
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an effective bi-functional Ni–CaO catalyst-sorbent for the sorption-enhanced water gas shift reaction through structural optimization and the controlled deposition of a stabilizer by atomic layer deposition†
Sung Min
Kim
 ,
Andac
Armutlulu
,
Agnieszka M.
Kierzkowska
,
Davood
Hosseini
,
Felix
Donat
and
Christoph
Müller
*
,
Andac
Armutlulu
,
Agnieszka M.
Kierzkowska
,
Davood
Hosseini
,
Felix
Donat
and
Christoph
Müller
*
Laboratory of Energy Science and Engineering, Department of Mechanical and Process Engineering, ETH Zürich, Leonhardstrasse 21, 8092 Zürich, Switzerland. E-mail: muelchri@ethz.ch
First published on 13th November 2019
Abstract
The integration of a CaO-based CO2 sorbent into catalytic schemes to remove CO2 from the product stream provides an effective means to reduce greenhouse gas emissions of chemical processes and to improve the yield and purity of the desired product. A key requirement for such so-called sorbent-enhanced processes is the availability of cyclically stable CO2 sorbent. To this end, we have developed CaO-based CO2 sorbents that combine favourable structural features and a high thermal stability by introducing a thin, conformal layer of Al2O3 (forming Ca3Al2O6 with CaO upon calcination) by atomic layer deposition. The structure and pore volume of the sorbent were found to play a key role in its CO2 capture. Functionalizing such CO2 sorbents with Ni nanoparticles yielded a highly effective bi-functional material for the sorption-enhanced water-gas shift (SE-WGS) reaction. The material showed a high yield of hydrogen of high purity and minimal CO slip over several cycles of repeated SE-WGS/regeneration operation.
Introduction
The key contributor to global warming is the increasing emission of anthropogenic carbon dioxide (CO2) into the atmosphere.1,2 To limit the global temperature increase to 2 °C compared to pre-industrial levels, the annual global CO2 emissions have to be reduced to 14 Gt CO2 by 2050.3 In this context, CO2 capture and storage (CCS) has the potential to provide a near-to midterm strategy to reduce anthropogenic CO2 emissions.3 In addition, CO2 capture and utilization (CCU) may offer a solution to omit CO2 storage and utilize CO2 as a carbon source for the production of value-added chemicals and/or fuels.4–8Currently, scrubbing relying on aqueous solutions of amines (e.g., monoethanolamine) is widely used to remove CO2 selectively from a gas stream, e.g., for the purification of natural gas. Yet amine scrubbing has several limitations such as high regeneration costs translating into high costs for CO2 capture and formation of hazardous side products, e.g., nitrosamines.9,10 Hence, recent research activities have focused primarily on developing alternative CO2 capture technologies that rely on solid sorbents, e.g., layered double oxides (LDO),11 zeolites,12,13 supported amines,14 activated carbons,15,16 metal organic frameworks (MOF),17,18 and alkaline earth metal oxides.19,20 Among these alternatives, high-temperature calcium looping (CaL) based on the reversible reaction of CaO with CO2 (i.e., CaO + CO2 ⇄ CaCO3, ΔH0298K = ±178 kJ mol−1) has attracted significant attention owing to a number of desirable characteristics of CaO that include its (i) high theoretical CO2 uptake capacity (0.78 gCO2 gCaO−1), (ii) large abundance of natural precursors (e.g., limestone and dolomite), and (iii) the fast kinetics of the CO2 capture and release reactions. Moreover, the CO2 capture costs of CaL are estimated to be in the range 12–32 € per t CO2, which is considerably lower compared to amine scrubbing (34–67 € per t CO2), pressure swing adsorption (26–33 € per t CO2) or membrane separation (31–36 € per t CO2).21
The main hurdle concerning the effective utilization of CaO derived from natural precursors (e.g., limestone) is the low Tammann temperature (TT) of pure CaCO3 (≈530 °C), which is below the operating temperature of calcium looping (650–900 °C)22 and thus, leads to a low cyclic stability of the material due to sintering. To alleviate the sintering-induced decay of the CO2 capture capacity of CaO-based CO2 sorbents, the incorporation of high-TT stabilizers into the CaO matrix has been proposed. Commonly used stabilizers can be classified into two different groups: (i) those forming a mixed oxide with CaO, such as Al2O3, SiO2, TiO2 and ZrO2,23–26 and (ii) inert stabilizers that do not react chemically with CaO under the relevant operating conditions, such as MgO, Y2O3, and ZnO.27–31 Furthermore, to make calcium looping a viable option at the industrial scale (e.g., operation in circulating fluidized beds) high CO2 uptakes within relatively short residence times are required. The carbonation of CaO is known to proceed in two reaction regimes,32,33viz., the kinetically controlled carbonation followed by a sluggish diffusion-limited carbonation regime (the diffusivity of CO2 in the CaCO3 product layer, DCaCO3 = 0.003 cm2 s−1, is two orders of magnitude smaller than that in CaO, DCaO = 0.3 cm2 s−1). The transition between these two carbonation regimes is believed to occur when the thickness of the CaCO3 product reaches ∼50 nm.32 Hence, nano-structuring CaO would minimize the diffusion lengths of CO2 through the CaCO3 product layer. In addition to nano-structuring of the sorbent, also the introduction of porosity plays a critical role owing to the large volumetric expansion of the material upon CO2 capture, as the molar volume of the product (CaCO3, 36.9 cm3 mol−1) is nearly twice as high as that of the reactant (CaO, 16.7 cm3 mol−1).
Hence, the ideal CaO-based sorbent would exhibit a nano-structured and highly porous morphology that is stabilized by a high-TT temperature material. Previous studies25,28,34–36 have revealed that the scale (micro-, nano- or even atomic level) at which the stabilizer is introduced into the matrix plays an important role for the overall performance of the sorbents. A number of techniques have been adopted to introduce a stabilizer into the CaO including wet-mixing,37 coprecipitation,38 hydration,39 flame spray pyrolysis,40 hydrolysis41 and sol–gel.42–44 However, these techniques offer only little control over morphological features, such as nanostructure and porosity. More recently, vapor-phase deposition techniques such chemical vapor deposition (CVD)45 and atomic layer deposition (ALD)25 have emerged as promising strategies to introduce a stabilizer to a CaO matrix. In particular, ALD offers the possibility of obtaining conformal coatings of a stabilizer, while controlling its thickness on the atomic level. Owing to these unique features of ALD, structurally well-controlled and -characterizable model materials can be realized to investigate the structure–performance relationships of CaO-based sorbents, which has not been conducted in detail yet.
Besides the capture of CO2, CaO-based sorbents have the potential to be integrated in processes that combine CO2 capture with a catalytic reaction, e.g., steam reforming46–50 and/or the water-gas shift reaction51–54 to yield high-purity H2. For example, a typical composition of the effluent gas of an industrial water-gas shift unit is approximately 76% H2, 17% CO2, 3% CO, and 4% CH4.55 The simultaneous removal of CO2 from this stream would not only increase substantially the purity of the H2 produced, but also shift the equilibrium to the product side according to Le Chatelier's principle. These sorption-enhanced processes reduce the operational complexity, since a nearly pure product stream is obtained in a single unit/reactor without further purification steps. Alkali metal-promoted hydrotalcite and Al2O3 have been employed for the sorption-enhanced water-gas shift reaction (SE-WGS),52,56–60 yet with the shortcomings of low CO2 capture capacities (0.01–0.02 gCO2 gsorbent−1) and poor carbonation kinetics. On the other hand, CaO-based CO2 sorbents are favorable candidates for CO2 capture purposes owing to their relatively high CO2 capture capacity (0.3–0.5 gCO2 gsorbent−1) and fairly rapid carbonation kinetics at the operating temperatures (i.e., 400–600 °C) of the SE-WGS (H2O + CO + CaO ⇆ H2 + CaCO3, ΔH0298K = −219 kJ mol−1) and SE-SMR (CH4 + 2H2O + CaO ⇆ 4H2 + CaCO3, ΔH0298K = −13 kJ mol−1) reactions. CaO-based CO2 sorbents combined with Ni-based catalysts have been employed predominantly for the sorption-enhanced steam methane reforming (SE-SMR). However, high loadings of Ni (i.e., 15–50 wt%) have been required to obtain an acceptable SMR activity, which leads to an appreciable reduction in the CO2 uptake capacity of the material. For SE-WGS, a relatively low Ni loading (3–10 wt%) has been shown to suffice for high CO conversions.61–64 Nevertheless, the sintering-induced deactivation of CaO renders them ineffective as a sorbent in a cyclic process.65,66 Therefore, the development of CaO-based sorbents with a high cyclic stability is vital for the SE-WGS reaction. In addition to a stable CO2 sorbent, a highly active and stable catalyst at the temperatures of interest is equally important to obtain high yields of high-purity hydrogen. Cu–Zn and Fe–Cr catalysts have been employed in industrial scale water-gas shift reaction units (WGS: H2O + CO ⇆ H2 + CO2, ΔH0298K = −41 kJ mol−1) at low (180–250 °C) and high (300–500 °C) temperatures, respectively.67–70 Due to the sintering-induced deactivation of Cu-based catalysts, Fe–Cr-based catalysts are often preferred. The strongly carcinogenic, water-soluble hexavalent chromium in Fe–Cr can be leached out under WGS conditions, leading to health and environmental concerns.71 Therefore, it is highly desirable to develop alternative catalyst formulations containing, e.g., Ni-, Pt-, Pd-, Ru- and Au-based materials for medium-temperature (250–350 °C) WGS.61 In particular, Ni-based catalysts are attractive as they combine a non-pyrophoric, water-tolerant active phase, relatively low cost and a high activity for CO conversion when compared to its precious metal counterparts, such as Pt, Pd or Ru.61,69,72
In the context of sorption-enhanced reactions, an important design consideration that needs to be taken into account is the effective interplay between the catalyst and the CO2 sorbent, which is not easily achieved by the physical mixing of the two components. In this regard, bi-functional catalyst-sorbents are attractive as they provide the means of enhancing both heat and mass transfer between the catalyst and sorbent sites.73,74 Bi-functional SE-WGS materials using hydrotalcites as the CO2 sorbent have been manufactured using co-precipitation approaches,52,75 a method which offers, however, only limited control over the textural and structural characteristics of the material. In addition, hydrotalcites feature a relatively low CO2 capture capacity when compared to CaO-based sorbents.76
Herein, we report the development of a bi-functional catalyst-sorbent containing CaO as the CO2-capture-active material. The structure of the CaO backbone is stabilized by Al2O3 that is introduced by ALD. CaO precursors with different morphologies are investigated, i.e., hollow CaCO3 realized through a sacrificial templating approach and benchmark materials in the form of commercial CaCO3, CaCO3 nanoparticles and limestone. To yield high purity hydrogen, the sorbent is decorated with Ni nanoparticles for SE-WGS operation.
Experimental
Materials
Calcium nitrate tetrahydrate (Acros, 99%), D-(+)-glucose (Sigma-Aldrich, ≥ 99.5%), ethylene glycol (Sigma-Aldrich, 99.8%), trimethyl aluminum (TMA, Pegasus Chemicals, electronic grade), boric acid (Fisher, laboratory reagent grade) CaCO3 (Fisher) and CaCO3 nanoparticles (American elements) were used without any further treatment.Preparation of carbon spheres
Carbon spheres were prepared via the carbonization of D-(+)-glucose under hydrothermal conditions. Specifically, aqueous glucose solutions (2.2 M) were transferred into a teflon-lined stainless steel autoclave. The hydrothermal treatment was carried out at 180 °C for 12 h (autogenous pressure). The resulting material was collected, washed with de-ionized (DI) water and anhydrous ethanol, alternatingly 5 times, and finally dried at 80 °C in an oven overnight.Preparation of hollow CaCO3
Dried carbon spheres (1.0 g) were dispersed in an aqueous solution of 10 mmol calcium nitrate and 50 mmol urea (100 ml). The carbon spheres were dispersed by sonication (10 min) followed by vigorous stirring (1 h). Urea hydrolysis to deposit CaCO3 onto the carbon spheres was performed at 90 °C for 6 h under reflux conditions. The resulting material was collected and washed thoroughly with DI water 5 times. After drying at 120 °C in an oven, hollow CaCO3 was obtained by the thermal removal of the carbon template in a muffle furnace at 600 °C (5 °C min−1, 2 h).Atomic layer deposition of Al2O3
An Al2O3 overcoat was deposited onto non-porous, commercial CaCO3 and hollow CaCO3 using a commercial ALD reactor (Picosun R-200) equipped with a system for the coating of powders. For the deposition of Al2O3, electronic grade TMA and DI water were used as precursors, and high-purity N2 served as both carrier and purge gas. The pulse and purge times were kept constant at 0.1 s/15 s/0.1 s/15 s for TMA/N2/H2O/N2. The deposition took place at 300 °C, and the precursor temperatures were fixed at 20 °C. Use of high-resolution TEM allowed us to determine a deposition rate of ∼0.1 nm/ALD cycle, which is in good agreement with the thickness of Al2O3 grown simultaneously on a Si wafer and measured by means of ellipsometry (SENTECH SE850).Bifunctional Ni–CaO catalyst preparation
Bi-functional Ni–CaO materials were prepared by wet impregnation with a solution of nickel acetylacetonate (Ni(acac)2) dissolved in tetrahydrofuran. Specifically, 0.14 g of Ni(acac)2 and 1 g of the sorbent, i.e., Al2O3-coated or non-coated hollow CaO sorbents were mixed with 50 ml of tetrahydrofuran. After vigorous stirring for 5 h, the materials were dried at 80 °C in an oven overnight, followed by calcination at 900 °C (5 °C min−1) for 2 h in a muffle furnace. The nominal nickel loading was 3.0 wt% as per determined by inductively coupled plasma optical emission spectrometry (ICP-OES).Material characterization
The elemental composition was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Agilent 5100 VDV).Dried hollow CaCO3 and as-synthesized Al2O3-coated, hollow CaCO3 (after one half and one full ALD cycle) were collected and characterized by diffuse reflectance FTIR spectroscopy (DRIFT) in the same glove box as the ALD reactor using an Alpha II spectrophotometer (Bruker).
N2 physisorption was measured using a NOVA 4000e analyzer (Quantachrome Instruments). The adsorption and desorption of N2 were determined at −196 °C after degassing each sample at 250 °C under vacuum for 2 h. The specific surface area and pore size distribution were calculated using the Brunauer–Emmett–Teller (BET) and the Barrett–Joyner–Halenda (BJH) models, respectively.
The crystallinity and chemical composition of the sorbents were investigated using powder X-ray diffraction (PANalytical Empyrean equipment) with Cu Kα radiation (λ = 1.5418 Å, 45 mA and 40 kV). Each sample was measured in the 2θ range of 10–90° using a step size of 0.016° with a time duration per step of 0.8 s. In the in situ XRD experiments, ∼0.1 g of the CO2 sorbent was placed in a reaction chamber (XRK 900, Anton Paar). The reaction gases (CO2 and N2) were controlled via mass flow controllers (Bronkhorst EL-FLOW MFC). The diffraction patterns during the cyclic carbonation and calcination experiments (following the procedure of the TGA experiments) were recorded in the 2θ range of 10–60° using a step size of 0.02°.
The morphology and textural features of the sorbents were analyzed by high-resolution scanning electron microscopy (HR-SEM, FEI Magellan 400 FEG), and focused ion beam scanning electron microscopy (FIB-SEM, Zeiss, NVision 40). Prior to SEM analysis, the samples were sputter-coated (MED 010) with an ∼5 nm layer of platinum. Transition electron microscopy (TEM) and scanning transmission electron microscopy (STEM) were performed on a FEI Talos F200X operated at 200 kV, which was equipped with a SuperX EDX consisting of four SDD detectors.
CO2 capture test
The cyclic CO2 uptake of the samples was measured in a thermogravimetric analyzer (TGA, Mettler Toledo TGA/DSC 3+). In a typical experiment, ∼10 mg of the calcined sorbent was loaded into an alumina crucible (70 μl) and heated to 900 °C (50 °C min−1 heating rate) in N2, using a flowrate of 150 ml min−1 (measured at ambient temperature and pressure), including a constant N2 flow (25 ml min−1) as a purge gas over the microbalance. After reaching the reaction temperature, the temperature was maintained for 4 min to completely calcine the material to CaO. Subsequently, the temperature was decreased to 650 °C (50 °C min−1 cooling rate) and the carbonation reaction was performed for 20 min in 20 vol% CO2 (150 ml min−1) balanced by N2. Subsequently, the temperature was increased to 900 °C (50 °C min−1 heating rate) in a flow of 30 ml min−1 of CO2 (25 ml min−1 of N2 purge gas over the micro-balance) and the sample was calcined for 10 min to regenerate the CO2 sorbent. The carbonation and calcination reactions were repeated for a given number of cycles for each sorbent.The cyclic CO2 capture performance under SE-WGS mimicking conditions (with regards to the temperature and the duration of the carbonation and regeneration reaction), i.e., carbonation at 400 °C using 70 ml min−1 of 20 vol% CO2 in N2 for 50 min and regeneration at 800 °C using 50 ml min−1 N2 for 15 min, was evaluated in the TGA. At the beginning of the experiment the calcined material was heated to 800 °C (10 °C min−1) in N2 (70 ml min−1) followed by reduction at 500 °C (10 °C min−1) for 2 h in 10 vol% H2/N2 (100 ml min−1). Ten repeated cycles of carbonation and regeneration were performed.
Sorption-enhanced water-gas shift reaction (SE-WGS)
The cyclic SE-WGS reaction was carried out in a fixed-bed quartz reactor (∼10.5 mm of internal diameter and 400 mm of length; the bed was placed in the middle of the reactor). In a typical experiment, 0.25 g of the calcined material diluted with SiC (weight ratio of 1 to 10) was used. Prior to the activity test, the material was heated at 800 °C (10 °C min−1) for 1 h in N2 (100 ml min−1) to ensure the full calcination to CaO. Subsequently, the material was reduced at 500 °C for 2 h in 10 vol% H2/N2 (100 ml min−1). The SE-WGS was performed at 400 °C using a total flow rate of 70 ml min−1 (5 ml min−1 CO, 20 ml min−1 H2O and 45 ml min−1 N2; GHSV = 16.8 L g−1 h−1 of GHSV). After 50 min of the SE-WGS, the material was heated to 800 °C (20 °C min−1 ramp) and regenerated for 15 min in N2 (70 ml min−1). In total ten SE-WGS/regeneration cycles were performed. The composition of the off-gas (after the condensation of unreacted steam) was analyzed using a micro-GC (C2V-200, Thermo Scientific) equipped with a thermal conductivity detector (TCD) and using molecular sieve 5A and U-plot cartridges.Estimation of CO2 uptake at the kinetically controlled regime
The CO2 uptake in the kinetically controlled reaction stage was estimated following a model by Pacciani et al. that assumes that the CO2 capture in the kinetically controlled reaction stage is completed once the volume available in small pores (dpore < 100 nm) is filled by CaCO3:33,77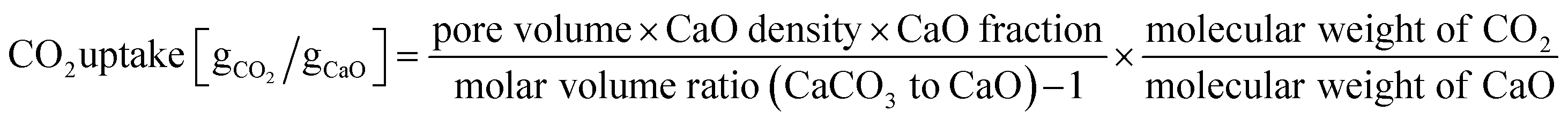
Results and discussion
Synthesis of the CO2 sorbents
The synthesis procedure to obtain Al2O3-coated, hollow CaO spheres is described schematically in Fig. 1a. Carbon nanospheres (CNS) serving as a sacrificial template were prepared by the hydrothermal treatment of an aqueous solution of glucose (0.22 M) at 180 °C. CaCO3 was deposited onto carbon spheres by precipitation of an aqueous solution (0.1 M) of Ca(NO3) with hydrolysed urea at 80 °C, viz., Ca(NO3)2 + CO(NH2)2 + 2H2O → CaCO3 + 2NH4NO3, (ΔH0298K = −184 kJ mol−1).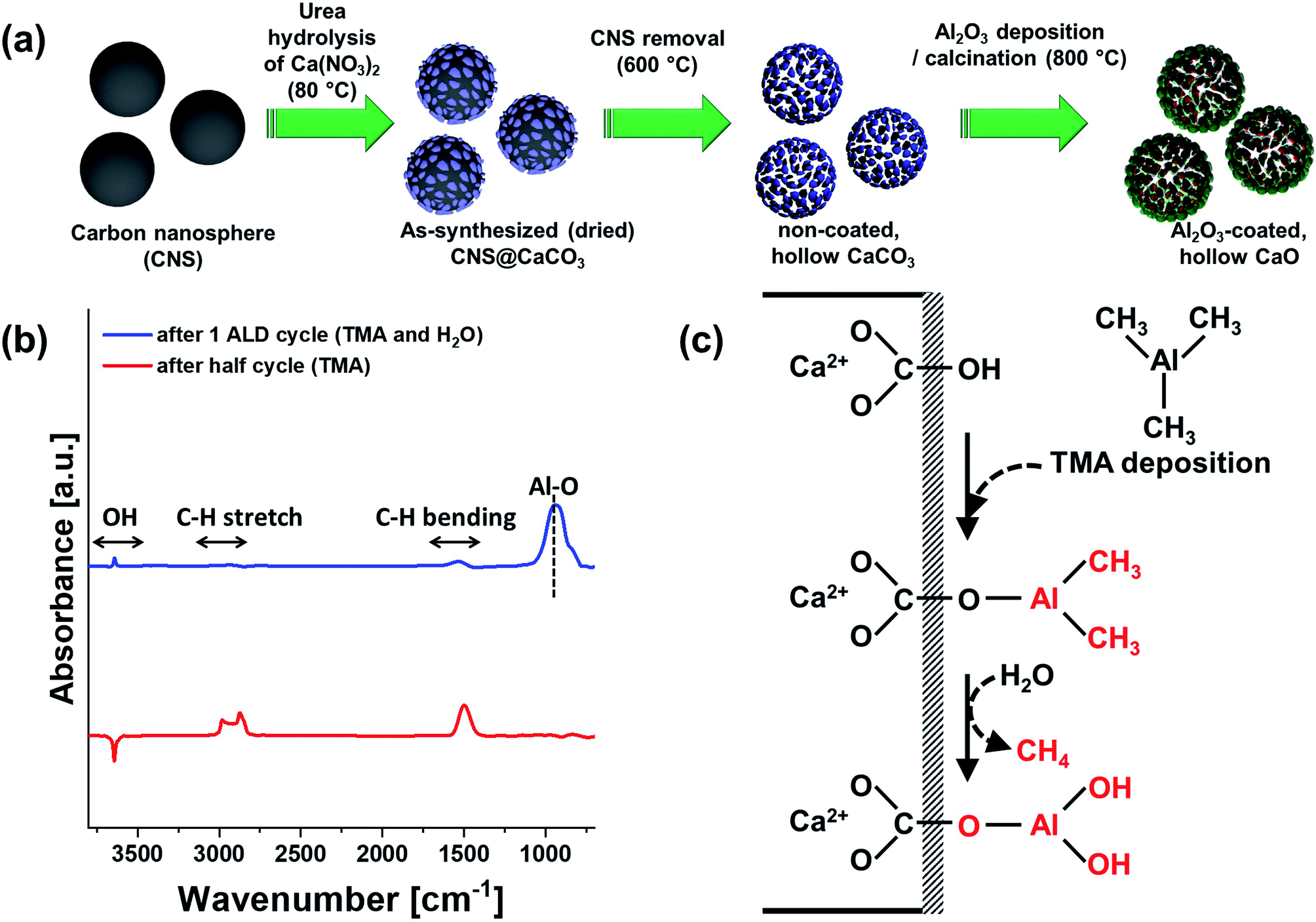 | ||
| Fig. 1 Al2O3-coated, hollow CaO spheres: (a) template-assisted synthesis procedure. (b) DRIFT difference spectra (700–3800 cm−1) after one half and one full ALD cycle. (c) Reaction schematic for the deposition of Al2O3 onto CaCO3via ALD. | ||
The thermal decomposition profile (in air) of as-synthesized CNS and CaCO3 coated CNS were assessed by TGA (Fig. S1†). In air, the carbon template was removed completely at 500 °C, yielding hollow CaCO3 structures. Due to the low TT of CaCO3, it is important to remove the template at the lowest temperature possible to avoid sintering. After template removal, a thin layer of Al2O3 was deposited onto the hollow CaCO3 spheres via ALD (using alternating pulses of trimethylaluminum (TMA) and steam at 300 °C for a given number of cycles). The final material, i.e., Al2O3-coated, hollow spheres of CaO, was obtained by calcination in air at 900 °C.
To probe how the deposition of an Al2O3 overcoat onto CaCO3via ALD proceeds, DRIFT spectra were collected after one half (only TMA pulse injection) and a full (TMA and subsequent steam pulse injection) ALD cycle (Fig. S2†). As the Al2O3 precursor (i.e., TMA) is highly reactive with air, the DRIFT analysis was performed in a glovebox. The infrared spectrum of hollow CaCO3 shows the typically signatures of CO32−, viz.; (i) out of plane bending (971 cm−1), (ii) symmetric stretching (1550 cm−1), and (iii) combination band of stretch and out of plane vibration (2387–2659 cm−1). The presence of bands (2750–3075 cm−1) due to C–H bonds indicates that there were some organic residues of the carbon sphere. The presence of hydroxyl groups on the surface of the hollow CaCO3 spheres was confirmed by the presence of O–H bending (1676 cm−1) and O–H stretching (3642 cm−1) vibrations. The O–H stretching band at 3642 cm−1 has been ascribed to hydroxyl ions on interstitial defects of the CaCO3 lattice framework.78Fig. 1b plots the difference DRIFT spectra after one half and one full ALD cycle compared to pristine, hollow CaCO3. After one half cycle the intensity of the C–H stretching (2750–3075 cm−1) and C–H bending vibrations (1480 cm−1) increased due to the methyl groups of surface-adsorbed (or chemisorbed) TMA. The reduced intensity of the O–H stretching vibrations indicates that, as expected, TMA reacted with surface O–H groups. After the subsequent injection of a pulse of steam, Al–O vibrations (940 cm−1) appeared and the intensity of the C–H vibrations reduced, indicative of the transformation of TMA to hydroxylated aluminium oxide. According to our DRIFT experiments, the following mechanism for the deposition of Al2O3 on CaCO3via ALD can be drawn (Fig. 1c): First TMA reacts with surface hydroxyl groups that are present on CaCO3. After the exposure to steam, the methyl groups on the surface are hydrolyzed with releasing methane, leading to the formation of a layer of hydroxylated Al2O3. Repeating the ALD cycle multiple times leads to the formation of a layer of Al2O3 (the surface is hydroxylated) on hollow CaCO3 spheres.
To label the different sorbents synthesized, the following nomenclature is used throughout the manuscript: “Al (theoretical thickness of the ALD-grown Al2O3 layer)–Ca containing phase (either CaCO3 or CaO)_X′′, where X refers to the CaCO3 structure that was coated by ALD, viz., hollow CaCO3 spheres, limestone, commercial, bulk CaCO3, or nanoparticles (NP) of CaCO3. For example, the material that contains a 2 nm thick overcoat of Al2O3 onto hollow CaCO3 is referred to as “Al (2 nm)–CaCO3_hollow”.
Morphological and structural characterization
Fig. 2 visualizes the morphology of the hollow CaCO3 spheres at different stages of the synthesis process. Scanning electron microscopy (SEM) of the CNS template revealed an average particle diameter of 780 ± 25 nm (Fig. 2a). After the deposition of CaCO3 by precipitation and subsequent calcination at 600 °C in static air (to remove the CNS template), hollow CaCO3 spheres were obtained (Fig. 2b). Increasing the calcination temperature to 900 °C in a second step converted CaCO3 to CaO further, yet lead to the collapse of the hollow sphere structure (Fig. 2c). High-resolution transmission electron microscopy (HR-TEM) of Al2O3 coated hollow CaCO3 spheres, i.e., Al (2 nm)–CaCO3_hollow (calcined at 600 °C) confirmed that ALD deposited a conformal Al2O3 overcoat on the shell-comprising CaCO3 nanometer-sized grains. The thickness of the Al2O3 layer was 2.0 ± 0.3 nm (grown by 20 ALD cycles, Fig. 2e). The high porosity of the hollow CaCO3 spheres ensured a rapid diffusion of the ALD precursors (TMA and steam) resulting in a homogeneous, conformal Al2O3 overcoat on all of the shell-comprising nanometre-sized grains. After calcination at 900 °C in air, the initially clear boundary between CaCO3 and the Al2O3 overcoat disappeared (Fig. 2f). This was due to the formation of a solid solution between Al and Ca, i.e., Ca3Al2O6 (discussed in more detail in the following section). Nevertheless, the hollow structure of Al (2 nm)–CaO_hollow was preserved during calcination at 900 °C (Fig. 2d). Imaging focused ion beam (FIB) cuts of these structures by SEM confirmed that the hollow structure was conserved after high temperature calcination at 900 °C (Fig. 2g and h). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) with energy-dispersive X-ray spectroscopy (EDX) analysis of Al (2 nm)–CaO_hollow (Fig. 2i–l) revealed a homogeneous distribution of Al and Ca in the material, i.e., after calcination the stabilizer Al2O3 was not localized as an overcoat anymore. | ||
| Fig. 2 Morphological characterization of Al2O3-coated, hollow spheres: SEM images of (a) the carbon sphere template, (b) hollow CaCO3, (c) calcined Al (0 nm)–CaO_hollow and (d) calcined Al (2 nm)–CaO_hollow. HR-TEM images of (e) Al (2 nm)–CaCO3_hollow and (f) calcined Al (2 nm)–CaO_hollow. (g and h) FIB-SEM and (i–l) HAADF-STEM with EDX analysis of calcined Al (2 nm)–CaO_hollow. | ||
The benchmark materials, Al (2 nm)–CaO_limestone and Al (2 nm)–CaO_bulk, featured rather coarsely-structured morphologies (Fig. 3a–c and d–f, respectively). Similarly, the benchmark CaCO3 NP, in the absence of an Al2O3 coating, i.e., Al (0 nm)–CaCO3_NP, grew substantially in particle size upon exposure to 900 °C in air (Fig. 3h), whereas the original structure of the spherical nanoparticles was preserved to a large extent when coated by Al2O3, viz., Al (1 nm)–CaO_NP (Fig. 3i–l). However, after the calcination of Al (1 nm)–CaO_NP the distribution of Al became more heterogeneous, forming small clusters (Fig. 3j–l). When the thickness of the Al2O3 coating was increased further to 12 nm, the core-shell-like morphology of as-synthesized Al (12 nm)–CaCO3_NP (Fig. 3m and n) transformed into a hollow-sphere-structured morphology after calcination at 900 °C, i.e., Al (12 mn)–CaO_NP (Fig. 3o and p). The formation of these hollow structures was due to the interdiffusion of CaO and Al2O3 (Kirkendall effect) upon calcination,79 forming the mixed oxide Ca3Al2O6.80 The accumulation of voids at the interface through interdiffusion yields small pores.81,82
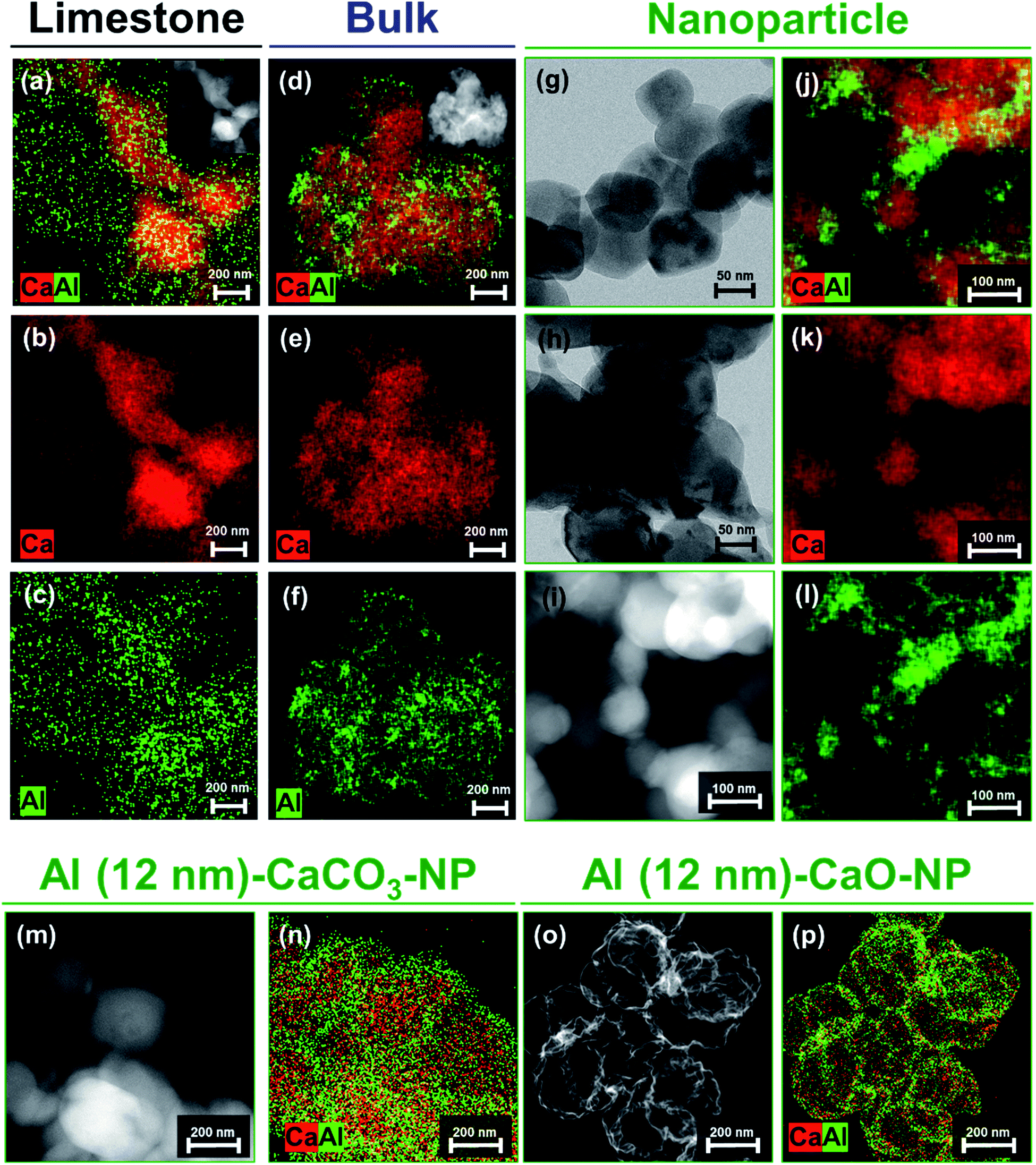 | ||
| Fig. 3 Morphological characterization of the benchmark sorbents: STEM EDX analysis of (a–c) Al (2 nm)–CaO_limestone and (d–f) Al (2 nm)–CaO_bulk. The insets in (a) and (d) represent HAADF-STEM images of Al (2 nm)–CaO_limestone and Al (2 nm)–CaO_bulk, respectively. TEM images of (g) Al (0 nm)–CaCO3_NP and (h) Al (0 nm)–CaO_NP. (i) HAADF-STEM with (j–l) EDX analysis of Al (2 nm)–CaO_NP, HAADF-STEM with EDX analysis of (m and n) Al (12 nm)–CaCO3_NP and (o and p) Al (12 nm)–CaO_NP. | ||
To obtain additional information on the structure and composition of the sorbents, in situ XRD experiments were performed on hollow CaCO3 in the presence or absence of an Al2O3 coating. In situ diffraction patterns were acquired during heating in air. For both Al (0 nm)–CaCO3_hollow and Al (2 nm)–CaCO3_hollow (Fig. 4a and S3†), the conversion of CaCO3 to CaO was completed at ∼700 °C; this is in good agreement with the decomposition profile obtained in the TGA (Fig. S1†). Al (2 nm)–CaCO3_hollow did not show any signatures of crystalline Al-containing phases in the temperature range 600–800 °C (Fig. 4a). The as-synthesized, ALD-grown Al2O3 overcoat was initially amorphous. Diffraction peaks due to Ca3Al2O6 appeared at 820 °C and intensified with increasing temperatures (Fig. 4b).
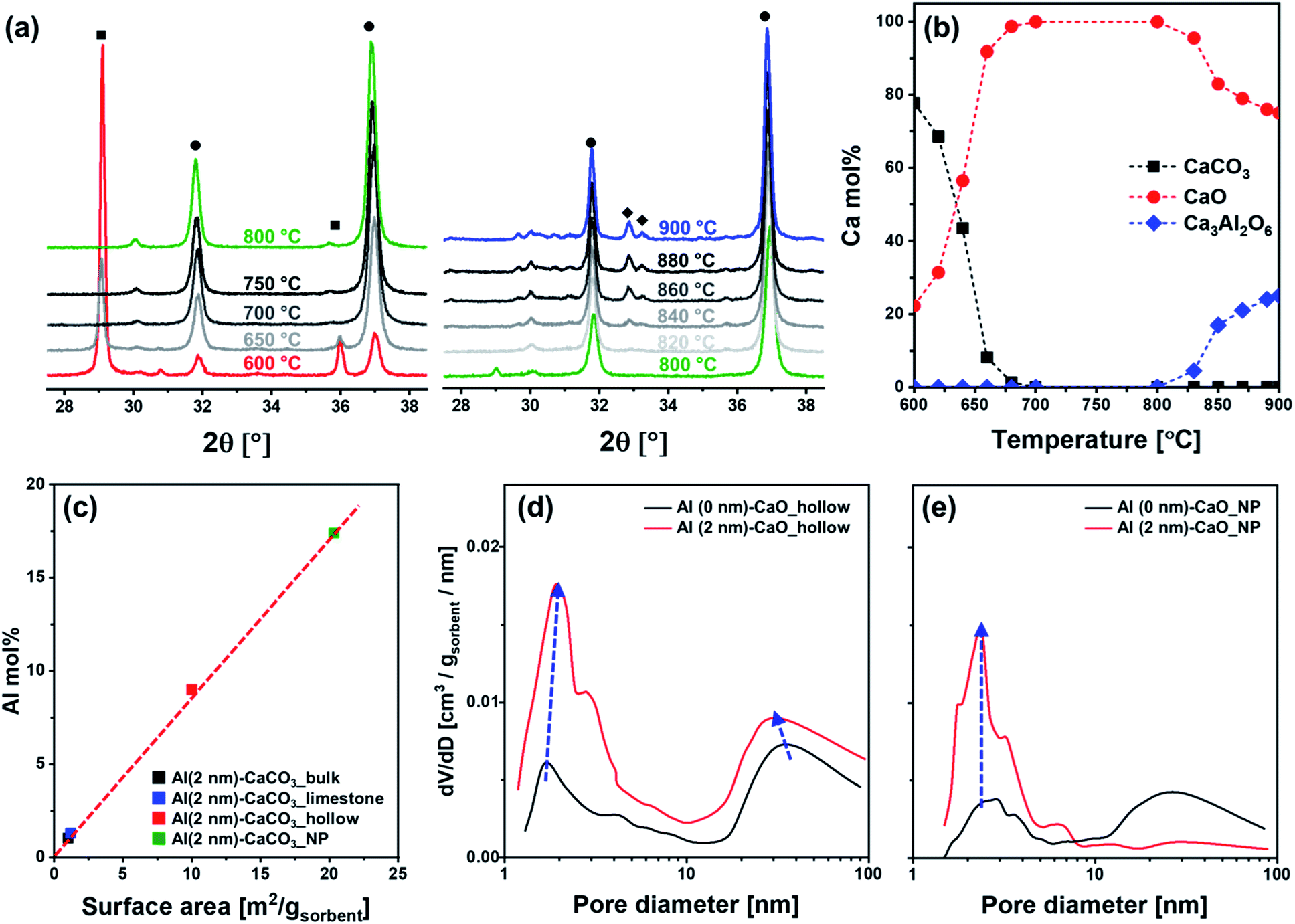 | ||
| Fig. 4 Physicochemical characterization: (a) in situ XRD diffractograms of Al (2 nm)–CaCO3 in the temperature range of 600–900 °C. (b) The Ca mol% of CaCO3, CaO and Ca3Al2O6 in the material calculated by Rietveld refinement as a function of temperature. The symbols represent (■) CaCO3, (●) CaO and (◆) Ca3Al2O6. (c) Mol% of Al3+ in different materials that have been coated by a 2 nm thick layer of Al2O3 (determined by ICP-OES) as a function of the surface area of the original sorbent. BJH pore size distribution of (d) CaO_hollow, (e) CaO_NP in the absence, i.e., Al (0 nm) and presence of an ALD-grown Al2O3 coating of 2 nm thickness. The arrow indicates the change in the pore volume with increasing thickness of the Al2O3 overcoat. | ||
The diffractograms of benchmark CaCO3 (limestone, bulk and NP) and the calcined derivatives in the absence or presence of an Al2O3 coating were acquired by ex situ XRD (Fig. S4†). In the case of the as-synthesized benchmark CaCO3 (limestone, bulk and NP) with a 2 nm-thick Al2O3 overcoat, only CaCO3 was identified, pointing to an amorphous nature of the as-synthesized Al2O3 overcoat. After calcination, Bragg reflections due to CaO and Ca(OH)2 were typically observed in all of the calcined benchmark sorbents regardless of the presence of an Al2O3 coating owing to the decomposition of CaCO3 to CaO and the formation of Ca(OH)2 due to the highly hygroscopic nature of CaO. Additionally, Ca3Al2O6 was observed for the calcined, Al2O3-coated benchmark sorbents (i.e., limestone, bulk and NP). The observations made by electron microscopy analysis and XRD measurements point to the formation of Ca3Al2O6 through the Kirkendall effect.
For Al (2 nm)–CaO sorbents (hollow, limestone, bulk and NP), the intensity of the diffraction peaks of Ca3Al2O6 increased with the thickness of the Al2O3 layer (Fig. S5a†). Rietveld analysis of the calcined, Al-containing samples confirmed a linear relationship between the amount of Ca3Al2O6 formed and the quantity of Al determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) (Fig. S5b†). This implies that all Al introduced as a stabilizer formed a Ca–Al mixed oxide with CaO, viz., Ca3Al2O6. It is also important to note that the same thickness of the Al2O3 overcoat (2 nm) translates to a different quantity of Al in the synthetic and benchmark sorbents owing to different surface areas (Table 1 and S5b†). A higher surface area translates into a higher fraction of Al2O3 in the as-synthesized sorbent, as ALD is a surface-dependent, self-limiting deposition process (Fig. 4c).
| Sorbent | Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al (molar ratio) :Al (molar ratio) |
Surface area [m2 gsorbent−1] | Pore volume [cm3 gsorbent−1] | |
|---|---|---|---|---|
| Hollow CaCO3 | 100:0 |
9 | 0.018 | |
| Al (2 nm)–CaCO3_hollow | 90:10 |
5 | 0.009 | |
| Calcined | Al (0 nm) | 100:0 |
18 | 0.210 |
| Al (2 nm) | 90:10 |
22 | 0.220 | |
| Limestone | 100:0 |
1 | 0.008 | |
| Calcined | Al (0 nm) | 100:0 |
11 | 0.123 |
| Al (2 nm) | 99:1 |
12 | 0.185 | |
| Al (10 nm) | 89:11 |
18 | 0.190 | |
| Bulk CaCO3 | 100:0 |
1 | 0.002 | |
| Calcined | Al (0 nm) | 100:0 |
11 | 0.112 |
| Al (2 nm) | 98:2 |
13 | 0.187 | |
| Al (10 nm) | 88:12 |
17 | 0.195 | |
| CaCO3 nanosphere | 100:0 |
19 | 0.175 | |
| Calcined | Al (0 nm) | 100:0 |
9 | 0.021 |
| Al (1 nm) | 90:10 |
22 | 0.175 | |
| Al (2 nm) | 82:18 |
27 | 0.182 | |
The textural properties of the sorbents were characterized by N2 physisorption. For sorbents without an Al2O3 overcoat, calcination of CaCO3 (hollow, limestone and bulk) lead to an increase in both surface area and pore volume owing to the release of CO2 during the decomposition of CaCO3 (Table 1). An exception was the calcination of the benchmark CaCO3 NP (Al (0 nm)–CaCO3_NP), which lead to a reduction in both the surface area and pore volume from 19 m2 gsorbent−1 and 0.175 cm3 gsorbent−1 to, respectively, 9 m2 gsorbent−1 and 0.012 cm3 gsorbent−1. As confirmed by electron microscopy, calcination of Al (0 nm)–CaCO3_NP resulted in a sintered, non-porous structure.
The deposition of Al2O3 onto structures of CaCO3 affected the textural properties of the materials after calcination (compared to the Al2O3-free counterparts). Fig. 4d plots the BJH pore size distribution of hollow CaCO3 with and without an Al2O3 overcoat. After calcination, the surface area and pore volume of Al (2 nm)–CaO_hollow (22 m2 gsorbent−1 and 0.223 cm3 gsorbent−1) were higher than that of Al (0 nm)–CaO_hollow (18 m2 gsorbent−1 and 0.210 cm3 gsorbent−1). In particular, the pore volume in pores with dpore < 10 nm increased significantly when an Al2O3 overcoat was deposited. For Al (2 nm)–CaO_NP, the increase in pore volume occured mostly in small pores with dpore < 10 nm (Fig. 4e). Similarly, the volume in pores with dpore < 10 nm increased appreciably for Al (2 nm)–CaO_limestone and Al (2 nm)–CaO_bulk when compared to their overcoat-free counterparts (Fig. S6†). The observations made by N2 physisorption in combination with the results of the XRD measurements and electron microscopy indicate that the interdiffusion of CaO and Al2O3 induced by the formation of Ca3Al2O6 upon calcination results in the formation of small pores (dpore < 10 nm).
CO2 capture performance of CaO-based CO2 sorbents
The cyclic CO2 capture performance of the different CaO-based CO2 sorbents was assessed in a TGA using industrially relevant operation conditions, i.e., calcination was performed at 900 °C in a CO2-rich atmosphere. The CO2 sorbent with a hollow sphere structure without the addition of an Al2O3 overcoat (Al (0 nm)–CaO_hollow) showed a significant reduction in its CO2 uptake with cycle number. The CO2 uptake decreased from 0.61 gCO2 gsorbent−1 in the first cycle to 0.26 gCO2 gsorbent−1 in the 10th cycle (Fig. S7a†). The other extreme, Al (12 nm)–CaO_hollow, which is almost pure Ca3Al2O6, showed no CO2 uptake as Ca3Al2O6 does not react with CO2 under the conditions studied here. Nevertheless, the deposition of an Al2O3 overcoat (forming Ca3Al2O6 upon calcination) stabilized the cyclic CO2 uptake of the material, despite the small quantity of Al2O3 added (overcoat thicknesses of 0.5 nm and 1.0 nm correspond to an Al3+ molar fraction of 2.5% and 5.1% in the sorbent, respectively, as determined by ICP-OES). After ten cycles, Al (0.5 nm)–CaO_hollow and Al (1 nm)–CaO_hollow retained 65% (0.38 gCO2 gsorbent−1) and 77% (0.44 gCO2 gsorbent−1) of their initial CO2 uptake capacity, respectively, compared to 42% for the unstabilized sample Al (0 nm)–CaO_hollow. When the thickness of the Al2O3 overcoat was increased to 2 nm and 8 nm, the retention value for the CO2 uptake over ten cycles was further improved to 93% and 97%, respectively, but the overall CO2 uptake capacity was much lower than with thinner coatings. Fig. 5a plots the CO2 uptake of hollow-sphere sorbents at the 10th cycle as a function of the thickness of the ALD-grown Al2O3 layer. The parabolic behaviour of the CO2 uptake, where a sharp increase is followed by a gradual decrease with an increasing thickness of the Al2O3 overcoat, suggests that there is a trade-off between the cyclic stability of a sorbent and the availability of CO2-capture-active CaO. Based on our measurements, the optimal thickness of the Al2O3 overcoat was 2 nm (corresponding to 10 mol% of Al3+ in the sorbent).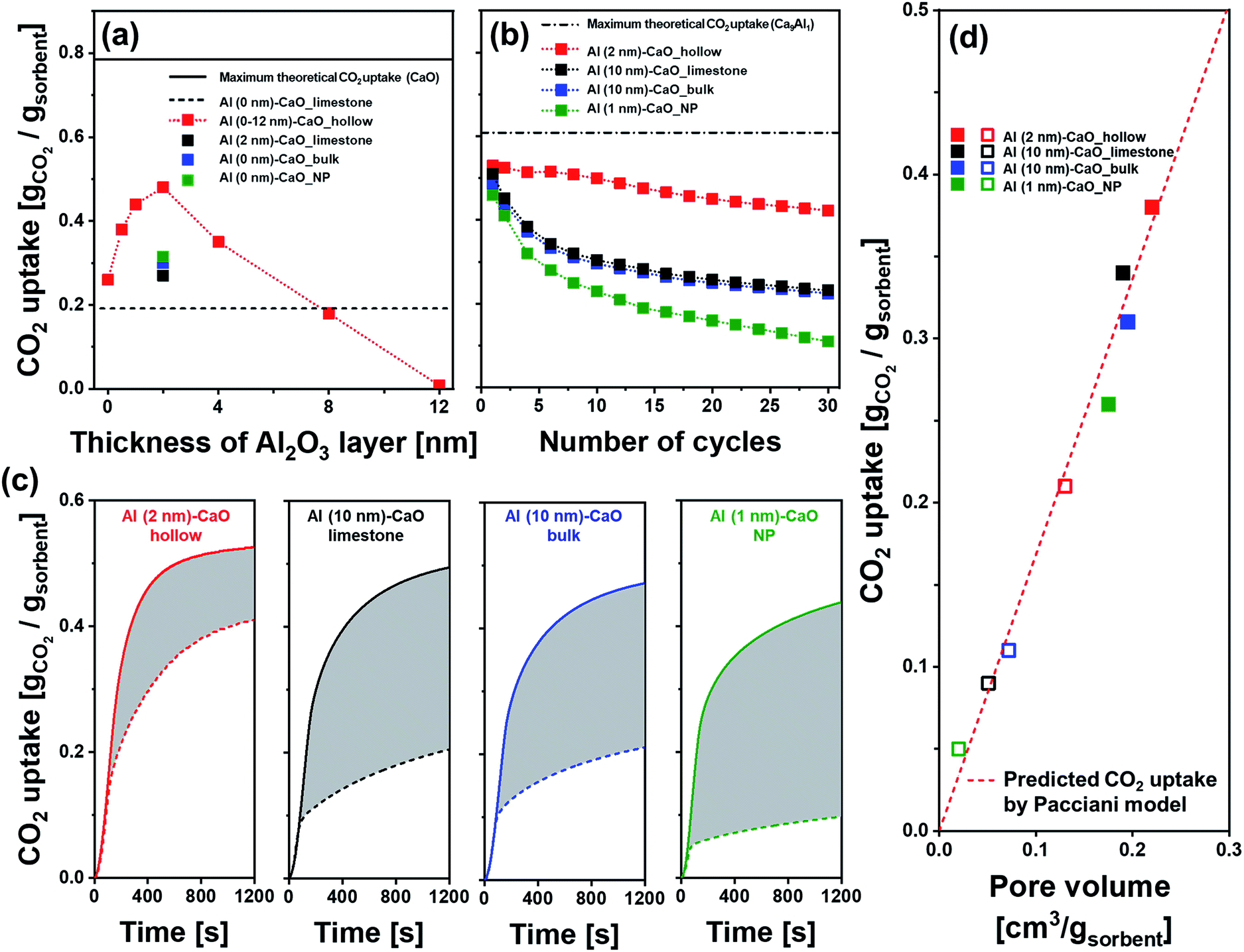 | ||
| Fig. 5 CO2 capture performance: (a) CO2 uptake of Al2O3-coated, hollow-spheres after the 10th cycle as a function of the thickness of the ALD-grown Al2O3 layer. The dashed line represents the cyclic CO2 uptake of limestone, i.e., Al (0 nm)–CaO_limestone, in the 10th cycle, and the solid line gives the maximum theoretical CO2 uptake of pure CaO. (b) CO2 uptake of Al2O3-coated, CaO-based CO2 sorbents as a function of the number of carbonation–regeneration cycles. The dashed line represents the maximum theoretical CO2 uptake of a sorbent with a molar ratio of Ca2+:Al3+ = 9:1 (i.e., 25 wt% of Ca3Al2O6) assuming the full conversion of all of the CO2 capture-active CaO in the material. (c) Temporally resolved CO2 uptake profiles of the sorbents studied as a function of carbonation time. The solid and dashed lines represent the 1st and 30th carbonation cycle, respectively. (d) CO2 uptake in the kinetically controlled carbonation stage as a function of pore volume in the 1st (■) and 30th (□) cycle. The dashed line represents the CO2 uptake in the kinetically controlled carbonation stage as predicted by the Pacciani et al. model.33,77 The fraction of Al3+ in Al (2 nm)–CaO_hollow, Al (10 nm)–CaO_limestone, Al (10 nm)–CaO_bulk and Al (1 nm)–CaO_NP was determined as 10 mol% by ICP-OES. | ||
For reference, Fig. S7b† plots the CO2 uptake of the benchmark sorbents (limestone, bulk and NP) that were also coated with a 2 nm-thick Al2O3 layer. The initial CO2 uptake and stability varied significantly with the type of CaO/CaCO3 matrix of the backbone. Specifically, the following values were observed for the initial CO2 uptake and retention of the CO2 uptake over 30 cycles: (i) Al (2 nm)–CaO_hollow (0.54 gCO2 gsorbent−1 and 80%), (ii) Al (2 nm)–CaO_limestone (0.55 gCO2 gsorbent−1 and 30%), (iii) Al (2 nm)–CaO_bulk (0.56 gCO2 gsorbent−1 and 46%), and (iv) Al (2 nm)–CaO_NP (0.53 gCO2 gsorbent−1 and 58%). Owing to differences in the surface areas of the different CaO/CaCO3 structures that have been coated with a 2 nm-thick layer of Al2O3, the molar fraction of Al3+ in the material varied between 1–18 mol%. Hence, the number of ALD cycles had to be adjusted to obtain a fixed quantity of 10 mol% of Al3+ (as determined by ICP-OES, Table 1) in the benchmark materials, yielding Al (10 nm)–CaO_limestone, Al (10 nm)–CaO_bulk and Al (1 nm)–CaO_NP. The weight fractions of Ca3Al2O6 in the calcined materials, as calculated by the Rietveld analysis, were as follows (Fig. S5b†): Al (2 nm)–CaO_hollow (25%), Al (10 nm)–CaO_limestone (26%), Al (10 nm)–CaO_bulk (24%) and Al (1 nm)–CaO_NP (24%). All of these values were very close to the theoretically expected weight fraction of 25%, which is based on the assumption that all of the Al available is present in the phase Ca3Al2O6. All of these sorbents (both hollow and benchmark morphologies) that contained 10 mol% of Al3+ showed nearly identical CO2 uptakes in the first cycle (Fig. 5b), i.e., in the range 0.49–0.53 gCO2 gsorbent−1 (Fig. 5b). However, the decay behavior of the sorbents differed largely. The initial CO2 uptake and decay rate per cycle were determined as follows: Al (2 nm)–CaO_hollow (0.53 gCO2 gsorbent−1 and 0.7%/cycle), Al (10 nm)–CaO_limestone (0.51 gCO2 gsorbent−1 and 1.8%/cycle), Al (10 nm)–CaO_bulk (0.50 gCO2 gsorbent−1 and 1.8%/cycle) and Al (1 nm)–CaO_NP (0.48 gCO2 gsorbent−1 and 2.5%/cycle).
To understand better the effect of the structure of the sorbent on the CO2 uptake, the temporally resolved carbonation profiles were acquired (Fig. 5c). The two reaction regimes, i.e., kinetically controlled and diffusion-limited, can be approximated by linearly increasing CO2 uptakes with time, whereby the intersection of the linear approximations can be used to determine the transition point between the two regimes. The CO2 uptake of Al (2 nm)–CaO_hollow in the kinetically controlled stage of the carbonation reaction reached 0.38 gCO2 gsorbent−1 in the 1st cycle (corresponding to 71% of the overall CO2 uptake), which reduced to 0.21 gCO2 gsorbent−1 in the 30th cycle (55% retention). For the benchmark sorbents (limestone, bulk and NP), 56–67% of the total CO2 uptake was captured in the kinetically controlled carbonation regime in the 1st cycle, i.e., comparable to the sorbent that possessed a hollow-structured morphology. Despite similar quantities of Al3+ (10 mol%) in all of the sorbents tested, the benchmark sorbents experienced a significant drop in their CO2 uptake in the kinetically controlled carbonation regime when compared to Al (2 nm)–CaO_hollow. For all sorbents investigated there appeared to be a linear relationship between the quantity of CO2 captured in the kinetically controlled carbonation regime and the BJH pore volume for dpore < 100 nm (Table 1 and Fig. 5d). The measured CO2 uptake agreed well with the one predicted by the model of Pacciani et al.33,77 providing further evidence for the assumption that the CO2 uptake in the kinetically controlled carbonation regime was governed by the filling of the small pores with dpore < 100 nm.33,77
Morphological characterization of the cycled CaO-based sorbents
To evaluate changes in the structure of the sorbent over multiple CO2 capture and regeneration cycles, the cycled CO2 sorbents were analyzed by electron microscopy. After 30 cycles, the grain size in all of the CO2 sorbents investigated increased appreciably (Fig. 6a). The growth in grain size (increase compared to the initial grain size) was in the following order: Al (1 nm)–CaO_NP (324%) ≫ Al (10 nm)–CaO_limestone (181%) ≈ Al (10 nm)-CaO_bulk (171%) > Al (2 nm)–CaO_hollow (130%). The trend in the growth of the size of the CaO grains is in line with the rate of decay in the CO2 uptake, i.e., a higher growth rate of the grain size leads to a higher decay rate in the CO2 uptake. This is indicative of a sintering-induced decay of the CO2 uptake in all of the sorbents tested. Furthermore, HAADF-STEM imaging of Al (2 nm)–CaO_hollow (Fig. 6b) reveals that the central void of the hollow spheres was preserved during carbonation and possibly acted as a buffer for the volumetric changes during carbonation and regeneration. Nevertheless, the size of the central void was reduced gradually over repeated cycles of carbonation and regeneration and finally disappeared after 20 cycles. Over 20 cycles, the overall diameter of the hollow spheres reduced from 780 ± 20 nm to 610 ± 15 nm. EDX analysis confirms the aggregation and enrichment of Al-containing phases on the surface of cycled Al (2 nm)–CaO_hollow, in particular from cycle number 10 onwards (Fig. 6b). The segregation and enrichment of Al-containing phases on the surface was also observed in the benchmark materials (Fig. 6c). The surface migration of the stabilizer (Al2O3 or Ca–Al mixed oxides) probably triggeried the sintering of the material.83 | ||
| Fig. 6 Morphological characterization of the cycled CO2 sorbents: (a) SEM images of freshly calcined and cycled CO2 sorbents. (b) HAADF-STEM with EDX analysis of Al (2 nm)–CaO_hollow after carbonation as a function of cycle number. (c) HAADF-STEM with EDX analysis of cycled benchmark materials. The molar ratio of Al in the benchmark materials is 10 mol% as determined by ICP-OES. All of the cycled benchmark materials are in the calcined state. | ||
Sorption-enhanced water-gas shift reaction of bi-functional Ni–CaO catalyst
CaO-based CO2 sorbents have the potential to be incorporated in catalytic processes, yet the possible interaction between the catalyst and the CO2 sorbent (both stabilizer and CaO) with respect to the formation of a catalytically inactive solid solution needs to be taken into consideration. Here, a bi-functional catalyst-sorbent was prepared by impregnating Ni onto the most promising CO2 sorbent, i.e., Al (2 nm)–CaO_hollow, and Al (0 nm)–CaO_hollow for reference. The large miscibility gap owing to an appreciable difference in the effective ionic radii of Ca2+ (100 pm) and Ni2+ (69 pm) in an octahedral coordination84 restricts the formation of a CaO–NiO solid solution.85 Concerning the interaction between Ni and Al2O3 under high-temperature calcination conditions NiAl2O4 can form.86 The bi-functional material was characterized in detailed after calcination at 900 °C in air. H2-TPR of both Ni/Al (2 nm)–CaO_hollow and Ni/Al (0 nm)–CaO_hollow showed a single reduction peak at 460–470 °C (Fig. 7a). This low-temperature reduction is ascribed to the reduction of α-type NiO that has only a weak interaction with the support.87,88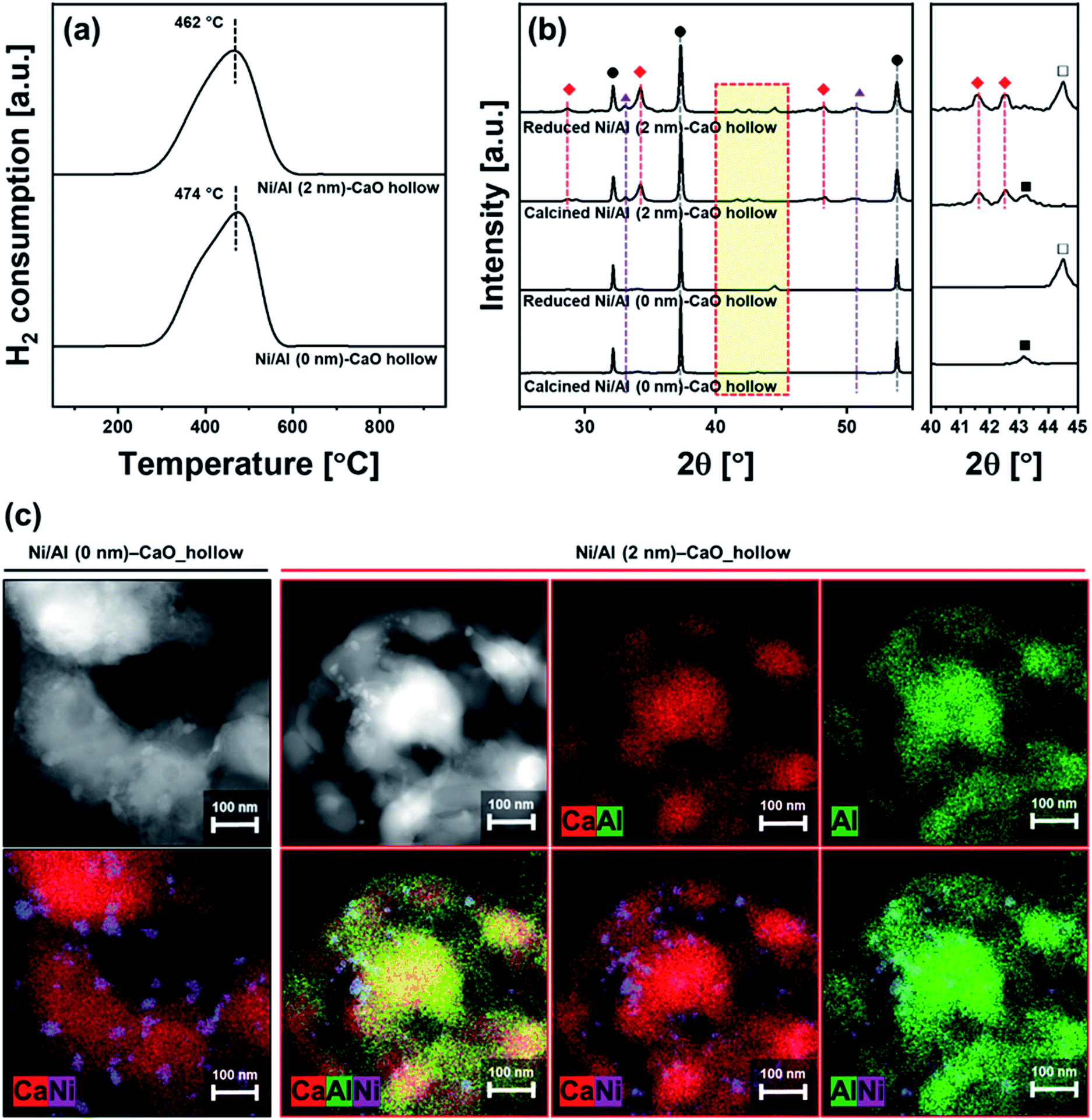 | ||
| Fig. 7 Characterization of bi-functional Ni/Al (0 nm)–CaO_hollow and Ni/Al (2 nm)–CaO_hollow: (a) H2-TPR, (b) XRD diffractograms of the calcined and reduced materials. The symbols represent (●) CaO, (◆) Ca3Al2O6, (▲) Ca(OH)2, (■) NiO, and (□) Ni. (c) HAADF-STEM images of the reduced materials coupled with EDX mappings of Ca, Ni, and Al. | ||
The chemical composition and structure of the calcined and reduced materials were characterized further by XRD. The following phases were identified in calcined Ni/Al (0 nm)–CaO_hollow and Ni/Al (2 nm)–CaO_hollow: NiO, CaO and Ca(OH)2. In Ni/Al (2 nm)–CaO_hollow, also Ca3Al2O6 was detected by XRD. After reduction, Bragg reflections due to Ni were observed in both Ni-containing materials (Fig. 7b). XRD did not provide any evidence that Ni interacted with CaO or Ca3Al2O6, which is in agreement with the findings of the H2-TPR experiments.
The reduced materials were visualized by HAADF-STEM coupled with EDX analysis (Fig. 7c). After reduction, Ni nanoparticles with a diameter of 11 ± 3.7 nm and 9.7 ± 2.2 nm were observed on Ni/Al (0 nm)–CaO_hollow and Ni/Al (2 nm)–CaO_hollow, respectively. It is important to note that the hollow-sphere structure of Ni/Al (2 nm)–CaO and the homogeneous distribution of Ca and Al were preserved over wet impregnation, calcination and reduction. However, in the absence of the stabilizer Al2O3, i.e., Ni/Al (0 nm)–CaO_hollow, the hollow structure collapsed.
The catalytic activity of the bi-functional materials for the cyclic SE-WGS reaction was assessed in a fixed-bed reactor at 400 °C using a total flow rate of 70 ml min−1 (5 ml min−1 CO, 20 ml min−1 H2O and 45 ml min−1 N2; GHSV = 16.8 L g−1 h−1 of GHSV). After 50 min of time on stream (TOS), the material was heated to 800 °C for regeneration (15 min in 50 ml min−1 of N2). Fig. 8a plots the mole fractions of H2, CH4, CO and CO2 (dry, N2-free basis) measured in the effluent stream during the first cycle. As the CO2 capture part is a transient process due to the finite CO2 capture capacity of the bifunctional material, the overall reaction proceeds in three distinct stages: (i) pre-breakthrough stage, producing high-purity H2 with full CO conversion; (ii) breakthrough stage, reducing H2 purity owing to the decreasing CO2 uptake capacity (and the kinetics) of the sorbent; and (iii) post-breakthrough stage, yielding an off-gas composition close to the conventional WGS process as the CO2 sorbent has reached its full capacity. High-purity H2 that requires hardly any additional purification steps is produced only in the pre-breakthrough stage. Hence, a material yielding a long pre-breakthrough period is highly desirable. Using Ni/Al (0 nm)–CaO_hollow, the effluent gas contained 98% H2 in the pre-breakthrough stage (t = 0–29 min), with negligible quantities of CH4, CO2 and CO, yielding 100% CO conversion and 25.3 mmol g−1 of high-purity H2. This observation confirms that the carbonation reaction reaches equilibrium rapidly, leading to an almost complete uptake of the CO2 produced in the WGS. In the breakthrough stage (t = 29–40 min), the molar fraction of H2 decreased gradually, whereas the mole fraction of CO2 increased. This was due to fact that the carbonation reaction transitioned into the slow, diffusion-limited reaction regime, limiting the uptake of CO2 in the given residence time of the reactor. At the end of the breakthrough stage, the mole fractions of H2 and CO2 were 0.45 and the mole fraction of CO increased from <0.01 to 0.06. The CO conversion was reduced from ≈100% to 94%. For Ni/Al (2 nm)–CaO_hollow, the overall trend was similar to Ni/Al (0 nm)–CaO_hollow; yet, the duration of the pre-breakthrough stage was shorter (t = 0–24 min) when compared to Ni/Al (0 nm)–CaO_hollow (t = 0–29 min). The CO conversion and H2 yield in the pre-breakthrough of Ni/Al (2 nm)–CaO_hollow were 100% and 21.0 mmol g−1, respectively. This was due to the formation of Ca3Al2O6 in Ni/Al (2 nm)–CaO_hollow, which reduced the quantity of CO2-capture-active CaO in the material (75%). Concerning the cyclic stability of the bi-functional materials, we observed that the duration of the pre-breakthrough period gradually decreased with number of repeated SE-WGS and regeneration cycles (Fig. 8b). Nevertheless, Ni/Al (2 nm)–CaO_hollow exhibited a good cyclic stability (reduction of the duration of the pre-breakthrough period from 24 to 21 min only over ten cycles), whereas the pre-breakthrough period of Ni/Al (0 nm)–CaO_hollow reduced from 29 min to 10 min. Indeed, the cyclic CO2 capture capacity of Ni/Al (0 nm)–CaO_hollow and Ni/Al (2 nm)–CaO_hollow measured during SE-WGS was in very good agreement with the values determined in a TGA under SE-WGS-mimicking conditions (carbonation at 400 °C using 70 ml min−1 of 20 vol% CO2 in N2 and regeneration at 800 °C in 70 ml min−1 N2; Fig. 8c). However, the CO2 uptake under SE-WGS conditions (carbonation a 400 °C) is lower than the value determined under CO2 capture conditions (carbonation at 650 °C), most likely due to slower carbonation kinetics. Concerning the window of the reaction temperature of the SE-WGS (∼400 °C), MgO-based CO2 sorbents might be more favorable.51,89 The particle size of Ni in Ni/Al (0 nm)–CaO_hollow and Ni/Al (2 nm)–CaO_hollow increased by a factor of, respectively, 3.9 (43 ± 7 nm) and 2.5 (24 ± 5 nm) over ten cycles of the SE-WGS (Fig. S9†). Nonetheless, both materials maintained a high CO conversion during pre-breakthrough (≈100%) and post-breakthrough (≈94%) over the ten cycles tested. A significant reduction was observed in the pore volume of Ni/Al (0 nm)–CaCO3_hollow from 0.175 cm3 gsorbent−1 to 0.021 cm3 gsorbent−1 after ten cycles of the SE-WGS and regeneration, whereas Ni/Al (0 nm)–CaCO3_hollow possessed a much higher pore volume of 0.120 cm3 gsorbent−1 (Table S2†). The reduction of the available pore volume in the material is closely linked to the decay of the CO2 uptake, leading to a poor SE-WGS stability of Ni/Al (0 nm)–CaO_hollow.
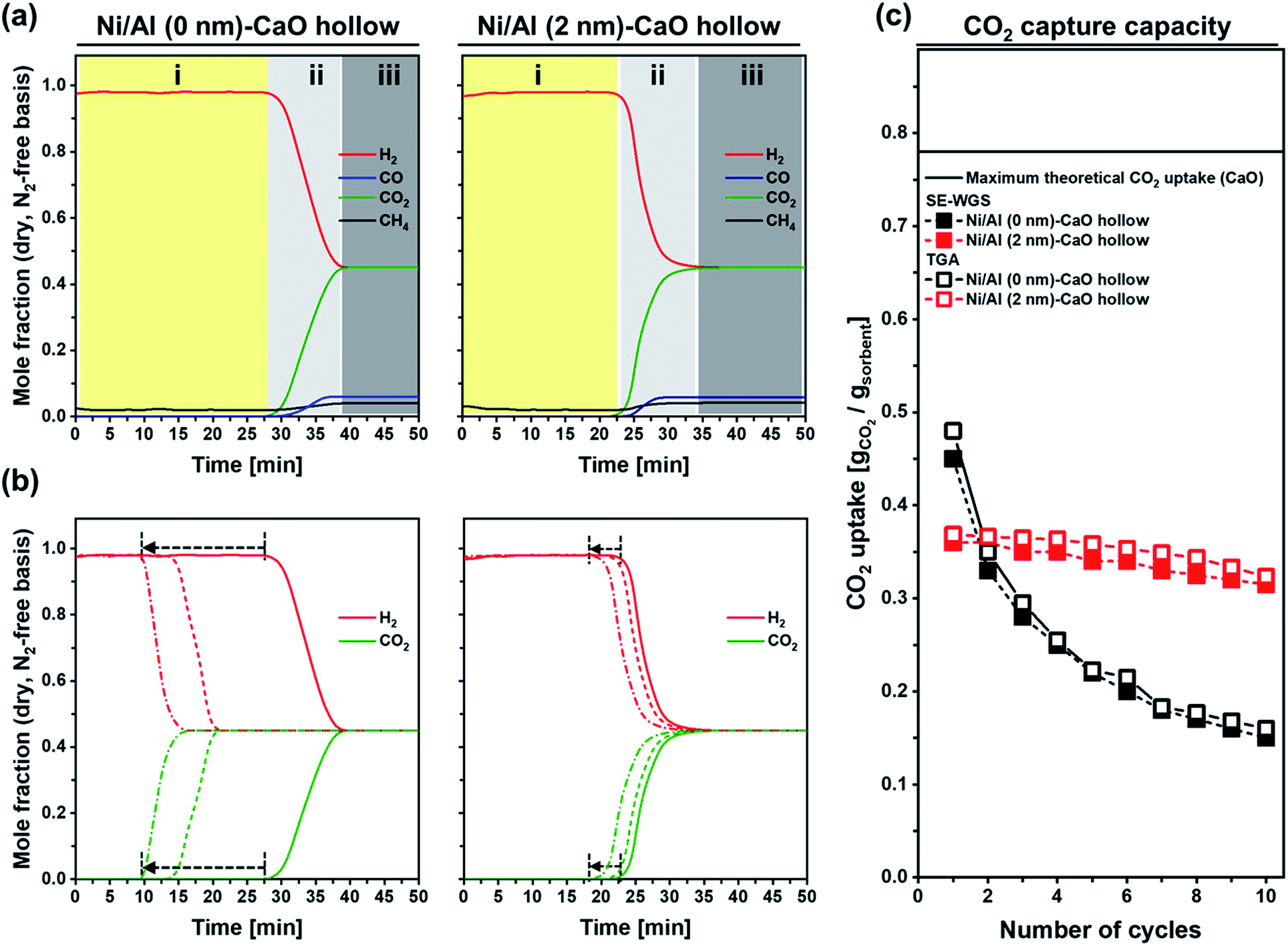 | ||
| Fig. 8 SE-WGS performance of Ni/Al (0 nm)–CaO_hollow and Ni/Al (2 nm)–CaO_hollow: (a) composition of the effluent during the 1st cycle. Three reaction stages are observed: (i) pre-breakthrough, (ii) breakthrough, and (iii) post-breakthrough. (b) Breakthrough curves of H2 (red) and CO2 (green) during the 1st (solid line), 5th (dashed line) and 10th (dotted line) cycle. The arrows indicate the shift of breakthrough with cycle number. (c) CO2 uptake as determined in a TGA and during SE-WGS conditions. The solid line gives the maximal theoretical CO2 uptake of pure CaO. | ||
The SE-WGS performance of Ni/Al (2 nm)–CaO_hollow was investigated further in the kinetic regime, i.e., far from the equilibrium CO conversion, using different steam-to-carbon (S/C) ratios and different temperatures (400–600 °C). When increasing the S/C ratio from 1 to 4 an improved WGS performance was observed, i.e., an increase in the CO conversion from 40.5% to 67.4% in the CO2-free pre-breakthrough stage (Fig. 9a). Furthermore, the CO conversion was increased from 55.8% to 69.5% when the temperature was increased from 400 °C to 500 °C under a total flow rate of 120 ml min−1 (10 ml min−1 CO, 20 ml min−1 H2O and 90 ml min−1 N2; GHSV = 28.8 L gcat−1 h−1). A further increase of the temperature to 600 °C resulted in a decrease of the CO conversion to 61.3% (Fig. 9b). The reduced CO conversion is mostly ascribed to the equilibrium limitation of the SE-WGS above 600 °C (Fig. 9b). The H2 yield in the CO2-free pre-breakthrough stage showed an insignificant change with S/C ratio (Fig. 9a), but increased when the temperature was increased from 400 °C to 600 °C (Fig. 9b). This increase is linked to the increase of the CO2 capture capacity of the sorbent from 0.36 gCO2 gsorbent−1 at 400 °C to 0.53 gCO2 gsorbent−1 at 600 °C (Fig. 9c). These findings are in line with the linear correlation of the H2 yield with CO2 uptake in the pre-breakthrough stage (Fig. 9d). It is noteworthy that the effective CO2 uptake capacity of Ni/Al (2 nm)–CaO_hollow (0.36–0.53 gCO2 gsorbent−1 at 400–600 °C) outperforms alkali metal-promoted hydrotalcite and Al2O3 materials that have been studied for the SE-WGS (i.e., 0.01–0.02 gCO2 gsorbent−1).52,56–60
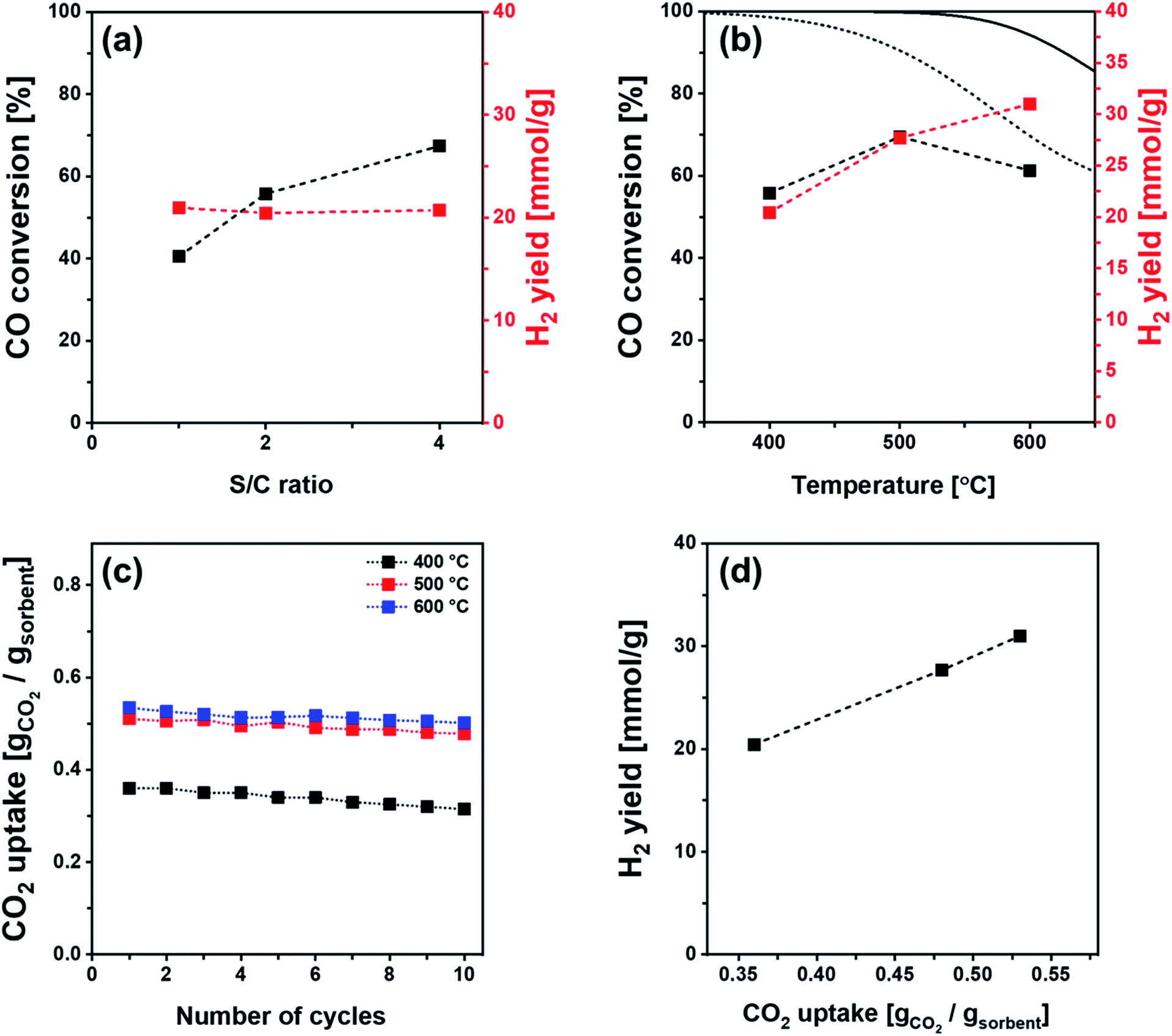 | ||
| Fig. 9 CO conversion and H2 yield of Ni/Al (2 nm)–CaO_hollow in the pre-breakthrough stage of SE-WGS as a function of (a) the steam-to-carbon ratio (400 °C using a total flow rate of 110–140 ml min−1 composed of 10 ml min−1 CO, 10–40 ml min−1 H2O and 90 ml min−1 N2; GHSV = 26.4–36.8 L gcat−1 h−1) and (b) the reaction temperature (400–600 °C using a total flow rate of 120 ml min−1 composed of 10 ml min−1 CO, 20 ml min−1 H2O and 90 ml min−1 N2; GHSV = 28.8 L gcat−1 h−1). The dotted and solid lines represent the equilibrium CO conversion of WGS and SE-WGS, respectively. (c) CO2 uptake of Ni/Al (2 nm)–CaO_hollow at different carbonation temperatures of 400, 500 and 600 °C as a function of the number of carbonation–regeneration cycles. (d) H2 yield in the pre-breakthrough stage of the SE-WGS (using a total flow rate of 120 ml min−1 composed of 10 ml min−1 CO, 20 ml min−1 H2O and 90 ml min−1 N2; GHSV = 28.8 L gcat−1 h−1) as a function of the CO2 uptake of Ni/Al (2 nm)–CaO_hollow at 400, 500 and 600 °C. | ||
The SE-WGS performance of bifunctional Ni–CaO is mostly affected by the catalytic activity for achieving a high CO conversion and on the CO2 capture capacity for obtaining a long pre-breakthrough stage during which high-purity hydrogen is produced. The development of bifunctional catalyst-sorbents thus aims at reducing the quantity of catalyst and stabilizer in the material while providing still a high CO conversion and an effective stabilization of the CaO against sintering. Utilizing inert stabilizers that do not form mixed oxides with CaO under the relevant operation conditions, such as Y2O3, MgO and ZnO27–31 typically allows for the production of materials with higher amounts of CaO, active for CO2 capture. For higher CO conversions, the development of Ni-based catalysts forming alloys with Cu, Fe, Co and Zn, or with an improved Ni dispersion on the surface have shown promising results and could thus be considered in the further improvement of bifunctional catalyst-sorbent.64
Conclusions
To summarize, we have employed atomic layer deposition (ALD) to deposit conformal layers of Al2O3 onto different structures of CaCO3, i.e., hollow CaCO3 spheres, natural limestone, bulk and nanoparticle-sized CaCO3. The interaction of the ALD-grown layer of Al2O3 with CaO resulted in the formation of CO2 capture-inactive Ca3Al2O6. Ca3Al2O6 acted as a stabilizer to maintain the original CaCO3 structure during calcination, in particular for hollow spheres. We found that the CO2 uptake capacity depends critically on the structure of the sorbent, whereby the volume available in pores with dpore < 100 nm dictates the CaO conversion in the kinetically controlled carbonation regime. Among the sorbents tested, hollow CaCO3 spheres coated with a 2 nm thick layer of Al2O3 yielded the highest CO2 uptake over 30 cycles. Functionalizing such hollow CaCO3 spheres with Ni nanoparticles lead to an effective bi-functional material for the sorption-enhanced water-gas shift reaction (SE-WGS). The newly developed bi-functional material showed a cyclically stable pre-breakthrough period in which high-purity hydrogen was produced with minimal CO slip.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge ETH (ETH 57 12-2) and the Swiss National Science Foundation (200020_156015) for financial support. We would like to thank the Scientific Center for Optical and Electron Microscopy (ScopeM) and the Center for Micro- and Nanoscience (FIRST) at ETH Zurich for providing training and access to electron microscopes.References
- IPCC, Climate Change 2014: Synthesis Report, The Intergovernmental Panel on Climate Change, IPCC, Geneva, 2014 Search PubMed.
- E. Levina, S. Bennett and S. McCoy, Technology Roadmap: Carbon capture and storage, IEA, Paris, 2013 Search PubMed.
- IEA, Energy Technology Perspectives 2016: Towards Sustainable Urban Energy Systems, IEA, Paris, 2016 Search PubMed.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- S. M. Kim, P. M. Abdala, T. Margossian, D. Hosseini, L. Foppa, A. Armutlulu, W. van Beek, A. Comas-Vives, C. Copéret and C. Müller, J. Am. Chem. Soc., 2017, 139, 1937–1949 CrossRef CAS PubMed.
- S. M. Kim, P. M. Abdala, M. Broda, D. Hosseini, C. Copéret and C. Müller, ACS Catal., 2018, 8, 2815–2823 CrossRef CAS.
- M. Zhao, A. I. Minett and A. T. Harris, Energy Environ. Sci., 2013, 6, 25–40 RSC.
- P. Fennell, in Calcium and Chemical Looping Technology for Power Generation and Carbon Dioxide (CO2) Capture, ed. P. Fennell and B. Anthony, Woodhead Publishing, Cambridge, 2015, pp. 39–48 Search PubMed.
- N. D. Hutson, S. A. Speakman and E. A. Payzant, Chem. Mater., 2004, 16, 4135–4143 CrossRef CAS.
- T.-H. Bae, M. R. Hudson, J. A. Mason, W. L. Queen, J. J. Dutton, K. Sumida, K. J. Micklash, S. S. Kaye, C. M. Brown and J. R. Long, Energy Environ. Sci., 2013, 6, 128–138 RSC.
- M. M. Lozinska, E. Mangano, J. P. Mowat, A. M. Shepherd, R. F. Howe, S. P. Thompson, J. E. Parker, S. Brandani and P. A. Wright, J. Am. Chem. Soc., 2012, 134, 17628–17642 CrossRef CAS.
- T. Tetsuo, F. Tsuyoshi, T. Yasushi and S. Takeo, Chem. Lett., 1992, 21, 2161–2164 CrossRef.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS.
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS.
- A. O. z. r. Yazaydın, R. Q. Snurr, T.-H. Park, K. Koh, J. Liu, M. D. LeVan, A. I. Benin, P. Jakubczak, M. Lanuza and D. B. Galloway, J. Am. Chem. Soc., 2009, 131, 18198–18199 CrossRef.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef.
- A. Perejón, L. M. Romeo, Y. Lara, P. Lisbona, A. Martínez and J. M. Valverde, Appl. Energy, 2016, 162, 787–807 CrossRef.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- S. Tian, J. Jiang, Z. Zhang and V. Manovic, Nat. Commun., 2018, 9, 4422 CrossRef PubMed.
- R. Barker, J. Appl. Chem. Biotechnol., 1973, 23, 733–742 CrossRef CAS.
- M. Broda and C. R. Müller, Fuel, 2014, 127, 94–100 CrossRef CAS.
- M. Broda and C. R. Müller, Adv. Mater., 2012, 24, 3059–3064 CrossRef CAS.
- A. Armutlulu, M. A. Naeem, H.-J. Liu, S. M. Kim, A. Kierzkowska, A. Fedorov and C. R. Müller, Adv. Mater., 2017, 29, 1702896 CrossRef PubMed.
- H. J. Yoon and K. B. Lee, Chem. Eng. J., 2019, 355, 850–857 CrossRef CAS.
- R. Filitz, A. M. Kierzkowska, M. Broda and C. R. Müller, Environ. Sci. Technol., 2012, 46, 559–565 CrossRef CAS PubMed.
- M. A. Naeem, A. Armutlulu, Q. Imtiaz, F. Donat, R. Schäublin, A. Kierzkowska and C. R. Müller, Nat. Commun., 2018, 9, 2408 CrossRef.
- H. Liu and S. Wu, Energy Fuels, 2019, 33, 7626–7633 CrossRef CAS.
- V. S. Derevschikov, A. I. Lysikov and A. G. Okunev, Ind. Eng. Chem. Res., 2011, 50, 12741–12749 CrossRef CAS.
- X. Zhang, Z. Li, Y. Peng, W. Su, X. Sun and J. Li, Chem. Eng. J., 2014, 243, 297–304 CrossRef CAS.
- D. Alvarez and J. C. Abanades, Ind. Eng. Chem. Res., 2005, 44, 5608–5615 CrossRef CAS.
- J. S. Dennis and R. Pacciani, Chem. Eng. Sci., 2009, 64, 2147–2157 CrossRef CAS.
- N. H. Florin, J. Blamey and P. S. Fennell, Energy Fuels, 2010, 24, 4598–4604 CrossRef CAS.
- F.-Q. Liu, W.-H. Li, B.-C. Liu and R.-X. Li, J. Mater. Chem. A, 2013, 1, 8037–8044 RSC.
- S. F. Wu, Q. H. Li, J. N. Kim and K. B. Yi, Ind. Eng. Chem. Res., 2008, 47, 180–184 CrossRef CAS.
- W. Liu, B. Feng, Y. Wu, G. Wang, J. Barry and J. C. Diniz da Costa, Environ. Sci. Technol., 2010, 44, 3093–3097 CrossRef CAS PubMed.
- L. Li, D. L. King, Z. Nie and C. Howard, Ind. Eng. Chem. Res., 2009, 48, 10604–10613 CrossRef CAS.
- N. Phalak, N. Deshpande and L.-S. Fan, Energy Fuels, 2012, 26, 3903–3909 CrossRef CAS.
- R. Koirala, G. K. Reddy and P. G. Smirniotis, Energy Fuels, 2012, 26, 3103–3109 CrossRef CAS.
- R. Pacciani, C. Müller, J. Davidson, J. Dennis and A. Hayhurst, Can. J. Chem. Eng., 2008, 86, 356–366 CrossRef CAS.
- M. Broda, A. M. Kierzkowska and C. R. Müller, ChemSusChem, 2012, 5, 411–418 CrossRef CAS PubMed.
- A. M. Kierzkowska and C. R. Müller, ChemPlusChem, 2013, 78, 92–100 CrossRef CAS.
- S. M. Kim, A. M. Kierzkowska, M. Broda and C. R. Müller, Energy Procedia, 2017, 114, 220–229 CrossRef CAS.
- R. Han, J. Gao, S. Wei, Y. Su and Y. Qin, J. Mater. Chem. A, 2018, 6, 3462–3470 RSC.
- C. Zhao, Z. Zhou, Z. Cheng and X. Fang, Appl. Catal., B, 2016, 196, 16–26 CrossRef CAS.
- M. Shokrollahi Yancheshmeh, H. R. Radfarnia and M. C. Iliuta, Chem. Eng. J., 2016, 283, 420–444 CrossRef CAS.
- C. Wang, Y. Chen, Z. Cheng, X. Luo, L. Jia, M. Song, B. Jiang and B. Dou, Energy Fuels, 2015, 29, 7408–7418 CrossRef CAS.
- C. Dang, H. Wang, H. Yu and F. Peng, Appl. Catal., A, 2017, 533, 9–16 CrossRef CAS.
- M. Shokrollahi Yancheshmeh, H. R. Radfarnia and M. C. Iliuta, ACS Sustainable Chem. Eng., 2017, 5, 9774–9786 CrossRef CAS.
- K. Wang, Y. Zhao, P. T. Clough, P. Zhao and E. J. Anthony, Chem. Eng. J., 2019, 372, 886–895 CrossRef CAS.
- T. Noor, M. V. Gil and D. Chen, Appl. Catal., B, 2014, 150–151, 585–595 CrossRef CAS.
- F. Ni and H. S. Caram, AlChE J, 2017, 63, 5452–5461 CrossRef CAS.
- K. Coenen, F. Gallucci, P. Cobden, E. van Dijk, E. Hensen and M. van Sint Annaland, Chem. Eng. J., 2016, 293, 9–23 CrossRef CAS.
- S. G. Mayorga, J. R. Hufton, S. Sircar and T. Gaffney, Sorption enhanced reaction process for production of hydrogen, Phase 1 final report, Report DOE/GO/10059-T1, Air Products and Chemicals, Inc., Allentown, PA 1997 Search PubMed.
- Z. Yong, V. Mata and A. r. E. Rodrigues, Sep. Purif. Technol., 2002, 26, 195–205 CrossRef CAS.
- C. H. Lee and K. B. Lee, Appl. Energy, 2017, 205, 316–322 CrossRef CAS.
- H. M. Jang, K. B. Lee, H. S. Caram and S. Sircar, Chem. Eng. Sci., 2012, 73, 431–438 CrossRef CAS.
- E. Ochoa-Fernández, C. Lacalle-Vilà, T. Zhao, M. Rønning and D. Chen, in Stud. Surf. Sci. Catal., ed. F. Bellot Noronha, M. Schmal and E. Falabella Sousa-Aguiar, Elsevier, 2007, vol. 167, pp. 159–164 Search PubMed.
- P. D. Cobden, P. van Beurden, H. T. J. Reijers, G. D. Elzinga, S. C. A. Kluiters, J. W. Dijkstra, D. Jansen and R. W. van den Brink, Int. J. Greenhouse Gas Control, 2007, 1, 170–179 CrossRef CAS.
- C. Wheeler, A. Jhalani, E. J. Klein, S. Tummala and L. D. Schmidt, J. Catal., 2004, 223, 191–199 CrossRef CAS.
- M. L. Ang, U. Oemar, E. T. Saw, L. Mo, Y. Kathiraser, B. H. Chia and S. Kawi, ACS Catal., 2014, 4, 3237–3248 CrossRef CAS.
- K.-R. Hwang, C.-B. Lee and J.-S. Park, J. Power Sources, 2011, 196, 1349–1352 CrossRef CAS.
- J. Ashok, M. H. Wai and S. Kawi, ChemCatChem, 2018, 10, 3927–3942 CrossRef CAS.
- C. Han and D. P. Harrison, Chem. Eng. Sci., 1994, 49, 5875–5883 CrossRef CAS.
- Y. Liu, Z. Li, L. Xu and N. Cai, Ind. Eng. Chem. Res., 2012, 51, 11989–11997 CrossRef CAS.
- C. Rhodes, B. P. Williams, F. King and G. J. Hutchings, Catal. Commun., 2002, 3, 381–384 CrossRef CAS.
- D. S. Newsome, Catal. Rev.: Sci. Eng., 1980, 21, 275–318 CrossRef CAS.
- C. Rhodes, G. J. Hutchings and A. M. Ward, Catal. Today, 1995, 23, 43–58 CrossRef CAS.
- A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, J. Am. Chem. Soc., 2008, 130, 1402–1414 CrossRef CAS PubMed.
- D.-W. Lee, M. S. Lee, J. Y. Lee, S. Kim, H.-J. Eom, D. J. Moon and K.-Y. Lee, Catal. Today, 2013, 210, 2–9 CrossRef CAS.
- D. C. Grenoble, M. M. Estadt and D. F. Ollis, J. Catal., 1981, 67, 90–102 CrossRef CAS.
- M. S. Yancheshmeh, H. R. Radfarnia and M. C. Iliuta, Chem. Eng. J., 2016, 283, 420–444 CrossRef.
- B. Dou, C. Wang, Y. Song, H. Chen, B. Jiang, M. Yang and Y. Xu, Renewable Sustainable Energy Rev., 2016, 53, 536–546 CrossRef CAS.
- T. Noor, Doctoral thesis, Norwegian University of Science and Technology, 2013 Search PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- R. Pacciani, C. R. Müller, J. F. Davidson, J. S. Dennis and A. N. Hayhurst, Can. J. Chem. Eng., 2008, 86, 356–366 CrossRef CAS.
- S. I. Kuriyavar, R. Vetrivel, S. G. Hegde, A. V. Ramaswamy, D. Chakrabarty and S. Mahapatra, J. Mater. Chem., 2000, 10, 1835–1840 RSC.
- N. S. Yüzbasi, A. Armutlulu, P. M. Abdala and C. R. Müller, ACS Appl. Mater. Interfaces, 2018, 10, 37994–38005 CrossRef PubMed.
- A. D. Smigelskas and E. O. Kirkendall, Trans. Aime, 1947, 171, 130–142 Search PubMed.
- P. Y. Hou, in Reference Module in Materials Science and Materials Engineering, Elsevier, 2016 Search PubMed.
- B. Pieraggi, in Developments in High Temperature Corrosion and Protection of Materials, ed. W. Gao and Z. Li, Woodhead Publishing, 2008, pp. 9–35 Search PubMed.
- S. M. Kim, W.-C. Liao, A. M. Kierzkowska, T. Margossian, D. Hosseini, S. Yoon, M. Broda, C. Copéret and C. R. Müller, Chem. Mater., 2018, 30, 1344–1352 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- V. Prostakova, J. Chen, E. Jak and S. A. Decterov, CALPHAD: Comput. Coupling Phase Diagrams Thermochem., 2012, 37, 1–10 CrossRef CAS.
- L. Zhou, L. Li, N. Wei, J. Li and J.-M. Basset, ChemCatChem, 2015, 7, 2508–2516 CrossRef CAS.
- Y. Xu, H. Long, Q. Wei, X. Zhang, S. Shang, X. Dai and Y. Yin, Catal. Today, 2013, 211, 114–119 CrossRef CAS.
- D. Hu, J. Gao, Y. Ping, L. Jia, P. Gunawan, Z. Zhong, G. Xu, F. Gu and F. Su, Ind. Eng. Chem. Res., 2012, 51, 4875–4886 CrossRef CAS.
- Y. Hu, H. Cui, Z. Cheng and Z. Zhou, Chem. Eng. J., 2018, 377, 119823 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00619b |
| This journal is © The Royal Society of Chemistry 2020 |