
Hybrid photoanodes for water oxidation combining a molecular photosensitizer with a metal oxide oxygen-evolving catalyst
Marie-Noëlle
Collomb
 *a,
Daniela V.
Morales
a,
Catalina N.
Astudillo
a,
Baptiste
Dautreppe
ab and
Jérôme
Fortage
*a
*a,
Daniela V.
Morales
a,
Catalina N.
Astudillo
a,
Baptiste
Dautreppe
ab and
Jérôme
Fortage
*a
aUniv. Grenoble Alpes, CNRS, DCM, 38000 Grenoble, France. E-mail: marie-noelle.collomb@univ-grenoble-alpes.fr; jerome.fortage@univ-grenoble-alpes.fr
bUniv. Grenoble Alpes, CNRS, CEA, IRIG, SyMMES, 38000 Grenoble, France
First published on 30th September 2019
Abstract
Visible light-driven water splitting performed within a photoelectrochemical cell (PEC) is one of the most investigated processes to produce hydrogen in a sustainable manner. Developing a photoanode for the water oxidation reaction (oxygen evolving reaction, OER) is considered more challenging than designing a photocathode for the water reduction reaction (hydrogen evolving reaction, HER) because the OER requires a higher overpotential and displays a higher activation barrier. Since the work of Fujishima and Honda in 1972, intense research has been devoted to the development of photoelectrodes solely based on inorganic semiconductor materials. Nevertheless, the large majority of photoanodes using inorganic semiconductors weakly absorb in the visible spectrum and present poor long-term stability. An alternative design, more recently explored, is to decouple the light absorption and the catalytic function by adsorbing a molecular photosensitizer on an n-type semiconducting electrode (generally TiO2), to promote visible light absorption, and an oxygen-evolving catalyst to promote the OER. Herein, we provide a comprehensive review of such hybrid photoanodes that associate with a molecular photosensitizer and an oxygen-evolving catalyst based on metal oxide nanoparticles. For these hybrid photoanodes, tris(bipyridine) ruthenium complexes and organic dyes such as perylenes, free-base porphyrins, polyheptazine and π-conjugated naphthalene benzimidazole polymers have been employed as molecular photosensitizers in combination with IrOx or CoOx OER catalysts. The preparation of these photoanodes, the evaluation of their photocatalytic performance for the OER, and the key factors that govern their efficiency and stability are highlighted.
Introduction
Light-driven water splitting into molecular hydrogen and oxygen has been the subject of extensive research since it is considered as a sustainable way to produce the “clean” fuel hydrogen, one of the best alternatives to fossil fuels.1–15 Since the pioneering work of Fujishima and Honda16 in the early 70's, demonstrating photoelectrochemical water splitting with a TiO2 photoanode and a Pt counter electrode under UV light, the construction of photoelectrochemical cells (PECs) to perform light-driven water splitting has been extensively explored.13 Three main configurations of water-splitting PECs were reported and have been extensively discussed in several reviews:1,2,6,7,17,18 (i) cells where the two compartments containing respectively the photoanode for water oxidation to oxygen (oxygen evolving reaction, denoted as OER) and the photocathode for water reduction to hydrogen (hydrogen evolving reaction, denoted as HER) are separated by a membrane or a frit (the two photoelectrodes being connected by a wire), (ii) cells where the photoanode and the photocathode are joined back to back via an ohmic contact, delimiting the boundary between the two compartments (i.e. a wireless design), and (iii) cells where either a p–n semiconductor junction layer is introduced between a photoanode and a cathode or two p–n junction layers are arranged in series between an anode and a cathode (respectively denoted as photoanode/photovoltaic (PV)–PEC and dual PV–PEC). Among these configurations, tandem PECs combining two photoelectrodes are very attractive since they have the advantage of absorbing over a wide range of wavelengths of the visible spectrum and thus presenting a realistic theoretical solar-to-hydrogen (STH) conversion efficiency in the order of 15% (ref. 2 and 19) (provided that the photosensitive material of each photoelectrode absorbs in wavelength domains that are different from each other and complementary). In practice and to simplify their study, photoanodes and photocathodes are often optimized separately and investigated along with a simple platinum counter electrode, requiring therefore the supply of electricity (i.e. a bias) to operate.Intense research is devoted to the development of photoelectrodes solely based on inorganic semiconductor (SC) materials,2,7,11,15 but they face the difficulty of having a SC that can both effectively absorb visible light and display conduction and valence bands (denoted as CB and VB, respectively) with adequate energy levels to catalyze the OER for photoanodes and the HER for photocathodes. Some PECs using SCs based on metal oxides (WO3, BiVO4, Fe2O3 and CuBi2O4)4,7,20–24 or oxynitrides (TaON, LaTiO2N and SrNbO2N)25–28 for the photoanodes and p-type chalcogenides (CdTe and CuIn1−xGaxSe2)29,30 or metal oxides (p-Cu2O)31–34 for the photocathodes do operate under visible light. Nevertheless, their STH conversion efficiency remains below 10%, the threshold for commercial applications. A few PECs using InP, GaAs/GaInP2 and AlGaAs/Si SCs can exceed 10% STH conversion efficiency under visible irradiation, but they suffer from poor stability and employ In and Ga which are rare and expensive metals.35–38
An alternative approach is to design hybrid systems by coupling SCs with a molecular photosensitizer (denoted as PS and also called dye) for visible light absorption and an oxygen- and hydrogen-evolving catalyst to promote the OER or HER.4,9,10,12–14,39–42 This molecular approach for the construction of water splitting PEC cells (or dye-sensitized-PECs, denoted as DS-PECs) is an emerging line of research inspired by the technology of photovoltaic cells (i.e. dye-sensitized solar cells, denoted as DSSCs).43 Herein, the OER and HER catalysts, which can be inorganic metal-based nanoparticles (NPs) or molecular metal complexes, are either bound or co-adsorbed with the molecular PS on the SC surface.9,10,14,44,45 Within the water splitting process, the OER remains the bottleneck even when catalyzed by such metal-based catalysts, due to its slow kinetics (i.e. with a high activation barrier) and its large anodic overpotential, and is still more challenging than the HER.45
In this review article, we will focus on “hybrid photoanodes” which combine a molecular PS and a metal oxide (MOx) OER catalyst. These hybrid photoelectrodes associate the great tunability of the photophysical and electronic properties of molecular photosensitizers with the high catalytic activity and stability of inorganic catalysts. Since the seminal paper by Mallouk and co-workers published in 2009,46 which reports a semi-conducting TiO2 electrode sensitized by a trisbipyridine ruthenium complex coupled to an iridium oxide catalyst, about fifteen examples have been reported. In addition to trisbipyridine ruthenium complexes,46–53 organic photosensitizers based on polyheptazine,54–60 perylenes,61–63 π-conjugated naphthalene benzimidazole polymers64 and free-base porphyrins51 have been employed in association with IrOx or CoOx OER catalysts. Basically, these hybrid photoanodes comprise a transparent doped semi-conducting electrode (Indium Tin Oxide (ITO) or Fluoride Tin oxide (FTO)) covered with an n-type SC such as TiO2, SnO2 or WO3 on which the molecular PS is adsorbed, generally in a mono-layer, as well as the MOx (nano)particles deposited in a chemical or photoelectrochemical way.
The electrochemical potential diagram of such DS-PECs for water oxidation is schematized in Fig. 1, along with the redox potentials of SCs and organic polymers and those of molecular photosensitizers at the ground and excited states. The light absorption by the dye molecule leads to an excited state (dye*), followed by a fast injection of the excited electron into the conduction band (CB) of the nanostructured n-SC, which then migrates into its Fermi level, resulting in an oxidized dye (dye+). In the absence of a photocathode within the DS-PEC, the application of a bias between the photoanode and the cathode (generally Pt) is required in order to increase the energy of the electrons injected into the n-SC, so that they move towards the cathode via the external circuit to promote the reduction of protons to H2. If the lifetime of the charge separated state (dye+/e− in the CB of the n-SC) is sufficiently long, an electron transfer from the MOx nanoparticle catalyst to the oxidized dye occurs. Since water oxidation requires four electrons, the catalyst needs to accumulate four holes in order to oxidize water. Catalytic water oxidation is typically slow (millisecond timescale) even when catalyzed by oxide (nano)particles, and a major problem in such photoanodes is the kinetic competition with the back electron transfer reactions which occur on a submillisecond time scale. Thus, low quantum yields for water splitting – typically 1–2% – are a consequence of the fast kinetics of charge recombination, which competes effectively with the catalytic oxidation of water.14 In this review article, different hybrid photoanodes are described as a function of the nature of the molecular dye. Hybrid photoanodes employing trisbipyridine ruthenium derivatives are described first, and then those using organic dyes are examined. The preparation of the photoanodes, the evaluation of their photocatalytic performance for the OER, and the key factors that govern their efficiency and stability are highlighted. Table 1 summarizes the photocatalytic performance of these hybrid photoanodes.
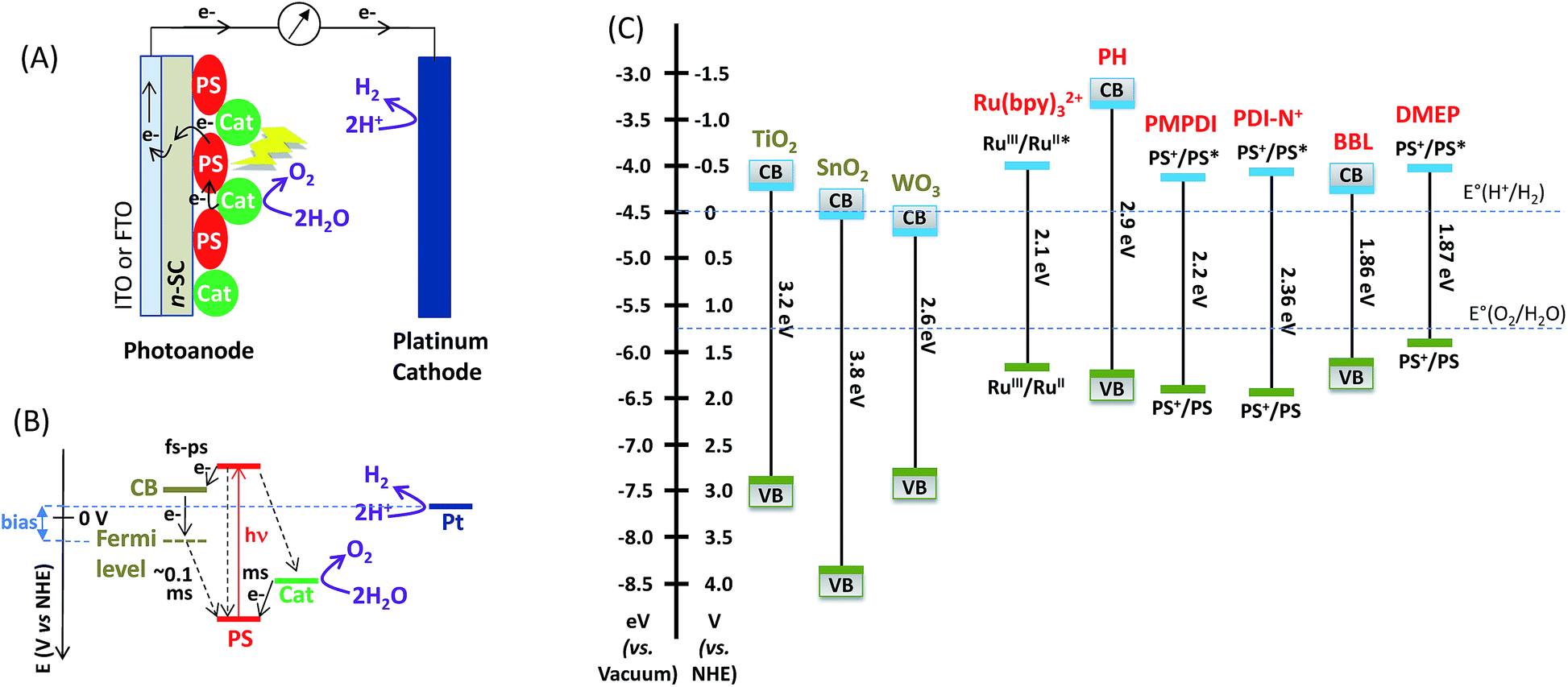 | ||
| Fig. 1 Schematic representation (A) and electrochemical potential diagram (B) of a water-splitting DS-PEC based on a “hybrid photoanode” combining a molecular photosensitizer (PS) and a metal oxide (nano)particle OER catalyst (Cat) coupled to a Pt counter-electrode. The conduction band (CB) and the Fermi level of the n-type semiconducting electrode (n-SC) are displayed. Solid and dashed lines represent respectively the forward and the back electron transfer. (C) Potential diagram of CB and CV bands of n-type SCs (TiO2, SnO2, and WO3) and organic polymers (polyheptazine (PH) and poly[benzimidazobenzophenanthroline] (BBL)), and oxidation potentials at the ground and excited states of molecular PSs: Ru(bpy)32+, N,N′-bis(phosphonomethyl)-3,4,9,10-perylenediimide (PMPDI), (N,N′-bis(2-(trimethylammonium)ethylene) perylene 3,4,9,10-tetracarboxylic acid diimide (PDI-N+), and 10-[2-(4-carboxyphenyl)ethenyl]-5,15-bis(2,4,6-trimethylphenyl)porphyrin (DMEP)). The dotted lines represent the thermodynamic potentials for water oxidation and reduction. | ||
| Entry | Substrate/SC/sensitizer/OER catalysta, mode of adsorption of the sensitizer and catalyst | Steady-state photocurrent, applied biasb, stability, experimental conditions | Faradaic efficiency for the OER | Corresponding authorsRef. |
|---|---|---|---|---|
| a Ru1–5 = tris-bipyridine ruthenium complexes with various substituents on the bipyridine ligands; BIP = benzimidazole-phenol; CEPA = 2-carboxyethylphosphonic acid; PH = poly-heptazine (g-C3N4); PMPDI = N,N′-bis(phosphonomethyl)-3,4,9,10-perylenediimide; PDI-N+ = (N,N′-bis(2-(trimethylammonium)ethylene) perylene 3,4,9,10-tetracarboxylic acid diimide); BBL = poly[benzimidazobenzophenanthroline]; DMEP = 10-[2-(4-carboxyphenyl)ethenyl]-5,15-bis(2,4,6-trimethylphenyl)porphyrin. b Potential of the applied bias has been converted to the RHE reference electrode: E(V vs. RHE) = E(V vs. Ag/AgCl 3 M KCl/NaCl) + 0.210 + 0.0592 × pH; E(V vs. RHE) = E(V vs. Ag/AgCl sat) + 0.197 + 0.0592 × pH; E(V vs. RHE) = E(V vs. SCE) + 0.241 + 0.0592 × pH. | ||||
| 1 | FTO/TiO2/Ru1–IrOx·nH2O, Ru1–IrOx assembly deposited | 10–30 μA cm−2 at 0 V vs. Ag/AgCl (0.55 V vs. RHE), stability: decayed over a period of 4 h, aqueous Na2SiF6–NaHCO3 buffer (pH 5.75), Xe lamp (>410 nm) | 20% | Mallouk46 |
| 2 | FTO/TiO2/ZrO2/Ru1–IrOx·nH2O, Ru1–IrOx assembly deposited | 40–50 μA cm−2 at 0 V vs. Ag/AgCl (0.55 V vs. RHE), stability: decreases within 40 s to stabilize at ca. 15 μA cm−2, aqueous Na2SiF6–NaHCO3 buffer (pH 5.8), Xe lamp (150 W, >410 nm) | ≈100% | Mallouk47 |
| 3 | FTO/TiO2/Ru2/BIP–IrOx·nH2O–CEPA, Co-adsorbed with a BIP mediator | 80 μA cm−2 at 0 V vs. Ag/AgCl (0.55 V vs. RHE), (25 μA cm−2 without a BIP mediator), stability: over 100 s, aqueous Na2SiF6–NaHCO3 buffer (pH 5.8), Xe lamp (150 W, >410 nm) | 85% (with or without BIP) | Mallouk48 |
| 4 | FTO/TiO2/Ru3/IrO2 sintered, successively deposited | 80 μA cm−2 at 0.1 V vs. Ag/AgCl (0.71 V vs. RHE), stability: decreases to 10 μA cm−2 after 600 s, phosphate buffer (pH 6.8), solar simulator (AM 1.5, >410 nm) | 98% | Mallouk49,50 |
| 5 | FTO/nanoITO/TiO2/Ru4/IrO2, successively deposited, FTO/nanoITO/TiO2/Ru4/TiO2/IrO2, successively deposited | 150 μA cm−2 (TiO2 6.6 nm) at 0.3 V vs. Ag/AgCl (0.84 V vs. RHE), stability: decreases to 50 μA cm−2 after 2 h, 150 μA cm−2 (TiO2 6.6 nm) at 0.3 V vs. Ag/AgCl, stability: current stable at 110 μA cm−2 over 2 h, aqueous Na2SiF6–NaHCO3 buffer (pH 5.8), LED (450 nm, 14.5 mW cm−2) | Not determined | Murray52 |
| 6 | FTO/TiO2/Ru5/Co3O4, successively deposited | 135 μA cm−2 at 0.3 V vs. Ag/AgCl (0.90 V vs. RHE), (15 μA cm−2 without Co3O4), stability: decreases to 50 μA cm−2 after 3 cycles of 20 s illumination, phosphate buffer (pH 6.8), Xe lamp (100 mW cm−2, >400 nm) | Not determined | Na53 |
| 7 | ITO or FTO/TiO2/PH/IrO2, successively deposited | 100 μA cm−2 at 0.5 V vs. Ag/AgCl (1.12 V vs. RHE), stability: over 90 min, phosphate buffer (pH 7), Xe lamp (150 W, >420 nm) | 19% | Beranek54,55 |
| 8 | ITO or FTO/TiO2/PH/Co–Pi, successively deposited, Co–Pi photoelectrochemically deposited | 190 μA cm−2 at 1.12 V vs. RHE, phosphate buffer (pH 7.0), stability: decreases to 40 μA cm−2 after 1 h 30 min, 120 μA cm−2 at 1.12 V vs. RHE, borate buffer (pH 7.7), stability: decreases to 40 μA cm−2 after 2 h, Xe lamp (150 W, >420 nm) | 17% | Beranek60 |
| 9 | ITO or FTO/TiO2/PH/CoO(OH)x, successively deposited, CoO(OH)x chemically deposited | 200 μA cm−2 at 1.12 V vs. RHE, Phosphate buffer (pH 7.0), stability: decreases to 40 μA cm−2 after 2 h 30 min, 160 μA cm−2 at 1.12 V vs. RHE, Borate buffer (pH 7.7), stability: decreases to 110 μA cm−2 after 4 h, Xe lamp (>420 nm) | 34% | Beranek60 |
| 10 | ITO/PMPDI/CoOx, successively deposited, CoOx photoelectrochemically deposited | 150 μA cm−2 at 1.0 V vs. Ag/AgCl (1.61 V vs. RHE) (η = 0.38 V), 125 μA cm−2 at 0.9 V vs. Ag/AgCl (1.51 V vs. RHE) (η = 0.28 V), stability: decreases to 100 μA cm−2 over 300 s at +0.9 V vs. Ag/AgCl, phosphate buffer (pH 7.0), Xe lamp (100 mW cm−2, 315–710 nm) | 80 ± 15% at +0.9 V vs. Ag/AgCl | Finke61 |
| 11 | FTO/SnO2/PMPDI/CoOx, successively deposited, CoOx photoelectrochemically deposited | ∼10 μA cm−2 between 0.2 and +0.9 V vs. Ag/AgCl (0.81–1.51 V vs. RHE), (40 μA cm−2 without CoOx which decreases to 20 μA cm−2 over 5 min), stability: not determined, phosphate buffer (pH 7.0), Xe lamp (100 mW cm−2, 315–710 nm) | 31 ± 7% | Finke62 |
| 12 | FTO/WO3/PDI-N+/IrO2, Co-deposited | 70 μA cm−2 at 0.55 V vs. Ag/AgCl (0.92 V vs. RHE), (∼17 μA cm−2 without IrO2), stability: not determined, H2O (pH 3.0), solar simulator (AM 1.5, >435 nm) | Not determined | Bignozzi63 |
| 13 | FTO/BBL/TiO2/Ni–Co, successively deposited | ∼30 μA cm−2 between 0.85 and 1.4 V vs. RHE, (∼15–20 μA cm−2 without Ni–Co), stability: not determined, phosphate buffer (pH 7.0), solar simulator (100 mW cm−2) | 82 ± 16% | Sivula64 |
| 14 | FTO/TiO2/DMEP/IrO2 sintered, successively deposited | ∼40 μA cm−2 at 0.1 V vs. Ag/AgCl (0.71 V vs. RHE), stability: decreases to 10 μA cm−2 after 600 s, phosphate buffer (pH 6.8), solar simulator (AM 1.5, >410 nm or >590 nm) | 102 ± 5% | Mallouk51 |
Hybrid photoanodes with a Ru-trisbipyridine derivative as a photosensitizer
The first example of a hybrid photoanode using a ruthenium tris-bipyridine derivative as a photosensitizer was published by Mallouk and coworkers46 in 2009. This group designed a heteroleptic trisbipyridine ruthenium complex (denoted as Ru1) with one bipyridine substituted by two phosphonate groups and another one by a bidentate carboxylic acid group (i.e. malonate) (Fig. 2).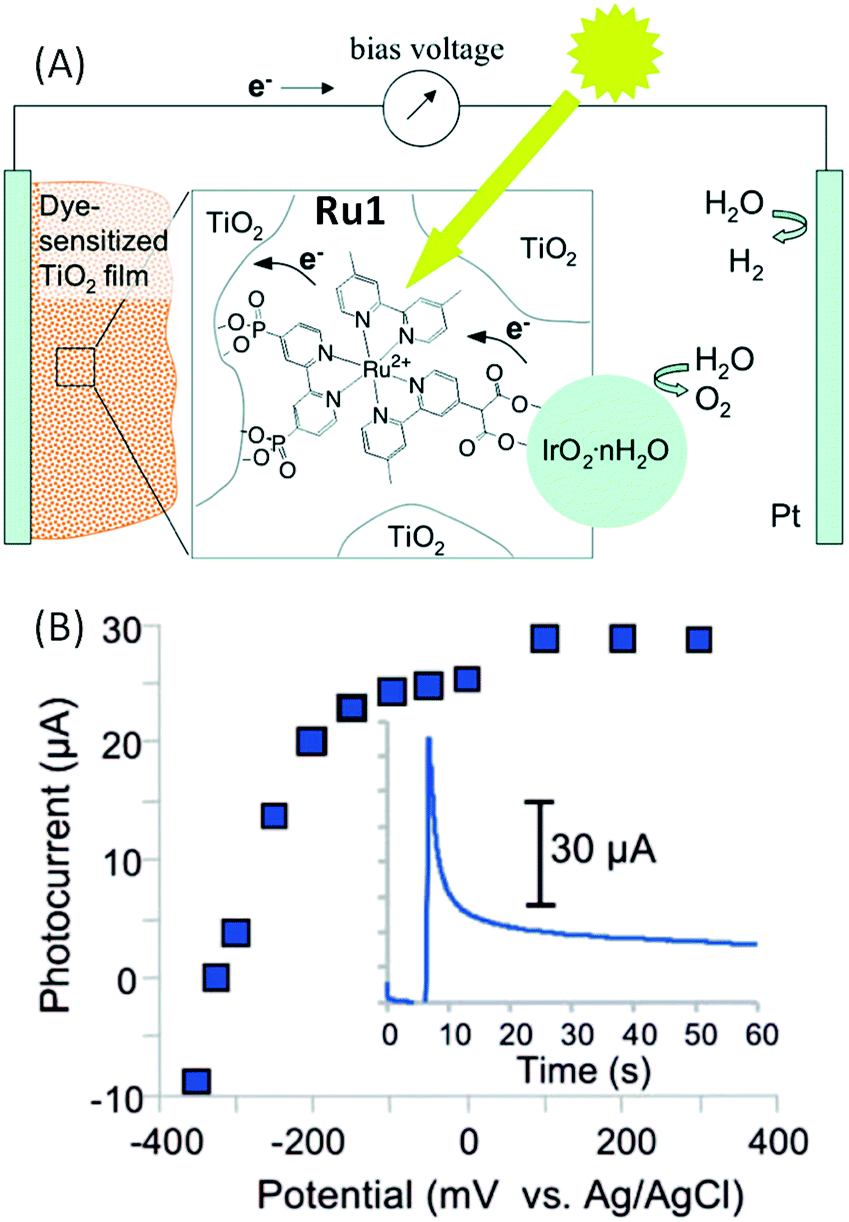 | ||
| Fig. 2 (A) Schematic representation of the water-splitting dye sensitized solar cell developed by Mallouk and coworkers46 based on a hybrid photoanode FTO/TiO2/Ru1–IrO2·nH2O. (B) Corresponding steady-state photocurrent as a function of applied potential and transient photocurrent recorded at 0 V vs. Ag/AgCl as a function of time (inset) in an aqueous buffered solution at pH 5.75 (reproduced from ref. 46, with permission from the American Chemical Society, Copyright 2009). | ||
The phosphonate groups of the dye allow its adsorption onto porous nanocrystalline TiO2 semi-conductors (SCs) while the malonate group links the dye to the hydrated iridium oxide nanoparticles IrO2·nH2O, which were used as efficient water oxidation catalysts.65 The Ru1–IrO2·nH2O assembly was first chemically synthetized from Ru1 and the K2IrCl6 salt in water, and then adsorbed onto TiO2. The malonate group of Ru1 acts as a stabilizer to control the size and polydispersity of the IrO2·nH2O nanoparticles, resulting in well dispersed particles of ∼2 nm diameter. The photoelectrochemical performance of the FTO/TiO2/Ru1–IrO2·nH2O photoanode was evaluated with a Pt wire as the counter electrode and a Ag/AgCl reference electrode, immersed in an aqueous buffered solution at pH 5.75 (Fig. 2(A)). Irradiation of the photoanode with visible light (λ > 410 nm) produced a measurable photocurrent at potentials more positive than −325 mV vs. Ag/AgCl (Fig. 2(B)). However, when a bias of 0 V vs. Ag/AgCl is applied, the observed anodic current spike decayed rapidly into a steady current (typically 10–30 μA vs. 1–2 μA with unsensitized TiO2) (Fig. 2(B) and Table 1, Entry 1). Under steady illumination, the current decayed over a period of approximately 4 hours. The decay in photocurrent is coupled to a bleaching of the visible absorbance of the dye grafted on TiO2. A small bias to achieve overall water splitting is needed in such a system, because electrons in trap states below of the TiO2 conduction band edge are not reductive enough to generate H2 and rapidly recombine with holes located on the photo-oxidized dyes.
Finally, the photocurrent of 12.7 μA cm−2 measured using 450 nm light at 7.8 mW cm−2 intensity corresponds to an internal quantum yield of ∼0.9%. The production of hydrogen and oxygen was confirmed by gas chromatography and, in the case of oxygen, the amount of gas was also estimated by using a pseudo-Clark electrode. The faradaic efficiency for the photoanodic oxygen generation was ∼20%. After approximately 4 h, the total photocurrent produced corresponds to a turnover of 16 per dye molecule. The low quantum yield was attributed to slow electron transfer from the IrO2·nH2O nanoparticles to the oxidized dye (2.2 ms), which was measured to be six times slower than the back-electron transfer (0.37 ms) from the trap state of the TiO2 SC to the photo-oxidized dye.
The strategy developed by the group of Mallouk to decrease the charge recombination was to introduce a thick insulating layer of ZrO2 or Nb2O5 (1–3 nm) between the Ru photosensitizer and TiO2.47 Core–shell FTO/TiO2/ZrO2 and FTO/TiO2/Nb2O5 were prepared by soaking FTO/TiO2 electrodes in an alcohol solution of zirconium(IV) butoxide or niobium(V) chloride, washed with water and sintered at high temperature (>400 °C). The Ru1–IrO2·nH2O assembly was then chemisorbed on these core–shell electrodes by soaking. Transient absorption spectroscopy under open-circuit conditions revealed that the back electron transfer from TiO2 to oxidized Ru1 is faster within TiO2/Ru1–IrO2·nH2O than within TiO2/ZrO2/Ru1–IrO2·nH2O and TiO2/Nb2O5/Ru1–IrO2·nH2O. The back electron transfer within TiO2/Ru1–IrO2·nH2O is multiphasic with two first-order decay times of 0.18 ms (42%) and 0.01 ms (55%). By contrast, the back electron transfer in the presence of insulating layers of ZrO2 (2 layers) or Nb2O5 (1 layer) is monophasic and slower by a factor 2–3. The authors assume that the slower charge recombination in the presence of ZrO2 and Nb2O5 layers occurs via the tunnel effect. This was also correlated with more negative potentials of the conduction bands of ZrO2 (−1.58 V vs. SCE)66,67 and Nb2O5 (−0.84 V)68 at pH 5.8, compared to that of TiO2 (−0.76 V).69,70 As a direct consequence of the slower back electron transfer, the photocurrent at −250 mV vs. Ag/AgCl for the core–shell photoanodes (25 and 19 μA cm−2 Nb2O5 and ZrO2, respectively) is higher than that of the photoanode without an insulating layer (14 μA cm−2). Besides, with a bias of 0 V vs. Ag/AgCl, the photocurrent of the core–shell photoanodes can reach 40–50 μA cm−2 (Table 1, Entry 2). Nevertheless, this photocurrent decreases rapidly within 40 s to stabilize at ca. 15 μA cm−2, a value similar to that obtained with the FTO/TiO2/Ru1–IrO2·nH2O photoanodes without insulating layers.46 Since the fast degradation of the ruthenium dye was ruled out, the authors ascribed this photocurrent decay to the presence of protons generated by water oxidation that induces a positive shift of the water oxidation potential and then a decrease of its driving force. This study highlights the fact that high and stable catalytic photocurrents could be obtained if the accumulation of protons on the electrode surface is reduced or even suppressed.
Another approach adopted by the group of Mallouk48 to improve the performance of such Ru/IrO2 photoanodes was to incorporate an electron transfer mediator between the IrOx nanoparticles and the ruthenium dye,47 mimicking the tyrosine–histidine pair present in photosystem II (Fig. 3(A)).71 To achieve the structure, colloidal IrOx·nH2O nanoparticles were first bound to the benzimidazole-phenol (BIP) electron mediator and to 2-carboxyethylphosphonic acid (CEPA) which is also used as an anchoring molecule onto the TiO2 electrode. Characterization by UV-visible spectroscopy of CEPA capped IrOx·nH2O without (1–IrOx·nH2O) and with the BIP electron mediator (2–IrOx·nH2O) showed a broad absorbance at 580 nm typical of Ir(IV) oxide nanoparticle signatures. For both 1–IrOx·nH2O and 2–IrOx·nH2O, irregular nanoparticles of 2 nm were observed by TEM. The functionalized IrOx nanoparticles with and without BIP were then co-adsorbed on TiO2 with the ruthenium photosensitizer containing three bipyridine ligands substituted by one phenyl phosphonate group (Ru2) (Fig. 3(A)). Different molar ratios of sensitizer and catalyst were investigated and the highest photocurrents were obtained with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio of Ru2:IrOx·nH2O with and without BIP. Under such conditions, at pH 5.8 and for a bias of 0 V vs. Ag/AgCl (overpotential (η) of −0.69 V), the steady-state photocurrent density reaches up to 80 μA cm−2 in the presence of the BIP mediator, with stability over 100 s (Fig. 3(B) and Table 1, Entry 3). In the absence of the mediator, a photocurrent of only 25 μA cm−2 was obtained (around three times lower), a comparable value to that previously obtained with the heteroleptic Ru1 dye.46 In addition, without BIP, the photocurrent is less stable. This result is in agreement with faster electron transfer between the IrOx·nH2O nanoparticles and Ru2 when the catalyst nanoparticles are covered by the BIP mediator. Finally, the steady state photocurrent densities measured using 450 nm of light at 4.5 mW cm−2 intensity were respectively 29.1 and 10.1 μA cm−2 with and without the mediator, corresponding to an internal quantum yield of 2.3% and 0.8% , respectively. Thus, the quantum yield has been enhanced by more than two rimes, in the presence of the redox mediator. A faradaic efficiency of more than 85% for oxygen evolution was obtained by Clark electrode measurements for both 1–IrOx·nH2O and 2–IrOx·nH2O. One drawback of the co-adsorption is the absence of control of the juxtaposition of the photosensitizer and BIP-catalyst onto the TiO2 surface (Fig. 3(A)). The electron transfer rate between the BIP mediator and Ru2 dye is thus not well controlled, due to the fact that both molecules were separately adsorbed onto the TiO2 electrode surface.48
:5 ratio of Ru2:IrOx·nH2O with and without BIP. Under such conditions, at pH 5.8 and for a bias of 0 V vs. Ag/AgCl (overpotential (η) of −0.69 V), the steady-state photocurrent density reaches up to 80 μA cm−2 in the presence of the BIP mediator, with stability over 100 s (Fig. 3(B) and Table 1, Entry 3). In the absence of the mediator, a photocurrent of only 25 μA cm−2 was obtained (around three times lower), a comparable value to that previously obtained with the heteroleptic Ru1 dye.46 In addition, without BIP, the photocurrent is less stable. This result is in agreement with faster electron transfer between the IrOx·nH2O nanoparticles and Ru2 when the catalyst nanoparticles are covered by the BIP mediator. Finally, the steady state photocurrent densities measured using 450 nm of light at 4.5 mW cm−2 intensity were respectively 29.1 and 10.1 μA cm−2 with and without the mediator, corresponding to an internal quantum yield of 2.3% and 0.8% , respectively. Thus, the quantum yield has been enhanced by more than two rimes, in the presence of the redox mediator. A faradaic efficiency of more than 85% for oxygen evolution was obtained by Clark electrode measurements for both 1–IrOx·nH2O and 2–IrOx·nH2O. One drawback of the co-adsorption is the absence of control of the juxtaposition of the photosensitizer and BIP-catalyst onto the TiO2 surface (Fig. 3(A)). The electron transfer rate between the BIP mediator and Ru2 dye is thus not well controlled, due to the fact that both molecules were separately adsorbed onto the TiO2 electrode surface.48
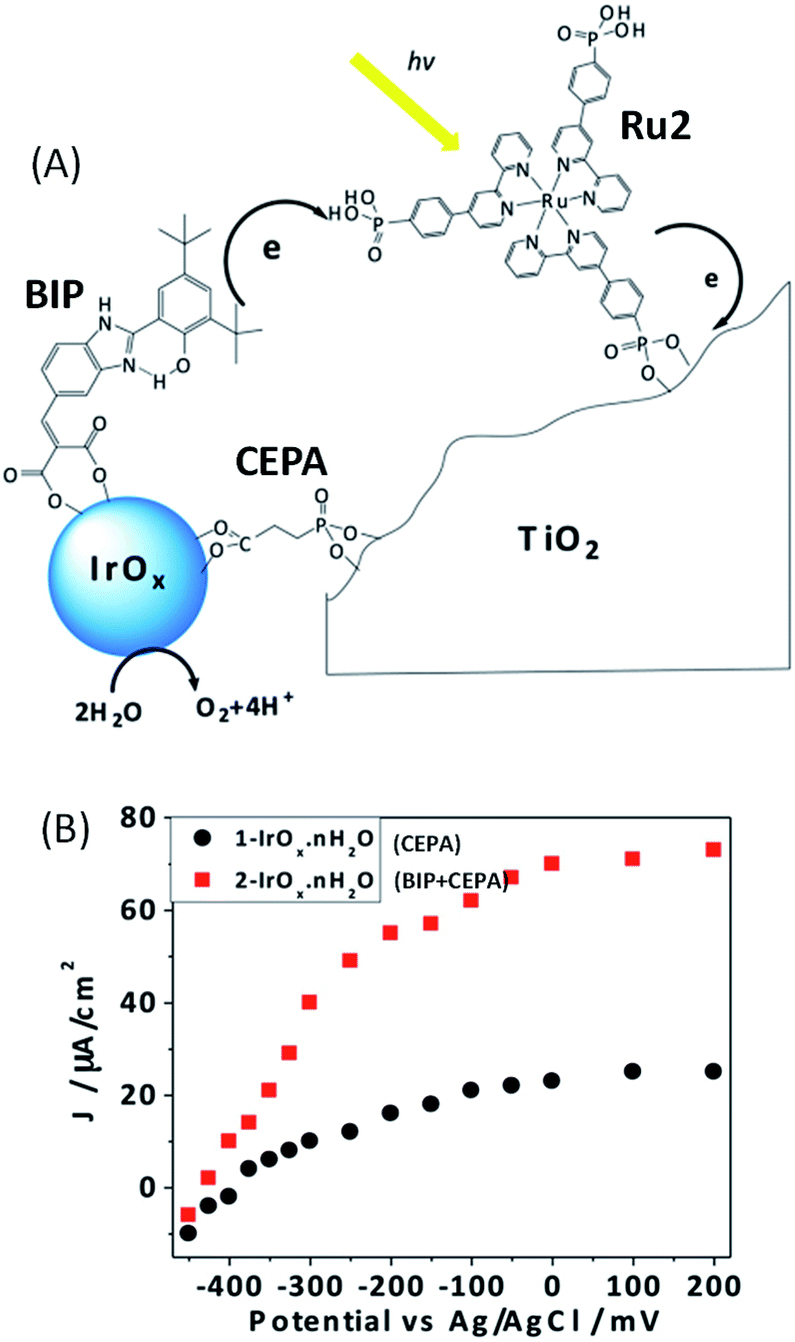 | ||
| Fig. 3 (A) Schematic representation of the electron transfer reactions in the mediator-based, dye-sensitized FTO/TiO2 photoanode developed by Mallouk and coworkers48 showing the molecular structures of the benzimidazole-phenol (BIP) mediator, Ru2 dye and 2-carboxyethylphosphonic acid (CEPA) anchoring group. (B) Corresponding steady-state photocurrent density as a function of anode potential for co-adsorbed Ru2 and 1–IrOx·nH2O (CEPA-capped IrOx) or 2–IrOx·nH2O (BIP- and CEPA-capped IrOx) in an aqueous buffered solution at pH 5.8 (reproduced from ref. 48, with permission from the National Academy of Sciences of the United states of America, Copyright 2012). | ||
Another approach investigated by Mallouk and coworkers49,50,72 was to sinter crystalline IrO2 nanoparticles (2 nm) directly onto a porous TiO2 film, instead of using amorphous IrOx nanoparticles with capping ligands. The rutile IrO2 nanoparticles were deposited by soaking FTO/TiO2 electrodes in a solution of colloidal IrOx capped with citrate, followed by treatment at 450 °C. Rutile IrO2 is known to exhibit a higher catalytic activity per Ir atom than amorphous IrOx. Then, the Ru photosensitizer comprising one bipyridine group functionalized by two phosphonate anchoring groups (denoted as Ru3) is grafted on the FTO/TiO2/IrO2 electrode by soaking at room temperature (Fig. 4). The FTO/TiO2/Ru3/IrO2 photoanode displayed an anodic photocurrent spike of 225 μA cm−2 with a bias of 0.1 V vs. Ag/AgCl which rapidly decayed to 80 μA cm−2 after 30 s and then to 10 μA cm−2 after 600 s (Fig. 4(A) and Table 1, Entry 4).50 Besides, the photocurrent and the open-circuit photovoltage of the photoanode are strongly dependent on the quantity of deposited IrO2: they reach a maximum at 0.50 pmol cm−2 of IrO2 and drop off at higher loadings. This non-monotonic behavior was ascribed to rapid charge recombination (also called electron scavenging (kscav), Fig. 4(B)) between the photo-injected electrons within the TiO2 conduction band and the IrO2 particle, the latter competing with the back electron transfer to the oxidized Ru3 (krecomb, Fig. 4(B)). This assumption was confirmed by electrochemical impedance spectroscopy (EIS) since the lifetime of electrons photoinjected within the TiO2 CB decreases from 6.25 ms without IrO2 to 4 ms in the presence of IrO2 (1.79 pmol cm−2).
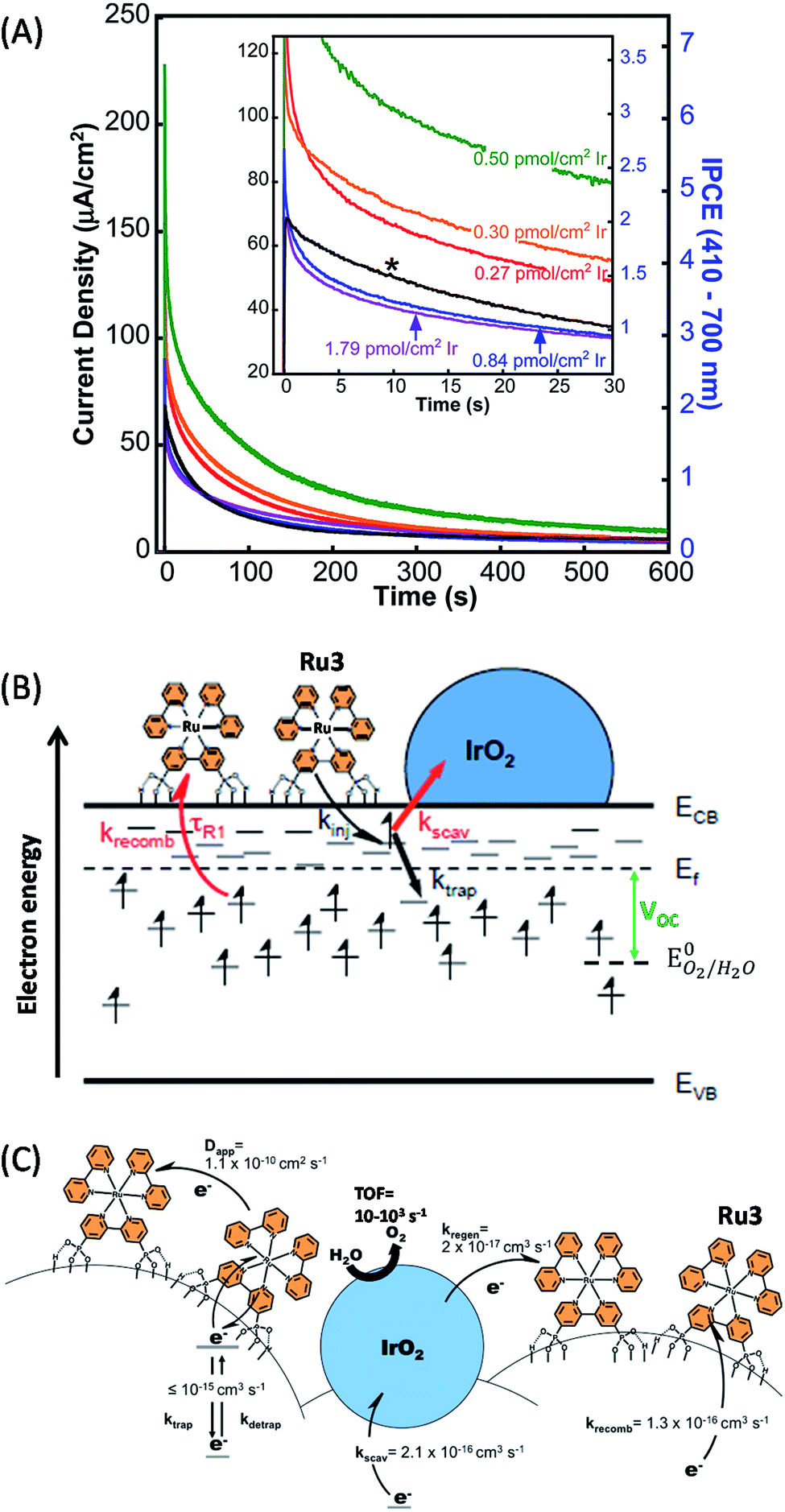 | ||
| Fig. 4 (A) Transient photocurrent recorded at 0.1 V vs. Ag/AgCl as a function of time in an aqueous buffered solution at pH 6.8 with hybrid photoanodes FTO/TiO2/Ru3/Cat using sintered IrO2 (Ir loading between 0.27 and 1.79 pmol cm−2) or citrate-capped IrOx (0.5 pmol cm−2, denoted by *) particles as a catalyst, developed by Mallouk and coworkers (reproduced from ref. 50 with permission from the American Chemical Society, Copyright 2014). (B) Energy diagram displaying the conduction (ECB) and valence (EVB) bands and the Fermi level (Ef) of TiO2, along with the different electron transfer processes occurring within the hybrid photoanode of FTO/TiO2/Ru3/IrO2, such as the photo-injection of electron into the low-lying surface states (kinj), the trapping of the injected electrons into states below the Fermi level (ktrap), the scavenging of these electrons by IrO2 (kscav), the charge recombination between the trap states and the oxidized Ru3 (krecomb), the electron transfer from IrO2 to the oxidized Ru3 inducing its regeneration (kregen), and the cross-surface electron diffusion (or the hole transport) between dyes with the Dapp coefficient. (C) Kinetics of electron transfer involved in the photocatalytic activity of the photoanode (reproduced from ref. 72, with permission from the American Chemical Society, Copyright 2015). | ||
The group of Mallouk also showed that the nature and the acidity of solvent used for the dye grafting strongly influence the OER performances of the photoanode.49 When an acidic aqueous solvent is used for the dye deposition, weaker photocurrents and open-circuit photovoltages are obtained compared with organic solvents such as DMSO or ethanol. Based on an EIS study, the authors proposed that the proton intercalation into TiO2 induces the formation of TiIII sites which promotes the charge recombination with the photoinjected electrons and thus the loss of photocurrent. This hypothesis can also explain the rapid decay of the photocurrent during the light-driven water oxidation under acidic conditions which releases protons at the surface of the photoanode.
Finally, Mallouk and co-workers72 drew a complete kinetic picture of the electron transport occurring within such hybrid photoanodes under an open-circuit regime by correlating EIS measurements with transient photocurrent and photovoltage (Fig. 4(C)). Since the electron transport occurs in three dimensions, the authors calculated second-order rate constants with the units of cm3 s−1, which are equivalent to the M−1 s−1 units employed for bimolecular electron transfer in solution. It is well known that the first event in these hybrid photoanodes, the electron injection from excited Ru3 dyes into TiO2, displays a high quantum yield and takes place on a time scale from femtoseconds to hundreds of picoseconds.73,74
Nevertheless, they showed via their model that only 2% of photo-injected electrons are trapped in the deep states below the Fermi level and contribute to the generation of the photocurrent. The other 98% remain in the shallow states just below the CB (i.e. above the Fermi level, see Fig. 4(B)) and recombine rapidly with the oxidized Ru3. The second-order rate constant of electron trapping (ktrap) in deep states was approximated between 10−14 and 10−15 cm3 s−1, which is 80 times faster than the charge recombination between these deeply trapped electrons and the oxidized Ru3 (krecomb = 1.3 × 10−16 cm3 s−1), but also much slower than the charge recombination from the shallow states. Once oxidized, the Ru3 dye is able to oxidize the IrO2 catalyst with an estimated kinetics (kregen) of 2 × 10−17 cm3 s−1, corresponding to a low bimolecular rate constant of 1.2 × 104 M−1 s−1. The slow electron transfer from IrO2 to the oxidized Ru3 was explained by the fact that the dye is chemically grafted on the semiconducting electrode promoting the charge recombination with TiO2, while Ru3 is not in proximity to the catalyst particles. When oxidized, the IrO2 particles can in turn oxidize water to O2. However the catalysis can also be short-circuited by the fast scavenging of electrons lying in the shallow states by IrO2 with a rate constant of (kscav) 2.1 × 10−16 cm3 s−1. This study confirms that the slow electron transfer between the molecular photosensitizer and the metal oxide catalyst is the weak point of such hybrid photoanodes, and that the slow dye regeneration by IrO2 competes directly with very fast recombination and scavenging processes, limiting their photocatalytic performance. In other words, the quantum yield for water-splitting with hybrid TiO2/Ru/IrO2 photoanodes is low since the charge recombination reaction at its surface is faster than the oxidation of the metal oxide catalyst or the catalytic four-electron oxidation of water.
Murray's group52 has made a significant improvement to the hybrid photoanodes developed by Mallouk by using a FTO semiconducting electrode coated with a nanoparticle film of ITO on which a thin layer of TiO2 (3.7 or 6.6 nm) was deposited by atomic layer deposition (ALD) (denoted as nanoITO/TiO2; 3.2 μm thickness) (Fig. 5(A)). When a Ru tris-bipyridine phosphonate dye (Ru4) and IrOx nanoparticles are successively deposited on the core/shell FTO/nanoITO/TiO2 electrode by soaking, a clear increase of the photocurrent efficiency is observed by a factor of 2.4 and 2.7 for a TiO2 layer of 3.7 nm and 6.6 nm thickness, respectively, in comparison to a photoanode with a regular mesoporous TiO2 film without nanoITO (i.e. FTO/TiO2/Ru4/IrOx). The marked difference of photocurrent with and without nanoITO was ascribed to slower back electron transfer between oxidized Ru4 and TiO2 within the core/shell photoanode, which could be due to fast electron transport via the thin TiO2 layer to nanoITO and then to the counter electrode. In addition, the authors demonstrated that the photocurrent intensity of FTO/nanoITO/TiO2/Ru4/IrOx depends on both the thickness of the TiO2 layer and the applied bias (Fig. 5(B)). With a bias of 0 V vs. Ag/AgCl at pH 5.8, the core/shell photoanode displays a modest photocurrent of ca. 30 and 50 μA cm−2 respectively with a TiO2 layer of 3.7 and 6.6 nm thickness. Increasing the bias until +0.5 V allows increasing the respective photocurrents up to ca. 60 and 200 μA cm−2 (Table 1, Entry 5), the latter being the highest photocurrent obtained with a hybrid photoanode using a ruthenium dye. It is noteworthy that the increase of photocurrent with the increase of bias cannot be attributed to the direct electro-oxidation of water on IrOx, since the latter needs a potential of +0.9 V vs. Ag/AgCl at pH 5.8 to catalyze this reaction.
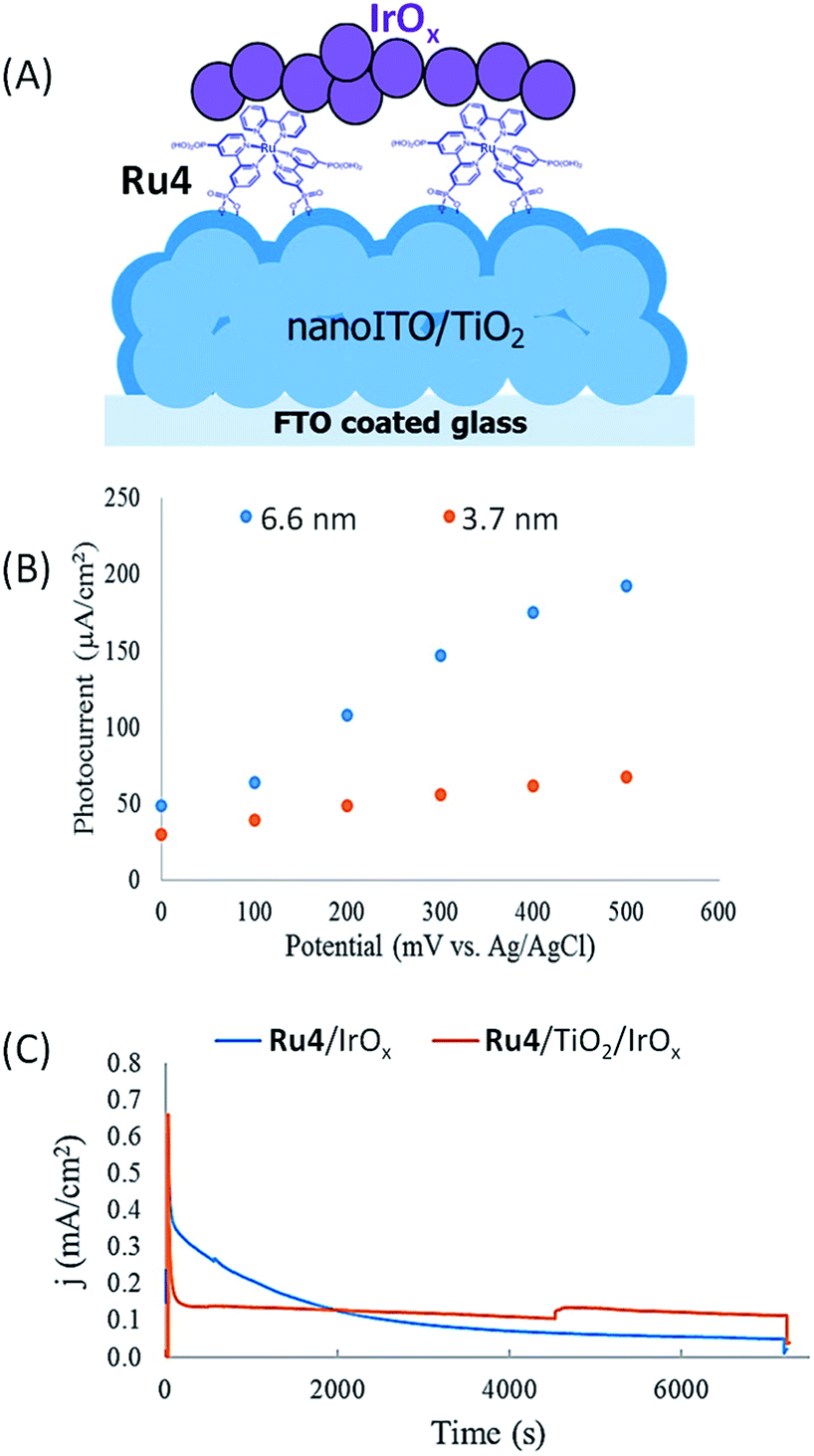 | ||
| Fig. 5 (A) Schematic representation of the FTO/nanoITO/TiO2/Ru4/IrOx photoanode developed by Murray and coworkers.52 (B) Photocurrent density as a function of anodic potential for FTO/nanoITO/TiO2/Ru4/IrOx photoanodes with a TiO2 thickness of 3.7 (red) and 6.6 nm (blue) under illumination at 455 nm, in an aqueous buffered solution at pH 5.8. (C) Chronoamperograms of FTO/nanoITO/TiO2/Ru4–IrOx (blue) and FTO/nanoITO/TiO2/Ru4/TiO2/IrOx (red) with a TiO2 thickness of 6.6 nm under illumination at 455 nm, in an aqueous buffered solution at pH 5.8 and with a bias of +0.3 V vs. Ag/AgCl (reproduced from ref. 52, with permission from the American Chemical Society, Copyright 2015). | ||
Nevertheless, during long-term photolysis, the photocurrent decrease with FTO/nanoITO/TiO2/Ru4/IrOx is rapid and significant because the Ru4 dye is released into solution owing to the hydrolysis of the phosphonate groups at near neutral pH (Fig. 5(C)). To avoid the detachment of the dye from the electrode surface, a very thin layer of TiO2 (1 nm) was coated by ALD on FTO/nanoITO/TiO2/Ru4 before the deposition of IrOx nanoparticles. The stabilized FTO/nanoITO/TiO2/Ru4/TiO2/IrOx electrode sustains a photocurrent of 110 μA cm−2 during photolysis for 2 h with an applied bias of +0.3 V vs. Ag/AgCl (pH 5.8), displaying remarkable longevity for a hybrid photoanode (Fig. 5(C) and Table 1, Entry 5).
In contrast to these above reported examples of photoanodes involving iridium oxide,46–48,51,52 Na and co-workers53 developed sensitized ruthenium TiO2 photoanodes with an earth abundant metal oxide as a water oxidation catalyst, consisting of functionalized cobalt oxide nanoparticles (Co3O4) (Fig. 6(A)). In this work, a new strategy to introduce a nanoparticle water oxidation catalyst into a water-splitting DS-PEC was investigated. Indeed, nanoparticles of Co3O4 were first synthetized and subsequently surface-modified with 3-aminopropyltriethoxysilane (APTES). Then, the functionalized Co3O4 nanoparticles were integrated into the ruthenium dye sensitized (Ru5) photoelectrode by a fast Schiff base reaction with 4-formylbenzoic acid which was co-adsorbed with Ru5 on TiO2, the Ru5 dye being primarily adsorbed onto mesoporous TiO2via two phosphonate groups. Fig. 6(B) displays the photocurrent density as a function of time under visible light irradiation (400 nm) at pH 6.8 in a phosphate buffer solution. The inset shows that the optimum bias applied is +0.3 V vs. Ag/AgCl (η = −0.33 V) producing a transient photocurrent density of 135 μA cm−2. Irradiation of the TiO2/Ru5 electrode without the cobalt catalyst produces a photocurrent density lower than 15 μA cm−2 due to fast charge transfer from TiO2 to Ru5 (Table 1, Entry 6), which is the direct consequence of the very slow water oxidation by the oxidized Ru5.
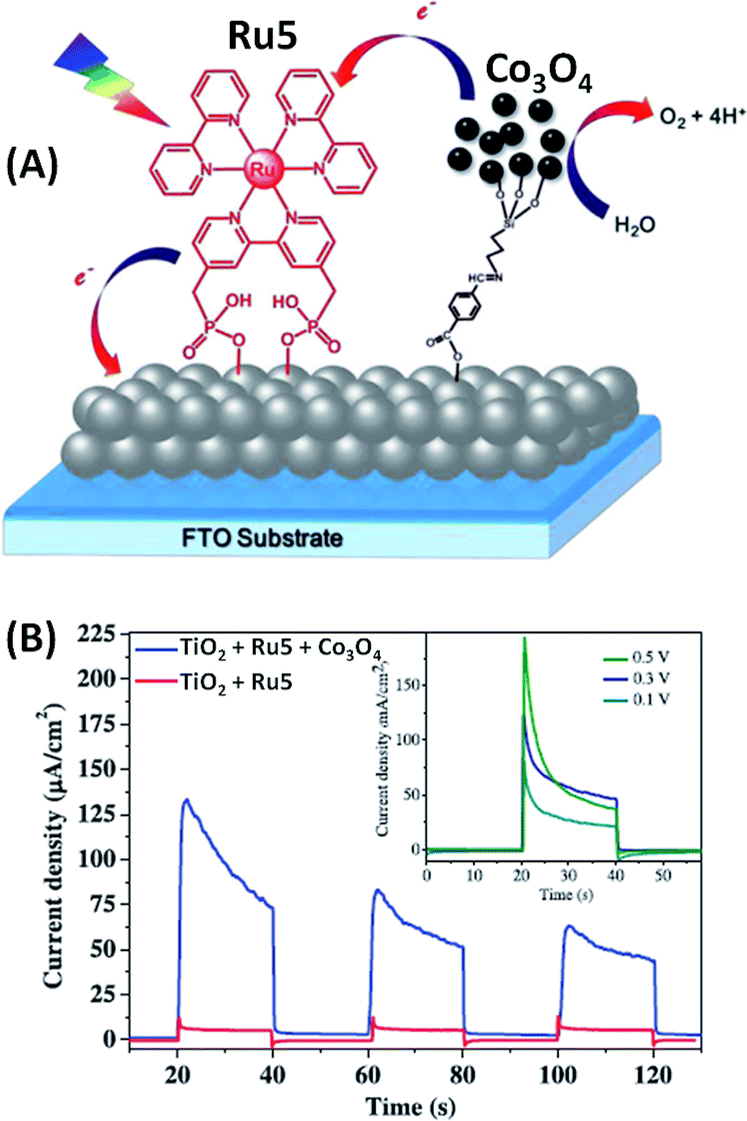 | ||
| Fig. 6 (A) Schematic representation of the FTO/TiO2/Ru5/Co3O4 photoanode developed by Na and coworkers.53 (B) Corresponding transient photocurrent in a phosphate buffer solution at pH 6.8 at different bias vs. Ag/AgCl as a function of time (reproduced from ref. 53, with permission from Elsevier, Copyright 2016). | ||
Thus, the combination of Ru5 with the modified Co3O4 catalyst increases the photocurrent density of the photoanode by a factor of eight compared to that of a photoanode without Co3O4, due to fast electron transfer from Co3O4 to the photooxidized Ru5 dye. However, with a decrease of the photocurrent density to 50 μA cm−2 (i.e. 40% of its initial value) after 3 cycles of 20 s ON–OFF illumination, the stability of this photoanode is not promising.53 This may be due to the low kinetics of water oxidation catalyzed by the modified Co3O4 particles, which therefore cannot give sufficient reducing equivalents to the oxidized dye, resulting in its decomposition or fast charge recombination between TiO2 and Ru5.
In summary, ruthenium sensitized TiO2 hybrid photoanodes display photocatalytic activities at negative overpotentials which is greatly desired, but the photocurrents are weak (<100 μA cm−2 at a bias of 0 V vs. Ag/AgCl (η = −0.69 V)) and generally not stable. Only Murray's photoanode FTO/nanoITO/TiO2/Ru4/TiO2/IrOx using thin layers of an n-type semiconductor, one between the FTO electrode and TiO2 and the other one between the Ru dye and the IrOx catalyst, displays a photocurrent higher than 100 μA cm−2 along with remarkable stability (110 μA cm−2 over 2 h of photolysis) but in that case the applied bias is also higher (+0.3 V vs. Ag/AgCl (η = −0.39 V)).
Hybrid photoanodes with an organic chromophore
Polyheptazine chromophores
In 2011, Beranek and coworkers54 studied the photo-oxidation of water to oxygen under visible light irradiation using a hybrid photoanode based on a novel class of inorganic/organic hybrid materials – nanocrystalline TiO2 modified at the surface with a thin layer (<1–3 nm) of polyheptazine (TiO2–PH) and loaded with IrO2 as an oxygen-evolving co-catalyst (Fig. 7(A)). PH, also called graphitic carbon nitride (g-C3N4), is a very attractive organic photosensitizer for light-driven water oxidation. Indeed, polyheptazine is extremely chemically stable and presents a strong absorption in the visible spectrum and its high conjugation promotes the electron transport through a graphite-like structure. In this work, polyheptazine sheets were first generated on an ITO/TiO2 electrode by pyrolysis of urea at high temperature (400 °C). Then, IrO2 particles were deposited on the ITO/TiO2–PH electrode by soaking in a colloidal solution of iridium oxide, which was prepared by hydrolysis of an aqueous solution of Na2IrCl6. In a phosphate buffer solution (pH 7) and with a bias of +0.5 V vs. Ag/AgCl, the ITO/TiO2–PH/IrO2 photoanode displayed a light-driven evolution of O2 with a significant photocurrent of ca. 100 μA cm−2, along with remarkable stability over 90 min of irradiation (Fig. 7(B) and Table 1, Entry 7).54,55 In this photoanode, after light absorption the PH film injects electrons into the CB of TiO2 and the holes generated in the PH film are transferred to IrO2 to catalyze water oxidation. TiO2 photoelectrodes modified with IrO2 nanoparticles exhibit only negligible photocurrents and do not show any oxygen evolution (Fig. 7(B)). The authors have shown that a strong electronic coupling between TiO2 and PH allows a direct optical electron transfer from the VB of PH to the CB of TiO2 following light absorption (Fig. 7(C)).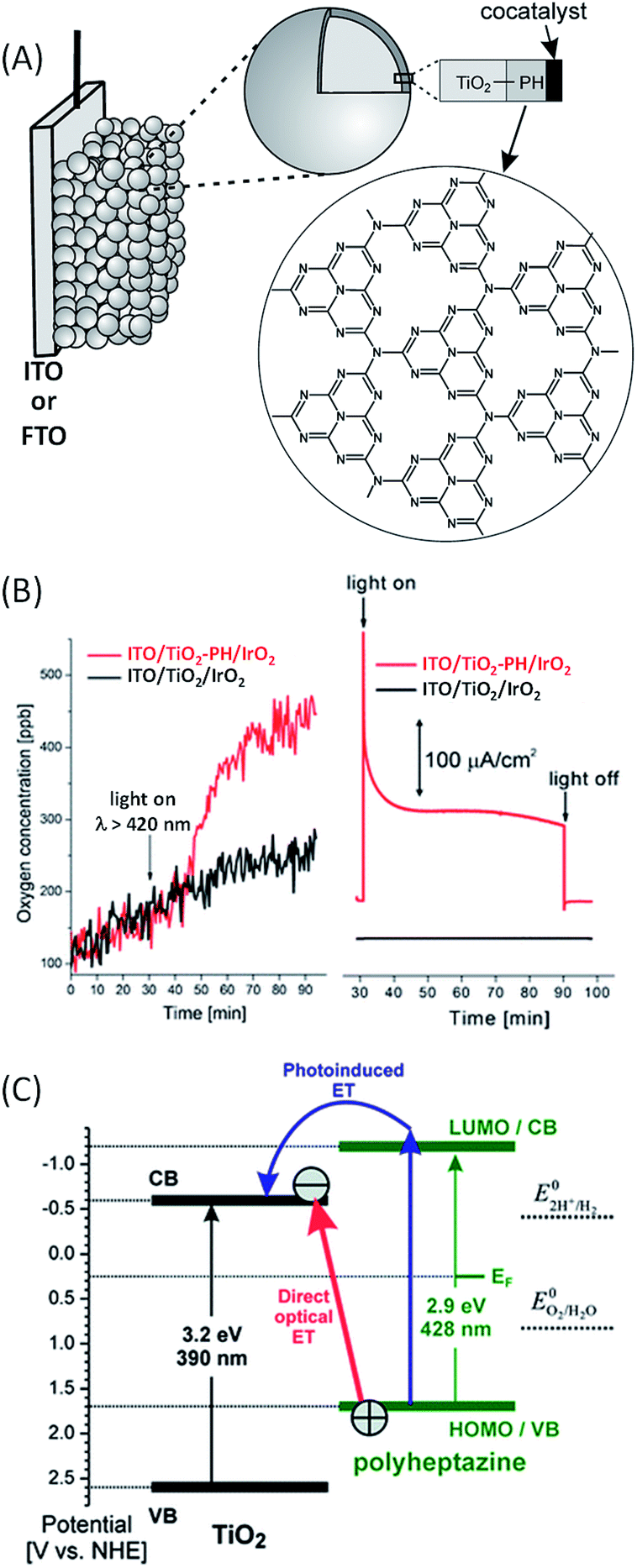 | ||
| Fig. 7 (A) Schematic representation of the photoanodes developed by Beranek and coworkers54,57,60 consisting of ITO or FTO/TiO2 loaded with a polyheptazine (PH) film and a co-catalyst (e.g. IrO2, Co–Pi, and CoO(OH)x) (reproduced from ref. 57, with permission from Wiley, Copyright 2012). (B) Oxygen evolution (left) and corresponding transient photocurrent density (right) as a function of time, measured with ITO/TiO2/IrO2 (black) and ITO/TiO2–PH/IrO2 (red) photoanodes under intermittent irradiation at λ > 420 nm, with a bias of +0.5 V vs. Ag/AgCl in a deaerated phosphate buffer at pH 7. (C) Potential diagram of the TiO2–PH interface (pH 7) showing the two photoexcitation pathways: photo-induced electron transfer (blue) and direct optical electron transfer (red) (reproduced from ref. 54, with permission from the Royal Society of Chemistry, Copyright 2011). | ||
Such a process is correlated with an additional optical absorption band above 430 nm (until ∼700 nm), where polyheptazine and TiO2 do not absorb. Basically, the valence band of PH, estimated at +1.7 V vs. NHE (i.e. +1.5 V vs. Ag/AgCl, pH 7) by UV/Vis diffuse reflectance spectroscopy, is positive enough to induce water oxidation (+0.82 V vs. NHE, pH 7) from the IrO2 co-catalyst. The role of IrO2 is also to reduce the recombination of photogenerated charges at the interface of TiO2–PH at a low bias potential. In other words, IrO2 particles reduce the hole accumulation within PH, which is very important to reduce the need for an external bias at hybrid photoanodes for the water splitting reaction. The photoconversion efficiency can be enhanced by increasing the number of effective catalytic sites for water oxidation on the surface of the hybrid photoanode.
Beranek and coworkers56–58 subsequently investigated hybrid photoanodes of TiO2–polyheptazine (TiO2–PH) with an earth abundant co-catalyst, cobalt oxide deposited in phosphate buffer, denoted as Co–Pi, which was initially reported by Nocera and co-workers.75–78 In this example, Co–Pi was photoelectrochemically deposited at +0.4 V vs. Ag/AgCl in a phosphate buffer (pH 7) from a Co2+ salt, in order to ensure its preferential generation at the sites with the highest concentration of photogenerated holes. The other advantage of such a photoelectrochemical process is to fine tune the amount of Co–Pi deposited by monitoring the deposition progress over time with the photocurrent change. Fig. 8 displays the linear sweep voltammogram of ITO/TiO2–PH with and without the Co–Pi catalyst under on–off monochromatic irradiation at 450 nm. The ITO/TiO2–PH/Co–Pi photoanode produces a photocurrent density of ∼100 μA cm−2 at 0.5 V vs. Ag/AgCl (i.e. 1.12 V vs. RHE (η = −0.43 V)), higher than that of a simple ITO/TiO2–PH photoanode. The photocurrent density of ITO/TiO2–PH/Co–Pi increases to 190 μA cm−2 by irradiating with a polychromatic visible light above 420 nm with a potential of +0.5 V vs. Ag/AgCl at neutral pH, but the latter decreases to 40 μA cm−2 after 1 h 30 min of irradiation (Table 1, Entry 8). Under these experimental conditions, the ITO/TiO2–PH/Co–Pi photoanode is able to produce O2 consistently for 1 h 40 min, attesting to the relatively good stability of the photoelectrode.57 Besides, no oxygen evolution was detected with the ITO/TiO2–PH photoelectrodes without the cocatalyst (Fig. 9(A)), as well as with TiO2 photoelectrodes loaded only with Co–Pi.
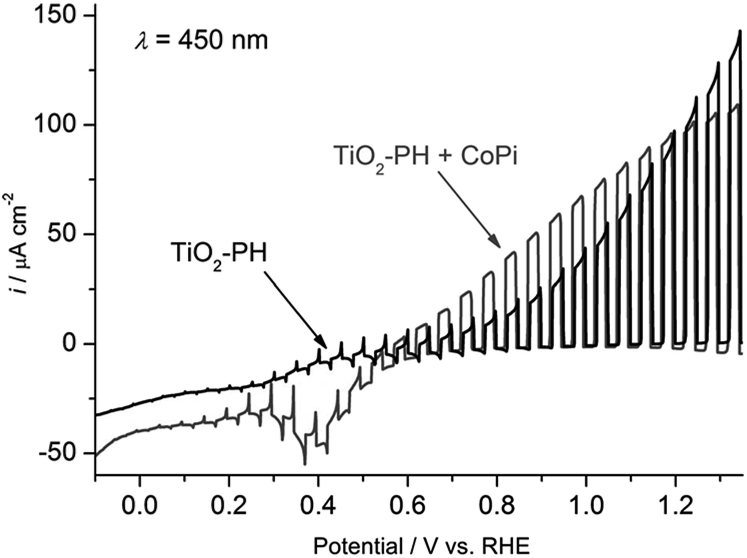 | ||
| Fig. 8 Transient photocurrent density measured at ITO/TiO2–PH photoelectrodes with and without Co–Pi, under intermittent irradiation at λ = 450 nm (5 s light and 5 s dark), at a cathodic potential sweep rate of 5 mV s−1 in a deaerated phosphate buffer at pH 7 (reproduced from ref. 57, with permission from Wiley, Copyright 2012). | ||
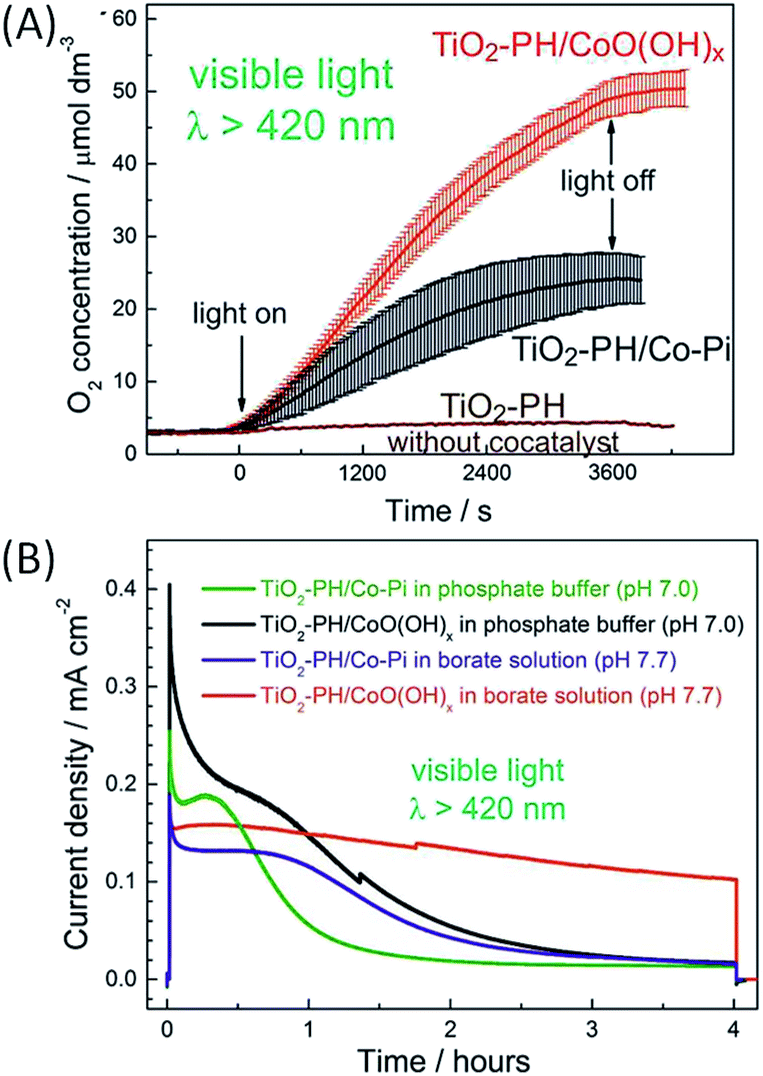 | ||
| Fig. 9 (A) Photocatalytic oxygen evolution as a function of time under visible light irradiation in a phosphate buffer (pH 7) at +1.12 V vs. RHE of FTO/TiO2–PH/CoO(OH)x (red), FTO/TiO2–PH/Co–Pi (black) and FTO/TiO2–PH (brown) photoanodes developed by Beranek and coworkers.60 (B) Chronoamperograms of FTO/TiO2–PH/CoO(OH)x (black and red) and FTO/TiO2–PH/Co–Pi (green and purple) under visible light irradiation in phosphate (pH 7) and borate (pH 7.7) buffers at +1.12 V vs. RHE (reproduced from ref. 60, with permission from the American Chemical Society, Copyright 2017). | ||
The performance (activity and stability) of this hybrid photoanode was further improved by substituting Co–Pi particles (size of 5 nm) with smaller particles of CoO(OH)x (∼1–2 nm).60 In that case, the CoO(OH)x co-catalyst was chemically deposited on the FTO/TiO2–PH electrode by successive immersion in a solution of Co(NO3)2 and then in a solution of weakly basic aqueous ammonia. In a phosphate buffer (pH 7), the resulting FTO/TiO2–PH/CoO(OH)x photoanodes outperformed photoanodes loaded with Co–Pi in terms of faradaic yield of O2 evolution (34 vs. 17% respectively) and stability under long term irradiation (Fig. 9). This improved photocatalytic performance was ascribed to the higher loading of CoO(OH)x on FTO/TiO2–PH compared to Co–Pi and the greater electroactive surface area of the photoanodes due to the small particles of CoO(OH)x. The improved performance also stems from the higher transparency of the CoO(OH)x film owing to its ultrasmall nanoparticles, which does not hinder the excitation of TiO2–PH and promotes the photocatalytic process. The stability of FTO/TiO2–PH/CoO(OH)x photoanodes was further significantly enhanced by using a borate buffer (pH 7.7) instead of a phosphate buffer (pH 7) (Fig. 9(B)). Indeed, the photocurrent remains higher than 110 μA cm−2 over a period of 4 hours under visible light irradiation at +0.5 V vs. Ag/AgCl (Table 1, Entry 9).
From a study by X-ray absorption spectroscopy (XAS), the authors related the higher stability in borate electrolyte to an increase in the structural order of CoO(OH)x after photoelectrocatalysis, which corresponds to the partial oxidation of the initially highly disordered CoO(OH)x nanoparticles. Besides, DFT calculations suggest that this order is governed by the chemical interactions between the electrolyte anions and the cobalt ions at the highest oxidation states generated during photoelectrocatalysis. Thus, borate anions bind to cobalt cations much less covalently than phosphate anions and the ionic bonds between borate and CoO(OH)x are more flexible due to their lower activation barrier for redirection, promoting a structural order within the nanoparticles. This study showed the strong influence of the structure and the optical properties of co-catalysts as well as the nature of the electrolyte on the performance and stability of photoanodes devoted to water oxidation. To sum up, hybrid photoanodes using a polyheptazine film co-deposited on a TiO2 electrode with a metal oxide catalyst display significant photocurrents (>100 μA cm−2) at a slightly negative overpotential (η ∼ −0.1 V, bias of 0.5 V vs. Ag/AgCl) along with good stability for up to 4 hours with CoO(OH)x nanoparticles in borate buffer. This promising performance could be related to the robustness of polyheptazine even at the oxidized state and the good mobility of holes within the polyheptazine film that competes with the charge recombination between TiO2 and PH, and thus contributes to a sustained catalytic photocurrent.
Perylene chromophores
Some examples of hybrid photoanodes with perylene diimide (PDI) chromophores have been recently published.61–63PDIs are interesting chromophores owing to their robustness with good thermal and photochemical stability, their strong absorption in the visible spectrum and their relatively low cost.Many PDIs also exhibit a highest occupied molecular orbital (HOMO) with a potential positive enough to oxidize water or to activate a water oxidation catalyst, a metal oxide or a molecular complex.79 All of these features make PDIs interesting candidates as photosensitizers for water-splitting reactions. However, the implementation of a PDI in dye-sensitized semiconducting electrodes remains limited due to its low oxidation potential at the excited state preventing efficient charge injection into the CB of many semi-conductors.63PDIs employed within photoanodes have thus to be carefully designed in order to adjust the energy levels of their molecular orbitals to those of the CB of the SC and of the catalytic onset of water oxidation by the co-catalyst.
The first example of a hybrid photoanode for water oxidation based on perylene dye was published by Finke and co-workers61 in 2014 (Fig. 10(A)). To this aim, a novel PDI dye functionalized with two phosphonate groups at the imide positions (N,N′-bis(phosphonomethyl)-3,4,9,10-perylenediimide, denoted as PMPDI) was synthetized. Thin films of PMPDI were deposited onto ITO electrodes by spin coating in basic aqueous solution and then the phosphonate groups were protonated by immersion in acidic solution to make the film insoluble in neutral or acidic medium. In order to promote water oxidation, CoOx particles were then photoelectrochemically deposited by applying a potential of +0.70 V vs. Ag/AgCl for 5 min to the ITO/PMPDI electrode immersed in a phosphate buffer (pH 7) containing 1 mM Co(NO3)2 (illumination with a Xe lamp, 100 mW cm−2). The photoelectrochemical process was chosen to preferentially deposit the catalyst at high concentration in areas where the holes are photogenerated, as previously described.57,80 With the ITO/PMPDI/CoOx electrodes, visible-light assisted water oxidation was obtained with a photocurrent density of about 150 μA cm−2 at an applied bias of +1.0 V vs. Ag/AgCl (η = 0.38 V) in a phosphate buffer aqueous solution at pH 7, with stability over 300 s (Table 1, Entry 10). Fig. 10(B) shows the linear sweep voltammogram of the ITO/PMPDI electrode with and without CoOx and the bare ITO, under on–off cycles of light irradiation. Almost no photocurrent was measured for bare ITO and relatively small photocurrents were measured for ITO/PMPDI (<10 μA cm−2) (Fig. 10(B)). The oxidation of water was confirmed by the direct detection of O2 obtained at an applied bias of +0.90 V vs. Ag/AgCl, which corresponds to a faradaic efficiency of 80 ± 15%. The quantum efficiency of this hybrid photoanode for water oxidation was ∼1%. When analogous photoanodes were prepared with a perylene diimide derivative having two alkyl groups in place of the phosphonate groups of PMPDI, the photocurrent obtained was about one order of magnitude smaller than that of the typical ITO/PMPDI/CoOx. This is due to the fact that, with this dye, only traces of cobalt were deposited as shown by XPS measurements. These results provide evidence that the phosphonate groups of PMPDI strongly bond the CoOx particles, and this could accelerate the electron transfer from the co-catalyst to the organic dye and minimize the charge recombination at the photoanode surface.
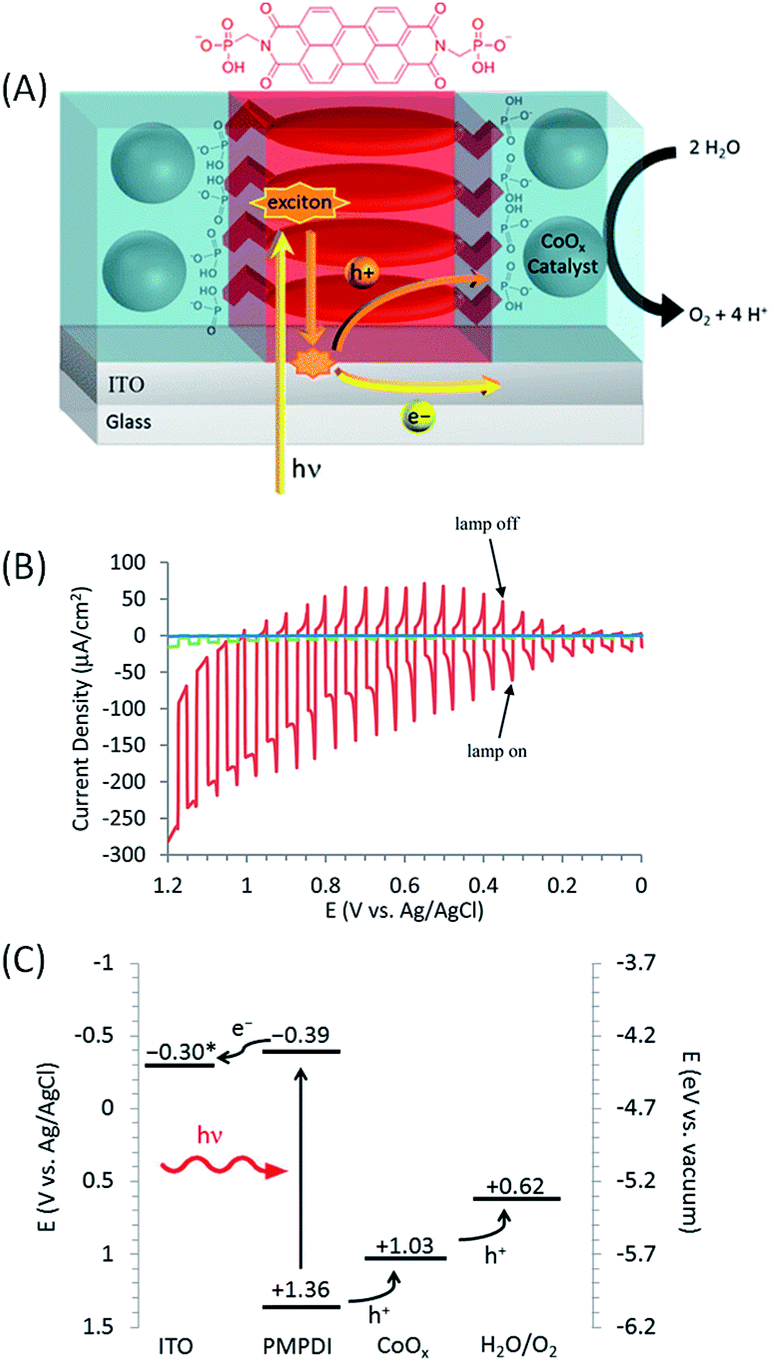 | ||
| Fig. 10 (A) Schematic representation of the ITO/PMPDI/CoOx photoanode developed by Finke and coworkers.61 (B) Corresponding transient photocurrent density as a function of applied potential measured with ITO/PMPDI/CoOx (red line), ITO/PMPDI (green line) and ITO bare (blue line) under intermittent irradiation (5 s light and 5 s dark), using a potential sweep rate of 5 mV s−1 in a deaerated phosphate buffer at pH 7. (C) Band diagram for the ITO/PMPDI/CoOx photoanode with the estimated energy levels (given vs. Ag/AgCl); the actual potential * of ITO (−0.3 V) corresponds to the bias applied in this study (reproduced from ref. 61, with permission from the American Chemical Society, Copyright 2014). | ||
Although the ITO/PMPDI/CoOx anodes are capable of performing light-driven water oxidation, their efficiency is limited by low light harvesting (only about 12% of incident light is adsorbed by the thin PMPDI films) and low charge transfer efficiency as well as effective charge recombination. However, thicker films absorb more light but exhibit lower photocurrents, due to the low exciton diffusion lengths of this organic material. Despite these limitations, this study showed for the first time that water oxidation can be achieved photochemically by using a thin film composed of a monolayer of molecular organic dye coupled to a water oxidation catalyst.
In order to improve the performance of their photoanode, Finke and co-workers62 introduced an inorganic semi-conductor, consisting of a nanostructured metal oxide of TiO2, SnO2 or WO3, to design a dye-sensitized photoelectrochemical cell (DS-PEC) (see Fig. 1). These semiconductors, deposited onto FTO electrodes, were photosensitized with the PMPDI dye (Fig. 11(A)). The dye-sensitized mesoporous semiconducting electrode (i.e. FTO/SC/PMPDI) would absorb more light due to its higher surface area compared to the simple deposition of PMPDI dye on flat ITO (ITO/PMPDI), by keeping the thickness of the dye monolayer constant. To increase the stability, PMPDI was chemisorbed via one of the phosphonate groups onto the SC surfaces, and not spin-coated as in their previous work.61
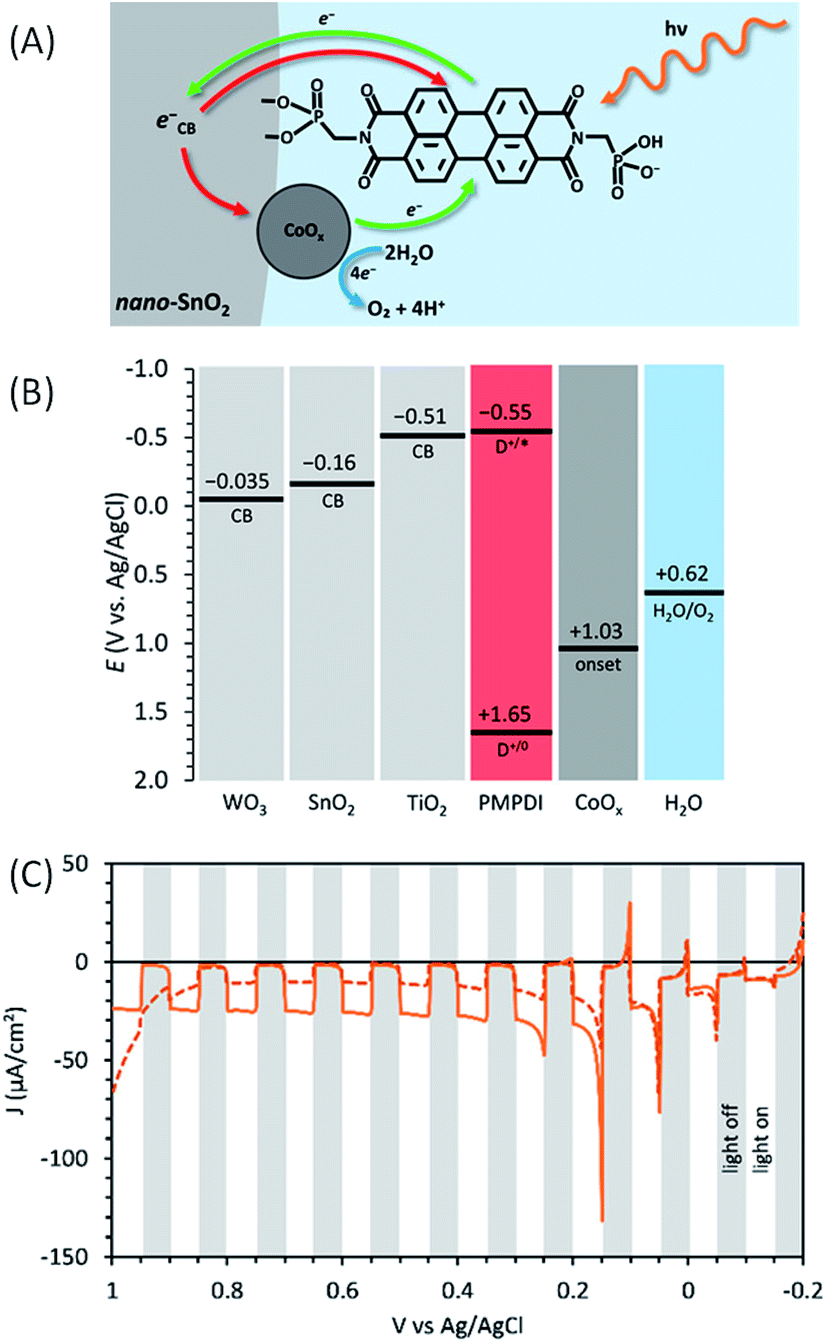 | ||
| Fig. 11 (A) Schematic representation of the FTO/SnO2/PMPDI/CoOx photoanode developed by Finke and coworkers62 (red arrows correspond to the charge recombination). (B) Estimated energy diagram for the SC/PMPDI/CoOx system at pH 7, where the SC was either WO3 or SnO2 or TiO2. (C) Corresponding transient photocurrent density as a function of applied potential for FTO/SnO2/PMPDI before (solid line) and after (dashed line) photoelectrochemical deposition of CoOx under intermittent irradiation (5 s white light transients), using a potential sweep rate of 10 mV s−1 in a deaerated phosphate buffer at pH 7 (reproduced from ref. 62, with permission from the American Chemical Society, Copyright 2017). | ||
The driving force for the electron injection from the photoexcited dye (PMPDI*) into the conduction band of the different SCs increases in the order TiO2 < SnO2 < WO3 (Fig. 11(B), the CB energies being estimated by the electrochemical photocurrent method). Consequently, it is expected that the photocurrent of the corresponding photoanodes using these SCs will follow the same trend. However, the chemisorption of PMPDI dye was less efficient on SnO2 than on TiO2 and not at all effective on WO3, due to the increasing acidity of the SC surface in the order TiO2 < SnO2 < WO3, which disfavors the coordination of the phosphonate group on the metal.
Then, the relative efficiency of the electron transfer occurring on the surface of the FTO/TiO2/PMPDI and FTO/SnO2/PMPDI photoelectrodes before the deposition of the CoOx particles was estimated by the measuring the photocurrent in the presence of a sacrificial reductant such as hydroquinone (denoted as H2Q) at 20 mM in aqueous phosphate buffer at pH 7. With a bias of +0.2 V vs. Ag/AgCl, the photocurrent produced by FTO/TiO2/PMPDI with H2Q was much lower than that measured with FTO/SnO2/PMPDI, the latter reaching 1100 μA cm−2 with a light-harvesting efficiency of 99% at 490 nm, the λmax of the dye.
Due to the higher photocurrent obtained with the SnO2 substrate indicating a more efficient electron injection from the excited PMPDI dye into the SC, the CoOx water oxidation catalyst was then photoelectrochemically deposited on FTO/SnO2/PMPDI (Fig. 11(A)), following a similar procedure used in their previous work61 (see above) except that a lower potential is applied in the presence of SnO2 (i.e. +0.2 V vs. Ag/AgCl). This potential is not positive enough to directly oxidize Co(II) but sufficiently positive compared to the CB energy level of SnO2 to collect photoinjected electrons. Unfortunately, the transient photocurrent density measured with the FTO/SnO2/PMPDI/CoOx photoanode (∼10 μA cm−2 between +0.2 and +0.9 V vs. Ag/AgCl (i.e. η = −0.42 to +0.28 V)) at pH 7 decreases by a factor of 2–3 compared to that obtained without CoOx deposition (40 μA cm−2 which decreases to 20 μA cm−2 over 5 min) (Fig. 11(C) and Table 1, Entry 11). The faradaic yield of FTO/SnO2/PMPDI/CoOx was estimated to be ∼31% from the quantification of O2 evolution using a generator–collector method.50,81 This yield is lower than that of a previous ITO/PMPDI/CoOx photoanode61 (80%) due to enhanced charge recombination between the photoinjected electrons in SnO2 and holes within CoOx (also called the “scavenging” process by Mallouk, see ref. 51), which is most likely directly deposited on the SnO2 surface in the case of FTO/SnO2/PMPDI/CoOx.
This SnO2/PMPDI/CoOx system was the second example in the literature of a DS-PEC composed only of earth-abundant materials to successfully oxidize water (the first example was TiO2–PH/Co–Pi published in 2012 by Beranek,57 see above). From these results, the authors underline the importance of maintaining the water oxidation catalyst far away from the SC surface and not directly adsorbed on it, to prevent charge recombination reactions.
In 2015, Bignozzi and coworkers63 reported a water-oxidation DS-PEC consisting of a nanocrystalline WO3 film photosensitized with a cationic PDI-N+ dye ((N,N′-bis(2-(trimethylammonium)-ethylene) perylene 3,4,9,10-tetracarboxylic acid bisimide)(PF6)2) with co-deposited IrO2 as a water oxidation catalyst (Fig. 12(A)). This dye is oxidized at a potential of +1.7 V vs. SCE in acetonitrile with a spectroscopic band gap of 2.36 eV resulting in an oxidation potential of −0.66 V vs. SCE at the excited state. The redox properties of PDI-N+ at the ground and excited states allowed an efficient light-driven electron injection into WO3 and an effective hole transfer to IrO2. A spontaneous adsorption of this dye on the surface of the SC via aggregation and hydrophobic forces was observed. The IrO2 nanoparticles were co-deposited by different methods (Fig. 12(B)), namely drop casting (b), spin coating (c), spin coating and drying in warm air (d) and soaking (e). Under irradiation of visible light at λ > 435 nm, all FTO/WO3/PDI-N+–IrO2 photoelectrodes exhibit a photocurrent at a bias of +0.5 V vs. SCE in aqueous solution at pH 3, which increases in the order a < b < c < d < e (type a, photoelectrode without IrO2), consistent with electron transfer to the photogenerated oxidized state of the PDI-N+ dye from IrO2, which in turn catalyzes water oxidation (Fig. 12(B)). The photocurrent can reach ∼70 μA cm−2 (Table 1, Entry 12), at a potential close to +0.5 V vs. SCE (i.e. +0.55 V vs. Ag/AgCl; η = −0.3 V at pH 3), for the best photoelectrodes (types c, d and e). This corresponds to an enhancement of about 4-fold compared to the photoelectrode without IrO2 (type a). The enhancement in photoanodic current is due to an efficient hole transfer from the oxidized dye to IrO2. Besides, the transient photocurrents rapidly decrease due to the charge recombination process between WO3 and the oxidized PDI-N+, which competes with hole transfer from oxidized PDI-N+ to IrO2.
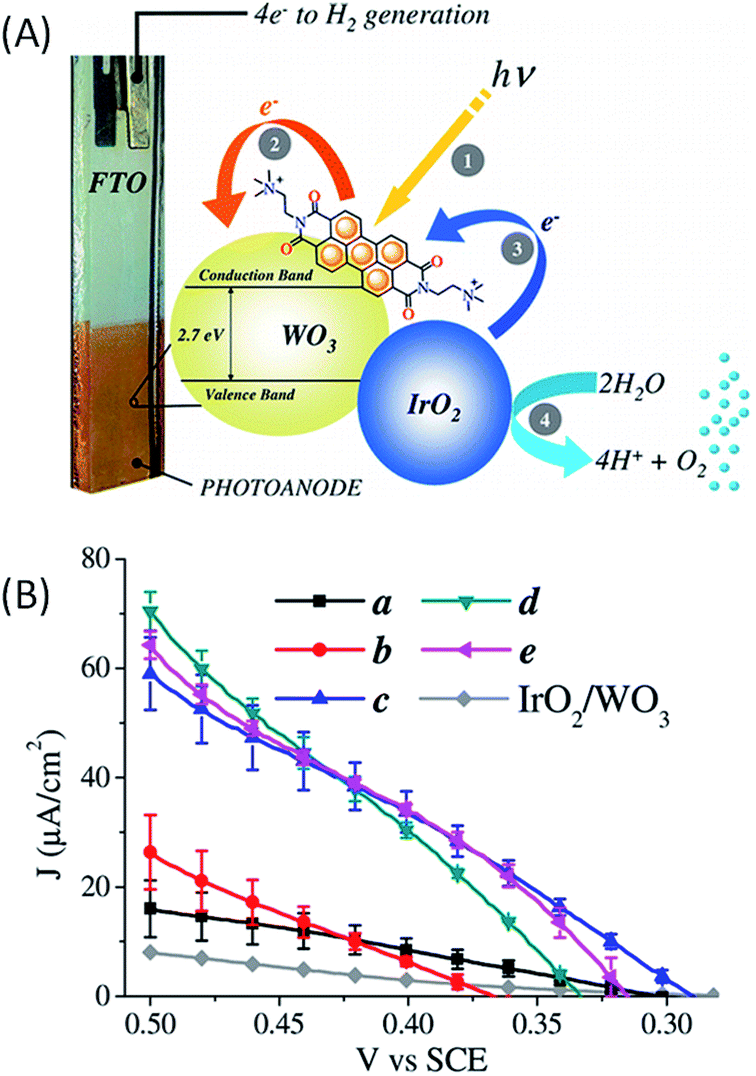 | ||
| Fig. 12 (A) Schematic representation the FTO/WO3/PDI-N+–IrO2 photoanode developed by Bignozzi and coworkers.63 (B) Corresponding photocurrent as a function of applied potential for photoelectrodes a–e, with visible light irradiation and a 435 nm cut off in aqueous 0.1 M NaClO4 solution at pH 3. Photoelectrodes with different deposition methods of IrO2 nanoparticles on FTO/WO3/PDI-N+: a-without IrO2 NPs, b-drop casting, c-spin coating, d-spin coating and drying with warm air, and e-soaking (reproduced from ref. 63, with permission from the American Chemical Society, Copyright 2015). | ||
The investigation of the charge transfer kinetics at 430 nm with the FTO/WO3/PDI-N+ photoelectrode (without IrO2) revealed that light-driven electron injection into the conduction band of WO3 from the excited PDI-N+ occurs in a few nanoseconds (rate constant of 0.4 × 109 s−1) and is 103 times faster than the charge recombination which takes place on the microsecond scale. When IrO2 is deposited on FTO/WO3/PDI-N+, the lifetime of the photooxidized PDI-N+ is just divided by two, until 0.5 μs, since the hole transfer kinetics to IrO2 occurs on the same time scale as that of the charge recombination process between WO3 and the oxidized dye. Once again, the kinetic study of the electron transfer involved in hybrid photoanodes (i.e. SC/molecular dyes/metallic catalyst) highlights the importance of minimizing the charge recombination processes in order to promote the activation of the catalyst and to ensure high photocatalytic performance.
In summary, hybrid photoanodes with perylene dyes need a significant overpotential (η ≥ +0.28 V, bias ≥ 0.9 V vs. Ag/AgCl) or to be associated with a low band gap semiconductor (such as WO3) for obtaining photocurrents of around 70 μA cm−2.
π-Conjugated naphthalene polymer based chromophores
In 2015, Sivula and coworkers64 investigated an n-type π-conjugated naphthalene benzimidazole polymer as an organic dye, also called poly[benzimidazobenzophenanthroline] (denoted as BBL), to be used in a direct semi-conductor-liquid junction configuration for solar water oxidation (Fig. 13(A)). BBL is known for its exceptional stability and good electron mobility. BBL films were prepared on FTO electrodes by a spray deposition method from an aqueous BBL nanofiber dispersion. Under intermittent simulated solar illumination in buffered aqueous solution at pH 7, a photocurrent density of up to 230 μA cm−2 at 1.23 V vs. RHE was obtained in the presence of a hole scavenger (SO32−), with an optimized nanostructured film thickness of 120 nm. The SO32− oxidation allows the investigation of the PEC properties of the BBL dye with minimum kinetic limitations. The FTO/BBL film was also investigated without SO32− in aqueous solution at different pH from 3.5 to 10.5. It was found that the photocurrent density increases with increasing pH (from ∼10 to 30 μA cm−2 at +1.23 V vs. RHE), but no O2 evolution was detected and no evidence of semi-conductor oxidation after 30 min was observed. Instead, the photooxidation reaction was found to be a two-electron oxidation of water by the bare film to H2O2 which in turn is reduced at the anode to produce the hydroxyl radical (˙OH). The latter was quantified by a fluorescence test in the presence of coumarin in solution.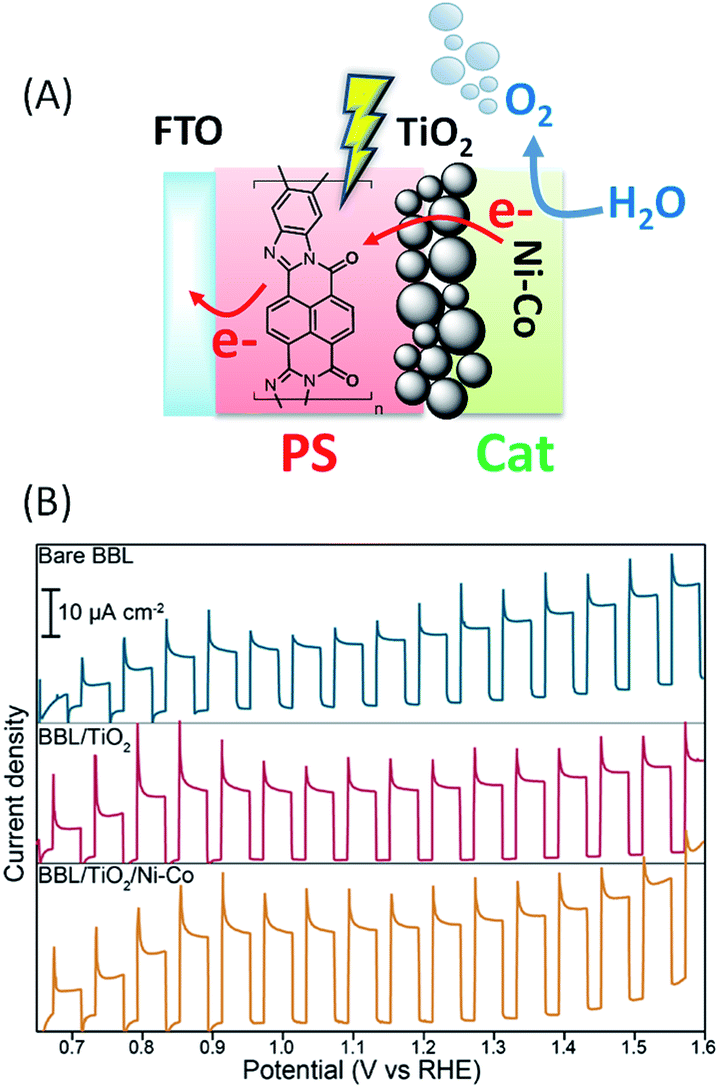 | ||
| Fig. 13 (A) Schematic representation of the FTO/BBL/TiO2/Ni–Co (BBL = poly(benzimidazobenzophenanthroline)) photoanode developed by Sivula and coworkers.64 (B) Corresponding transient photocurrent density as a function of applied potential for the spray deposited film of BBL (230 nm thickness), before and after TiO2 deposition, and after co-deposition of the Ni–Co catalyst in aqueous phosphate buffer at pH 7 (reproduced from ref. 64, with permission from the American Chemical Society, Copyright 2015). | ||
In order to drive the four electron oxidation of water, an OER catalyst consisting of Ni–Co nanoparticles has been directly attached to the BBL surface by a solvothermal method from an aqueous solution of NiSO4·6H2O and Co(NO3)2·6H2O. However no O2 was produced due to the poor catalyst attachment.
The successful attachment of the catalyst was obtained when the BBL film is pretreated with a thin layer of TiO2 (∼1 nm) deposited by ALD and acting as a tunnel junction. Even if the application of TiO2 did not result in a significant change of the observed photocurrent density (from ∼15 to 20 μA cm−2), a slight increase (up to ∼30 μA cm−2) was observed upon application of the catalyst layer (Fig. 13(B) and Table 1, Entry 13). Molecular O2 was confirmed by gas chromatography during constant illumination chronoamperometry measurements (j ∼ 10 μA cm−2) with a faradaic yield of 82 ± 16%.
In the same vein as Finke61,62 and Beranek's54,57,60 work, this study showed that robust π-conjugated organic semi-conductors are suitable for direct PEC water oxidation opening new opportunities for the design and optimization of photoanodes for solar water splitting.
Free-base porphyrin chromophores
In 2015, Mallouk and co-workers51 reported the first examples of functional photoanodes devoted to water oxidation using free-base porphyrins as an organic photosensitizer (Fig. 14). Crystalline IrO2 particles used as a catalyst were first deposited on a semiconducting FTO/TiO2 electrode by a solvothermal method from a solution of citrate-capped IrOx, followed by treatment at 450 °C, as previously described.50 Then, the porphyrins were chemisorbed via a carboxylate substituent on the resulting FTO/TiO2/IrO2 electrodes by soaking.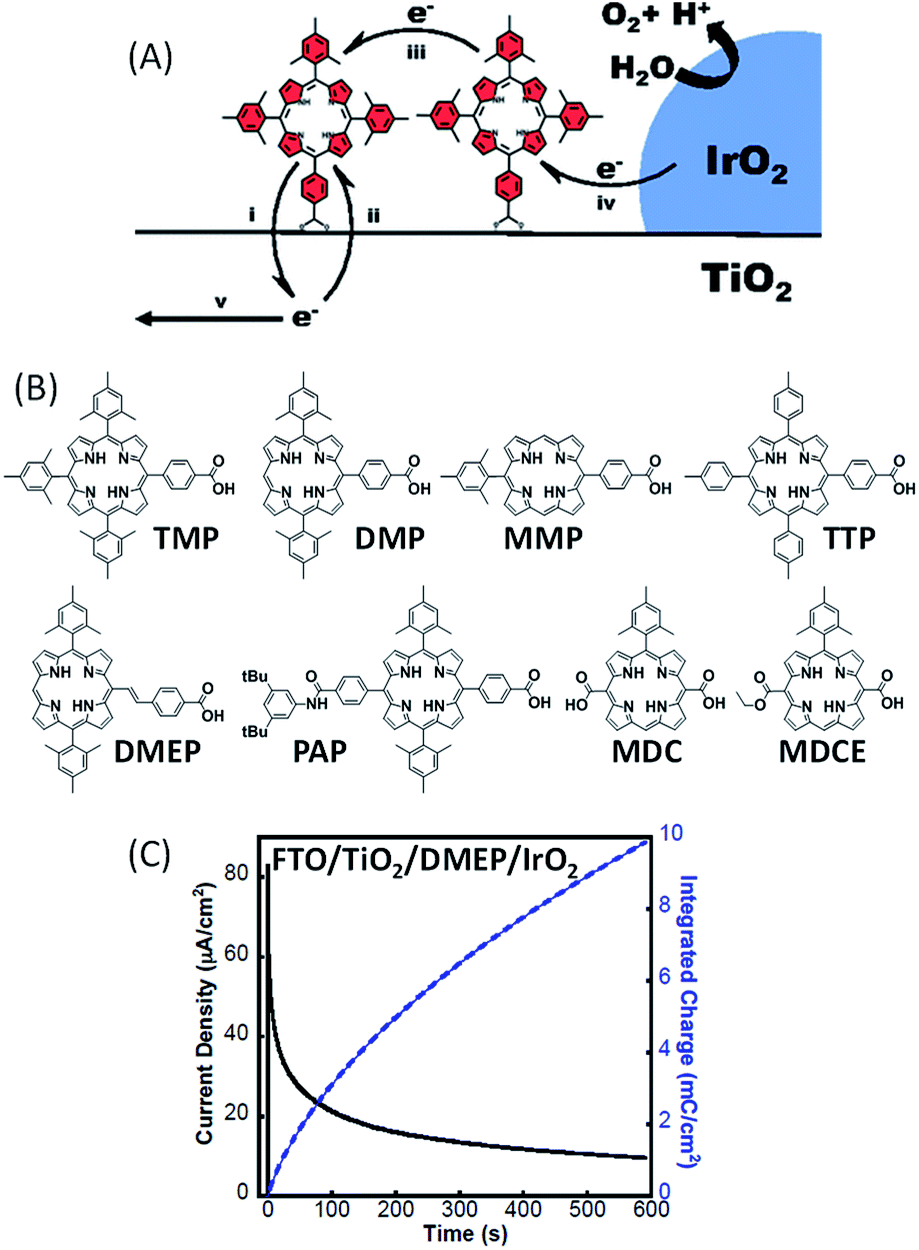 | ||
| Fig. 14 (A) Schematic representation of the FTO/TiO2/porphyrin/IrO2 photoanode developed by Mallouk and coworkers;51 the five steps of the electron transfer process in such a water-splitting DS-PEC are: (i) electron injection, (ii) charge recombination, (iii) hole transfer, (iv) regeneration of the neutral photosensitizer and (v) electron transport to the FTO electrode. (B) Series of free-base porphyrins to implement hybrid photoanodes. (C) Representative photocurrent density and integrated charge as a function of time for a FTO/TiO2/DMEP/IrO2 photoanode under illumination at λ ≥ 410 nm in phosphate buffer at pH 6.8 under an applied potential of +0.1 V vs. Ag/AgCl, and associated with a platinum mesh counter electrode (reproduced from ref. 51, with permission from the National Academy of Sciences of the United states of America, Copyright 2015). | ||
A series of metal-free porphyrins with different substituents were synthesized in order to tune their redox and photophysical properties (Fig. 14(B)). They exhibit Soret bands at ∼410 nm with high extinction coefficients (ε > 105 M−1 cm−1), along with intense Q bands between 500 and 650 nm (1600 < ε < 22500 M−1 cm−1), offering the possibility to activate the photoanodes sensitized by these porphyrins at two different wavelengths in the visible spectrum (410 and 590 nm). When chemisorbed on TiO2, the oxidation of these porphyrins is electrochemically irreversible in aqueous solution with an anodic peak (Epa) located between +1.02 and +1.29 V vs. Ag/AgCl, although some of them present a reversible system in non-aqueous electrolytes. These potentials are positive enough to activate the IrO2 catalyst for water oxidation. Besides, considering the energy level of their singlet excited state (1.87 < E00 < 1.96 eV), the oxidation potential of the porphyrins at the excited state become sufficiently negative for most of these photosensitizers (−0.94 < 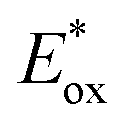 < −0.62 V vs. Ag/AgCl) to inject an electron into the conduction band of TiO2, which is estimated at −0.81 V vs. Ag/AgCl at pH 6.8.
< −0.62 V vs. Ag/AgCl) to inject an electron into the conduction band of TiO2, which is estimated at −0.81 V vs. Ag/AgCl at pH 6.8.
Despite the variability of the spectral and electrochemical properties of these organic dyes, the majority of FTO/TiO2/porphyrin/IrO2 photoanodes exhibit a non-negligible photocurrent with quite similar intensity under full visible light illumination (>410 nm) or red light illumination (>590 nm). Direct observance of O2 with a faradaic yield close to 100% demonstrates that the photocurrent is related to water oxidation. For instance, the photoanode sensitized with DMEP displayed an anodic photocurrent spike of 80 μA cm−2 with a bias of 0.1 V vs. Ag/AgCl (Fig. 14(C)). However, the photocurrent rapidly decreases reaching ∼10 μA cm−2 after 10 min of visible irradiation (Table 1, Entry 14). The poor stability of the photocurrent is not due to the dye degradation, since the UV-visible spectra of the electrodes before and after photoelectrolysis generally show no change, confirming the stability of free-base porphyrin dyes. Otherwise, these photoelectrodes sensitized with free-base porphyrins generated roughly half of the photocurrent of analogous photoanodes sensitized with [Ru(bpy)2(4,4′-(PO3H2)(bpy))] (Ru2) under visible irradiation (>410 nm), although the latter are not active under red light illumination (>590 nm). The authors attributed the poor photocatalytic performance of the FTO/TiO2/porphyrin/IrO2 photoanodes to two factors: slow electron injection into TiO2 and weak cross-surface hole transport between photosensitizers (respectively the steps (i) and (iii) in Fig. 14(A)).
Short circuit absorbed photon-to-current efficiency (APCE) measurements, recorded with FTO/TiO2/dye (without the IrO2 catalyst) and within the first few milliseconds of illumination, demonstrated that the light-driven electron injection from porphyrin dyes into TiO2 is much less efficient than that from the Ru dye, with APCE values between 2.4 and 7.4% for porphyrins and 20.9% for the Ru dye. The weak electron injection into the semiconducting electrode leads to a poor photocurrent during photocatalysis. Another factor affecting the generation of photocurrent is the efficiency of the hole transport at the surface of TiO2, which requires a great cross-surface electron diffusion coefficient (Dapp) of the photosensitizer and slow charge recombination (associated with the time constant τr) between the holes on the oxidized dye and the electrons within the CB of TiO2. In spite of the fact that the porphyrins on TiO2 display slower charge recombination (25 < τr < 59 ms) than that of Ru dye (4 ms) and surface coverages (0.42 × 10−7 < Γ < 2.8 × 10−7 mol cm−2) similar to that of Ru dye (1 × 10−7 mol cm−2), the hole transport over the surface with porphyrins remains inefficient with low Dapp values (<10−11 cm2 s−1). The Dapp values of porphyrin dyes are clearly inferior to those of Ru polypyridyl dyes which generally show values between 10−9 and 10−11 cm2 s−1 and slightly higher photocurrents (see above) when they are deposited on TiO2.
In summary, as for the π-conjugated polymer DS-PEC published by Sivula,64 these hybrid photoanodes sensitized with free base porphyrins do not exhibit significant photocurrents (<40 μA cm−2) and present poor stability over time. Nevertheless, the latter open new opportunities to implement efficient photoelectrochemical water-splitting devices, since they can use red light to drive the water-splitting reaction, thereby extending the absorption of the device across the solar spectrum which could significantly improve the overall energy conversion efficiency.
Summary and outlook
This review focusses on the few examples of hybrid photoanodes reported for light-driven water oxidation which are based on an n-type semiconducting electrode sensitized by a molecular dye in association with a metal oxide as a water oxidation catalyst. Since the introduction of the first example published by Mallouk in 2009, based on TiO2/trisbipyridine Ru–IrOx, efforts have been devoted to the re-design of photoanodes in order to improve their photocatalytic performance and to implement more earth abundant elements, by substituting the polypyridyl Ru photosensitizers with metal-free organic dyes and the iridium oxide catalyst with cobalt oxide.All these hybrid photoanodes are active in neutral or weakly acidic medium, demonstrating their potential for the water oxidation reaction. However, they still suffer from low stability over time under photocatalytic conditions (limited to a few hours in the best case) and exhibit fairly low photocurrents (10–200 μA cm−2; Table 1). The relatively modest photocurrents have been mainly attributed to the poor hole diffusion on the SC surface through PSs and the fast kinetics of back electron transfer (essentially the charge recombination between PS and SC), which competes effectively with the slow electron transfer kinetics between the oxidized PS and the inorganic metal oxide catalyst and the slow catalytic oxidation of water. It is worth noting that the intensity of the photocurrent depends on the bias applied. It is therefore difficult to really compare the performance of different photoanodes because they are not evaluated at the same bias. The bias can vary from 0.55 V to 1.61 V vs. RHE which corresponds to an overpotential varying between −0.68 V and 0.38 V.
Various strategies have been investigated to improve the photocatalytic performance (photocurrent and stability) of TiO2/Ru/IrOx hybrid photoanodes. Among them, the addition of a redox mediator to accelerate the electron transfer between the oxidized Ru dye and IrOx and the introduction of a thin insulating layer of a metal oxide (ZrO2 or Nb2O5) between Ru dye and TiO2 to slow down the back electron transfer were not successful. The approach that considerably increased the photocurrent (by a factor of 2.7) was to deposit a thin layer of TiO2 onto a nanostructured ITO film coated onto a FTO electrode, in order to promote fast electron transport through TiO2 to nanoITO and decrease the charge recombination between the Ru dye and TiO2. Nevertheless, the photocurrent was unstable once again owing to the PS release from the SC surface by hydrolysis of the anchoring groups. To avoid this PS release, a very thin layer of TiO2 was deposited on the Ru dye, before the deposition of IrOx particles, resulting in a stable photocurrent of 100 μA cm−2 over 2 h.
In addition to the detachment or degradation of the PS, the decline of the photocurrent for such Ru-based photoanodes was also ascribed to the drop of pH at the photoanode surface induced by the accumulation of protons (under acidic or neutral conditions) or the consumption of hydroxide anions (under basic conditions) during the photo-oxidation of water. The acidification of the medium close to the surface induces a positive shift of the standard potential of water oxidation, resulting in a decrease of photocurrent at a fixed bias. In order to display a sustained and high photocurrent, future hybrid photoanodes should be able to maintain a neutral or basic pH in proximity to the electrode surface.
Regarding non-noble metal based photoanodes, Beranek's group designed effective and stable hybrid photoanodes using a robust organic polymer film, polyheptazine, to photosensitize a TiO2 electrode. Combined with a metal oxide catalyst such as IrO2, Co–Pi or CoO(OH)x particles, relatively high photocurrents (above 100 μA cm−2) were obtained along with a relatively good stability (up to 4 hours) compared to the other hybrid photoanodes reviewed herein. This promising performance could be related to the robustness of polyheptazine and the good hole mobility within this conjugated polymer which competes effectively with the charge recombination between oxidized polyheptazine and TiO2. This work paves the way for the development of new robust and highly efficient photoanodes devoted to water oxidation employing conjugated polymers as photosensitizers.
Moreover, it should be stressed that in most of these published water-splitting PEC examples, the amount of photosensitizers and metal oxide catalysts deposited on the surface of the electrodes is low and not well-controlled. Photosensitizers are generally adsorbed as a monolayer and the amount of catalyst is rarely evaluated. To go one step further, it will be very interesting to explore novel methods of fabrication and deposition of photosensitizers/metal oxide nanoparticles on semi-conducting electrodes in order to evaluate the effect of the amount and the PS/MOx ratio of the deposited material and its nano-structuration on the performance of the photoanodes (i.e. intensity of the catalytic photocurrent and stability). To this end, a strategy could be incorporating photosensitizers and metal oxide particles into a polymer matrix, by controlling the formation and the size of the particles and the amount of the photosensitizer deposited. The polymer matrix will promote the stability of the MOx particles by precluding their corrosion during photocatalysis.82,83 Our group recently reported such a nanostructured material electrogenerated at the surface for a hybrid photocathode for water reduction to H2 based on a polypyrrole film of Ru tris-bipyridine containing MoSx NPs.41 This photosensitive material, which strongly absorbs visible light due to its thick layer of the Ru dye, exhibits a significant photocatalytic activity owing to the high PS/Cat ratio of 2 and the small (60 nm) and non-agglomerated MoSx particles, which confer a large catalytically active surface area. Such a strategy could be implemented for the preparation of efficient hybrid photoanodes for water oxidation.
Regarding photosensitizers, the published examples of hybrid photoanodes for water oxidation already employed the main families of molecular photosensitizers with adequate absorption, photophysical and redox properties to promote visible-light water oxidation through a metal oxide catalyst. If other dye families potentially more resistant to the degradation in aqueous media have to be explored in order to improve the global stability of the photoelectrodes, the bottleneck of these hybrid photoanodes is not so much the dye, but rather the slow kinetics and high overpotential of the oxidation of water. Up to now, only three families of metal oxides (IrO2, Co–Pi or CoO(OH)x) have been tested while plenty of inorganic OER catalysts with lower overpotentials have been reported in the last few decades such as, for example, alloys of NiFeOx or CoFeOx.45,84,85 Using such inorganic OER catalysts with lower overpotentials should be another way to significantly improve the performance of hybrid photoanodes.
Finally, these hybrid photoanodes could also be used to drive photo-induced oxidation of organic or inorganic substrates, as an alternative to the OER, within a PEC cell geared toward H2 production. Basically, the OER, the bottleneck of the water splitting reaction, could be replaced with more kinetically facile oxidation of organic (e.g. 2,5-hydroxymethylfurfural (HMF)) or halide substrates (e.g. chloride) to produce more valorized chemicals than O2 (respectively 2,5 difuran carboxylic acid (FDCA) and Cl2 gas).86 Since metal oxide materials have been identify as catalysts for the oxidation of alternate substrates,86 the use of hybrid photoanodes combining metal oxide catalysts and molecular PSs would enable defining new paradigms for solar-driven organic chemistry and access to new PECs cleanly producing H2 at the photocathode and value-added chemicals at the photoanode from cheap and renewable feedstocks.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Becas-Chile (Conicyt) “Programa de formación de capital humano avanzado” for the PhD fellowship of DVM (No. 72150091), CAN (No. 72170399), the ECOS-CONICYT exchange program (C13E01), and the COST CM1202 program (PERSPECT H2O) are acknowledged. This work has been partially supported by Labex Arcane and CBH-EUR-GS (ANR-17-EURE-0003).Notes and references
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946 CrossRef CAS PubMed.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357 RSC.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC.
- K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., Int. Ed., 2013, 52, 10426 CrossRef CAS PubMed.
- M. S. Prévot and K. Sivula, J. Phys. Chem. C, 2013, 117, 17879 CrossRef.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407 CrossRef CAS.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760 RSC.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90 CrossRef CAS.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560 RSC.
- P. Xu, N. S. McCool and T. E. Mallouk, Nano Today, 2017, 14, 42 CrossRef CAS.
- A. Eftekhari, V. J. Babu and S. Ramakrishna, Int. J. Hydrogen Energy, 2017, 42, 11078 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- T. Bosserez, J. Rongé, J. van Humbeeck, S. Haussener and J. Martens, Oil Gas Sci. Technol., 2015, 70, 877 CrossRef CAS.
- M. Crespo-Quesada and E. Reisner, Energy Environ. Sci., 2017, 10, 1116 RSC.
- M. F. Weber and M. J. Dignam, J. Electrochem. Soc., 1984, 131, 1258 CrossRef CAS.
- L. L. Gao, F. Li, H. G. Hu, X. F. Long, N. Xu, Y. P. Hu, S. Q. Wei, C. L. Wang, J. T. Ma and J. Jin, ChemSusChem, 2018, 11, 2502 CrossRef CAS PubMed.
- F. Li, J. Li, L. Gao, Y. Hu, X. Long, S. Wei, C. Wang, J. Jin and J. Ma, J. Mater. Chem. A, 2018, 6, 23478 RSC.
- C. Wang, X. Long, S. Wei, T. Wang, F. Li, L. Gao, Y. Hu, S. Li and J. Jin, ACS Appl. Mater. Interfaces, 2019, 11, 29799 CrossRef CAS PubMed.
- L. Gao, X. Long, S. Wei, C. Wang, T. Wang, F. Li, Y. Hu, J. Ma and J. Jin, Chem. Eng. J., 2019, 378, 122193 CrossRef CAS.
- N. Xu, F. Li, L. Gao, H. Hu, Y. Hu, X. Long, J. Ma and J. Jin, Int. J. Hydrogen Energy, 2018, 43, 2064 CrossRef CAS.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- A. Kasahara, K. Nukumizu, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2003, 107, 791 CrossRef CAS.
- A. Kasahara, K. Nukumizu, G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. A, 2002, 106, 6750 CrossRef CAS.
- D. Yokoyama, T. Minegishi, K. Maeda, M. Katayama, J. Kubota, A. Yamada, M. Konagai and K. Domen, Electrochem. Commun., 2010, 12, 851 CrossRef CAS.
- K. Ohashi, K. Uosaki and J. O. Bockris, Int. J. Energy Res., 1977, 1, 25 CrossRef CAS.
- W. Siripala, A. Ivanovskaya, T. F. Jaramillo, S.-H. Baeck and E. W. McFarland, Sol. Energy Mater. Sol. Cells, 2003, 77, 229 CrossRef CAS.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456 CrossRef CAS PubMed.
- A. Paracchino, N. Mathews, T. Hisatomi, M. Stefik, S. D. Tilley and M. Grätzel, Energy Environ. Sci., 2012, 5, 8673 RSC.
- C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482 RSC.
- E. Aharon-Shalom and A. Heller, J. Electrochem. Soc., 1982, 129, 2865 CrossRef CAS.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425 CrossRef CAS PubMed.
- S. Licht, B. Wang, S. Mukerji, T. Soga, M. Umeno and H. Tributsch, Int. J. Hydrogen Energy, 2001, 26, 653 CrossRef CAS.
- E. Verlage, S. Hu, R. Liu, R. J. R. Jones, K. Sun, C. Xiang, N. S. Lewis and H. A. Atwater, Energy Environ. Sci., 2015, 8, 3166 RSC.
- W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966 CrossRef CAS PubMed.
- H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Sci., 2018, 5, 1700684 CrossRef PubMed.
- Y. Lattach, J. Fortage, A. Deronzier and J.-C. Moutet, ACS Appl. Mater. Interfaces, 2015, 7, 4476 CrossRef CAS PubMed.
- C. E. Castillo, M. Gennari, T. Stoll, J. Fortage, A. Deronzier, M. N. Collomb, M. Sandroni, F. Légalité, E. Blart, Y. Pellegrin, C. Delacote, M. Boujtita, F. Odobel, P. Rannou and S. Sadki, J. Phys. Chem. C, 2015, 119, 5806 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788 CrossRef PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337 RSC.
- W. J. Youngblood, S. H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926 CrossRef CAS PubMed.
- S.-H. A. Lee, Y. Zhao, E. A. Hernandez-Pagan, L. Blasdel, W. J. Youngblood and T. E. Mallouk, Faraday Discuss., 2012, 155, 165 RSC.
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612 CrossRef CAS PubMed.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber and T. E. Mallouk, J. Am. Chem. Soc., 2014, 136, 10974 CrossRef CAS PubMed.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber, M. E. Strayer, N. M. Vargas-Barbosa and T. E. Mallouk, J. Phys. Chem. C, 2014, 118, 17046 CrossRef CAS.
- J. R. Swierk, D. D. Méndez-Hernández, N. S. McCool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V. Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1681 CrossRef CAS PubMed.
- K. E. Michaux, A. A. Gambardella, L. Alibabaei, D. L. Ashford, B. D. Sherman, R. A. Binstead, T. J. Meyer and R. W. Murray, J. Phys. Chem. C, 2015, 119, 17023 CrossRef CAS.
- P. Wei, B. Hu, L. Zhou, T. Su and Y. Na, J. Energy Chem., 2016, 25, 345 CrossRef.
- M. Bledowski, L. Wang, A. Ramakrishnan, O. V. Khavryuchenko, V. D. Khavryuchenko, P. C. Ricci, J. Strunk, T. Cremer, C. Kolbeck and R. Beranek, Phys. Chem. Chem. Phys., 2011, 13, 21511 RSC.
- L. Wang, M. Bledowski, A. Ramakrishnan, D. König, A. Ludwig and R. Beranek, J. Electrochem. Soc., 2012, 159, H616 CrossRef CAS.
- M. Bledowski, L. Wang, A. Ramakrishnan and R. Beranek, MRS Proceedings, 2012, 1442, mrss12 CrossRef.
- M. Bledowski, L. Wang, A. Ramakrishnan, A. Bétard, O. V. Khavryuchenko and R. Beranek, ChemPhysChem, 2012, 13, 3018 CrossRef CAS PubMed.
- M. Bledowski, L. Wang, A. Ramakrishnan and R. Beranek, J. Mater. Res., 2013, 28, 411 CrossRef CAS.
- O. V. Khavryuchenko, L. Wang, D. Mitoraj, G. H. Peslherbe and R. Beranek, J. Coord. Chem., 2015, 68, 3317 CrossRef CAS.
- L. Wang, D. Mitoraj, S. Turner, O. V. Khavryuchenko, T. Jacob, R. K. Hocking and R. Beranek, ACS Catal., 2017, 7, 4759 CrossRef CAS.
- J. T. Kirner, J. J. Stracke, B. A. Gregg and R. G. Finke, ACS Appl. Mater. Interfaces, 2014, 6, 13367 CrossRef CAS PubMed.
- J. T. Kirner and R. G. Finke, ACS Appl. Mater. Interfaces, 2017, 9, 27625 CrossRef CAS PubMed.
- F. Ronconi, Z. Syrgiannis, A. Bonasera, M. Prato, R. Argazzi, S. Caramori, V. Cristino and C. A. Bignozzi, J. Am. Chem. Soc., 2015, 137, 4630 CrossRef CAS PubMed.
- P. Bornoz, M. S. Prévot, X. Yu, N. Guijarro and K. Sivula, J. Am. Chem. Soc., 2015, 137, 15338 CrossRef CAS PubMed.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795 RSC.
- A. Kay and M. Grätzel, Chem. Mater., 2002, 14, 2930 CrossRef CAS.
- E. Palomares, J. N. Clifford, S. A. Haque, T. Lutz and J. R. Durrant, J. Am. Chem. Soc., 2003, 125, 475 CrossRef CAS PubMed.
- W. L. Hoffeditz, M. J. Pellin, O. K. Farha and J. T. Hupp, Langmuir, 2017, 33, 9298 CrossRef CAS PubMed.
- R. Beranek, Adv. Phys. Chem., 2011, 2011, 20 Search PubMed.
- K. Kalyanasundaram and M. Grätzel, Coord. Chem. Rev., 1998, 177, 347 CrossRef CAS.
- B. Gerey, E. Gouré, J. Fortage, J. Pécaut and M.-N. Collomb, Coord. Chem. Rev., 2016, 319, 1 CrossRef CAS.
- J. R. Swierk, N. S. McCool and T. E. Mallouk, J. Phys. Chem. C, 2015, 119, 13858 CrossRef CAS.
- Z. Huang, Y. V. Geletii, D. Wu, C. L. Anfuso, D. G. Musaev, C. L. Hill and T. Lian, ed. Y. Tachibana, in Interfacial Charge Transfer Dynamics in TiO2-Sensitizer-Ru4POM Photocatalytic Systems for Water Oxidation, SPIE, Bellingham, WA, 2011, vol. 8109, p. 810903 Search PubMed.
- J. B. Asbury, Y.-Q. Wang, E. Hao, H. N. Ghosh and T. Lian, Res. Chem. Intermed., 2001, 27, 393 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- Y. Surendranath, M. Dinca and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615 CrossRef CAS PubMed.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109 RSC.
- Y. Surendranath, D. A. Lutterman, Y. Liu and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6326 CrossRef CAS PubMed.
- S. K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513 CrossRef CAS.
- E. M. P. Steinmiller and K.-S. Choi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20633 CrossRef CAS PubMed.
- B. D. Sherman, M. V. Sheridan, C. J. Dares and T. J. Meyer, Anal. Chem., 2016, 88, 7076 CrossRef CAS PubMed.
- Z. B. Shifrina, V. G. Matveeva and L. M. Bronstein, Chem. Rev., 2019 DOI:10.1021/acs.chemrev.9b00137.
- D. V. Morales, C. N. Astudillo, Y. Lattach, B. F. Urbano, E. Pereira, B. L. Rivas, J. Arnaud, J.-L. Putaux, S. Sirach, S. Cobo, J.-C. Moutet, M.-N. Collomb and J. Fortage, Catal. Sci. Technol., 2018, 8, 4030 RSC.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549 CrossRef CAS.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |