
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thiazolo[5,4-d]thiazole-based organic sensitizers with improved spectral properties for application in greenhouse-integrated dye-sensitized solar cells†
Alessio
Dessì
 *a,
Massimo
Calamante
ab,
Adalgisa
Sinicropi
acd,
Maria Laura
Parisi
c,
Luigi
Vesce
e,
Paolo
Mariani
e,
Babak
Taheri
e,
Manuela
Ciocca
e,
Aldo
Di Carlo
e,
Lorenzo
Zani
a,
Alessandro
Mordini
ab and
Gianna
Reginato
*a
*a,
Massimo
Calamante
ab,
Adalgisa
Sinicropi
acd,
Maria Laura
Parisi
c,
Luigi
Vesce
e,
Paolo
Mariani
e,
Babak
Taheri
e,
Manuela
Ciocca
e,
Aldo
Di Carlo
e,
Lorenzo
Zani
a,
Alessandro
Mordini
ab and
Gianna
Reginato
*a
aInstitute of Chemistry of Organometallic Compounds (CNR-ICCOM), Via Madonna del Piano 10, 50019 Sesto Fiorentino, Italy. E-mail: a.dessi@iccom.cnr.it; gianna.reginato@iccom.cnr.it
bDepartment of Chemistry “U. Schiff”, University of Florence, Via della Lastruccia 13, 50019 Sesto Fiorentino, Italy
cDepartment of Biotechnology, Chemistry and Pharmacy, University of Siena, Via A. Moro 2, 53100 Siena, Italy
dCSGI, Consorzio per lo Sviluppo dei Sistemi a Grande Interfase, Via della Lastruccia 3, 50019 Sesto Fiorentino, Italy
eCenter for Hybrid and Organic Solar Energy (C.H.O.S.E.), Department of Electronic Engineering, University of Rome Tor Vergata, Via del Politecnico 1, 00133 Rome, Italy
First published on 25th February 2020
Abstract
Organic photosensitizers especially designed for producing semitransparent dye-sensitized solar cells (DSSCs) for greenhouse integration were prepared by introduction of different heterocyclic moieties into the thiazolo[5,4-d]thiazole molecular scaffold. The aim was that of improving their light absorption capability in the green part of the visible spectrum while maintaining a good transparency in the blue and red regions, where the photosynthetic response is maximized. A short and efficient synthetic approach, featuring two consecutive C–H activation reactions in a one-pot procedure as key steps, was used. Based on their spectroscopic and electrochemical characterization, two of the dyes prepared appeared especially suitable for greenhouse-integrated photovoltaics. The corresponding semitransparent DSSCs yielded 5.6–6.1% power conversion efficiencies, which were largely superior to those provided by other organic dyes previously proposed for the same application.
Introduction
Widespread energy production from renewable sources is one of the main goals our society has to reach to tackle the continuous increase in global energy needs and satisfy the urgency to reduce CO2 emissions and global warming.1 Among renewable energy sources, solar radiation appears especially attractive since it is abundant, widely distributed, free and practically inexhaustible.2 As a consequence, technologies used to exploit it have been constantly improving in recent years, resulting in the production of very efficient devices. Despite the continuous increase in the global installed photovoltaic (PV) capacity (which reached 505 GW at the end of 2018, accounting for more than 2% of world’s electricity production),3 there is still a tremendous need for further expansion. To expand the use of PV technologies, reduction of their manufacturing and installation costs and improvement of their power conversion efficiencies will obviously be crucial, but extension to novel applications can play a very important role as well.4In this regard, the possibility of integrating photovoltaics into agricultural settings, also known as “agrivoltaics”,5 recently became a relevant subject of research. To ensure a sustained food supply, farming requires a significant energy input, especially due to the recent progress of controlled environment agriculture, employing greenhouses and hangars to grow plants all year round.6 In particular, greenhouses consume an enormous amount of expensive electricity for regulating temperature, lighting, fans, and monitoring systems, which currently comes almost entirely from fossil fuels. To address this problem, traditional opaque silicon modules have been experimented successfully on greenhouse roofs; however, the lack of light transmission greatly affected plant development and crop productivity, allowing only a partial coverage of the greenhouse and thus reducing the maximum achievable energy production.7 Accordingly, it has been estimated that to have an optimal balance between electricity production and crop harvest, the roof coverage with crystalline Si-modules should not exceed 30%,8,9 although for crops of high economic value such as tomato, or in cases where food yield is considered a priority, a much smaller value of <10% was indicated.10,11
The above problem could be overcome by using semitransparent PV cells, able to transmit sunlight in a wavelength range compatible with plant growth. Indeed, in the region of the spectrum utilized by plants (400–700 nm), often referred to as the “photosynthetically active region” (PAR), not all light is converted with the same efficiency, with two peaks occurring in the blue (425–475 nm) and red (625–750 nm) parts of the spectrum,12 corresponding to those regions where the most widespread forms of chlorophyll, A and B, as well as red and far-red phytochromes, are active.13–15 Green light, on the other hand, is not absorbed by chlorophylls and thus is less important for photosynthesis itself, although it has been reported that monochromatic irradiation with green light can alter chlorophyll composition in higher plants,16 improve plant growth and morphology17–19 and influence the production of various metabolites.20 Nevertheless, it has been shown that crops can be successfully cultivated under artificial red/blue light,21 suggesting that fabrication of solar cells capable of absorbing visible light mostly in the green region while significantly transmitting blue and red wavelengths can constitute a promising approach towards development of a greenhouse-integrated PV system.
Some studies have already focused on the greenhouse application of organic solar cells containing polymers or small molecules absorbing in the near-IR region (such as PMDPP3T with λmax ≈ 830 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and IEICO-4F with λmax ≈ 860 nm),23 but mostly outside the phytochrome absorption range (having maxima at around 670–730 nm in green plants),24,25 as well as tandem photonic crystals.26 Dye-sensitized solar cells (DSSCs),27 on the other hand, have also been recently proposed as an ideal solution due to their advantageous characteristics.28,29 The working principle of a DSSC is inspired from natural photosynthesis and relies on the use of a sensitizer, which is adsorbed on a thin layer of a large band-gap semiconductor (such as TiO2) and mediates the light harvesting and charge separation processes.30 Thus, the cell’s light absorption profile can be efficiently tuned by modifying the structure of the sensitizer. In addition, the working electrodes of such devices can be made from glass or plastic, enabling the development of colored, lightweight and transparent cells, which might work under a wide array of lighting conditions without suffering from angular dependence of incident light.31 For these reasons DSSCs can be easily integrated in buildings and can be installed in a diverse range of shaded and diffuse light locations. Furthermore, the use of highly transparent TiO2 layers allows partial transmission of light in the UV region, which is not used in photosynthesis, but is involved in signaling pathways ensuring correct plant growth and development.32
22 and IEICO-4F with λmax ≈ 860 nm),23 but mostly outside the phytochrome absorption range (having maxima at around 670–730 nm in green plants),24,25 as well as tandem photonic crystals.26 Dye-sensitized solar cells (DSSCs),27 on the other hand, have also been recently proposed as an ideal solution due to their advantageous characteristics.28,29 The working principle of a DSSC is inspired from natural photosynthesis and relies on the use of a sensitizer, which is adsorbed on a thin layer of a large band-gap semiconductor (such as TiO2) and mediates the light harvesting and charge separation processes.30 Thus, the cell’s light absorption profile can be efficiently tuned by modifying the structure of the sensitizer. In addition, the working electrodes of such devices can be made from glass or plastic, enabling the development of colored, lightweight and transparent cells, which might work under a wide array of lighting conditions without suffering from angular dependence of incident light.31 For these reasons DSSCs can be easily integrated in buildings and can be installed in a diverse range of shaded and diffuse light locations. Furthermore, the use of highly transparent TiO2 layers allows partial transmission of light in the UV region, which is not used in photosynthesis, but is involved in signaling pathways ensuring correct plant growth and development.32
Accordingly, there is a considerable opportunity in developing DSSCs containing sensitizers able to enhance cell transparency, but also filter incident light, allowing plant growth and electricity generation at the same time. Despite that, to date only two reports have appeared on the preparation of new sensitizers for semitransparent DSSCs for greenhouse integration, describing, respectively, two simple derivatives of the well-known Ru-based dye N71933 and a series of diketopyrrolopyrrole-derived organic dyes.34 In both cases, promising optical properties but relatively low power conversion efficiencies (≤2.24%) were reported.
Among the huge number of organic compounds successfully applied as sensitizers in DSSCs, D–π–A architectures, in which a donor group (D) is connected to an acceptor/anchoring group (A) through a conjugated bridge (π), offer many advantages, in particular allow an easy and fine tuning of the spectral and electrochemical properties by simple modification of the D, π and A moieties.35
In recent years, we have been engaged in the design and synthesis of D–π–A dyes for DSSCs, and devised a new family of thiazolo[5,4-d]thiazole-based dyes (TTZ3–7) displaying broad and intense absorption spectra in the visible region, with exceptional molar absorptivities up to 9.67 × 104 M−1 cm−1, especially suitable for the fabrication of thin layer devices. The best sensitizer was dye TTZ5 (Fig. 1) which could be used to build dye-sensitized solar cells with good power conversion efficiencies (up to 7.71%) and stability.36–38
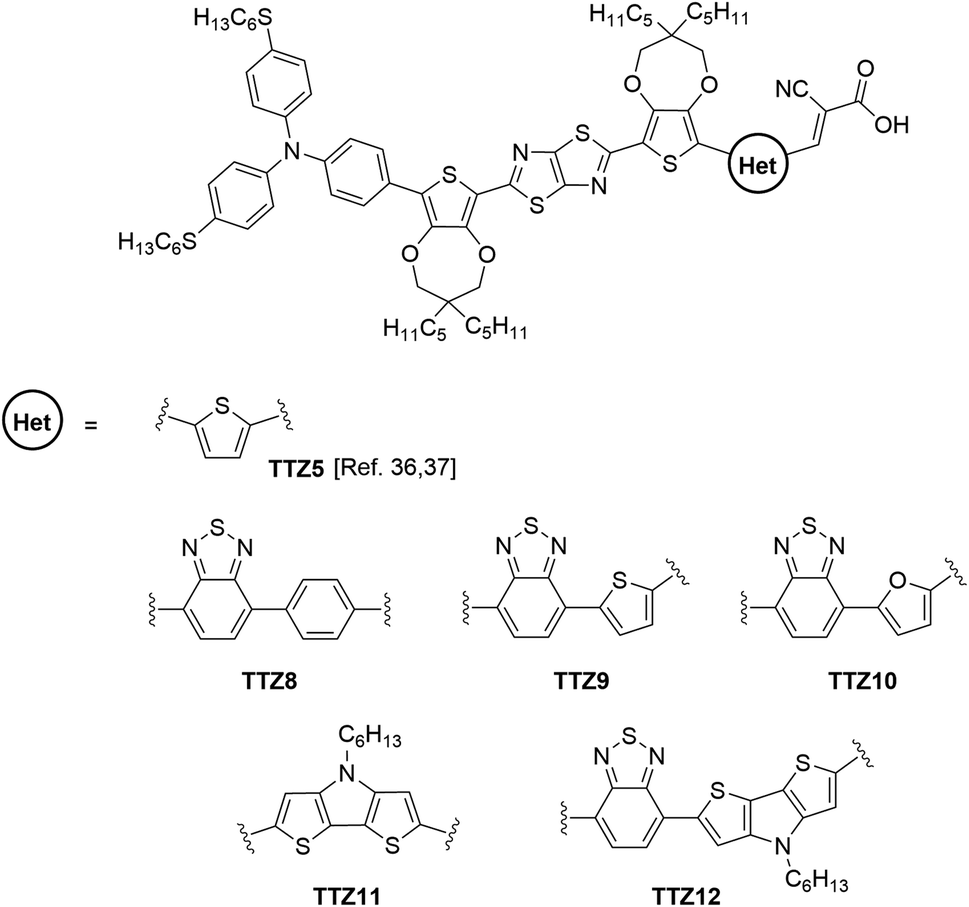 | ||
| Fig. 1 Structure of parent sensitizer TTZ5 and of the new dyes TTZ8–12 studied in this work. | ||
In this paper, we report our approach to extend the above studies towards the preparation of new D–π–A organic photosensitizers specifically designed for application in greenhouse buildings. Using the structure of dye TTZ5 as a starting point, we designed five new thiazolothiazole-based sensitizers with extended conjugated scaffolds (TTZ8–12, Fig. 1), which were investigated by means of Density Functional Theory (DFT) and Time-Dependent DFT (TD-DFT) calculations39 in order to simulate their spectroscopic properties and evaluate their ability to transmit the solar radiation required for plant photosynthesis. The dyes were then synthesized, their optical properties were experimentally verified and the performances of the corresponding semi-transparent DSSCs were measured and compared with those of the parent compound.
Results and discussion
We selected dye TTZ536,37 as our reference structure. In THF solution, this dye is characterized by an intense light absorption around 510 nm with a very high molar extinction coefficient (ε) of 9.41 × 104 M−1 cm−1, and shows no absorption over 600 nm (Fig. 2). Such a high ε value, while beneficial for maximizing light harvesting and power conversion efficiency of thin-film DSSCs, could not be optimal for farming applications, since it could lead to a decrease of device transparency.
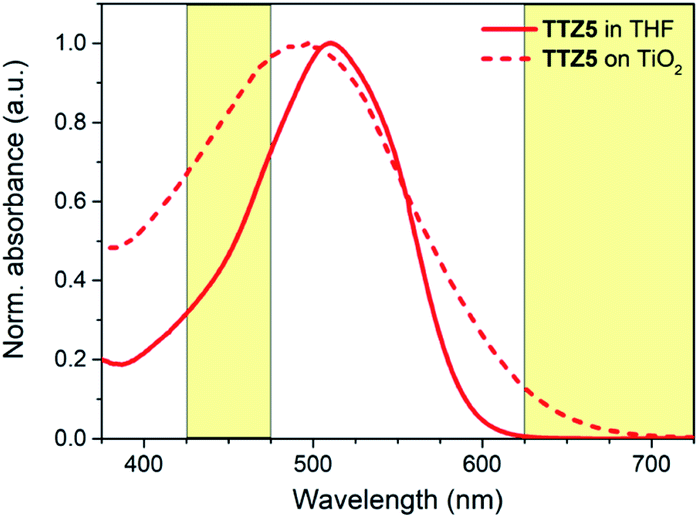 | ||
| Fig. 2 Normalized absorption spectra of sensitizer TTZ5 in THF solution (solid line) and when adsorbed on TiO2 (dash line). Photosynthetically active spectral regions are highlighted in yellow. | ||
Furthermore, the spectral region between 425 and 475 nm is significantly shielded by the dye absorption, which is worsened by the fact that TTZ5 presents a significant blue shift upon adsorption on TiO2 (λmax = 487–497 nm).37,40 For the above reasons, we supposed that dye TTZ5 could not be optimal for employment in greenhouses, and designed new structures in an effort to modulate the intensity of dye absorption and shift it to the middle of the green region of the visible spectrum.
The new dyes were designed by replacing the terminal thiophene of TTZ5 with different carbo- and heterocyclic moieties having distinct structural and electronic properties (Fig. 1). In particular, compounds TTZ8–10 featured a benzo-2,1,3-thiadiazole (BTD) unit as an auxiliary acceptor, which in turn is connected to different (hetero)aromatic cycles (benzene, thiophene and furan, respectively); such a modification was expected to cause a red-shift in the absorption spectra of organic D–π–A dyes,41–43 which was anticipated to be more or less pronounced depending on the compound’s planarity.44,45 Dye TTZ11 was instead endowed with a dithieno[3,2-b:2′,3′-d]pyrrole (DTP) moiety; in this case a better π-conjugation was expected due to the coplanar bridged π-system, which could possibly lead to an extension of the light absorption range of the sensitizer.46–48 Finally, compound TTZ12 presented both the BTD and the DTP groups on the same molecular scaffold, a combination that was shown to enhance photovoltaic performances compared to dyes presenting the DTP unit alone.49
Computational analysis of dyes TTZ8–12
To obtain a first evaluation of their optoelectronic properties, the structures of dyes TTZ8–12 were subjected to computational analysis by means of DFT and TD-DFT calculations, performed using the Gaussian09, Revision C.01 suite of programs.50 Geometry optimization was carried out in vacuo using the B3LYP functional51,52 and the standard 6-31G* basis set for all atoms, and considering methyl groups in place of the alkyl chains on the donor and ProDOT groups to reduce the computational cost. All compounds presented a relatively linear scaffold, with a flat middle region comprising the two ProDOT and the central thiazolothiazole rings (Fig. S1, ESI†), and a fairly constant angle between the ProDOT and the donor triarylamine (23–24°).Small dihedral angles were found between the other ProDOT ring and the benzothiadiazole unit in compounds TTZ8–10 and TTZ12 (8.0–10.0°), and an even smaller angle with the dithienopyrrole moiety in TTZ11 (2.3°). The main geometrical difference within the series was observed for the angle between the benzothiadiazole unit and the neighboring benzene, thiophene, furan and dithienopyrrole rings in compounds TTZ8, 9, 10 and 12, respectively. While the latter three were once again quite small (0–7°), in TTZ8 a relatively large angle of 34° was obtained, which could reduce conjugation along its entire structure, causing a blue-shift of its UV-Vis absorption spectrum.
The spatial distribution and energies of frontier molecular orbitals were then calculated for all dyes. In all cases, the HOMO−1 was located almost over the entire conjugated scaffold, while the HOMO was mostly localized on the donor moiety, as could be expected from the dyes’ structure (Fig. S2†). As for the unoccupied orbitals at the ground state, both the LUMO and the LUMO+1 levels were essentially distributed over the acceptor group, with some contribution of the auxiliary accepting and donating moieties benzothiadiazole and dithienopyrrole, respectively. Orbital energies are shown in Fig. 3, and compared with those of reference dye TTZ5:37 while HOMO levels were almost identical for all compounds, reflecting the fact that the same donor group was always employed, significant differences were observed for the LUMO positions. In particular, introduction of the additional electron-withdrawing group benzothiadiazole caused a significant LUMO stabilization going from TTZ5 to TTZ8, which was even more pronounced for dyes TTZ9–10, probably owing to their more extended conjugation. Conversely, in the case of compound TTZ11 the presence of an electron-donating moiety such as dithienopyrrole caused an increase in the energy of the LUMO, which was even superior to that of TTZ5. Finally, TTZ12, comprising both the benzothiadiazole and the dithienopyrrole groups, was in a somewhat intermediate situation, both in terms of LUMO energy and HOMO–LUMO gap. To better understand the photoexcitation mechanism of the designed dyes, their absorption maxima (λamax), vertical excitation energies (Eexc) and oscillator strengths (f) in CH2Cl2 solution were calculated on the minimized structures via time-dependent DFT (TD-DFT) at the CAM-B3LYP53/6-311G(d,p) level of theory (Table S1†), except for TTZ12, which was calculated in THF to compare it with the experimental values, since it was not soluble in CH2Cl2 (see below). Solvent effects were included by using the polarizable continuum model (PCM).54 The obtained data were also used to simulate the corresponding UV-Vis spectra, considering a Gaussian distribution and an arbitrary line width of 0.03 eV using GaussSum 3.0 (Fig. 4).55 For all dyes, the most intense absorption band was predicted to be in the desired region of the spectrum with a maximum above 500 nm, resulting mainly from mixed HOMO → LUMO and HOMO−1 → LUMO transitions. A significant red shift was predicted going from TTZ8 (507 nm) to TTZ9, 10 (550–555 nm), in agreement with the decreasing HOMO–LUMO gaps shown above. Interestingly, however, also dye TTZ11 displayed a bathochromic shift (maximum at 533 nm) compared to TTZ8, despite its larger frontier orbital gap: this could be explained by the different composition of its main absorption band, comprising a larger fraction of HOMO → LUMO transition compared to the other dyes (Table S1†). Finally, thanks to its extended conjugated structure, TTZ12 displayed the most red-shifted computed absorption band of all sensitizers, with a maximum of 575 nm. For all compounds the presence of other absorption bands, due to localized π–π* transitions, was predicted in the near-UV/UV region of the spectrum (with reduced intensity in the case of TTZ11), possibly leading to partial coverage of the “forbidden” spectral interval between 425 and 475 nm, which is part of the photosynthetically active radiation. Nevertheless, since the computational analysis suggested that all dyes had potentially the right spectroscopic and electronic properties to be used in greenhouse-integrated DSSCs, they were prepared and fully characterized.
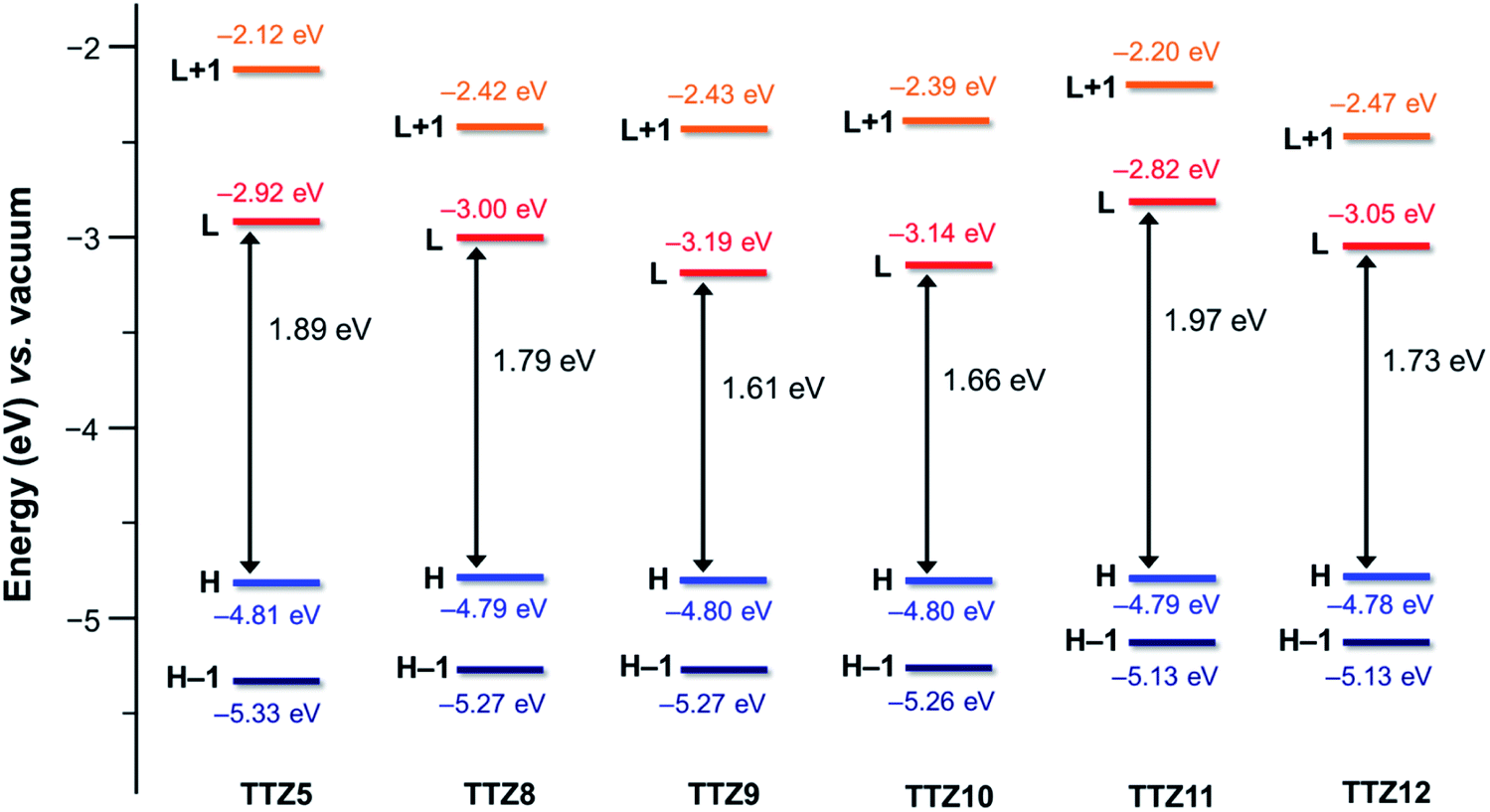 | ||
| Fig. 3 Computed (B3LYP/6-31G*) energies for DFT orbitals of compounds TTZ5 (ref. 37) and TTZ8–12 in CH2Cl2 solution. | ||
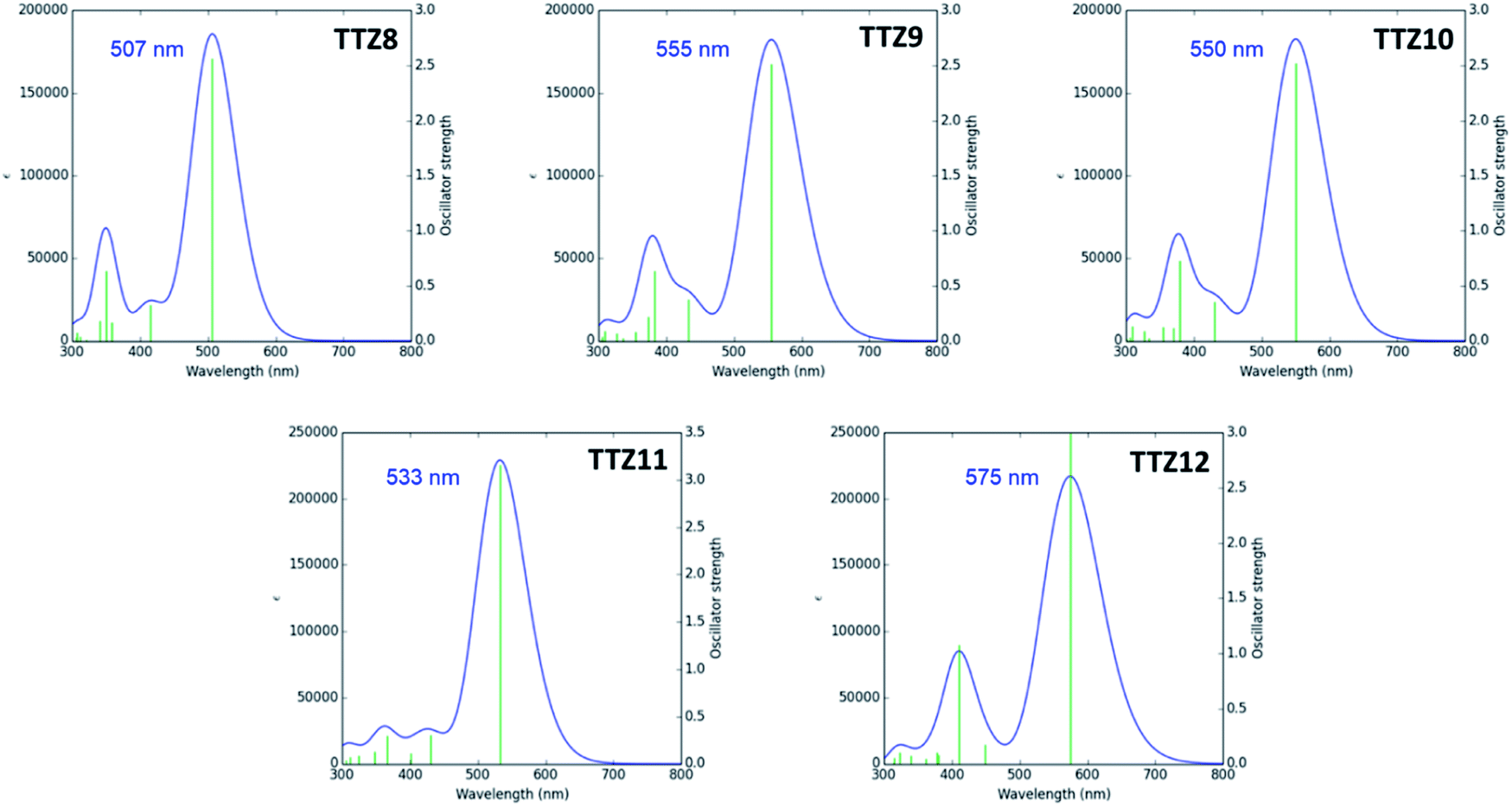 | ||
| Fig. 4 Simulated UV-Vis absorption spectra of compounds TTZ8–11 in CH2Cl2 solution (and of dye TTZ12 in THF solution). | ||
Synthesis of dyes TTZ8–12
Our aim was to accomplish the synthesis of dyes TTZ8–12 extending the scope of our new one-pot direct arylation protocol, previously designed for the gram-scale synthesis of the parent compound TTZ5.56 Such a procedure allows the introduction of both donor and acceptor moieties onto the thiazolothiazole-based skeleton 1 in just one step through two consecutive Pd-catalyzed C–H activation reactions (Scheme 1).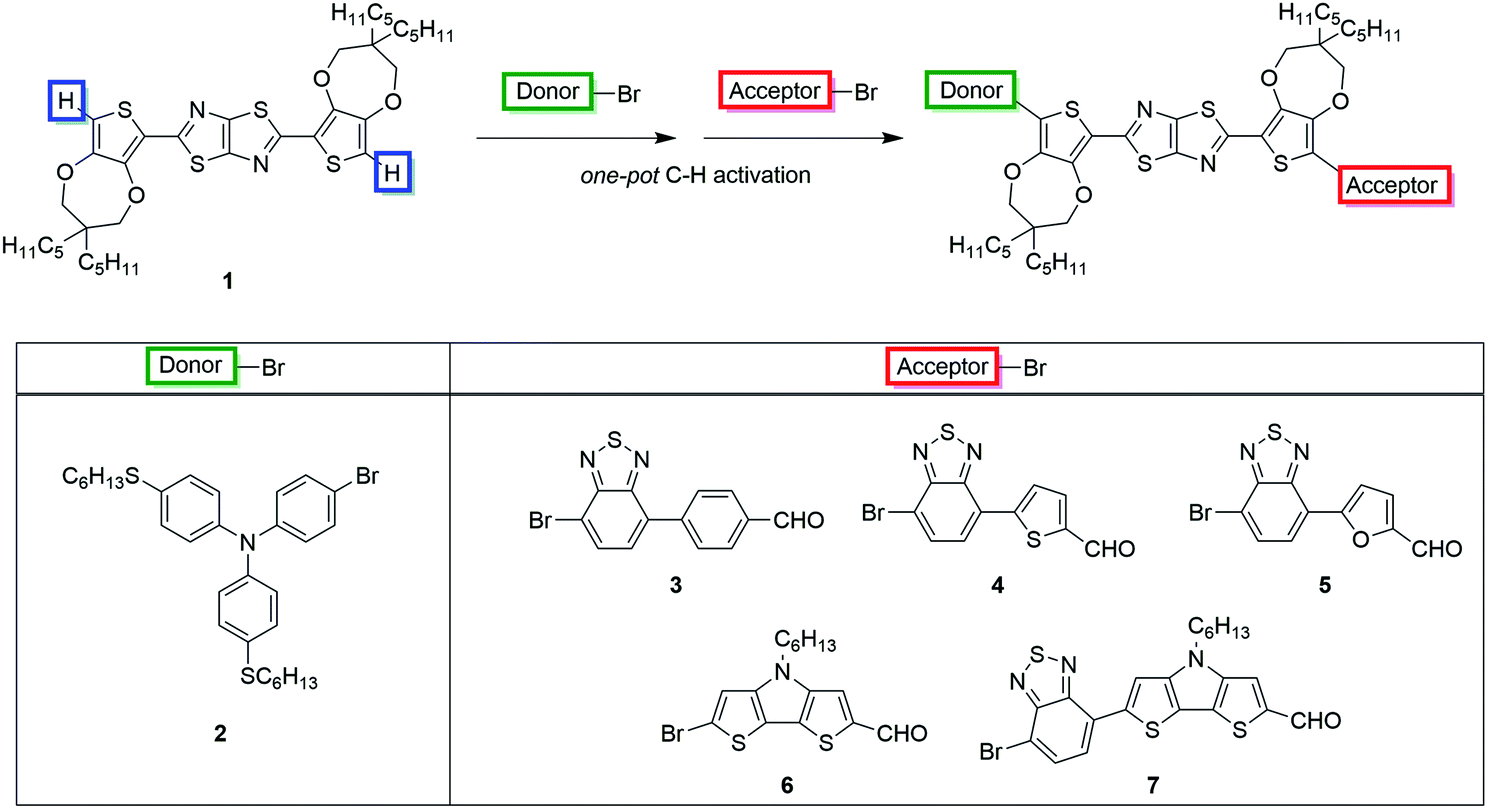 | ||
| Scheme 1 One-pot C–H activation strategy for the synthesis of D–π–A TzTz-derivatives. | ||
While the proper donor fragment 2 was prepared according to a previously reported procedure,36 appropriate synthetic procedures had to be devised for each of the five necessary bromine-containing (hetero)aryl aldehydes 3–7. Remarkably, we found out that a different protocol was required in each case to enhance the final yield of the product (Scheme 2 and 3). In particular, the first derivative containing a simple phenyl ring as a spacer (3) was synthesized through a Suzuki–Miyaura reaction starting from commercially available boronic acid 8 and dibromide 9 under standard conditions:57 the reaction proceeded smoothly and the desired building block 3 was isolated with a good yield (Scheme 2). In the case of building block 4, on the other hand, the direct arylation of 2-thiophenecarboxaldehyde 10 with an excess of dibromide 9 proved more efficient to obtain the desired product than the Suzuki–Miyaura coupling.
 | ||
| Scheme 2 Synthesis of building blocks 3–5. | ||
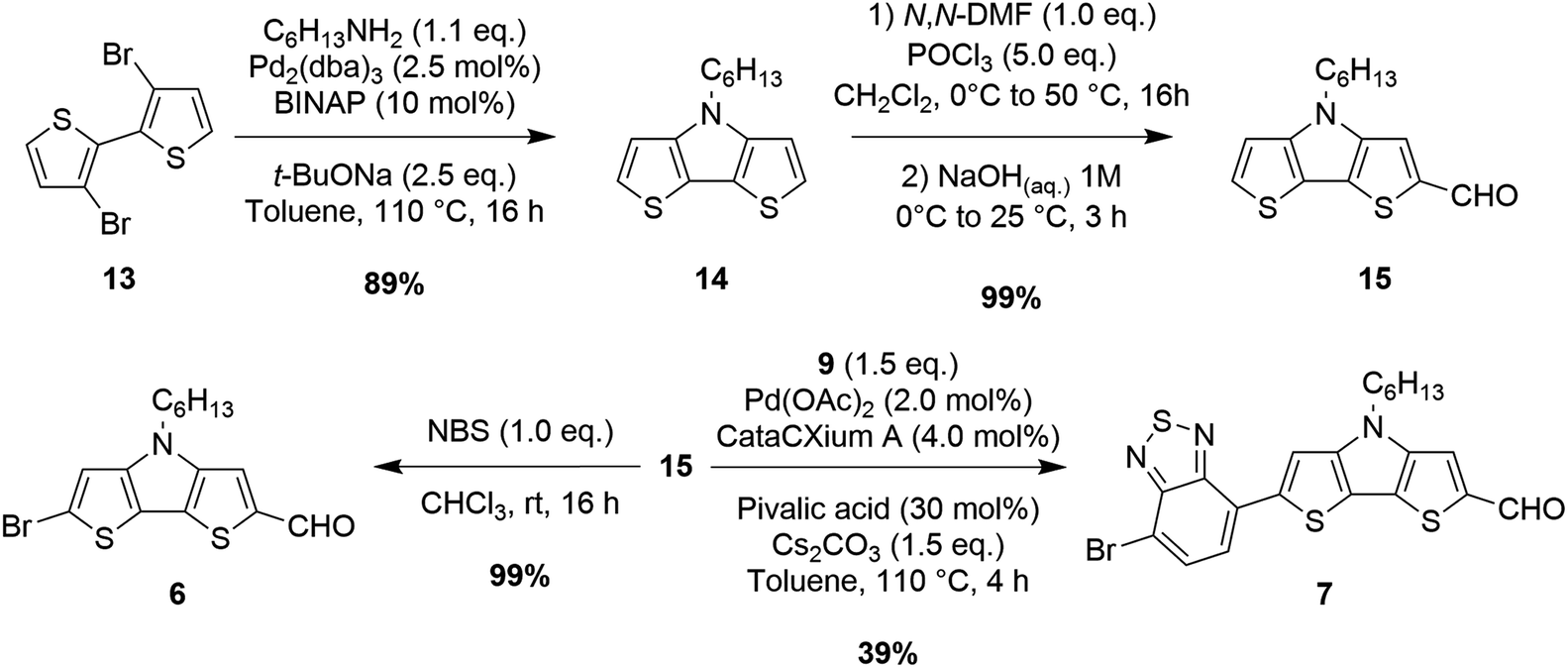 | ||
| Scheme 3 Synthesis of building blocks 6–7. | ||
Unfortunately, however, the latter was still obtained in a relatively low yield since its further reaction with dibromide 9 under direct arylation conditions could not be completely suppressed. Since furfural (11) can be prone to decomposition under basic and/or acidic reaction conditions, the synthesis of compound 5 was instead performed by means of a Stille–Migita cross-coupling, which, requiring milder conditions, usually tolerates the presence of sensitive functional groups. First, the formyl moiety of furfural (11) was protected in situ by reaction with lithium N,O-dimethylhydroxylamide, then lithiation with n-BuLi and transmetalation with tributyltin chloride afforded stannane 12 in a good yield. Finally, the Stille–Migita coupling of compound 12 and dibromide 9 in stoichiometric ratio produced the desired furan-containing building block 5 in a moderate yield. Building block 6 could be prepared starting from commercially available 3,3′-dibromo-2,2′-bithiophene (13) following a literature procedure58 (Scheme 3). Intermediate 15 was instead converted to derivative 7 once again through a C–H activation protocol involving reaction with dibromide 9 in the presence of Pd(OAc)2.
Several ligands were tested to optimize this transformation: both P(t-Bu)3 and dppp were not able to promote any conversion of the starting materials; however using P(Cy)3, XantPhos or CataCXium® A we were able to obtain the desired product with yields of 25, 29 and 32%, respectively. Replacement of toluene with a more polar solvent like N,N-DMF increased the conversion of the starting materials, but gave a lower yield of compound 7 due to the formation of higher amounts of by-products. Finally, the use of Cs2CO3 instead of K2CO3 as the base in toluene allowed the increase of the yield from 32 to 39%.
With all the brominated (hetero)aryl aldehydes 3–7 in hand, we then attempted to apply the direct arylation protocol shown in Scheme 1 to the synthesis of advanced intermediates 16–20. Accordingly, a first direct arylation with electron-rich bromide 2 was performed, using Pd(OAc)2 and CataCXium® A as the catalytic system, acetic acid as the additive and potassium carbonate as the base, followed by a second direct arylation with bromides 3–7 under very similar conditions (Scheme 4). The two reactions were carried out in one pot, without isolation of the intermediate monoarylated compounds.
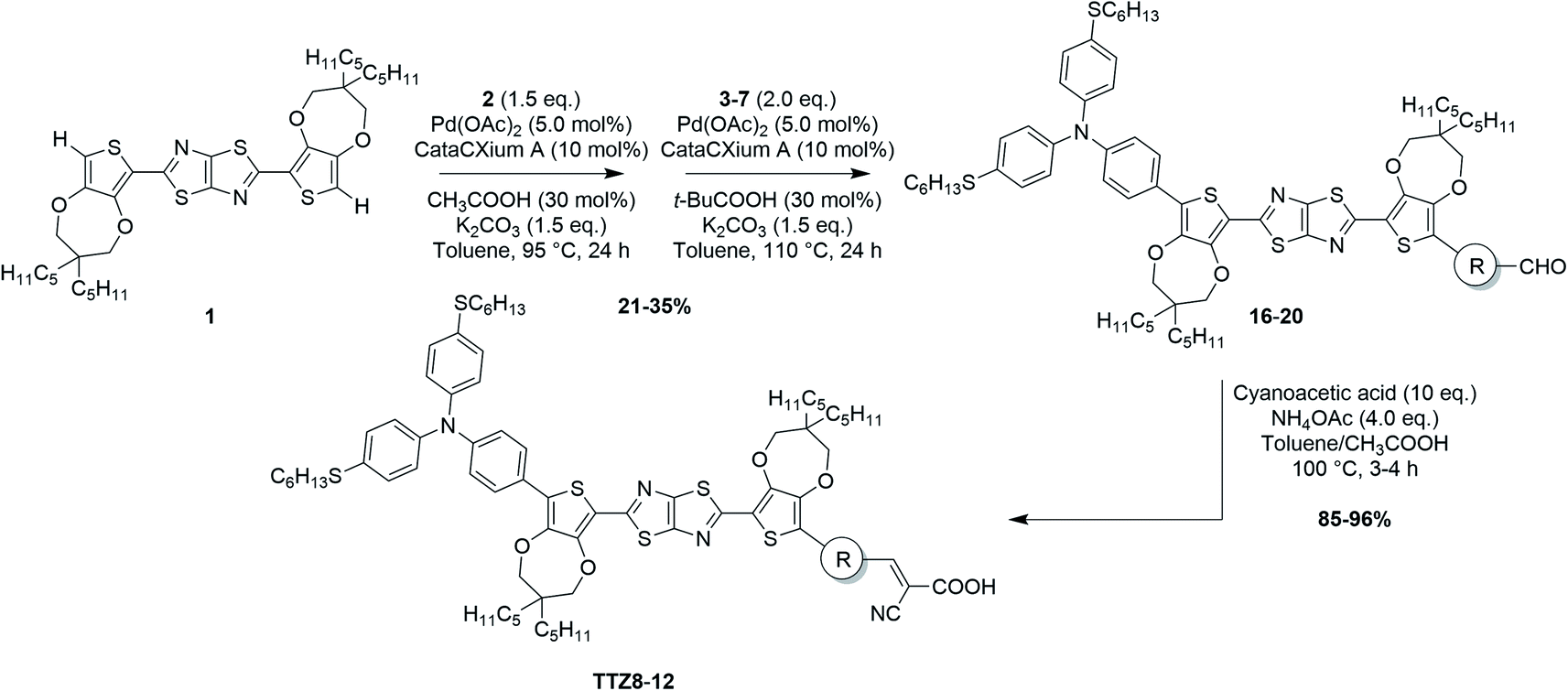 | ||
| Scheme 4 One-pot direct arylation protocol and synthesis of dyes TTZ8–12. | ||
Remarkably, our procedure appeared robust and tolerated all the chemical modifications introduced to obtain the target compounds. Replacement of acetic acid with pivalic acid and the increase of the reaction temperature in the second step allowed the enhancement of the conversion of the intermediate resulting from the first C–C bond formation, allowing access to the desired product. Better yields (31–35%) were obtained with electron-poor bromides 3–5, while the reagents containing the electron-rich dithienopyrrole unit (6–7) seemed less reactive, leading to more complex reaction crudes and, consequently, to lower yields of the desired products (21–25%), in agreement with literature precedents on direct arylation reactions.57 Finally, all the advanced intermediates 16–20 were efficiently converted to the final products TTZ8–12 through a typical Knoevenagel condensation with cyanoacetic acid.
Spectroscopic and electrochemical characterization of dyes TTZ8–12
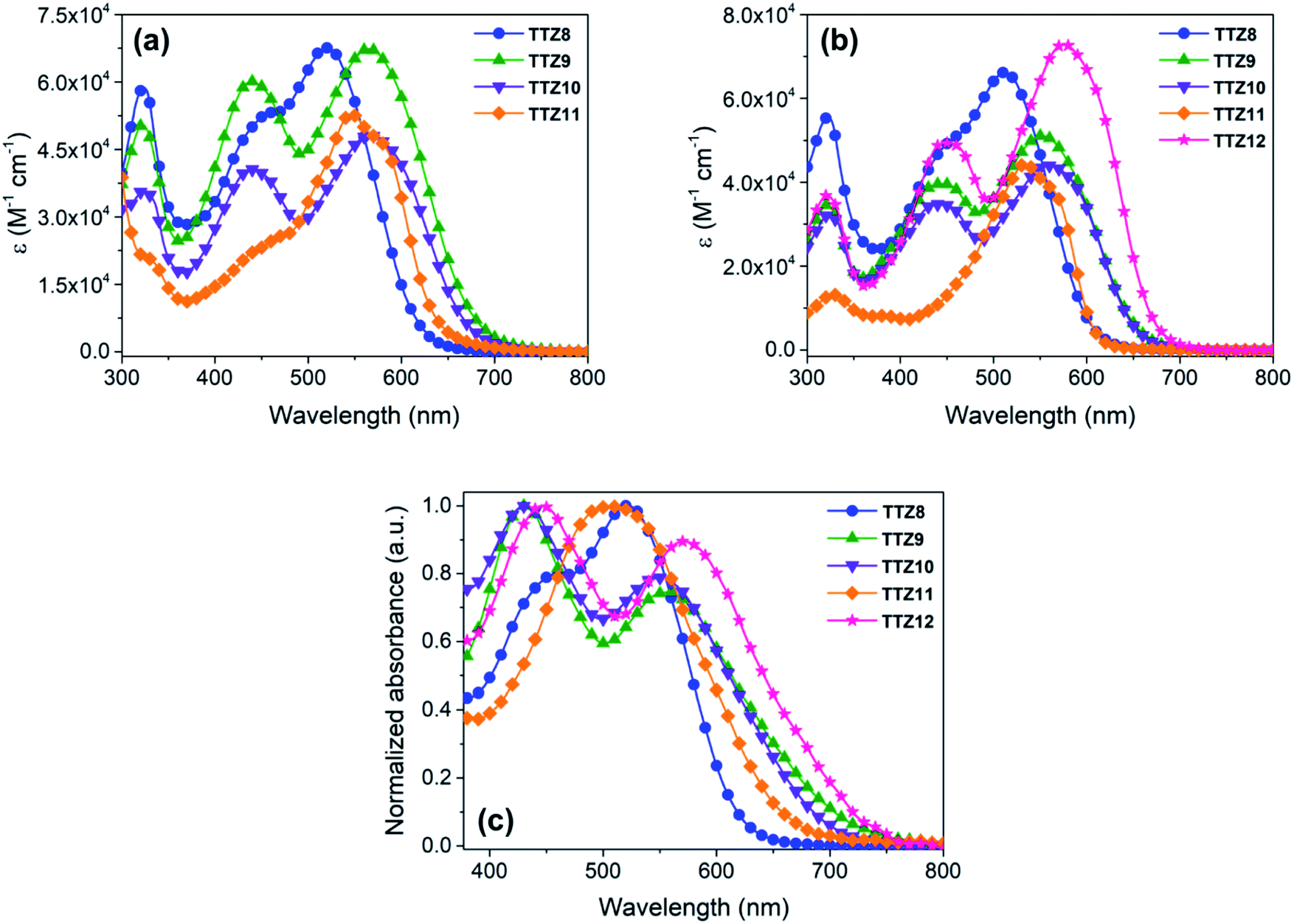
The new dyes were subjected to a comprehensive spectroscopic and electrochemical characterization, and the results are reported in Table 1. First, their UV-Vis spectra were measured in CH2Cl2 solution, to compare them with the results of the computational analysis shown above; unfortunately, we realized that compound TTZ12 was not soluble enough in CH2Cl2 to record a meaningful absorption spectrum, and therefore its spectroscopic analysis was carried out in a different solvent. All dyes exhibited a low energy absorption band above 500 nm, with a progressive bathochromic shift in the order TTZ8 < TTZ11 < TTZ9 ≤ TTZ10 (Fig. 5a). This trend was in good agreement with the results of the computational analysis, as confirmed by the difference between the experimental values of the vertical excitation energies (2.19–2.38 eV) and the computed values (2.23–2.45), which was inferior to 0.1 eV in every case. Molar extinction coefficients were relatively high (4.38–6.68 × 104 M−1 cm−1), indicating good light harvesting capability of the compounds, but still lower than that of dye TTZ5, which could prove useful in enhancing the transparency of the final devices. Compounds TTZ8 and TTZ11 had an absorption onset around 650 nm and displayed a shoulder peak in the 400–500 nm region, which was much less pronounced in the case of TTZ11, once again in agreement with TD-DFT calculations (see above, Fig. 4). In contrast, the main peaks of compounds TTZ9–10 had onsets above 700 nm and were accompanied by relatively intense, higher energy peaks centered around 440 nm, an undesired feature in view of the foreseen application on greenhouse rooftops.| Dye | λ max CH2Cl2a [nm] | λ max THFa [nm] | E 0–0 [eV] | λ max TiO2 [nm] | E S+/S [V] | E S+/S* , [V] | Γ (×10−7) [mol cm−2] | WTe [%] |
|---|---|---|---|---|---|---|---|---|
| a Values in parentheses refer to molar extinction coefficients [×104 M−1 cm−1]. b Calculated from the data in THF solution, based on the corresponding Tauc plots (Fig. S3). Values in parentheses refer to the E0–0 calculated considering the onset of the lowest energy absorption band. c Values vs. NHE, obtained using ferrocene as an external standard and assuming a reduction potential of +0.63 V for the Fc+/Fc couple vs. NHE.59 d Calculated using the expression ES+/S* = ES+/S − E0–0. e Weighted transparency against the absorption spectra of plant photoreceptors (chlorophyll a, chlorophyll b, beta-carotene). f Value taken from ref. 37. | ||||||||
| TTZ5 | — | 510 (9.41)f | 2.16f (2.10) | 487f | +0.91f | −1.25f | 1.19f | 27 |
| TTZ8 | 521 (6.45) | 512 (6.49) | 2.15 (2.06) | 522 | +0.90 | −1.25 | 1.47 | 41 |
| TTZ9 | 564 (6.68), 438 (6.00) | 551 (5.22), 443 (4.05) | 1.98 (1.90) | 559, 431 | +0.87 | −1.11 | 1.15 | 18 |
| TTZ10 | 566 (4.38), 440 (3.70) | 559 (4.33), 439 (3.48) | 1.97 (1.90) | 550, 432 | +0.85 | −1.12 | 1.46 | 38 |
| TTZ11 | 547 (5.19) | 533 (4.45) | 2.08 (2.04) | 505 | +0.90 | −1.18 | 1.27 | 52 |
| TTZ12 | — | 576 (7.26), 449 (4.89) | 1.91 (1.85) | 570, 447 | +0.90 | −1.01 | 1.56 | 13 |
 | ||
| Fig. 5 UV-Vis absorption spectra of compounds TTZ8–12 in CH2Cl2 solution (a), THF solution (b) and when adsorbed on nanocrystalline TiO2 films (c). | ||
The UV-Vis absorption spectra of all compounds were recorded also in THF (Fig. 5b), allowing analysis of dye TTZ12 as well. In general, absorption maxima were shifted to lower wavelengths than those found in CH2Cl2, while molar extinction coefficients remained almost unchanged or decreased slightly. In the case of dye TTZ12, an intense absorption band at 576 nm was observed, which was accompanied by an additional, higher energy band around 440 nm also observed for dyes TTZ9–10. Once again, the experimental result was in very good agreement with TD-DFT calculations, with a difference of only 0.01 eV between the computed and measured vertical excitation energies for the most red-shifted transition. Optical band gaps were estimated from the dyes spectra in THF by drawing the corresponding Tauc plots (Fig. S3†): they were all in the 1.91–2.15 eV range, confirming the excellent ability of the new sensitizers to absorb light in the visible region.
To have a more realistic depiction of the dyes’ absorption properties under the conditions of use, they were adsorbed on a thin transparent TiO2 film, similar to those later employed for the construction of DSSCs (see the Experimental section for details), and the corresponding normalized UV-Vis spectra were recorded in transmission mode (Fig. 5c). Interestingly, all dyes containing the BTD auxiliary acceptor unit (TTZ8–10 and TTZ12) displayed only small shifts of their absorption maxima compared to those observed in THF solution, either to longer (TTZ8 and TTZ9, 8–10 nm) or shorter wavelengths (TTZ10 and TTZ12, 6–9 nm). A different behavior was observed for dye TTZ11, which showed a significant hypsochromic shift of 28 nm compared to the spectrum in solution in analogy with what was previously found with dye TTZ5 (a blue shift of 23 nm going from THF solution to TiO2), which is also devoid of the BTD unit.37 Despite that, the maximum absorption wavelength of TTZ11 was still significantly red-shifted compared to that previously found for TTZ5. Remarkably dyes TTZ8 and TTZ11 had absorption onsets below 700 nm, which should make them the most suitable for agrivoltaics applications. On the other hand, the broader profiles showed by the remaining dyes could penalize cell transparency in the spectral regions relevant for photosynthesis. The density of adsorbed dyes on TiO2 was measured by comparing the absorbance of a standard solution of dye before and after sensitization (see the Experimental section for details); in general, the new sensitizers were adsorbed in quite similar amounts on TiO2 films (1.15–1.56 × 10−7 mol cm−2), which were also comparable to the analogous value previously obtained for TTZ5.
The redox properties of the new dyes were investigated by means of cyclic voltammetry in chloroform solution, using 0.1 M TBAPF6 as the supporting electrolyte (Fig. S4a†). All compounds presented quasi-reversible first oxidation peaks; since the spectroscopic analysis highlighted the dyes TTZ8 and TTZ11 as the most suitable for agrivoltaics applications, the quasi-reversible behavior of the oxidation process of these two dyes was further confirmed by recording their voltammograms at different rates (Fig. S4b†). Since the CV spectra did not allow an accurate estimation of the oxidation potentials (ES+/S) for all the dyes, differential pulse voltammetry (DPV) experiments were run as well. All the values of ES+/S were very similar (+0.85 − +0.90 V vs. NHE), as could be expected from the fact that all dyes bear the same thioalkyl-substituted triphenylamine donor. In the case of dye TTZ8, the first oxidation peak was visible as a shoulder of a second oxidation, which was observed for all dyes at potentials between +1.08 V and +1.44 V vs. NHE. In general, ground-state oxidation potentials (ES+/S) appeared fully compatible with regeneration by the I−/I3− redox shuttle traditionally used in DSSCs (E° = +0.35 V vs. NHE).60 Excited-state oxidation potentials (ES+/S*) were also obtained from the ES+/S values by subtracting the optical bandgap: the values obtained are in the −1.01 to −1.25 V range vs. NHE, which ensured a large overpotential for fast injection of excited electrons into the TiO2 conduction band (ECB (TiO2) = −0.5 V vs. NHE).27
The characterization of the new compounds was completed by recording the transmission spectra of the corresponding thin (5 μm), transparent TiO2 films, analogous to those employed for the construction of the final devices (Fig. 6a). Films dyed with compounds TTZ8 and TTZ11 were highly transparent above 625 nm, with T% values above 88% and 83%, respectively; in addition, they also displayed a reasonable transparency in the 425–475 nm region, in the range of 26–32% for TTZ8 and 37–40% for TTZ11. In general, compound TTZ8 showed also a lower transmittance in the green spectral region than TTZ11, likely due to its higher molar absorptivity. Due to their red-shifted absorption spectra and relatively high molar extinction coefficients, dyes TTZ9 and TTZ12, in contrast, exhibited extended absorption tails above 625 nm, resulting in much lower transmittance values in the red region of the spectrum; furthermore, the presence of a second absorption band around 430–440 nm caused also a significant decrease of transparency in the 425–475 nm region, with T% values lower than 15% in both cases. Finally, the film sensitized with dye TTZ10 exhibited a transmission spectrum qualitatively similar to that of dye TTZ9, but with higher transmittance levels, probably due to its lower molar absorptivity: however, despite having sufficient transparency in the 425–475 nm range (28–37%), it still showed a significant screening effect above 625 nm, which was much more pronounced than that of dyes TTZ8 and TTZ11. As previously demonstrated by Yip et al. in the case of organic PV cells,23 it was also possible to evaluate the impact on plant growth by calculating the weighted transmittance of our semitransparent film sensitized with compounds TTZ5 and TTZ8–12 against the absorption spectra of the most common photoreceptors present in green plants, namely chlorophyll a, chlorophyll b and beta-carotene (WT%, Table 1, see the Experimental section for details on the calculation).23 Results showed that the WT% values decreased in the order TTZ11 > TTZ8 > TTZ10 > TTZ9 > TTZ12 with the highest value being 52%, suggesting that compounds TTZ8 and TTZ11 possessed the most appropriate optical properties for application in greenhouses. To further support such a statement, we plotted the transmittance curve of TTZ8 and TTZ11 together with the absorption spectra of the above-mentioned photoreceptors, showing transmittance properties reasonably well-matched with the characteristic absorption peaks of plants (Fig. S5†). In addition, we also compared the transmission spectra of dyes TTZ8 and TTZ11 with those of parent dye TTZ5, recorded under the same conditions, showing that both new dyes presented superior transparency in the photosynthetically relevant regions (Fig. 6b).
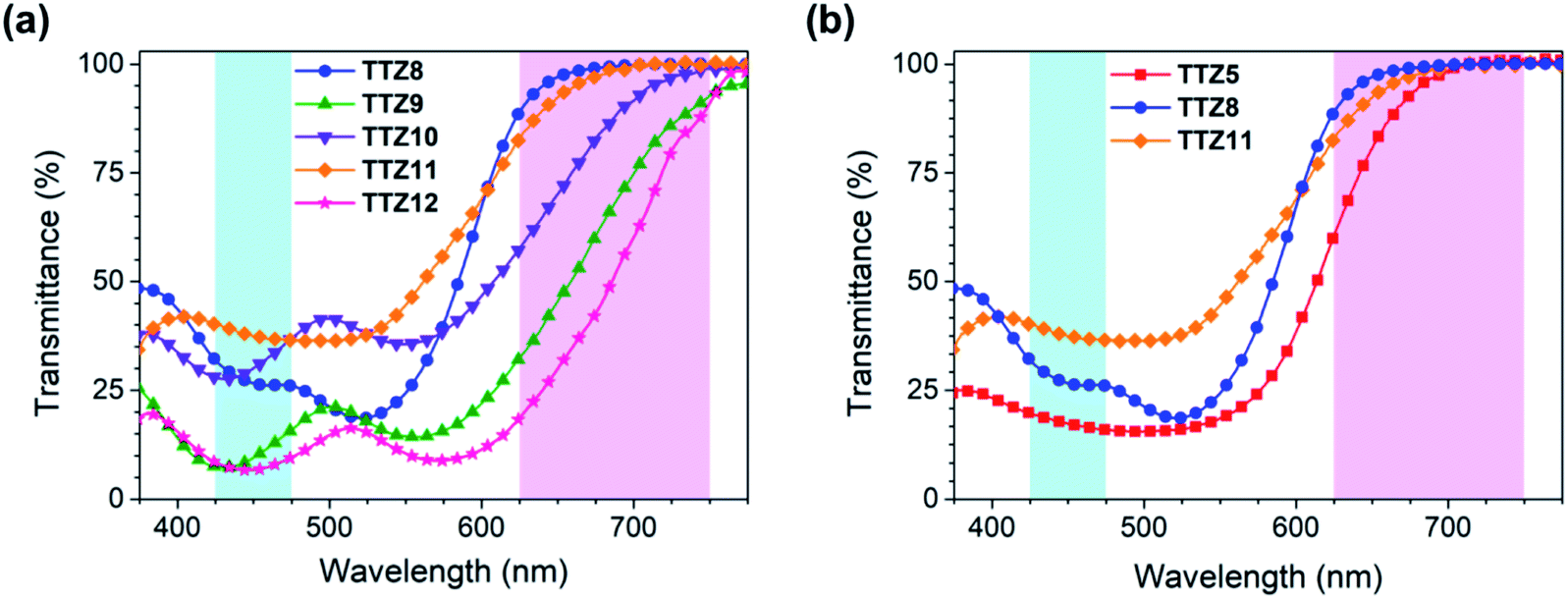 | ||
| Fig. 6 Transmission spectra of thin (5 μm) transparent TiO2 films sensitized with dyes TTZ8–12 (a), and comparison with the transmission spectra of compound TTZ5 (b). Photosynthetically relevant regions are marked in light blue (425–475 nm) and pink (625–750 nm). | ||
Photovoltaic characterization of DSSCs built with dyes TTZ8–12
Following their spectroscopic and electrochemical characterization, dyes TTZ8–12 were then used to build small scale (0.25 cm2) dye-sensitized solar cells, using a thin and transparent TiO2 semiconductor layer (5.5 μm, TiO2 18-NRT, Greatcell Solar) and a commercial electrolyte composition based on the I−/I3− redox couple (EL-HPE, Greatcell Solar). The cell fabrication procedure was purposely kept as simple as possible (see the Experimental section), so as to make the results obtained from the laboratory tests more relevant in view of the possible scale-up of the final devices;61 accordingly, no compact TiO2 blocking layer was formed either on the conductive glass substrate or on the nanocrystalline semiconductor film by exposure to aq. TiCl4, and no light-scattering layer was deposed on the photoanode. Photovoltaic performances were compared with those of the parent dye TTZ5 to see how modulation of the electronic and optical properties of the dyes affected electricity production by the corresponding dye-sensitized solar cells. J/V curves of the best-performing cells for each dye are reported in Fig. 7a, while the relevant figures-of-merit (obtained from the average values of at least four devices) are indicated in Table 2. Compound TTZ8 was the best performing among the new sensitizers, yielding an average power conversion efficiency of 6.1%, which was very close to that provided by parent sensitizer TTZ5 (6.3%, Table 1). Compared to TTZ5, TTZ8 yielded basically the same photocurrent and fill factor, but a slightly diminished open-circuit voltage, resulting in the lower PCE. The second best dye was TTZ11 which, compared to TTZ8, produced a lower photocurrent while maintaining good values of VOC and fill factor, resulting in an average PCE of 5.6%. All other dyes gave largely inferior efficiencies (2.0–4.1%), due both to lower short-circuit currents and open-circuit voltages. The average short-circuit current values were in good agreement with the results of IPCE measurements (Fig. 7b), showing relatively broad curves for all thiazolothiazole-based dyes. In particular, parent dye TTZ5 had the highest IPCE values in the center of the visible region, reaching a maximum of 88% at around 480 nm, consistent with its maximum absorption wavelength on TiO2. Dye TTZ8, on the other hand, presented a much broader IPCE spectrum, which was also the most intense among the new sensitizers in the 350–600 nm region. In agreement with the spectroscopic analysis discussed above, dyes TTZ9–12 had red-shifted IPCE onsets compared to TTZ8, reaching values around 780 nm, but their spectra were less intense in the visible range, explaining the lower photocurrent values observed for them.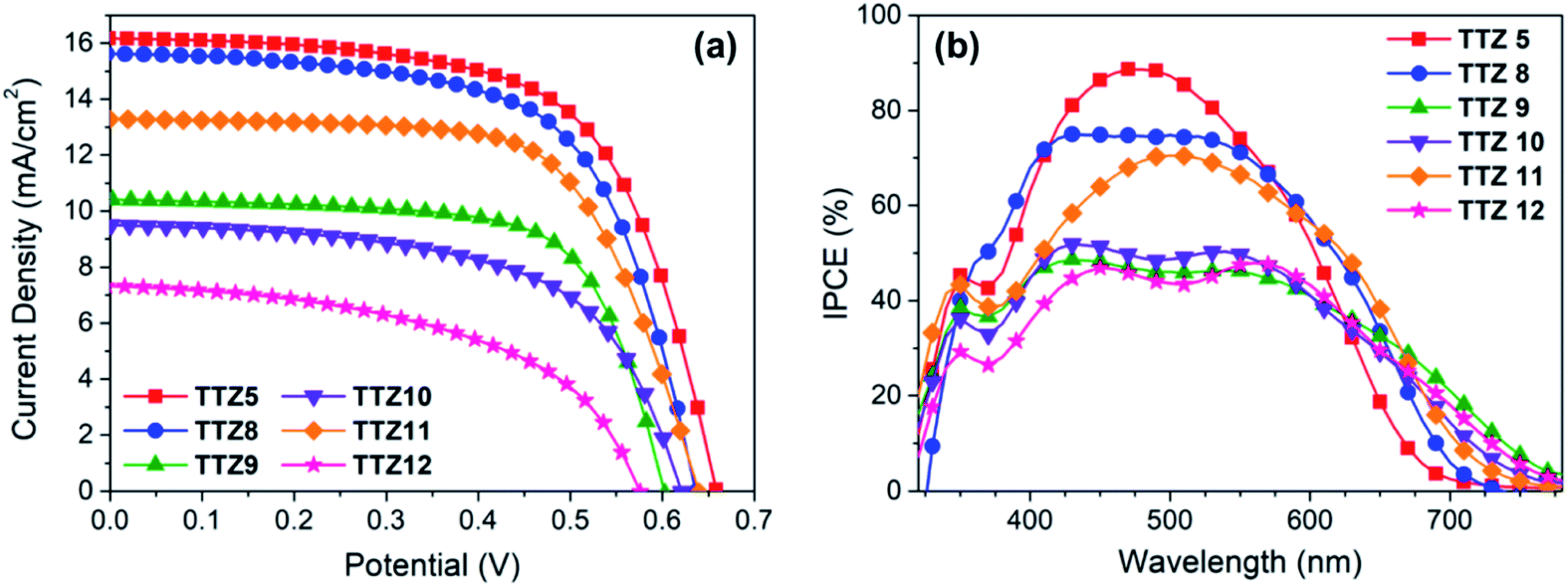 | ||
| Fig. 7 J/V curves (a) and IPCE spectra (b) of the best-performing transparent cells built with dyes TTZ5 and TTZ8–12. | ||
| Compound | J SC [mA cm−2] | V OC [V] | ff [%] | η [%] |
|---|---|---|---|---|
| a Average values of five different devices, except for TTZ10 (four devices), together with the corresponding standard deviations. Active area: 0.25 cm2, photoanode thickness 5 μm. | ||||
| TTZ5 | 15.0 ± 1.0 | 0.67 ± 0.02 | 61.8 ± 2.0 | 6.3 ± 0.4 |
| TTZ8 | 15.2 ± 0.5 | 0.65 ± 0.01 | 62.0 ± 1.1 | 6.1 ± 0.2 |
| TTZ9 | 10.0 ± 0.4 | 0.58 ± 0.03 | 67.2 ± 1.2 | 4.1 ± 0.2 |
| TTZ10 | 9.6 ± 0.1 | 0.62 ± 0.01 | 57.7 ± 2.8 | 3.4 ± 0.2 |
| TTZ11 | 13.2 ± 0.6 | 0.64 ± 0.02 | 66.0 ± 3.0 | 5.6 ± 0.3 |
| TTZ12 | 7.1 ± 0.2 | 0.55 ± 0.02 | 50.1 ± 1.6 | 2.0 ± 0.2 |
To better understand the results of the efficiency measurements, we also carried out Electrochemical Impedance Spectroscopy (EIS) studies on the DSSCs built with the new dyes. Experiments for TTZ5 and TTZ8–12-containing cells were performed in the dark at −0.60 V forward bias over the 0.1 Hz to 100 kHz frequency range: the corresponding Nyquist plots (i.e. the plots showing the minus imaginary part of the impedance −Z′′ vs. the real part Z′ when sweeping the frequency) are shown in Fig. 8a, while the equivalent circuit used for data fitting is shown in Fig. 8b. In the Nyquist plot, the mid-frequency semicircle is associated with charge transfer processes taking place at the electrolyte–dye–TiO2 interface, and by measuring its diameter the corresponding interfacial resistance (RCT) can be estimated.62,63
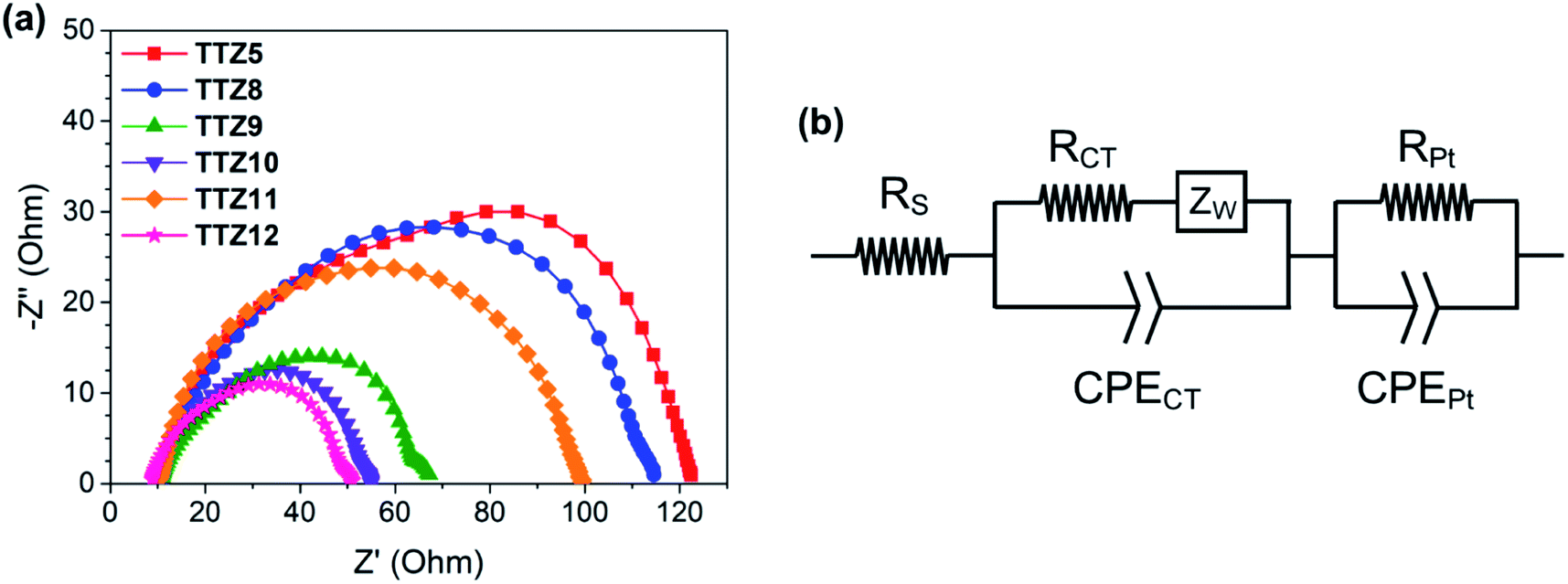 | ||
| Fig. 8 Nyquist plots for DSSCs built with dyes TTZ5 and TTZ8–12 (a), and the equivalent circuit used for data fitting (b): RS = series resistance; RCT = charge transfer resistance at the electrolyte–dye–TiO2 interface; RPt = resistance at the platinum counter-electrode. | ||
As can be seen from the figure, dyes TTZ5, TTZ8 and TTZ11, giving the highest values of VOC (0.64–0.67 V), displayed also the largest mid-frequency semicircles (TTZ5: 65.1 Ω; TTZ8: 74.4 Ω; TTZ11: 67.5 Ω), while for the other three dyes much lower RCT values were calculated (TTZ9: 40.8 Ω; TTZ10: 24.7 Ω; TTZ12: 31.1 Ω). The results suggest that for the latter three dyes a faster charge recombination was taking place at the anode, explaining the lower photovoltages recorded with the corresponding cells.
The above findings can be understood by considering the geometry of the dyes around the heterocyclic spacers and the anchoring groups present in their structures. The three dyes presenting the lowest recombination resistances and thus also giving the worst VOC values, namely TTZ9, 10 and TTZ12, are characterized by the presence of an electron-withdrawing benzothiadiazole (BTD) group separated from the anchoring cyanoacrylic moiety by a five-membered ring (thiophene in TTZ9 and furan in TTZ10) or a fused heterocyclic moiety composed of five-membered rings (TTZ12). All these sections are almost perfectly flat (Fig. S1†), as opposed to what is observed for dye TTZ8, featuring a benzene spacer, whose optimized structure features a dihedral angle of approx. 34° between BTD and the benzene ring. As previously observed by Kwon and co-workers,64 sensitizers presenting a twisted π-spacer between the BTD unit and the anchoring group are characterized by a slower back-electron transfer (BET) of injected electrons compared to those with a planar π-spacer, or with the BTD group directly connected to the cyanoacrylic acid,65,66 thanks to a lower hole delocalization on the acceptor group of the oxidized dye. Consequently, charge recombination is expected to be less severe for dye TTZ8 than for TTZ9, 10 and TTZ12, giving rise to a higher RCT value and a superior photovoltage. Similarly, a slower BET would be expected also for dyes TTZ5 and TTZ11, lacking the electron-withdrawing BTD moiety on their backbone and therefore having a lower driving force for charge recombination.
To complement our studies, we also tried to maximize the photovoltaic efficiency of the best new dye by optimizing the cell structure. Accordingly, TTZ8 was used to fabricate a series of opaque DSSCs featuring a thicker, multilayer TiO2 film. In this case, photoanodes were prepared by double deposition of a transparent titania film (Greatcell Solar 18NR-T) followed by deposition of a TiO2 scattering layer (Greatcell Solar WER2-O), to obtain a total film thickness of approximately 13 μm. In addition, the photoanode was also treated with an aqueous solution of TiCl4 in order to form a compact TiO2 blocking layer to minimize charge recombination (see the Experimental section for details). The same counter-electrode and electrolyte mixture were used as in the experiments described above. Under such conditions, dye TTZ8 provided a maximum efficiency of 7.63% (Fig. S6†), especially due to a higher photocurrent (17.40 mA cm−2), obviously deriving from the pre- and post-TiCl4 treatment67 and from the scattering effect of the opaque layer.68
Conclusions
In this work we have described the design, synthesis and characterization of thiazolothiazole-containing organic sensitizers for DSSCs with potential application in photovoltaic greenhouses. Starting from the already known structure of sensitizer TTZ5, the spectroscopic properties of the new compounds were systematically tuned by introduction of different electron-withdrawing and electron-donating heterocyclic spacers in place of the simple thiophene ring present in the parent compound. Structural modification was guided by computational analysis at the DFT/TDDFT level in order to reduce light harvesting in the photosynthetically active region of plants and to enhance the molar absorptivity of the dyes.Following synthesis and characterization of five new sensitizers (TTZ8–12), we identified compounds TTZ8 (featuring a benzothiadiazole-phenyl spacer) and TTZ11 (having a dithienopyrrole spacer) as being the most promising for the supposed application in greenhouses. In particular, dyes TTZ8–11 guaranteed moderate-to-high transparency in the photosynthetically relevant regions of the spectrum (26–40% between 425 and 475 nm, and >83% above 625 nm, respectively), coupled with good photovoltaic efficiencies (5.6–6.1%), which were close to that of parent dye TTZ5 under the same conditions and superior to those reported for other organic or metal–organic sensitizers developed for greenhouse-integrated DSSCs.33,34
Optimized, opaque dye-sensitized solar cells built with dye TTZ8 and a thicker photoanode provided a higher photovoltaic efficiency up to 7.63%, highlighting how an optimal trade-off between optical properties and power conversion efficiencies will be crucial to support the possible application of greenhouse-integrated DSSCs. Based on the results reported in this work, we are currently investigating how the light absorption properties of different dye-sensitized semiconductor films affect plant development, chemical composition and photosynthetic efficiency. The results of these studies will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Fondazione Cassa di Risparmio di Pistoia e Pescia (“FOTOSER” project) and Regione Toscana (“ARIADNE” project, POR-FESR 2014-2020) for financial support. Dr Jonathan Filippi (CNR-ICCOM) is acknowledged for his help in conducting the DPV experiments. M. L. P. and A. S. acknowledge MIUR Grant – Department of Excellence 2018-2022.References
- IRENA, Global energy transformation: a roadmap to 2050, International Renewable Energy Agency, Abu Dhabi, 2019 edn, 2019 Search PubMed.
- Q. Schiermeier, J. Tollefson, T. Scully, A. Witze and O. Morton, Nature, 2008, 454, 816–823 CrossRef CAS PubMed.
- REN21, Renewables 2019 Global Status Report, 2019 Search PubMed.
- A. K. Pandey, V. V. Tyagi, J. A. Selvaraj, N. A. Rahim and S. K. Tyagi, Renewable Sustainable Energy Rev., 2016, 53, 859–884 CrossRef.
- C. Dupraz, H. Marrou, G. Talbot, L. Dufour, A. Nogier and Y. Ferard, Renewable Energy, 2011, 36, 2725–2732 CrossRef.
- R. Shamshiri, F. Kalantari, K. C. Ting, K. R. Thorp, I. A. Hameed, C. Weltzien, D. Ahmad and Z. M. Shad, Int. J. Agric. Biol. Eng., 2018, 11, 1–22 Search PubMed.
- M. Cossu, L. Murgia, L. Ledda, P. A. Deligios, A. Sirigu, F. Chessa and A. Pazzona, Appl. Energy, 2014, 133, 89–100 CrossRef.
- H. Marrou, J. Wery, L. Dufour and C. Dupraz, Eur. J. Agron., 2013, 44, 54–66 CrossRef.
- M. Kadowaki, A. Yano, F. Ishizu, T. Tanaka and S. Noda, Biosyst. Eng., 2012, 111, 290–297 CrossRef.
- R. Ureña-Sánchez, Á. J. Callejón-Ferre, J. Pérez-Alonso and Á. Carreño-Ortega, Sci. Agric., 2012, 69, 233–239 CrossRef.
- J. Pérez-Alonso, M. Pérez-García, M. Pasamontes-Romera and A. J. Callejón-Ferre, Renewable Sustainable Energy Rev., 2012, 16, 4675–4685 CrossRef.
- K. Inada, Plant Cell Physiol., 1976, 365, 355–365 Search PubMed.
- K. J. McCree, Agricultural Meteorology, 1971, 9, 191–216 CrossRef.
- C. S. Brown, A. C. Schuerger and J. C. Sager, J. Am. Soc. Hortic. Sci., 1995, 120, 808–813 CAS.
- S.-J. Kim, E.-J. Hahn, J.-W. Heo and K.-Y. Paek, Sci. Hortic., 2004, 101, 143–151 CrossRef.
- Z. Materová, R. Sobotka, B. Zdvihalová, M. Oravec, J. Nezval, V. Karlický, D. Vrábl, M. Štroch and V. Špunda, Plant Physiol. Biochem., 2017, 116, 48–56 CrossRef.
- M. Johkan, K. Shoji, F. Goto, S. Hahida and T. Yoshihara, Environ. Exp. Bot., 2012, 75, 128–133 CrossRef CAS.
- Z. Bian, Q. Yang, T. Li, R. Cheng, Y. Barnett and C. Lu, Physiol. Plant., 2018, 164, 226–240 CrossRef CAS PubMed.
- H. Liu, Y. Fu, D. Hu, J. Yu and H. Liu, Sci. Hortic., 2018, 236, 10–17 CrossRef CAS.
- A. Viršilė, A. Brazaitytė, V. Vaštakaitė-Kairienė, J. Miliauskienė, J. Jankauskienė, A. Novičkovas, K. Laužikė and G. Samuolienė, Food Chem., 2020, 310, 125799 CrossRef PubMed.
- N. C. Yorio, G. D. Goins, H. R. Kagie and R. M. Wheeler, HortScience, 2001, 36, 380–383 CAS.
- C. J. M. Emmott, J. A. Röhr, M. Campoy-Quiles, T. Kirchartz, A. Urbina, N. J. Ekins-Daukes and J. Nelson, Energy Environ. Sci., 2015, 8, 1317–1328 RSC.
- H. Shi, R. Xia, G. Zhang, H. L. Yip and Y. Cao, Adv. Energy Mater., 2019, 9(1–8), 1803438 CrossRef.
- R. A. Sharrock, Genome Biol., 2008, 9(1–7), 230 CrossRef.
- A. Rivadossi, F. M. Garlaschi, A. P. Casazza, G. Zucchelli and R. C. Jennings, Photochem. Photobiol. Sci., 2008, 7, 986–990 RSC.
- F. Yang, Y. Zhang, Y. Hao, Y. Cui, W. Wang, T. Ji, F. Shi and B. Wei, Appl. Opt., 2015, 54, 10232 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- C. S. Allardyce, C. Fankhauser, S. M. Zakeeruddin, M. Grätzel and P. J. Dyson, Sol. Energy, 2017, 155, 517–522 CrossRef.
- N. Roslan, M. E. Ya’acob, M. A. M. Radzi, Y. Hashimoto, D. Jamaludin and G. Chen, Renewable Sustainable Energy Rev., 2018, 92, 171–186 CrossRef.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- L. Dominici, L. Vesce, D. Colonna, F. Michelotti, T. M. Brown, A. Reale and A. Di Carlo, Appl. Phys. Lett., 2010, 96, 10–13 CrossRef.
- V. C. Galvão and C. Frankhauser, Curr. Opin. Neurobiol., 2015, 34, 46–53 CrossRef PubMed.
- J. J. Kim, M. Kang, O. K. Kwak, Y. J. Yoon, K. S. Min and M. J. Chu, Int. J. Photoenergy, 2014, 376315, DOI:10.1155/2014/376315.
- N. Duvva, D. Raptis, C. V. Kumar, E. N. Koukaras, L. Giribabu and P. Lianos, Dyes Pigm., 2016, 134, 472–479 CrossRef CAS.
- J.-M. Ji, H. Zhou and H. K. Kim, J. Mater. Chem. A, 2018, 6, 14518–14545 RSC.
- A. Dessì, M. Calamante, A. Mordini, M. Peruzzini, A. Sinicropi, R. Basosi, F. F. De Biani, M. Taddei, D. Colonna, A. Di Carlo, G. Reginato and L. Zani, Chem. Commun., 2014, 50, 13952–13955 RSC.
- A. Dessì, M. Calamante, A. Mordini, M. Peruzzini, A. Sinicropi, R. Basosi, F. Fabrizi De Biani, M. Taddei, D. Colonna, A. Di Carlo, G. Reginato and L. Zani, RSC Adv., 2015, 5, 32657–32668 RSC.
- G. Reginato, A. Mordini, L. Zani, M. Calamante and A. Dessì, Eur. J. Org. Chem., 2016, 233–251 CrossRef CAS.
- C. Bernini, L. Zani, M. Calamante, G. Reginato, A. Mordini, M. Taddei, R. Basosi and A. Sinicropi, J. Chem. Theory Comput., 2014, 10, 3925–3933 CrossRef CAS PubMed.
- A. Dessì, M. Monai, M. Bessi, T. Montini, M. Calamante, A. Mordini, G. Reginato, C. Trono, P. Fornasiero and L. Zani, ChemSusChem, 2018, 11, 793–805 CrossRef PubMed.
- W. Zhu, Y. Wu, S. Wang, W. Li, X. Li, J. Chen, Z. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756–763 CrossRef CAS.
- L. Han, X. Zu, Y. Cui, H. Wu, Q. Ye and J. Gao, Org. Electron., 2014, 15, 1536–1544 CrossRef CAS.
- K. D. Seo, I. T. Choi and H. K. Kim, Chem.–Eur. J., 2015, 21, 14804–14811 CrossRef CAS.
- Y. Liu, J. He, L. Han and J. Gao, J. Photochem. Photobiol., A, 2017, 332, 283–292 CrossRef CAS.
- L. Han, J. Liu, Y. Liu and Y. Cui, J. Mol. Struct., 2019, 1180, 651–658 CrossRef CAS.
- L. E. Polander, A. Yella, J. Teuscher, R. Humphry-Baker, B. F. E. Curchod, N. Ashari Astani, P. Gao, J.-E. Moser, I. Tavernelli, U. Rothlisberger, M. Grätzel, M. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 2642–2648 CrossRef CAS.
- Z. Wang, M. Liang, L. Wang, Y. Hao, C. Wang, Z. Sun and S. Xue, Chem. Commun., 2013, 49, 5748–5750 RSC.
- H. Dong, M. Liang, C. Zhang, Y. Wu, Z. Sun and S. Xue, J. Phys. Chem. C, 2016, 120, 22822–22830 CrossRef CAS.
- Y. Xie, W. Wu, H. Zhu, J. Liu, W. Zhang, H. Tian and W.-H. Zhu, Chem. Sci., 2016, 7, 544–549 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, 2010 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- M. L. Parisi, A. Dessì, L. Zani, S. Maranghi, S. Mohammadpourasl, M. Calamante, A. Mordini, R. Basosi, G. Reginato and A. Sinicropi, Front. Chem. Search PubMed , submitted.
- L. Zani, A. Dessì, D. Franchi, M. Calamante, G. Reginato and A. Mordini, Coord. Chem. Rev., 2019, 392, 177–236 CrossRef CAS.
- P.-Y. Ho, Y. Wang, S.-C. Yiu, W.-H. Yu, C.-L. Ho and S. Huang, Org. Lett., 2017, 19, 1048–1051 CrossRef CAS PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS PubMed.
- L. Vesce and R. Riccitelli, Prog. Photovoltaics, 2012, 20, 960–966 CAS.
- J. Bisquert, J. Phys. Chem. B, 2002, 106, 325–333 CrossRef CAS.
- F. Fabregat-Santiago, J. Bisquert, G. Garcia-Belmonte, G. Boschloo and A. Hagfeldt, Sol. Energy Mater. Sol. Cells, 2005, 87, 117–131 CrossRef CAS.
- D.-H. Roh, K. M. Kim, J. S. Nam, U.-Y. Kim, B.-M. Kim, J. S. Kim and T.-H. Kwon, J. Phys. Chem. C, 2016, 120, 24655–24666 CrossRef CAS.
- W. Ma, Y. Jiao and S. Meng, Phys. Chem. Chem. Phys., 2013, 15, 17187–17194 RSC.
- P. Li, C. Song, Z. Wang, J. Li and H. Zhang, New J. Chem., 2018, 42, 12891–12899 RSC.
- L. Vesce, R. Riccitelli, G. Soscia, T. M. Brown, A. Di Carlo and A. Reale, J. Non-Cryst. Solids, 2010, 356, 1958–1961 CrossRef CAS.
- Z. S. Wang, H. Kawauchi, T. Kashima and H. Arakawa, Coord. Chem. Rev., 2004, 248, 1381–1389 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0se00124d |
| This journal is © The Royal Society of Chemistry 2020 |