
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic foldamers as scaffolds for metal second coordination sphere design†
Antoine
Meunier‡
a,
Michael L.
Singleton‡§
a,
Brice
Kauffmann
 b,
Thierry
Granier
a,
Guillaume
Lautrette
a,
Yann
Ferrand
*a and
Ivan
Huc
*ac
b,
Thierry
Granier
a,
Guillaume
Lautrette
a,
Yann
Ferrand
*a and
Ivan
Huc
*ac
aUniversité de Bordeaux, CNRS, Bordeaux Institut National Polytechnique, CBMN (UMR 5248), IECB, 2 Rue Robert Escarpit, 33600 Pessac, France. E-mail: y.ferrand@iecb.u-bordeaux.fr
bUniversité de Bordeaux, CNRS, INSERM, Institut Européen de Chimie et Biologie (UMS 3033), 2 Rue Robert Escarpit, 33600, Pessac, France
cDepartment of Pharmacy, Centre for Integrated Protein Science, Ludwig-Maximilians-Universität, Butenandtstraße 5-13, D-81377 Munich, Germany. E-mail: ivan.huc@cup.lmu.de
First published on 12th October 2020
Abstract
As metalloproteins exemplify, the chemical and physical properties of metal centers depend not only on their first but also on their second coordination sphere. Installing arrays of functional groups around the first coordination sphere of synthetic metal complexes is thus highly desirable, but it remains a challenging objective. Here we introduce a novel approach to produce tailored second coordination spheres. We used bioinspired artificial architectures based on aromatic oligoamide foldamers to construct a rigid, modular and well-defined environment around a metal complex. Specifically, aza-aromatic monomers having a tethered [2Fe–2S] cluster have been synthesized and incorporated in conical helical foldamer sequences. Exploiting the modularity and predictability of aromatic oligoamide structures allowed for the straightforward design of a conical architecture able to sequester the metal complex in a confined environment. Even though no direct metal complex–foldamer interactions were purposely designed in this first generation model, crystallography, NMR and IR spectroscopy concurred to show that the aromatic oligoamide backbone alters the structure and fluxional processes of the metal cluster.
Introduction
The influence of second sphere interactions on the physical and chemical properties of metal complexes is well established. Half of all proteins existing in nature are metalloproteins.1 Their active site is often composed of a metal and its ligands (i.e. the first coordination sphere) surrounded by an array of amino acids precisely arranged in space by protein folding. These amino acids modulate the position, electrostatic and redox properties of the ligands of the first coordination sphere and thus of the metal center. More often than not, a metal complex that only mimics the first coordination sphere of a metalloprotein does not possess the same activity.2–4 Significant efforts have thus been devoted to rationally introduce second sphere interactions in metal complexes. For example, functional groups covalently appended to the first sphere ligands may engage in useful second sphere interactions.5–13 In other approaches, a cavity has been built around the metal center to surround some first sphere ligands14 using for example calixarenes15–17 or cyclodextrins.18,19 As an extension, an entire metal complex, that is, including its first sphere ligands, may be encapsulated within the cavity of a synthetic receptor based, for example, on resorcinarenes20,21 or self-assembled metallo-organic cages.22,23 In recent years, great progress has been made in the incorporation of synthetic metal co-factors into proteins that then only serve as a second coordination sphere.24–29Ab initio metallo-protein design including second sphere control has also witnessed remarkable advances.30–35Because of their biological inspiration, metallofoldamers,36–42i.e. synthetic folded oligomers incorporating metal ions, may have potential for implementing second sphere interactions as they occur in proteins. In the following, we propose to use aromatic oligoamide foldamers (AOFs)43,44 to surround a metal complex and act as a tailored second coordination sphere. AOFs may fold into stable and largely predictable shapes in which sites for molecular recognition can be designed.45 The folding information contained in each monomer determines local strand curvature and may give rise to cavity-containing molecules such as capsules,46–48 curved sheets49 or, as introduced here, helical cones, singular architectures in which the primary sequence encodes a gradual increase of helix diameter. Within these structures, the projection of recognition groups at precise positions in space allows for the molecular recognition of organic guests with a certain degree of predictability.47,48 Furthermore, AOFs are inherently modular. Straightforward fine-tuning of their folded conformations can be achieved via additions, deletions or mutations of monomers. Thus, structure-based iterative improvements have been implemented to selectively recognize polar guests.46–48
As an extension to molecular recognition, we surmised that second sphere coordination might also be tailored within AOF scaffolds. Earlier studies showed that a foldamer capsule may restrict the first coordination sphere of a copper or silver cation coordinated to the inner wall of the capsule cavity,39 and that hydrated calcium or magnesium ions could be encapsulated solely via second sphere–first sphere interactions.40 In this work, we explore a novel strategy to prepare an aromatic helical foldamer cone able to control the orientation of an entire [2Fe–2S] cluster (Fig. 1A), namely (μ-SCH2(NR)CH2S-)[Fe(CO)3]2 (with R representing an amine substituent) a metallic complex known to be a good model of the native hydrogenase enzyme cluster.50,51 Interest for this metal centre stemmed from the properties of hydrogenase enzymes. Their structure based on iron, nickel, or both involve usually toxic cyanide and carbon monoxide ligands as essential parts of their active site. Recent studies indicate that specific amino acid residues in the second coordination sphere are required for the metal complex geometry and catalytic properties.2–4 Until now, few attempts have been made to include both first and second coordination sphere features in models of [2Fe–2S] clusters.52
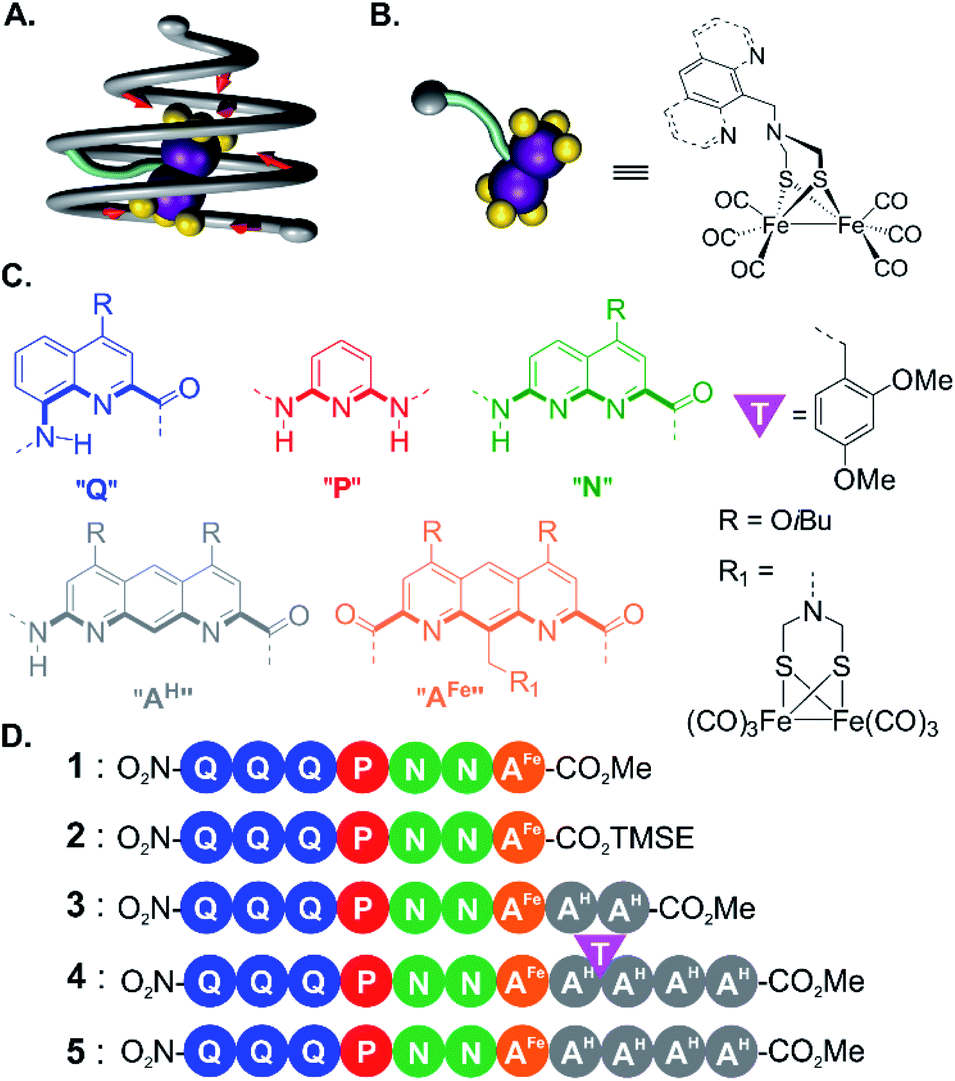 | ||
| Fig. 1 (A) Schematic representation of a helical foldamer cone (grey tube) surrounding a metal complex (purple and yellow spheres) covalently attached to the cone (light blue tube). The red arrows indicate potential interactions between the helix inner rim and the complex. (B) Schematic representation (left) and developed structure (right) of the metal complex (μ-SCH2(NR)CH2S-)[Fe(CO)3]2 attached to a benzylic R group. (C) Color-coded formulas and associated letter code of amino acid, diamino and diacid aromatic monomers. The inner rim of the helix is marked by thick bonds on the monomer formulas. T indicates a 2,4-dimethoxybenzyl tertiary amide group. (D) Sequences of the metallofoldamers studied in this manuscript. | ||
For this initial investigation, we synthesized an aromatic monomer with a covalent link to a biomimetic diiron cluster (Fig. 1B) in such a way that the cluster would locate at the inner rim of a foldamer helix. From this first unit, we elongated the foldamer into a cone using monomers coding for a large diameter so as to fully surround the metal cluster. In order to make the synthesis convergent, an approach was developed that takes advantage of temporary disruptors of helicity in foldamer segments that would otherwise be prone to aggregation. After each elongation of the sequence, the molecular structures were assessed using NMR and IR spectroscopy and X-ray crystallography. Helical folding of the foldamer and sequestration of the metal complex in the helix cavity were validated. The environment of the metal center was shown to become more and more restricted despite the relative flexibility of the covalent foldamer–metal center linker. Even though no direct metal complex–foldamer interactions were purposely designed in this first generation model, crystallography, NMR and IR spectroscopy concurred to show that the aromatic oligoamide backbone alters the structure and fluxional processes of the metal cluster. The final structure constitutes an advanced stage on which to base second coordination sphere design and rational iterative modifications directed by structural elucidation to allow for the potential emergence of catalytic functions.
Results and discussion
Functionalization of the diazaanthracene monomer
For incorporation of the complex in foldamer sequences, monomer AFe containing (μ-SCH2(NR)CH2S-)[Fe(CO)3]2 (Fig. 1C and 2A) was synthesized through derivatization of the 9-position of dimethyl 1,8-diaza-2,7-anthracenedicarboxylate (letter code A). Monomer 6, was obtained as previously described53,54 through benzylic bromination of dimethyl 9-methyl-1,8-diaza-2,7-anthracenedicarboxylate, followed by substitution of the bromide with di-tert-butylimidocarbonate, and treatment with Mg(ClO4)2. Boc deprotection using HCl in dioxane was performed to afford the benzylic ammonium chloride salt of the diazaanthracene. Monomer 9 (AFe) was then obtained in 40% yield by condensation of the amine with (SH)2[Fe(CO)3]2 (ref. 55) and formaldehyde. The solid state structure of AFe was elucidated by crystallographic analysis of single crystals obtained from the slow diffusion of hexane in a chloroform solution (Fig. 2B and C). The diazaanthracene group is oriented away from the [Fe(CO)3]2 fragment and no significant interactions are observed. The Fe–Fe bond length (2.52 Å) was found to be almost identical to that reported for the unfunctionalized (μ-SCH2(NR)CH2S-)[Fe(CO)3]2.55 The torsion angle between the two apical carbonyl groups (Cap1–Fe1–Fe2–Cap2) was measured to be γ = 16.8°, deviating from the fully eclipsed conformation (γ = 0°) observed in the structure of (μ-SCH2(NR)CH2S-)[Fe(CO)3]2. We note that the catalytically active complex also has a twisted conformation.56 However, the distortion observed here appears to be due to a close-packing interaction between one of the carbonyl groups and the methyl group of an ester of another AFe unit. As such, the presence of the diazaanthracene unit alone appears to have little impact on the structure of the diiron moiety. As shown below, more significant effects are observed upon incorporation of AFe into the oligomers.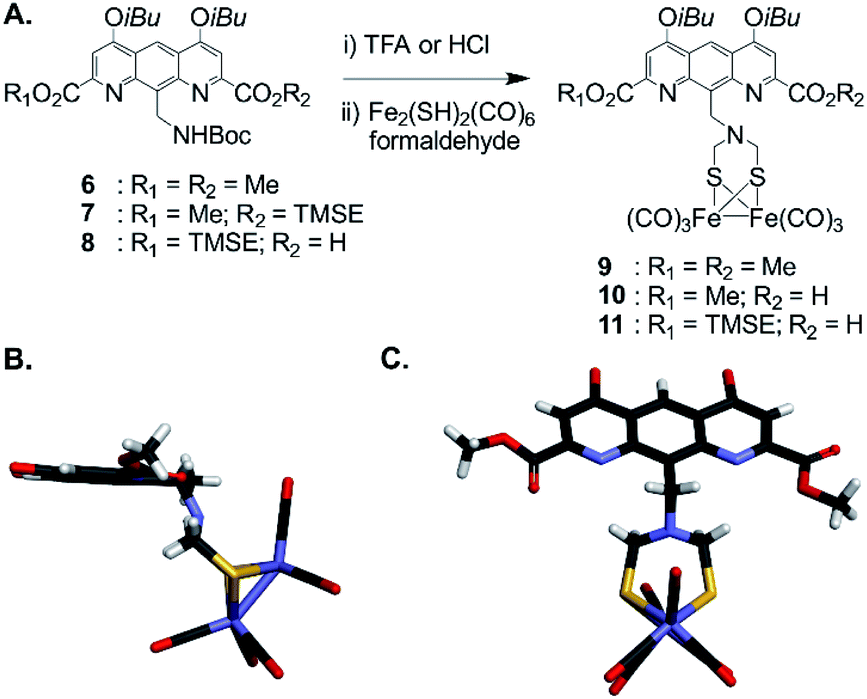 | ||
| Fig. 2 (A) Synthesis of diazaanthracenyl-methyl monomers with appended (μ-SCH2(N)CH2S-)[Fe(CO)3]2 complexes. TMSE: 2-(trimethylsilyl)ethyl. (B) Side view and (C) view from above of the crystal structure of AFe monomer 9 (CCDC # 2031550†). The structure is shown in tube representation. Solubilizing isobutyl side chains have been omitted for clarity. | ||
Elongation of the conical sequence
For the initial elongation of the sequence, the AFe monomer was coupled to a conical aromatic oligoamide segment. Conical segments composed of monomers having gradually decreasing contributions to helix curvature are key components of aromatic capsules for the recognition of chiral organic acids46,48 or carbohydrates.47 These cones consist of a single molecular strand with a narrow diameter at one end and a larger diameter at the other end as illustrated in Fig. 1A. For example, hexamer sequence 12 (Q3PN2, Fig. 3A) is composed of a quinoline trimer Q3 that constitutes an end-cap and that prevents undesired self-assembly in multi-stranded helices, and of a PN2 segment having a weaker curvature that thus generates a cavity around the helix axis. In the case of the (μ-SCH2(NR)CH2S-)[Fe(CO)3]2 group, it was surmised that this would lead to at least partial surrounding of the complex by the foldamer backbone.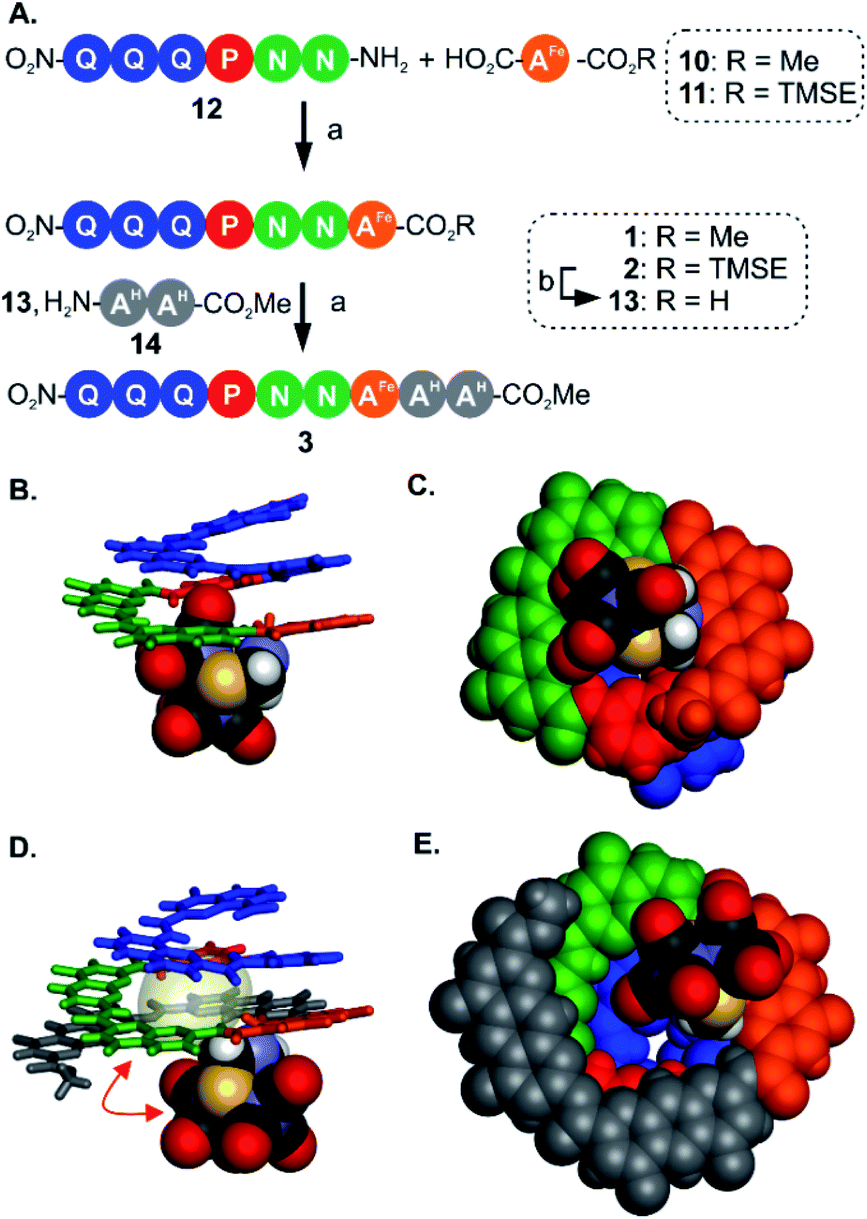 | ||
| Fig. 3 (A) Synthesis of metallofoldamers 1–3: (a) PyBOP, DIEA, room temperature. (b) TFA. (B) Side view and (C) view from below of the crystal structure of 1 (CCDC # 2031551†). (D) Side view and (E) view from below of the crystal structure of 3 (CCDC # 2031556†). In (B and D) the aromatic backbone is shown in color coded tube representation whereas the metal complex is show in CPK representation. In (C and E) the helix and the metal complex are shown in CPK representation. In (D) the space occupied by a chloroform molecule in the cavity of 3 is shown in transparent surface and a red double-headed arrow indicates how the metal complex has moved with respect to its position in (B). Side chains and included solvent molecules have been omitted for clarity. | ||
We proceeded with the synthesis of conical heptaamide 1 from the amine of 12 and the acid of 10 using PyBOP, a coupling agent compatible with the metal complex (59% yield). Oligomer 1 proved to be stable on silica gel, which allowed for a straightforward chromatographic purification. Sequence 1 was characterized by 1H and 13C NMR, mass spectrometry and infrared spectroscopy (see below and in the ESI†). Single crystals suitable for X-ray analysis were grown by slow diffusion of hexane into a chloroform solution of 1. The crystal structure was solved in the centrosymmetrical C2/c space group. The structure shows that the oligomer is well-folded in the desired conical shape. The [2Fe–2S] cluster occupies a central position with the two iron atoms aligned parallel with the helix axis. In this conformation, one of the apical carbonyl groups (Cap1) was found buried within the nascent cavity of the foldamer (Fig. 3B and C), surrounded by the pyridine and naphthyridine units of the Q3PN2 sequence on three sides, and the anthracene of AFe unit on the fourth.
This asymmetric environment provided by the helical nature of the foldamer sequence is interesting for its potential to provide multiple types of interactions. Nevertheless, with the small size of the cavity in this sequence a large part of the complex remains exposed and has limited interaction with the scaffold.
Thus, to further elongate sequence 1 and surround the [2Fe–2S] cluster, we considered to append two 1,8-diazaanthracene amino acids (AH in Fig. 1C) that code for a large helix diameter. For 1, the metal complex proved to be unstable under the basic (LiOH) or nucleophilic (LiI) conditions required to cleave the methyl ester. As result, the analogous sequence 2 was prepared (Fig. 3A), for which the (trimethylsilyl)ethyl (TMSE) ester of 2 can be readily removed using trifluoroacetic acid (TFA), a reagent fully tolerated by the diiron complex. Thus, amine 12 was coupled to AFe monomer 11 using PyBOP. The resulting heptaamide 2 was treated with TFA to give heptamer acid 13 in 94% yield. Finally, 13 was coupled to AH dimer amine 14 to give nonamer 3 in 74% yield.
Again, structural information was gathered using X-ray crystallography. As for 1, the solid state structure revealed a well-folded conical foldamer showing a gradual increase of diameter from the quinoline to the diazaanthracene units (Fig. 3D and E). However, unlike the structure of 1 and despite the longer foldamer sequence, the metal complex was found expelled out of the cone's cavity with its Fe–Fe bond perpendicular to the helical axis. A molecule of chloroform fills the cavity instead of the metal complex. This result reflects the dynamic conformation behavior of the flexible linkage between the foldamer and the [2Fe–2S] cluster. The crystal structures of 1 and 3 are two snapshots of several possible conformations and show that additional modifications are required to further restrict the position of the metal cluster.
Segment coupling strategy using transient tertiary amide disruption of aromatic helix folding
The views from below of the crystal structures of 1 and 3 (Fig. 3C and E) suggest that elongating the AH segment of 3 to a full helix turn should eventually lock the metal complex in the helix cavity by creating a steric bulk below the AFe diazaanthracenyl moiety, where the complex is found in the structure of 3. Sequence 5 (Fig. 1D) was designed for this purpose. This sequence could in principle be obtained by producing an analogue of 3 with a cleavable terminal ester and adding two more AH monomers. Yet, we realized that such additions could not be iterated multiple times without a considerable drop in overall yield: even though the metal centre is quite robust, it remains the most fragile part of the molecule and it is desirable not to submit it to several coupling and deprotection cycles. A convergent method was thus optimized to couple longer diazaanthracene oligomers in one step to e.g. acid 13. However, there is an inherent difficulty in reacting longer diazaanthracene oligomers due to their propensity to self-assemble into multi-stranded helices. Sequences comprised of pyridine,57 naphthyridine58 or 8-fluoroquinoline59 monomers all form stable multiple helices. Diazaanthracene oligomers do not escape this trend and double and triple helices were observed both in solution and in the solid state.60 This aggregation causes severe steric hindrance of the strands' termini and may lead to very slow kinetics for the coupling steps. This normally prohibits a convergent approach and in the past has forced us to use longer synthetic approaches based on iterative addition of short segments.61 To circumvent this problem we devised that introducing TFA labile 2,4-dimethoxybenzyl (DMB) tertiary amides in the sequence would locally disrupt helicity and prevent the undesired aggregation of the strands, thus enhancing their availability for coupling reactions (Fig. 4A). The assumption that a tertiary amide would disrupt helicity was based on the well-documented preference of aryl–alkyl tertiary amides to prefer a cis conformation as opposed to the favored trans conformation of secondary aryl amides required for aromatic helix folding.43 Similar approaches based on tertiary amides have been previously proposed to elongate foldamer single helices62 and to prevent aggregation of para-amino-benzoic acid oligoamides.63 Also related is the use of pseudo-prolines in peptide synthesis.64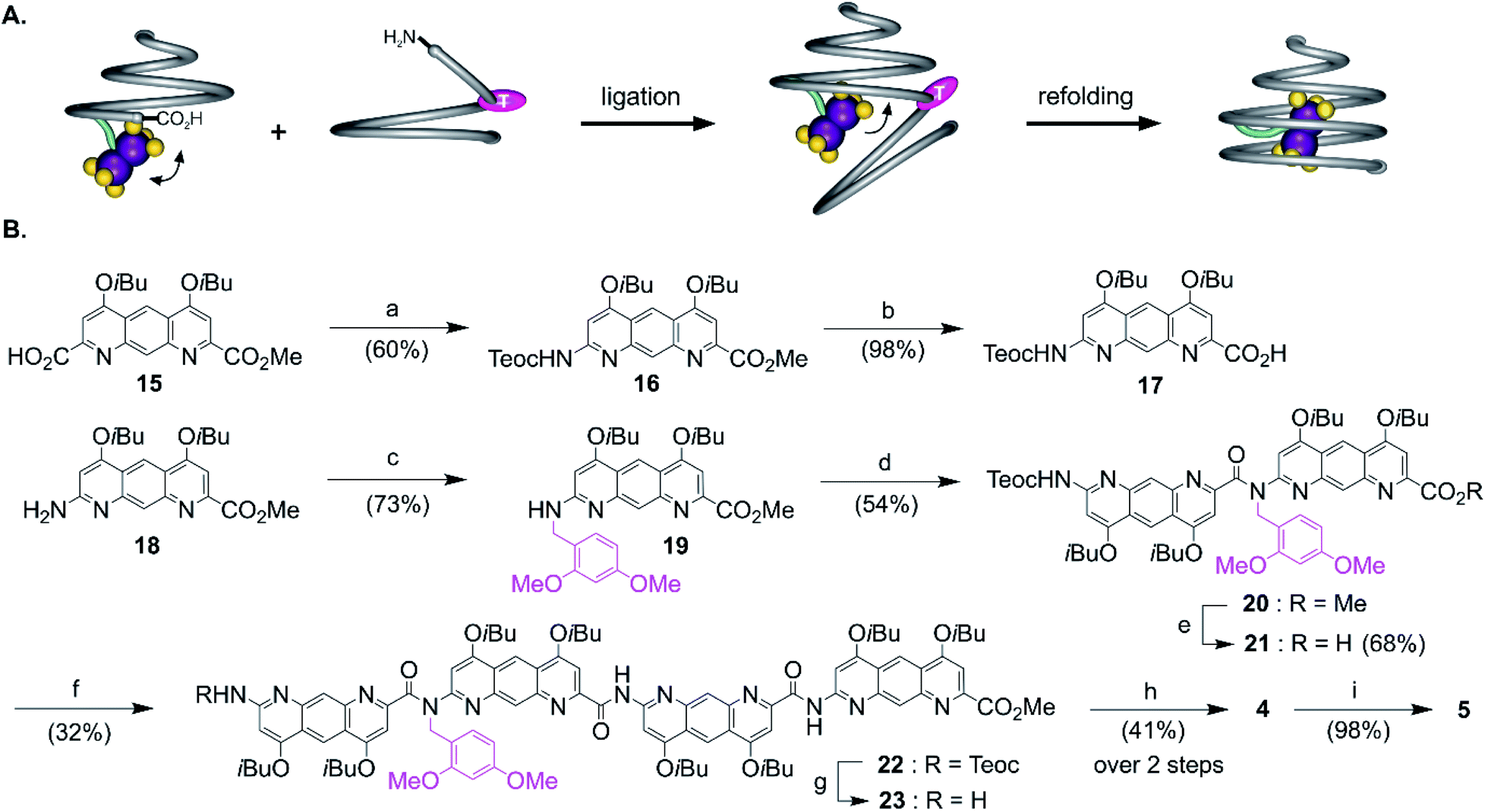 | ||
Fig. 4 (A) Cartoon representation of the amide bond formation between a conical metallofoldamer acid and a partially unfolded amine-terminated aromatic amide strand (left). The tertiary amide responsible for the unfolding of the amine-terminated strand is shown as a pink ovoid. Recovery of the secondary amide under acidic condition leads to the proper refolding of the oligoamide cone and to the incarceration of the complex in the helix cavity (right). (B) Synthetic pathway to cone 5: (a) diphenylphosphoryl azide, DIEA, 2-(trimethylsilyl)ethanol, toluene, 100 °C; (b) LiOH·H2O, THF/MeOH/H2O 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1; (c) 2,4-dimethoxybenzaldehyde, NaBH(OAc)3, 1,2-dichloroethane, 40 °C; (d) 17, PyBOP, DIEA, CH2Cl2; (e) LiI, EtOAc, 80 °C; (f) 14, PyBOP, DIEA, CHCl3; (g) TBAF, succinic acid, THF/DMF; (h) 13, PyBOP, DIEA, CHCl3; (i) TFA, CH2Cl2, room temperature. Teoc: 2-(trimethylsilyl)ethoxycarbonyl. :1:1; (c) 2,4-dimethoxybenzaldehyde, NaBH(OAc)3, 1,2-dichloroethane, 40 °C; (d) 17, PyBOP, DIEA, CH2Cl2; (e) LiI, EtOAc, 80 °C; (f) 14, PyBOP, DIEA, CHCl3; (g) TBAF, succinic acid, THF/DMF; (h) 13, PyBOP, DIEA, CHCl3; (i) TFA, CH2Cl2, room temperature. Teoc: 2-(trimethylsilyl)ethoxycarbonyl. | ||
The synthesis of the diazaanthracene tetramer 23 with one tertiary amide bond near its N-terminus is depicted in Fig. 4B. The DMB functionalized dimer 20 was obtained by coupling the PyBOP activated acid of monomer 17 to the secondary amine of monomer 19. The nucleophilic deprotection of the methyl ester of 20 using LiI yielded acid 21 which was then coupled to dimer 14 to yield the DMB functionalized tetramer 22. The 1H NMR spectrum of 22 shows a concentration independent single set of sharp resonances indicating that it exists as a well-defined species and is not subject to aggregation. In addition, the signal of the 2,4-dimethoxybenzyl methylene did not show any anisochronicity, in contrast with typical diastereotopic patterns observed in the spectra of helically folded oligomers, suggesting the disruption of the aromatic helix (Fig. S4†). Tetramer amine 23 was finally obtained upon Teoc deprotection with tetrabutyl ammonium fluoride (TBAF). This step required to be buffered with a weak acid such as succinic acid to remove the silylated carbamate protecting group (Teoc). This weak acid was used to prevent the hydrolysis of the tertiary amide bond which proved to be hydrolysed under even mildly basic conditions. Then, the convergent coupling of conical heptamer acid 13 and tetramer amine 23 proceeded normally to yield DMB protected undecamer 4, which was purified by GPC. The 1H NMR spectrum of 4 showed an AB spin system (5.4–4.7 ppm) assigned to the DMB benzylic methylene under the influence of chiral environment generated by the helical cone (Fig. S4†). At last, the DMB group was cleaved with TFA in quantitative yield to recover a fully folded conical conformation in sequence 5.
Structural evidence for metal complex inclusion in the foldamer cavity
Oligomers 4 and 5 were also structurally characterized using 13C NMR, mass spectrometry, IR spectroscopy, and X-ray crystallography. Single crystals of both sequences were obtained from the slow liquid–liquid diffusion of hexane into chloroform or chlorobenzene solutions, respectively. X-ray structure determination yielded high resolution (<1 Å) information concerning the position of the [2Fe–2S] cluster as well as the conformation of the aromatic strand. The structure of 4 revealed that, in spite of the tertiary amide linkage, the undecamer can adopt a typical aromatic helix fold. The tertiary amide was found in a trans conformation contrary to the expected cis conformation (Fig. 5A, C and E). In the trans conformation, the benzyl group points towards the unhindered open ended extremity of the cone and the terminal three AH units stack flat on top of the aromatic helix. The enthalpic gain associated with this extensive aromatic stacking may provide the necessary energy for the trans conformation to prevail instead of a less ordered folding in a cis conformation. Yet the (AH)4 segment could not tightly wrap around the complex, a void being observed between the cavity wall and the complex (Fig. 5E). Nevertheless, the Fe–Fe cluster is positioned as desired, that is vertically with its metal–metal axis parallel to the helix axis as observed for sequence 1.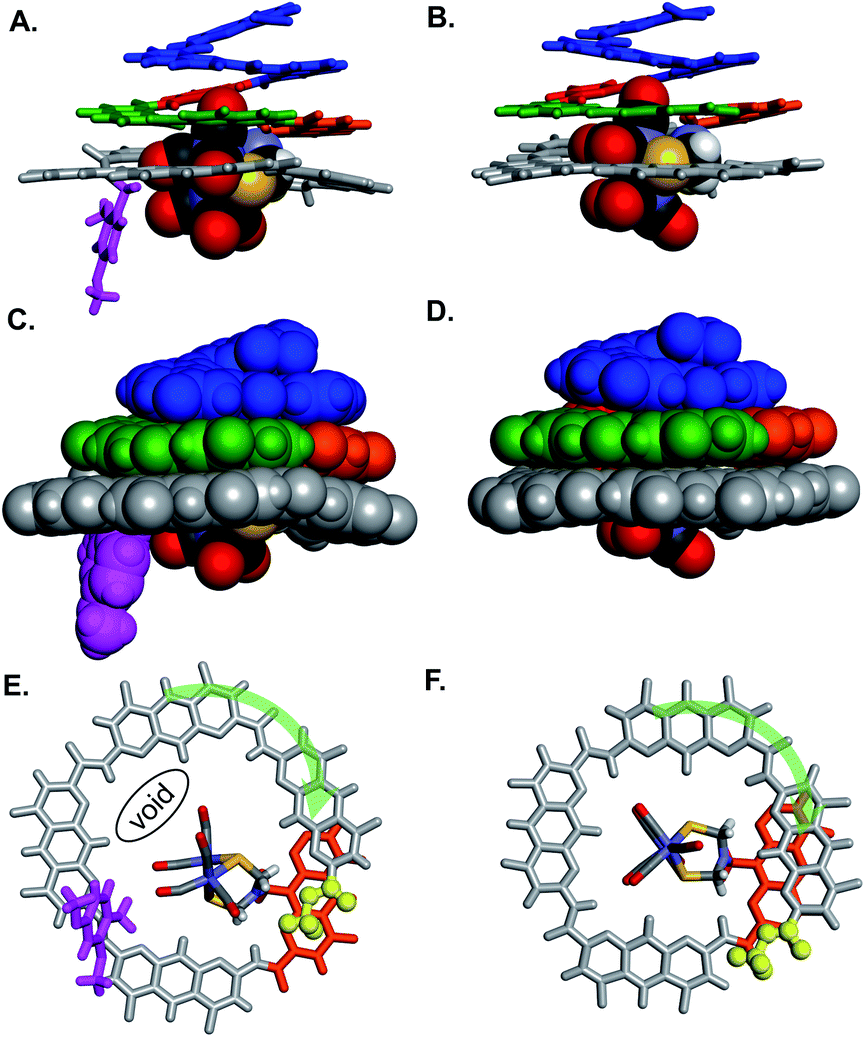 | ||
| Fig. 5 Side views of the crystal structure of (A) oligomer 4 (CCDC # 2031552†) and (B) oligomer 5 (CCDC # 2031555†) shown as tube representation for the aromatic backbone whereas the metal complex is show in CPK representation. (C and D) Similar views of the same structures with the aromatic backbones also shown in CPK representation. Views from below of the crystal structure of (E) oligomer 4 and (F) oligomer 5. Both the complex and the aromatic backbone are shown in tube representation. Q3PN2 segments have been removed for clarity. The terminal methyl ester is shown as yellow balls. The transparent green arrows indicate the curvature direction. Monomers in A–F are color coded as in Fig. 1. Side chains and solvent molecules have been omitted for clarity. | ||
The structure of 5 also exhibits interesting features (Fig. 5B, D and F). The recovered secondary amide bond favours a local increase of helix curvature as compared to the tertiary amide due to the pinching associated with a five-membered hydrogen-bonded pyridine-carboxamide ring. The terminal (AH)4 segment thus almost spans a full turn (Fig. 5Evs.5F) and winds tightly around the cluster suppressing the void observed in the structure of 4 and allowing only a small part of the complex to protrude (Fig. 5D). The terminal AH is then located just behind the [2Fe–2S] cluster below the AFe monomer (in orange in Fig. 5F) and the cluster is locked in vertical orientation with the Fe–Fe bond parallel to the helical axis. The structure of 5 thus provides a proof-of-concept of the use of an AOF backbone to design a shell that wraps around a large dinuclear metal complex, controls its orientation and isolates it from the solvent.
The structure of 5 was also elucidated in solution and confirmed the position of the [2Fe–2S] cluster with respect to the helical backbone observed in the solid state. The 1H NMR of 5 exhibits sharp signals, sharper than the signals of 4, reflecting a better defined folding of the helix. Assignment of the aromatic backbone proton resonances was made possible by a combination of 1H–13C HSQC, 1H–13C HMBC and 1H–1H NOESY pulse sequences. Chemical shift values are reported in the ESI (Fig. S1–S7 and Table S1).† The clearest evidence for positioning the metal complex was shown by a dipolar coupling between the proton in position 9 of the terminal AH monomer (H9) and the methylene resonances (Hb) of the azadithiolate bridge (Fig. 6). Additional NOE correlations involving some amide resonances of the neighbouring diazaanthracene and naphthyridine units were also observed and correlated well with the vertical position of the cluster in the cone (Fig. 6C).
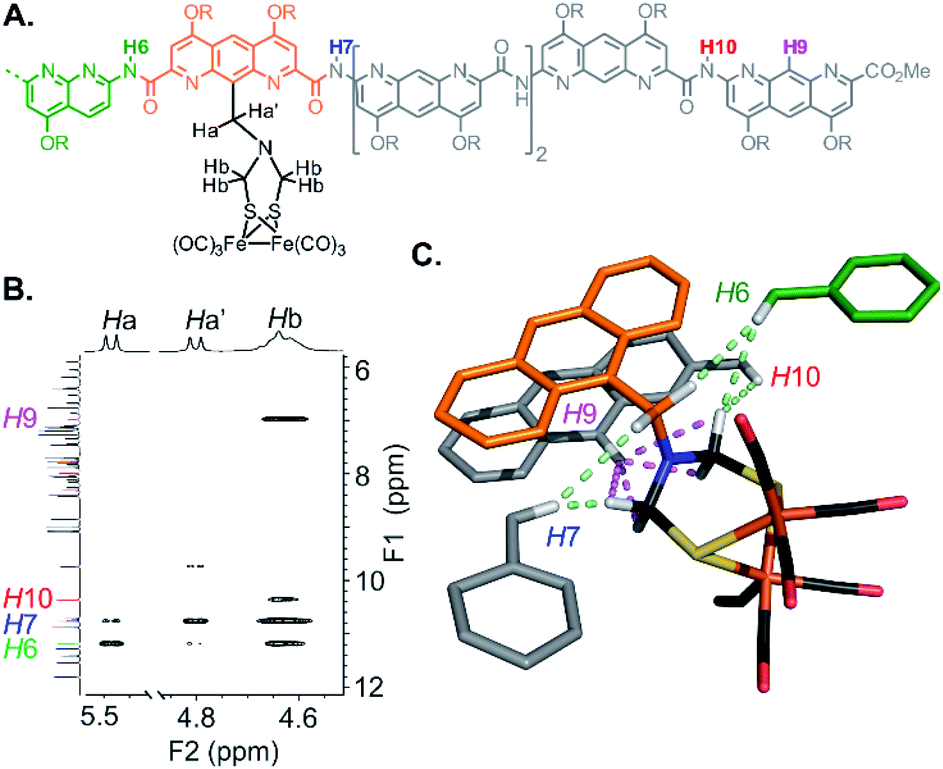 | ||
| Fig. 6 NMR evidence for the positioning of the metal complex within the cavity of 5. (A) Formula and numbering of selected hydrogen atoms of 5. (B) Excerpt of the 1H–1H NOESY of 5 at 8 mM in CDCl3 (700 MHz, 298 K). (C) Representation of NOE correlations observed in solution between the aromatic oligoamide backbone and the (μ-SCH2(NR)CH2S-)[Fe(CO)3]2 complex of 5 on its solid state structure. Dipolar couplings measured between the H9 proton of the terminal diazaanthracene and the azadithiolate methylene groups of the metal complex are shown in magenta. Dipolar couplings between some NH amides of the conical foldamer and the complex are shown in light green. | ||
Influence of the foldamer second coordination sphere on the [2Fe–2S] cluster
The proof-of-concept foldamer backbone of 5 was not designed to engage in any specific interactions with its metal complex (as the red arrows in Fig. 1A). Yet the extensive surrounding of the first coordination sphere hinted at the possibility of observable effects. Indeed, a comprehensive comparison of the series of crystal structures described above provided important information on the influence of the foldamer backbone over the [2Fe–2S] cluster conformation. A feature of the (μ-SCH2(NR)CH2S-)[Fe(CO)3]2 complex that varies in the different structures is its propensity to adopt a chair- or a boat-like conformation with respect to one or the other iron atom (Fig. 7C and D). A boat-like conformation of the cycle involving Fe1 was observed in monomer 9 and in sequences 1, 4 and 5, i.e. every time the complex locates in the helix cavity, whereas a chair conformation was observed in the structure of 3. In other words, the two iron atoms of 5 are discriminated not just because Fe1 is located deeper in the foldamer cavity than Fe2 but also because the peripheral Fe2 is in a chair-like ring whereas Fe1 is in a boat-like ring. Concomitant with this change, the bridgehead amine nitrogen was found to be less pyramidal in sequences 1, 4 and 5 (β values markedly closer to 360°, see Fig. 7B, table).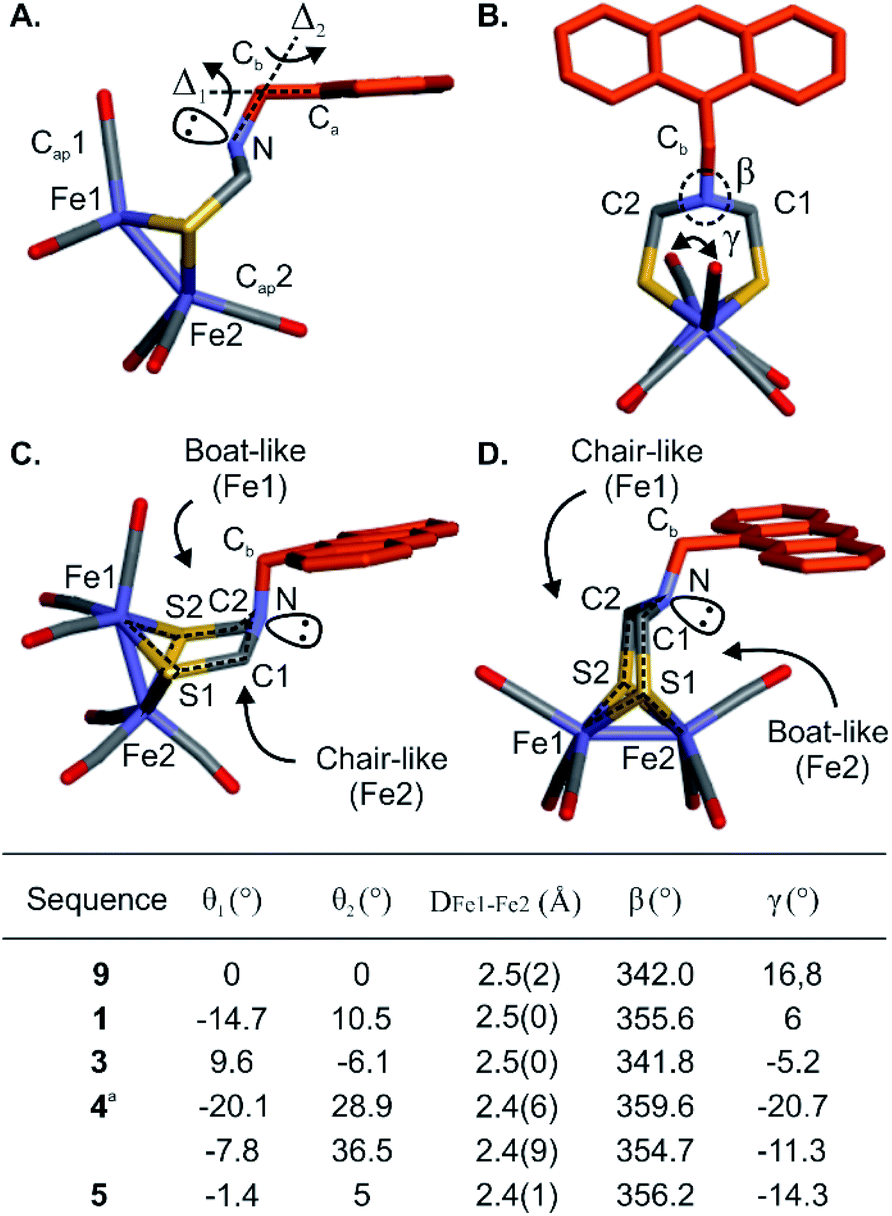 | ||
| Fig. 7 Description of the conformations and orientations of the different (μ-SCH2(NR)CH2S-)[Fe(CO)3]2 complexes in the solid state. (A) Side view of the complex appended diazaanthracene monomer 9, showing the orientation Δ1 and Δ2 axes. θ1 and θ2 are defined here as the rotation angles around Δ1 and Δ2 axes, respectively. (B) Front view of 9 showing the sum of bending angles, β, around the nitrogen atom (i.e. the extent to which it is pyramidal) and the dihedral angle, γ, between Cap1, Fe1, Fe2 and Cap2. (C and D) Side views showing the boat- and chair-like conformations for sequences 1 and 3, respectively. aAngles and distance measured on the two independent molecules observed in the crystal lattice. | ||
The torsion angle γ defined by the Cap1–Fe1–Fe2–Cap2 atoms constitutes a valuable probe of the conformation of the metal complex. This angle reflects the overall torsion of the complex and its ability to position a carbonyl ligand in a bridging position between Fe1 and Fe2, a feature considered important to the catalytic activity of the native enzyme. Among the different solid state structures, γ varies within a range of ∼40° spanning from +17° to −21° for monomer 9 and sequence 4, respectively (Fig. 7B, table). In addition, slight variations of the Fe1–Fe2 bond length were also observed (Fig. 7B, table). The longest distance (2.5 Å) was observed for the functionalized diazaanthracene monomer 9, a value almost identical to that of synthetic models lacking a second coordination sphere.50–52 A shorter bond length was measured at 2.4 Å for sequence 5. It goes along with a shortening of the Fe–CO bonds (1.6–1.7 Å) and a lengthening of the FeC–O (1.3–1.4 Å) bonds relative to monomer 9 and oligomer 3 (1.8 for Fe–CO and 1.1–1.2 Å for FeC–O). These changes hint at increased π-backbonding within the foldamer when the complex is inside the foldamer scaffold. However, no evidence of this is observed in the solution IR studies (see below).
The electrochemical properties of the diiron centre were another obvious parameter to investigate. A cyclic voltammogram of monomer 9 measured in CH2Cl2 showed the expected FeIFeI/FeIFe0 reduction wave at a normal value of −1.81 V, indicating a lack of influence of the anthracene moiety. However, FeIFeI/FeIFeII oxidation is usually not seen in CH2Cl2 and limited solubility of the series in more polar solvents (MeCN or DMF) prevented a full assessment. Furthermore, for oligomers 1 and 3, a reductive event (−1.62 and −1.69 V, respectively) was assigned to a reduction of the terminal NO2 group (data not shown). This made it complicated to assign any effect of the foldamer on the redox properties of the complex and electrochemical studies were not pushed further with these compounds.
We then engaged a systematic comparison between monomer 9 and sequences 1 and 5 using 13C NMR, 1H NMR and IR spectroscopy. The interactions between the [2Fe–2S] cluster and the foldamer backbone were first investigated by monitoring the resonances of the carbonyl ligand using natural abundance 13C NMR. Variable temperature experiments in CDCl3 of compounds 9, 1 and 5 were carried out to compare the influence of the second sphere of coordination on the metal complex fluxional processes (Fig. 8A and S8–S10†). To estimate the dynamics of the CO ligands we measured the full width at half maximum (FWHM) of the carbonyl 13C signals (Fig. 8B). At 298 K, a sharp singlet is observed for all compounds indicating a magnetic equivalence of the six CO due to fast exchange on the NMR time scale. The FWHMs were similar, i.e. 13, 18 and 21 Hz for 9, 1 and 5, respectively. Decreasing the temperature to 273 K led to a peak widening for sequence 1 (32 Hz) and 5 (43 Hz) whereas the carbonyl ligands of monomer 9 were not much affected (17 Hz). Signal broadening for sequence 5 was even more significant at 243 K (165 Hz) while the resonance observed for the carbonyl groups of monomer 9 remained relatively well resolved (65 Hz). In addition, the 1H NMR spectra of sequence 5 at room temperature highlighted the effect of the vicinity between the first coordination sphere of the metal complex and the helical foldamer. While the four protons of the azadithiolate methylenes were found to be equivalent for 9 (singlet at 4.2 ppm), they exhibit a complex downfield-shifted pattern near 4.6 ppm in the case of 5 (Fig. 8C) as a consequence of the foldamer environment.
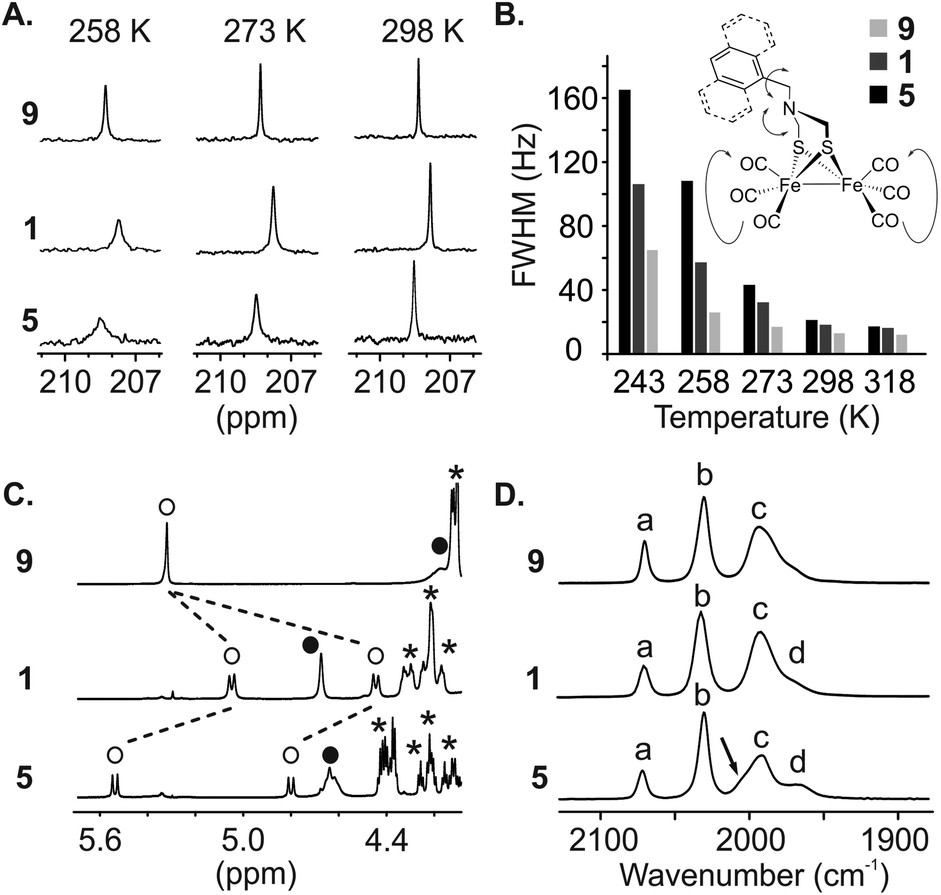 | ||
| Fig. 8 Solution characterization of 1, 5 and 9. (A) Excerpts of the variable temperature 13C NMR at 8 mM in CDCl3 (176 MHz) showing the broadening of the carbonyl resonances of the metal complex. (B) Full Width at Half Maximum (FWHM) of the carbonyl ligand signal extracted from the variable temperature 13C NMR measurement at 8 mM in CDCl3 (176 MHz). Structure of the complex showing the possible fluxional processes. (C) Excerpts of the 1H NMR spectra of 1, 5 and 9 in CDCl3 (700 MHz, 298 K) showing the benzylic protons of AFe (Cb, empty circle) and the methylene protons of the metal complex (C1–C2, filled circle). Stars (*) denote aliphatic resonances of isobutoxy side chains. (D) Parts of IR spectra (CH2Cl2, 298 K) showing three main bands (a, b, c) for the carbonyl ligand vibrations; one should note the shift of vibration (d) for 5 and the desymmetrization of (c) (black arrow). | ||
The dependence of metal–CO stretching frequencies on their local electrostatic environments makes IR an interesting tool for probing the effect of the foldamer structures on the complexes.65 The (ν)CO region of the IR spectra of 9, 1 and 5 are shown in Fig. 8D. Both 9 and 1 show IR spectra similar to previously reported (μ-SRS)Fe2(CO)6 complexes,66,67 with two sharp bands (a) and (b) at 2070 and 2031 cm−1 respectively and a broader band (c) consisting of three overlapping CO stretching bands around 1993 cm−1. This overlap can be evidenced by the asymmetric appearance of the band and notably the shoulder present at lower wave-numbers. The appearance of the three stretching modes comprising (c) has been shown to be dependent on the solvent/local electrostatic environment around the complex. Taking for example (μ-SCH2CH2S)Fe2(CO)6, three fully resolved signals appear in non-polar solvents such as hexane. With increased solvent polarity these broaden and shift towards each other giving the appearance of a single band.66,67 Considering this, it is proposed that the complexes in 9 and 1 experience a similar local electrostatic environment, likely dominated by interaction with solvent.
In the spectrum of 5, band (c) has a different appearance. The low intensity shoulder observed around 1969 cm−1 in 9 and 1 appears as a more distinct peak (labelled (d) in Fig. 8D), and the larger band has some asymmetry with a shoulder at higher wavenumbers. Interestingly, while the broadness and position of the bands changes in (μ-SRS)Fe2(CO)6, the order of the bands with regards to their intensity (when visible) has been reported to stay the same across most solvents;66,67 the most intense stretching mode in (c) is at higher wavenumbers and the intensity of the bands generally decreases with decreasing wavenumbers. This trend is inverted for 5, i.e. the lower intensity shoulder is found at higher wavenumbers. Such effects have not been reported for diiron hexacarbonyl complexes and indicate that the foldamer scaffold of 5 provides a local electrostatic environment different from common solvents. The reasons for this are difficult to define. In the crystal structure, the two basal carbonyls of the Fe(CO)3 fragment deepest in the cavity are oriented towards amide N–H groups. Despite the long CO–N distance (3.5–3.6 Å), a weak electrostatic interaction between these groups may influence certain vibrational modes. Nevertheless, the changes observed in 5 may come from a difference in the intensity of the signals or from a significant shift leading to change in the order of the signals. As such, it is difficult to use previous DFT-based assignments of the vibrational modes to state which CO groups are affected.
Conclusion
We have introduced an approach to design and synthesize aromatic foldamer shells around a bioinspired [2Fe–2S] cluster. The synthetic methodology is versatile and can, in principle, be adapted to a variety of metal complexes. These aromatic foldamers have been previously shown to fold in almost any solvent. Their conformations are particularly stable in protic medium.68 The proposed approach may thus be applied to a broad range of contexts. Although the proposed model system was not designed to engage in specific second coordination sphere interactions, structural and spectroscopic studies show that the metal cluster of 5 is significantly influenced by the foldamer shell. Evident effects include its desymmetrization and the confinement of part of its first coordination sphere within the foldamer cavity. The detailed structural information provided by the crystal structures should make it possible to design specific coordination sphere interactions. The partial surrounding of the complex may also allow for selective modifications of the first coordination sphere, e.g. the selective substitution of carbon monoxide ligands. Undertaking advanced calculations and establishing structure–property correlations will be of interest at this stage. Another aspect that now deserves to be studied is to measure the actual catalytic activity of the foldamer–metal cluster constructs. The chemistry required to anchor these molecules on electrodes is being investigated and will be reported in due course.Materials and methods
Details are in ESI† Materials and methods, which includes detailed methods for synthetic procedures and spectroscopic data. Supplementary dataset comprised crystallographic information files for 1, 2, 4, 5 and 9 (CIF).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by ANR-DFG joint project FOLDHYD (ME 1378/15-1 and ANR-14-CE35-0016), the Seventh Framework Programme of the European Union through Marie Curie Actions (PIIF-GA-2010-275209, post-doctoral fellowship to M. L. S.) and by the Fondation Simone et Cino Del Duca de l'Institut de France (postdoctoral fellowship to M. L. S.). This work has benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform at IECB, CNRS UMS3033, INSERM US001, Bordeaux University, France.Notes and references
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855 CrossRef CAS.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66 CAS.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607 CrossRef CAS.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489 CrossRef.
- J. P. Collman, R. R. Gagne, C. A. Reed, T. R. Halbert, G. Lang and W. T. Robinson, J. Am. Chem. Soc., 1975, 97, 1427 CrossRef CAS.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561 CrossRef CAS.
- S. Sahu, L. R. Widger, M. G. Quesne, S. P. de Visser, H. Matsumura, P. Moënne-Loccoz, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2013, 135, 10590 CrossRef CAS.
- V. Yadav, J. B. Gordon, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2019, 141, 10148 CrossRef CAS.
- S. K. Barman, J. R. Jones, C. Sun, E. A. Hill, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2019, 141, 1114 CrossRef.
- V. F. Oswald, J. L. Lee, S. Biswas, A. C. Weitz, K. Mittra, R. Fan, J. Li, J. Zhao, M. Y. Hu, E. E. Alp, E. L. Bominaar, Y. Guo, M. T. Green, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2020, 142, 11804 CrossRef CAS.
- D. Z. Zee, M. Nippe, A. E. King, C. J. Chang and J. R. Long, Inorg. Chem., 2020, 59, 5206 CrossRef CAS.
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 4504 CAS.
- E. Haviv, D. Azaiza-Dabbah, R. Carmieli, L. Avram, J. M. L. Martin and R. Neumann, J. Am. Chem. Soc., 2018, 140(39), 12451 CrossRef CAS.
- J.-N. Rebilly, B. Colasson, O. Bistri, D. Over and O. Reinaud, Chem. Soc. Rev., 2015, 44, 467 RSC.
- S. Blanchard, L. Le Clainche, M.-N. Rager, B. Chansou, J.-P. Tuchagues, A. F. Duprat, Y. Le Mest and O. Reinaud, Angew. Chem., Int. Ed., 1998, 37, 2732 CrossRef CAS.
- R. Cacciapaglia, A. Casnati, L. Mandolini, D. N. Reinhoudt, R. Salvio, A. Sartori and R. Ungaro, J. Am. Chem. Soc., 2006, 128, 12322 CrossRef CAS.
- D. Coquière, A. de la Lande, S. Martí, O. Parisel, T. Prangé and O. Reinaud, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10449 CrossRef.
- P. Zhang, J. Meijide Suárez, T. Driant, E. Derat, Y. Zhang, M. Ménand, S. Roland and M. Sollogoub, Angew. Chem., Int. Ed., 2017, 56, 10821 CrossRef CAS.
- M. Jouffroy, R. Gramage-Doria, D. Armspach, D. Sémeril, W. Oberhauser, D. Matt and L. Toupet, Angew. Chem., Int. Ed., 2014, 53, 3937 CrossRef CAS.
- A. Cavarzan, A. Scarso, P. Sgarbossa, G. Strukul and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 2848 CrossRef CAS.
- S. Korom and P. Ballester, J. Am. Chem. Soc., 2017, 139, 12109 CrossRef CAS.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746 CrossRef CAS.
- C. J. Brown, G. M. Miller, J. W. Johnson, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 11964 CrossRef CAS.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142 CrossRef CAS.
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Acc. Chem. Res., 2019, 52, 585 CrossRef CAS.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5(7), 1120 CrossRef CAS.
- S. Wu, Y. Zhou, J. G. Rebelein, M. Kuhn, H. Mallin, J. Zhao, N. V. Igareta and T. R. Ward, J. Am. Chem. Soc., 2019, 141, 15869 CrossRef CAS.
- S. I. Mann, T. Heinisch, A. C. Weitz, M. P. Hendrich, T. R. Ward and A. S. Borovik, J. Am. Chem. Soc., 2016, 138(29), 9073 CrossRef CAS.
- S. I. Mann, T. Heinisch, T. R. Ward and A. S. Borovik, J. Am. Chem. Soc., 2017, 139(48), 17289 CrossRef CAS.
- K.-H. Lee, C. Cabello, L. Hemmingsen, E. N. G. Marsh and V. L. Pecoraro, Angew. Chem., Int. Ed., 2006, 45, 2864 CrossRef CAS.
- L. Ruckthong, A. Deb, L. Hemmingsen, J. E. Penner-Hahn and V. L. Pecoraro, J. Biol. Inorg Chem., 2018, 23, 123 CrossRef CAS.
- L. Ruckthong, J. A. Stuckey and V. L. Pecoraro, Chem.–Eur. J., 2019, 25, 6773 CrossRef CAS.
- A. Lombardi, C. M. Summa, S. Geremia, L. Randaccio, V. Pavone and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6298 CrossRef CAS.
- S. Geremia, L. Di Costanzo, L. Randaccio, D. E. Engel, A. Lombardi, F. Nastri and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 17266 CrossRef CAS.
- L. N. Slope, M. G. Hill, C. F. Smith, P. Teare, F. J. de Cogan, M. M. Britton and A. F. A. Peacock, Chem. Commun., 2020, 56, 3729 RSC.
- Z. Dong, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2007, 129, 11850 CrossRef CAS.
- S. Chowdhury, G. Schatte and H.-B. Kraat, Angew. Chem., Int. Ed., 2008, 47, 7056 CrossRef CAS.
- S. Tashiro, K. Matsuoka, A. Minoda and M. Shionoya, Angew. Chem., Int. Ed., 2012, 51, 13123 CrossRef CAS.
- M. Horeau, G. Lautrette, B. Wicher, V. Blot, J. Lebreton, M. Pipelier, D. Dubreuil, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2017, 56, 6823 CrossRef CAS.
- P. Mateus, B. Wicher, Y. Ferrand and I. Huc, Chem. Commun., 2017, 53, 9300 RSC.
- G. Maayan and M. Albrecht, Metallofoldamers: Supramolecular Architectures from Helicates to Biomimetics, Wiley, 2013 Search PubMed.
- T. Ghosh, N. Fridman, M. Kosa and G. Maayan, Angew. Chem., Int. Ed., 2018, 57, 7703 CrossRef CAS.
- I. Huc, Eur. J. Org. Chem., 2004, 17 CrossRef CAS.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271 CrossRef CAS.
- J. Garric, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2005, 44, 1954 CrossRef CAS.
- Y. Ferrand, A. M. Kendhale, B. Kauffmann, A. Grélard, C. Marie, V. Blot, M. Pipelier, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2010, 132, 7858 CrossRef CAS.
- N. Chandramouli, Y. Ferrand, G. Lautrette, B. Kauffmann, C. D. Mackereth, M. Laguerre, D. Dubreuil and I. Huc, Nat. Chem., 2015, 7, 334 CrossRef CAS.
- G. Lautrette, B. Wicher, B. Kauffmann, Y. Ferrand and I. Huc, J. Am. Chem. Soc., 2016, 138, 10314 CrossRef CAS.
- A. Lamouroux, L. Sebaoun, B. Wicher, B. Kauffmann, Y. Ferrand, V. Maurizot and I. Huc, J. Am. Chem. Soc., 2017, 139, 14668 CrossRef CAS.
- T. B. Rauchfuss, Acc. Chem. Res., 2015, 48, 2107 CrossRef CAS.
- C. Tard, X. Liu, S. K. Ibrahim, M. Bruschi, L. De Gioia, S. C. Davies, X. Yang, L.-S. Wang, G. Sawers and C. J. Pickett, Nature, 2005, 433, 610 CrossRef CAS.
- M. L. Singleton, J. H. Reibenspies and M. Y. Darensbourg, J. Am. Chem. Soc., 2010, 132, 8870 CrossRef CAS.
- C. M. Hall, J. B. Wright, H. G. Johnson and A. J. Taylor, J. Med. Chem., 1977, 20, 1337 CrossRef CAS.
- M. L. Singleton, N. Castellucci, S. Massip, B. Kauffmann, Y. Ferrand and I. Huc, J. Org. Chem., 2014, 79, 2115 CrossRef CAS.
- J. D. Lawrence, H. Li, T. B. Rauchfuss, M. Bénard and M.-M. Rohmer, Angew. Chem., Int. Ed., 2001, 40, 1768 CrossRef CAS.
- M. L. Singleton, N. Bhuvanesh, J. H. Reibenspies and M. Y. Darensbourg, Angew. Chem., Int. Ed., 2008, 47, 9492 CrossRef CAS.
- B. Baptiste, J. Zhu, D. Haldar, B. Kauffmann, J.-M. Léger and I. Huc, Chem.–Asian J., 2010, 5, 1364 CAS.
- Y. Ferrand, A. M. Kendhale, J. Garric, B. Kauffmann and I. Huc, Angew. Chem., Int. Ed., 2010, 49, 1778 CrossRef CAS.
- Q. Gan, C. Bao, B. Kauffmann, A. Grélard, J. Xiang, S. Liu, I. Huc and H. Jiang, Angew. Chem., Int. Ed., 2008, 47, 1715 CrossRef CAS.
- M. L. Singleton, G. Pirotte, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 13140 CrossRef CAS.
- C. Bao, Q. Gan, B. Kauffmann, H. Jiang and I. Huc, Chem.–Eur. J., 2009, 15, 11530 CrossRef CAS.
- A. Zhang, J. S. Ferguson, K. Yamato, C. Zheng and B. Gong, Org. Lett., 2006, 8, 5117 CrossRef CAS.
- H. M. König, R. Abbel, D. Schollmeyer and A. F. M. Kilbinger, Org. Lett., 2006, 8, 1819 CrossRef.
- T. Haack and M. Mutter, Tetrahedron Lett., 1992, 33, 1589 CrossRef CAS.
- S. D. Fried and S. G. Boxer, Acc. Chem. Res., 2015, 48, 998 CrossRef CAS.
- M. G. I. Galinato, C. M. Whaley and N. Lehnert, Inorg. Chem., 2010, 49, 3201 CrossRef CAS.
- P. A. Eckert and K. J. Kubaruch, J. Phys. Chem. A, 2017, 121, 608 CrossRef CAS.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic protocols, detailed NMR and crystallographic studies, characterization of new compounds. CCDC 2031550–2031552, 2031555 and 2031556. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05143h |
| ‡ A. M. and M. L. S. contributed equally to this work. |
| § Current address: Institute of Condensed Matter and Nanosciences, Université Catholique de Louvain, Place Louis Pasteur 1, Louvain-la-Neuve, B1348, Belgium. |
| This journal is © The Royal Society of Chemistry 2020 |