.57,58 Similarly, the following transition from Stage 3 to Stage 2L, which appearance is temperature-dependent57 and for which the definite structure is still discussed,57,59–61 remains to be completely understood. The next transition from Stage 2L to Stage 2 includes an increasing lithium content within the same intercalant layers (i.e., every second in the graphite lattice), accompanied by an enhanced in-plane ordering.57,59,60 The final (first-order) transition to Stage 1, leading to the formation of LiC6 and providing the largest capacity fraction, is probably the best understood one.62 Generally – as also depicted in Fig. 4 – the occurrence of these phase transitions can be well monitored electrochemically by the appearance of a sequence of galvanostatic potential plateaus57 or redox peak couples when performing cyclic voltammetry,63 while a series of complementary techniques, including in situ XRD,57,64–66in situ neutron diffraction,58,67,68 or in situ Raman spectroscopy,69 has been used to provide additional insights. Nonetheless, the detailed de-/lithiation mechanism and, particularly, the transition between the elevated stages remains to be fully elucidated and is, thus, still under debate.58,70–72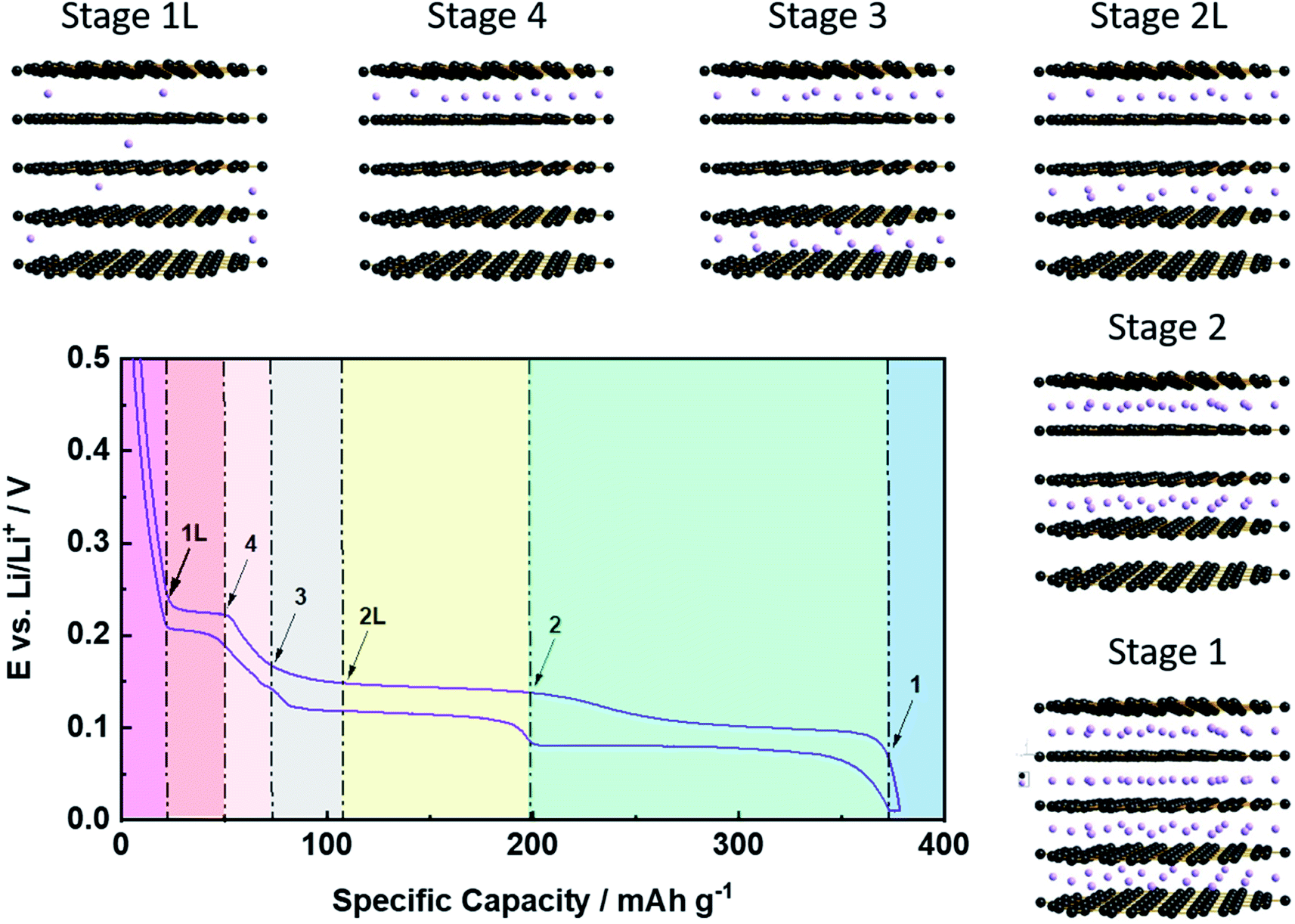 | ||
| Fig. 4 Illustration of the staging mechanism for the electrochemical de-/intercalation of lithium cations (purple spheres) into graphite. The given representative potential profile refers to the 2nd dis-/charge cycle in half-cell configuration, i.e., vs. metallic lithium as counter electrode. | ||
2.4 The solid electrolyte interphase (SEI) as key for the reversible Li+ de-/intercalation
The key for the present and ongoing success of graphite as state-of-the-art lithium-ion anode, beside the potential to reversibly host a large amount of lithium cations, in fact, has been the identification of a suitable electrolyte composition in order to overcome an intrinsic challenge associated with the use of graphite as active material in a LIB: the continuous exfoliation of graphene layers as a result of the solvent co-intercalation and the reductive electrolyte decomposition at the electrode/electrolyte interface, as common organic liquid electrolytes are not sufficiently stable towards such low potentials.18,26,73,74 We may elaborate on this in more detail in the following. The electrochemical stability window (ESW) of an electrolyte is related to its LUMO (lowest unoccupied molecular orbital) and HOMO (highest occupied molecular orbital) energy. Hence, if the electrochemical potential μ of the relevant active material for the anode (μa) and cathode (μc) is lower than the LUMO or higher than the HOMO energy of the electrolyte, respectively, the electrolyte is theoretically stable for the operation of such a cell (from a thermodynamic point of view).74 The intercalation of lithium cations into graphite, however, occurs between 0.25 and 0.01 V vs. Li/Li+, which is well below the electrochemical stability of, e.g., organic carbonate-based electrolytes, as these commonly decompose at about 0.8 V vs. Li/Li+.18,26,73,74 While such a low operational potential is highly advantageous with regard to the energy density of the full-cell – as will be discussed in the subsequent chapter – the consequence is that the electrolyte is continuously decomposed at the (lithiated) graphite surface. In 1990, however, Dahn and co-workers15 showed that Li+ can be reversibly intercalated into graphite in liquid electrolytes comprising EC as (co-)solvent rather than the earlier used propylene carbonate (PC),75 for which massive graphite exfoliation had been observed as a result of the co-intercalation of the Li+ cation along with its solvation shell.76 Differently, when using a sufficient amount of EC as (co-)solvent, the electrolyte decomposition occurring during the initial charge (i.e., lithium intercalation) – commonly continuing for a few cycles – leads to the formation of a solid layer of the corresponding organic and inorganic decomposition products on the graphite particle surface, accompanied by an irreversible capacity loss, as illustrated by the comparison of the 1st and 5th dis-/charge cycle in Fig. 5. This layer prevents the direct (electronic) contact of the graphite particles with the electrolyte and, thus, kinetically suppresses the continuous electrolyte decomposition kinetically16,77 and inhibits the solvent co-intercalation,78 while allowing for lithium cation conduction. Accordingly, it has been termed solid electrolyte interphase (SEI),16,79 as it is an additional (in situ formed) phase that is acting essentially like a solid electrolyte at the interface between the graphite active material and the liquid electrolyte. As this layer is essential for the reversible operation of a lithium-ion cell and greatly determines its properties, including the rate capability, i.e., the power density, and the initial charge loss, i.e., the energy density, there have been tremendous efforts undertaken to unveil its chemistry, functionality, and formation mechanism. These studies have been comprehensively reviewed in several articles18,76,80–83 and we will, hence, just provide a brief overview herein for the sake of completeness. It has been proposed that the SEI has a mosaic-like structure, consisting of multiple inorganic and organic grains and layers.84 Close to the graphite particle, a dense layer of inorganic compounds like Li2O, Li2CO3, and LiF is formed, i.e., compounds which are thermodynamically stable against lithium. This inorganic “layer” is covered by (rather) organic decomposition products such as semicarbonates and polyolefins.84–86 Such general composition of the SEI has been confirmed by a great variety of complementary experimental techniques, including spectroscopic85,87–90 and microscopic91–95 studies as well as theoretical investigations.96–99 In fact, the formation of a thin, but dense internal layer of inorganic compounds like Li2CO3 and LiF rather than organic metastable compounds like ROLi and ROCO2Li (with R being a low-molecular alkyl group) is considered to limit the initial charge loss by rapidly suppressing electrolyte decomposition.77,81,100 The ion conduction mechanism in these two different phases, however, appears to be different, as proposed by Shi et al.101 employing DFT calculations for their studies of the Li+ transport in the SEI. According to their work, the Li+ diffusion is fast in the outer organic layer and following a relatively slower repetitive “knock-of” mechanism in the dense inner inorganic layer, largely consisting of (nanocrystalline) Li2CO3. Using highly oriented pyrolytic graphite (HOPG) as a model, it was shown that the composition and the thickness of the SEI, however, at the edge planes and at the basal planes differ significantly.89,90,102 On the basal planes, the fraction of organic species is higher, while it mainly comprises inorganic compounds, resulting from the lithium salt decomposition, on the edge planes. Moreover, TOF-SIMS (time-of-flight secondary ion mass spectrometry) studies revealed that the basal SEI with a common thickness of about 7 nm is around three to five times thinner than the SEI on the edge plane with around 35 nm.103In situ AFM (atomic force microscopy) studies indicated, slightly differently, a smaller difference in the thickness of the basal and edge SEI. Nonetheless, it was also shown that the thickness of the SEI on the basal plane continues to grow upon cycling, whereas the thickness of the SEI at the edge plane does not change significantly after the first cycle.91,104 Some studies suggested that these differences are related to the fact that the Li+ ions are desolvated at the edge planes before being intercalated in-between the graphene layers. Accordingly, the anions of the Li-salt decompose at the edges into inorganic species, while there is essentially no lithium intercalation across the basal planes in absence of defects.86,105 In addition and/or alternatively, the different electrochemical reaction and charge transfer kinetics at the edge planes and the basal planes may play a role for the different behavior towards the reductive electrolyte decomposition.86,106,107 In fact, these differences in SEI composition and morphology somehow also reflect the different functionality, as the SEI on the basal planes does not have to serve as Li+ conductor, if there is no Li+ intercalation across these planes. Strictly speaking, it is thus not an SEI, though it still prevents the continuous electrolyte decomposition at these surfaces.76 While the precise composition and formation mechanism as well as the lithium transport through the SEI and across the interfaces with the electrolyte and the graphite particles still remains to be fully elucidated, also the important difference between EC and PC is not completely understood, yet. Chemically speaking, the two molecules differ only by an additional methyl group in PC, but only the presence of EC allows for the formation of a stable SEI that suppresses solvent co-intercalation and graphite exfoliation, as mentioned earlier. Based on the consideration that the Li(solvent)x complexes are initially intercalated at the edge planes, followed the subsequent decomposition of the ternary Li(solvent)x-GICs,78 Shkrob et al.108,109 investigated the radical intermediates formed during the reduction of organic carbonate-based electrolytes. They found that the one-electron reduction of EC generates secondary radical intermediates, which are forming a branched 3D polymer network. In contrast, the formation of ternary radicals in PC leads either to the formation of substituted olefins via disproportionation or to linear polymer chains via anionic polymerization. These different decomposition products were considered the reason for the successful graphite passivation in case of EC and the continuous electrolyte decomposition and graphite exfoliation in case of PC.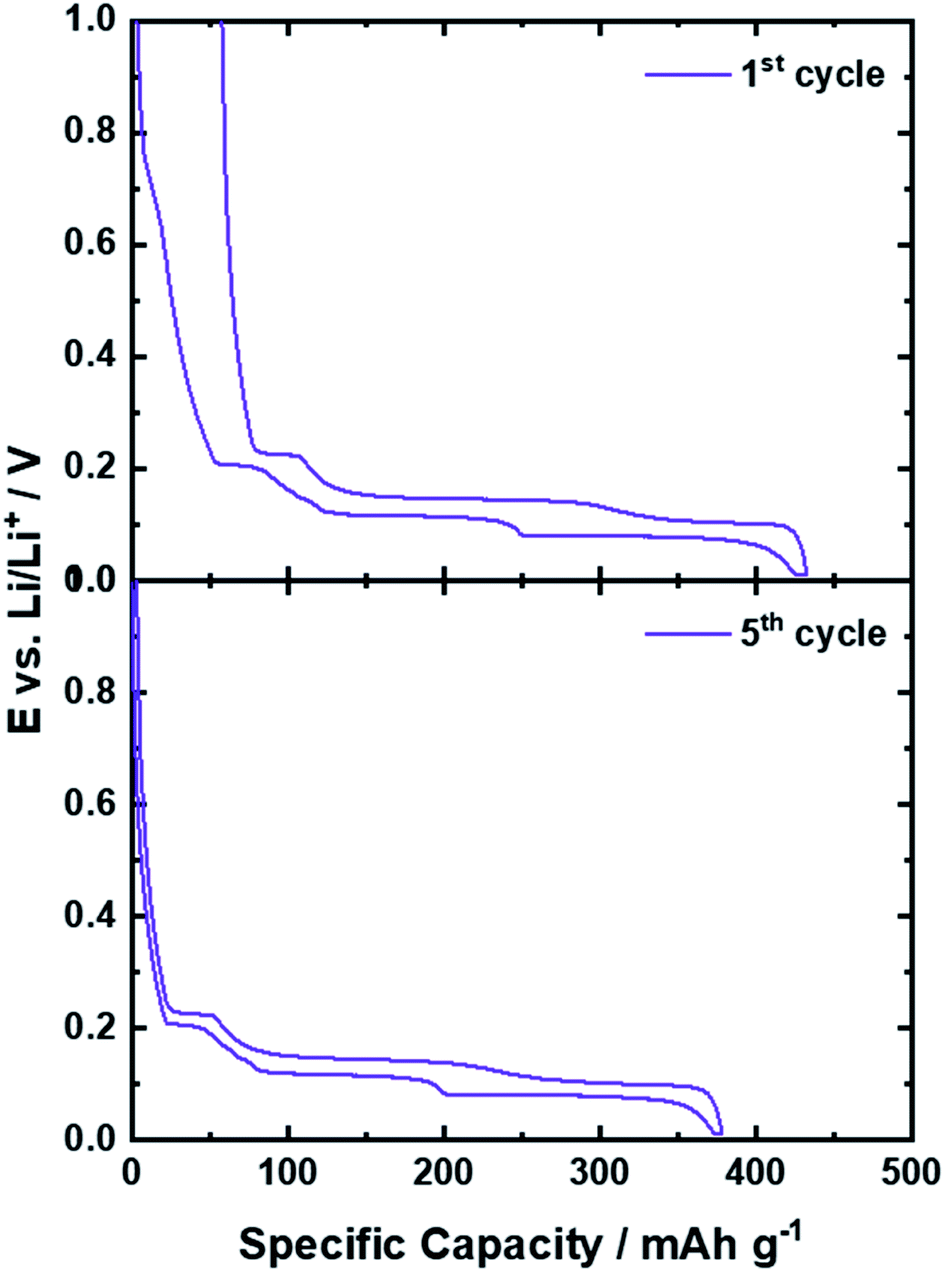 | ||
| Fig. 5 Comparison of the potential profile of graphite half-cells for the 1st (upper panel) and 5th (lower panel) dis-/charge cycle. | ||
Xing et al.110 proposed recently a different explanation based on the different solvation energy of the two solvents. For EC, the solvation energy is significantly smaller compared to PF6−, which leads to the co-intercalation of Li+ along with a large fraction of PF6− and the latter substantially contributes to the SEI. In contrast, the solvation energy of PC is only slightly smaller than for PF6−. Hence, the fraction of co-intercalated PF6− is smaller and more solvent molecules are co-intercalated, resulting in a rather organic decomposition layer that does not effectively suppress further degradation reactions at the electrode/electrolyte interface.
2.5 The low de-/lithiation potential and its advantages
The theoretical specific capacity of graphite is 372 mA h g−1, higher than the capacity of most common cathode materials, but lower than the capacity of conversion- or alloying-type anodes as the most promising alternatives.22 Nevertheless, an aspect that is frequently overlooked is the final energy density at the full-cell level, which depends not only on the capacity, but also on the cell voltage. In this regard, graphite has a clear advantage compared to these alternatives, since it benefits from the lowest average de-/lithiation potential (i.e., 0.2 V vs. Li/Li+) except metallic lithium. Nonetheless, the simplicity of determining the specific capacity compared to the rather complex determination of the energy density – especially if determined experimentally in suitable full-cells – render this common procedure more favorable. In fact, the evaluation of the gravimetric and volumetric energy density in full-cells is rather challenging, since one has to consider the balancing between the anode and the cathode, the dis-/charge rate and its impact on the achievable capacity of both electrodes, as well as the volume changes upon de-/lithiation – even if only considering the energy density on the basis of the comprised active materials and, thus, ignoring the impact of the inactive cell components, i.e., the binder, conductive additives, the current collector, the packaging, or the amount of electrolyte. In a recent study, Andre et al.6 compared different anode materials and their effect on the practical full-cell energy densities in full-cells. According to this study, most alternative anode materials would provide lower energy densities than graphite, which explains why it is still used in most commercial lithium-ion batteries. Another reason is the frequently neglected energy efficiency, i.e., the ratio of energy that is stored relative to the amount of energy that is released upon discharge, which is directly linked to the difference between the charge and discharge potential (if considering a constant coulombic efficiency). For graphite, this difference, also referred to as voltage hysteresis, is relatively small, rendering it additionally favorable compared to most alternatives investigated so far,111 although some progress has been reported very recently for (pre-lithiated) conversion/alloying-type materials when limiting the de-/lithiation to a rather narrow potential range.1123. Remaining (intrinsic) challenges
3.1 Rate capability
One of the major challenges for graphite anodes in LIBs is the limited rate capability, especially for the lithiation process, i.e., the charging of the full-cell, and the associated risk of lithium metal plating on the electrode surface, potentially resulting in a short-circuiting of the cell or at least rapid ageing and accelerated cell fading.113,114 While it has been reported that the rate performance is directly related to the ambient temperature,115 the particle size and morphology116,117 as well as the electrode engineering,118 other studies revealed that the Li+ diffusion in-between the graphene sheets is, in principle, extremely fast. Kühne et al.,119 for instance, investigated the Li+ diffusion in bilayer graphene via in situ time-dependent Hall voltage measurements and found a diffusion coefficient as high as 7 × 10−5 cm2 s−1. Similarly, Persson et al.120 measured the diffusion of lithium ions in HOPG and found a high lithium-ion diffusivity of 10−7 to 10−6 cm2 s−1 parallel to the graphene planes and a sluggish ion transport (10−11 cm2 s−1) perpendicular to them. Based on these findings it would be possible to de-/lithiate graphite with a typical crystalline domain size of 45 nm in less than 0.2 ms. Nevertheless, this is in contrast to the experimental observation that for standard electrodes the lithiation process is limited to charge rates of maximum 1C, while it is possible to delithiate such electrodes at discharge rates as high as 10C.118 According to the study by Persson et al.,120 the lithium-ion diffusion coefficient was calculated to be ∼10−7 cm2 s−1 for the Stages 1 and 2 in presence of many vacancies, but decreased to ∼5 × 10−9 cm2 s−1 for both stages in the fully lithiated state. A similar trend was also observed by Levi et al.,121 who determined the Li+ diffusion coefficient of graphite at elevated temperatures by the potentiostatic intermittent titration technique (PITT). Additionally, they found an increased lithium-ion diffusion coefficient (by a factor of 10) for the liquid-like stages compared to fully lithiated planes. Recently, Guo et al.122 reported an all-optical investigation of the lithium intercalation in a single-crystal graphite disc-electrode. They found that the liquid-like stages are homogeneously formed across the disc. This indicates that the Li+ diffusion from the edge to the center of the particle is fast compared to the Li+ “injection” at the edge. However, the Stage 2 phase nucleates at the boundaries of the disc electrode, followed by the nucleation of Stage 1 almost immediately after Stage 2 forms. This indicates a slower Li+ transport through Stage 2 and Stage 1 compared to the diffusion in Stage 1L. Furthermore, the system must be out of equilibrium when three phases are observed, since in equilibrium maximum two phases exist simultaneously in the phase diagram for the lithiation of graphite.57 Heß and Novák62 studied the kinetic performance of highly crystalline graphite particles in thin layer electrodes of ca. 1 μm thickness. They found that the Li+ diffusion coefficient in the liquid-like stages substantially differs from the densely lithiated stages. During lithiation, the transition from Stage 2L to Stage 2 cannot deliver its full capacity at high current rates. Consequently, the overpotential increases due to diffusion limitations until the next stage, i.e., Stage 1 nucleates at the edge planes of the graphite particle (Fig. 6). Since the different stages propagate into the particle like the annuli of a tree, they named this phenomenon the “shrinking annuli model”. Upon delithiation, the diffusion-limited transition from Stage 1 to Stage 2 takes place first. Once the overpotential is high enough, Stage 2L nucleates at the edge of the particle. Due to the higher diffusion coefficient for this stage, the phase boundary between Stage 2 and Stage 2L propagates rapidly into the particle. As a result, according to this model, the liquid stages with a high diffusion coefficient can balance the slow lithium-ion diffusion of the densely lithiated stages only during the delithiation of graphite. Beside the limited maximum overpotential for the lithiation process (due to the generally low lithiation potential), the shrinking annuli mechanism thereby can explain the asymmetry of the rate capability for the charge and discharge process. Recently, the shrinking annuli mechanism was confirmed by Bauer et al.,123 who found that the dilatation behavior of graphite varies with the dis-/charge rate applied and that this deviation is due to the formation of over-next GICs, especially at elevated states of charge (SOCs). Moreover, the relaxation behavior differs for the charge and discharge step. | ||
| Fig. 6 Schematic illustration of the “shrinking annuli model” for a single graphite particle. The golden, red, and purple color indicate the formation of Stage 1, Stage 2, and Stage 2L, respectively. The figure has been reproduced from Heß and Novák62 with permission from Elsevier. | ||
While these studies essentially focused on the Li+ transport within the graphite particle, another limiting factor, in fact, is considered to be the initial Li+ intercalation step. Electrochemical impedance spectroscopy (EIS) measurements of lithium-ion cells at low temperatures showed that the cell impedance is mostly dominated by the charge transfer resistance.124 This finding is in line with the study by Wang et al.125 who investigated the low-temperature performance of graphite and mesocarbon microbeads (MCMBs) by microperturbation and found that at room temperature the phase transitions are the rate limiting step, whereas at −30 °C the resistance of the SEI becomes limiting. In fact, the charge transfer can be separated in different processes: first, the solvated Li+ ions, present in the liquid electrolyte, are desolvated, before, second, entering into the SEI layer and further into the graphite particle. Simultaneously, an e− is injected at the electrode/SEI interface, as described in the following equations:126
| Li+(Solvent)n → Li+ + nSolvent |
| Li+ → Li+SEI |
| xLi+ + xe− + C6 → LixC6 |
To further elucidate the impact of the different contributions, Abe et al.127 investigated the Li+ charge transfer process at the interphase between a HOPG-based electrode and the electrolyte by alternating current (AC) EIS. When a 1 M solution of LiCF3SO3 in 1,2-dimethoxyethane (DME) was used, the solvated Li+ ions can be rather directly intercalated into HOPG, as there is no substantial SEI formation. For this process, they found an activation energy of only 25 kJ mol−1. Differently, the activation energy increases to 53–59 kJ mol−1 when a 1 M solution of LiClO4 in a mixture of EC and diethyl carbonate (DEC) was used, since such electrolyte composition leads to the formation of a relevant SEI and Li+ needs to desolvate first before diffusing through the SEI. Therefore, the authors concluded that the desolvation of Li+ at the SEI surface prior to the intercalation into the graphite particle is the rate limiting step. This conclusion was supported by the finding that the lithium-ion transfer resistance at a model interface, consisting of a lithium-ion conductive ceramic and a liquid electrolyte, correlates with the interaction between the lithium ions and the nature of the solvent in the liquid electrolyte.128 Nevertheless, since the desolvation of Li+ ions occurs at both the anode and the cathode, one would expect similar activation energies for both electrodes.126 Accordingly, Jow et al.129 investigated two different full-cell chemistries by EIS, i.e., graphite//LiFePO4 (LFP) and graphite//LiNi0.80Co0.15Al0.05O2 (NCA), with an electrolyte consisting of 1 M LiPF6 in a mixture of EC, dimethyl carbonate (DMC), and methyl butyrate (MB) as well as vinylene carbonate (VC) as additive. The activation energies for the graphite/electrolyte, NCA/electrolyte and LFP/electrolyte interfacial charge transfer were calculated to be 67 kJ mol−1, 50 kJ mol−1, and 33 kJ mol−1, respectively. The authors concluded that the large differences in activation energy cannot be explained by the desolvation as the predominant step for limiting the kinetics only. Instead the results suggest, that the different nature of the corresponding electrode/electrolyte interfaces is influencing the Li+ charge transfer kinetics. This was supported by the finding that the activation energy is greatly influenced by incorporating different additives into the electrolyte,130 which would theoretically not have a significant effect on the solvation energy of the electrolyte. Moreover, the pre-formation of the SEI on graphite leads to a great variation of the activation energy for the Li+ transfer, which further supports the conclusion that the Li+ ion transfer kinetics are substantially influenced by the SEI composition.131
In sum, the rate capability of graphite, even though not fully understood yet, appears to be the complex interplay of all potential contributions, i.e., the particle size and morphology, the electrode architecture, the phase transitions occurring, the chemistry and composition of the SEI (and, accordingly, the electrolyte composition), the desolvation prior to entering the SEI layer, and the ambient temperature.
3.2 First cycle irreversible capacity
Apart from the rather poor rate capability, another drawback of graphite anodes in LIBs is the first cycle irreversible capacity (Cirr) due to the reductive electrolyte decomposition and, thus, the consumption of Li+ as the charge carrier. One strategy to reduce the Cirr, that has been pursued since the commercialization of graphite-based LIBs, is the modification of the graphite particles' surface (Fig. 7a). Such surface modification can employ the treatment of the graphite particles by either using (oxidative) solutions, ceramic materials, or gases, commonly followed by a heat treatment. As a result, both basal and edge planes are chemically altered to enhance the electrochemical performance. Another approach relies on the introduction of electrolyte additives that kinetically favor the SEI formation in a more efficient way and, thus, allow for increasing the initial coulombic efficiency (ICE). These two different approaches will be discussed in more detail in the following two paragraphs.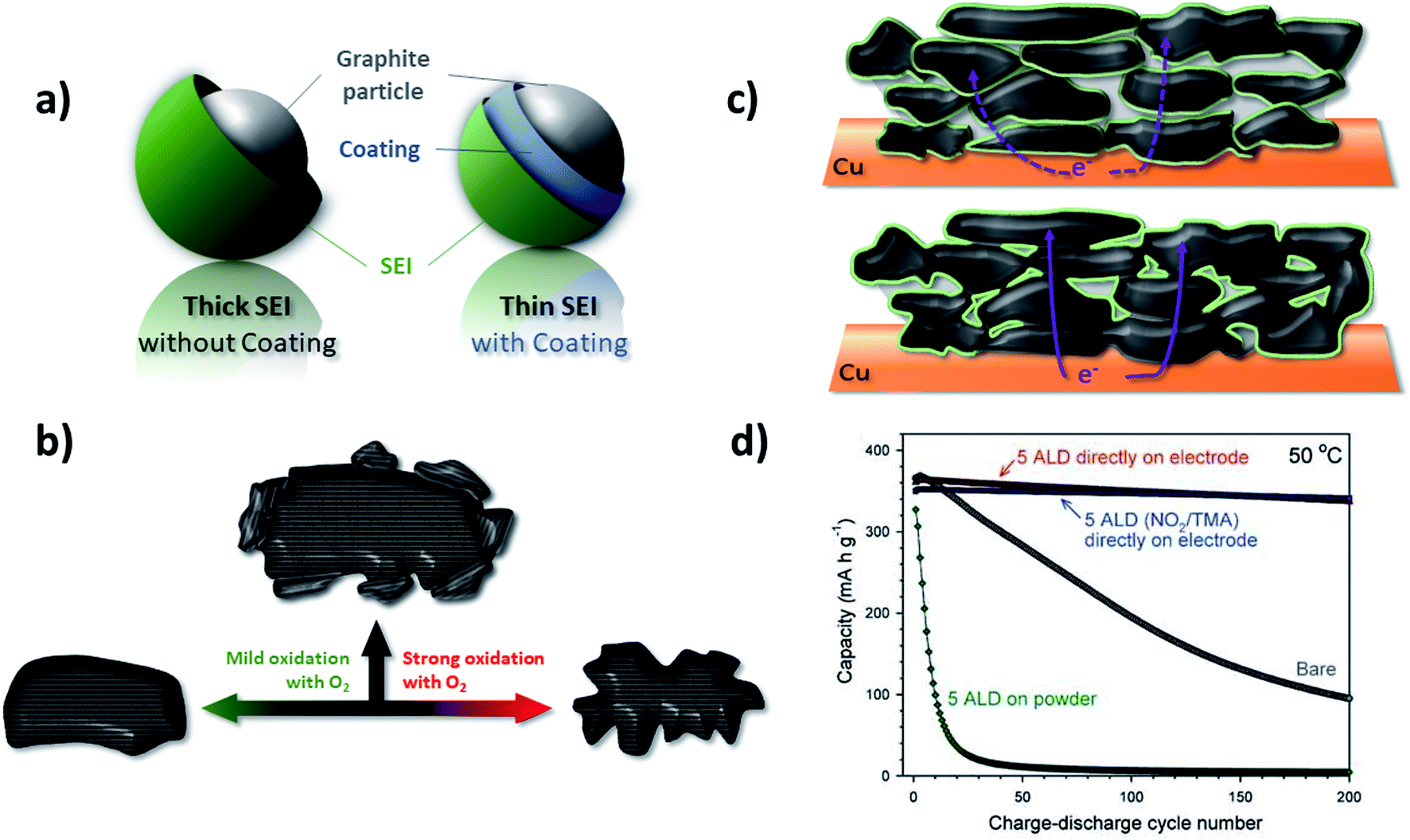 | ||
| Fig. 7 Different approaches to optimize the electrochemical performance of graphite by surface modification: (a) the general target of achieving a more efficient SEI layer formation and the consequent realization of thinner SEI films. (b) Schematic illustration of the mild oxidation proposed by Peled et al.,132 resulting in a “smoothening and cleaning” of the particle surface, in contrast to the application of a strong oxidation, leading to particle degradation (figure redrawn from ref. 360). (c) Illustration of the difference between powder coating (upper panel) and electrode coating (lower panel) approaches; the latter one resulting in the formation of less electronically insulating surfaces and interfaces in case of, e.g., Al2O3 coatings (figure redrawn from ref. 145). (d) Comparison of the galvanostatic cycling of bare graphite electrodes and those coated with Al2O3 at the powder and electrode level (figure reproduced from Jung et al.145 with permission from Wiley). | ||
Apart from these pioneering works, the surface modifications investigated can be mainly categorized into inorganic, organic, and carbonaceous coatings. Yu et al.,134,135 for instance, reported an improved electrochemical performance in PC-based electrolytes when nano-sized Ni-composite particles were deposited on the graphite surface. The thus limited direct contact with the electrolyte reduced the solvent co-intercalation, mitigated PC reduction, and decreased the charge transfer resistance. For the optimum ratio of such coating (ca. 10 wt%), the ICE and reversible discharge capacity were improved from 59% to 84% and by about 30–40 mA h g−1, respectively. Besides Ni, the incorporation of a series of other metals such as Ag, Au, Al, In, Bi, Pd, Pb, Sn, Zn, and Cu has been studied as either by forming composites or applying them as coating materials for graphite.136–138 In general, the application of such metal films facilitates the SEI formation and leads to higher reaction kinetics for the lithium de-/intercalation due to an enhanced charge transfer and reduced SEI resistance, originating from the reduced activation energy for the Li+ desolvation and subsequent migration through the SEI towards the intercalation sites.139 This improvement is particularly pronounced at low temperatures, at which a drastic polarization is observed otherwise. When the metal itself is electrochemically active, i.e., able to alloy with lithium, just like for Sn, it was found that the impact on the electrochemical performance is highly dependent on the metal concentration. For instance, in case of rather small amounts of Sn (up to 20 wt%), its incorporation strongly favors the reaction kinetics, hence, contributing to significantly enhanced specific capacities.140 For larger amounts, however, the presence of the alloying metal increases the volume expansion upon lithiation and, as a consequence, reduces the ICE compared to pristine graphite.141 In case the metal does not alloy electrochemically with lithium (and is ideally characterized by a different crystal structure – such as Ni or Cu), such coatings may, moreover, address the issue of lithium metal plating at elevated current densities (and low temperatures – see Section 3.3), thanks to an increase of the lithium metal deposition overpotential.142
In addition to the potential introduction of metals, also metal fluorides and oxides have been investigated in order to reduce the initial irreversible capacity loss. For example, the application of a thin AlF3 layer on graphite demonstrated improved cycling stability and capacity retention; nonetheless, with a slightly negative effect on the ICE.143 Differently, Al2O3, characterized by a suitable bandgap, serves as SEI formation inhibitor and, thus, enables an increased ICE by up to 5%.144 The application of an Al2O3 coating on the readily made electrode rather than on the graphite powder by atomic layer deposition (ALD), thus avoiding the extensive introduction of electronically insulating surfaces and interfaces (Fig. 7c), has an even higher impact on the electrochemical performance (Fig. 7d).145 The authors did not only record a higher ICE and enhanced capacity retention, but moreover such coating allowed for the use of PC as electrolyte solvent.
Similarly to inorganic coatings, also the utilization of polymer coatings, including inter alia polyaniline,146,147 (poly)acrylonitrile,148 polypyrrole,149 polymerized thiophene,150 or nitrophenyl,151 has been reported as an effective strategy to improve the ICE. Compared with the use of inorganic compounds, the implementation of polymers as the coating material exhibits two salient advantages which are high chemical diversity and mechanical flexibility. For example, Li et al.152 demonstrated that the application of a polyether-based coating drastically increased the ICE and enhanced the rate capability if the ether has a phenyl-based terminal group, as this enables a good adhesion to the graphite surface due to the π–π interaction. Furthermore, they were able to strengthen the mechanical stability of the electrode by crosslinking the ether molecules via the additional introduction of an allyl-amine polymer.
The mostly investigated and presently most cost-efficient surface coating material, though, is probably carbon – in fact, not only for graphite, but for essentially every LIB active material.153–159 In one of the very early studies, Yoshio et al.160 investigated the impact of different carbon coating amounts, applied via thermal vapor deposition from toluene as precursor, on the electrochemical performance of natural graphite using different electrolyte compositions based on 1 M LiPF6 in various organic carbonates. They found that in their case a carbon coating content of 17.6 wt% marked the optimum concerning the ICE, resulting in an improvement from 87.2% to 94.5% and from 76.6% to 92.4% for an electrolyte composition of EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC (1:2) and PC:DMC (1:4), respectively. An important drawback of such (rather large amount of) carbonaceous coating, however, is its commonly lower density than graphite, accompanied by significantly inferior contribution to the overall capacity, which leads to significantly reduced gravimetric and volumetric energy densities at the full-cell level. This issue could be overcome by employing spherical graphite particles and the consequent reduction of the required carbon coating content to only 3 wt% while still offering superior electrochemical performance161 and even enhanced thermal stability.162 It should be noted, though, that the application of carbonaceous coatings might also have a negative impact on the irreversible electrolyte decomposition if it leads to an increased surface area of the active material.163 Hence, particular attention has to be paid on the choice of the carbon coating precursor and its content, the graphite particle morphology, and the general processing to achieve an enhanced electrochemical performance regarding the long-term cycling stability, the initial charge loss, and the eventual energy density.
:DMC (1:2) and PC:DMC (1:4), respectively. An important drawback of such (rather large amount of) carbonaceous coating, however, is its commonly lower density than graphite, accompanied by significantly inferior contribution to the overall capacity, which leads to significantly reduced gravimetric and volumetric energy densities at the full-cell level. This issue could be overcome by employing spherical graphite particles and the consequent reduction of the required carbon coating content to only 3 wt% while still offering superior electrochemical performance161 and even enhanced thermal stability.162 It should be noted, though, that the application of carbonaceous coatings might also have a negative impact on the irreversible electrolyte decomposition if it leads to an increased surface area of the active material.163 Hence, particular attention has to be paid on the choice of the carbon coating precursor and its content, the graphite particle morphology, and the general processing to achieve an enhanced electrochemical performance regarding the long-term cycling stability, the initial charge loss, and the eventual energy density.
Generally speaking, electrolyte additives that are improving the ICE are either contributing to a more efficient SEI by being simply electrochemically reduced (e.g., prior to the base electrolyte itself), inducing the reduction of other electrolyte components in a favorable way, or reductively polymerizing – or a combination of these mechanisms. Ideally, the thus formed surface layer inhibits further extensive electrolyte decomposition and, by this, leads to an increased ICE. Classic examples are fluoroethylene carbonate (FEC)166,167 and VC.168–171 The latter has been reported to form a polymeric film on the graphite anode following a ring-opening reaction prior to the decomposition of EC and, thus, stabilizing the electrode/electrolyte interface.169 Moreover, it was reported that the addition of VC allows for effectively deactivating the oxides at defective sites and at the edge planes of (HOP) graphite and that the resulting modified SEI would be extremely thin, i.e., only a few nm.168 Remarkably, the use of VC as electrolyte additive enables even the utilization of PC as electrolyte solvent and successfully suppresses the PC co-intercalation and subsequent graphite exfoliation.171,172 Additionally, several studies reported a beneficial effect of VC also for suppressing the irreversible reactions occurring at the cathode,173–175 rendering it particularly advantageous. Nevertheless, it has been reported as well that the incorporation of VC leads to an increased irreversible capacity at elevated temperatures,176,177 which has been assigned to its rather poor thermal stability.178 As a potential alternative, vinyl ethylene carbonate (VEC) was found to be more stable than VC owing to its electron rich double bond. According to Hu et al.172 the stable passivation layer formed due to the reductive decomposition of VEC on the graphite surface contributed to an improved battery performance and prevented PC co-intercalation into the graphite anode.
A slightly different mechanism was reported for additives such as ethylene sulfite179 and propylene sulfite,180 as their reduced products are adsorbed on the catalytically active sites of graphite, resembling somehow the poisoning of a catalyst and, by this, suppressing the electrolyte decomposition that would be preferably occurring at these sites. Nonetheless, their relatively low stability towards oxidation strictly limits the amount that might be added to the electrolyte in lithium-ion cells.164
In combination these different, but complementary approaches to reduce the first cycle irreversibility have led to coulombic efficiencies as high as 90–96% for commercial graphite anodes, thus providing the benchmark for any alternative negative electrode active material.
3.3 Ageing and the resulting safety concerns
An issue that essentially concerns all battery materials, but is particularly important for graphite as a result of the low de-/lithiation potential close to the plating of metallic lithium, is ageing – induced by both usage (cycling) and storage (calendar ageing).181,182 Generally, ageing processes are very complicated – not least due to the long time frames that have to be considered and the fact that a large variety of different electro-/chemical processes and their interactions have to be considered.182,183 In fact, the ageing mechanisms at the negative and positive electrode significantly differ from each other and while we will focus herein on those occurring at the graphite anode, also the impact from the cathode has to be kept in mind.The cathode-induced ageing that has been most investigated so far is related to the dissolution of transition metal cations from the cathode and their diffusion to and deposition on the anode, as comprehensively reviewed recently by Zhan et al.184 and Li.185 The most prominent example in this regard is the manganese dissolution from spinel-structured LiMn2O4 (ref. 186–192) or LiNi0.5Mn1.5O4 (ref. 112, 193 and 194) due to the formation of soluble Mn2+, resulting from the Jahn–Teller distortion.195,196 As a consequence, the manganese dissolution does not only lead to capacity and power fading at the cathode, but also influences the SEI composition at the anode.184 In fact, an analysis of aged LiMn2O4/graphite cells via EIS revealed that the increased impedance at the graphite anode is largely contributing to the capacity fading of such lithium-ion cells.187 It was reasoned that – due to the low de-/lithiation potential of graphite, well below the reduction potential of Mn2+ – metallic manganese is deposited either via electrochemical197 or chemical192 reduction at the anode surface, causing an increased electrolyte decomposition.198,199 An alternative mechanism was reported by Zhan et al.,200 who showed that the oxidation state of manganese deposited on the surface is 2+ and postulated an ion exchange model in which Mn2+ accumulates in the SEI by an ion-exchange reaction with Li+ and, thus, blocks lithium ion diffusion. Apparently, these two mechanisms do not exclude each other and further research will be needed to fully understand the ongoing ageing phenomena.
Another cathode-induced ageing mechanism for graphite anodes is related to the decomposition of the electrolyte at elevated potentials (and temperatures) at the cathode surface and the migration of soluble decomposition species to the anode, where they can further react – also referred to as “cross-talk” species and reactions.201,202 Sahore et al.201 developed very recently a two-compartment lithium-ion cell, in which they separated the two electrodes by a solid electrolyte interlayer to retrieve and segregate such species before they can further react at the negative (or positive) electrode. They suggested that these species are likely fluorinated carbons and compounds with carbon–carbon double bonds, as proposed also earlier.203–205
Focusing essentially on the anode, Vetter et al.181 comprehensively summarized the potential ageing mechanisms for graphite and provided a general classification, differentiating ageing processes with respect to (i) the (long-term) instability of the SEI (which also includes the aforementioned cross-talk phenomena), (ii) lithium plating, and (iii) the contribution of electrochemically inactive electrode components such as the binder, conductive additives, and the current collector. Among these, they considered the former two most relevant – also with regard to potential safety issues.
As mentioned above, the formation of the SEI occurs mainly during the first cycles – but not exclusively. Upon cycling and storage, the morphology and composition of the SEI further develop. This temperature-related evolution of the SEI induces a continuous loss of lithium, consumption of the electrolyte, and, as a result, an increasing resistance. Beside the impact on the performance, however, the occurring changes also affect the overall safety of the battery cell. According to Feng et al.206 the destructive interactions between the graphite anode and the electrolyte can be summarized into three characteristic stages: the first stage is the heat-induced SEI decomposition. This heat may evolve from severe abuse conditions such as a deep overcharge, (too) high and/or highly inhomogeneous current distribution, or external heat sources. The onset of the SEI decomposition has been reported to occur at ca. 90 °C, but might even be as low as ca. 57 °C, depending on individual system.207,208 During the second stage (at ca. 120–250 °C), the SEI decomposition and regeneration occur simultaneously, leading to a rather thick, but stabilizing SEI. Nevertheless, if the temperature increases further, i.e., above 250 °C, the third stage occurs – the breakdown of the SEI due to the structural collapse of graphite206 and/or the decomposition of the electrolyte.209 This series of reactions marks the origin of the thermal runaway of the battery cell, including the extensive decomposition of the electrolyte, the melting of the separator, and the degradation of the cathode active material, potentially causing severe safety issues such as inflammation and even the explosion of the cell.206,210 In fact, to inhibit this series of destructive reactions and, hence, prevent the cell from thermal runaway, enormous efforts have been devoted towards the development of suitable surface modifications for the graphite anode and SEI forming additives, as also discussed earlier in the Sections 3.2.1 and 3.2.2, targeting not only an improved electrochemical performance, but also enhanced thermal stability of the SEI and, thus, pushing its decomposition towards higher temperatures.
Another severe safety issue is the potential plating of metallic lithium at the graphite electrode surface due to the low lithiation potential close to the deposition of metallic lithium. Accordingly, any increased overpotential for the lithiation process, resulting from, e.g., a sufficiently low operational temperature or elevated currents, especially in combination with an (ageing-induced) increase in resistance, might cause lithium plating, as depicted in Fig. 8a.211 Waldmann et al.212 demonstrated in a post mortem study that the rate of lithium plating increases when the temperature is decreased from 25 °C to −20 °C as a consequence of the slower Li+ diffusion. Moreover, in a recent study by Lüders et al.,213 it was shown that the degree of lithium plating correlates with the applied current; by this limiting the possibility to rapidly charge the lithium-ion cell (as discussed above in Section 3.1). Interestingly, though, it appears that the rate of lithium plating is decreasing upon cycling, which was assigned to the fact that the potential (or SOC) at which plating commonly occurs is not reached anymore due to the (also plating-induced) capacity fading upon long-term cycling. Hence, it has been stated that lithium plating “counteracts with its own occurrence”.214 Precisely, it has been found that the plated metallic lithium reacts with the electrolyte, thus, forming its “own SEI” on top of the graphite electrode at elevated SOCs and is partially consumed during this process. Upon the subsequent stripping, the metallic lithium is partially reoxidized and reinserted into the cathode, while electronically insulated lithium continues reacting with the electrolyte, leading to the formation of so-called “dead” lithium species (see also Fig. 8b).214,215 In line with these findings, Fleischhammer et al.216 correlated the ageing history of commercial cells with their safety behavior by means of accelerating rate calorimetry and simultaneous thermal analysis and observed that the self-heating rate and extent of thermal runaway were much more severe for cells that were aged at low temperature (i.e., with lithium plating) due to reaction of the electrolyte with metallic lithium, while cells aged under high current conditions did not show significant differences regarding their safety compared to unaged cells. As a consequence, temperature is a highly decisive parameter for ensuring long-term stable electrochemical performance and, eventually, the evaluation of the cells concerning their suitability for second-life applications such as stationary storage.
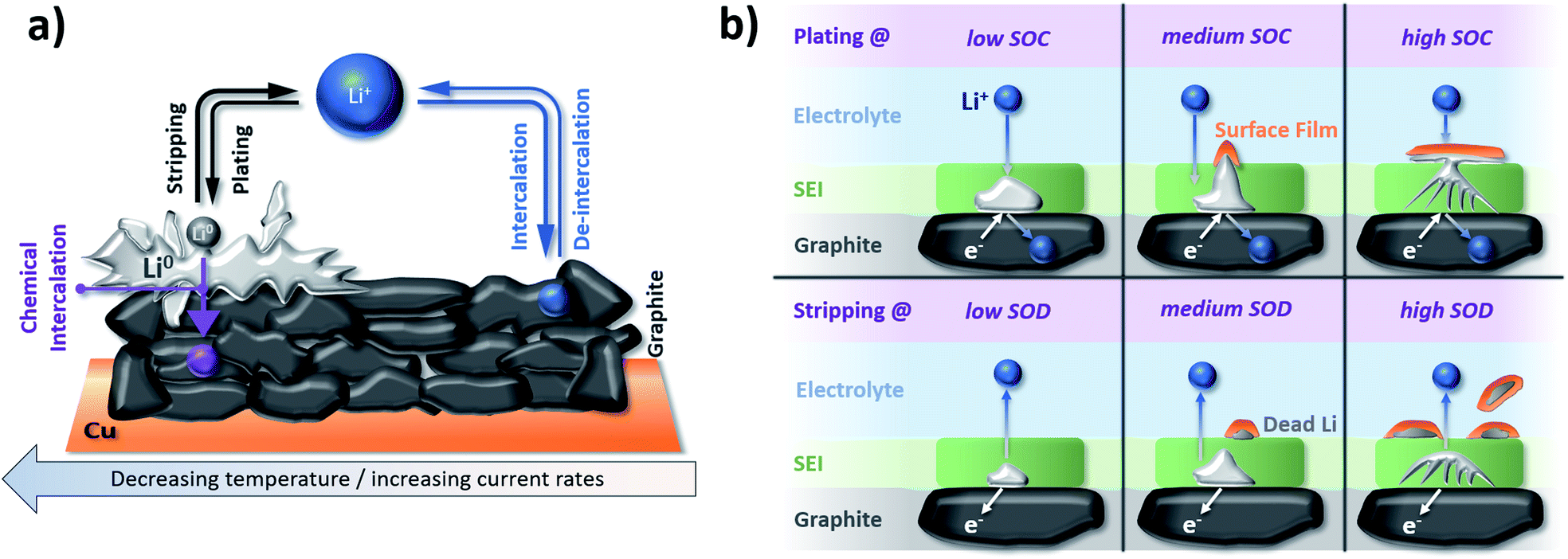 | ||
| Fig. 8 (a) Schematic illustration of lithium plating/stripping on graphite electrodes in competition with the de-/intercalation of lithium cations into graphite depending on the applied current and temperature (figure redrawn from ref. 211). (b) The behavior of plated lithium metal on top of a graphite electrode as a function of the ‘state of charge’ (SOC) and ‘state of discharge’ (SOD) upon plating and stripping, respectively (figure redrawn from ref. 215). | ||
3.4 Recycling
The recycling of spent LIBs has focused so far mostly on the recovery of (heavy) metals, i.e., on the elements comprised in the cathode and the current collectors, and little attention has been paid on the recycling of the graphite anode. The latter aspect has been dealt with only by very few studies.217–219 As a matter of fact, the recycling rate of graphite in 2010 was 0% and even by 2014 the recycling of graphite was still of low (economic) priority.220 Nonetheless, natural graphite has been classified as a critical resource by the European Union (EU) – not least as there are no natural resources in the EU.221 Thus, the recovery of graphite might become more important in the future along with an increasing electrification of the transport sector and the consequently rising number of spent LIBs.Generally, LIB recycling can be categorized into pyro-metallurgical, hydro-metallurgical, and physical processes. However, it has been shown that both pyro-metallurgical processes (due to the high temperature needed) and hydro-metallurgical processes (due to the aggressive chemicals involved) fail in recycling graphite in a reusable way.219 Differently, the physical processing allows for the recycling of the current collectors, the cathode active material (in form of suitable precursors), and the graphite anode, as schematically illustrated in Fig. 9. Hanisch et al.222 described the Adhesion Neutralization via Incineration and Impact Liberation (ANVIIL) process which can be adapted to the process depicted in Fig. 9 after shredding the cell and electrolyte extraction. This process employs moderate heat treatment to decompose the PVdF binder and, thus, lower the adhesion between the electrode coating and the current collector. Subsequently, the metal foils and electrode coatings are separated in an air jet separator and the active material agglomerates are broken down to primary particles through mechanical impact. However, during the separation and de-agglomeration most of the graphite particles get lost. Bertuol et al.223 were able to separate cell components by physically shredding commercial LIBs and separate the components, i.e., the separator, metals, and active materials, by spouted bed elutriation. The fraction of recovered active materials was 42.7 wt% (containing both LiCoO2 and graphite), which is certainly remarkable already, but still insufficient with respect to the continuously increasing importance of LIBs and the need for efficient recycling technologies. In 2016, Nowak and co-workers217 studied possible routes towards the recycling of graphite by means of different electrolyte extraction and SEI removal strategies. Both aged and non-aged commercial cells were discharged, opened in a glove box, and the jelly rolls were extracted. After several steps to obtain lab-scale electrodes, the graphite anode was subjected to electrolyte extraction via subcritical CO2 or static supercritical CO2, followed by a subsequent simple heat treatment. The results showed that, by subcritical CO2-assisted electrolyte extraction, the re-used graphite provided superior performance compared to commercial synthetic graphite. Interestingly, all re-used graphite samples from aged cells outperformed the ones obtained from non-aged cells, suggesting that such treatment appears generally favorable.
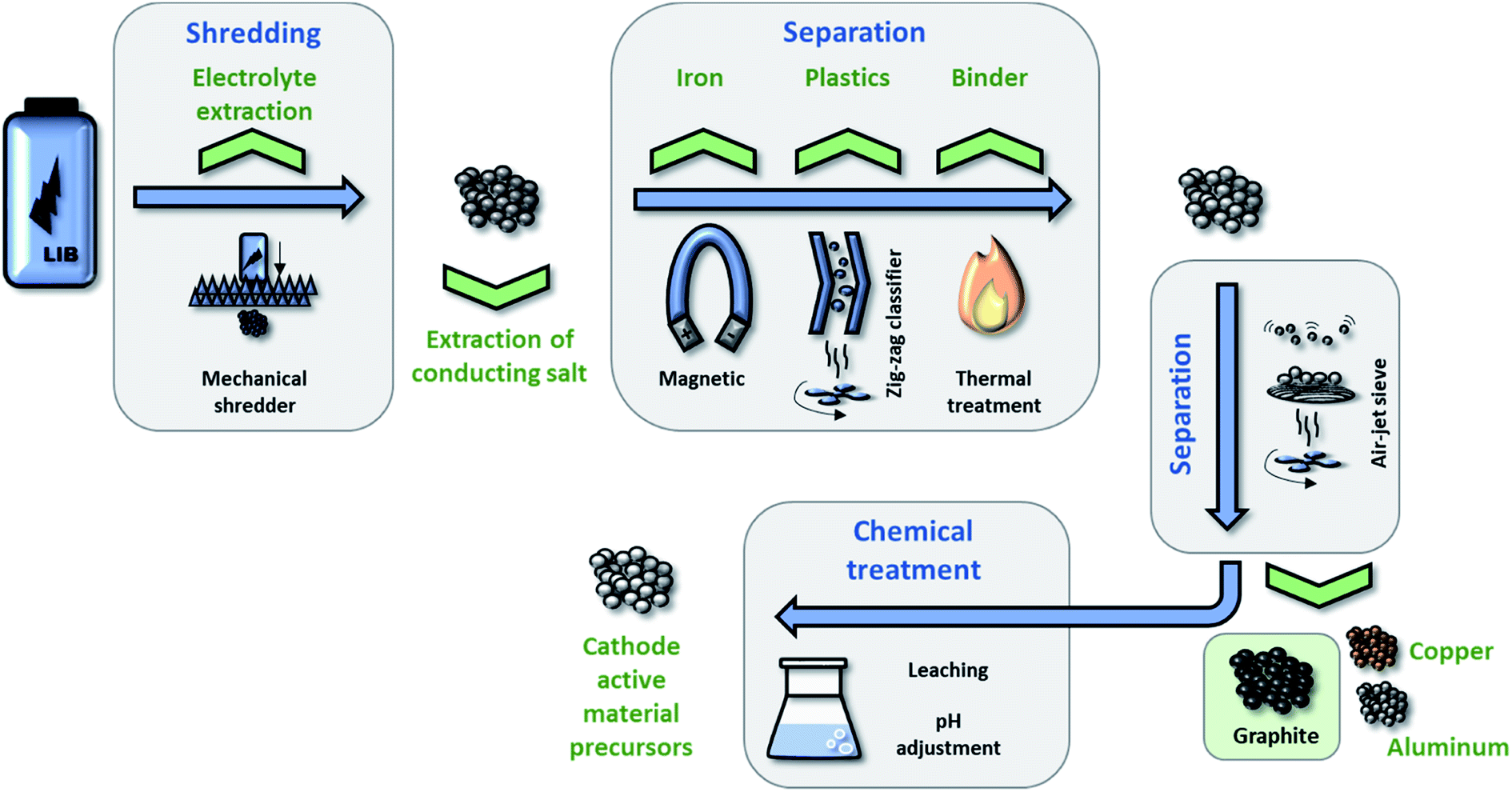 | ||
| Fig. 9 Schematic illustration of the ‘physical recycling’ process for LIBs, enabling the recycling of the metallic current collectors, the cathode active material (in form of suitable precursors), and the graphite anode (figure redrawn from ref. 217). | ||
As an alternative, after having shredded the lithium-ion cells, graphite can also be recovered via wet chemistry, e.g., by Fenton reagent (Fe2+/H2O2) assisted flotation. He et al.218 showed that treatment of graphite and LiCoO2 with the Fenton agent allows for removing the organic surface layer (as validated by XPS) on both the cathode and the anode active material. Subsequently, the two materials can be simply separated by flotation when taking into account the different hydrophility. Nevertheless, their study also revealed that the optimum recovery rate is achieved only with high excess of the Fenton agent, which limits the commercial applicability of this process for the moment.
4. Recent developments
4.1 Natural vs. synthetic graphite
There are essentially two kinds of graphite used in state-of-the-art LIBs – natural graphite (NG) and synthetic graphite (SG). In both cases, the particles are polycrystalline and composed of numerous single-crystalline domains (depending on the size of these domains, one differentiates between micro- and macro-crystalline graphite).224–226 In NG, though, these domains are commonly oriented in the same direction, while their orientation in SG is more random, as also apparent from Fig. 10. In fact, for obtaining such spherical particle shape (also referred to as “potato shape”), the originally flake-like NG particles and the frequently randomly shaped SG particles have to be subjected to an additional mechanical treatment after the mining process, the spheroidization.227 This process aims at homogenizing the particle size (ideally, 8–30 μm) and morphology and, thus, improving the packing density in the electrode coating layer for enhanced volumetric capacities. In case of NG, this modification of the particle morphology allows moreover for avoiding the parallel-to-the-current-collector orientation of the flake-like material, which would hinder rapid lithium cation de-/intercalation.161 In both cases, this mechanical treatment is frequently followed by applying a thin carbon coating to reduce the surface area and, hence, improve the ICE by reducing the reductive electrolyte decomposition and further suppressing solvent co-intercalation.228 | ||
| Fig. 10 Comparison of SEM micrographs of sphere-shaped natural graphite (NG; after several processing steps) and synthetic graphite (SG). | ||
While the eventual performance of any specific graphite material eventually depends on a variety of different parameters, there are several general characteristics for the two different kinds: SG is basically produced by heating unsaturated carbon, commonly derived from petroleum, to 2500 °C for a certain period up to several days in some cases.36,224 This high-temperature treatment renders SG relatively expensive compared to NG (ca. 8 US$ kg−1vs. around 13 US$ kg−1, respectively229), but offers the advantages of higher purity, lower thermal expansion, and better thermal stability.230 Additionally, the possibility to synthesize graphite, in ideal case from sustainable resources (though this is highly challenging given the required reproducibility, quality, and availability of precursors with a suitable structure, frequently resulting in rather amorphous carbons231), provides the advantage of being independent of natural graphite resources and the rather isotropic orientation of the crystalline domains within each particles, accompanied by a relatively larger fraction of edge-planes at the particle surface. This allows for superior de-/lithiation kinetics, although also frequently higher irreversibility in the initial cycles.36,71,225 Simultaneously, the smaller domain size and the, thus, relatively larger fraction of domain interfaces leads to lower capacities compared to NG, characterized by larger, rather anisotropically oriented crystalline domains.71
For such reason, both materials are presently used for commercial LIBs;32,227,232 SG especially in EV applications (due to the higher purity and quality, despite the higher cost) and NG typically for portable electronics, as these applications do not have the requirement of very long cycle life. Accordingly, it is expected that the demand for both NG and SG will further increase with the increasing importance of LIBs, while the fact that NG is considered a critical raw material is anticipated to further trigger the development of high-performance SG.32,227,233
4.2 Aqueous electrode processing
An aspect about graphite anodes that is nowadays very well established (also in industry), but has happened, in fact, only about ten years ago, is the use of water-soluble binders for the electrode preparation, providing substantial advantages concerning cost and sustainability. Since this has very recently been reviewed very comprehensively for LIBs in general,234 we may just provide a brief overview herein, highlighting several findings that are particularly relevant for graphite. One of its key advantages in this regard is its chemical stability towards water, which has triggered extensive efforts for finding alternative polymeric binders to replace relatively costly PVdF as well as harmful and teratogen NMP.235–239 Among the first (and presently used) are styrene butadiene rubber (SBR) in combination with sodium carboxymethyl cellulose (CMC), polyacrylic acid (PAA), or gelatine.229,240–246 Especially CMC does not only add to the binding functionality, but moreover enables the realization of stable suspensions of the hydrophobic graphite particles in water thanks to the coordinative interaction between the carboxylic groups and, e.g., hydroxyl groups at the graphite surface.234 The use of CMC only, however, does not allow for mechanically stable high mass loading electrodes due to its rather brittle nature. Hence, SBR is added as well, providing a highly suitable combination of properties. Remarkably, the utilization of such water-soluble binding agents does not only reduce the cost for the electrode processing and adds to the environmental friendliness of LIBs,234 but also presents beneficial electrochemical characteristics such as a decreased initial capacity loss and a stabilized SEI.240,247 Furthermore, the replacement of PVdF by CMC and PAA, for instance, enhances the thermal stability of the resulting electrodes and suppresses the thermal runaway246 thanks to a lower reactivity with lithiated graphite.248–251 In some cases, the water-soluble alternatives even provide enhanced charge transfer kinetics for the lithium de-/intercalation,249 rendering this development advantageous from essentially every perspective and without any apparent performance loss or other significant disadvantage compared to the earlier used PVdF in NMP.4.3 Introducing silicon for enhanced energy densities
A rather recent (industrial) development is the introduction of small amounts (up to ca. 8 wt%252) of silicon or understoichiometric silicon oxide (SiOx, with x < 2) to graphite composite electrodes,253,254 since pure graphite anodes have essentially reached their maximum performance concerning energy density thanks to the intensive research and development efforts in the past decades. The impact of further increasing the specific capacity of the anode on the total lithium-ion cell capacity is illustrated in Fig. 11 for a few selected cathode material candidates, ranging from state-of-the-art LiCoO2 with a specific capacity of 140 mA h g−1 (in black), next-generation layered lithium-rich transition metal oxides (LR-MO) with an anticipated capacity of 250 mA h g−1 (in red), an oxygen cathode with a limited specific capacity of 500 mA h g−1 (in yellow), and a sulfur cathode with a limited capacity of 1000 mA h g−1 (in green). While an increase in capacity for the anode has a beneficial effect in combination with all these cathodes, this effect is particularly pronounced for increasing cathode capacities well beyond the state of the art. Nevertheless, it is also apparent from Fig. 11 that for a given cathode capacity the total full-cell capacity reaches a plateau at a certain anode capacity, suggesting that especially for the state-of-the-art and next-generation cathodes an anode capacity of about 1000–1500 mA h g−1 will be sufficient – at least from the total gravimetric capacity point of view.255 Accordingly, the incorporation of reasonable amounts of high-capacity anode materials like abundant and environmentally friendly silicon with a theoretical capacity of 3578 mA h g−1, in fact the alloying element with the highest capacity known (Fig. 12), and an average delithiation potential of ca. 0.4 V vs. Li/Li+ in graphite-based composites appears as a very suitable approach for the realization of next-generation lithium-ion anodes.21,256,257 | ||
| Fig. 11 Illustration of the impact of increasing anode capacities on the total capacity of lithium-ion full-cells in combination with different cathodes, i.e., varying cathode capacities, here exemplarily presented for state-of-the-art LiCoO2 (140 mA h g−1, in black), a next-generation layered lithium-rich transition metal oxide cathode (LR-MO, 250 mA h g−1, in red), an oxygen cathode with a limited specific capacity of 500 mA h g−1 (in yellow), and a capacity-limited sulphur cathode (1000 mA h g−1, in green). The values represent the ideal maximum that can theoretically be achieved for a specific combination neglecting full-cell balancing and oversizing effects. | ||
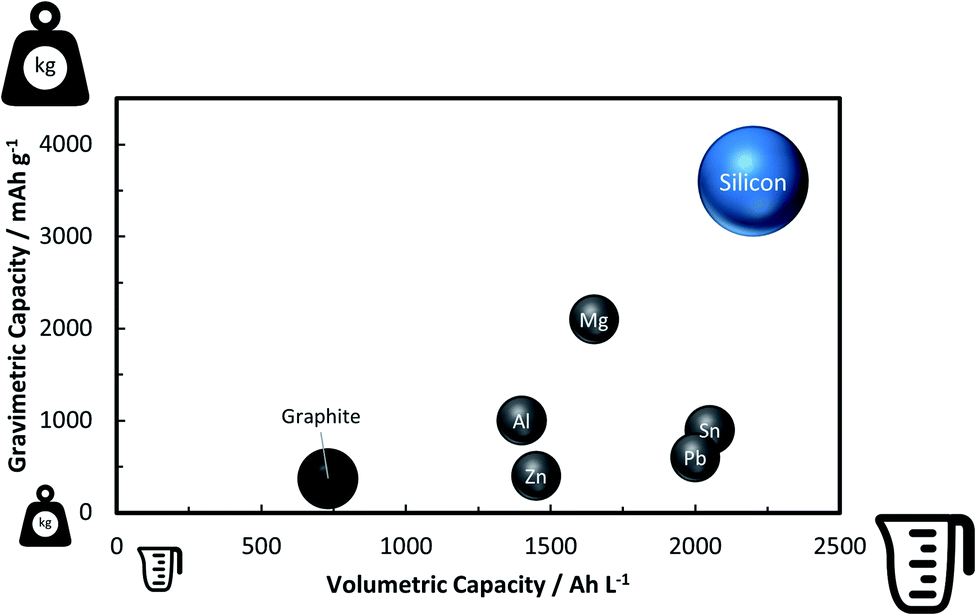 | ||
| Fig. 12 Comparison of the gravimetric and volumetric capacity of graphite and selected alloying elements such as zinc, aluminum, lead, magnesium, tin, and silicon (the volumetric values refer to the fully-lithiated state, according to ref. 21 and 361, in order to be of practical relevance, but do not consider other important factors such as the volume of the dislocated electrolyte362). | ||
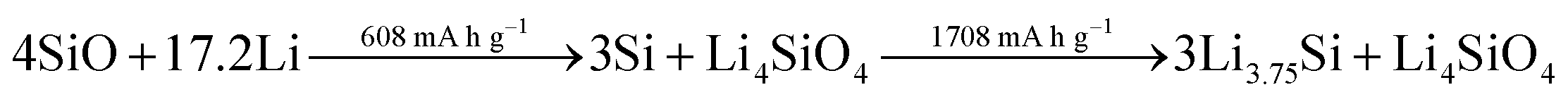
While the high initial irreversibility is certainly an issue, the higher density matrix of Li4SiO4 (2.39 g cm−3 – compared to 1.18 g cm−3 for Li3.75Si) acts as an effective buffer for the occurring volume changes, limiting it to about 160% compared to almost 300% for elemental silicon.262,265 Moreover, the presence of the lithium silicate phase may enhance the lithium diffusion into the nano silicon domains by creating additional phase boundaries within the mixed structure.265 Taking these considerations into account, the major strategies to overcome the intrinsic limitations of adding SiOx to graphite composite electrodes are: (1) the design of elaborated nanostructures, including the incorporation of secondary carbon phases, to improve the electrochemical performance and buffer the volume changes, (2) the development of industrially relevant pre-lithiation techniques to tackle the low ICE, and (3) the development and optimization of additional cell components such as the binding agent and the electrolyte composition, targeting enhanced mechanical properties of the electrode and a stabilized electrode/electrolyte interphase (see also Section 3.2.2).
4.3.1.1 Design of elaborated nanostructures. To mitigate the negative impact of the large volume changes of the added SiOx, the reduction of the particle size to the nanoscale is considered a very viable approach. Nonetheless, several issues have to be addressed with respect to a meaningful application of nano-sized materials: (i) the high surface area leads to an increased electrolyte decomposition and lithium loss; (ii) the low tap density results in decreased volumetric energy densities; (iii) the handling of nanomaterials at the commercial scale provides severe processing and safety issues; and (iv) the overall cost for the synthesis and processing, including the final incorporation into the graphite-based electrodes, have to be kept as low as possible.266–270 In an attempt to address these issues (at least partially), Doh et al.,271 for instance, reported a 1
:1 ball-milled mixture of graphite and SiO comprising moreover a carbon precursor that was subsequently carbonized at elevated temperatures. Such composite provided a capacity of 500 mA h g−1 after 100 cycles with a capacity retention of about 82%.272 Similarly, Lu et al.273 reported carbon-coated Si–SiO2–C composites by ball-milling a mixture of SiO, graphite, and coal pitch (serving as carbon source), followed by a high temperature treatment which leads to the formation of an amorphous carbon coating around the composite particles. Such material provided stable cycling for 90 cycles with a capacity of more than 700 mA h g−1. Using furfuryl alcohol as precursor for the carbon coating, scientists from Toshiba published a detailed analysis of a similar ball-milled SiO/graphite composite. Mixed with graphite such composite exhibited a reversible capacity of about 700 mA h g−1.274 The dispersion of nanometric (5–10 nm) silicon clusters throughout the sample was considered essential for maintaining the structural integrity of the isotropic composite particles with a size of 5–20 μm, while the combined silicate/carbon matrix effectively buffered the volume changes upon de-/lithiation, resulting in a capacity retention of 85% after 200 cycles and a CE approaching 100% after several cycles. Following a simple sol–gel synthesis and subsequent carbon coating based on glucose as carbon precursor, other groups prepared hierarchically structured carbon-coated SiOx/C composites.275–277 Such materials revealed specific capacities between ca. 675 mA h g−1 (ref. 275 and 276) and about 800 mA h g−1 (ref. 277) after 100 cycles, in all cases corresponding to a capacity retention of about 84% with an average CE of above 99%, suggesting that the external carbon shell stabilized the interface with the electrolyte. In 2014, Park et al.278 followed up the approach from Toshiba and employed a scalable sol–gel process for the synthesis of a SiOx/C composite in which silicon nanodots were embedded into the SiOx matrix. The confinement of nanometric Si into the matrix could buffer the severe volume changes as proven by cross-sectional SEM analysis of the cycled electrodes, the height of which only changed by 37% after 100 cycles. Simultaneously, the uniform carbon coating layer on the nanospheres allowed for an excellent rate capability with more than 800 mA h g−1 delivered at 2C, i.e., more than 90% of the capacity at 0.05C. The stabilized electrode/electrolyte interphase in combination with the buffered volume changes enabled an average CE above 99.5% and a good capacity retention of 78% after 100 cycles with a reversible capacity of 740 mA h g−1.
To briefly summarize, the carbon coating fulfills several functions: (i) it acts as a buffer for the volume variations occurring; (ii) it stabilizes the electrode/electrolyte interphase; (iii) it serves as an electronically conductive, percolating network throughout the electrode (in addition to/combination with the subsequently added conductive agent).
Given the aforementioned studies, the application of such coating in combination with the realization of nanostructured SiOx/carbon composites appears indispensable for the successful implementation of SiOx in commercially viable electrodes. In fact, it has been reported that carbon coating content needs to be as high as 50% to achieve stable cycling performance with a reversible capacity above 500 mA h g−1,279–281 while nanometric carbon coatings cannot effectively suppress the formation and propagation of particle cracking and a continuous SEI reformation.282 The latter issue is frequently overlooked in half-cell studies, where the lithium counter electrode provides a large reservoir for additional lithium cations, but becomes immediately apparent when studying new anode materials in full-cells. This effect is illustrated also in Fig. 13 for different CEs (from 99.0% to 99.995%) on the full-cell level, i.e., with a limited amount of lithium in the cathode. In literature, however, only a few full-cell studies have been reported. Yamada et al.283 prepared negative electrodes based on a mixture of 50 wt% SiOx, 21 wt% graphite, and 9 wt% carbon fibers, subsequently coated by 20 wt% of carbon via physical vapor deposition (PVD), and compared this elaborated mixture with a simple 1:1 blend of SiOx and graphite. The combination of the elaborated mixture with blended LiNi1/3Co1/3Mn1/3O2/LiCoO2 (NCM/LCO) cathodes revealed an anode capacity of ca. 700 mA h g−1 after 100 cycles, corresponding to a capacity retention of remarkable 85%. On the contrary, the simple anode blend showed only 20% capacity retention after just 20 cycles in the same lithium-ion configuration. The large amounts of carbon, though, allow for only limited improvements in energy density. Hence, Kajita et al.284 investigated the use of carbon-coated SiOx as sole anode active material in full-cells with LiNi0.8Co0.15Al0.05O2 (NCA) cathodes. Despite an impressive capacity retention of >80% after 500 cycles in an operational voltage range of 2.5 V to 4.2 V, the ICE remained rather low with less than 65%. Such low ICE, however, would exclude any meaningful commercial application,266,285 as the extra lithium comes with a lot of additional mass in the commonly used lithium transition metal oxide cathodes.
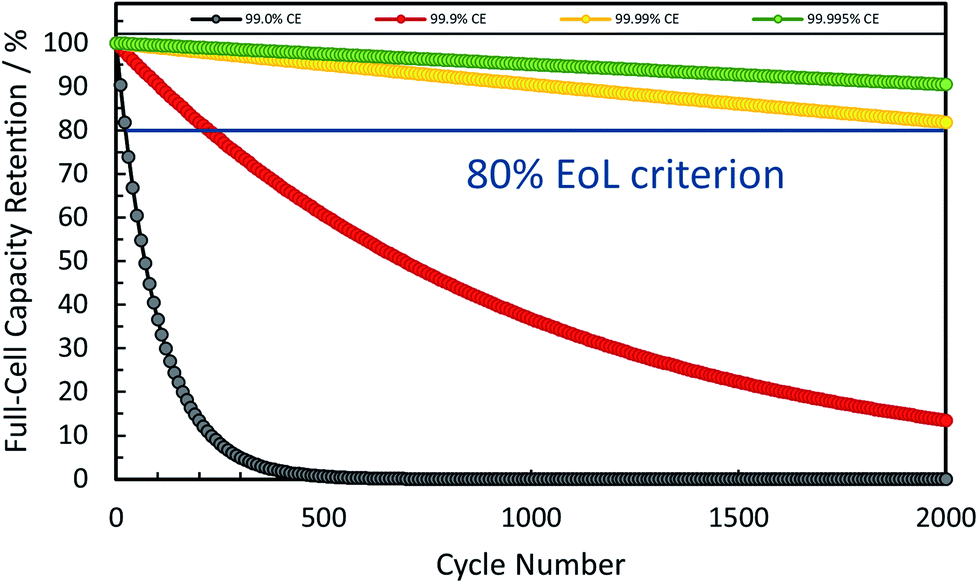 | ||
| Fig. 13 Illustration of the impact of the average CE for the full-cell capacity retention by varying the average CE from 99.0% (in black) over 99.9% (in red) and 99.99% (in yellow) to 99.995% (in green). The horizontal blue line indicates the common end-of-life (EoL) criterion of 80% capacity retention. | ||
4.3.1.2 Pre-lithiation strategies. To overcome the limitation in energy density due to the rather low ICE, different strategies have been developed to either compensate this initial loss by providing some additional lithium source inside the cell (in addition to the cathode) or to address this issue prior to the cell assembly by forming partially lithiated phases already during the synthesis or at the electrode level. Following the “synthesis approach”, Veluchamy et al.286 pre-lithiated SiOx by blending it with LiOH and subjected this blend to 550 °C before ball-milling the resulting mixture with graphite. The partial formation of Li4SiO4 in the pretreated material helped to reduce the first cycle irreversible capacity from 55% for the non-prelithiated composite to 44% and allowed for an improved cycling stability. Another rather simple, but effective “synthesis approach” relies on the in situ mechano-chemical reduction of SiOx by ball-milling it together with lithium metal.287 As a result, the ICE was increased to 81% with a reversible capacity of around 770 mA h g−1 at a dis-/charge rate of 0.1C and the CE was approaching 99% after 50 cycles. Nevertheless, the rate capability of such composite remained improvable; for instance, at 1C, the reversible capacity was less than 360 mA h g−1, presumably due to an insufficient active material utilization.
Work on the “electrode pre-lithiation approach”, similar to the frequently employed lab-scale approach of first pre-lithiating the electrode in half-cell configuration prior to the full-cell assembly (Fig. 14a),112,288–291 was inter alia reported by Kim et al.,292 who developed a scalable roll-to-roll process for the pre-lithiation of the electrode prior to the full-cell assembly by spraying an electrolyte solution onto the electrode tape and bringing it in contact with lithium foil with a separator in-between (Fig. 14b), followed by the application of an external short circuit. The incorporation of a resistor in the external circuit allowed for the controlled lithiation of the SiOx-based electrode. To confirm the suitability of this approach, the authors assembled a pre-lithiated SiOx//NCA full-cell with electrodes that had a commercially relevant areal capacity of >2.0 mA h cm−2. Such cells showed a remarkable ICE of 95% and a specific energy above 500 W h kg−1 (1080 W h L−1), thus, exceeding the internal graphite-based reference by more than 10%. In a very similar way, the company Nanoscale Components designed a pre-lithiation bath, in which negative electrodes are pre-lithiated from a lithium-salt containing solution, potentially via a roll-to-roll process.293,294 To demonstrate the general feasibility of this approach, Chevrier et al.295 realized 2 Ah 18650-type cylindrical cells with large fractions (up to 55 wt%) of 3 M Si alloy negative electrodes (see also Section 4.3.2) with precisely defined lithium reservoirs. As a result, they could achieve a gain in energy density as high as 11% over their baseline cells with a capacity retention of 80% after 500 cycles. It should be noted that such approaches are generally applicable also to any other active material and can lead to further enhanced energy densities even for standard graphite-based electrodes. Nonetheless, they require the installation of relatively large space with a protected atmosphere (either noble gases or at least dry air – depending on the reactivity of the pre-lithiated compounds) and adds another processing step to the cell assembly.
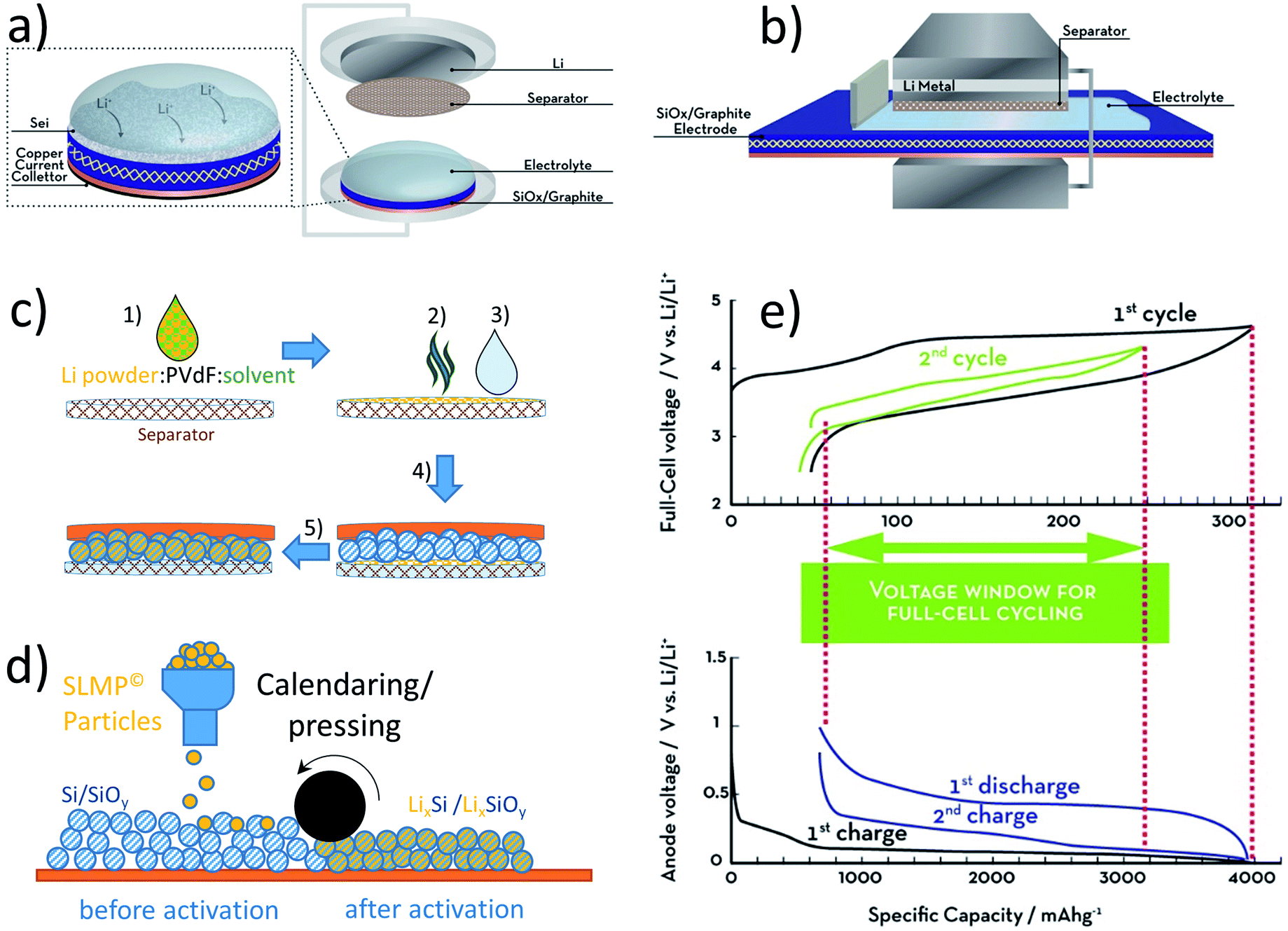 | ||
| Fig. 14 Pre-lithiation approaches for SiOx-based anodes (though generally applicable also to other active materials): (a) lab-scale pre-lithiation in coin-type half-cells using a lithium metal counter electrode. (b) Continuous electrochemical pre-lithiation on large-scale via a roll-to-roll process employing a quasi-half-cell configuration with the SiOx-based anode wetted by an electrolyte and moving continuously across a stationary Li–metal counter electrode with a stationary separator; the application of an external short circuit or generally a current allows for a controlled pre-lithiation of the electrode to a specified potential and/or capacity (figures redrawn from ref. 292). (c) Deposition of lithium–metal powder, comprised in a PVdF solution, onto the polymer separator (1), followed by the evaporation of the solvent (2) and wetting with the electrolyte (3). Subsequently, the separator is placed on the readily cast electrode (4) and the lithium powder reacts with the SiOx-based electrode coating (5) (figure inspired by ref. 296). (d) Addition of stabilized lithium metal powder (SLMP©) onto the readily made electrode and its activation by calendaring/pressing (figure redrawn from ref. 299). (e) Compensation of the initial irreversibility of SiOx-based anodes by coupling them with carefully balanced LR-NCM cathodes in order to utilize the initially released Li2O along the first cycle high-voltage activation step upon charge (figure redrawn from ref. 302). | ||
Similarly, also the “compensation approach” is generally independent of the active material chemistry. Hwang and Yoon296 developed such a methodology by dip coating a polypropylene separator with a thin film of lithium powder dispersed in PVdF to pre-lithiate SiOx/graphite (1:1) electrodes (Fig. 14c). The subsequent characterization in lithium-ion cells with LCO as cathode active material revealed that the ICE could be increased from 66% in case of the uncoated separator to an impressive value of 89% for an optimized lithium content in the separator coating layer. In fact, such improvement was observed not only for SiOx-based electrodes, but also pure graphite (93% vs. 89%) and anodes based on elemental silicon (87% vs. 67%). Nonetheless, the handling of the lithium-containing separator is certainly a challenge for industry – not only with regard to its high reactivity, but even more the severe safety issues in case the separator gets damaged upon processing. A method that is presumably easier to be implemented at industrial level is the pre-lithiation approach reported by Gao et al.,297 employing the incorporation of commercially available stabilized lithium metal powder (SLMP® (ref. 298)). As depicted in Fig. 14d, the SLMP® particles can be simply added onto the electrode – either directly as powder or in form of a suspension – and activated by calendaring/pressing to break the stabilizing coating.299,300 Thanks to the pre-lithiation of the anode, full-cells with an NCM cathode provided an improved energy density by up to 30% compared to reference full-cells comprising pure (and non-prelithiated) graphite as anode.299 In a more recent study, the application of SLMP® for a SiOx/graphite demonstrated an impressive ICE as high as 98.5% (vs. 68% without pre-lithiation) and highly stable half-cell cycling with an average CE of 99.4% and capacity retention of 95% of the initial 905 mA h g−1 after 200 cycles.300 Nevertheless, it has to be kept in mind that during this process metallic lithium is spontaneously reacting with the electrode active material, thus, requiring strict safety measures such as working under dry air/protective atmosphere. Additionally, achieving a precise control concerning the homogeneity of the pre-lithiation is certainly not trivial, which calls for additional safety measures to prevent a local overcharge.301
While these pre-lithiation strategies are anticipated to be of great importance for the development of next-generation anodes in combination with, e.g., Ni-rich NCM or even future cathodes such as oxygen or sulfur (if not using metallic lithium),22 another potential cathode candidate for next-generation LIBs can, indeed, serve as internal lithium source itself (without simply losing capacity): LR-NCM. In this regard, Yamano et al.302 reported a well-conceived study on SiOx//LR-NCM full-cells, attempting to compensate the anode and cathode irreversible capacity simultaneously by making use of the initial Li2O removal from the LR-NCM structure (Fig. 14e).303,304 Carefully adjusting the mass loading of the two electrodes by taking into consideration the initial irreversible capacity allowed for compensating the first cycle irreversible capacity on the anode side and the resulting full-cells demonstrated very stable cycling performance. The same concept also applies for the overlithiated spinel-structured lithium nickel manganese oxide, as demonstrated by Axmann and co-workers,305 where the extra-lithium introduced during solid-state synthesis can be used to compensate the irreversible loss of, e.g., silicon-containing carbon composites with a net-gain of 25% in specific energy as compared to regular graphite/LiNi0.5Mn1.5O4 (LNMO) full-cells.
Eventually, any approach that shall be commercially implemented will have to be cost-efficient and safe (at least as safe as the current technology). Therefore, especially the approaches involving the use of metallic lithium will have to be critically assessed – even though industry might benefit of the experiences towards the development of lithium–metal batteries, which is presently attracting great attention again.306–308 Insightful review articles on this subject have recently been published by Aravindan et al.309 and Holtstiege et al.,310 to which the interested reader is kindly referred at this point.
4.3.1.3 Development of advanced binding agents. Similar as for pure graphite electrodes (see Section 4.2), the development of advanced binding agents has been intensively studied in the past years also for Si-containing composites. In contrast to pure graphite, however, these composites are much more demanding due to the extensive volume variation upon de-/lithiation. A suitable candidate (at least for rather low mass loadings) turned out to be CMC with promising results reported for both Si- and SiOx-based anodes.247,311 Following these results, the search for suitable binders was further extended towards other water-processable polymers such as PAA,312,313 poly(vinyl alcohol) (PVA),314 and alginate.315 Nowadays, the development of enhanced binders has become a research field on its own, covering the development of complex functional polymers and their interaction with the active material surface316–318 as well as the (carbon-coated) current collector,319,320 as comprehensively reviewed in detail very recently by Li et al.321 and Bresser et al.234
Accordingly, we would like to just briefly highlight herein that an ideal binder for Si- and SiOx-based anodes should fulfill the following requirements: (i) display only weak interactions with the electrolyte in terms of swelling and changes of the mechanical properties, while providing high (electro-)chemical stability; (ii) maintain efficient Li+ diffusion pathways to the particle surface without forming blocking layers; (iii) provide potential binding sites to firmly attach to the active material particles, e.g., by hydrogen bonding between functional groups such as carboxylate and hydroxyl groups, as well as the current collector – ideally accompanied by self-healing properties to re-establish the contact between the volume-changing particles and the current collector.
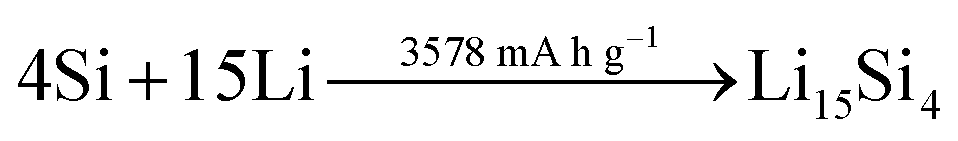
Nevertheless, the extensive volume variation of up to 280%, inducing great mechanical stress on the composite electrode, the electrode/electrolyte interface and interphase, and the whole cell, provides a great challenge that still remains to be overcome.21,336 Since these issues, however, are essentially the same as for SiOx and Si–metal alloys, the strategies to address them are basically the same, i.e., (1) the design of smart nanostructures, (2) the development of pre-lithiation strategies, and (3) the development and optimization of advanced binding agents and electrolyte additives (see also Section 4.3.1). Accordingly, we will focus in the following particularly on those studies dealing with the interaction of silicon and graphite and the continuous improvement of such composites, though taking into account, of course, also the overall electrode composition and their fabrication.
Generally, a limitation of the silicon content to maximum 30–50% accompanied by a limitation of the lithium concentration within the silicon phase was reported to be advantageous with respect to cycling stability.337 In addition, a reduction of the silicon particle size below 150 nm helps to address the detrimental impact of the large volume variation, as there is no significant particle cracking observed anymore, though simultaneously the overall specific surface area of the composite has to be kept low to avoid extensive electrolyte decomposition at the electrode/electrolyte interface.338,339 At the same time, the synthesis and particle design are ideally kept rather simple to facilitate the commercial application of such materials at reasonable cost and allow for achieving suitable electrode mass loadings. This trade-off is schematically illustrated in Fig. 15. Trying to address these aspects, Lee et al.340 mixed silicon nanoparticles with an average size of about 100 nm and NG flakes (∼5 μm) in a pitch containing solution via ball milling to obtain a spheroidized carbon matrix after carbonization. While the silicon nanoparticles were distributed randomly in the composite, the thin graphite flakes tended to align in parallel, which provided an enhanced structural stability and to some extent buffered the volume variation within the spherical composite particles. However, such a rather uncontrolled and random composition might lead to a pronounced inhomogeneity of the different compounds that can potentially induce local structural damages and/or voltage differences, causing electrode cracking and lithium plating in the worst case, respectively.338 In fact, the choice of the mixing method (and conditions) is decisive for the eventual structure of such silicon/graphite composites. For instance, the mechano-chemical milling of silicon and graphite in a ratio of 20:80 was demonstrated to result in the surface decoration of silicon on graphite, while a treatment by rotational impact blending yielded silicon encapsulated graphite particles.341 And although the ICE was relatively high for both composites with about 86%, the electrochemical performance was rather different. The one with silicon decorated on the graphite surface exhibited significantly better capacity retention and overall electrode stability. The inferior performance of the other sample was ascribed to the damage of the graphite particles and inter-particle cleavage due to the large expansion of the encapsulated silicon aggregates. In fact, the synthesis of such Si/carbon/graphite multicomponent structures does not need to be complicated, following the results reported by Kim et al.342 Based on commercial silicon nanoparticles and NG microspheres, composites with a varying silicon content of 10 wt%, 15 wt%, and 20 wt% were prepared via a simple and cost-efficient synthesis route. These composites exhibited an ICE of ca. 80%, stable cycling at capacities of about 600 mA h g−1, 650 mA h g−1, and 700 mA h g−1, respectively, and excellent rate performance with approximately 88% of the initial capacity at a dis-/charge rate of 5C (3250 mA g−1). The authors ascribed this superior electrochemical performance to the hierarchical structure of the composite with the silicon nanoparticles homogeneously covering the graphite particle surface and the realization of an amorphous carbon layer on top, which effectively accommodates the volume expansion and simultaneously maintains good electrical contact throughout the composite. Comparable results were obtained for similar composites within which the silicon nanoparticles (∼100 nm) were uniformly distributed on the micrometric graphite particle surface, both together encapsulated by an additional carbon coating,343,344 suggesting that such composite architecture is generally advantageous for achieving good electrochemical performance. However, care has to be taken that this carbon coating does not get too thick and that the graphite particles do not block the lithium transport to the silicon nanoparticles, as these are commonly particularly active at elevated dis-/charge rates.345 Nonetheless, a comparison of such composites blended with commercial graphite and taking into account commercially relevant electrode characteristics, i.e., an electrode density of >1.6 g cm−3, an areal capacity of >3 mA h cm−2, and a low binder content of <3 wt%, revealed that such composites can indeed meet the requirements of industry if the silicon and carbon surface layer on the graphite particles is sufficiently thin and if the composite particles have a similar size, tap density, and surface area as the pure graphite used for obtaining the blend. The best performing composite displayed a high ICE of 91.6% and retained 97.2% (420 mA h g−1) of the initial capacity in half-cells after 100 cycles. When evaluated in prototype full-cells with LCO as the cathode active material, an ICE of 89.7% was recorded and the capacity retention after 400 cycles was 67%.345
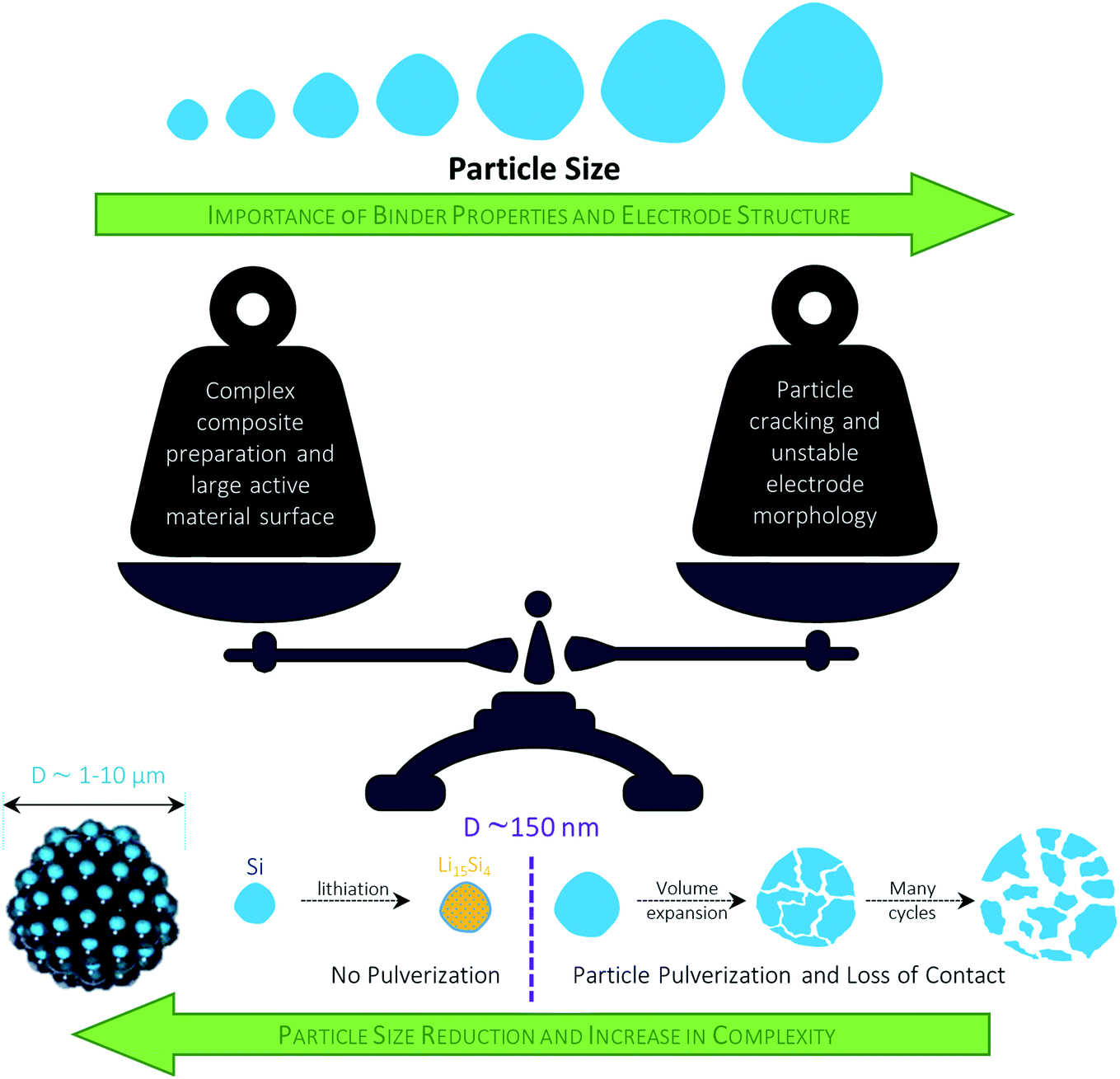 | ||
| Fig. 15 Schematic summary of the trade-off between the advantages of a reduced particle size for the silicon active material and the accompanying challenges with respect to particle pulverization, active material surface, electrolyte decomposition at the electrode/electrolyte interface, mechanical properties of the electrode, and packing density (figure inspired by and redrawn from ref. 338 and 339). | ||
Further improvement was suggested to be achievable by optimizing the bonding between the carbonaceous species and the silicon particles346,347 as well as the overall electrode composition and the nature of the binder. To optimize the overall electrode composition, Beattie et al.348 designed a theoretical model based on geometrical considerations, i.e., the packing density and the volume variations occurring, targeting the realization of electrodes with minimized particle movement in order to achieve high active material mass loadings. This model predicts that electrodes with a silicon content of <20 vol% may cycle stably for hundreds of cycles with specific capacities of around 660 mA h g−1 despite a very high binder content of up to 56 wt%. Experimental data showed that even higher capacities could be achieved by increasing the silicon content to 33 vol%, which could not be predicted by the model, as it does not consider electrode porosity. However, the electrode porosity and its reduction by calendaring play a critical role, especially, when it comes to the evaluation of energy densities, which – in most cases – is inferior to that of highly densified graphite anodes.21,332 Nevertheless, also Bridel et al.349 reported superior performance for rather high binder contents with an optimum ratio of 1:1:1 for the silicon/carbon/polymer composition. Their study, moreover, revealed the importance of the chemical nature of the binder by comparing CMC, PAA, amylose, and chitosan; precisely, the interaction of the binder with the silicon active material. According to their results, hydrogen bonding was advantageous over ester-type covalent bonding, as it enables self-healing properties, i.e., a reversible bonding to the silicon surface upon the extensive volume variations. The best performance was achieved with long-chain CMC, delivering almost the full capacity of silicon for more than 100 cycles with an average CE of 99.9%. However, the porosity of such electrodes was very high, leading to rather low volumetric capacities. In a follow-up study, the CMC binder was further optimized to promote hydrogen bonding over covalent bonding by adjusting the slurry pH with hydrochloric acid. Despite the still rather high porosity of around 60%, the authors could nearly double the volumetric capacity compared to classical graphite electrodes with only 25% porosity.350 Following these findings, Yim et al.351 performed a detailed investigation on practical full-cells with silicon/graphite blend negative electrodes. Based on their own theoretical model, they firstly optimized the silicon:graphite:binder ratio in order to limit the composite volume expansion to 40%, which was chosen as the available free “void” within a commercial battery composite. As a result, they obtained a practical ratio of 9:81:10 with a theoretical reversible capacity of 692 mA h g−1. Subsequently, they also calculated the energy density improvement in full-cells comprising NCM, LCO, or LNMO as positive electrode active material in dependence of the amount of silicon in the composite. For the optimum amount of silicon in the anode composite (9 wt%), theoretical improvements of 18%, 23% and 34%, respectively, could be achieved. However, the model fails to take into account also the irreversible capacity, which substantially varies with the silicon content. Thus, an accordingly optimized full-cell with the theoretically almost ideal ratio retained only 63% of the initial capacity after 50 cycles. Following a purely experimental approach, Schott et al.352 studied the influence of the lithiation procedure and particle size of commercial silicon nanoparticles as an additive to enhance the specific capacity of graphite electrodes starting from a silicon content of 5 wt%. They found that stable cycling can be achieved for relatively small particles with a diameter of 30–50 nm and applying a slightly elevated cut-off potential of 50 mV for the lithiation process when using a CCCV (constant current, constant voltage) protocol – presumably due to the only partial utilization of graphite and silicon. In fact, the application of a lower lithiation potential of 5 mV did not result in any significant improvement despite the increased risk of lithium metal plating, while the complete lithiation also leads to a more severe volume expansion when forming the fully lithiated Li15Si4 phase.353 After an extrapolation of the expected capacity values for higher silicon contents, Schott et al.352 also studied electrodes with a ratio similar to that suggested previously by Yim et al.351 The resulting electrodes containing 10 wt% of silicon were able to compensate for the incomplete utilization of graphite when applying the higher cut-off potential of 50 mV, with silicon contributing with more than 50% to the total specific electrode capacity of ca. 650 mA h g−1.352 Eventually, the authors pointed out that stable cycling with a capacity of more than 600 mA h g−1 could be expected for electrodes containing only 8 wt% of silicon, while further improvement was anticipated for electrodes comprising a different matrix than graphite that would also be electrochemically active in such slightly elevated potential region.352
Another approach to enhance the electrochemical performance relies on the earlier discussed pre-lithiation of the anode (see Section 4.3.1). This has been demonstrated, for instance, for a composite of hard carbon (40 wt%) and silicon nanoparticles (15 wt%) mixed with graphite (45 wt%).354 Without pre-lithiation, this composite showed stable cycling above 600 mA h g−1 with a capacity retention of 92% after 150 cycles. The ICE, however, was only 80%. Introducing 7.5 wt% or 8.3 wt% of SLMP© into the composite electrode (see Fig. 14d) allowed for increasing the ICE to remarkable 86% and 93%, respectively. Finally, by introducing FEC as a suitable electrolyte additive, the authors realized high-areal capacity anodes (>2.0 mA h cm−2), pointing towards the possibility to scale-up the process and meet commercial requirements.
Focusing on the optimization of the electrolyte composition for silicon-comprising anodes, Abraham et al.355 conducted a detailed EIS analysis of full-cells based on 15 wt% silicon/graphite blend negative electrodes and NCM532 positive electrodes. The comparative investigation of different electrolyte additives revealed that the incorporation of 10 wt% FEC or VC led to an improvement in capacity retention after 50 cycles from 40.2% to 65.4% and 67.5%, respectively. The introduction of these additives enabled the formation of a stabilized SEI, showing hardly any increase in impedance upon cycling (different from the additive-free electrolyte), which better accommodates the volume changes and facilitates rapid ion transport, presumably originating from the rather high Li+ concentration in the SEI. Moreover, the conducted dQ/dV analysis showed that the addition of VC and FEC suppressed the loss of cyclable lithium and electrochemically active silicon. Nevertheless, in all cases, significant capacity fading was observed due to the continuous shift of both the anode and cathode voltage window to higher values upon cycling. For the negative electrode, the eventual shift above 0.15 V vs. Li/Li+ rendered the lower potential plateaus of graphite no longer accessible, which largely contributed to the fading recorded. Interestingly, Dupré et al.356 showed that this fading is not necessarily related to active material degradation, as the single electrodes, after a comparable full-cell study, revealed stable capacities as high as the initial values in subsequent half-cells tests. Hence, the authors concluded that the major reason for the fading was the continuous loss of cyclable lithium due to parasitic reactions at the anode/electrolyte interface. This finding is in line with a study by Dahn and co-workers who observed a continuous consumption of the (10 wt%) FEC additive upon cycling and, once it had been completely consumed after about 400 cycles, the full-cells were fading rapidly.357 Based on these results they concluded that the presence of FEC prevents the transesterification of the EMC co-solvent, which in absence of FEC leads to steady increase in cell polarization.
5. Conclusive summary and perspective
Graphite is and will remain to be an essential component of commercial lithium-ion batteries in the near- to mid-term future – either as sole anode active material or in combination with high-capacity compounds such as understoichiometric silicon oxide, silicon–metal alloys, or elemental silicon. As a matter of fact, important EV manufacturers,358 material suppliers,285 and cell producers359 have recently announced that such graphite-containing composites will mark the state-of-the-art for next-generation lithium-ion batteries, providing significantly enhanced energy densities compared to the current technology.Nonetheless, despite the extensive research in the past decades, there are still several aspects that remain to be understood concerning, e.g., the detailed de-/lithiation mechanism, the reactions occurring at the electrode/electrolyte interface, and the impact factors that limit the rapid intercalation of lithium cations into the graphite lattice. In fact, very recent and so far unpublished data indicate that the desolvation energy at the SEI plays a decisive role and its careful control by modifying the electrode/electrolyte interface might enable substantial improvements for the fast charging of the resulting lithium-ion cell. Besides, the recycling of graphite as potentially critical raw material still needs to be enhanced as well, while synthetic graphite as alternative provides several advantages, but also shortcomings. Accordingly, there is plenty of room for further improvement despite the commonly achieved theoretical capacity – especially also with regard to the initial capacity loss. In combination with the still inferior cycling stability of graphite composites including silicon, the latter aspect, i.e., the rather low ICE, marks a highly industry-relevant challenge for the near-term future, although the rapid evolution of potential pre-lithiation strategies appears to provide suitable approaches to address this issue. Another remaining key issue is the identification of suitable electrolyte compositions that sustain stable electrode/electrolyte interfaces and interphases and are not continuously consumed upon cycling. While this aspect has only briefly been touched in this review, the finding of the stabilizing effect of EC for pure graphite presents an excellent example for its outstanding importance.
We may, thus, anticipate that the further development and improvement of lithium-ion batteries comprising graphite – at least as one of the anode components – has not yet reached an end and will continue for the next several years at minimum. In fact, the better we will understand the underlying mechanisms, the more room for further advances we will identify and be able to explore.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Vector Foundation and the German Federal Ministry of Education and Research (BMBF) for financial support within the NEW E2 and the Li-EcoSafe project (03X4636D), respectively. Moreover, financial support by the Helmholtz Association is acknowledged. Additionally, the authors would like to thank SGL Carbon for providing exemplary natural and synthetic graphite samples.References
- Y. Nishi, J. Power Sources, 2001, 100, 101–106 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- D. Andre, H. Hain, P. Lamp, F. Maglia and B. Stiaszny, J. Mater. Chem. A, 2017, 5, 17174–17198 RSC.
- D. Bresser, K. Hosoi, D. Howell, H. Li, H. Zeisel, K. Amine and S. Passerini, J. Power Sources, 2018, 382, 176–178 CrossRef CAS.
- D. Bresser, E. Paillard and S. Passerini, in Advances in Batteries for Medium and Large-Scale Energy Storage, Elsevier, 2014, pp. 125–211 Search PubMed.
- D. Guerard and A. Herold, Carbon, 1975, 13, 337–345 CrossRef CAS.
- M. Zanini, S. Basu and J. E. Fischer, Carbon, 1978, 16, 211–212 CrossRef CAS.
- S. Basu, C. Zeller, P. J. Flanders, C. D. Fuerst, W. D. Johnson and J. E. Fischer, Mater. Sci. Eng., 1979, 38, 275–283 CrossRef CAS.
- A. N. Dey and B. P. Sullivan, J. Electrochem. Soc., 1970, 117, 222–224 CrossRef CAS.
- J. O. Besenhard, Carbon, 1976, 14, 111–115 CrossRef CAS.
- R. Yazami and Ph. Touzain, J. Power Sources, 1983, 9, 365–371 CrossRef CAS.
- R. Fong, U. von Sacken and J. R. Dahn, J. Electrochem. Soc., 1990, 137, 2009 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS.
- D. Guyomard and J. M. Tarascon, J. Electrochem. Soc., 1993, 140, 3071–3081 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- D. Larcher, S. Beattie, M. Morcrette, K. Edstrom, J.-C. Jumas and J.-M. Tarascon, J. Mater. Chem., 2007, 17, 3759–3772 RSC.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- D. Bresser, S. Passerini and B. Scrosati, Energy Environ. Sci., 2016, 9, 3348–3367 RSC.
- Y.-G. Guo, J.-S. Hu and L.-J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- N. Loeffler, D. Bresser, S. Passerini and M. Copley, Johnson Matthey Technol. Rev., 2015, 59, 34–44 CrossRef CAS.
- S. Fang, D. Bresser and S. Passerini, Adv. Energy Mater., 2020, 10, 1902485 CrossRef CAS.
- M. Winter and J. O. Besenhard, in Handbook of Battery Materials, Wiley-VCH, Weinheim, Germany, 1999, vol. 1, pp. 383–418 Search PubMed.
- M. Winter, K.-C. Möller and J. O. Besenhard, in Lithium batteries - Science and technology, Springer, USA, 1st edn, 2003, pp. 144–194 Search PubMed.
- M. Inagaki, in Carbons for Electrochemical Energy Storage and Conversion Systems, CRC Press, Taylor & Francis Group, Boca Raton, London, New York, 1st edn, 2010, pp. 37–76 Search PubMed.
- P. Novák, D. Goers and M. E. Spahr, in Carbons for Electrochemical Energy Storage and Conversion Systems, CRC Press, Taylor & Francis Group, Boca Raton, London, New York, 1st edn, 2010, pp. 263–328 Search PubMed.
- E. A. Olivetti, G. Ceder, G. G. Gaustad and X. Fu, Joule, 2017, 1, 229–243 CrossRef.
- D. D. L. Chung, J. Mater. Sci., 2002, 37, 1475–1489 CrossRef CAS.
- J. D. Bernal, Proc. R. Soc. A, 1924, 106, 749–773 CAS.
- H. Lipson and A. R. Stokes, Nature, 1942, 149, 328 CrossRef CAS.
- Z. Ogumi and H. Wang, in Lithium-Ion Batteries - Science and Technologies, Springer-Verlag, New York, U.S.A., 1st edn, 2009, pp. 49–73 Search PubMed.
- H. Shi, J. Barker, M. Y. Saïdi and R. Koksbang, J. Electrochem. Soc., 1996, 143, 3466 CrossRef CAS.
- H. O. Pierson, Handbook of Carbon, Graphite, Diamonds and Fullerenes, Noyes Publications, New Jersey, USA, 1st edn, 1994 Search PubMed.
- I. L. Spain and D. A. Young, Philos. Trans. R. Soc., A, 1967, 262, 345–386 CrossRef CAS.
- M. Smalc, G. Shives, G. Chen, S. Guggari, J. Norley and R. A. Reynolds, in ASME 2005 Pacific Rim Technical Conference and Exhibition on Integration and Packaging of MEMS, NEMS, and Electronic Systems collocated with the ASME 2005 Heat Transfer Summer Conference, American Society of Mechanical Engineers, 2005, pp. 79–89 Search PubMed.
- T. Tran and K. Kinoshita, J. Electroanal. Chem., 1995, 386, 221–224 CrossRef.
- C. Schafhaeutl, J. Prakt. Chem., 1840, 21, 129–157 CrossRef.
- T. Nakajima, A. Mabuchi and R. Hagiwara, Carbon, 1988, 26, 357–361 CrossRef CAS.
- Y. Kita, N. Watanabe and Y. Fujii, J. Am. Chem. Soc., 1979, 101, 3832–3841 CrossRef CAS.
- U. Hofmann and W. Rüdorff, Trans. Faraday Soc., 1938, 34, 1017–1021 RSC.
- W. Rüdorff and U. Hofmann, Z. Anorg. Allg. Chem., 1938, 238, 1–50 CrossRef.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- N. Daumas and A. Herold, C. R. Hebd. Seances Acad. Sci., Ser. C, 1969, 268, 373 CAS.
- S. H. A. Axdal and D. D. L. Chung, Carbon, 1987, 25, 377–389 CrossRef CAS.
- G. Kirczenow, Phys. Rev. Lett., 1985, 55, 2810–2813 CrossRef CAS PubMed.
- J. M. Thomas, G. R. Millward, R. F. Schlögl and H. P. Boehm, Mater. Res. Bull., 1980, 15, 671–676 CrossRef CAS.
- R. Levi-Setti, G. Crow, Y. L. Wang, N. W. Parker, R. Mittleman and D. M. Hwang, Phys. Rev. Lett., 1985, 54, 2615–2618 CrossRef CAS PubMed.
- A. M. Dimiev, G. Ceriotti, N. Behabtu, D. Zakhidov, M. Pasquali, R. Saito and J. M. Tour, ACS Nano, 2013, 7, 2773–2780 CrossRef CAS PubMed.
- E. R. White, J. J. Lodico and B. C. Regan, Nat. Commun., 2017, 8, 1969 CrossRef PubMed.
- W. R. McKinnon and R. R. Haering, in Modern Aspects of Electrochemistry, Springer US, Boston, MA, 1983, pp. 235–304 Search PubMed.
- X. Y. Song, K. Kinoshita and T. D. Tran, J. Electrochem. Soc., 1996, 143, L120 CrossRef CAS.
- J. R. Dahn, Phys. Rev. B, 1991, 44, 9170–9177 CrossRef CAS PubMed.
- A. Senyshyn, O. Dolotko, M. J. Mühlbauer, K. Nikolowski, H. Fuess and H. Ehrenberg, J. Electrochem. Soc., 2013, 160, A3198–A3205 CrossRef CAS.
- D. Billaud, F. X. Henry, M. Lelaurain and P. Willmann, J. Phys. Chem. Solids, 1996, 57, 775–781 CrossRef CAS.
- D. Billaud and F. X. Henry, Solid State Commun., 2002, 124, 299–304 CrossRef CAS.
- M. Drüe, M. Seyring and M. Rettenmayr, J. Power Sources, 2017, 353, 58–66 CrossRef.
- M. Heß and P. Novák, Electrochim. Acta, 2013, 106, 149–158 CrossRef.
- M. D. Levi and D. Aurbach, J. Electroanal. Chem., 1997, 421, 79–88 CrossRef CAS.
- M. D. Levi, E. A. Levi and D. Aurbach, J. Electroanal. Chem., 1997, 421, 89–97 CrossRef CAS.
- A. Missyul, I. Bolshakov and R. Shpanchenko, Powder Diffr., 2017, 32, S56–S62 CrossRef CAS.
- N. A. Cañas, P. Einsiedel, O. T. Freitag, C. Heim, M. Steinhauer, D.-W. Park and K. A. Friedrich, Carbon, 2017, 116, 255–263 CrossRef.
- A. Senyshyn, M. J. Mühlbauer, K. Nikolowski, T. Pirling and H. Ehrenberg, J. Power Sources, 2012, 203, 126–129 CrossRef CAS.
- X.-L. Wang, K. An, L. Cai, Z. Feng, S. E. Nagler, C. Daniel, K. J. Rhodes, A. D. Stoica, H. D. Skorpenske, C. Liang, W. Zhang, J. Kim, Y. Qi and S. J. Harris, Sci. Rep., 2012, 2, 747 CrossRef PubMed.
- C. Sole, N. E. Drewett and L. J. Hardwick, Faraday Discuss., 2014, 172, 223–237 RSC.
- S. Krishnan, G. Brenet, E. Machado-Charry, D. Caliste, L. Genovese, T. Deutsch and P. Pochet, Appl. Phys. Lett., 2013, 103, 251904 CrossRef.
- D. Allart, M. Montaru and H. Gualous, J. Electrochem. Soc., 2018, 165, A380–A387 CrossRef CAS.
- T. Zheng and J. R. Dahn, Phys. Rev. B, 1996, 53, 3061–3071 CrossRef PubMed.
- X. Zhang, R. Kostecki, T. J. Richardson, J. K. Pugh and P. N. Ross, J. Electrochem. Soc., 2001, 148, A1341–A1345 CrossRef CAS.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef CAS PubMed.
- Y. Nishi, Chem. Rec., 2001, 1, 406–413 CrossRef CAS PubMed.
- M. Winter, Z. Phys. Chem., 2009, 223, 1395 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- J. O. Besenhard, M. Winter, J. Yang and W. Biberacher, J. Power Sources, 1995, 54, 228–231 CrossRef CAS.
- E. Peled, J. Power Sources, 1983, 9, 253–266 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- E. Peled, D. Golodnitsky and G. Ardel, J. Electrochem. Soc., 1997, 144, L208 CrossRef CAS.
- D. Aurbach, B. Markovsky, A. Shechter, Y. Ein-Eli and H. Cohen, J. Electrochem. Soc., 1996, 143, 3809 CrossRef CAS.
- X. Yu and A. Manthiram, Energy Environ. Sci., 2018, 11, 527–543 RSC.
- D. Aurbach, B. Markovsky, I. Weissman, E. Levi and Y. Ein-Eli, Electrochim. Acta, 1999, 45, 67–86 CrossRef CAS.
- K. Edström, M. Herstedt and D. P. Abraham, J. Power Sources, 2006, 153, 380–384 CrossRef.
- D. Bar-Tow, E. Peled and L. Burstein, J. Electrochem. Soc., 1999, 146, 824 CrossRef CAS.
- E. Peled, D. Bar Tow, A. Merson, A. Gladkich, L. Burstein and D. Golodnitsky, J. Power Sources, 2001, 97–98, 52–57 CrossRef CAS.
- S.-K. Jeong, M. Inaba, T. Abe and Z. Ogumi, J. Electrochem. Soc., 2001, 148, A989–A993 CrossRef CAS.
- G. Zampardi, F. La Mantia and W. Schuhmann, RSC Adv., 2015, 5, 31166–31171 RSC.
- H. Bülter, F. Peters, J. Schwenzel and G. Wittstock, Angew. Chem., Int. Ed., 2014, 53, 10531–10535 CrossRef PubMed.
- A. v. Cresce, S. M. Russell, D. R. Baker, K. J. Gaskell and K. Xu, Nano Lett., 2014, 14, 1405–1412 CrossRef CAS PubMed.
- T. Kranz, S. Kranz, V. Miß, J. Schepp and B. Roling, J. Electrochem. Soc., 2017, 164, A3777–A3784 CrossRef CAS.
- G. Ramos-Sanchez, F. A. Soto, J. M. Martinez de la Hoz, Z. Liu, P. P. Mukherjee, F. El-Mellouhi, J. M. Seminario and P. B. Balbuena, J. Electrochem. Energy Convers. Storage, 2016, 13, 031002 CrossRef.
- R. N. Methekar, P. W. C. Northrop, K. Chen, R. D. Braatz and V. R. Subramanian, J. Electrochem. Soc., 2011, 158, A363 CrossRef CAS.
- F. Single, B. Horstmann and A. Latz, Phys. Chem. Chem. Phys., 2016, 18, 17810–17814 RSC.
- F. Single, B. Horstmann and A. Latz, J. Electrochem. Soc., 2017, 164, E3132–E3145 CrossRef CAS.
- D. Aurbach, M. D. Levi, E. Levi and A. Schechter, J. Phys. Chem. B, 1997, 101, 2195–2206 CrossRef CAS.
- S. Shi, P. Lu, Z. Liu, Y. Qi, L. G. Hector, H. Li and S. J. Harris, J. Am. Chem. Soc., 2012, 134, 15476–15487 CrossRef CAS PubMed.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed.
- E. Peled, D. Bar Tow, A. Merson and L. Burnstein, J. New Mater. Electrochem. Syst., 2000, 3, 321–328 Search PubMed.
- Y. Domi, M. Ochida, S. Tsubouchi, H. Nakagawa, T. Yamanaka, T. Doi, T. Abe and Z. Ogumi, J. Phys. Chem. C, 2011, 115, 25484–25489 CrossRef CAS.
- A. Funabiki, M. Inaba and Z. Ogumi, J. Power Sources, 1997, 68, 227–231 CrossRef CAS.
- K. R. Kneten and R. L. McCreery, Anal. Chem., 1992, 64, 2518–2524 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. Abraham, J. Phys. Chem. C, 2013, 117, 19255–19269 CrossRef CAS.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. Abraham, J. Phys. Chem. C, 2013, 117, 19270–19279 CrossRef CAS.
- L. Xing, X. Zheng, M. Schroeder, J. Alvarado, A. von Wald Cresce, K. Xu, Q. Li and W. Li, Acc. Chem. Res., 2018, 51, 282–289 CrossRef CAS PubMed.
- P. Meister, H. Jia, J. Li, R. Kloepsch, M. Winter and T. Placke, Chem. Mater., 2016, 28, 7203–7217 CrossRef CAS.
- A. Birrozzi, J. Asenbauer, T. E. Ashton, A. R. Groves, D. Geiger, U. Kaiser, J. A. Darr and D. Bresser, Batteries Supercaps, 2020, 3, 284–292 CrossRef CAS.
- K. G. Gallagher, S. E. Trask, C. Bauer, T. Woehrle, S. F. Lux, M. Tschech, P. Lamp, B. J. Polzin, S. Ha, B. Long, Q. Wu, W. Lu, D. W. Dees and A. N. Jansen, J. Electrochem. Soc., 2016, 163, A138–A149 CrossRef CAS.
- T. Waldmann, B.-I. Hogg and M. Wohlfahrt-Mehrens, J. Power Sources, 2018, 384, 107–124 CrossRef CAS.
- X.-G. Yang, T. Liu, Y. Gao, S. Ge, Y. Leng, D. Wang and C.-Y. Wang, Joule, 2019, 3, 3002–3019 CrossRef CAS.
- K. Zaghib, F. Brochu, A. Guerfi and K. Kinoshita, J. Power Sources, 2001, 103, 140–146 CrossRef CAS.
- J. C. Arrebola, A. Caballero, L. Hernán and J. Morales, J. Electrochem. Soc., 2009, 156, A986 CrossRef CAS.
- H. Buqa, D. Goers, M. Holzapfel, M. E. Spahr and P. Novák, J. Electrochem. Soc., 2005, 152, A474 CrossRef CAS.
- M. Kühne, F. Paolucci, J. Popovic, P. M. Ostrovsky, J. Maier and J. H. Smet, Nat. Nanotechnol., 2017, 12, 895–900 CrossRef PubMed.
- K. Persson, V. A. Sethuraman, L. J. Hardwick, Y. Hinuma, Y. S. Meng, A. van der Ven, V. Srinivasan, R. Kostecki and G. Ceder, J. Phys. Chem. Lett., 2010, 1, 1176–1180 CrossRef CAS.
- M. D. Levi, C. Wang, E. Markevich, D. Aurbach and Z. Chvoj, J. Solid State Electrochem., 2003, 8, 40–43 CrossRef CAS.
- Y. Guo, R. B. Smith, Z. Yu, D. K. Efetov, J. Wang, P. Kim, M. Z. Bazant and L. E. Brus, J. Phys. Chem. Lett., 2016, 7, 2151–2156 CrossRef CAS PubMed.
- M. Bauer, M. Wachtler, H. Stöwe, J. V. Persson and M. A. Danzer, J. Power Sources, 2016, 317, 93–102 CrossRef CAS.
- S. S. Zhang, K. Xu and T. R. Jow, Electrochim. Acta, 2004, 49, 1057–1061 CrossRef CAS.
- C. Wang, A. J. Appleby and F. E. Little, J. Electrochem. Soc., 2002, 149, A754 CrossRef CAS.
- T. R. Jow, S. A. Delp, J. L. Allen, J.-P. Jones and M. C. Smart, J. Electrochem. Soc., 2018, 165, A361–A367 CrossRef CAS.
- T. Abe, H. Fukuda, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1120 CrossRef CAS.
- T. Abe, F. Sagane, M. Ohtsuka, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2005, 152, A2151 CrossRef.
- T. R. Jow, M. B. Marx and J. L. Allen, J. Electrochem. Soc., 2012, 159, A604 CrossRef CAS.
- J.-P. Jones, M. C. Smart, F. C. Krause, B. V. Ratnakumar and E. J. Brandon, ECS Trans., 2017, 75, 1–11 CrossRef CAS.
- Y. Yamada, Y. Iriyama, T. Abe and Z. Ogumi, Langmuir, 2009, 25, 12766–12770 CrossRef CAS PubMed.
- E. Peled, C. Menachem, D. Bar-Tow and A. Melman, J. Electrochem. Soc., 1996, 143, L4–L7 CrossRef CAS.
- Y. Ein-Eli and V. R. Koch, J. Electrochem. Soc., 1997, 144, 2968 CrossRef CAS.
- P. Yu, J. A. Ritter, R. E. White and B. N. Popov, J. Electrochem. Soc., 2000, 147, 1280–1285 CrossRef CAS.
- P. Yu, J. A. Ritter, R. E. White and B. N. Popov, J. Electrochem. Soc., 2000, 147, 2081–2085 CrossRef CAS.
- T. Takamura, K. Sumiya, J. Suzuki, C. Yamada and K. Sekine, J. Power Sources, 1999, 81–82, 368–372 CrossRef CAS.
- F. Nobili, S. Dsoke, T. Mecozzi and R. Marassi, Electrochim. Acta, 2005, 51, 536–544 CrossRef CAS.
- F. Nobili, S. Dsoke, M. Mancini, R. Tossici and R. Marassi, J. Power Sources, 2008, 180, 845–851 CrossRef CAS.
- F. Nobili, M. Mancini, P. E. Stallworth, F. Croce, S. G. Greenbaum and R. Marassi, J. Power Sources, 2012, 198, 243–250 CrossRef CAS.
- B. Veeraraghavan, A. Durairajan, B. Haran, B. Popov and R. Guidotti, J. Electrochem. Soc., 2002, 149, A675 CrossRef CAS.
- I. T. Kim, J. Lee, J. C. An, E. Jung, H. K. Lee, M. Morita and J. Shim, Int. J. Electrochem. Sci., 2016, 11, 5807–5818 CrossRef CAS.
- K. R. Tallman, B. Zhang, L. Wang, S. Yan, K. Thompson, X. Tong, J. Thieme, A. Kiss, A. C. Marschilok, K. J. Takeuchi, D. C. Bock and E. S. Takeuchi, ACS Appl. Mater. Interfaces, 2019, 11, 46864–46874 CrossRef CAS PubMed.
- F. Ding, W. Xu, D. Choi, W. Wang, X. Li, M. H. Engelhard, X. Chen, Z. Yang and J.-G. Zhang, J. Mater. Chem., 2012, 22, 12745 RSC.
- T. Feng, Y. Xu, Z. Zhang, X. Du, X. Sun, L. Xiong, R. Rodriguez and R. Holze, ACS Appl. Mater. Interfaces, 2016, 8, 6512–6519 CrossRef CAS PubMed.
- Y. S. Jung, A. S. Cavanagh, L. A. Riley, S. H. Kang, A. C. Dillon, M. D. Groner, S. M. George and S. H. Lee, Adv. Mater., 2010, 22, 2172–2176 CrossRef CAS PubMed.
- M. Gaberscek, Electrochem. Solid-State Lett., 1999, 3, 171 CrossRef.
- M. Gaberšček, M. Bele, J. Drofenik, R. Dominko and S. Pejovnik, J. Power Sources, 2001, 97–98, 67–69 CrossRef.
- K. Guo, Q. Pan and S. Fang, J. Power Sources, 2002, 111, 350–356 CrossRef CAS.
- B. Veeraraghavan, J. Paul, B. Haran and B. Popov, J. Power Sources, 2002, 109, 377–387 CrossRef CAS.
- T. Doi, K. Takeda, T. Fukutsuka, Y. Iriyama, T. Abe and Z. Ogumi, Carbon, 2005, 43, 2352–2357 CrossRef CAS.
- Q. Pan, H. Wang and Y. Jiang, J. Mater. Chem., 2007, 17, 329–334 RSC.
- F. S. Li, Y. S. Wu, J. Chou, M. Winter and N. L. Wu, Adv. Mater., 2014, 27, 130–137 CrossRef PubMed.
- K. Zaghib, A. Guerfi, P. Hovington, A. Vijh, M. Trudeau, A. Mauger, J. B. Goodenough and C. M. Julien, J. Power Sources, 2013, 232, 357–369 CrossRef CAS.
- R. Dominko, M. Gaberscek, M. Bele, D. Mihailovic and J. Jamnik, J. Eur. Ceram. Soc., 2007, 27, 909–913 CrossRef CAS.
- J. Wang and X. Sun, Energy Environ. Sci., 2012, 5, 5163–5185 RSC.
- L. Zhao, Y.-S. Hu, H. Li, Z. Wang and L. Chen, Adv. Mater., 2011, 23, 1385–1388 CrossRef CAS PubMed.
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC.
- D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513–523 CrossRef CAS.
- D. Bresser, E. Paillard, E. Binetti, S. Krueger, M. Striccoli, M. Winter and S. Passerini, J. Power Sources, 2012, 206, 301–309 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, Y. Hara and Y. Adachi, J. Electrochem. Soc., 2000, 147, 1245 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, T. Umeno, T. Abe and Z. Ogumi, J. Mater. Chem., 2004, 14, 1754–1758 RSC.
- Y.-S. Park, H. J. Bang, S.-M. Oh, Y.-K. Sun and S.-M. Lee, J. Power Sources, 2009, 190, 553–557 CrossRef CAS.
- V. Sharova, A. Moretti, G. Giffin, D. Carvalho and S. Passerini, C, 2017, 3, 22 Search PubMed.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- W. Zhao, Y. Ji, Z. Zhang, M. Lin, Z. Wu, X. Zheng, Q. Li and Y. Yang, Curr. Opin. Electrochem., 2017, 6, 84–91 CrossRef CAS.
- R. Mogi, M. Inaba, S.-K. Jeong, Y. Iriyama, T. Abe and Z. Ogumi, J. Electrochem. Soc., 2002, 149, A1578–A1583 CrossRef CAS.
- I. A. Profatilova, S.-S. Kim and N.-S. Choi, Electrochim. Acta, 2009, 54, 4445–4450 CrossRef CAS.
- O. Matsuoka, A. Hiwara, T. Omi, M. Toriida, T. Hayashi, C. Tanaka, Y. Saito, T. Ishida, H. Tan, S. S. Ono and S. Yamamoto, J. Power Sources, 2002, 108, 128–138 CrossRef CAS.
- H. Ota, Y. Sakata, A. Inoue and S. Yamaguchi, J. Electrochem. Soc., 2004, 151, A1659–A1669 CrossRef CAS.
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423–1439 CrossRef CAS.
- R. W. Schmitz, P. Murmann, R. Schmitz, R. Müller, L. Krämer, J. Kasnatscheew, P. Isken, P. Niehoff, S. Nowak, G.-V. Röschenthaler, N. Ignatiev, P. Sartori, S. Passerini, M. Kunze, A. Lex-Balducci, C. Schreiner, I. Cekic-Laskovic and M. Winter, Prog. Solid State Chem., 2014, 42, 65–84 CrossRef CAS.
- Y. Hu, W. Kong, H. Li, X. Huang and L. Chen, Electrochem. Commun., 2004, 6, 126–131 CrossRef CAS.
- J. C. Burns, N. N. Sinha, D. J. Coyle, G. Jain, C. M. VanElzen, W. M. Lamanna, A. Xiao, E. Scott, J. P. Gardner and J. R. Dahn, J. Electrochem. Soc., 2011, 159, A85–A90 CrossRef.
- H.-C. Wu, C.-Y. Su, D.-T. Shieh, M.-H. Yang and N.-L. Wu, Electrochem. Solid-State Lett., 2006, 9, A537–A541 CrossRef CAS.
- D. Takamatsu, Y. Orikasa, S. Mori, T. Nakatsutsumi, K. Yamamoto, Y. Koyama, T. Minato, T. Hirano, H. Tanida, H. Arai, Y. Uchimoto and Z. Ogumi, J. Phys. Chem. C, 2015, 119, 9791–9797 CrossRef CAS.
- D. Xiong, J. C. Burns, A. J. Smith, N. Sinha and J. R. Dahn, J. Electrochem. Soc., 2011, 158, A1431–A1435 CrossRef CAS.
- N. N. Sinha, J. C. Burns and J. R. Dahn, J. Electrochem. Soc., 2013, 160, A709–A714 CrossRef CAS.
- X. Wang, H. Naito, Y. Sone, G. Segami and S. Kuwajima, J. Electrochem. Soc., 2005, 152, A1996–A2001 CrossRef CAS.
- G. H. Wrodnigg, J. O. Besenhard and M. Winter, J. Electrochem. Soc., 1999, 146, 470–472 CrossRef CAS.
- G. H. Wrodnigg, T. M. Wrodnigg, J. O. Besenhard and M. Winter, Electrochem. Commun., 1999, 1, 148–150 CrossRef CAS.
- J. Vetter, P. Novak, M. R. Wagner, C. Veit, K.-C. Möller, J. O. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, J. Power Sources, 2005, 147, 269–281 CrossRef CAS.
- M. R. Palacín, Chem. Soc. Rev., 2018, 47, 4924–4933 RSC.
- A. Barré, B. Deguilhem, S. Grolleau, M. Gérard, F. Suard and D. Riu, J. Power Sources, 2013, 241, 680–689 CrossRef.
- C. Zhan, T. Wu, J. Lu and K. Amine, Energy Environ. Sci., 2018, 11, 243–257 RSC.
- W. Li, J. Electrochem. Soc., 2020, 167, 90514 CrossRef.
- S. C. Park, Y. M. Kim, S. C. Han, S. Ahn, C. H. Ku and J. Y. Lee, J. Power Sources, 2002, 107, 42–47 CrossRef CAS.
- K. Amine, J. Liu, S. Kang, I. Belharouak, Y. Hyung, D. Vissers and G. Henriksen, J. Power Sources, 2004, 129, 14–19 CrossRef CAS.
- A. Blyr, C. Sigala, G. Amatucci, D. Guyomard, Y. Chabre and J. -M. Tarascon, J. Electrochem. Soc., 1998, 145, 194–209 CrossRef CAS.
- Y. Terada, Y. Nishiwaki, I. Nakai and F. Nishikawa, J. Power Sources, 2001, 97–98, 420–422 CrossRef CAS.
- D. H. Jang, Y. J. Shin and S. M. Oh, J. Electrochem. Soc., 1996, 143, 2204–2211 CrossRef CAS.
- S. R. Gowda, K. G. Gallagher, J. R. Croy, M. Bettge, M. M. Thackeray and M. Balasubramanian, Phys. Chem. Chem. Phys., 2014, 16, 6898–6902 RSC.
- H. Tsunekawa, A. Tanimoto Satoshi, R. Marubayashi, M. Fujita, K. Kifune and M. Sano, J. Electrochem. Soc., 2002, 149, A1326–A1331 CrossRef CAS.
- J. Asenbauer, J. R. Binder, F. Mueller, M. Künzel, D. Geiger, U. Kaiser, S. Passerini and D. Bresser, ChemSusChem, 2020 DOI:10.1002/cssc.202000559.
- J. Betz, J.-P. Brinkmann, R. Nölle, C. Lürenbaum, M. Kolek, M. C. Stan, M. Winter and T. Placke, Adv. Energy Mater., 2019, 9, 1900574 CrossRef.
- J. Cho and M. M. Thackeray, J. Electrochem. Soc., 1999, 146, 3577–3581 CrossRef CAS.
- M. M. Thackeray, L. A. de Picciotto, A. de Kock, P. J. Johnson, V. A. Nicholas and K. T. Adendorff, J. Power Sources, 1987, 21, 1–8 CrossRef CAS.
- J. M. Tarascon, W. R. McKinnon, F. Coowar, T. N. Bowmer, G. Amatucci and D. Guyomard, J. Electrochem. Soc., 1994, 141, 1421–1431 CrossRef CAS.
- S. Komaba, N. Kumagai and Y. Kataoka, Electrochim. Acta, 2002, 47, 1229–1239 CrossRef CAS.
- C. Delacourt, A. Kwong, X. Liu, R. Qiao, W. L. Yang, P. Lu, S. J. Harris and V. Srinivasan, J. Electrochem. Soc., 2013, 160, A1099–A1107 CrossRef CAS.
- C. Zhan, J. Lu, A. J. Kropf, T. Wu, A. N. Jansen, Y.-K. Sun, X. Qiu and K. Amine, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- R. Sahore, F. Dogan and I. D. Bloom, Chem. Mater., 2019, 31, 2884–2891 CrossRef CAS.
- B. Michalak, B. B. Berkes, H. Sommer, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2017, 121, 211–216 CrossRef CAS.
- S. Wiemers-Meyer, M. Winter and S. Nowak, Phys. Chem. Chem. Phys., 2017, 19, 4962–4966 RSC.
- M. Gruetzke, W. Weber, M. Winter and S. Nowak, RSC Adv., 2016, 6, 57253–57260 RSC.
- M. Tochihara, H. Nara, D. Mukoyama, T. Yokoshima, T. Momma and T. Osaka, J. Electrochem. Soc., 2015, 162, A2008–A2015 CrossRef CAS.
- X. Feng, M. Ouyang, X. Liu, L. Lu, Y. Xia and X. He, Energy Storage Materials, 2018, 10, 246–267 CrossRef.
- Q. Wang, J. Sun, X. Yao and C. Chen, Thermochim. Acta, 2005, 437, 12–16 CrossRef CAS.
- Z. Chen, Y. Qin, Y. Ren, W. Lu, C. Orendorff, E. P. Roth and K. Amine, Energy Environ. Sci., 2011, 4, 4023–4030 RSC.
- J. Yamaki, H. Takatsuji, T. Kawamura and M. Egashira, Solid State Ionics, 2002, 148, 241–245 CrossRef CAS.
- Q. Wang, P. Ping, X. Zhao, G. Chu, J. Sun and C. Chen, J. Power Sources, 2012, 208, 210–224 CrossRef CAS.
- T. Waldmann and M. Wohlfahrt-Mehrens, Electrochim. Acta, 2017, 230, 454–460 CrossRef CAS.
- T. Waldmann, M. Wilka, M. Kasper, M. Fleischhammer and M. Wohlfahrt-Mehrens, J. Power Sources, 2014, 262, 129–135 CrossRef CAS.
- C. von Lüders, V. Zinth, S. V. Erhard, P. J. Osswald, M. Hofmann, R. Gilles and A. Jossen, J. Power Sources, 2017, 342, 17–23 CrossRef.
- M. Petzl, M. Kasper and M. A. Danzer, J. Power Sources, 2015, 275, 799–807 CrossRef CAS.
- M. Petzl and M. A. Danzer, J. Power Sources, 2014, 254, 80–87 CrossRef CAS.
- M. Fleischhammer, T. Waldmann, G. Bisle, B. I. Hogg and M. Wohlfahrt-Mehrens, J. Power Sources, 2015, 274, 432–439 CrossRef CAS.
- S. Rothermel, M. Evertz, J. Kasnatscheew, X. Qi, M. Gruetzke, M. Winter and S. Nowak, ChemSusChem, 2016, 9, 3473–3484 CrossRef CAS PubMed.
- Y. He, T. Zhang, F. Wang, G. Zhang, W. Zhang and J. Wang, J. Cleaner Prod., 2017, 143, 319–325 CrossRef CAS.
- B. Moradi and G. G. Botte, J. Appl. Electrochem., 2016, 46, 123–148 CrossRef CAS.
- X. Wang, G. Gaustad, C. W. Babbitt, C. Bailey, M. J. Ganter and B. J. Landi, J. Environ. Manage., 2014, 135, 126–134 CrossRef PubMed.
- E. Tzimas, SETIS Magazine, 2015, 9, 26–29 Search PubMed.
- C. Hanisch, T. Loellhoeffel, J. Diekmann, K. J. Markley, W. Haselrieder and A. Kwade, J. Cleaner Prod., 2015, 108, 1–11 CrossRef.
- D. A. Bertuol, C. Toniasso, B. M. Jiménez, L. Meili, G. L. Dotto, E. H. Tanabe and M. L. Aguiar, J. Power Sources, 2015, 275, 627–632 CrossRef CAS.
- A. V. Tamashausky, NLGI Conf. Proc., NLGI Paper#0505, 2005, pp. 1–16 Search PubMed.
- S. L. Glazier, J. Li, A. J. Louli, J. P. Allen and J. R. Dahn, J. Electrochem. Soc., 2017, 164, A3545–A3555 CrossRef CAS.
- I. Mochida, Y. Korai, Y.-G. Wang and S.-H. Hong, in World of Carbon, Gordon and Breach Science Publishers, Amsterdam, Netherlands, 1st edn, 2001, pp. 221–248 Search PubMed.
- M. Mundszinger, S. Farsi, M. Rapp, U. Golla-Schindler, U. Kaiser and M. Wachtler, Carbon, 2017, 111, 764–773 CrossRef CAS.
- H. Zhao, J. Ren, X. He, J. Li, C. Jiang and C. Wan, Solid State Sci., 2008, 10, 612–617 CrossRef CAS.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- P. P. Magampa, N. Manyala and W. W. Focke, J. Nucl. Mater., 2013, 436, 76–83 CrossRef CAS.
- X. Dou, I. Hasa, D. Saurel, C. Vaalma, L. Wu, D. Buchholz, D. Bresser, S. Komaba and S. Passerini, Mater. Today, 2019, 23, 87–104 CrossRef CAS.
- K. Zaghib, X. Song, A. Guerfi, R. Rioux and K. Kinoshita, J. Power Sources, 2003, 119–121, 8–15 CrossRef CAS.
- C. Ma, Y. Zhao, J. Li, Y. Song, J. Shi, Q. Guo and L. Liu, Carbon, 2013, 64, 553–556 CrossRef CAS.
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Energy Environ. Sci., 2018, 11, 3096–3127 RSC.
- W. Zhu, D. R. Schmehl, C. A. Mullin and J. L. Frazier, PLoS One, 2014, 9, e77547 CrossRef PubMed.
- F. M. Courtel, S. Niketic, D. Duguay, Y. Abu-Lebdeh and I. J. Davidson, J. Power Sources, 2011, 196, 2128–2134 CrossRef CAS.
- T. S. Poet, C. R. Kirman, M. Bader, C. van Thriel, M. L. Gargas and P. M. Hinderliter, Toxicol. Sci., 2010, 113, 468–482 CrossRef CAS PubMed.
- K. P. Lee, N. C. Chromey, R. Culik, J. R. Barnes and P. W. Schneider, Fundam. Appl. Toxicol., 1987, 9, 222–235 CrossRef CAS PubMed.
- D. E. Malek, L. A. Malley, T. W. Slone, G. S. Elliott, G. L. Kennedy, W. Mellert, K. Deckardt, C. Gembardt, B. Hildebrand, S. R. Murphy, D. B. Bower and G. A. Wright, Drug Chem. Toxicol., 1997, 20, 63–77 CrossRef CAS PubMed.
- J. Drofenik, M. Gaberšček, R. Dominko, M. Bele and S. Pejovnik, J. Power Sources, 2001, 94, 97–101 CrossRef CAS.
- J. Drofenik, M. Gaberscek, R. Dominko, F. W. Poulsen, M. Mogensen, S. Pejovnik and J. Jamnik, Electrochim. Acta, 2003, 48, 883–889 CrossRef CAS.
- S. F. Lux, F. Schappacher, A. Balducci, S. Passerini and M. Winter, J. Electrochem. Soc., 2010, 157, A320–A325 CrossRef CAS.
- C. Forestier, S. Grugeon, C. Davoisne, A. Lecocq, G. Marlair, M. Armand, L. Sannier and S. Laruelle, J. Power Sources, 2016, 330, 186–194 CrossRef CAS.
- J.-H. Lee, S. Lee, U. Paik and Y.-M. Choi, J. Power Sources, 2005, 147, 249–255 CrossRef CAS.
- J.-P. Yen, C.-M. Lee, T.-L. Wu, H.-C. Wu, C.-Y. Su, N.-L. Wu and J.-L. Hong, ECS Electrochem. Lett., 2012, 1, A80–A82 CrossRef CAS.
- Y.-S. Park, E.-S. Oh and S.-M. Lee, J. Power Sources, 2014, 248, 1191–1196 CrossRef CAS.
- H. Buqa, M. Holzapfel, F. Krumeich, C. Veit and P. Novák, J. Power Sources, 2006, 161, 617–622 CrossRef CAS.
- M. W. Verbrugge and B. J. Koch, J. Electrochem. Soc., 2003, 150, A374 CrossRef CAS.
- S. S. Zhang, K. Xu and T. R. Jow, J. Power Sources, 2004, 138, 226–231 CrossRef CAS.
- S. S. Zhang and T. R. Jow, J. Power Sources, 2002, 109, 422–426 CrossRef CAS.
- H. Maleki, G. Deng, I. Kerzhner-Haller, A. Anani and J. N. Howard, J. Electrochem. Soc., 2000, 147, 4470 CrossRef CAS.
- L. K. Willenberg, P. Dechent, G. Fuchs, D. U. Sauer and E. Figgemeier, Sustainability, 2020, 12, 557 CrossRef.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- Y. Zhang, Y. Zhu, L. Fu, J. Meng, N. Yu, J. Wang and Y. Wu, Chin. J. Chem., 2017, 35, 21–29 CrossRef CAS.
- M. Yoshio, T. Tsumura and N. Dimov, J. Power Sources, 2005, 146, 10–14 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- J. K. Lee, C. Oh, N. Kim, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2016, 4, 5366–5384 RSC.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Y. Xie and O. Yamamoto, Solid State Ionics, 2002, 152–153, 125–129 CrossRef CAS.
- K. Schulmeister and W. Mader, J. Non-Cryst. Solids, 2003, 320, 143–150 CrossRef CAS.
- R. J. Temkin, J. Non-Cryst. Solids, 1975, 17, 215–230 CrossRef CAS.
- H. Yamamura, K. Nobuhara, S. Nakanishi, H. Iba and S. Okada, J. Ceram. Soc. Jpn., 2011, 119, 855–860 CrossRef CAS.
- M. A. Al-Maghrabi, J. Suzuki, R. J. Sanderson, V. L. Chevrier, R. A. Dunlap and J. R. Dahn, J. Electrochem. Soc., 2013, 160, A1587–A1593 CrossRef CAS.
- K. Yasuda, Y. Kashitani, S. Kizaki, K. Takeshita, T. Fujita and S. Shimosaki, J. Power Sources, 2016, 329, 462–472 CrossRef CAS.
- S. C. Jung, H. J. Kim, J. H. Kim and Y. K. Han, J. Phys. Chem. C, 2016, 120, 886–892 CrossRef CAS.
- M. Yamada, A. Inaba, A. Ueda, K. Matsumoto, T. Iwasaki and T. Ohzuku, J. Electrochem. Soc., 2012, 159, A1630–A1635 CrossRef CAS.
- U.S. Department of Energy, Prog. Rep. Adv. Battery Mater. Res. BMR Program, 2017 Search PubMed.
- X. Chen, C. Li, M. Grätzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Adv. Energy Mater., 2014, 4, 1300882 CrossRef.
- A. S. Aricò, P. Bruce, B. Scrosati, J. M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 RSC.
- C.-H. Doh, C.-W. Park, H.-M. Shin, D.-H. Kim, Y.-D. Chung, S.-I. Moon, B.-S. Jin, H.-S. Kim and A. Veluchamy, J. Power Sources, 2008, 179, 367–370 CrossRef CAS.
- C.-H. Doh, H.-M. Shin, D.-H. Kim, Y.-C. Ha, B.-S. Jin, H.-S. Kim, S.-I. Moon and A. Veluchamy, Electrochem. Commun., 2008, 10, 233–237 CrossRef CAS.
- Z. Lu, L. Zhang and X. Liu, J. Power Sources, 2010, 195, 4304–4307 CrossRef CAS.
- T. Morita and N. Takami, J. Electrochem. Soc., 2006, 153, A425 CrossRef CAS.
- W. Wu, J. Shi, Y. Liang, F. Liu, Y. Peng and H. Yang, Phys. Chem. Chem. Phys., 2015, 17, 13451–13456 RSC.
- P. Lv, H. Zhao, C. Gao, Z. Du, J. Wang and X. Liu, J. Power Sources, 2015, 274, 542–550 CrossRef CAS.
- P. Lv, H. Zhao, C. Gao, T. Zhang and X. Liu, Electrochim. Acta, 2015, 152, 345–351 CrossRef CAS.
- M.-S. Park, E. Park, J. Lee, G. Jeong, K. J. Kim, J. H. Kim, Y.-J. Kim and H. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 9608–9613 CrossRef CAS PubMed.
- D. J. Lee, M.-H. Ryou, J.-N. Lee, B. G. Kim, Y. M. Lee, H.-W. Kim, B.-S. Kong, J.-K. Park and J. W. Choi, Electrochem. Commun., 2013, 34, 98–101 CrossRef CAS.
- J.-H. Kim, H.-J. Sohn, H. Kim, G. Jeong and W. Choi, J. Power Sources, 2007, 170, 456–459 CrossRef CAS.
- Y. Yao, J. Zhang, L. Xue, T. Huang and A. Yu, J. Power Sources, 2011, 196, 10240–10243 CrossRef CAS.
- K. W. Kim, H. Park, J. G. Lee, J. Kim, Y.-U. Kim, J. H. Ryu, J. J. Kim and S. M. Oh, Electrochim. Acta, 2013, 103, 226–230 CrossRef CAS.
- M. Yamada, A. Ueda, K. Matsumoto and T. Ohzuku, J. Electrochem. Soc., 2011, 158, A417 CrossRef CAS.
- T. Kajita, R. Yuge, K. Nakahara, J. Iriyama, H. Takahashi, R. Kasahara, T. Numata, S. Serizawa and K. Utsugi, J. Electrochem. Soc., 2013, 160, A1806–A1810 CrossRef CAS.
- T. Chen, J. Wu, Q. Zhang and X. Su, J. Power Sources, 2017, 363, 126–144 CrossRef CAS.
- A. Veluchamy, C.-H. Doh, D.-H. Kim, J.-H. Lee, D.-J. Lee, K.-H. Ha, H.-M. Shin, B.-S. Jin, H.-S. Kim, S.-I. Moon and C.-W. Park, J. Power Sources, 2009, 188, 574–577 CrossRef CAS.
- X. Yang, Z. Wen, X. Xu, B. Lin and S. Huang, J. Power Sources, 2007, 164, 880–884 CrossRef CAS.
- J. Hassoun, J. Kim, D.-J. Lee, H.-G. Jung, S.-M. Lee, Y.-K. Sun and B. Scrosati, J. Power Sources, 2012, 202, 308–313 CrossRef CAS.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- J. Hassoun, H.-G. Jung, D.-J. Lee, J.-B. Park, K. Amine, Y.-K. Sun and B. Scrosati, Nano Lett., 2012, 12, 5775–5779 CrossRef CAS PubMed.
- A. Varzi, D. Bresser, J. von Zamory, F. Mueller and S. Passerini, Adv. Energy Mater., 2014, 4, 1400054 CrossRef PubMed.
- H. J. Kim, S. Choi, S. J. Lee, M. W. Seo, J. G. Lee, E. Deniz, Y. J. Lee, E. K. Kim and J. W. Choi, Nano Lett., 2016, 16, 282–288 CrossRef CAS PubMed.
- R. W. Grant, M. Sweetland and A. M. Acharige, US Pat., US 10,128,487 B2, 2016.
- R. W. Grant, M. Sweetland and A. M. Acharige, US Pat., US 9,598,789 B2, 2012.
- V. L. Chevrier, L. Liu, R. Wohl, A. Chandrasoma, J. A. Vega, K. W. Eberman, P. Stegmaier and E. Figgemeier, J. Electrochem. Soc., 2018, 165, A1129–A1136 CrossRef CAS.
- S. W. Hwang and W. Y. Yoon, J. Electrochem. Soc., 2014, 161, A1753–A1758 CrossRef CAS.
- Y. Gao and J. Burba III, US Pat., No. 6,706,447, 2004.
- FMC Lithium, Stabilized Lithium Metal Powder (SLMP®), http://www.fmclithium.com/energy/Products/SLMP.aspx, accessed May 1, 2018 Search PubMed.
- H. Zhao, Z. Wang, P. Lu, M. Jiang, F. Shi, X. Song, Z. Zheng, X. Zhou, Y. Fu, G. Abdelbast, X. Xiao, Z. Liu, V. S. Battaglia, K. Zaghib and G. Liu, Nano Lett., 2014, 14, 6704–6710 CrossRef CAS PubMed.
- Q. Pan, P. Zuo, T. Mu, C. Du, X. Cheng, Y. Ma, Y. Gao and G. Yin, J. Power Sources, 2017, 347, 170–177 CrossRef CAS.
- M.-T. F. Rodrigues, J. A. Gilbert, K. Kalaga and D. P. Abraham, J. Phys.: Energy, 2020, 2, 024002 Search PubMed.
- A. Yamano, M. Morishita, M. Yanagida and T. Sakai, J. Electrochem. Soc., 2015, 162, A1730–A1737 CrossRef CAS.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S.-H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- J. Wang, X. He, E. Paillard, N. Laszczynski, J. Li and S. Passerini, Adv. Energy Mater., 2016, 6, 1600906 CrossRef.
- G. Gabrielli, M. Marinaro, M. Mancini, P. Axmann and M. Wohlfahrt-Mehrens, J. Power Sources, 2017, 351, 35–44 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- Y. Guo, H. Li and T. Zhai, Adv. Mater., 2017, 29, 1700007 CrossRef PubMed.
- M. Marinaro, D. Bresser, E. Beyer, P. Faguy, K. Hosoi, H. Li, J. Sakovica, K. Amine, M. Wohlfahrt-Mehrens and S. Passerini, J. Power Sources, 2020, 459, 228073 CrossRef CAS.
- V. Aravindan, Y.-S. Lee and S. Madhavi, Adv. Energy Mater., 2017, 7, 1602607 CrossRef.
- F. Holtstiege, P. Baermann, R. Noelle, M. Winter and T. Placke, Batteries, 2018, 4, 4–42 CrossRef.
- J. Li, R. B. Lewis and J. R. Dahn, Electrochem. Solid-State Lett., 2007, 10, A17–A20 CrossRef CAS.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004–3010 CrossRef CAS PubMed.
- J. Zhang, C. Zhang, Z. Liu, J. Zheng, Y. Zuo, C. Xue, C. Li and B. Cheng, J. Power Sources, 2017, 339, 86–92 CrossRef CAS.
- S. Komaba, K. Shimomura, N. Yabuuchi, T. Ozeki, H. Yui and K. Konno, J. Phys. Chem. C, 2011, 115, 13487–13495 CrossRef CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- U. S. Vogl, P. K. Das, A. Z. Weber, M. Winter, R. Kostecki and S. F. Lux, Langmuir, 2014, 30, 10299–10307 CrossRef CAS PubMed.
- B. Lestriez, S. Bahri, I. Sandu, L. Roué and D. Guyomard, Electrochem. Commun., 2007, 9, 2801–2806 CrossRef CAS.
- D. Mazouzi, Z. Karkar, C. Reale Hernandez, P. Jimenez Manero, D. Guyomard, L. Roué and B. Lestriez, J. Power Sources, 2015, 280, 533–549 CrossRef CAS.
- D. Reyter, S. Rousselot, D. Mazouzi, M. Gauthier, P. Moreau, B. Lestriez, D. Guyomard and L. Roué, J. Power Sources, 2013, 239, 308–314 CrossRef CAS.
- M. Kuenzel, D. Bresser, G.-T. Kim, P. Axmann, M. Wohlfahrt-Mehrens and S. Passerini, ACS Appl. Energy Mater., 2020, 3, 218–230 CrossRef CAS.
- J.-T. Li, Z.-Y. Wu, Y.-Q. Lu, Y. Zhou, Q.-S. Huang, L. Huang and S.-G. Sun, Adv. Energy Mater., 2017, 7, 1701185 CrossRef.
- Z. Du, J. Li, C. Daniel and D. L. Wood, Electrochim. Acta, 2017, 254, 123–129 CrossRef CAS.
- M.-S. Park, S. Rajendran, Y.-M. Kang, K.-S. Han, Y.-S. Han and J.-Y. Lee, J. Power Sources, 2006, 158, 650–653 CrossRef CAS.
- M.-S. Park, Y.-J. Lee, Y.-S. Han and J.-Y. Lee, Mater. Lett., 2006, 60, 3079–3083 CrossRef CAS.
- H. Jung, Y.-U. Kim, M.-S. Sung, Y. Hwa, G. Jeong, G.-B. Kim and H.-J. Sohn, J. Mater. Chem., 2011, 21, 11213–11216 RSC.
- S.-B. Son, S.-C. Kim, C.-S. Kang, T. A. Yersak, Y.-C. Kim and C.-G. Lee, Adv. Energy Mater., 2012, 2, 1226–1231 CrossRef CAS.
- K. J. Lee, S.-H. Yu, J.-J. Kim, D.-H. Lee, J. Park, S. S. Suh, J. S. Cho and Y.-E. Sung, J. Power Sources, 2014, 246, 729–735 CrossRef CAS.
- S.-S. Suh, W. Y. Yoon, C.-G. Lee, S.-U. Kwon, J.-H. Kim, Y. Matulevich, Y.-U. Kim, Y. Park, C.-U. Jeong, Y.-Y. Chan and S.-H. Kang, J. Electrochem. Soc., 2013, 160, A751–A755 CrossRef CAS.
- T. Song, K. C. Kil, Y. Jeon, S. Lee, W. C. Shin, B. Chung, K. Kwon and U. Paik, J. Power Sources, 2014, 253, 282–286 CrossRef CAS.
- M. N. Obrovac, L. Christensen, D. Le and J. Singh, ECS Meeting Abstracts, 2009, 542 Search PubMed.
- V. L. Chevrier, L. Liu, D. B. Le, J. Lund, B. Molla, K. Reimer, L. J. Krause, L. D. Jensen, E. Figgemeier and K. W. Eberman, J. Electrochem. Soc., 2014, 161, A783–A791 CrossRef CAS.
- Z. Du, R. A. Dunlap and M. N. Obrovac, J. Electrochem. Soc., 2014, 161, A1698–A1705 CrossRef CAS.
- M. Marinaro, D. hwan Yoon, G. Gabrielli, P. Stegmaier, E. Figgemeier, P. C. Spurk, D. Nelis, G. Schmidt, J. Chauveau, P. Axmann and M. Wohlfahrt-Mehrens, J. Power Sources, 2017, 357, 188–197 CrossRef CAS.
- V. L. Chevrier, J. W. Zwanziger and J. R. Dahn, Can. J. Phys., 2009, 87, 625–632 CrossRef CAS.
- R. A. Sharma and R. N. Seefurth, J. Electrochem. Soc., 1976, 123, 1763–1768 CrossRef CAS.
- L. Y. Beaulieu, T. D. Hatchard, A. Bonakdarpour, M. D. Fleischauer and J. R. Dahn, J. Electrochem. Soc., 2003, 150, A1457–A1464 CrossRef CAS.
- N. Dimov, S. Kugino and M. Yoshio, J. Power Sources, 2004, 136, 108–114 CrossRef CAS.
- M. Yoshio, T. Tsumura and N. Dimov, J. Power Sources, 2006, 163, 215–218 CrossRef CAS.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- J.-H. Lee, W.-J. Kim, J.-Y. Kim, S.-H. Lim and S.-M. Lee, J. Power Sources, 2008, 176, 353–358 CrossRef CAS.
- Y. N. Jo, Y. Kim, J. S. Kim, J. H. Song, K. J. Kim, C. Y. Kwag, D. J. Lee, C. W. Park and Y. J. Kim, J. Power Sources, 2010, 195, 6031–6036 CrossRef CAS.
- S. Y. Kim, J. Lee, B.-H. Kim, Y.-J. Kim, K. S. Yang and M.-S. Park, ACS Appl. Mater. Interfaces, 2016, 8, 12109–12117 CrossRef CAS PubMed.
- M. Pan, X. Liu, H. Liu and Y. Chen, Mater. Lett., 2016, 178, 252–255 CrossRef CAS.
- J. Li, J. Wang, J. Yang, X. Ma and S. Lu, J. Alloys Compd., 2016, 688, 1072–1079 CrossRef CAS.
- S. Chae, N. Kim, J. Ma, J. Cho and M. Ko, Adv. Energy Mater., 2017, 7, 1700071 CrossRef.
- Y. Zhang, K. Hu, J. Ren, Y. Wu, N. Yu, A. Feng, Z. Huang, Z. Jia and G. Wu, Dalton Trans., 2019, 48, 17683–17690 RSC.
- Y. Zhang, K. Hu, Y. Zhou, Y. Xia, N. Yu, G. Wu, Y. Zhu, Y. Wu and H. Huang, Nanomaterials, 2019, 9, 1624 CrossRef CAS PubMed.
- S. D. Beattie, D. Larcher, M. Morcrette, B. Simon and J.-M. Tarascon, J. Electrochem. Soc., 2008, 155, A158 CrossRef CAS.
- J. S. Bridel, T. Azaïs, M. Morcrette, J. M. Tarascon and D. Larcher, Chem. Mater., 2010, 22, 1229–1241 CrossRef CAS.
- J.-S. Bridel, T. Azaïs, M. Morcrette, J.-M. Tarascon and D. Larcher, J. Electrochem. Soc., 2011, 158, A750 CrossRef CAS.
- C.-H. Yim, S. Niketic, N. Salem, O. Naboka and Y. Abu-Lebdeh, J. Electrochem. Soc., 2017, 164, A6294–A6302 CrossRef CAS.
- T. Schott, R. Robert, P. A. Ulmann, P. Lanz, S. Zürcher, M. E. Spahr, P. Novák and S. Trabesinger, J. Phys. Chem. C, 2017, 121, 18423–18429 CrossRef CAS.
- T. Schott, R. Robert, S. Pacheco Benito, P. A. Ulmann, P. Lanz, S. Zürcher, M. E. Spahr, P. Novák and S. Trabesinger, J. Phys. Chem. C, 2017, 121, 25718–25728 CrossRef CAS.
- S. Jeong, X. Li, J. Zheng, P. Yan, R. Cao, H. J. Jung, C. Wang, J. Liu and J.-G. Zhang, J. Power Sources, 2016, 329, 323–329 CrossRef CAS.
- M. Klett, J. A. Gilbert, S. E. Trask, B. J. Polzin, A. N. Jansen, D. W. Dees and D. P. Abraham, J. Electrochem. Soc., 2016, 163, A875–A887 CrossRef CAS.
- N. Dupré, P. Moreau, E. De Vito, L. Quazuguel, M. Boniface, A. Bordes, C. Rudisch, P. Bayle-Guillemaud and D. Guyomard, Chem. Mater., 2016, 28, 2557–2572 CrossRef PubMed.
- R. Petibon, V. L. Chevrier, C. P. Aiken, D. S. Hall, S. R. Hyatt, R. Shunmugasundaram and J. R. Dahn, J. Electrochem. Soc., 2016, 163, A1146–A1156 CrossRef CAS.
- Benchmark Mineral Intelligence, Elon Musk: Our lithium ion batteries should be called, http://benchmarkminerals.com/elon-musk-our-lithium-ion-batteries-should-be-called-nickel-graphite/, accessed April 28, 2018 Search PubMed.
- R. Nagai, F. Kita, M. Yamada and H. Katayama, Hitachi Rev., 2011, 60, 28–32 Search PubMed.
- T. Placke, V. Siozios, R. Schmitz, S. F. Lux, P. Bieker, C. Colle, H.-W. Meyer, S. Passerini and M. Winter, J. Power Sources, 2012, 200, 83–91 CrossRef CAS.
- M. N. Obrovac, L. Christensen, D. B. Le and J. R. Dahn, J. Electrochem. Soc., 2007, 154, A849–A855 CrossRef CAS.
- S. A. Freunberger, Nat. Energy, 2017, 2, 17091 CrossRef.
Footnote |
| † These authors have contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |