
A photo-crosslinkable bis-triarylamine side-chain polymer as a hole-transport material for stable perovskite solar cells†
Marie-Hélène
Tremblay‡
a,
Kelly
Schutt‡
b,
Yadong
Zhang
a,
Jongchul
Lim
 b,
Yen-Hung
Lin
b,
Jonathan H.
Warby
b,
Stephen
Barlow
a,
Henry J.
Snaith
b and
Seth R.
Marder
*a
b,
Yen-Hung
Lin
b,
Jonathan H.
Warby
b,
Stephen
Barlow
a,
Henry J.
Snaith
b and
Seth R.
Marder
*a
aSchool of Chemistry and Biochemistry, Center for Organic Photonics and Electronics (COPE), Georgia Institute of Technology GA, Atlanta 30332-0400, USA. E-mail: seth.marder@chemistry.gatech.edu
bClarendon Laboratory, Department of Physics, University of Oxford, Parks Road, Oxford OX1 3PU, UK
First published on 12th September 2019
Abstract
A crosslinkable acrylate random copolymer with both hole-transporting bis(triarylamine) and photocrosslinkable cinnamate side chains is compared to the widely used poly(4-butyl-triphenylamine-4′,4′′-diyl) (PolyTPD) as a hole-transport material (HTM) in positive–intrinsic–negative (p–i–n) perovskite solar cells (PSCs). The crosslinked films of this HTM exhibit improved wettability by precursor solutions of the perovskite relative to PolyTPD; this facilitates high-quality full film coverage by the subsequently deposited perovskite layer on smooth substrates, which is difficult to achieve with PolyTPD without the use of additional interlayers. PSCs fabricated using undoped and crosslinked copolymer achieve steady-state power outputs that are comparable to those of cells incorporating p-doped PolyTPD (with interlayers) as the HTM. The devices made with this material also exhibited improved initial stability under high-intensity ultraviolet LED irradiation, in comparison to those with the PolyTPD analogue. Remarkably, after 3000 h of aging in an oven at 85 °C in a nitrogen-filled glovebox, device efficiency showed no degradation; the SPO was comparable to the initial performance.
Introduction
The efficiency of lead-halide perovskite solar cells (PSCs) has improved significantly since their debut in the literature in 2009.1–5 A transition to polycrystalline thin films,6,7 improvements in film quality,8–10 organic and metal contacts,11,12 doping of charge extraction layers,13,14 and band-gap tuning15,16 have all contributed to the achievement of power conversion efficiency (PCE) values greater than 20%. Nevertheless, the stability of these solar cells must be improved for commercialization of this technology,17,18 and often the most efficient cells are not the most stable.19 Among ways to achieve improved stability, one can use mixed-cation perovskites20,21 or a quasi two-dimensional perovskite,22–27 and modify the chemical structure of the hole-28–31 and electron-transport materials32–34 (HTMs and ETMs, respectively) and of the dopants used to enhance their conductivity.HTMs play a crucial role in PSCs by reducing the charge recombination that occurs at direct perovskite/hole-collecting electrode interfaces. Organic molecules and polymers, as well as various inorganic materials, can be used as HTMs in PSCs.35 The development of crosslinkable HTMs and ETMs for multilayer organic electronic devices, such as light-emitting diodes (OLEDs), was motivated by the need to avoid possible dissolution of the first layer during solution deposition of subsequent layers. Once crosslinked (thermally, photochemically, or by acid), the material becomes insoluble, allowing the subsequent layer to be easily processed on top of it and protecting the layer underneath. Crosslinking offers similar advantages for the HTMs of so-called positive-intrinsic-negative (p–i–n) PSCs, potentially allowing the lead-halide perovskite to be solution-processed without damage to the HTM layer. Easily processed and insolubilized HTMs are also ideal candidates for tandem cells, where many more layers are required to be sequentially processed. A few crosslinkable HTMs and ETMs have been reported to show promise in PSCs applications. Thermal crosslinking has been achieved using HTMs with two or more styrene,36–38 or other vinyl,39 moieties or by reacting styrene-functionalized HTMs with oligo(thiol) derivatives.40 While respectable photo and thermal stability, and performance have been achieved with those materials, the high temperature and crosslinking time needed for alkene polymerization, and the multiple ways in which the thiols used in the ene/thiol approach can react, are not optimal for multilayer printable electronics.
Lithium bis(trifluoromethanesulfonyl)imide (Li-TFSI), in combination with oxygen exposure, has been the frequently used dopant for HTMs in PSCs, and is typically used in conjunction with 4-tert-butylpyridine (tBP), which helps prevent phase segregation.41 Unfortunately, the use of both Li-TFSI and tBP have been shown to degrade performance in long term stability tests, since Li-TFSI is hygroscopic and tBP is volatile.18,42 While alternative dopants have not been as heavily investigated, F4-TCNQ is commonly utilized in p–i–n PSCs, but has been found to migrate into adjacent layers in device stacks due to its relatively high diffusivity, and is also highly volatile.43,44 A dopant-free HTM or one in which dopant diffusion is minimized is clearly desirable for long-term stability. Crosslinking could potentially also help in this regard.
Here we report the use of a photo-crosslinkable HTM, P1-2 (Fig. 1A), in PSCs. We investigate the impact of a crosslinked copolymer of acrylate monomers with bis(triarylamine) and cinnamate side groups45 on the stability of PSCs. By careful optimization of p–i–n PSCs, we achieve a stable, dopant-free HTM with comparable performance to state-of-the-art p–i–n cells employing F4-TCNQ-doped poly(4-butyl-triphenylamine-4′,4′′-diyl) (PolyTPD, Fig. 1B) as the HTM.
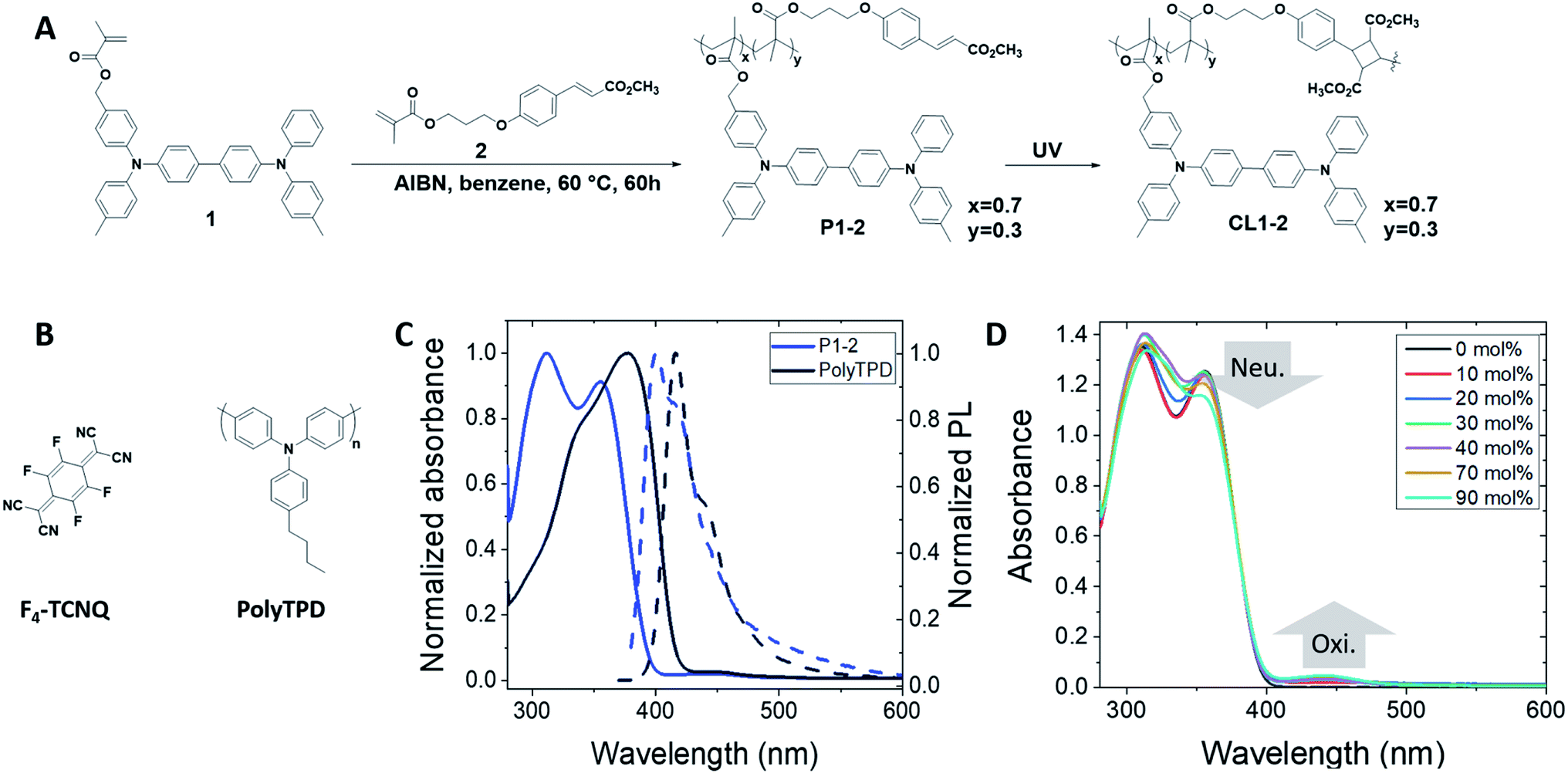 | ||
| Fig. 1 (A) Synthesis of copolymer P1-2 from the bis(triarylamine) 1 and the cinnamate 2. The crosslinked material CL1-2 can be obtained after illumination with UV light. (B) Chemical structure of PolyTPD and F4-TCNQ, the dopant usually used to dope triarylamine-based HTMs. (C) Comparison of the absorbance (solid line) and photoluminescence (dashed line) spectra of P1-2 and PolyTPD in toluene (with 20 mol% F4-TCNQ per triarylamine unit). (D) UV-vis spectrum showing the doping in toluene solution of P1-2 with F4-TCNQ (where Neut. and Ox. indicate signals attributable to the neutral and oxidized bis(diarylamino)biphenyl units, respectively, and z mol% is relative to the number of triarylamine units, i.e. corresponds to 2z molar equivalents per 100 moles of monomer). | ||
Results and discussion
Synthesis and characterization of the HTM
HTMs for PSCs often contain the bis(diarylamino)biphenyl moiety. For example, the widely used spiro-OMeTAD consists of two such moieties linked by a bridging carbon atom. More recently, polymeric materials containing the same moiety, such as PolyTPD and PTAA (poly(2,4,6-trimethyltriphenylamine-4′,4′′-diyl)), have shown great potential as HTMs in so-called “inverted” (i.e., p–i–n) PSCs,46–48 where the HTM is the first layer deposited. Cells using PolyTPD have achieved a record of 19.1% PCE.14 The same bis(diarylamino)biphenyl motif has also been widely used in OLEDs and some of us have previously reported a series of papers45,49–52 in which random copolymers of monomers with bis(diarylamino)biphenyl functionalization and monomers with cinnamate side chains have been used as solution-processible materials that can subsequently be insolubilized through brief UV irradiation to induce a [2 + 2] cycloaddition of the cinnamate groups. This approach is modular in that the properties of the polymer can be adjusted through varying the HTM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :crosslinker ratio, the ionization energy (IE) of the polymer can be varied through the substitution pattern of the HTM monomer, and other polymer properties can be varied through the choice of the polymer backbone. We chose one of these previously reported polymers – P1-2, the copolymer of a bis(diarylamino)biphenyl-acrylate monomer (1) and a cinnamate-acrylate monomer (2)45 (Fig. 1A) – as a candidate HTM for investigation in PSCs.
:crosslinker ratio, the ionization energy (IE) of the polymer can be varied through the substitution pattern of the HTM monomer, and other polymer properties can be varied through the choice of the polymer backbone. We chose one of these previously reported polymers – P1-2, the copolymer of a bis(diarylamino)biphenyl-acrylate monomer (1) and a cinnamate-acrylate monomer (2)45 (Fig. 1A) – as a candidate HTM for investigation in PSCs.
Monomers 1 and 2 and copolymer P1-2 were synthesized as previously described.45 Films of the polymer were insolubilized using a short illumination time at 356 nm, which does not significantly degrade the HTM unit of the polymer. From the point of view of PSC fabrication, this facile photocrosslinking is potentially advantageous over alternative thermal crosslinker approaches that require high temperatures >150 °C since the process can be done at ambient temperature on plastic substrates. Moreover, for n–i–p structures, the low power UV-lamp used would not degrade the underlying perovskite layer, whereas the temperatures required for many thermal crosslinking reactions would.
P1-2 has a slightly higher energy onset of optical absorption than PolyTPD (Fig. 1C, Table 1), which could be advantageous in allowing more of the solar spectrum to reach the active layer, potentially reducing parasitic absorption and maximizing current density. Electrochemical measurements on the monomer 1 in CH2Cl2/0.1 M Bu4NPF6 indicate a reversible oxidation at +0.26 V vs. FeCp2+/0 (see Fig. S1†), from which an IE of 5.1 eV is estimated, similar to that of PolyTPD (Table 1). Addition of F4-TCNQ to solutions of either PolyTPD or P1-2 leads to only moderate changes in the optical spectra, suggesting inefficient doping with this oxidant. This is further illustrated by Fig. 1D, where the effects of successive dopant addition are shown for the case of P1-2. The low yield of oxidized HTM in each case is expected given the redox potential of F4TCNQ (+0.15 V in the CH2Cl2/0.1 M Bu4NPF6).53,54
| PolyTPD | P1-2 | |
|---|---|---|
| a The spectra were measured in toluene. b Measured at 0.1 absorbance. c Estimated according to IE (eV) = 4.8 + E1,1/2 (V), where the E1,1/2 is relative to FeCp2+/0. d Electron affinity estimated from EA = IE − Eopt,gap. e Optical gap calculated using the crossing of normalized absorbance and fluorescence spectra. f Measured by 4-point probe of the undoped films. g According to literature: PolyTPD,55P1-2 (prior to crosslinking).45 h From ref. 55. i Estimated from E1/2 of monomer 1. j Measured on the crosslinked film CL1-2. | ||
| λ max,abs /nm | 377 (338) | 313 (356) |
| λ max,PL , /nm | 417 (441) | 400 (416) |
| IEc/eV | 5.2h | 5.1i |
| EAd/eV | 2.1 | 1.8 |
| E opt,gap /eV | 3.06 | 3.23 |
| σ /S m−1 | 0.010 ± 0.001 | 0.0141 ± 0.0007j |
| T g /°C | 230 | 137 |
Characterization of HTM films and perovskite growth
Atomic force microscopy of thin films of CL1-2 and PolyTPD on glass substrates showed similar homogeneity and smoothness (see Fig. S2†). Perovskite films crystallized on top of the two HTM films on FTO substrates also appeared similar by scanning electron microscopy (SEM), although we observed a larger distribution of bright color grains on the CL1-2 film than on PolyTPD (Fig. S3†); it is unclear what the cause of this observation is, or whether it has any device consequence, although excess PbI2 has been found to be beneficial in previous PSC studies.56,57 The powder X-ray diffraction (PXRD) of the perovskite films on top of the two different HTMs are also similar, indicating comparable perovskite growth on both materials when FTO is used as the substrate (see Fig. S4†).The conductivity of the HTMs was characterized using four-point probe measurements with and without F4-TCNQ. Slightly lower conductivity was measured for pristine PolyTPD compared to pristine CL1-2 (see Fig. 2) after annealing at 130 °C (the temperature at which the HTMs were annealed – see experimental in the ESI†). In both cases, doping the HTM with F4-TCNQ p-dopant increased the conductivity of the HTM by similar amounts; however, as discussed in the following section, PSC device performance was comparable for undoped and doped CL1-2, while the performance increased upon doping of PolyTPD (see device data later). Although the active layers of the PSCs fabricated in this work (see below) are annealed at only 100 °C, some perovskite materials necessitate high annealing temperature (e.g., often >200 °C for inorganic CsPbX3 perovskites).58,59 Accordingly, we also investigated the effect of higher temperatures on both doped CL1-2 and doped uncrosslinked P1-2. After annealing at 180 °C, the doped CL1-2 conductivity was found to be higher than that of doped uncrosslinked P1-2, which is consistent with crosslinking perhaps helping to retain the volatile F4-TCNQ within the film. When films were further annealed at 250 °C, the conductivity of CL1-2 actually increased, while the integrity of the PolyTPD film was damaged, and the conductivity of PolyTPD decreased dramatically, perhaps due to film damage occurring above the Tg of the latter polymer. This enhanced dopant retention of doped CL1-2 may, therefore, make it a suitable HTM for PSCs in which subsequently deposited layers require high annealing temperatures.
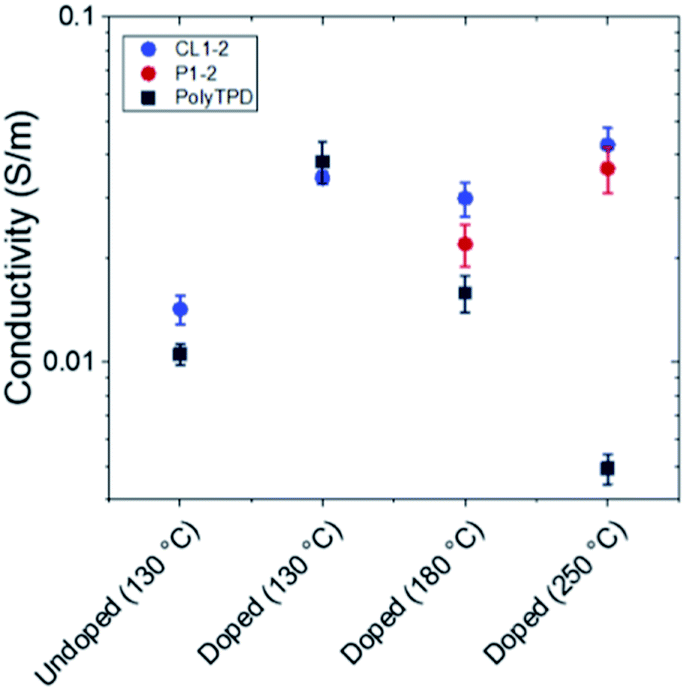 | ||
| Fig. 2 4-point probe conductivity measurement of PolyTPD and P1-2/CL1-2. Films are 10 nm thick. The samples are first annealed 130 °C, which correspond to the processing temperature in the solar cells, and further annealed at the indicated temperature for 10 min. The HTMs are both doped with 20 mol% F4-TCNQ in solution prior to film deposition. | ||
Finally, to investigate the charge transfer at the perovskite and the HTM interface, we performed steady-state photoluminescence (PL) measurements (Fig. S5†). The doped CL1-2 showed similar PL quantum yield quenching compared to that of PolyTPD, indicating comparable hole-extraction capability of both HTMs at the perovskite/HTM interface, but different PL lifetimes (see below).
Device fabrication and characterization
The photovoltaic performance of CL1-2 and PolyTPD as HTMs in PSCs was compared using the architecture: FTO/HTM/Cs0.05(FA0.85MA0.15)0.95Pb(I0.9Br0.1)3/PCBM/BCP/Ag (or Cr/Au for thermal stability tests), where FTO = fluorine-doped tin oxide, MA = methylammonium, FA = formamidinium, PCBM = phenyl-C61-butyric acid methyl ester, and BCP = bathocuproine. The two HTM layers had approximately the same thickness (∼6–8 nm as determined by profilometry). Both pristine and doped CL1-2 showed similar performance to PolyTPD. The current-density–voltage (J–V) characteristics were measured under a simulated AM 1.5G (100 mW cm−2) sunlight (see Fig. 3 and S6, S7 and Table S1†).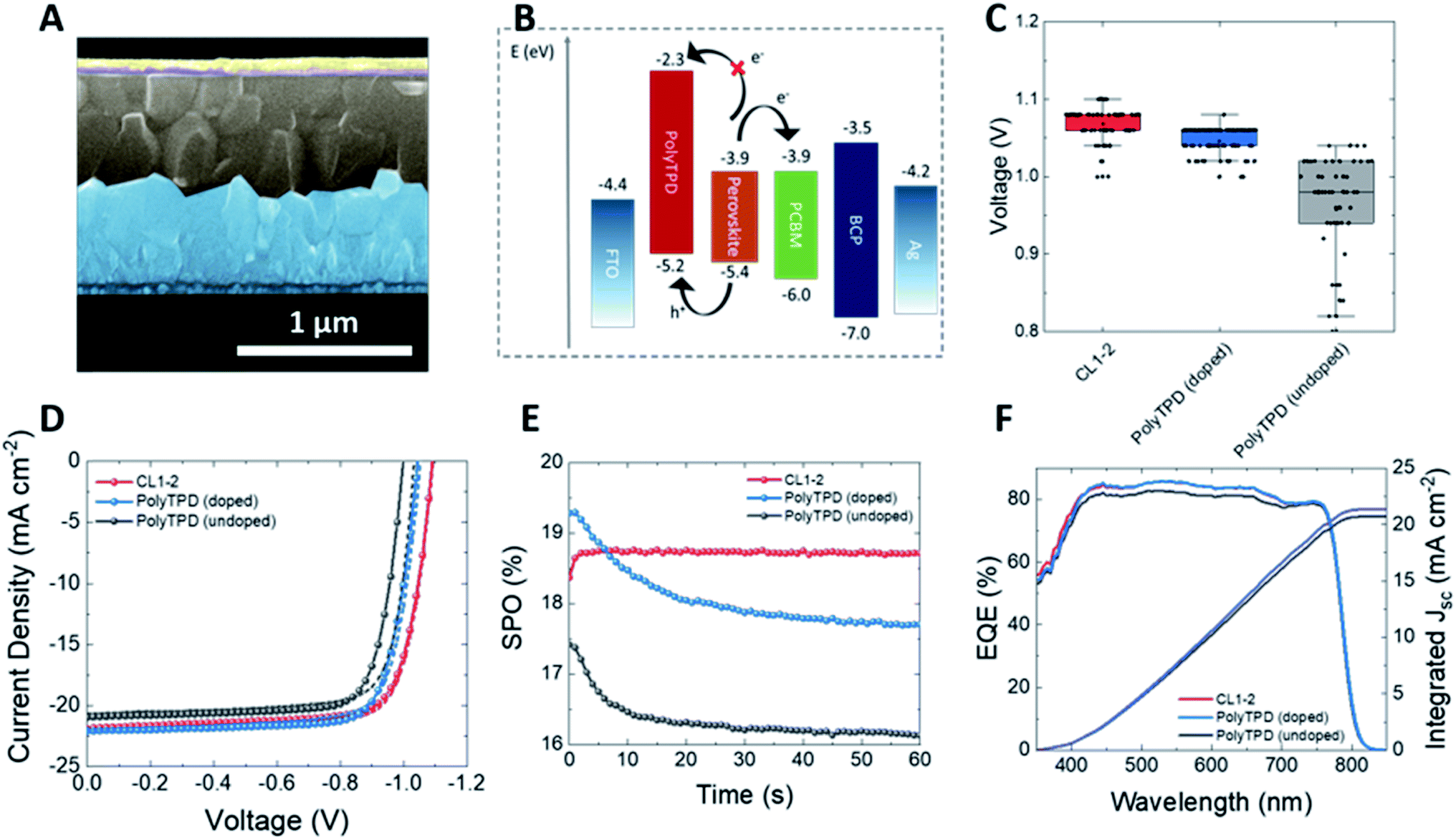 | ||
| Fig. 3 (A) SEM device cross section with FTO (blue), CL1-2, Cs0.05(FA0.85MA0.15)0.95Pb(I0.9Br0.1)3 (gray), PCBM and BCP (purple), and Ag electrode (yellow). (B) Schematic representation of the inverted “p–i–n” PSCs. (C) Comparison of Voc for undoped CL1-2, doped PolyTPD, and undoped PolyTPD. (D) J–V characteristics for champion devices employing undoped CL1-2, doped PolyTPD, and undoped PolyTPD collected under AM 1.5G simulated sunlight. (E) Steady-state power output. (F) External quantum efficiency (EQE). | ||
Only a slight improvement was noticed when CL1-2 was doped using F4-TCNQ (see Fig. S8 and Table S2†), while a significant improvement was obtained for PolyTPD devices. The devices using undoped CL1-2 in its optimal condition (see ESI Fig. S9, S10 and Tables S3, S4† for optimization of CL1-2 concentration and annealing temperature), shows a PCE of (16 ± 2)%, which is similar to the control device with doped PolyTPD ((16 ± 2)%; Table S1†). Fig. S11† shows the low hysteresis present in the cells presented here. The SPO provides a more complete view of solar cell performance under operating conditions and avoids the ambiguities associated with hysteresis in the J–V characteristics. CL1-2 achieves 18.7% SPO, matching its scanned efficiency, while PolyTPD achieves 17.7% and exhibits a transient decay. Typically, when SPO is lower than the scanned efficiency it is related to hysteresis induced by ion migration and unbalanced charge extraction rates at selective contacts;60,61 however, with doped PolyTPD we observe SPO decay without accompanying J–V hysteresis. This behavior may be related to a photo instability, such as the evolution of photo-induced trap states, rather than to mobile ions and unbalanced charge extraction. In such a case, a J–V scan collected over a few seconds may not reflect the longer term SPO. This possibility is consistent with photoluminescence quantum efficiency for half devices (FTO/PolyTPD/perovskite), collected at comparable time scales to the SPO, and the intermediate term UV stability of full devices. Both measurements reflect an illumination- and time-dependent decay similar to that observed in the SPO, as discussed further in the device stability section. The performance of the cells over time is consistent with the trend seen on the J–V curves. Despite the longer wavelength onset of absorption seen for PolyTPD, all devices gave similar external quantum efficiency (EQE) spectra, which is consistent with the similar Jsc measured in the J–V scans. Moreover, PSCs made with FA0.83Cs0.17PbI0.83Br0.17 also showed comparable performance with CL1-2 and PolyTPD (see Fig. S12 and Table S5†), showing that this HTM also performs well with this more stable double-cation perovskite.
One way that the solar-cell performance of cells with PolyTPD and CL1-2 HTMs differ is that the latter exhibit somewhat larger Voc values (Fig. 3C). It has previously been shown that Lewis base-containing HTMs or small molecules can passivate the surface traps and electronic disorder at the surface of the perovskite layer;31,62–64 in the present case coordination of the multiple ester moieties present in CL1-2 to the perovskite surface might act in a similar way, thus reducing recombination and increasing Voc (Fig. 3C). PL lifetime experiments on films of perovskite in contact with undoped CL1-2 or doped PolyTPD (Fig. S5†) are consistent, the former exhibiting longer lifetimes associated with bimolecular recombination. Correlations between PL decay and Voc have been reported in the literature.65–67 In addition, several previous studies have found esters can play a useful role in perovskite growth and/or passivation: poly(methyl methacrylate) has been codeposited with the active layer of n–i–p cells,68 and used as an interlayer between perovskite and HTM in both n–i–p69 and p–i–n70 cells.
Perovskite growth using more planar substrates
While no major differences were noticed on the perovskite film on top of FTO/HTMs by SEM and PXRD, a major difference in the perovskite growth is noticeable when glass or tin-doped indium oxide (ITO), both of which are considerably smoother substrates than FTO, are used as the substrate. ITO has achieved slightly higher transmittance in the visible spectrum and can offer somewhat lower resistivity than FTO, making it attractive when maximum current density and fill factor are desired.71,72 The CL1-2 film displays a slightly more hydrophilic surface, with a water contact angle, θ, of (82 ± 3)°, than the PolyTPD film (θ = 93 ± 6)°, which could explain why it is easier to spin-coat perovskite on smooth CL1-2 films on glass or ITO, while it is not possible or difficult for PolyTPD films on glass and ITO. Stronger perovskite–HTM interactions could also be the cause for the improved wettability of CL1-2 relative to PolyTPD. Indeed, the esters and ether on CL1-2 could possibly hydrogen bond with the cations of the perovskite to facilitate its deposition, or the carbonyl group could form an intermediate adduct with PbI2,68 and hypotheses such as this will be probed in the future. While PolyTPD can be treated with UV Ozone to improve its wettability, sufficient treatment time to ensure full perovskite film coverage of the substrate substantially degrades photovoltaic device performance.73 In order to make PSCs on ITO using PolyTPD, we tested a polyelectrolyte that has previously been used to improve the wettability and performance of PTAA in p–i–n PSCs, as shown in Fig. S13, S14 and Table S6†.67,74 We find that (9,9-bis(3-(N,N-dimethylamino)propyl)fluorene-2,7-diyl)-alt-(9,9-dioctylfluoren-2,7-diyl) (PFN) treatment on PolyTPD degrades fill factor and the SPO of the PolyTPD devices.Another consequence of the improved growth of perovskites on CL1-2 on a smooth substrate relative to that on PolyTPD is shown in Fig. 4. Here we show that a layer of MAPbI3 can be successively coated onto CL1-2 deposited onto FA0.83Cs0.17Pb(I0.90Br0.10)3 (itself grown on FTO), whereas it does not coat an analogous structure using a PolyTPD interlayer. This result suggests cross-linked organic semiconductors may help facilitate solution processing of tandem solar cells, where the crosslinking approach could potentially be extended to allow for a recombination layer that is itself a multilayer composed of sequentially deposited and crosslinked materials with separate hole- and electron-transport functions and potentially with and without dopants. Although multilayer recombination layers of this kind have been fabricated using organic semiconductors,75 they generally require vacuum evaporation to avoid the deposition of one layer leading to dissolution of another. An alternative solution-processed approach involves the use of PEDOT:PSS and ITO nanoparticles,76 but the former may lead to incorporation of traces of water in the device, and both can impair transmission.
 | ||
| Fig. 4 Left: SEM cross section showing a “tandem” structure FTO/FACs/CL1-2/MAPbI3 (FACs = FA0.83Cs0.17Pb(I0.90Br0.10)3). Center: photographs from the top of structures such as those shown left with PolyTPD and CL1-2 interlayers, indicating a poorer coverage of MAPbI3 when using PolyTPD. Right: absorption spectra of the films shown in the center, compared to those of films of the two perovskite materials deposited separately. These data show use of CL1-2 leads to a structure that is much more strongly absorbing than FACs alone due to successful deposition of MAPbI3, whereas use of PolyTPD does not. | ||
Device stability studies
The photo- and thermal-stability of the Cs0.05(FA0.85MA0.15)0.95Pb(I0.9Br0.1)3 PSCs incorporating CL1-2 and PolyTPD were studied without encapsulation in a N2-filled glovebox. The devices were periodically removed to ambient atmosphere for measurements under an AM 1.5G solar simulator, where the stabilized power output was measured after 50 s. Fig. 5A shows the stability in an oven at 85 °C; CL1-2 showed similar stability to PolyTPD devices. Remarkably, the devices show an improvement in performance over the first 1000 hours of aging, and retain greater than their initial efficiency until after 3000 h, which to the best of our knowledge is the longest 85 °C thermal stability timescale reported in the literature (see Fig. S15† for picture of the device after 3000 h of aging).41,77–79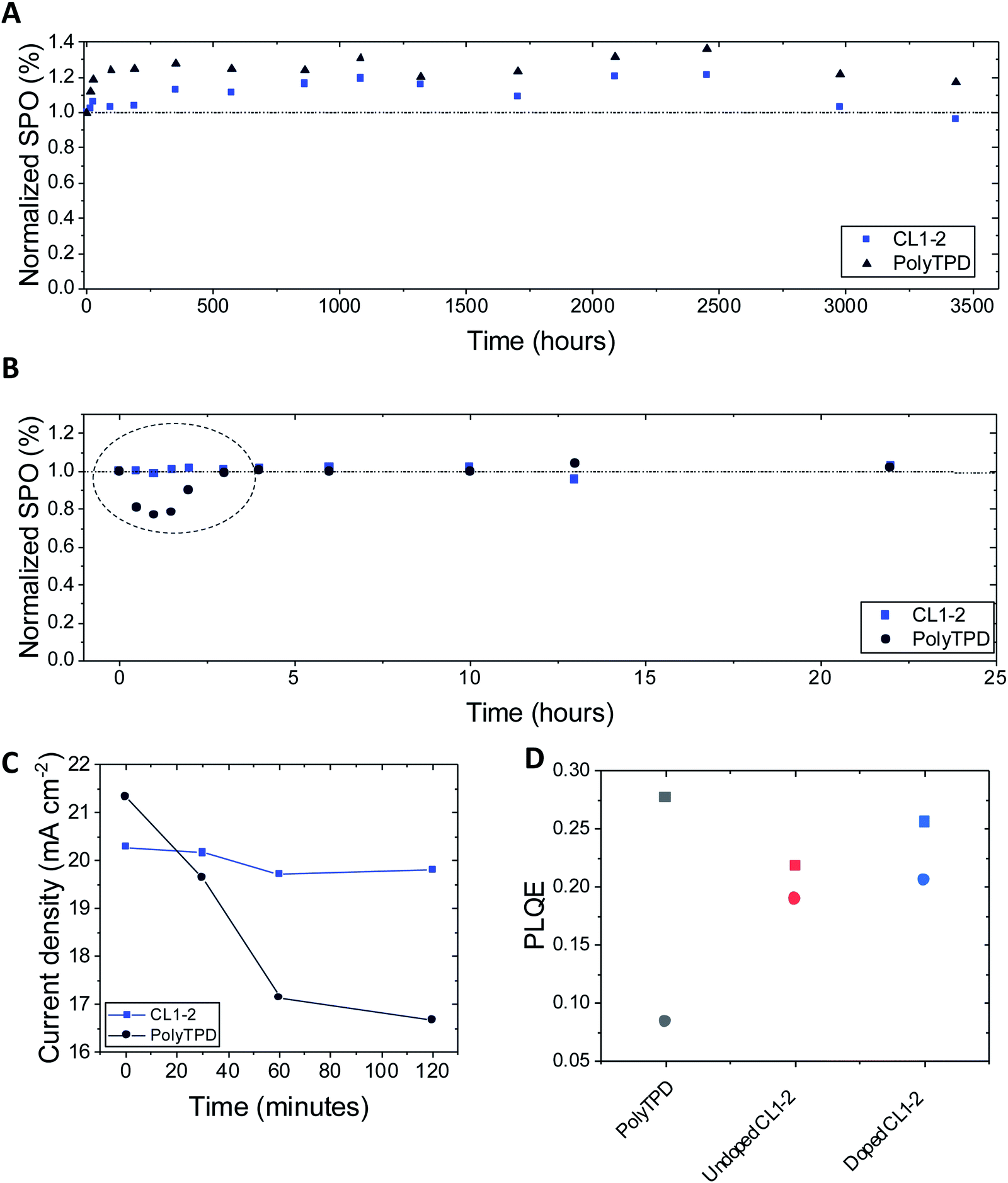 | ||
| Fig. 5 Normalized stabilized power output (SPO) over 50 s after (A) aging the devices at 85 °C in an oven inside a N2-filled glovebox without encapsulation, (B) aging the devices with a UV LED light inside a N2-filled glovebox without encapsulation. The devices were periodically removed to atmosphere for measurements. Both HTMs are doped with F4-TCNQ. (C) Evolution of the current density of CL1-2 and PolyTPD devices when aged under UV LED light (same experiments as B). (D) Initial (square) and stabilized (circle) PLQE of the half devices (FTO/HTM/perovskite). See Fig. S16 and S17† for plots showing the temporal evolution of key solar-cell parameters for the experiments shown in (A and B). | ||
Photostability was studied under a high intensity LED array (365 nm LED, equivalent to 12 suns UV component of the AM 1.5G spectrum). Initially, a drop in SPO is seen for the PolyTPD devices, while the CL1-2 devices stayed approximately at the same initial SPO (Fig. 5B). The current density decreased at a greater extent for PolyTPD devices (Fig. 5C). In order to study this instability, we measured the photoluminescence quantum efficiency (PLQE) over time of the perovskite film crystallized on top of the HTM layer on FTO. We observe a 70% drop in PolyTPD PLQE after stabilization (2 min), while the CL1-2 doped and pristine PLQE retained 81% and 87% of their initial PLQE respectively (Fig. 5D and Table S7†).
Conclusion
In summary, we have described the first use of a photo-crosslinked HTM in PSCs. The crosslinkable acrylate copolymer with bis(triarylamine) and cinnamate side chains presented here is promising since it has similar performance to PolyTPD, a widely used HTM, combined with outstanding 85 °C thermal stability and improved PSC UV photo stability. Solar cells using CL1-2 match the state-of-the-art HTM PolyTPD in terms of both performance and stability under 85 °C. We also demonstrated multiple advantages of CL1-2 over PolyTPD: it does not need to be doped, while PolyTPD does; we achieved highly reproducible photovoltaic performance across multiple batches of devices; the HTM is transparent in the visible region, while PolyTPD is not; and the perovskite is easier to process on top of the CL1-2 HTM, which together suggest that the crosslinkable polymeric CL1-2 is a promising candidate for single junction and tandem solar cells.Conflicts of interest
HJS is co-founder and Chief Scientific Officer of Oxford PV Ltd.Acknowledgements
This work was supported by NSERC (ES. D. Scholarship for MHT), the AFOSR (FA9550-18-1-0499), the Marshall Aid Commemoration Commission, and EPSRC UK.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-b. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- N. Arora, M. I. Dar, A. Hinderhofer, N. Pellet, F. Schreiber, S. M. Zakeeruddin and M. Grätzel, Science, 2017, 358, 768–771 CrossRef CAS PubMed.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2016, 28, 5778–5793 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739–1743 RSC.
- A. Baltakesmez, M. Biber and S. Tüzemen, J. Appl. Phys., 2017, 122, 085502 CrossRef.
- M. Saliba, J.-P. Correa-Baena, M. Grätzel, A. Hagfeldt and A. Abate, Angew. Chem., Int. Ed., 2018, 57, 2554–2569 CrossRef CAS PubMed.
- F. Huang, M. Li, P. Siffalovic, G. Cao and J. Tian, Energy Environ. Sci., 2019, 12, 518–549 RSC.
- J. You, L. Meng, T.-B. Song, T.-F. Guo, Y. Yang, W.-H. Chang, Z. Hong, H. Chen, H. Zhou, Q. Chen, Y. Liu, N. De Marco and Y. Yang, Nat. Nanotechnol., 2015, 11, 75 CrossRef PubMed.
- M. Kaltenbrunner, G. Adam, E. D. Głowacki, M. Drack, R. Schwödiauer, L. Leonat, D. H. Apaydin, H. Groiss, M. C. Scharber, M. S. White, N. S. Sariciftci and S. Bauer, Nat. Mater., 2015, 14, 1032 CrossRef CAS PubMed.
- F. Giordano, A. Abate, J. P. Correa Baena, M. Saliba, T. Matsui, S. H. Im, S. M. Zakeeruddin, M. K. Nazeeruddin, A. Hagfeldt and M. Graetzel, Nat. Commun., 2016, 7, 10379 CrossRef CAS PubMed.
- J. T.-W. Wang, Z. Wang, S. Pathak, W. Zhang, D. W. deQuilettes, F. Wisnivesky-Rocca-Rivarola, J. Huang, P. K. Nayak, J. B. Patel, H. A. Mohd Yusof, Y. Vaynzof, R. Zhu, I. Ramirez, J. Zhang, C. Ducati, C. Grovenor, M. B. Johnston, D. S. Ginger, R. J. Nicholas and H. J. Snaith, Energy Environ. Sci., 2016, 9, 2892–2901 RSC.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 RSC.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, Nano Lett., 2014, 14, 3608–3616 CrossRef CAS PubMed.
- N.-G. Park, M. Grätzel, T. Miyasaka, K. Zhu and K. Emery, Nat. Energy, 2016, 1, 16152 CrossRef CAS.
- G. Niu, X. Guo and L. Wang, J. Mater. Chem. A, 2015, 3, 8970–8980 RSC.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 7843–7850 CrossRef CAS PubMed.
- Y. Chen, Y. Sun, J. Peng, W. Zhang, X. Su, K. Zheng, T. Pullerits and Z. Liang, Adv. Energy Mater., 2017, 7, 1700162 CrossRef.
- C. Ma, D. Shen, T.-W. Ng, M.-F. Lo and C.-S. Lee, Adv. Mater., 2018, 30, 1800710 CrossRef PubMed.
- L. Mao, W. Ke, L. Pedesseau, Y. Wu, C. Katan, J. Even, M. R. Wasielewski, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 3775–3783 CrossRef CAS PubMed.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312 CrossRef CAS PubMed.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, Nano Lett., 2014, 14, 5561–5568 CrossRef CAS PubMed.
- K. Rakstys, S. Paek, P. Gao, P. Gratia, T. Marszalek, G. Grancini, K. T. Cho, K. Genevicius, V. Jankauskas, W. Pisula and M. K. Nazeeruddin, J. Mater. Chem. A, 2017, 5, 7811–7815 RSC.
- D. Bi, B. Xu, P. Gao, L. Sun, M. Grätzel and A. Hagfeldt, Nano Energy, 2016, 23, 138–144 CrossRef CAS.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. De Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- J. Cao, B. Wu, R. Chen, Y. Wu, Y. Hui, B.-W. Mao and N. Zheng, Adv. Mater., 2018, 30, 1705596 CrossRef PubMed.
- C. Liu, K. Wang, P. Du, T. Meng, X. Yu, S. Z. D. Cheng and X. Gong, ACS Appl. Mater. Interfaces, 2015, 7, 1153–1159 CrossRef CAS PubMed.
- Z. Zhu, Y. Bai, X. Liu, C.-C. Chueh, S. Yang and A. K.-Y. Jen, Adv. Mater., 2016, 28, 6478–6484 CrossRef CAS PubMed.
- Z. Yu and L. Sun, Adv. Energy Mater., 2015, 5, 1500213 CrossRef.
- T.-Y. Chiang, G.-L. Fan, J.-Y. Jeng, K.-C. Chen, P. Chen, T.-C. Wen, T.-F. Guo and K.-T. Wong, ACS Appl. Mater. Interfaces, 2015, 7, 24973–24981 CrossRef CAS PubMed.
- J. Xu, O. Voznyy, R. Comin, X. Gong, G. Walters, M. Liu, P. Kanjanaboos, X. Lan and E. H. Sargent, Adv. Mater., 2016, 28, 2807–2815 CrossRef CAS PubMed.
- C.-C. Chang, J.-H. Tao, C.-E. Tsai, Y.-J. Cheng and C.-S. Hsu, ACS Appl. Mater. Interfaces, 2018, 10, 21466–21471 CrossRef CAS PubMed.
- Y. Zhang, C. Kou, J. Zhang, Y. Liu, W. Li, Z. Bo and M. Shao, J. Mater. Chem. A, 2019, 7, 5522–5529 RSC.
- Z. a. Li, Z. Zhu, C.-C. Chueh, J. Luo and A. K.-Y. Jen, Adv. Energy Mater., 2016, 6, 1601165 CrossRef.
- T. H. Schloemer, J. A. Christians, J. M. Luther and A. Sellinger, Chem. Sci., 2019, 10, 1904–1935 RSC.
- W. Li, H. Dong, L. Wang, N. Li, X. Guo, J. Li and Y. Qiu, J. Mater. Chem. A, 2014, 2, 13587–13592 RSC.
- J. Li, C. W. Rochester, I. E. Jacobs, S. Friedrich, P. Stroeve, M. Riede and A. J. Moulé, ACS Appl. Mater. Interfaces, 2015, 7, 28420–28428 CrossRef CAS PubMed.
- Z. Q. Gao, B. X. Mi, G. Z. Xu, Y. Q. Wan, M. L. Gong, K. W. Cheah and C. H. Chen, Chem. Commun., 2008, 117–119, 10.1039/b713566a.
- Y.-D. Zhang, R. D. Hreha, G. E. Jabbour, B. Kippelen, N. Peyghambarian and S. R. Marder, J. Mater. Chem., 2002, 12, 1703–1708 RSC.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897 CrossRef CAS PubMed.
- S. Ryu, J. H. Noh, N. J. Jeon, Y. Chan Kim, W. S. Yang, J. Seo and S. I. Seok, Energy Environ. Sci., 2014, 7, 2614–2618 RSC.
- D. Zhao, M. Sexton, H.-Y. Park, G. Baure, J. C. Nino and F. So, Adv. Energy Mater., 2015, 5, 1401855 CrossRef.
- B. Domercq, R. D. Hreha, Y.-D. Zhang, A. Haldi, S. Barlow, S. R. Marder and B. Kippelen, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2726–2732 CrossRef.
- R. D. Hreha, A. Haldi, B. Domercq, S. Barlow, B. Kippelen and S. R. Marder, Tetrahedron, 2004, 60, 7169–7176 CrossRef CAS.
- B. Domercq, R. D. Hreha, Y.-D. Zhang, N. Larribeau, J. N. Haddock, C. Schultz, S. R. Marder and B. Kippelen, Chem. Mater., 2003, 15, 1491–1496 CrossRef CAS.
- A. Kimyonok, B. Domercq, A. Haldi, J.-Y. Cho, J. R. Carlise, X.-Y. Wang, L. E. Hayden, S. C. Jones, S. Barlow, S. R. Marder, B. Kippelen and M. Weck, Chem. Mater., 2007, 19, 5602–5608 CrossRef CAS.
- Y. Qi, T. Sajoto, S. Barlow, E.-G. Kim, J.-L. Brédas, S. R. Marder and A. Kahn, J. Am. Chem. Soc., 2009, 131, 12530–12531 CrossRef CAS PubMed.
- The redox potentials of the 1 monomer and F4TCNQ indicate the equilibrium constant for the reaction 1 + F4TCNQ = 1+ + F4TCNQ˙− is only 0.014, which corresponds to oxidation of only ca. 6.6% of the diamines at a 20 mol% dopant loading. This value may differ somewhat in different solvents (such as toluene in Fig. 1C and D) or in solid films.
- Q. Sun, G. Subramanyam, L. Dai, M. Check, A. Campbell, R. Naik, J. Grote and Y. Wang, ACS Nano, 2009, 3, 737–743 CrossRef CAS PubMed.
- D. H. Cao, C. C. Stoumpos, C. D. Malliakas, M. J. Katz, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, APL Mater., 2014, 2, 091101 CrossRef.
- B.-w. Park, N. Kedem, M. Kulbak, D. Y. Lee, W. S. Yang, N. J. Jeon, J. Seo, G. Kim, K. J. Kim, T. J. Shin, G. Hodes, D. Cahen and S. I. Seok, Nat. Commun., 2018, 9, 3301 CrossRef PubMed.
- B. Yu, H. Zhang, J. Wu, Y. Li, H. Li, Y. Li, J. Shi, H. Wu, D. Li, Y. Luo and Q. Meng, J. Mater. Chem. A, 2018, 6, 19810–19816 RSC.
- A. Ho-Baillie, M. Zhang, C. F. J. Lau, F.-J. Ma and S. Huang, Joule, 2019, 3, 938–955 CrossRef CAS.
- Y. Zhang, M. Liu, G. E. Eperon, T. C. Leijtens, D. McMeekin, M. Saliba, W. Zhang, M. de Bastiani, A. Petrozza, L. M. Herz, M. B. Johnston, H. Lin and H. J. Snaith, Mater. Horiz., 2015, 2, 315–322 RSC.
- V. W. Bergman, S. A. L. Weber, F. J. Ramos, M. K. Nazeeruddin, M. Grätzel, D. Li, A. L. Domanski, I. Lieberwirth, S. Ahmad and R. Berger, Nat. Commun., 2014, 5, 5001 CrossRef PubMed.
- H. Zhang, L. Xue, J. Han, Y. Q. Fu, Y. Shen, Z. Zhang, Y. Li and M. Wang, J. Mater. Chem. A, 2016, 4, 8724–8733 RSC.
- B. Chaudhary, A. Kulkarni, A. K. Jena, M. Ikegami, Y. Udagawa, H. Kunugita, K. Ema and T. Miyasaka, ChemSusChem, 2017, 10, 2473–2479 CrossRef CAS PubMed.
- M. L. Petrus, K. Schutt, M. T. Sirtl, E. M. Hutter, A. C. Closs, J. M. Ball, J. C. Bijleveld, A. Petrozza, T. Bein, T. J. Dingemans, T. J. Savenije, H. Snaith and P. Docampo, Adv. Energy Mater., 2018, 8, 1801605 CrossRef.
- H. Li, L. Tao, F. Huang, Q. Sun, X. Zhao, J. Han, Y. Shen and M. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 38967–38976 CrossRef CAS PubMed.
- J. Peng, Y. Wu, W. Ye, D. A. Jacobs, H. Shen, X. Fu, Y. Wan, T. Duong, N. Wu, C. Barugkin, H. T. Nguyen, D. Zhong, J. Li, T. Lu, Y. Liu, M. N. Lockrey, K. J. Weber, K. R. Catchpole and T. P. White, Energy Environ. Sci., 2017, 10, 1792–1800 RSC.
- M. Stolterfoht, C. M. Wolff, J. A. Márquez, S. Zhang, C. J. Hages, D. Rothhardt, S. Albrecht, P. L. Burn, P. Meredith, T. Unold and D. Neher, Nat. Energy, 2018, 3, 847–854 CrossRef CAS.
- D. Bi, C. Yi, J. Luo, J.-D. Décoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Grätzel, Nat. Energy, 2016, 1, 16142 CrossRef CAS.
- F. Wang, A. Shimazaki, F. Yang, K. Kanahashi, K. Matsuki, Y. Miyauchi, T. Takenobu, A. Wakamiya, Y. Murata and K. Matsuda, J. Phys. Chem. C, 2017, 121, 1562–1568 CrossRef CAS.
- X. Liu, Y. Cheng, C. Liu, T. Zhang, N. Zhang, S. Zhang, J. Chen, Q. Xu, J. Ouyang and H. Gong, Energy Environ. Sci., 2019, 12, 1622–1633 RSC.
- W.-J. Choi, D.-J. Kwak, C.-S. Park and Y.-M. Sung, Nanosci. Nanotechnol., 2012, 44, 1458–1461 Search PubMed.
- M. A. Aouaj, R. Diaz, A. Belayachi, F. Rueda and M. Abd-Lefdil, Mater. Res. Bull., 2009, 44, 1458–1461 CrossRef CAS.
- X. Xu, C. Ma, Y. Cheng, Y.-M. Xie, X. Yi, B. Gautam, S. Chen, H.-W. Li, C.-S. Lee, F. So and S.-W. Tsang, J. Power Sources, 2017, 360, 157–165 CrossRef CAS.
- S. Hong, J. Lee, H. Kang, G. Kim, S. Kee, J.-H. Lee, S. Jung, B. Park, S. Kim, H. Back, K. Yu and K. Lee, Sci. Adv., 2018, 4, eaat3604 CrossRef CAS PubMed.
- D. Forgács, L. Gil-Escrig, D. Pérez-Del-Rey, C. Momblona, J. Werner, B. Niesen, C. Ballif, M. Sessolo and H. J. Bolink, Adv. Energy Mater., 2017, 7, 1602121 CrossRef.
- D. P. McMeekin, S. Mahesh, N. K. Noel, M. T. Klug, J. C. Lim, J. H. Warby, J. M. Ball, L. M. Herz, M. B. Johnston and H. J. Snaith, Joule, 2019, 3, 387–401 CrossRef CAS.
- J. A. Christians, S. N. Habisreutinger, J. J. Berry and J. M. Luther, ACS Energy Lett., 2018, 3, 2136–2143 CrossRef CAS.
- F. Arabpour Roghabadi, M. Alidaei, S. M. Mousavi, T. Ashjari, A. S. Tehrani, V. Ahmadi and S. M. Sadrameli, J. Mater. Chem. A, 2019, 7, 5898–5933 RSC.
- E. Raza, F. Aziz and Z. Ahmad, RSC Adv., 2018, 8, 20952–20967 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional characterization of HTM film and perovskite growth, device characterization and stability data. See DOI: 10.1039/c9se00513g |
| ‡ Authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |