
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen hydrogen from anion exchange membrane water electrolysis: a review of recent developments in critical materials and operating conditions
Hamish Andrew
Miller
 *a,
Karel
Bouzek
b,
Jaromir
Hnat
b,
Stefan
Loos
c,
Christian Immanuel
Bernäcker
c,
Thomas
Weißgärber
c,
Lars
Röntzsch
c and
Jochen
Meier-Haack
d
*a,
Karel
Bouzek
b,
Jaromir
Hnat
b,
Stefan
Loos
c,
Christian Immanuel
Bernäcker
c,
Thomas
Weißgärber
c,
Lars
Röntzsch
c and
Jochen
Meier-Haack
d
aIstituto di Chimica dei Composti Organometallici (CNR-ICCOM), Via Madonna del Piano 10, 50019 Sesto Fiorentino, Firenze, Italy. E-mail: hamish.miller@iccom.cnr.it
bUniversity of Chemistry and Technology, Prague, Department of Inorganic Technology, Technická 5, 166 28, Prague 6, Czech Republic
cFraunhofer Institute for Manufacturing Technology and Advanced Materials IFAM, Branch Lab Dresden, Winterbergstraße 28, 01277, Dresden, Germany
dLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Straße 6, D-01069 Dresden, Germany
First published on 5th March 2020
Abstract
Hydrogen production using water electrolysers equipped with an anion exchange membrane (AEM), a pure water feed and cheap components such as platinum group metal-free catalysts and stainless steel bipolar plates (BPP) can challenge proton exchange membrane (PEM) electrolysis systems as the state of the art. For this to happen the performance of the AEM electrolyzer must match the compact design, stability, H2 purity and high current densities of PEM systems. Current research aims at bringing AEM water electrolysis technology to an advanced level in terms of electrolysis cell performance. Such technological advances must be accompanied by demonstration of the cost advantages of AEM systems. The current state of the art in AEM water electrolysis is defined by sporadic reports in the academic literature mostly dealing with catalyst or membrane development. The development of this technology requires a future roadmap for systematic development and commercialization of AEM systems and components. This will include basic and applied research, technology development & integration, and testing at a laboratory scale of small demonstration units (AEM electrolyzer shortstacks) that can be used to validate the technology (from TRL 2–3 currently to TRL 4–5). This review paper gathers together recent important research in critical materials development (catalysts, membranes and MEAs) and operating conditions (electrolyte composition, cell temperature, performance achievements). The aim of this review is to identify the current level of materials development and where improvements are required in order to demonstrate the feasibility of the technology. Once the challenges of materials development are overcome, AEM water electrolysis can drive the future use of hydrogen as an energy storage vector on a large scale (GW) especially in developing countries.
 Hamish Andrew Miller | Hamish Andrew Miller is a researcher at the ICCOM institute of the CNR based in Florence, Italy. After receiving his PhD in inorganic chemistry from the Queen's University of Belfast (UK) in 1999, he spent 10 years working in the chemical industry including 6 years developing fuel cells and electrolyzers. His major research interests involve nanotechnology and electrocatalysis in energy related fields. In particular developing PGM-free electrocatalysts for alkaline anion exchange membrane fuel cells and electrolysers, electroreforming of renewable alcohols for co-production of chemicals and hydrogen and investigating the electrochemical reduction of CO2 to fuels and chemicals. |
 Karel Bouzek | Karel Bouzek obtained his PhD from the University of Chemistry and Technology, Prague and currently he acts at the same University as a full professor at the Department of Inorganic Technology. His primary focus is on electrochemical engineering and technical electrochemistry. Currently he deals predominantly with hydrogen related processes, i.e. fuel cells and water electrolysers, including alkaline ones. Beside experimental study, his activities involve also the topic of mathematical modelling of electrochemical systems. |
 Jaromir Hnat | Jaromir Hnat finished his PhD in Inorganic technology at the University of Chemistry and Technology in Prague (UCTP) in 2015. Currently, he is in the position of Professor assistant at the Department of Inorganic Technology at UCTP. His research focuses primarily on the alkaline water electrolysis, particularly catalyst synthesis, anion exchange membrane testing, membrane electrode assembly preparation, cell and stack design and their characterisation. He is active in the field of the alkaline fuel cells, too. |
 Stefan Loos | Stefan Loos received his PhD in Physical Chemistry from Freiberg University of Mining and Technology in 2015 supervised by Professor Florian Mertens working on LiFePO4 cathodes for lithium ion batteries. As a postdoctoral researcher he did three years of research on catalytically active transition metal oxides/hydroxides for water oxidation in the group of Professor Holger Dau at Free University Berlin investigating catalytic mechanisms and catalyst structures by spectroelectrochemical methods. Currently he is doing applied research on electrode manufacturing and development at Fraunhofer IFAM in Dresden with focus on alkaline electrolysis and membrane electrolysis. |
 Christian Immanuel Bernäcker | Christian Immanuel Bernaecker received his PhD in Chemistry from Julius-Maximilians-Universität Würzburg (Germany) in 2011 supervised by Professor Christoph Lambert. The topic of the thesis was Physical Properties of Chromophore Funktionalized Gold Nanoparticles. In 2011, he joined the Hydrogen Technology group of Dr Lars Röntzsch at the Fraunhofer Institute for Manufacturing Technology and Advanced Materials IFAM in Dresden. Currently, the main research topics are applied electrochemistry and developing of scalable production routes for electrodes. |
 Jochen Meier-Haack | Dr Jochen Meier-Haack received his PhD in polymer chemistry from Hamburg University in 1992. He joined the Leibniz-Institut für Polymerforschung Dresden (IPF) in 1993 and became member of the membrane group in 1996. Since 2000 Jochen has been head of the Polymeric Membrane Materials group. His research interests are surface modification for fouling mitigation of membranes used in water treatment and development of ion-exchange membranes (AEM and CEM) for electrochemical applications (fuel cells, water electrolysers). |
1. Introduction
Hydrogen is a vital raw material used in industries such as ammonia synthesis for fertilisers, metallurgical reduction for steel refining and in the processing of crude oil.1,2 At present, globally around 70 MtH2 per year are used in a pure form while a further 45 MtH2 are used in industry without prior purification from other gases. H2 has long been proposed as an alternative energy vector to fossil fuels to generate power for domestic heating,3,4 industrial5 and transport sectors.6–10 In this sense, it has the potential to revolutionize the world's energy economy towards the predicted hydrogen economy/society.9,11,12 Hydrogen is contained in water and in hydrocarbons and is one of the most abundant elements available on our planet. It can be produced by a variety of methods (thermal, electrolytic and photolytic) from various sources like hydrocarbons, water or biomass. By far the most common method is steam reforming of methane or other hydrocarbons, which causes significant CO2 emissions. On the other hand, water electrolysis is a well-established mature technology used in special applications. Recently, H2 has been used to exploit renewable energy (wind, solar) to produce H2 as energy vector for grid balancing or power-to-gas and power-to-liquid processes.13–16 There are two main water electrolysis technologies that produce H2 at low temperatures, which can be distinguished by the electrolyte used in the electrolysis cell: alkaline electrolysis (AE) and proton exchange membrane (PEM) electrolysis. Additionally, the solid oxide high-temperature steam electrolysis is being developed due to the low cell voltages required, which would be especially useful if the required heat can be used from other exothermic industrial processes.2. Established water electrolysis technologies
2.1 Alkaline electrolysis
Alkaline electrolysis is a mature technology for H2 production up to the MW scale, and represents the most widely used electrolytic technology on a commercial level worldwide.13,17,18 The AE cell consist of two electrodes (anode and cathode) immersed in a highly concentrated aqueous alkaline electrolyte consisting of 20 to 30 mass% KOH. In traditional AE, the most commonly used anode and cathode materials are low-cost steel or nickel alloy-plated steel materials. The two electrodes are arranged in a zero-gap (or quasi zero-gap) formation using a thin diaphragm, which enables separation of the product gases. The diaphragm is permeable to hydroxide ions and water. Major challenges associated with AE are the handing of the corrosive electrolyte and limited current densities due to moderate OH− mobility. Furthermore, the diaphragm does not completely prevent the cross-over of gases from one half-cell to the other. The diffusion of oxygen into the cathode compartment reduces the efficiency of the electrolyser, reacting with the hydrogen present on the cathode side to form water. Additionally, extensive mixing (particularly hydrogen diffusion to the O2 evolution half-cell) also occurs and must be avoided for safety aspects. This is particularly problematic at low loads (<40%) where the O2 production rate decreases, thus increasing the H2 cross-over concentration to dangerous levels (lower explosion limit >4 mol% H2).2.2 PEM electrolysis
Proton exchange membrane (PEM) water electrolysis is young technology that has good performance and stability and has established itself in the market place in certain niche applications. In PEM electrolysis, the anode and cathode catalysts are typically IrO2 and Pt, respectively. An acidic membrane is used as solid electrolyte (perfluorosulfonic acid membranes) instead of a liquid electrolyte (Fig. 1). The membrane conducts H+ cations from the anode to the cathode and separates the H2 and O2 produced in the reaction. PEM electrolysers generally operate at a current density of 2 A cm−2 at 50–80 °C and approx. 2.1 V. The kinetics of the hydrogen evolution reaction (HER) in PEM electrolysis are faster than in alkaline electrolysis due to the low pH of the electrolyte and the high active metal surface of Pt electrodes. PEM electrolysis is also safer due to the absence of any caustic electrolyte. An additional advantage of PEM electrolysis is the possibility of using high pressure on the cathode side, while the anode can be operated at atmospheric pressure. The corrosive acidic cell operation environment requires the use of specialised materials. These materials must not only resist the harsh corrosive low pH condition, but also sustain the high applied over voltage at the anode (2 V), especially at high current densities. Corrosion resistance applies not only for the catalysts used, but also for current collectors and separator plates. Only a few materials can be selected that can perform in this harsh environment. This demands the use of scarce, expensive materials and components such as noble metal catalysts (e.g. platinum group metals (PGM) like Pt, Ir and Ru), Ti-based current collectors, and separator plates. Ir is one of the rarest elements in the Earth's crust, having an average mass fraction of 0.001 ppm in the Earth's crust (annual production of some tons only). Moreover, Ir use has recently increased due to its use in crucibles employed to fabricate LEDs for smartphones, tablets, televisions and automobiles. It can be expected that production of high volumes of PEM electrolysis units will considerably affect the demand for Ir and consequently the price.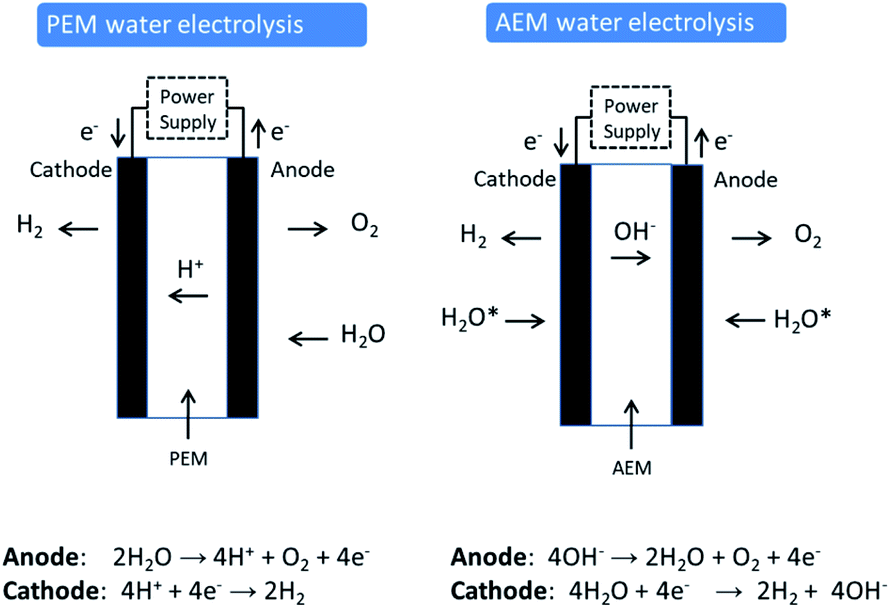 | ||
| Fig. 1 Comparison of water electrolysis cells and chemistries using either a Proton Exchange Membrane (PEM) or an Anion Exchange Membrane (AEM).* Current AEM electrolyzers use a water feed with added electrolyte (HCO3−/CO32− or dilute KOH) to obtain sufficient performance. AEM electrolyzers can operate with electrolyte feed at both electrodes or in anode feed only mode. | ||
The principle difference between the two technologies (PEM and AE) lies in the significantly higher current densities achievable by a PEM electrolyser that leads to higher production rates and more compact systems (see Table 1 for comparison). To achieve this high loadings of rare and expensive metals for catalysts and expensive corrosion resistant components such as bipolar plates based on high-quality Ti are required.
| Alkaline | PEM | AEM | |
|---|---|---|---|
| Electrolyte | Aqueous KOH (20–40 wt%) | Proton exchange ionomer (e.g. Nafion) | Anion exchange ionomer (e.g. AS-4) + optional dilute caustic solution |
| Cathode | Ni, Ni–Mo alloys | Pt, Pt–Pd | Ni and Ni alloys |
| Anode | Ni, Ni–Co alloys | RuO2, IrO2 | Ni, Fe, Co oxides |
| Half-cell separation | Diaphragm (Zirfon Perl 500 μm) | Nafion 117 (e.g. 180 μm) | AEM (20–100 μm) |
| Current density (A cm−2) | 0.2–0.4 | 0.6–2.0 | 0.2–1.0 |
| Cell voltage (V) | 1.8–2.4 | 1.8–2.2 | 1.8–2.2 |
| Cell area (m2) | <4 | <3 | Lab testing cells |
| Operating temperature (°C) | 60–80 | 50–80 | 50–60 |
| Operating pressure (bar) | 1–30 | 30–76 | 1–30 |
| Production rate (Nm3 h−1) | <760 | <40 | <1 |
| Gas purity (vol%) | >99.5 | >99.9999 | >99.99 |
| System response | Seconds | Milliseconds | na |
| Stack lifetime (h) | 60k to 100k | 20–60k | na |
| Technology status | Mature | Commercial | R&D |
AEM electrolyser technology, which is discussed in detail in this review, aims at combining the advantages of PEM (membrane separation, pure water feed) with the advantages of AE (cheap and abundant materials).
2.3 AEM electrolysis
The concept of AEM water electrolysis has been the subject of various reports in recent years in the academic literature although emphasis has been predominantly on the development of catalyst materials rather than AEM membranes or ionomers. There are very few reports on actual AEM water electrolysis cell performance especially when deionized water is used instead of liquid aqueous KOH. A search of the academic literature (Web of Science) with the words “anion exchange membrane water electrolysis” in the title yielded only 20 papers (over the period 2012–2019). The concept and chemistry of AEM water electrolysis is shown schematically in Fig. 1 and compared to PEM water electrolysis. Research coordination in this area has been boosted recently by significant US DOE efforts19 in developing AEM electrolysis and by the EU through its FCH-JU funding programme.20AEM electrolysers work with an alkaline environment at the membrane interface provided by the immobilized positively charged functional groups on the polymer backbone or on pendant polymeric side chains. While the largest impediments to the development of AEM systems are membrane stability and ionic conductivity, an improved understanding of how to integrate catalysts into AEM systems is necessary. Research on AEM systems to date has been limited to the laboratory scale with focus on developing electrocatalysts, membranes and understanding operational mechanisms with the general objective of obtaining a high efficiency, low cost and stable AEM devices. Table 2 lists the most significant recent literature reports on AEM systems. The most important materials (catalysts, membranes, and ionomers) and conditions (electrolyte, operating temperature) are listed along with the best voltage current performance reported.
| Membrane electrode assembly (MEA) components | Ionomer/binder | Water feed | Cell Voltage (V) | Current dens. A cm−2 | Cell temp. °C | Reference | ||
|---|---|---|---|---|---|---|---|---|
| Anode | Membrane | Cathode | ||||||
| IrO2 | A-201 Tokuyama | Pt black | AS-4 | Deionized water | 1.8 | 0.399 | 50 | 7 |
| CuCoO3 | A-201 Tokuyama | Ni/CeO2–La2O3/C | PTFE | 1% K2CO3/KHCO3 | 1.9 | 0.47 | 50 | 21 |
| Pb2Ru2O6.5 | Chloromethylated PSF | Pt black | PSF-TMA+Cl− | Ultrapure water | 1.8 | 0.4 | 50 | 22 |
| Ni–Fe | (xQAPS) | Ni–Mo | (xQAPS) | Ultrapure water | 1.85 | 0.4 | 70 | 33 |
| CuCoOx | A-201 Tokuyama | Pt/C | AS-4 | K2CO3 10% | 1.95 | 1 | 50 | 113 |
| IrO2 | FAA-3-50 | Pt/C | FAA-3-Br | 1 M KOH | 1.9 | 1.5 | 70 | 134 |
| NiCoOx:Fe | FAA-3 | Pt black | FAA-3 | Pure water | 2.25 | 0.8 | 50 | 53 |
| Ni | A-201 Tokuyama | Ni | — | 1 M KOH | 1.9 | 0.15 | 50 | 31 |
| CuCoO3 | LDPE-g-VBC | Ni/CeO2–La2O3/C | PTFE | 1% K2CO3/KHCO3 | 2.1 | 0.46 | 50 | 143 |
| Cu0.7CO2.3O4 | QPDTB | Nano Ni | Poly(DMAEMA-co-TFEMA-co-BM) | Deionized water | 1.9 | 0.1 | 50 | 132 |
| CuCoO3 | Mg–Al layered double hydroxide | Ni/CeO2–La2O3/C | PTFE | 1% K2CO3/KHCO3 | 2.2 | 0.28 | 70 | 136 |
| Ce0.2MnFe1.8O4 | FAA-3-PK-130 | Pt on Ti | — | Deionized water | 1.8 | 0.3 | — | 58 |
| Pd/TNTA web | A-201 Tokuyama | Pt/C | PTFE | 2 M NaOH | 2 | 2 | 80 | 163 |
| IrO2 | Piperidinium-poly(2,6-DPO) LSCPi | Pt/C | 5 wt% LSCPi | Deionized water | 1.8 | 0.5 | 50 | 90 |
| CuCoOx (on Ni foam) | A-201 Tokuyama, FAA-3,FAA-3-PP-75 | Ni/(CeO2–La2O3)/C on carbon paper | I2 | 1% K2CO3 | 1.95 | 0.5 | 60 | 55 |
| CuCoOx (on Ni foam) | A-901 Tokuyama | Ni/(CeO2–La2O3)/C on carbon paper | I2 | 1% K2CO3 | 2.1 | 0.5 | 50 | 54 |
| NiCo2O4 | Polyethylene based radiation grafted | Pt | Polystyrene-b-poly (ethylene/butylene)-b-polystyrene | 0.1 M KOH | 1.65 | 0.1 | 60 | 59 |
| NiCo2O4 on steel mesh | Homemade, not specified | Powder catalyst, Acta 4030 (PGM free) | Homemade, not specified | DI-water | 1.86 | 0.4 | 60 | 60 |
| CuxCo3−xO4 | PTFE based | Pt/C | q-ammonium polymethacrylate | DI-water | 1.6 (2.0) | 0.1 (0.4) | 22 | 57 |
| CuxCo3−xO4 | Developmental AEM with quaternary ammonium functional groups | Pt/C | — | 1 M KOH (DI-H2O) | 1.9 (2.0) | 1.4 (0.2) | 25 | 140 |
| NiFe2O4 | Sustainion 37–50 | NiFeCo | Nafion | 1 M KOH | 1.9 | 1 | 60 | 125 |
| NiAl | HTM-PMBI | NiAlMo | — | 1 M KOH | 2.1 | 2 | 60 | 129 |
3. Critical AEM components
In the following sections we describe recent developments in the critical components of AEM electrolysis (PGM free HER and OER catalysts and anion exchange membranes). Auxiliary cell components such as bipolar plates and current collectors are not covered as we believe these aspects lie outside the scope of this review.3.1 PGM-free electrocatalysts
The development of non-noble metal catalysts for both the HER and the OER is crucial to reducing the capital cost of AEM water electrolysis. The challenges lie in optimizing chemical composition, stability and activity of such materials. Because of the relatively low mass specific activity when compared with noble metals, large catalyst loadings are required and this leads to large Ohmic resistance losses.The HER in alkaline media proceeds by the initial dissociation of water and the formation of hydrogen intermediates (Had) in the Volmer step (eqn (1)):26
| H2O + e− ⇌ Had + OH− | (1) |
| H2O + Had + e− ⇌ H2 + OH− | (2) |
| 2Had ⇌ H2 | (3) |
The H2O dissociation (eqn (1)) is typically a slow reaction and hence it is generally accepted that initial water dissociation is the rate-determining step. Alkaline HER is also more complex than acid HER with Had, hydroxyl adsorption (OHad), and water dissociation all important species/processes to be optimized in developing catalyst materials.27,28 In a nutshell, the HER in alkaline media requires the breaking of strong covalent H–O bonds (in water) which is a difficult first-up reaction.29 This section discusses recent advances in the development of PGM-free catalysts for the HER under alkaline conditions. Emphasis will be given to materials tested in complete AEM electrolysis cells.
A large number of Ni-based HER electrocatalysts have been investigated in the literature as potential PGM-free materials. They generally show significantly inferior activity with respect to Pt benchmark catalysts. Commercial Ni nanopowder (2 mg cm−2) was used in an AEM water electrolyser with a pure water feed and produced 0.3 A cm−2 at 1.8 V.30 Other researchers used low loading Ni nanoparticles electrodeposited onto carbon paper (8.5 μg cm−2) as HER catalyst in an AEM electrolyser. With a 1 M KOH feed 0.15 A cm−2 was reached at 1.9 V cell potential.31 The activity and stability of Ni by itself is hence relatively poor. Combination with other transition metals or oxides or as sulphides, selenides, nitrides or phosphides has been used as strategy to obtain improved performance.
Ni supported on a mixed oxide and carbon material (CeO2–La2O3/C) has been employed in an AEM electrolyzer using a mild alkaline electrolyte (1% K2CO3/KHCO3) operating at pH 10–11.21 Cathode catalyst loading was shown to have the greatest influence on cell performance. Cathode catalyst loading was varied from 0.6 to 7.4 mg cm−2, resulting in cell potentials ranging between 2.01 and 1.89 V at 470 mA cm−2 (Tcell = 50 °C). The measured alternating-current (AC) resistance at 1 kHz varied between 0.218 and 0.132 Ω cm2 for catalyst loading ranging between 0.6 to 7.4 mg cm−2. The cell performance parameters were directly related to the cathode catalyst loading with the highest loading producing the best performance despite the thicker electrode layer.
In alkaline media Ni–Mo alloys have been reported to have the best activity of PGM-free catalysts.32 Zhuang and co-workers first reported an AEM water electrolysis cell working with pure water in 2012 with PGM-free electrocatalysts.33 A Ni–Mo composite catalyst was employed for the HER.34 The challenge when removing the electrolyte from the water feed is to have sufficiently high Ni–Mo loading to avoid high Ohmic losses due to the resulting thick electrode. A co-deposition procedure was used to fill a stainless steel skeleton with sufficient Ni–Mo catalyst precursor. Annealing at 500 °C in H2 led to the active catalyst that exhibited a very low HER overpotential (0.11 V at 0.4 A cm−2 in 1 M KOH).34 Recently, a mixed phased catalyst, composed of crystalline Ni-rich Ni–Mo alloy nanoparticles embedded in a Mo-rich oxide matrix was prepared by Patel and co-workers.35 This material has low activity toward hydrogen evolution. However, its activity markedly increased upon activation by postdeposition reductive annealing or by including carbon black as a catalyst support. These researchers concluded that the HER activity is limited not only by kinetics but also by electrical resistivity arising from thin oxide layers at the interfaces between the Ni–Mo alloy nanoparticles. On the other hand, it has been consistently reported that a mix of metallic and oxidic species at the catalyst surface is beneficial for HER/HOR, most probably related to the simultaneous need of adsorption sites for hydroxidic (oxidic) and hydride-type species.36,37
Zhang et al. prepared a MoNi4 electrocatalyst supported on MoO2 cuboids on Ni foam (MoNi4/MoO2@Ni).39 A reduced energy barrier of the Volmer step, was responsible for the high HER activity under alkaline conditions. The same authors made MoNi4/MoO3−x nanorod arrays with similarly high activity.38 Their combined results reveal that this class of alloy exhibits a near zero onset potential, a very small overpotential of 1 mV at 10 mA cm−2, and a Pt-like Tafel slope of 30 mV dec−1 in alkaline media, which are comparable to Pt and outperforms all other state-of-the-art Pt-free catalysts reported (Fig. 2). This catalyst was also shown to be stable during short constant current testing (Fig. 2c). The active sites were determined to be metallic MoNi4 and oxygen-deficient MoO3−x. After an electrochemical scan from 1.17 to 1.72 V, the catalyst lost HER activity (Fig. 2d) which was associated with irreversible oxidation of Mo0, Mo4+ and Mo5+ to Mo6+ and Ni0 to Ni2+ species conducting (from XPS).
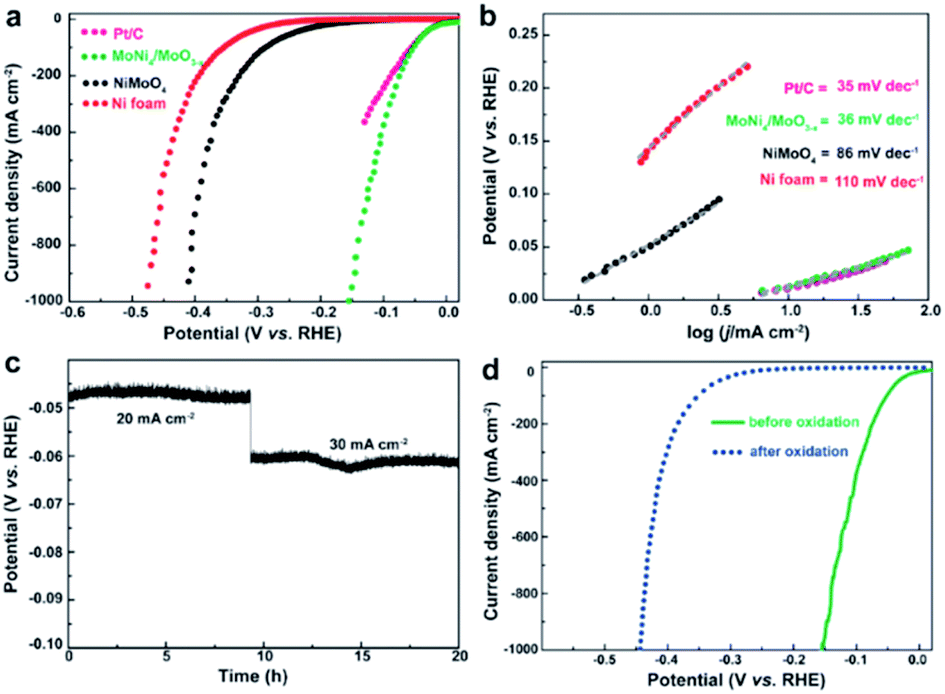 | ||
| Fig. 2 (a) LSV curves for Pt/C, Ni foam, NiMoO4, and MoNi4/MoO3−x. (b) The corresponding Tafel plots. (c) Short-term stability test of MoNi4/MoO3−x. (d) LSV curves of MoNi4/MoO3−x for HER before and after electrochemical oxidation.38 Reprinted from ref. 38 with permission from John Wiley and Sons. | ||
Poor conductivity of transition metal oxides makes them unsuitable for the HER in general. Some examples however have shown promise; Mo- or W-based oxides are examples. Porous MoO2 nanosheets prepared on Ni foam by Jin et al. were found to be highly active and stable for the HER in alkaline media.40 Ni-NiO nanostructures supported on CNTs prepared by Gong and co-workers showed excellent activity (100 mA cm−2 at 100 mV overpotential).41 The remarkable performance of this catalyst is likely to be due to a combination of synergistic effects of the nano-interfaces (Ni, NiO and CNT) and the high intrinsic conductivity of the carbon nanotubes. A water electrolyser was tested with this catalyst as cathode and achieved a current density of 20 mA cm−2 at a cell voltage of 1.5 V in 1 M KOH (membrane-less).
In summary, there are very few reports of PGM-free HER catalysts applied in complete AEM electrolysers. These are simple Ni-based or Ni-supported on mixed oxide-carbon supports. By comparison, there are many studies of HER catalysts with only half-cell electrochemical characterization. Of all of these, quite remarkable activity, approaching that of Pt, has been demonstrated with Ni–Mo alloyed materials making this class of HER catalyst the most promising for application in AEM electrolyser cells. Engineering rather than chemical solutions may be required to exploit successfully these materials in AEM electrolysis cells on a larger scale.
| 4OH− → 2H2O + O2 + 4e− | (4) |
| 2H2O + 2e− → 2OH− + H2 | (5) |
The OER requires the transfer of four electrons per O2 molecule whereas for the HER reaction only two electrons need to be transferred for the formation of a single H2 molecule. This gives rise to inherent sluggish OER kinetics, a significant contribution to the cell voltage and in many cases to a more complex mechanism as four OH− ions need to take part in the catalytic cycle. When the water feed contains an electrolyte such as KOH this aids the reaction by supplying OH− to the active sites. In the case of AEM technology the only source of OH− is the ion conducting ionomer.
Regarding the OER mechanism, many models have been proposed recently. For the active and alkaline stable (oxy)hydroxides of the 3d-transition metals such as Mn, Fe, Co, and Ni it is consensus that μ-oxo-bridged MO6-units play an important role to facilitate the OH− bonding, further oxidation and release of molecular O2.42,43
Excluding the vast amount of work which has been conducted in concentrated KOH the following catalysts are described as effective OER catalysts in dilute KOH, K2CO3/KHCO3 or deionized water (see also Table 2). Another important review has been collected by the Bessarabov group.44
3.1.2.1 Ni-based OER catalysts. Recent research has shown that Ni–Fe catalysts offer improved activity with respect to pure Ni catalysts.45 The best result using deionized water reported to date is still the fundamental work of Xiao et al. from 2012 with a Ni–Fe anode (xQAPS membrane, Ni–Mo cathode, 70 °C and pure water feed) produced by solid-state electrochemical reduction reaching a current density of 0.6 A cm−2 at 1.9 V cell voltage (Fig. 3).33 In addition to the ionomer used a PTFE binder was used for electrode preparation based on a Ni foam. On the other hand after running at 0.4 A cm−2 for eight hours a degradation of 50 mV was already observed. The authors suspect that the NiFe-anode might cause degradation as iron leaching can occur at the anode and be redeposited at the cathode.46 As such this system of catalysts worked equally well as it has been reported with Ir (anode) and Pt (cathode) for example by Leng et al. (also using deionized water, Tokuyama membrane A-201).47 The Fe is useful in order to enhance the catalytic activity towards O2 evolution and the mechanistic details and active species are heavily discussed in recent years.48–51 Indeed, simple physical mixing of Ni(OH)2 and Fe(OOH) leads to atomically intermixed Ni–Fe catalysts with unexpectedly high OER activity.52 Xu and co-workers investigated NiCoOx-based catalysts in AEM electrolysis and found excellent performance that was further improved by adding Fe species to the particle surface.53
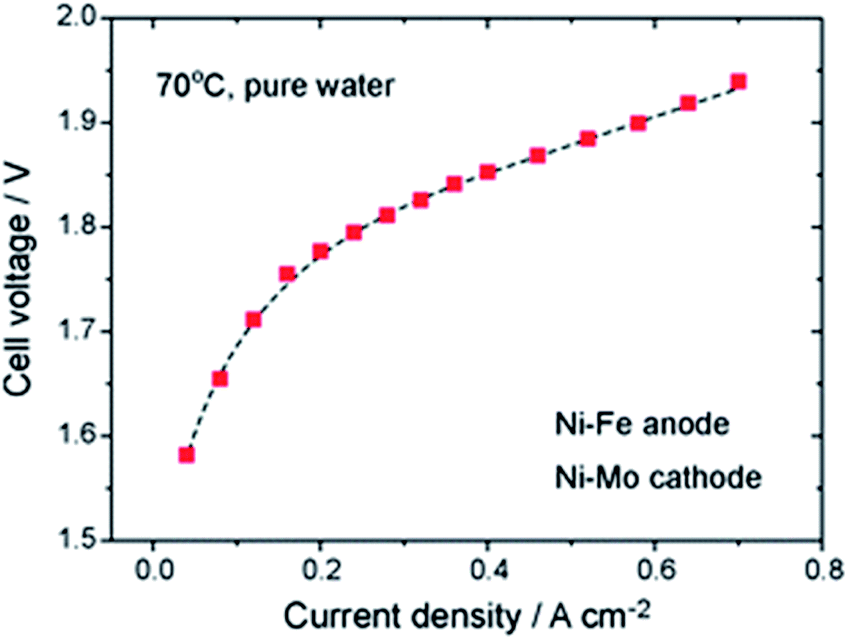 | ||
| Fig. 3 Cell performance of the first prototype of an AEM water electrolysis system using Ni–Fe anode and Ni–Mo cathode and working only with pure water.33 Reproduced from ref. 33 with permission. Copyright@2014. Royal Society of Chemistry. | ||
3.1.2.2 Other PGM-free-based OER catalysts. The CuxCo3−xO3 spinel anode catalysts described by Vincent et al. and several times by Wu and Roggan were shown to have optimized performance in combination with a Ni/(CeO2–La2O3)/C, 30 mg cm−2, cathode (often tested in combination with Pt) and most stable with the A201-Tokuyama membrane, but were also tested with FAA-3 membranes (Table 2).54–57 They used the I2 Acta Spa ionomer and tested the MEA for 200 hours in a 5 cm2 cell. Although performing quite well with PGM-free catalysts, the degradation rate was significant with an average voltage increase of 2.37 mV h−1. With the same type of OER catalyst Pavel et al.21 obtained similar performance in 2014. In this case the degradation in the cell test was estimated to be close to 0.2 mV h−1 (approximately 200 mV in a 1000 h cell test). An interesting class of OER catalyst based upon Ce incorporation into MnFe2O4 crystal lattice provided improved activity and conductivity.58 At 25 °C, the single cell with Ce0.2MnFe1.8O4 exhibited a current density of 300 mA cm−2 at 1.8 V. Notably, Ce0.2MnFe1.8O4 demonstrates a durability of >100 hours in continuous electrolysis.
The Bessarabov group using the CuxCo1−xO3 anode and a very thin (9 μm thick) A209 membrane showed stable performance for almost 200 h at ca. 2.1 V cell voltage at a current density of 500 mA cm−2 with a degradation rate of 0.2 mV h−1.54
Gupta et al. were investigating the NiCo2O4 catalyst as an anode at 60 °C in 0.1 M KOH (Pt cathode) with a reasonable performance of 100 mA cm−2 with a polyethylene based radiation grafted AEM and a polystyrene-based ionomer.59 Unfortunately, no tests in deionized water are shown and degradation studies on the long term have not been put forward.
In conclusion, the number of PGM-free catalysts used as anodes in AEM electrolysis with a deionized water feed is small. The very active Ni–Fe catalysts (layered double hydroxides or oxyhydroxides) as well as the Ni- and Cu/Co-mixed spinel-type oxides remain the most likely candidates for an efficient anode in AEM water electrolysis. The only publications focusing on a combination of deionized water and PGM-free catalyst materials in AEM electrolysis that obtained reasonable activity originate from the year 2012 (ref. 33) and 2018.60 It is clear that fast development of preparation methods and rational catalyst design principles together with the necessity to substitute Ir-based and Pt precious metal catalysts will drive AEM electrolysis.
A further challenge remains the preparation of solid polymer electrolyte membranes with fast OH− transport and low degradation at high temperatures. Additionally, the challenge remains to optimize the catalyst loadings and the catalyst/ionomer ratio in order to maximize the cell performance and the ionic contact between electrodes and anion exchange membrane.61 These aspects will be discussed in the following sections.
4. Anion exchange membranes and ionomers
Anion exchange membranes and ionomers are the fundamental core components of AEM electrolysis technology. Generally, they are formed by a polymer backbone with anchored cationic groups that confer anion selectivity.62 Most often, the anion exchange group consists of trialkyl quaternary ammonium salts attached to polymeric backbones like polystyrene, polysulfone, poly(ether sulfone) or poly(phenylene oxide) by benzylic methylene groups (see for example63 and references cited within).The major shortcoming of anion exchange materials is their limited thermal stability, especially at high pH.64 Two main mechanisms, namely Hofmann elimination and nucleophilic attack of hydroxide on N-alkyl groups (SN2 mechanism) lead to degradation of anion-exchange groups at high temperature under basic conditions.65 Other degradation mechanisms have also been recently identified such as the electrochemical oxidation of the adsorbed phenyl group (in the polymeric ionomer) on oxygen evolution catalysts.66
This limitation has important consequences for the long-term stability of AEM electrolyzer systems as well as the operational temperature limits. Consequently, extensive recent research has involved the development of new anion exchange materials with higher thermal/chemical stability in alkaline medium for the use in electrochemical applications (Fig. 4). Most AEMs have been developed for alkaline fuel cells (AEMFCs) (see reviews by Ran et al.,67 Hagesteijn et al.,68 Hickner et al.,69 Merle et al.,70 Couture et al.71 and Wang et al.72). The advantages of alkaline AEM-based systems when compared to CEM-based systems in electrochemical applications have been highlighted in review papers by Varcoe et al.,55 Gu et al.73 and Paidar.74 Bodner (general alkaline electrolysis) has paid special attention to alkaline electrolysis.75 However, less attention has been paid to membrane-based alkaline water electrolyzers (see review by Vincent and Bessarabov44).
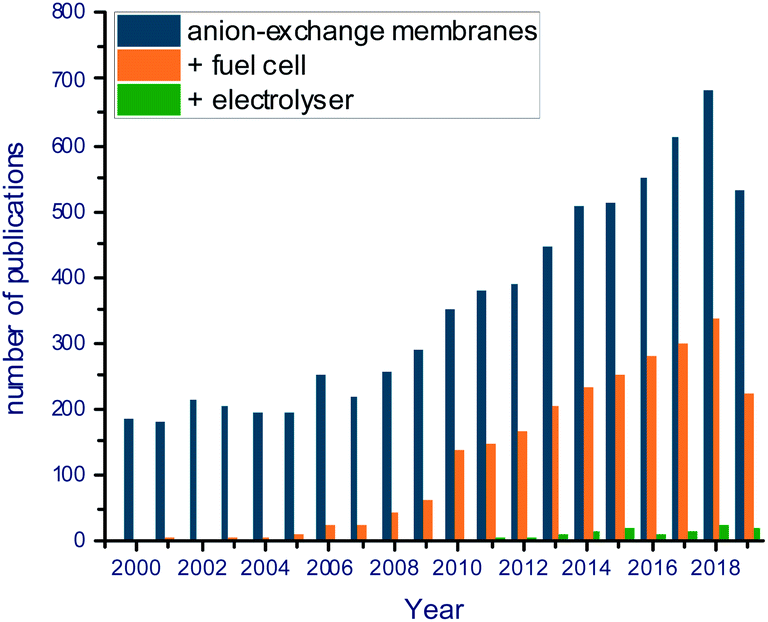 | ||
| Fig. 4 Development of annual number of publications in the field of anion exchange membranes (from Web of Science (access 18.10.2019)). Search terms in topic: anion AND exchange AND membrane (AND fuel AND cell; AND electrolyser). | ||
The following section reviews the development of new temperature and alkaline stable AEMs. The last section describes AEMs used specifically in AEM water electrolysers.
An unexpected high thermal stability in alkaline medium (20% NaOH, 100 °C) has been reported for spirocyclic bis-2,2′-biphenylylene ammonium iodide.76 Since the synthesis of this compound is not straightforward, simpler and easier to synthesize molecules have to be developed mimicking this structure. Marino and Kreuer reported on the thermal stability of quaternary ammonium salt model compounds.77 The most stable compound in this study was 6-azonia-spiro[5.5]undecane followed by N,N-dimethylpiperidinium salt, the former one having a similar structure to that described by Hellwinkel and Seiffert.76 A similar study was performed by Gu et al.,78 showing that 5-azoniaspiro[4.5]decane possesses the highest alkaline stability (2 N NaOH 80 °C, 168 h) among the tested compounds. Linear (water soluble) polymers bearing 5-azoniaspiro[4.4]nonane moieties in the backbone, obtained by cyclopolymerisation of N,N-diallylpyrrolidinium chloride showed no degradation after treatment in 2 N NaOH at 80 °C after 168 h.79 Even additional treatment at 120 °C for 18 h resulted in no decomposition. The Jannasch group at Lund University (Sweden) reported several approaches for the preparation of alkaline stable anion exchange materials based on spirocyclic quaternary ammonium salts.80–87 These approaches include incorporation of such functional groups into the polymer backbone80–83 or directly attached to the polymer backbone via benzylic methylene groups,86 as a sidechain with different spacer lengths85 and different ion exchange groups87 or as homopolymer in an interpenetrating network with brominated poly(phenylene oxide) as second component.84 Direct attachment of spirocyclic ammonium groups (piperidinium) was achieved by reacting tetrakis(bromomethyl)benzene units in poly(ether sulfone) with N-heterocycles of different ring size (5–7).86 Depending on the ring size, hydroxide conductivities were found to be in the range of 19 to 110 mS cm−1 (the smaller the ring the higher the conductivity). While the materials were stable in alkaline solution (1 N NaOH) at 20 °C, degradation of the spirocyclic quaternary ammonium group was observed after 7 days at 40 °C by 1H NMR spectroscopy. Treatment at 60 °C resulted in an additional degradation of the polymer backbone. The same chemistry, namely conversion of tetrakis(bromomethyl)benzenze with N-heterocycles (bipiperidine or trimethylenedipiperidine), was used to prepare anion exchange materials with the ion exchange group in the polymer backbone.83 Both materials showed high stability under alkaline conditions (1 N KOH) at 80 °C (no degradation after 672 h) and slight degradation after 336 h at 120 °C. For the trimethylenedipiperidine-based material, no degradation was observed even after 1896 h storage in 1 M KOH at 80 °C. Since these materials are water soluble, membranes were prepared from blends with poly(benzimidazole) (ionic crosslinking) containing 70–80 mass% of the respective ionomer. Hydroxide conductivities at 90 °C under fully hydrated conditions were in the range from 70 (80 mass% ionomer) to 120 mS cm−1 (70 mass% ionomer). This result was explained by increasing water uptake with increasing ionomer content in the blend (up to 450%). Another method to obtain anion exchange materials and membranes with six-membered heterocycles in the polymer backbone involves the polymerization of N-methyl-4-piperidone and aryl compounds (biphenyl, p-terphenyl) and 1,1,1-trifluoroacetone or 2,2,2-trifluoroacetophenone as comonomer and triflic acid as catalyst.80–82 Quaternary ammonium groups were obtained by reaction of the pendant piperidine moiety with halogenoalkanes82 or α,ω-bis-halogenoalkanes.81 The latter reaction results either in spirocyclic or crosslinked products.80 In all cases, products with high alkaline stabilities at elevated temperatures were obtained. Quaternizing the piperidine moiety with long alkyl chains resulted in a decreasing alkaline stability due to destabilization of the piperidinium ring and thus facilitating the degradation by ring opening elimination. Maximum ion conductivities (OH−-form) were in the range of 100 mS cm−1 at 80 °C under fully hydrated conditions.
In a comparative study Dang and Jannasch investigated the properties of different hetero cycloaliphatic quaternary ammonium groups.88 These groups were attached to the polymer backbone (PPO) via pentyl spacer chains. Cycloaliphatic quaternary ammoniums groups based on 5- and 6-membered rings and tetraalkyl ammonium groups linked to the polymer backbone via pentyl spacer chains showed no degradation under the applied test conditions (394 h, 90 °C, 1 M NaOH) (Fig. 5). Larger ring size, methyl substituents in o-position to the nitrogen, incorporation of hetero atoms into the ring as well as linkage of the ammonium groups via methylene spacers resulted in degradation by Hofmann elimination and/or nucleophilic substitution.
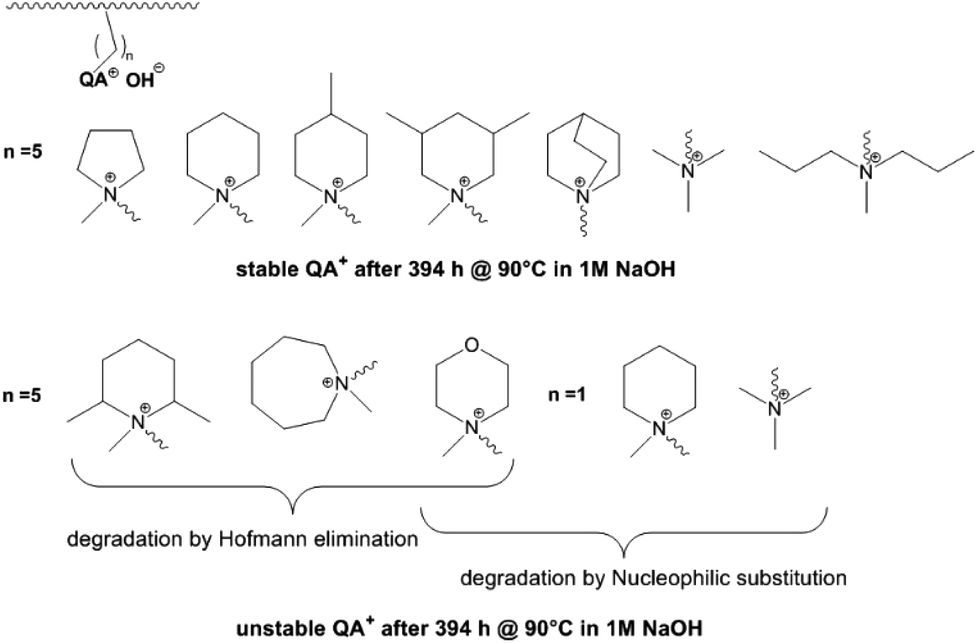 | ||
| Fig. 5 Stability and degradation mechanisms of different quaternary ammonium groups tethered to a polymer backbone (PPO) by different spacer lengths as revealed by 1H NMR spectroscopy. Degradation of PPO backbone in these experiments was not detected.88 | ||
Strasser et al. described alkaline stable multiblock copolymers based on polysulfone and diallylpiperidinium hydroxide.89 α,ω-fluorophenylsulfone terminated poly(diallyl piperidinium chloride) was used as macromonomer in the synthesis of polysulfone block copolymers. These block copolymers exhibited a phase-separated morphology as indicated by DSC and AFM measurements. Depending on the IEC (0.90 to 2.02 meq g−1) OH−-conductivities up to 102 mS cm−1 were recorded. More importantly, after thermal treatment in methanolic 1 N KOH for 42 days no decomposition of the ion exchange group was observed by 1H NMR spectroscopy.
Although this class of anion exchange materials has many promising properties with respect to the use in electrochemical processes, only a few papers have been published so far.
Gu et al. prepared anion exchange membranes by copolymerization of N,N-diallylpyrrolidinium bromide or N,N-diallylpiperidinium bromide or N,N-diallyl-N-hexamethylene iminium bromide, acrylonitrile and styrene.78 Divinylbenzene (3 mass%) was used as crosslinker. Membranes with a theoretical IEC of 1.2 meq g−1 (exp. 0.97–1.15 meq g−1) were obtained having a hydroxide conductivity in the range of 18.9 to 20.3 mS cm−1. As expected from investigations of model compounds in the same study, the highest alkaline stability (168 h, 80 °C, 1 N NaOH) was observed for the membrane based on N,N-diallylpiperidinium hydroxide.
Chen et al. synthesized a series of anion exchange membranes for alkaline membrane fuel cell applications, based on poly(biphenyl piperidinium) (PBP)/6-azaspiro[5.5]undecane functionalized polyphenyl ether (ASU-PPO).90 The advantages of both polymers were combined by crosslinking. Furthermore, the problem of high water uptake of PBP and the insufficient film-forming property of ASU-PPO were addressed. These crosslinked PBP-ASU-PPO membranes exhibit good ion conductivity (max. 128 mS cm−1 at 80 °C), durability, and mechanical properties, while the swelling was only 15.7%. The chlorine conductivity decreased only by 13.6% after alkaline treatment in 1 M NaOH at 80 °C for 2000 h. A maximum power density of 324 mW cm−2 at current density of 750 mA cm−2 was recorded in fuel cell tests. Chu et al. fixed N-methyl-N-alkyl-piperidinium moieties to a poly(phenylene oxide) backbone via copper catalysed azide alkyn dipolar cycloaddition.91 Anion exchange membranes prepared by this route exhibited superior alkaline stability over membranes with piperidinium units attached via methylene bridges to the polymer backbone (Fig. 5). A conductivity loss of 2% was noted after 560 h at 80 °C in 1 N NaOH. Furthermore, these membranes showed promising performance in fuel cells (H2/O2) operated at 60 °C (max. power density 116 mW at ca. 225 mA cm2; catalyst loading on each electrode was 0.5 mg cm−2 Pt) and water electrolysis (pure water) at 50 °C (300 mA cm−2 at 1.8 V) using IrO2 as anode catalyst and Pt/C as cathode catalyst (loading 1.5 mg cm−2 each).
Several attempts have been reported using KOH-doped polybenzimidazole membranes in alkaline water electrolysis.92–95 Diaz et al. prepared blend membranes composed of polyvinylalcohol (PVA) and two different polybenzimidazoles, namely PBI (prepared by condensation of isophthalic acid and 3,3′,4,4′-tetraaminobipheny) and ABPBI (prepared by polycondensation of 3,4-diaminobenzoic acid).92 The PVA content was varied between ca. 10 and ca. 33 mass%. The stability of these membranes was improved by crosslinking the PVA with glutaraldehyde (c-PBI, c-ABPBI). Best results in terms of through-plane conductivity at 90 °C were obtained with membranes with a PBI or ABPBI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PVA ratio of 4:1 after doping with 15 mass% KOH (PBI and ABPBI: 75 mS cm−1; c-PBI: <1 mS cm−1, c-ABPBI: 55 mS cm−1) or 30 mass% KOH (c-ABPBI: 90 mS cm−1). Short-term electrolysis using a c-ABPBI with 20 mass% PVA membrane doped with 15 mass% KOH showed good performance at 70 °C (360 mA cm−2) using Ni foam electrodes and 15 mass% KOH solution as feed. In another publication Diaz et al. prepared anion exchange membranes using ABPBI and 3-phenyl-6-methyl-3,4-dihydro-2H-1,3-benzoxazin as crosslinker.93 The membranes were doped by soaking in KOH solutions with a concentration ranging from 1.9 to 4.2 mol L−1. While non-crosslinked membranes were brittle after doping with 4.2 mol L−1 KOH, the crosslinked samples remained stable. The crosslinked membrane, doped with 4.2 mol L−1 KOH showed an ion-conductivity of 25 mS cm−1 at room temperature. A current density of 335 mA cm−2 was attained with a crosslinked membrane doped with 3 mol L−1 KOH at 70 °C and an applied constant voltage of 2 V. These tests were run in a zero-gap configuration using Ni foam electrodes and 3 mol L−1 KOH solution as feed. Marinkas et al. prepared anion exchange membranes by reacting brominated poly(phenylene oxide) (PPO) with 2-mesitylbenzimidazole.94 Only materials with an IEC of 1.9 meq g−1 yielded flexible and self-supporting membranes. An initial conductivity of 8 mS cm−1 at room temperature was detected. However, these membranes were only stable in 0.5 M KOH at 80 °C. In 1 M KOH a 70% loss in conductivity after 14 days and 30–40% mass loss after 21 days were observed. Water electrolysis experiments were carried out with 3 mg cm−2 IrO2 on Ti paper and 1.5 mg cm−2 Pt/C on carbon paper as cathode and anode catalysts, respectively. In both cases, PTFE was used as binder material. Furthermore, the anode was fed with 0.5 M KOH, while the cathode was kept dry. Current densities of 85 mA cm−2 at 1.55 V and 318 mA cm−2 at 1.8 V were achieved at 50 °C. By comparison, a commercial anion exchange membrane (fumatech FAA-3-PK-75) operated under the same conditions produced 86 mA cm−2 at 1.55 V and 524 mA cm−2 at 1.8 V. Differences were explained by the lower water diffusion through the PPO membrane. Blend membranes from mPBI and fumtatech FAA-3 with a mPBI content ranging from 67 to 100 mass% (PF21, PF31, PF41, PF51, PBI), where the number denotes the PBI:FAA-3 ratio, were reported.95 Doping was carried out by immersion in 10 to 30 mass% KOH solutions. The PF41 membrane turned out to be most stable regarding mechanical properties. However, the highest ion conductivity of 166 mS cm−1 (RT) was observed for the PF51 membrane doped with 25 mass% KOH solution (measurements were carried out in doping solution). Electrolysis experiments were run for example with the PF41 membranes at 60 °C employing different KOH concentrations (10, 15 and 20 mass%) in the supporting electrolyte solution. Ni foam was used as electrode material and catalyst. The polarization performance increased with increasing KOH concentration from ∼75 mA cm−2 at 2 V (10 mass% KOH) to ∼175 mA cm−2 at 2 V (20 mass% KOH). After cell operation at 200 mA cm−2 for 4 days, a slight increase in current density at 2 V to ∼200 mA cm−2 was detected. Running the electrolysis test with an open cathode resulted initially in a high current density of more than 470 mA cm−2 at 1.8 V. However, the current density dropped to 230 mA cm−2 due to leakage of anode electrolyte solution.
:PVA ratio of 4:1 after doping with 15 mass% KOH (PBI and ABPBI: 75 mS cm−1; c-PBI: <1 mS cm−1, c-ABPBI: 55 mS cm−1) or 30 mass% KOH (c-ABPBI: 90 mS cm−1). Short-term electrolysis using a c-ABPBI with 20 mass% PVA membrane doped with 15 mass% KOH showed good performance at 70 °C (360 mA cm−2) using Ni foam electrodes and 15 mass% KOH solution as feed. In another publication Diaz et al. prepared anion exchange membranes using ABPBI and 3-phenyl-6-methyl-3,4-dihydro-2H-1,3-benzoxazin as crosslinker.93 The membranes were doped by soaking in KOH solutions with a concentration ranging from 1.9 to 4.2 mol L−1. While non-crosslinked membranes were brittle after doping with 4.2 mol L−1 KOH, the crosslinked samples remained stable. The crosslinked membrane, doped with 4.2 mol L−1 KOH showed an ion-conductivity of 25 mS cm−1 at room temperature. A current density of 335 mA cm−2 was attained with a crosslinked membrane doped with 3 mol L−1 KOH at 70 °C and an applied constant voltage of 2 V. These tests were run in a zero-gap configuration using Ni foam electrodes and 3 mol L−1 KOH solution as feed. Marinkas et al. prepared anion exchange membranes by reacting brominated poly(phenylene oxide) (PPO) with 2-mesitylbenzimidazole.94 Only materials with an IEC of 1.9 meq g−1 yielded flexible and self-supporting membranes. An initial conductivity of 8 mS cm−1 at room temperature was detected. However, these membranes were only stable in 0.5 M KOH at 80 °C. In 1 M KOH a 70% loss in conductivity after 14 days and 30–40% mass loss after 21 days were observed. Water electrolysis experiments were carried out with 3 mg cm−2 IrO2 on Ti paper and 1.5 mg cm−2 Pt/C on carbon paper as cathode and anode catalysts, respectively. In both cases, PTFE was used as binder material. Furthermore, the anode was fed with 0.5 M KOH, while the cathode was kept dry. Current densities of 85 mA cm−2 at 1.55 V and 318 mA cm−2 at 1.8 V were achieved at 50 °C. By comparison, a commercial anion exchange membrane (fumatech FAA-3-PK-75) operated under the same conditions produced 86 mA cm−2 at 1.55 V and 524 mA cm−2 at 1.8 V. Differences were explained by the lower water diffusion through the PPO membrane. Blend membranes from mPBI and fumtatech FAA-3 with a mPBI content ranging from 67 to 100 mass% (PF21, PF31, PF41, PF51, PBI), where the number denotes the PBI:FAA-3 ratio, were reported.95 Doping was carried out by immersion in 10 to 30 mass% KOH solutions. The PF41 membrane turned out to be most stable regarding mechanical properties. However, the highest ion conductivity of 166 mS cm−1 (RT) was observed for the PF51 membrane doped with 25 mass% KOH solution (measurements were carried out in doping solution). Electrolysis experiments were run for example with the PF41 membranes at 60 °C employing different KOH concentrations (10, 15 and 20 mass%) in the supporting electrolyte solution. Ni foam was used as electrode material and catalyst. The polarization performance increased with increasing KOH concentration from ∼75 mA cm−2 at 2 V (10 mass% KOH) to ∼175 mA cm−2 at 2 V (20 mass% KOH). After cell operation at 200 mA cm−2 for 4 days, a slight increase in current density at 2 V to ∼200 mA cm−2 was detected. Running the electrolysis test with an open cathode resulted initially in a high current density of more than 470 mA cm−2 at 1.8 V. However, the current density dropped to 230 mA cm−2 due to leakage of anode electrolyte solution.
A completely different approach to prepare anion exchange membranes was described by Hnát et al.96 Here, a commercially available anion exchange resin (Dowex Marathon A; particle size 10–30 μm; IEC 3.9 meq g−1) was incorporated into a LDPE matrix by melt mixing. Membranes with a thickness of 300 μm were obtained by press-molding at 140 °C. These membranes were used to study the impact of liquid electrolyte solution composition on the performance in electrolysers. Trimethyl ammonium functionalized PPO was used as catalyst binder. The catalysts themselves were NiCo2O4 (2.5 mg cm−2) for anode and Pt/C (0.3 mg cm−2) for the cathode, each supported on a Ni foam electrode. For long-term electrolysis tests the anode catalyst loading was increased to 8 mg cm−2 of NiCo2O4. As expected, membranes in the OH−-form showed much higher ion conductivities at 70 °C (67 mS cm−1) than membranes in the CO32−-form (24 mS cm−1) or HCO3−-form (18 mS cm−1). These differences in conductivity are also reflected in the load curves of alkaline water electrolysis, recorded at 70 °C. At an applied voltage of 1.75 V (85% efficiency) a current density of 266 mA cm−2 was achieved for the membrane in the OH−-form (1 M KOH), while that of the membranes in the CO32−-form and HCO3−-form was 25 mA cm−2 and 36 mA cm−2, respectively. Long-term tests (100 h) with membranes in OH−-form (electrolyte 1 M KOH) and CO22−-form (electrolyte 0.5 M Na2CO3) were conducted at 70 °C and a current density of 300 mA cm−2. A third experiment was carried out at 50 °C and 300 mA cm−2 using a 1.95 M KOH. The initial voltages were 1.7 V (KOH, 70 °C), 1.8 V (KOH, 50 °C) and 1.97 V (Na2CO3, 70 °C). During the experiments at 70 °C, an increase of cell voltage of 0.4 mV h−1 (after an initial period of 40 h) and 0.6 mV h−1 was observed using KOH and Na2CO3, respectively. It should be further noted that in the case of KOH as electrolyte at 70 °C, the slope of voltage increase could be divided into two regions. The voltage increase at 50 °C was only 0.2 mV h−1. Membranes used at 70 °C in the electrolysis experiment showed both a decrease in IEC from 2.45 meq g−1 to 2.31 meq cm−1 (KOH) and 2.29 meq g−1 (Na2CO3), meaning that both membranes degraded to a certain extend at the applied temperature over 100 h. In contrast, the IEC barely changed for the samples under operation at 50 °C.
In another publication Hnát et al. described the preparation of anion exchange materials by conversion of chloromethylated polystyrene-block-poly(ethylene-ran-butylene)-block-polystyrene (SEBS) with 1,4-diazabicyclo[2.2.2]octane.97 This material was used as membrane and catalyst binder. Stability tests conducted at 30, 50 and 60 °C in 10 mass% KOH for one week showed no change in conductivity and IEC at 30 °C and a slight decrease by ca. 10% at 50 °C. Further increase of temperature to 60 °C resulted in a loss of IEC from 0.76 meq cm−1 to 0.4 meq cm−1 and the conductivity dropped from 75 mS cm−1 to 40 mS cm−1. Catalyst coated electrodes were prepared by spraying a catalyst ink, containing the binder in the chloromethylated form and the catalyst (NiCo2O4 (anode); NiFe2O4 (cathode)), onto the nickel foam electrode. After drying, the binder was converted into the anion exchange form by immersing the electrode in ethanolic DABCO solution. The binder and catalyst loadings were 1.11 mg cm−2 and 10 mg cm−2, respectively. Load curves of alkaline water electrolysis were recorded with KOH concentrations ranging from 1 to 15 mass% at 40 °C. The current density at 2 V increased from 70 mA cm−2 (1 wt% KOH) to 150 mA cm−2 (15 mass% KOH). Long-term electrolysis tests was performed with bare Ni foam electrodes at 50 °C under galvanostatic conditions at 300 mA cm−2 using a 10 mass% KOH solution as electrolyte. The dynamic nature of the electrolysis process resulted in a slightly fluctuating voltage around 2.27 V over a period of 150 h. An increase in voltage by 0.02 mV h−1 was observed during this test, mainly caused by degradation of the membrane. The IEC dropped from initially 0.76 meq cm−1 to 0.72 meq cm−1, which is comparable to results (0.72 meq cm−1 after 168 h) obtained from ex situ test performed under similar conditions. Žitka et al. quaternized SEBS with trimethylamine.98 This material with an IEC of 0.75 meq g−1 was used as membrane binder for the catalyst. SAXS measurements indicated a clear phase separation and a lamellar morphology with long periods in the range of 32–35 nm. Since the ion exchange groups are located in the polystyrene microdomains, a very high IEC of 2.7 meq cm−1 inside these domains and therefore in the ion conducting pathways were estimated from SAXS measurements and degree of functionalization. This high local IEC gives rise to high ion conductivities, which are in the range from 56 mS cm−1 to 79 mS cm−1 for 30 to 70 °C. MEAs were prepared with 8 mg cm−2 NiCo2O4 (anode) and different amounts of binder material ranging from 0.42 mg cm−2 to 2.67 mg cm−2. These were compared with MEAs prepared with a PTFE binder (2.67 mg cm−2) and bare Ni foam. The cathode catalyst consisted always of 0.3 mg cm−2 Pt and 0.05 mg cm−2 PTFE as binder. Although not perfect, highest current density (280 mA cm−1) at 70 °C and 1.74 V were obtained with 0.42 mg cm−2 quaternized SEBS as binder and 15 mass% KOH solution as electrolyte. Even with 1 mass% KOH electrolyte solution, membranes in combination with optimized MEA composition delivered comparable performance to industrial water electrolysers (120–320 mA cm−2 at 1.8–2 V). In a long-term electrolyser test at 50 °C and 10 mass% KOH, the investigated MEAs showed stable performance (300 mA cm−2; 1.78 V) over 800 h.
In summary, the search for stable anion exchange polymeric membranes for water electrolysis is dominated by the screening of the cationic ion selective groups. Enhanced stability has been shown for certain materials. The combination with catalysts materials to form MEAs for testing in cells is the subject of the next section of this review paper.
5. Membrane electrode assembly preparation approaches
Traditionally, in alkaline water electrolysis large scale electrodes are based typically on Ni at the anode side95,99–104 and on Ni or stainless steel on the cathode side of the cell.105 Various strategies have been employed to improve the efficiency of the electrolysis cell. Such as by increasing the electrode surface area, typically by using RANEY® Ni, or by applying suitable nanostructured electrocatalysts. Two principal approaches have been developed to prepare MEAs:1. Catalyst-coated substrate (CCS).
2. Catalyst-coated membrane (CCM).
Both techniques will be discussed here, although more attention will be paid to the CCM approach as it offers several advantages over the CCS one. Once all MEA components have been prepared, the MEA is typically assembled by pressing them together directly in the cell hardware. The hot-press approach commonly used with PEM systems is not a suitable option because the metal-based electrode substrates (e.g. metal foams) are used, which under such conditions damage the membrane.
Other critical components of the AEM cell like current feeders or bipolar plates are also important in determining overall system cost. The alkaline environment offers the advantage of the broader variety of less expensive materials for these components compared to PEM systems. In PEM water electrolysis, Ti or platinized Ti is the common choice.106 In alkaline water electrolysis cheaper materials like stainless steel,107,108 nickel109,110 or graphite111,112 have been employed. Despite this potential advantage many studies of AEM systems still utilize Ti materials even if working in alkaline media.113–115
5.1 Catalyst-coated substrate
The CCS approach benefits from robustness and stability of the catalyst layer.116 CCS is based on the deposition of the catalyst layer onto the surface of an appropriate substrate. The role of the substrate is to enable electron transfer, support the catalyst layer mechanically and to allow the efficient removal of gaseous products. Several different materials can be used as the substrate. For the anode side Ti paper,113,117–119 platinized Ti plates,120 stainless steel felt,91 Ni foam54,55,108,109,115,121,122 or even carbon cloth123 or carbon paper22,111,112,124 are reported as substrate materials. Ni is the standard material for alkaline water electrolysis while Ti is generally used for PEM water electrolysis. Nevertheless, both of these materials show high thermodynamic stability as anode support in alkaline water electrolysis. In alkaline environment, stainless steel generally passivates at anodic potentials, which ensures stability. At the same time, however, the passive layer reduces the electrical conductivity of the interface between the electrode and electrolyte. The long-term use of carbon materials as anode substrate can be ruled out due to instability under OER conditions. From a thermodynamic point of view, the stability of carbon under alkaline conditions is up to 18-folds lower than in the acidic environment,125 (see Pourbaix diagram) due the fact that HO− ions are excellent nucleophiles, which accelerate carbon degradation.57 Indeed, carbon is used as fuel at the anode for molten NaOH carbon air batteries.57On the cathode side carbon can be readily used,54,55,111,113,119,120,123,124,126 as well as Ni95,101,102,108,115,121,127,128 and Ti.117,118 The electrode preparation method is predominantly based on spraying a catalyst ink over the activated support surface.22,54,55,119,120,122,129 Other frequently used techniques include electrodeposition,109,111,112,121 magnetron sputtering,124 chemical electroless plating109,115 and screen printing.54,55,123 Plasma sprayed electrodes containing non PGMs (NiAl anode and NiAlMo cathode) have been prepared on stainless steel gradient porous metal frameworks.130 Combined with a HTM-PMBI membrane in an AEM electrolyzer fed with 1 M KOH this CCS approach produced 2 A cm−2 at 2.1 V (60 °C).
5.2 Catalyst-coated membrane
The CCM approach is based on depositing the catalyst directly onto the membrane surface. Hence, the main advantage is the resulting intimate contact of the catalyst with the polymer electrolyte membrane and thus improved ionic conductivity. This also enables a decrease in the catalyst loading131 while maintaining performance of the MEA. On the other hand, the electrical contact between the current collectors is worse. However, as the electron conductivity of the system is significantly higher than the ionic conductivity of the electrolyte, this seems to be a reasonable trade off. The main obstacle to a wide spread application of the CCM approach lies in the absence of suitable polymeric binder. Only recent advances in alkaline polymer electrolyte research has resulted in increasing interest in this approach.The most commonly applied method of CCM-MEA preparation is spray coating of a catalyst ink onto the surface of the polymer anion-selective membrane.131–136 Recently, Ito et al. compared the performance of CCM-MEAs prepared by spraying and doctor blade method. Better results were achieved using the spraying technique.116 The reason is lower resistivity of the cell prepared by this technique. Spraying also allowed easier and more precise control of the catalyst and binder loading.116 The decal method, which is commonly used in PEM water electrolysis or fuel cell technologies137 is not suitable for anion-selective polymer membranes. It is because of the hot-pressing step it includes. Anion-selective polymer membranes suffer from chemical instability when exposed to elevated temperatures,135,138 which precludes catalyst transfer under high temperature conditions.
Amongst the first papers dealing with the issue of the CCM-MEA for alkaline systems are reports by Wu and Scott.57,107,133,134,139 These works do not directly focus on the issue of the CCM-MEA preparation and characterisation. They are aimed more at the catalyst or membrane/polymer binder itself using the CCM-MEA based MEAs as an experimental testing technique. Using the radiation grafted anion exchange membrane the best achieved performance reached 980 mA cm−2 in 1 mol dm−3 KOH at 25 °C and cell voltage of 1.8 V.56 However, this MEA showed a degradation rate of 22.3 mV hour−1 (during 11 hours of the chronoamperometry experiment at 300 mA cm−2, at 30 °C, in deionized water).134 Three reasons were addressed by the authors to be responsible for the degradation: (i) drying of the membrane due to bubble evolution, (ii) corrosion of the anode components at cell voltages above 2 V and (iii) degradation of the ionomer in the membrane or binder.134
Simultaneously, Leng et al. published in 2012 work using commercial materials (Tokuyama A201 membrane and Tokuyama AS-4 polymer binder).132 The CCM-MEA prepared showed, however, only limited stability under the conditions of alkaline water electrolysis. After 27 h of operation at 200 mA cm−2 in deionized water (50 °C) feed into the cathode compartment only, the cell voltage together with resistivity of the cell increased sharply.132 The authors however observed recovery of the cell voltage to the initial value when the 1 mol dm−3 KOH was supplied to the anode chamber. Based on this, the authors concluded that degradation of CCM-MEA was mainly due to the degradation of the ionomer and/or membrane–electrode interface.132
Limited stability of the Tokuyama AS-4 polymer binder was recently observed also by Ito et al.116 who, due to the limited stability of the AS-4 polymer binder, prepared mixed MEAs utilizing the CCM approach for the cathode side and the CCS approach for the anode side. The CCS approach allows to use inert poly(tetrafluoroethylene) (PTFE) as binder of the catalyst layer. Ito et al. focused primarily on CCM-based MEAs utilizing PGM-free based catalysts.116 Subsequently, if the PGM-free cathode catalyst (CeO2–La2O3) was replaced by Pt (1.7 mg Pt cm−2) the performance of the AWE increased from 40 mA cm−2 to 300 mA cm−2 at 1.8 V cell voltage.114,116
Direct comparison of the CCS and CCM approaches was provided by Park et al.135 Better performance was in this case achieved using CCM approach (500 mA cm−2 at 1.8 V) when compare to CCS approach (210 mA cm−2 under identical conditions). The explanation given by the authors was due to negative effects of the CCS preparation method on the structure of the catalyst layer increasing mass transport losses demonstrated by electrochemical impedance spectroscopy analysis of the system.135 On the other hand, Gupta et al.59 observed the opposite result when the CCM achieved 200 mA cm−2 at 1.8 V when compared to 390 mA cm−2 for CCS approach under the same conditions.59 However, the results not adequately discussed by the authors and the reasons for this observation were not provided.
Comparison of CCS and CCM methods for MEA preparation is not straightforward. It is mainly due to the absence of standard testing protocols. Fig. 6 summarises the data obtained from the literature. Box plot showing the 25th and 75th percentile, median, average, error bars (showing 10th and 90th percentile) values and outlying points was chosen to present the data gathered. Obviously, higher performance of AEM water electrolysers are generally achieved, when PGM catalysts113,117,118,135 and elevated temperatures109,113,122 are used. Of course, there can be some exceptions e.g. work of Wu et al.139 who achieved 980 mA cm−2 at 1.8 V at 25 °C using Pt as catalyst on cathode and a PGM-free catalyst on anode side of the cell, Liu et al.126 who achieved 500 mA cm−2 (1.8 V) at 60 °C using PGM-free catalysts or Pavel et al.122 who achieved 485 mA cm−2 (1.8 V) with no-PGM catalysts at 43 °C. Further, it can be stated that none of the values above 90th percentile was measured with pure water as liquid electrolyte. The main conclusion resulting from Fig. 7 is that both CCM and CCS methods of the MEA construction lead in general to similar results.
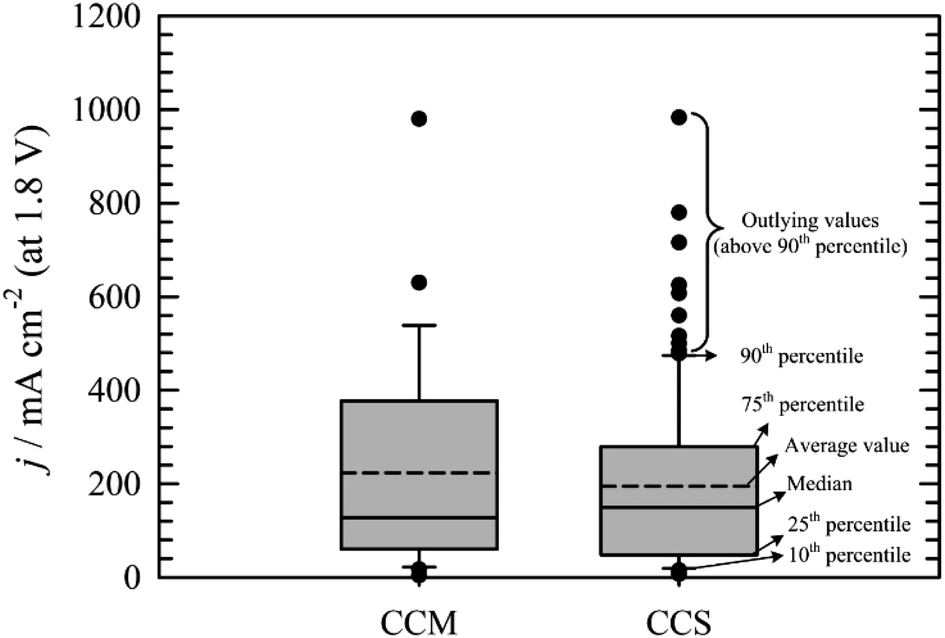 | ||
| Fig. 6 Comparison of the current densities achieved at cell voltage 1.8 V using the CCM57,59,106,107,114,116,131–136,139–141 or CCS22,54,55,95,101–104,108,109,111–113,115,117–124,126–129,142,143 method of MEA construction. | ||
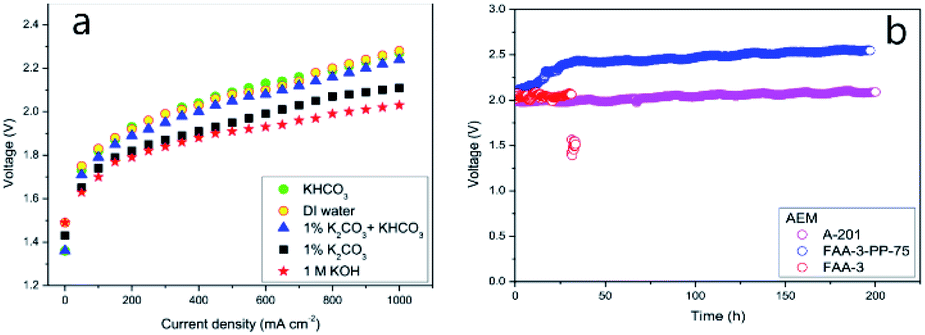 | ||
| Fig. 7 (a) Polarization curves recorded for different electrolytes and (b) stability of various MEAs: A201, FAA-3, and FAA-3-PP-75.55 Reprinted from ref. 55 with permission from Elsevier. | ||
6. Influence of the liquid electrolyte on cell performance
Traditionally in alkaline electrolysis a concentrated aqueous KOH solution of up to 30 mass% is circulated through the cell, thus guaranteeing sufficient ionic conductivity to the electrolyte. High cell temperatures are also used which increases electrode kinetics and facilitate separation of the gasses produced from the electrolyte solution. The negative consequences of using such a corrosive electrolyte regard safety issues and poor flexibility. As described above, in order to compete with PEM electrolysis in terms of cost and flexibility, simple demineralised water must be used in AEM. Hence, we will concentrate our discussion on systems that use only pure water or dilute KOH or sodium carbonate/bicarbonate solutions.6.1 Hydroxide solutions
The electrolyte that has been most commonly used in AEM electrolysers are hydroxide solutions (KOH and NaOH). A wide range of concentrations from 0.06 mass% (ref. 114) up to 30 mass% (ref. 127) is reported. This is mainly due to the variation of the types of the materials used as separators. Concentrations above 20 mass% are used in AE together with ion solvating membranes95,103,104,127 or diaphragm separators.104,109,128 With anion exchange polymer membranes, concentrations can range from 3 to 10 mass%.99–101,113,123,126,144 Generally, electrolysis performance improves with increasing concentration of the hydroxide solution.121,139,145,146 This improvement is explained by the decrease in the polarization resistance of the both electrodes and cell resistance at the same time.146 While the polarisation resistance decreases linearly with increasing electrolyte concentration,146 the cell resistance decreases at higher concentrations of the electrolyte less progressively than at the lower ones.136,146 At low concentrations, the cell resistance can be affected by CO2 contamination of the KOH solution. (Bi)carbonate ions contaminate the anion selective polymer electrolytes and decrease the ionic conductivity. As the concentration of the KOH solution becomes high enough, a buffer effect decreases the influence of the (bi)carbonates on the conductivity of the membrane and thus the cell resistance remains small.Only a few studies refer to the utilization of the NaOH as liquid electrolyte.59,141 NaOH is cheaper than KOH while KOH solutions show much higher values of conductivity147 when compare to NaOH solutions. More importantly the solubility of K2CO3 is significantly higher than Na2CO3 (ref. 148 and 149) which mitigates the problem precipitate formation and separator scaling. Moreover, KOH solutions are characterised by lower viscosity when compared to NaOH.150 Thus, KOH provides a more reliable and flexible electrolysis cell operation despite its higher cost.
6.2 Bicarbonate/carbonate
Dilute carbonate or bicarbonate solutions have a mildly alkaline pH (10–12) that retains sufficient ionic conductivity. Additionally, such conditions should provide enhanced stability of cell materials as well as the polymeric AEM/binders.114,122,143 The ionic conductivity of AEMs is significantly lower in the carbonate or bicarbonate (CO32−/HCO32−) form.151 The electrode reaction kinetics are also lower in carbonate or bicarbonate environment.151 It has been shown, however, that under high current load the OH− produced at the cathode is transported through the anion selective membrane and exchanges the OH−/CO32− ratio in its bulk. This ratio becomes current dependent.114,152 Nevertheless, CO32− ions acting as counter ions are present in certain amount in the membrane even under high current conditions.114 The membrane resistance thus tends to be always higher than it is in the case of hydroxide liquid solutions. Despite the fact that in the case of the liquid electrolytes utilizing the carbonates or bicarbonates the concentration of OH− ions is lower when compared to hydroxide solutions (pH is lower), the pH value in the bulk of the membrane can generally differ from those of the surrounding solution.153 Increased number of OH− under higher current load can thus still cause membrane degradation.151 Most studies use potassium salts, i.e. K2CO3 or KHCO3 solutions.54,55,106,114,116,122,143 These show commonly better performance than with Na2CO3 or NaHCO3.141,151 Regardless of the counter-cation (Na+ or K+), when the CO32− and HCO3− ions are compared, better results are obtained for CO32− solutions.55,122,141,1516.3 Water
The use of demineralised water is ultimately the most desired solution to make AEM electrolysis a competitive technology. Using pure water as feed, however, brings important new challenges. Ni represents, without any doubts, the standard material for electrode construction in AEM electrolysis. However, under anodic polarisation, it is stable only at pHs >9, ruling out the possibility of using a pure water feed.154 Ti-based porous materials115,118,120,132 and/or current collectors55,141 are suitable but more expensive.114 Another problem with using deionized water is the influence of CO2 on performance. Parrondo et al.22 observed a drop in current density at a cell voltage 1.8 V from 365 mA cm−2 to 135 mA cm−2 in just 30 min. This loss was associated with an increase in both high frequency resistance and charge transfer resistance. The authors explained that this phenomenon was due to CO2 contamination leading to dissolved carbonate and bicarbonate anions that decrease the ionic conductivity of the membrane and catalytic layer binders.22 The nature of the AEM ionomer/binder is an important aspect of water fed AEM electrolysis. This material must ensure the mechanical stability of the catalyst layer and at the same time provide ionic conductivity within the catalyst layer. The importance of the AEM binder has been highlighted in the literature. Current density (<15 mA cm−2 at 1.8 V) with no,115,124,142 or inert binder (e.g. PTFE).114,141 Using PTFE as a binder, Cho et al.118 were able to achieve a current density (60 mA cm−2 at 1.8 V) comparable with some of the works utilizing anion selective polymer binders.107,133,136 However, in the case of the Cho et al., pure water was fed only to the cathode compartment while the anode compartment was fed with 0.5 mol dm−3 KOH thus ensuring the ionic conductivity within the catalyst layer from the anode side. When an anion exchange polymeric binder is used in the catalyst layer, current densities higher than 90 mA cm−2 have been obtained.22,55,57,108,129,134 Obviously, a significant ionic conductivity of the AEM ionomer is required. In some cases poor activity (17–36 mA cm−2) was obtained even using ionomers.135,140 For example Cao et al. observed ionic conductivity of their material to be 1.5 S m−1 at 25 °C,140 which is less when compare to papers by Wu et al. who used materials with ionic conductivity of 1.8–4.0 S m−1 at 20 °C.57,1346.4 Comparison of the liquid electrolytes
Performance data of AEM electrolyzers reported in the literature using different electrolytes is summarised in Fig. 9. It clearly shows that using pure water cannot compete with utilization of dissolved ionic electrolytes. The average current density at a cell voltage of 1.8 V with a pure water feed was found to be 95 mA cm−2, 160 mA cm−2 for (bi)carbonates solutions and 220 mA cm−2 for KOH solutions (Fig. 9). The best performance reported for water and (bi)carbonate solutions are 450 (ref. 57) and 440 (ref. 122) mA cm−2 respectively. The best performance of 450 mA cm−2 for circulating water was achieved using CCM based MEA.57 The high outlying values are generally achieved using PGM catalysts120 and higher operating temperature,108 However, in one case at temperature of 70 °C the current density of 250 mA cm−2 was reached at 1.8 V utilizing PGM-free catalysts.108 Vincent and co-workers using PGM-free catalysts and commercial membrane/ionomer compared liquid electrolytes.54,55 With DI water, 1% KHCO3, and 1% (K2CO3 + KHCO3) performance was similar (Fig. 7). The voltages at 400 mA cm−2 were 2.04, 2.03, and 2.0 V, respectively. However, the performances achieved with electrolytes 1% K2CO3 and 1 M KOH were better than those achieved when using DI water, 1% KHCO3, and 1% (K2CO3 + KHCO3).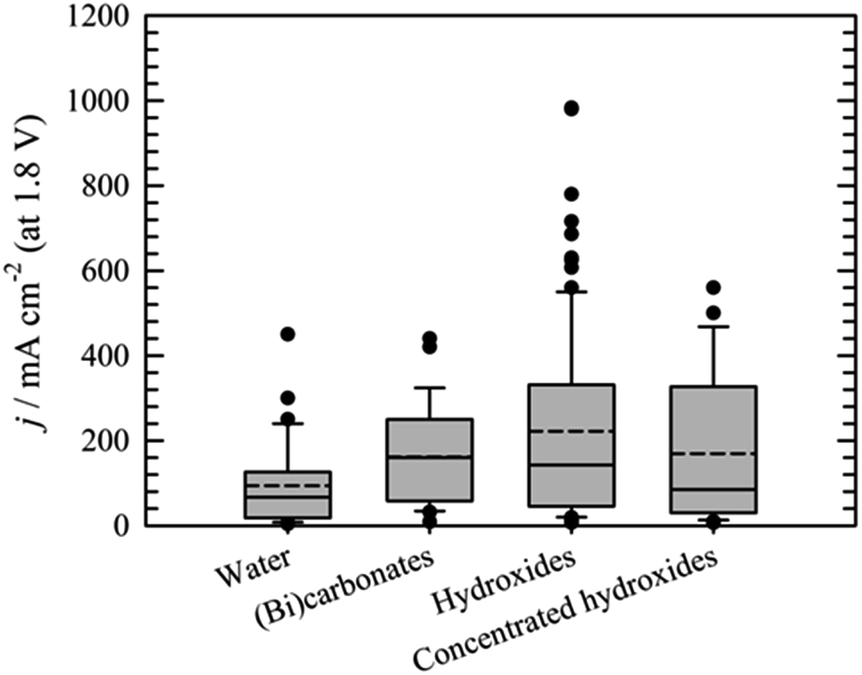 | ||
| Fig. 8 Comparison of the current densities achieved at cell voltage 1.8 V using water,22,55,57,107,108,114,115,118,120,124,129,132–136,139–142 (bi)carbonates54,55,96,106,114,116,122,141,143,151 or hydroxides55,59,93,95,101–104,109,111–114,117–119,121,123,124,126–128,132,135,136,139,141,145,146,155–159 as liquid electrolyte. Bar for “concentrated hydroxides represents KOH solutions >1 mol dm−3. | ||
In the case of the (bi)carbonates the best performance was achieved using PGM-free catalyst at 55 °C.122 Generally, in the case of (bi)carbonate solutions PGM-free catalysts are widely used.54,55,116,122,141,143 The important parameters found to influence the performance are catalyst load,122 temperature54,55,122 and concentration of the solution used.114
In the case of the hydroxide solution, situation is slightly different. The reason consists in the fact that hydroxide solutions represent standard test solution and many papers are thus using hydroxide solutions for extreme experiments. Outlying values above percentile 90th are thus e.g. due to high KOH concentration,109,121 high operating temperature,109,113,121 or high operating pressure.109 Under milder conditions the best performance is commonly achieved utilizing PGMs.117–119,132,135
However, as it is possible to see from the last bar in Fig. 8 showing the current densities achieved at a cell voltage of 1.8 V using concentrated KOH solutions (concentrations >1 mol dm−3), the high concentration of the liquid electrolyte by itself cannot guarantee high current density. The reason in this particular case lies probably in the type of the separator used. Highly concentrated KOH solutions are typically used when diaphragms102,160 or ion solvating membranes95,103,104,127 are utilized as the separator of the electrode compartments. In these cases, however, the current densities did not exceed 120 mA cm−2.
6.5 Arrangement of water or electrolyte circulation in the cell
In AEM electrolysis cells the liquid electrolyte may be circulated through (i) both anode and cathode compartment;59,126,131,134,161 (ii) anode compartment only54,55,113,116,117,122,143 and (iii) cathode compartment only.111,112 A special case involves feeding of both compartments with streams of different composition. Cathode fed with the water and anode with hydroxide or (bi)carbonate solution represents the typical case here.118,119,132In traditional industrial cells the liquid electrolyte flows through both electrode compartments. This is primarily given by utilizing porous diaphragm based separator. In such a case feeding of just one electrode compartment leads to significant electrolyte cross-over. If a dense polymer anion selective membrane is used as a separator, the situation differs. Liquid electrolyte can be fed only to one electrode compartment without significant cross-over. This can result in simplification of the liquid electrolyte circulation and even in simplification of the gas separation and processing due to the possibility to achieve higher purity of the produced gasses.110 In the literature, electrolyte flowing through both compartments represents the state-of-the-art.101,103,120,121,126 Leng et al.132 tested different feeding methods in 2012, observing that the best cell stability was achieved with the cathode compartment filled with water and water circulated through the anode side (stable cell voltage for more than 500 h). When water was circulated just through one of the electrode compartments, sharp increase of the cell resistance was observed after 100 h or 250 h of operation for water circulation only through the cathode or anode side respectively. Another direct comparison was recently provided by Park et al.135 Best performance was achieved for the case of the 1 mol dm−3 KOH solution circulating through both electrode compartments.
Cathode reaction is connected with H2 molecule formation under consumption of two H2O molecules and release of the two OH− ions. If liquid electrolyte is fed to the cathode compartment exclusively, OH− ions are transported to the anode compartment across the membrane, accompanied by the solvating shell formed by the water molecules. O2 evolution at the anode results in producing additional H2O molecules. Thus, environment pH in the anode compartment will be close to neutral and thus, it will not satisfy the condition of pH higher than 9 to ensure stability of the Ni-based anode.110 Therefore, this option does not ensure the corrosion stability of the cell.
Alternatively, the liquid electrolyte can be fed to the anode compartment only. In this case, H2, which is generally considered as the main product, can be obtained with higher purity.110 Such configuration of the cell also allows easier utilisation of the cell as an electrochemical compressor.143 Nature of the electrode reactions taking place ensures stable pH at both electrodes. Water supply to the cathode, where it is consumed, is ensured through the hydrophilic membrane. Under high current loads, this can be theoretically connected with partial drying out of the membrane. It, in turn, can cause membrane degradation.162 As it was shown, however, performance of the APEWE with liquid electrolyte fed only to anode compartment showed current density up to 485 mA cm−2 at cell voltage 1.8 V and stability over 1000 h.122 Up to 2 A cm−2 has also been achieved with no water feed to the cathode using Tokuyama A-201 membrane and 2 M NaOH fed to the anode compartment.163 So, the degradation of the polymer electrolyte membrane seems not to be a critical issue under such conditions.
Literature reveals that circulation of the liquid electrolyte through the both electrode compartments is beneficial from the APEWE performance point of view. However, sufficiently high current densities of about 500 mA cm−2 at 1.8 V can be reached even with circulation only through anode side.117,122 Due to the advantages coming from circulation of the liquid electrolyte only through one electrode chamber as discussed earlier the circulation of the liquid electrolyte only through anode side represents the candidate for the future state-of-the-art arrangement.
6.6 Long-term stability tests
Comparison of the long-term performance of AEM water electrolyser cells (LTE) published by different authors is difficult. Once again, the reason lies in the absence of standard testing protocols. Some experiments last only for a few hours120,121,123,124 or tens103 of hours. The longest experiment was probably performed by Choe et al. (more than 2000 hours).164 However, significant increase of the cell voltage was observed during this experiment due to degradation of the polymer backbone. This is probably due to the type of the polymer backbone used based on poly(arylene ether), which is well known to undergo the chemical degradation (via aryl-ether bond cleavage) in alkaline environment.165–167 This confirms that the choice of suitable polymer backbone plays an important role. In this case, however, solution exists and number of polymers stable under alkaline water electrolysis conditions was identified, e.g. heteroatom-free all-carbon backbones (like polyolefins or polyphenylenes).168 Functional groups, however, are considered more susceptible towards chemical degradation and clear solution does not exists here yet. A constant current experiment showing improved stability (approaching 2000 h) was published by Liu et al. (Fig. 9).125 The Sustainion 37–50 membrane maintained 1 A cm−2 with PGM-free catalysts (60 °C and 1 M KOH) although some instability in cell voltage can be noted during the test.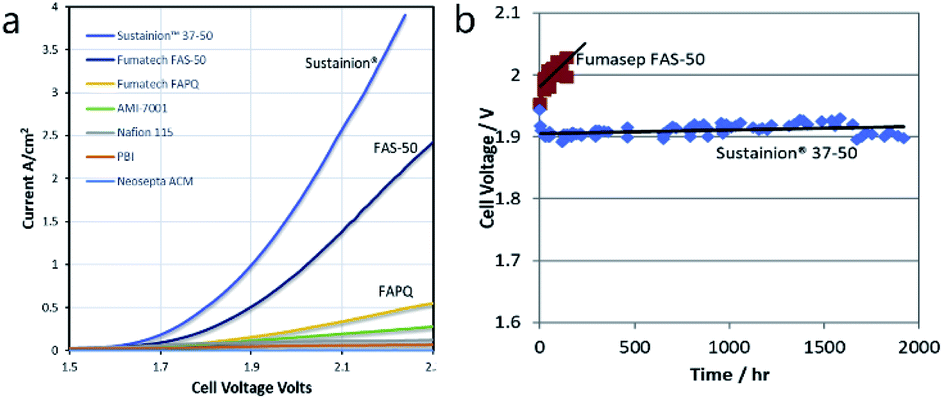 | ||
| Fig. 9 (a) Comparison of current–voltage response of AEM electrolysis with various membranes in ref. 119. (b) Steady state voltage performance at 1 A cm−2 with Sustainion 37–50 membrane and FAS-50 membrane (60 °C, 1 M KOH, NiFe2O4 anode and NiFeCo cathode). Reprinted from ref. 125 with permission from Elsevier.125 | ||
Another interesting feature is, that the V–I load curves, are usually obtained in the temperature range of 80 to 90 °C,55,113,142 while the LTE experiments are typically carried on at temperatures below 60 °C.122,126 It clearly indicates the poor stability of performance at higher temperatures. It is thus possible to conclude, that short-term performance figures have to be considered carefully, as they often are performed outside the long-term stability conditions window. Interestingly, all of the MEAs tested for more than 400 h were prepared by the CCS approach. The reason consists in the fact, that CCM-MEAs represent significantly more recent approaches to the MEA production and the number of data collected so far is still relatively small. Moreover, as already discussed, the main obstacle in studying this approach consists in the absence of sufficiently stable and conductive polymer electrolytes. In the case of the CCM approach one of the longest experiments lasted for approx. 100 h,136 which is still is significantly less when compared to the CCS approach. A positive aspect is, that the voltage of the CCM-based MEA in 1 mol dm−3 KOH at 50 °C and 400 mA cm−2 was in this case constant, without signs of degradation.136 Generally, the reasons of CCM-based MEA degradation mentioned in the literature are (i) delamination of the catalyst layer;132,134,136 (ii) membrane degradation;134,136 (iii) ionomer degradation;116,132,134 (iv) drying of the membrane due to gas phase evolution134 and (v) corrosion of anode components at cell voltage above 2 V.134 The first three reasons are clearly related to the insufficient stability of the anion selective polymer under conditions of alkaline water electrolysis as discussed above. It is also the reason for slow development in the field of CCM-MEA.
6.7 High pressure operation
An important feature of membrane electrolysis is that H2 inside the cell can be pressurized. In typical commercial PEM electrolyzers, H2 is pressurized to 15–30 bar. Pressurized operation of AEM electrolysers should be possible as AEMs have similar mechanical properties to PEMs. Ito and co-workers recently investigated the effect of high H2 pressure on electrolysis performance up to 8.5 bar.106 A Tokuyama A201 AEM was used with Pt/C and CuCoOx cathode and anode catalysts respectively. The cell showed pressurized operation (8.5 bar) at the cathode while the anode was operated at atmospheric pressure. Very low crossover of H2 to the anode was observed, 0.16 times that of PEM systems as well as low water content in the produced H2. This study confirms that high pressure operation of AEM electrolysis is possible and advantageous.7. Summary
Despite a flurry of recent research activity, AEM electrolysis technology remains at an early stage of development. In this review article we present a panoramic view of the current state of the technology. We have divided the critical components between (i) electrocatalysts for the HER and OER, (ii) membranes and ionomers and (iii) MEA preparation techniques. We have concentrated our review on low-cost materials, in particular, electrocatalysts based upon cheap and abundant metals (PGM-free). Together with these aspects, description of the MEA preparation techniques CCM and CCS are presented. Further, we have reviewed important cell parameters such as temperature, electrolyte composition and flow regime.7.1 Catalysts
Recent reports highlight the problem of the slow kinetics of PGM-free HER catalysts for AEM electrolysis. Low mass activity of Ni-based catalysts leads to high losses from thick catalyst layers in AEM test cells. Nevertheless, very promising candidates based on Ni–Mo alloyed materials have been identified. Electrochemical tests in half cells show HER behaviour similar to Pt benchmark catalysts. Regarding OER, high activity of transition metal mixed oxides has been shown in AEM electrolysis. The best performing materials are CuxCo3−xO4, NiCo2O4:Fe and Ni–Fe alloys used in AEM cells on Ni foam supports.7.2 Membranes and ionomers
The chemical stability of AEMs under alkaline conditions has improved markedly due to the development of stabilized functional groups on the polymer backbone. This allows the use of such membranes in AEM electrolysis at higher temperatures for long periods. Less emphasis has been given to the ionomer/binder material used to provide ionic conductivity within the catalyst layer, in part, because of the continued use of liquid electrolytes. Ultimately, a pure water feed will be necessary for AEM electrolysis and the ionomer/binder material will be very important.7.3 MEA preparation and cell performance
The physical and electrochemical characterization of the MEA prepared by either the CCS or the CCM method suggests that the CCM is preferable because improvements in ionic conductivity far outweigh any improvements in electronic conductivity. There are few studies of degradation mechanisms that link to MEA preparation methods. A review of AEM water electrolysis performance highlights the importance of liquid electrolyte used. Pure water feeds result in poor current densities while 1% K2CO3 or dilute KOH solutions give good results. A review of the literature results in the best performance with PGM-free catalysts (Ni–Fe, Ni–Mo, Ni/(CeO2–La2O3)/C and CuxCo3−xO4) and a commercial membrane (Tokuyama A201). The best electrolysis performance recorded was 500 mA cm−2 at 1.95 V at 60 °C with a 1% K2CO3 electrolyte by the Bessarabov group.54 The long-term durability of AEM water electrolyser cell performance has yet to be demonstrated and is a considerable challenge. The best data show performance approaching 2000 h. A clear understanding of the source(s) of degradation mechanisms (membrane, ionomer, catalyst or MEA) would help in developing more stable performance.8. Conclusions and recommendations
To date, the development of catalysts, membranes and ionomers for AEM electrolysis has been sporadic with little research on integration of the various components in MEAs and cell testing. As a result the best performance data shown in AEM cells has been obtained with commercially available materials. Hence, the development of these critical components of this technology requires a future road map for systematic development and commercialization of AEM systems and components. This should include the coordination of basic and applied research, technology development & integration, and testing at a laboratory scale of small demonstration units (AEM electrolyzer short stacks) that can be used to validate the technology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Ente Cassa di Risparmio di Firenze for funding (project EnergyLab) and the PRIN 2017 Project funded by the Italian Ministry MUIR Italy (No. 2017YH9MRK). The financial support of this research received from the Ministry of Industry and Trade of the Czech Republic under project No. FV10529 is gratefully acknowledged.Notes and references
- P. Nikolaidis and A. Poullikkas, Renewable Sustainable Energy Rev., 2017, 67, 597–611 CrossRef CAS.
- M. Momirlan and T. Veziroglu, Renewable Sustainable Energy Rev., 1999, 3, 219–231 CrossRef CAS.
- P. E. Dodds and S. Demoullin, Int. J. Hydrogen Energy, 2013, 38, 7189–7200 CrossRef CAS.
- P. E. Dodds, L. Staffell, A. D. Hawkes, F. Li, P. Grunewald, W. McDowall and P. Ekins, Int. J. Hydrogen Energy, 2015, 40, 2065–2083 CrossRef CAS.
- T. A. Napp, A. Gambhir, T. P. Hills, N. Florin and P. S. Fennell, Renewable Sustainable Energy Rev., 2014, 30, 616–640 CrossRef CAS.
- J. Tollefson, Nature, 2010, 464, 1262–1264 CrossRef CAS.
- A. Midilli, M. Ay, I. Dincer and M. A. Rosen, Renewable Sustainable Energy Rev., 2005, 9, 255–271 CrossRef CAS.
- A. Midilli, M. Ay, I. Dincer and M. A. Rosen, Renewable Sustainable Energy Rev., 2005, 9, 273–287 CrossRef.
- A. Demirbas, Energy Sources, Part B, 2017, 12, 172–181 CrossRef CAS.
- R. Shandarr, C. A. Trudewind and P. Zapp, J. Cleaner Prod., 2014, 85, 151–163 CrossRef.
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- J. O. M. Bockris, Int. J. Hydrogen Energy, 2013, 38, 2579–2588 CrossRef CAS.
- A. Ursua, E. L. Barrios, J. Pascual, I. S. Martin and P. Sanchis, Int. J. Hydrogen Energy, 2016, 41, 12852–12861 CrossRef CAS.
- J. G. G. Clua, R. J. Mantz and H. De Battista, Appl. Energy, 2011, 88, 1857–1863 CrossRef.
- A. Kovac, D. Marcius and L. Budin, Int. J. Hydrogen Energy, 2019, 44, 9841–9848 CrossRef CAS.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- K. Zeng and D. K. Zhang, Prog. Energy Combust. Sci., 2011, 37, 631 CrossRef.
- M. Schalenbach, O. Kasian and K. J. J. Mayrhofer, Int. J. Hydrogen Energy, 2018, 43, 11932–11938 CrossRef CAS.
- https://www.hydrogen.energy.gov/annual_review19_h2fuel.html#electrolysis .
- https://www.fch.europa.eu/page/call-2019 .
- C. C. Pavel, F. Cecconi, C. Emiliani, S. Santiccioli, A. Scaffidi, S. Catanorchi and M. Comotti, Angew. Chem., Int. Ed., 2014, 53, 1378–1381 CrossRef CAS.
- J. Parrondo, C. G. Arges, M. Niedzwiecki, E. B. Anderson, K. E. Ayers and V. Ramani, RSC Adv., 2014, 4, 9875–9879 RSC.
- G. Gardner, J. Al-Sharab, N. Danilovic, Y. B. Go, K. Ayers, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2016, 9, 184–192 RSC.
- J. Durst, A. Siebel, C. Simon, F. Hasche, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- W. C. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Nano Energy, 2016, 29, 29–36 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS.
- C. L. Hu, L. Zhang and J. L. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- E. S. Liu, J. K. Li, L. Jiao, H. T. T. Doan, Z. Y. Liu, Z. P. Zhao, Y. Huang, K. M. Abraham, S. Mukerjee and Q. Y. Jia, J. Am. Chem. Soc., 2019, 141, 3232–3239 CrossRef CAS PubMed.
- X. Wu and K. Scott, J. Power Sources, 2012, 214, 124–129 CrossRef CAS.
- S. H. Ahn, B. S. Lee, I. Choi, S. J. Yoo, H. J. Kim, E. Cho, D. Henkensmeier, S. W. Nam, S. K. Kim and J. H. Jang, Appl. Catal., B, 2014, 154, 197–205 CrossRef.
- J. Y. Huot, M. L. Trudeau and R. Schulz, J. Electrochem. Soc., 1991, 138, 1316–1321 CrossRef CAS.
- L. Xiao, S. Zhang, J. Pan, C. X. Yang, M. L. He, L. Zhuang and J. T. Lu, Energy Environ. Sci., 2012, 5, 7869–7871 RSC.
- X. Tang, L. Xiao, C. X. Yang, J. T. Lu and L. Zhuang, Int. J. Hydrogen Energy, 2014, 39, 3055–3060 CrossRef CAS.
- R. B. Patil, A. Mantri, S. D. House, J. C. Yang and J. R. McKone, ACS Appl. Energy Mater., 2019, 2, 2524–2533 CrossRef CAS.
- F. Juarez, D. Salmazo, P. Quaino and W. Schmickler, Electrocatalysis, 2019, 10, 584–590 CrossRef CAS.
- A. G. Oshchepkov, A. Bonnefont and E. R. Savinova, Electrocatalysis, 2019, 133–142 Search PubMed.
- Y. Y. Chen, Y. Zhang, X. Zhang, T. Tang, H. Luo, S. Niu, Z. H. Dai, L. J. Wan and J. S. Hu, Adv. Mater., 2017, 29, 170331–170338 Search PubMed.
- J. Zhang, T. Wang, P. Liu, Z. Q. Liao, S. H. Liu, X. D. Zhuang, M. W. Chen, E. Zschech and X. L. Feng, Nat. Commun., 2017, 8, 15437–15445 CrossRef CAS PubMed.
- Y. S. Jin, H. T. Wang, J. J. Li, X. Yue, Y. J. Han, P. K. Shen and Y. Cui, Adv. Mater., 2016, 28, 3785–3790 CrossRef CAS PubMed.
- M. Gong, W. Zhou, M. C. Tsai, J. G. Zhou, M. Y. Guan, M. C. Lin, B. Zhang, Y. F. Hu, D. Y. Wang, J. Yang, S. J. Pennycook, B. J. Hwang and H. J. Dai, Nat. Commun., 2014, 5, 4695–4701 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- I. Vincent and D. Bessarabov, Renewable Sustainable Energy Rev., 2018, 81, 1690–1704 CrossRef CAS.
- S. Klaus, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 18303–18316 CrossRef CAS.
- F. D. Speck, K. E. Dettelbach, R. S. Sherbo, D. A. Salvatore, A. X. Huang and C. P. Berlinguette, Chem, 2017, 2, 590–597 CAS.
- Y. J. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C. Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS.
- J. Y. C. Chen, L. N. Dang, H. F. Liang, W. L. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef CAS.
- D. Gonzalez-Flores, K. Klingan, P. Chernev, S. Loos, M. R. Mohammadi, C. Pasquini, P. Kubella, I. Zaharieva, R. D. L. Smith and H. Dau, Sustainable Energy Fuels, 2018, 2, 1986–1994 RSC.
- R. D. L. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. Gonzalez-Flores and H. Dau, Energy Environ. Sci., 2018, 11, 2476–2485 RSC.
- M. Gorlin, P. Chernev, P. Paciok, C. W. Tai, J. Ferreira de Araujo, T. Reier, M. Heggen, R. Dunin-Borkowski, P. Strasser and H. Dau, Chem. Commun., 2019, 55, 818–821 RSC.
- D. Y. Xu, M. B. Stevens, M. R. Cosby, S. Z. Oener, A. M. Smith, L. J. Enman, K. E. Ayers, C. B. Capuano, J. N. Renner, N. Danilovic, Y. G. Li, H. Z. Wang, Q. H. Zhang and S. W. Boettcher, ACS Catal., 2019, 9, 7–15 CrossRef CAS.
- I. Vincent, A. Kruger and D. Bessarabov, Int. J. Electrochem. Sci., 2018, 13, 11347–11358 CrossRef CAS.
- I. Vincent, A. Kruger and D. Bessarabov, Int. J. Hydrogen Energy, 2017, 42, 10752–10761 CrossRef CAS.
- X. Wu and K. Scott, J. Mater. Chem., 2011, 21, 12344–12351 RSC.
- X. Wu, K. Scott, F. Xie and N. Alford, J. Power Sources, 2014, 246, 225–231 CrossRef CAS.
- T. Pandiarajan, L. J. Berchmans and S. Ravichandran, RSC Adv., 2015, 5, 34100–34108 RSC.
- G. Gupta, K. Scott and M. Mamlouk, J. Power Sources, 2018, 375, 387–396 CrossRef CAS.
- L. Zeng, T. S. Zhao, R. H. Zhang and J. B. Xu, Electrochem. Commun., 2018, 87, 66–70 CrossRef CAS.
- R. I. Masel, Z. Liu and S. D. Sajjad, Polym. Electrolyte Fuel Cells, 2016, 75(16), 1143–1146 CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- C. Vogel and J. Meier-Haack, Desalination, 2014, 342, 156–174 CrossRef CAS.
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187–200 CrossRef CAS.
- J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399–400, 49–59 CrossRef CAS.
- D. G. Li, I. Matanovic, A. S. Lee, E. J. Park, C. Fujimoto, H. T. Chung and Y. S. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 9696–9701 CrossRef CAS PubMed.
- J. Ran, L. Wu, Y. He, Z. Yang, Y. Wang, C. Jiang, L. Ge, E. Bakangura and T. Xu, J. Membr. Sci., 2017, 522, 267–291 CrossRef CAS.
- K. F. L. Hagesteijn, S. Jiang and B. P. Ladewig, J. Mater. Sci., 2018, 53, 11131–11150 CrossRef CAS.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1727–1735 CrossRef CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS.
- G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36, 1521–1557 CrossRef CAS.
- Y. J. Wang, J. Qiao, R. Baker and J. Zhang, Chem. Soc. Rev., 2013, 42, 5768–5787 RSC.
- S. Gu, B. J. Xu and Y. S. Yan, in Annual Review of Chemical and Biomolecular Engineering, Vol 5, ed. J. M. Prausnitz, M. F. Doherty and R. A. Segalman, Annual Reviews, Palo Alto, 2014, vol. 5, pp. 429–454 Search PubMed.
- M. Paidar, V. Fateev and K. Bouzek, Electrochim. Acta, 2016, 209, 737–756 CrossRef CAS.
- M. Bodner, A. Hofer and V. Hacker, WIREs Energy Environ., 2015, 4, 365–381 CrossRef CAS.
- D. Hellwinkel and H. Seifert, Liebigs Ann. Chem., 1972, 762, 29–54 CrossRef CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS.
- L. Gu, H. Dong, Z. Sun, Y. Li and F. Yan, RSC Adv., 2016, 6, 94387–94398 RSC.
- C. Vogel, H. Komber and J. Meier-Haack, React. Funct. Polym., 2017, 117, 34–42 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, J. Membr. Sci., 2019, 578, 183–195 CrossRef CAS.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2018, 6, 16537–16547 RSC.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 10 CrossRef.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS PubMed.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2017, 50, 2784–2793 CrossRef CAS.
- H. S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 11924–11938 RSC.
- T. H. Pham and P. Jannasch, ACS Macro Lett., 2015, 4, 1370–1375 CrossRef CAS.
- H. S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 17138–17153 RSC.
- H. S. Dang and P. Jannasch, J. Mater. Chem. A, 2017, 5, 21965–21978 RSC.
- D. J. Strasser, B. J. Graziano and D. M. Knauss, J. Mater. Chem. A, 2017, 5, 9627–9640 RSC.
- N. Chen, C. Lu, Y. Li, C. Long and H. Zhu, J. Membr. Sci., 2019, 572, 246–254 CrossRef CAS.
- X. M. Chu, Y. Shi, L. Liu, Y. D. Huang and N. W. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC.
- L. A. Diaz, R. E. Coppola, G. C. Abuin, R. Escudero-Cid, D. Herranz and P. Ocon, J. Membr. Sci., 2017, 535, 45–55 CrossRef CAS.
- L. A. Diaz, J. Hnat, N. Heredia, M. M. Bruno, F. A. Viva, M. Paidar, H. R. Corti, K. Bouzek and G. C. Abuin, J. Power Sources, 2016, 312, 128–136 CrossRef CAS.
- A. Marinkas, I. Struzynska-Piron, Y. Lee, A. Lim, H. S. Park, J. H. Jang, H. J. Kim, J. Kim, A. Maljusch, O. Conradi and D. Henkensmeier, Polymer, 2018, 145, 242–251 CrossRef CAS.
- A. Konovalova, H. Kim, S. Kim, A. Lim, H. S. Park, M. R. Kraglund, D. Aili, J. H. Jang, H. J. Kim and D. Henkensmeier, J. Membr. Sci., 2018, 564, 653–662 CrossRef CAS.
- J. Hnat, M. Paidar, J. Schauer and K. Bouzek, Int. J. Hydrogen Energy, 2014, 39, 4779–4787 CrossRef CAS.
- J. Hnat, M. Plevova, J. Zitka, M. Paidar and K. Bouzek, Electrochim. Acta, 2017, 248, 547–555 CrossRef CAS.
- J. Zitka, J. Peter, B. Galajdova, L. Pavlovec, Z. Pientka, M. Paidar, J. Hnat and K. Bouzek, Desalin. Water Treat., 2019, 142, 90–97 CrossRef CAS.
- J. Hnat, M. Paidar, J. Schauer, J. Zitka and K. Bouzek, J. Appl. Electrochem., 2011, 41, 1043–1052 CrossRef CAS.
- J. Hnat, M. Paidar, J. Schauer, J. Zitka and K. Bouzek, J. Appl. Electrochem., 2012, 42, 545–554 CrossRef CAS.
- D. D. Tham and D. Kim, J. Membr. Sci., 2019, 139–149 CrossRef CAS.
- N. Lee, D. T. Duong and D. Kim, Electrochim. Acta, 2018, 271, 150–157 CrossRef CAS.
- D. Aili, A. G. Wright, M. R. Kraglund, K. Jankova, S. Holdcroft and J. O. Jensen, J. Mater. Chem. A, 2017, 5, 5055–5066 RSC.
- M. R. Kraglund, D. Aili, K. Jankova, E. Christensen, Q. Li and J. O. Jensen, J. Electrochem. Soc., 2016, 163, F3125–F3131 CrossRef CAS.
- P. Häussinger, R. Lohmüller and A. M. Watson, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- H. Ito, N. Kawaguchi, S. Someya and T. Munakata, Electrochim. Acta, 2019, 297, 188–196 CrossRef CAS.
- X. Wu and K. Scott, J. Power Sources, 2012, 214, 124–129 CrossRef CAS.
- L. Xiao, S. Zhang, J. Pan, C. Yang, M. He, L. Zhuang and J. Lu, Energy Environ. Sci., 2012, 5, 7869–7871 RSC.
- N. V. Kuleshov, V. N. Kuleshov, S. A. Dovbysh, S. A. Grigoriev, S. V. Kurochkin and P. Millet, Int. J. Hydrogen Energy, 2019, 29441–29449 CrossRef CAS.
- J. Hnát, R. Kodým, K. Denk, M. Paidar, J. Žitka and K. Bouzek, Chem.-Ing.-Tech., 2019, 91, 821–832 CrossRef.
- S. H. Ahn, S. J. Yoo, H. J. Kim, D. Henkensmeier, S. W. Nam, S. K. Kim and J. H. Jang, Appl. Catal., B, 2016, 180, 674–679 CrossRef CAS.
- S. H. Ahn, B. S. Lee, I. Choi, S. J. Yoo, H. J. Kim, E. Cho, D. Henkensmeier, S. W. Nam, S. K. Kim and J. H. Jang, Appl. Catal., B, 2014, 154–155, 197–205 CrossRef CAS.
- A. Lim, H. J. Kim, D. Henkensmeier, S. Jong Yoo, J. Young Kim, S. Young Lee, Y. E. Sung, J. H. Jang and H. S. Park, J. Ind. Eng. Chem., 2019, 76, 410–418 CrossRef CAS.
- H. Ito, N. Kawaguchi, S. Someya, T. Munakata, N. Miyazaki, M. Ishida and A. Nakano, Int. J. Hydrogen Energy, 2018, 43, 17030–17039 CrossRef CAS.
- S. Vengatesan, S. Santhi, S. Jeevanantham and G. Sozhan, J. Power Sources, 2015, 284, 361–368 CrossRef CAS.
- H. Ito, N. Miyazaki, S. Sugiyama, M. Ishida, Y. Nakamura, S. Iwasaki, Y. Hasegawa and A. Nakano, J. Appl. Electrochem., 2018, 48, 305–316 CrossRef CAS.
- A. Marinkas, I. Struźyńska-Piron, Y. Lee, A. Lim, H. S. Park, J. H. Jang, H. J. Kim, J. Kim, A. Maljusch, O. Conradi and D. Henkensmeier, Polymer, 2018, 145, 242–251 CrossRef CAS.
- M. K. Cho, H. Y. Park, H. J. Lee, H. J. Kim, A. Lim, D. Henkensmeier, S. J. Yoo, J. Y. Kim, S. Y. Lee, H. S. Park and J. H. Jang, J. Power Sources, 2018, 382, 22–29 CrossRef CAS.
- M. K. Cho, H. Y. Park, S. Choe, S. J. Yoo, J. Y. Kim, H. J. Kim, D. Henkensmeier, S. Y. Lee, Y. E. Sung, H. S. Park and J. H. Jang, J. Power Sources, 2017, 347, 283–290 CrossRef CAS.
- X. Chu, Y. Shi, L. Liu, Y. Huang and N. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC.
- S. Seetharaman, R. Balaji, K. Ramya, K. S. Dhathathreyan and M. Velan, Int. J. Hydrogen Energy, 2013, 38, 14934–14942 CrossRef CAS.
- C. C. Pavel, F. Cecconi, C. Emiliani, S. Santiccioli, A. Scaffidi, S. Catanorchi and M. Comotti, Angew. Chem., Int. Ed., 2014, 53, 1378–1381 CrossRef CAS PubMed.
- G. C. Chen, T. H. Wondimu, H. C. Huang, K. C. Wang and C. H. Wang, Int. J. Hydrogen Energy, 2019, 44, 10174–10181 CrossRef CAS.
- E. López-Fernández, J. Gil-Rostra, J. P. Espinós, A. R. González-Elipe, F. Yubero and A. de Lucas-Consuegra, J. Power Sources, 2019, 415, 136–144 CrossRef.
- B. K. Kakati, D. Sathiyamoorthy and A. Verma, Int. J. Hydrogen Energy, 2010, 35, 4185–4194 CrossRef CAS.
- Z. Liu, S. D. Sajjad, Y. Gao, H. Yang, J. J. Kaczur and R. I. Masel, Int. J. Hydrogen Energy, 2017, 42, 29661–29665 CrossRef CAS.
- D. Aili, M. K. Hansen, J. W. Andreasen, J. Zhang, J. O. Jensen, N. J. Bjerrum and Q. Li, J. Membr. Sci., 2015, 493, 589–598 CrossRef CAS.
- W. Ju, M. V. F. Heinz, L. Pusterla, M. Hofer, B. Fumey, R. Castiglioni, M. Pagani, C. Battaglia and U. F. Vogt, ACS Sustainable Chem. Eng., 2018, 6, 4829–4837 CrossRef CAS.
- E. J. Park, C. B. Capuano, K. E. Ayers and C. Bae, J. Power Sources, 2018, 375, 367–372 CrossRef CAS.
- L. Wang, T. Weissbach, R. Reissner, A. Ansar, A. S. Gago, S. Holdcroft and K. A. Friedrich, ACS Appl. Energy Mater., 2019, 2, 7903–7912 CrossRef CAS.
- J. Hnát, M. Plevova, R. A. Tufa, J. Zitka, M. Paidar and K. Bouzek, Int. J. Hydrogen Energy, 2019, 44, 17493–17504 CrossRef.
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C. Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS PubMed.
- X. Wu and K. Scott, J. Power Sources, 2012, 206, 14–19 CrossRef CAS.
- X. Wu and K. Scott, Int. J. Hydrogen Energy, 2013, 38, 3123–3129 CrossRef CAS.
- J. E. Park, S. Y. Kang, S. H. Oh, J. K. Kim, M. S. Lim, C. Y. Ahn, Y. H. Cho and Y. E. Sung, Electrochim. Acta, 2019, 295, 99–106 CrossRef CAS.
- X. Su, L. Gao, L. Hu, N. A. Qaisrani, X. Yan, W. Zhang, X. Jiang, X. Ruan and G. He, J. Membr. Sci., 2019, 283–292 CrossRef CAS.
- X. Liang, G. Pan, L. Xu and J. Wang, Fuel, 2015, 139, 393–400 CrossRef CAS.
- V. Vijayakumar and S. Y. Nam, J. Ind. Eng. Chem., 2019, 70, 70–86 CrossRef CAS.
- X. Wu and K. Scott, J. Mater. Chem., 2011, 21, 12344–12351 RSC.
- Y. C. Cao, X. Wu and K. Scott, Int. J. Hydrogen Energy, 2012, 37, 9524–9528 CrossRef CAS.
- L. Zeng and T. S. Zhao, Nano Energy, 2015, 11, 110–118 CrossRef CAS.
- G. Borisov, H. Penchev, K. Maksimova-Dimitrova, F. Ublekov, E. Lefterova, V. Sinigersky and E. Slavcheva, Mater. Lett., 2019, 240, 144–146 CrossRef CAS.
- M. Faraj, M. Boccia, H. Miller, F. Martini, S. Borsacchi, M. Geppi and A. Pucci, Int. J. Hydrogen Energy, 2012, 37, 14992–15002 CrossRef CAS.
- J. Žitka, M. Bleha, J. Schauer, B. Galajdová, M. Paidar, J. Hnát and K. Bouzek, Desalin. Water Treat., 2015, 56, 3167–3173 Search PubMed.
- L. A. Diaz, J. Hnát, N. Heredia, M. M. Bruno, F. A. Viva, M. Paidar, H. R. Corti, K. Bouzek and G. C. Abuin, J. Power Sources, 2016, 312, 128–136 CrossRef CAS.
- J. Hnát, M. Plevová, J. Žitka, M. Paidar and K. Bouzek, Electrochim. Acta, 2017, 248, 547–555 CrossRef.
- L. S. Darken and H. F. Meier, J. Am. Chem. Soc., 1942, 64, 621–623 CrossRef CAS.
- C. Thieme, in Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- H. Schultz, in Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- M. Schalenbach, A. R. Zeradjanin, O. Kasian, S. Cherevko and K. J. J. Mayrhofer, Int. J. Electrochem. Sci., 2018, 13, 1173–1226 CrossRef CAS.
- J. Hnát, M. Paidar, J. Schauer and K. Bouzek, Int. J. Hydrogen Energy, 2014, 39, 4779–4787 CrossRef.
- S. Suzuki, H. Muroyama, T. Matsui and K. Eguchi, Electrochim. Acta, 2013, 88, 552–558 CrossRef CAS.
- F. Helfferich, Ion exchange, Dover publications, Inc., New York, 1995 Search PubMed.
- B. Beverskog and I. Puigdomenech, Corros. Sci., 1997, 39, 969–980 CrossRef CAS.
- J. Schauer, J. Hnát, L. Brožová, J. Žitka and K. Bouzek, J. Membr. Sci., 2015, 473, 267–273 CrossRef CAS.
- D. Chanda, J. Hnát, T. Bystron, M. Paidar and K. Bouzek, J. Power Sources, 2017, 347, 247–258 CrossRef CAS.
- D. Chanda, J. Hnát, A. S. Dobrota, I. A. Pašti, M. Paidar and K. Bouzek, Phys. Chem. Chem. Phys., 2015, 17, 26864–26874 RSC.
- D. Chanda, J. Hnát, M. Paidar and K. Bouzek, Int. J. Hydrogen Energy, 2014, 39, 5713–5722 CrossRef CAS.
- D. Chanda, J. Hnát, M. Paidar, J. Schauer and K. Bouzek, J. Power Sources, 2015, 285, 217–226 CrossRef CAS.
- W. Ju, M. V. F. Heinz, L. Pusterla, M. Hofer, B. Fumey, R. Castiglioni, M. Pagani, C. Battaglia and U. F. Vogt, ACS Sustainable Chem. Eng., 2018, 6, 4829–4837 CrossRef CAS.
- M. Ďurovič, J. Hnát, C. I. Bernäcker, T. Rauscher, L. Röntzsch, M. Paidar and K. Bouzek, Electrochim. Acta, 2019, 306, 688–697 CrossRef.
- D. R. Dekel, I. G. Rasin and S. Brandon, J. Power Sources, 2019, 420, 118–123 CrossRef CAS.
- Y. X. Chen, A. Lavacchi, H. A. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 4036–4042 CrossRef CAS PubMed.
- Y.-K. Choe, C. Fujimoto, K.-S. Lee, L. T. Dalton, K. Ayers, N. J. Henson and Y. S. Kim, Chem. Mater., 2014, 26, 5675–5682 CrossRef CAS.
- C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423–424, 438–449 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y.-K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- K. M. Meek, J. R. Nykaza and Y. A. Elabd, Macromolecules, 2016, 49, 3382–3394 CrossRef CAS.
- W. You, K. J. T. Noonan and G. W. Coates, Prog. Polym. Sci., 2020, 100, 101177 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |