
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot thiol–amine bioconjugation to maleimides: simultaneous stabilisation and dual functionalisation†
Archie
Wall
a,
Alfie G.
Wills
a,
Nafsika
Forte
 a,
Calise
Bahou
a,
Lisa
Bonin
a,
Karl
Nicholls
b,
Michelle T.
Ma
c,
Vijay
Chudasama
*ad and
James R.
Baker
*a
a,
Calise
Bahou
a,
Lisa
Bonin
a,
Karl
Nicholls
b,
Michelle T.
Ma
c,
Vijay
Chudasama
*ad and
James R.
Baker
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: j.r.baker@ucl.ac.uk; v.chudasama@ucl.ac.uk
bAlbumedix Ltd, Mabel Street, Nottingham, NG2 3ED, UK
cSchool of Biomedical Engineering and Imaging Sciences, King's College London, St Thomas' Hospital, London SE1 7EH, UK
dResearch Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal
First published on 2nd October 2020
Abstract
Maleimide chemistry is widely used in the site-selective modification of proteins. However, hydrolysis of the resultant thiosuccinimides is required to provide robust stability to the bioconjugates. Herein, we present an alternative approach that affords simultaneous stabilisation and dual functionalisation in a one pot fashion. By consecutive conjugation of a thiol and an amine to dibromomaleimides, we show that aminothiomaleimides can be generated extremely efficiently. Furthermore, the amine serves to deactivate the electrophilicity of the maleimide, precluding further reactivity and hence generating stable conjugates. We have applied this conjugation strategy to peptides and proteins to generate stabilised trifunctional conjugates. We propose that this stabilisation-dual modification strategy could have widespread use in the generation of diverse conjugates.
Introduction
Maleimides represent one of the most widely used functional groups for conjugation chemistry.1–4 This is due to the extremely favourable kinetics of their reaction with thiols, which ensures high yields of the desired conjugates are generated rapidly whilst minimising competing side-reactions. This has led to their use in fields ranging from therapeutic bioconjugates,5–7 to multifunctional polymers and a diverse array of materials.8,9 Despite this widespread use, maleimides suffer several limitations which have been widely noted. Most significantly, the reaction generates succinimides 1 that are unstable over prolonged time in vivo, undergoing retro-conjugate additions and subsequent trapping with endogenous thiols such as glutathione.10–13 To overcome this, the succinimides must be hydrolysed post-conjugation, which can lead to a reduction in the yield of the conjugates,14,15 prolonged reaction times and undesirable exposure to high pH conditions. Furthermore, the hydrolysis generates two regioisomeric maleamic acids 2a and 2b, which when combined with the formation of a stereocenter in the initial conjugate addition, results in four isomeric products (Fig. 1A).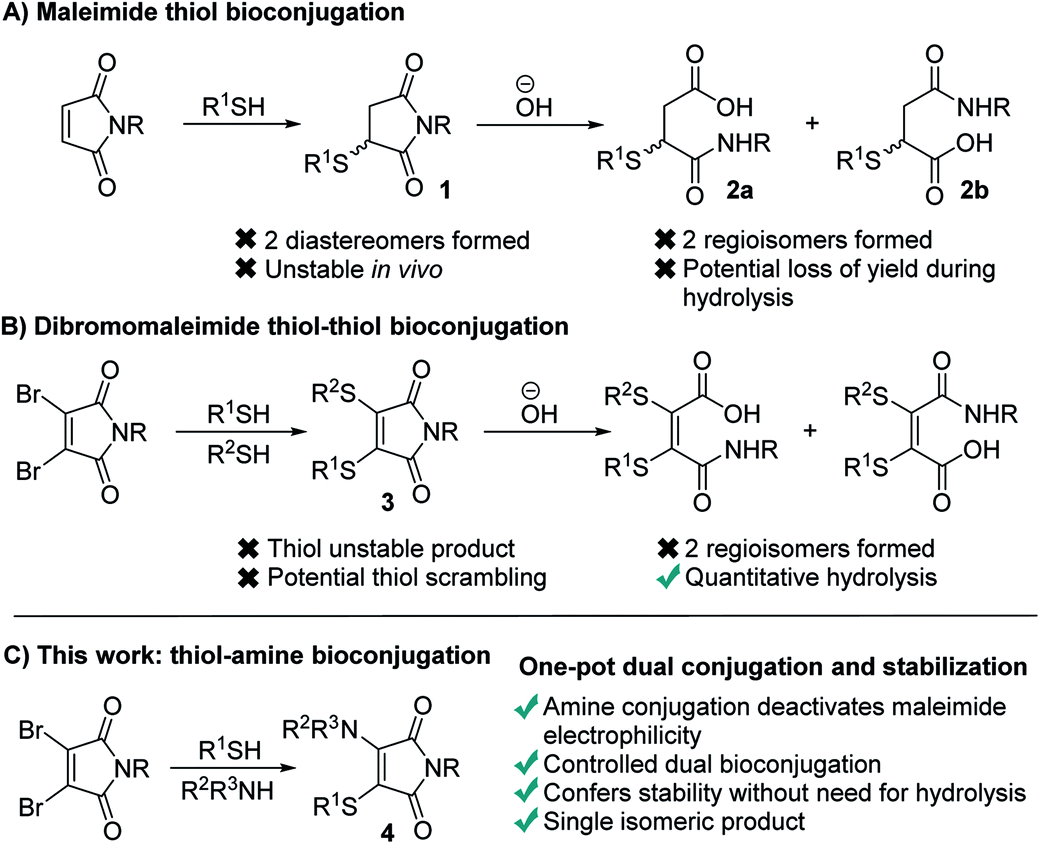 | ||
| Fig. 1 (A) Thiol conjugation with classical maleimides. (B) Dual thiol conjugation using Next Generation Maleimides (NGMs). (C) Dual thiol-amine conjugation with simultaneous stabilisation described herein. | ||
To overcome these limitations we, and others, have reported extensively on the use of dibromomaleimides (DBMs),16–23 and related substituted maleimides known collectively as next generation maleimides (NGMs).20,24 These reagents retain the favourable kinetics of maleimides25,26 whilst avoiding the formation of stereoisomers, and through the addition of two thiols allow the construction of dual conjugates (Fig. 1B). Whilst this bis-thiol conjugation has found widespread use in the bridging (also known as stapling) of disulfide bonds it has found less application to intermolecular conjugates, as the dithiomaleimide products 3 are still thiol reactive and thus the control of stoichiometry is challenging to prevent scrambling of the thioethers. Furthermore, hydrolysis post-conjugation is required to confer in vivo stability, which still generates a mixture of two regioisomeric maleamic acids.10,11
We hypothesized that a completely different mechanism for maleimide conjugate stabilization might prove viable, overcoming the limitations of imide hydrolysis whilst offering new opportunities for the construction of multifunctional conjugates. The addition of an amine to a dibromomaleimide-thiol conjugation reaction would afford an aminothiomaleimide 4 (Fig. 1C), in which the electron donating amino group would deactivate the maleimide to further nucleophilic attack. The product would thus be stable, precluding the requirement for post-conjugation hydrolysis and generating a single isomer (Fig. 1C). Furthermore, the amine would represent an additional site for functional attachment, thus representing an ideal scaffold for the construction of triconjugates.
It has previously been shown that dibromomaleimides (DBMs) react selectively with thiols over amines, hence the initial addition of thiol was expected to occur rapidly and selectively.16,21 Amine addition to dihalomaleimides has been reported to generate aminohalomaleimides, suggesting that whilst the reaction is slower than for thiols it is still viable.27 Interestingly, thiol addition to aminobromomaleimides has been found to occur only under high temperatures (80 °C),28 indicating that the amino group does indeed dramatically reduce the maleimide's electrophilicity. Whilst promising indicators for the desired reaction sequence, far milder reaction conditions would be required for this reaction to be broadly applicable as a bioconjugation strategy. For protein conjugates the reactions would have to take place at, or near, room temperature, at high dilution and be effective in aqueous buffered conditions in which most amines would be substantially protonated.
Results and discussion
Bromothiomaleimides 5 and 6 were synthesized as test substrates for initial tests of amine additions. Enticingly all amines trialled conjugated efficiently in methanol to generate the desired aminothiomaleimides 7–12 in excellent yields (Fig. 2), with both N-aryl and N-alkyl substituents. As expected, the more nucleophilic secondary alkyl amines, such as piperidine and pyrrolidine, reacted more efficiently than aryl amines (ca. 10 min vs. 24 h, see ESI Table S1† for more details).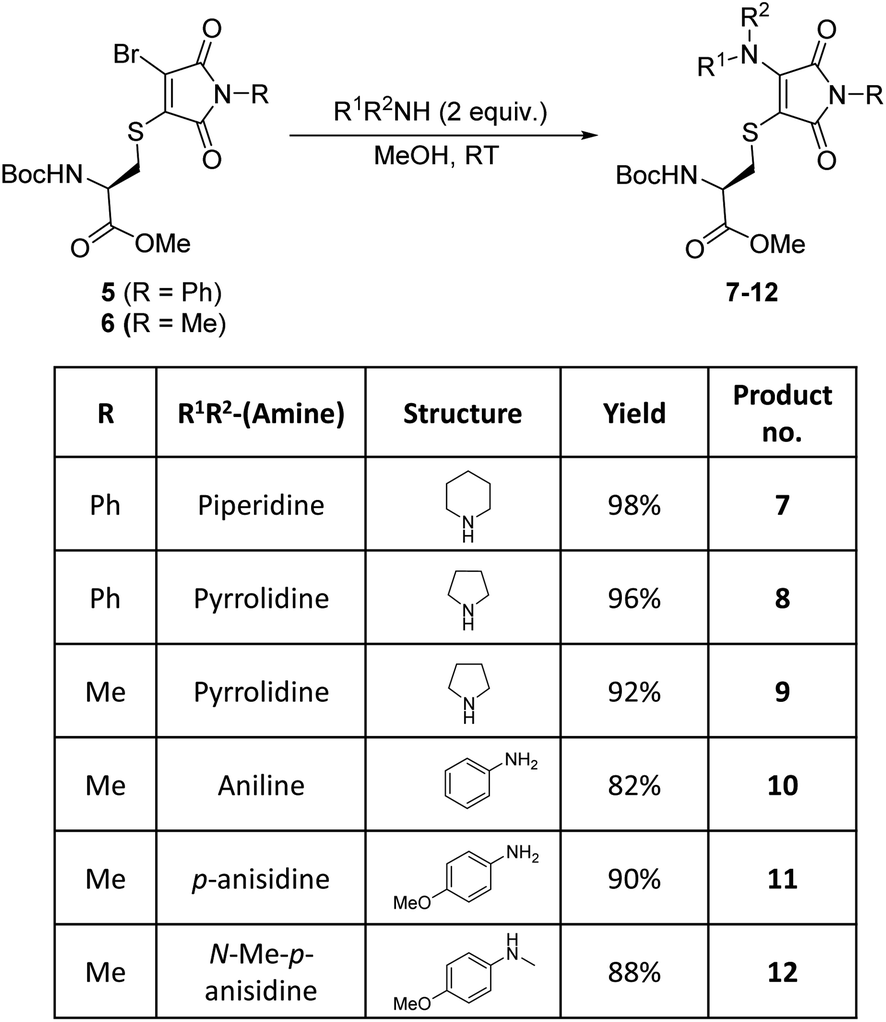 | ||
| Fig. 2 Reactions of bromothiomaleimides 5 and 6 with amines in methanol. | ||
Whilst this study shows the promising potential of this dual conjugation sequence in organic solvent, we moved on to test aqueous buffered conditions. Analysis of the relative rates of amine addition under aqueous conditions (pH 7.8, 4.7 mM, with 2 equiv. of amine) was carried out using NMR, via the integration of the starting material and product N-methyl peaks (Fig. 3). We observed that aniline (pKaH = 4.87) offered a substantial rate enhancement over pyrrolidine (pKaH = 11.31), confirming that protonation was substantially attenuating reactivity of the secondary amine. The pyrrolidine reaction was also observed to generate undefined side products by crude NMR analysis, and was therefore ruled out as a viable reagent. In contrast, incorporating a para-methoxy group onto the aniline, which subtly raises the pKaH (p-anisidine, pKaH = 5.36), afforded the most reactive amine under these conditions. p-Anisidine represented the best balance between nucleophilicity and pKa for undertaking these reactions at near neutral pH,29,30 yielding a single product and reaching completion in 185 min. It is notable that the aminothiomaleimide products were observed to be fluorescent in THF, which has been reported for related aminothiomaleimides by O'Reilly and coworkers and in which they also note that this fluorescence is not observed in protic solvents.28 The aminothiomaleimide 11 was then assessed for stability by prolonged incubation in: pH 7.4 buffer, pH 5 buffer and also 7 mM thiol solution (2-mercaptoethanol). The resulting NMR data confirmed the product as completely stable in all conditions (monitored for 6–8 days, see ESI Fig. S23–S26†). The p-anisidine conjugation to bromothiomaleimides was thus shown to be an extremely promising new bioconjugation reaction, occurring under mild aqueous conditions and generating stable products.
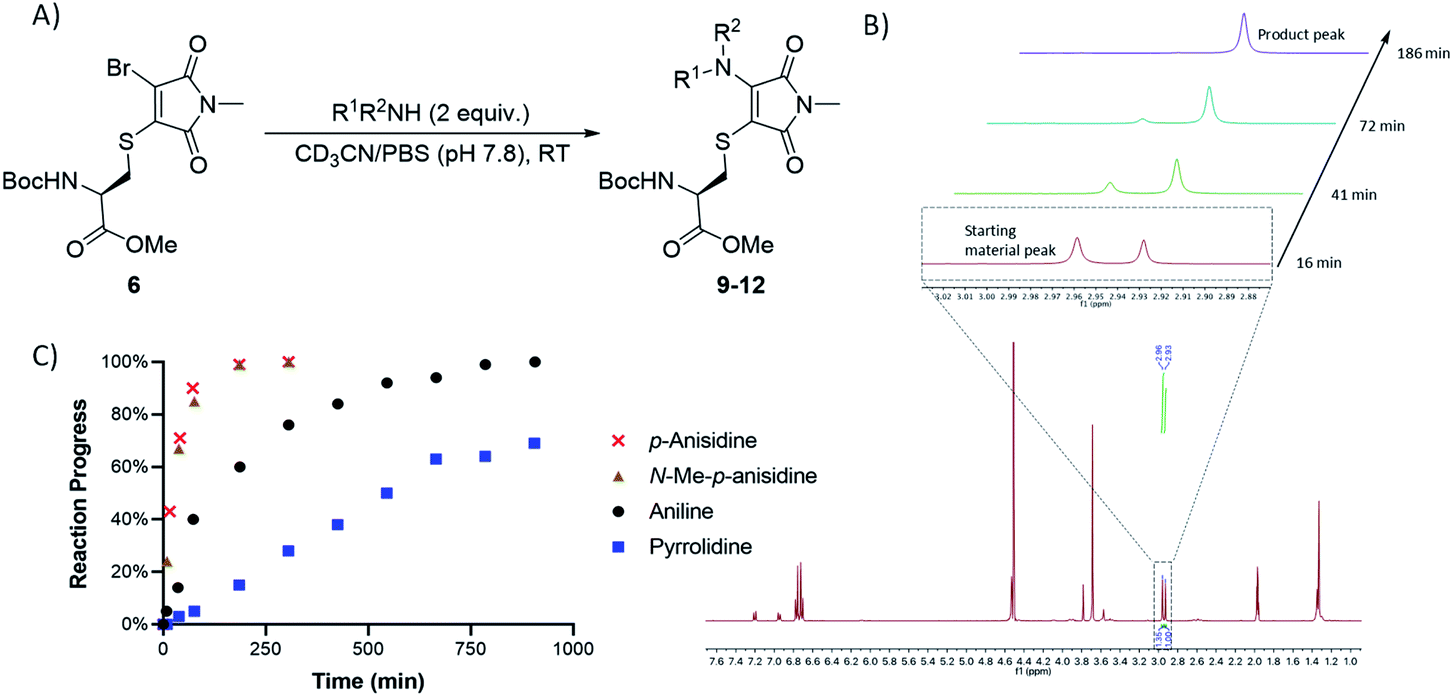 | ||
| Fig. 3 (A) General reaction scheme for NMR time course reactions. (B) Methyl peak development observed in time course 1H NMR study for reaction between p-anisidine and bromothiomaleimide 6. (C) Graph showing the progress of 4 key amine reactions under buffered conditions (see ESI Table S3†). | ||
To trial this strategy in protein bioconjugations, wild-type human serum albumin (HSA) was chosen as an initial target; it contains a single cysteine (Cys-34) and HSA-conjugates are of clinical interest as a leading platform for half-life extension.31–34 A useful feature of this chemistry is the ability to monitor the progress of both thiol and amine additions by UV-vis spectroscopy, as distinct λmax values are associated with the bromothiomaleimide (ε375 2910 M−1) and the red-shifted aminothiomaleimide products (ε415 4250 M−1). The conjugation of DBM 13 to the protein was complete after just 20 min, confirmed by UV (Fig. 4), and subsequently LCMS analysis (ESI Fig. S7†). p-Anisidine (50 mM) was then added to the reaction mixture and the absorbance was monitored at 415 nm, taking 180 min to plateau. LCMS analysis confirmed that the amine addition had taken place (Fig. 4). It was unsurprising to observe relatively long reaction times on HSA, with high concentrations of amine required to ensure the reaction went to completion; this is likely due to the buried nature of the Cys-34 cleft, and the fact that a proximal nucleophilic Tyr-84 has been proposed to intercept dihalomaleimide–HSA conjugates, as both halide atoms are lost from the backbone of the DBM during conjugation.20
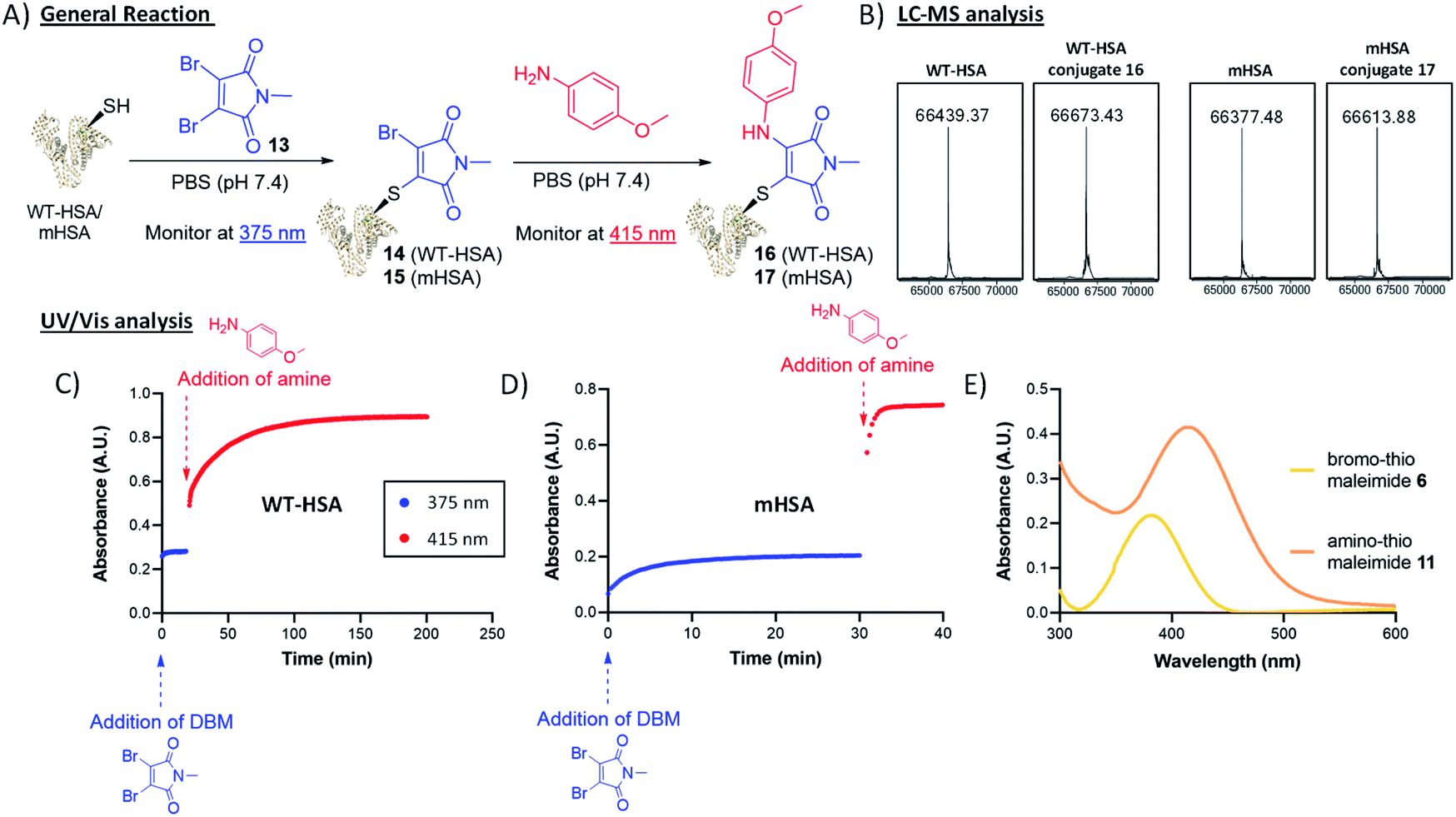 | ||
Fig. 4 Thiol–amine dual bioconjugation on wild type HSA (WT-HSA) and mutant HSA (mHSA); (A) general reaction scheme, (B) LCMS analysis of native WT-HSA, mHSA and final conjugates; expected mass for conjugate 16: 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 671 Da, observed mass: 66673 Da; expected mass for conjugate 17: 66613 Da, observed mass: 66614 Da, (C) and (D) UV/vis analysis of thiol–amine bioconjugation on WT-HSA and mHSA respectively; the absorbance of the resultant bromothiomaleimide was monitored at 375 nm and upon reaction completion, p-anisidine was added in the conjugation, where the progress was monitored at 415 nm, (E) UV/vis analysis of bromothiomaleimide 6 and amino-thiomaleimide 11. 671 Da, observed mass: 66673 Da; expected mass for conjugate 17: 66613 Da, observed mass: 66614 Da, (C) and (D) UV/vis analysis of thiol–amine bioconjugation on WT-HSA and mHSA respectively; the absorbance of the resultant bromothiomaleimide was monitored at 375 nm and upon reaction completion, p-anisidine was added in the conjugation, where the progress was monitored at 415 nm, (E) UV/vis analysis of bromothiomaleimide 6 and amino-thiomaleimide 11. | ||
To preclude these specific issues associated with wild-type HSA, we shifted our focus onto an HSA mutant (mHSA) with an alternative cysteine location (C34A + K93C), kindly provided by Albumedix.35 It was pleasing to observe in this case that the bromothiomaleimide was present in the LCMS analysis of the intermediate prior to amine addition, indicating that there were no reactions with other nucleophilic residues near the conjugation site (ESI Fig. S8†). Interestingly, p-anisidine addition was rapid (∼20-fold faster compared to WT HSA, see Fig. 4) for mHSA intermediates. A pseudo-first order analysis of the reaction with UV-vis spectroscopy was carried out and enabled calculation of the second order rate constant k2 for the p-anisidine addition to conjugate 15, with k2 = 0.5226 ± 0.0247 M−1 s−1 (ESI Fig. S36†). Interestingly, this was ∼16-fold faster than the k2 = 0.0367 ± 0.0059 M−1 s−1 calculated for the reaction of p-anisidine to single cysteine conjugate 6 using NMR analysis (ESI Fig. S33†). The reason for this rate acceleration on the protein is unclear, but could be postulated to be influenced by reduced organic solvent concentration in the protein reaction (7% vs. 42%), or an effect of the local amino-acid environment. Crucially, the amine addition possesses kinetics “on the protein” towards the faster end of the strain-promoted azide–alkyne cycloaddition (SPAAC) scale and more than two orders of magnitude faster than the Staudinger ligation.36–42
The high reactivity of the mHSA intermediate suggested that the amine could be reacted at much lower concentrations. Furthermore, we postulated that the rate difference between thiol and amine additions would enable an in situ reaction, in which DBM 13 (3 equiv.) was added to a mixture of the protein and the p-anisidine (5 equiv.). This proved effective after being left overnight at RT (ESI Fig. S13†), demonstrating this thiol–amine dual conjugation to be a rare example of an efficient multi-component reaction on a protein. Finally, conjugate 17 was assessed for thiol stability (glutathione, GSH). Treatment with 10 μM GSH (representative of extracellular concentrations),43–46 pH 7.4, 37 °C for 7 days, showed stability by LCMS analysis (ESI Fig. S29†). A further experiment was carried out to approximate the harsh intracellular GSH concentrations (4 mM GSH, pH 6.8, 37 °C)43–46 and showed little change after 24 h (ESI Fig. S31†). In summary, p-anisidine addition was shown to offer an alternative approach to hydrolysis for maleimide bioconjugate stabilisation. It offers the further advantages of taking place under mild conditions, can be accelerated simply by increasing the amine concentration and can be carried out step-wise or in situ.
A further enticing opportunity offered by this conjugation methodology was the possibility of generating triconjugates, if a heterobifunctional amine based on the p-anisidine core was used. To this end, two variants of p-anisidine, alkyne 18 and azide 22 were synthesised that would enable construction of clickable triconjugates. These scaffolds would then facilitate further modification as desired through the plethora of well-defined click chemistry currently available, depending on the nuances of both protein and probe in use. Importantly, functional analogues 18 and 22 displayed very similar reaction rates with the bromothiomaleimide 11 as p-anisidine (ESI Fig. S2†).
Our first test of this dual conjugation strategy was trialled on a peptide. Cell Penetrating Peptides (CPPs) have garnered interest as a means to achieve payload internalisation in a cell.47–49 P218R is an optimised cell-penetrating peptide designed by Dixon et al. and represents an extremely challenging modification target due to the abundance of lysine and arginine residues present (9 and 11 residues respectively) in a peptide of 3.8 kDa.50 Interestingly, the rapid kinetics of the thiol conjugation enabled efficient and rapid dual modification of the CPP via an in situ protocol (40 equiv. amine–alkyne 18, 1.2 equiv. DBM 19, 30 min at 37 °C, see Fig. 5), indicated by crude LCMS data (ESI Fig. S20†) and immediate colour change upon addition of DBM 19. Subsequent HPLC purification afforded conjugate 20 (Fig. 5). Work with modified analogues of 20 is underway for use as imaging agents.
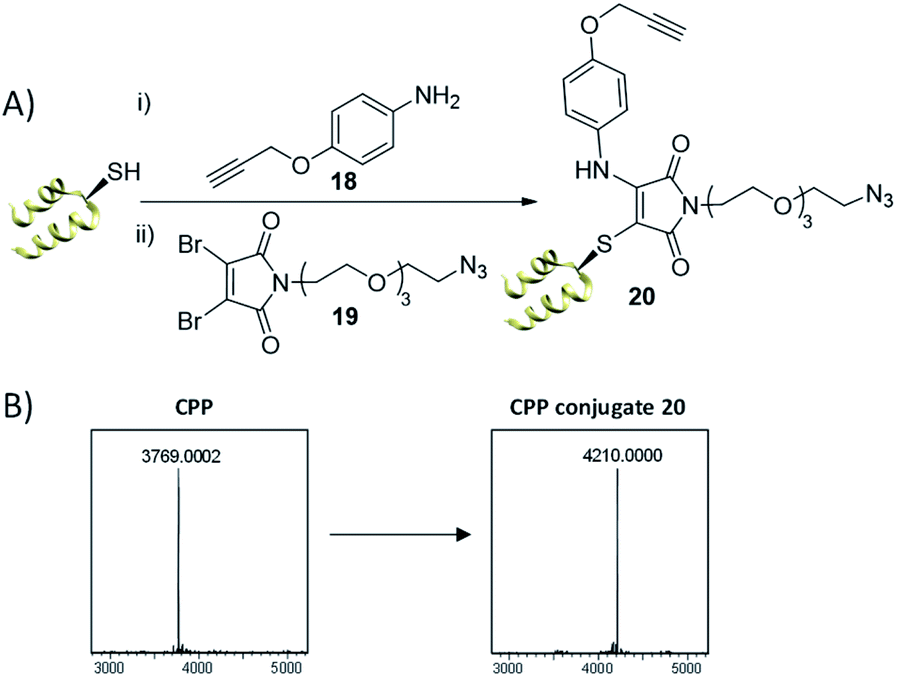 | ||
| Fig. 5 (A) Dual thiol–amine conjugation on CPP. CPP (0.9 mM) in PB (10 mM, pH 6.5), amine–alkyne 18 (40 equiv.), DBM 19 (1.2 equiv.) left at 37 °C for 30 min, (B) LCMS data; expected mass for conjugate 20: 4208 Da, observed mass: 4210 Da. See ESI Fig. S19–S22† for more detail. | ||
The creation of clickable triconjugates was also carried out on two different proteins. DBM 21, functionalised with biotin as a model handle (used widely in affinity purification and biological assays) was conjugated to mHSA, before amine–azide 22 (20 equiv.) was added and left at room temperature. The dual-functionalization occurred in 2 h at room temperature, with a small amount of unmodified and doubly modified HSA present as the only other detectable species (Fig. 6). Green Fluorescent Protein (GFP) was chosen as a second single cysteine containing protein to further appraise this dual conjugation strategy. Gratifyingly, conjugation of DBM 21 to the thiol was complete in just 5 minutes and amine–azide 22 in a further 95 minutes, with LCMS analysis showing a high degree of conversion (Fig. 6). To avoid the use of toxic copper in CuI-catalysed click reactions,36 modification of conjugates 23 and 25 was achieved with dibenzocyclooctyne–tetramethylrhodamine (DBCO–TAMRA) via SPAAC. This successfully yielded the desired fluorescent-biotinylated dual conjugates 24 and 26 (ESI Fig. S15 and S18†). These procedures serve as examples to show the simplicity of forming useful triconjugates with thiol–amine dual conjugation. Similar bifunctional biotin–TAMRA conjugates have recently been used to observe protein modification in human cells.51,52
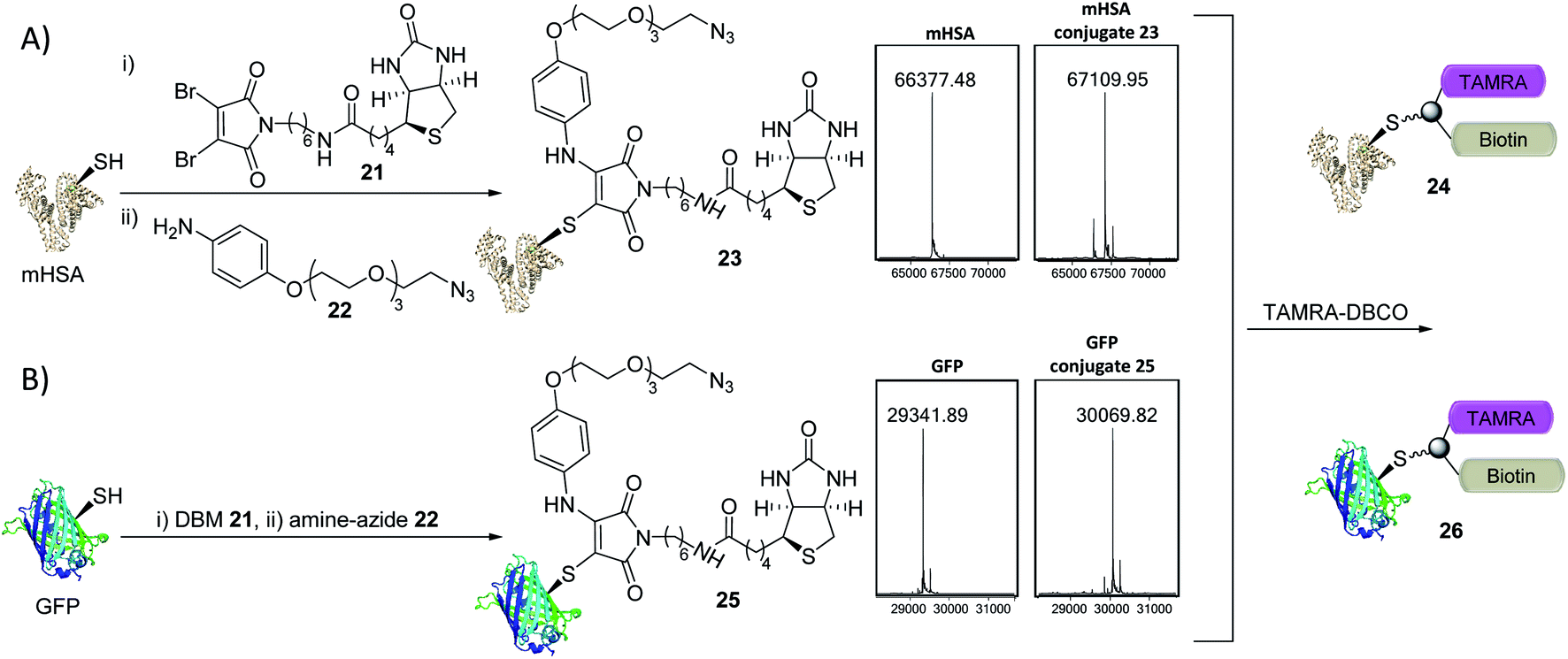 | ||
| Fig. 6 Trifunctional conjugate formation with mHSA and GFP (LCMS analysis shown). (A) (i) DBM 21 (1.5 equiv.), mHSA (100 μM) in PBS pH 7.4 at RT for 45 min; (ii) amine–azide 22 (20 equiv.), RT for 2 h; (iii) TAMRA–DBCO (10 equiv.), RT for 16 h. (B) (i) DBM 21 (2.0 equiv.), GFP (40 μM) in PBS pH 7.4 at 25 °C for 5 min; (ii) amine–azide 22 (30 equiv.), 25 °C for 95 min; (iii) TAMRA–DBCO (24 equiv.), RT for 5 h. See ESI Fig. S14–S18† for more detail. | ||
Conclusion
We have demonstrated a dual thiol–amine bioconjugation strategy that provides distinct advantages over the hydrolysis of maleimide conjugates. This method simultaneously confers thiol-stability and enables addition of an extra functional handle. Unlike maleimide conjugation and hydrolysis, this method does not form regio- or steroisomeric products. The dual conjugation reactions may be followed in real time using UV-vis spectroscopy owing to the formation of the aminothiomaleimide scaffold (ε415 4250 M−1). More specifically, we have demonstrated the use of p-anisidine-based functional amines that react with the bromothiomaleimide backbone and created trifunctional conjugates on both proteins and peptides. We have observed that the environment local to the cysteine of interest can affect the rate of amine addition and have shown that post-conjugation functionalisation of the aminothiomaleimide through click chemistry provides an efficient route to such triconjugates. This thiol–amine coupling is also demonstrated to be a rare example of an efficient multi-component bioconjugation reaction on a protein.Conflicts of interest
K. Nicholls is an employee of Albumedix ltd. V. Chudasama and J. R. Baker are Directors of the spinoff ThioLogics.Acknowledgements
The authors gratefully acknowledge EPSRC (CASE award and EP/R034621/1) and Albumedix, and LifeArc for funding and the EPSRC UK National Mass Spectrometry Facility (NMSF), Swansea. MTM is supported by Cancer Research UK (C63178/A24959) and Wellcome Trust (201959/Z/16/Z). In the Department of Chemistry, UCL, we also thank Kersti Karu and Malgorzata Puchnarewicz for assistance with mass spectrometry analysis, Rachael Dickman for HPLC assistance, and Abil Aliev for NMR analysis.References
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem.–Eur. J., 2019, 25, 43–59 CrossRef CAS.
- K. Renault, J. W. Fredy, P. Y. Renard and C. Sabot, Bioconjugate Chem., 2018, 29, 2497–2513 CrossRef CAS.
- P. Adumeau, S. K. Sharma, C. Brent and B. M. Zeglis, Mol. Imaging Biol., 2016, 18, 1–17 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, London, 3rd edn, 2013 Search PubMed.
- F. Li and R. I. Mahato, Mol. Pharm., 2017, 14, 1321–1324 CrossRef.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS.
- E. Dolci, V. Froidevaux, C. Joly-duhamel, R. Auvergne, B. Boutevin and S. Caillol, Polym. Rev., 2016, 56, 512–556 CrossRef CAS.
- Z. Yang, Y. Guo, S. L. Ai, S. X. Wang, J. Z. Zhang, Y. X. Zhang, Q. C. Zou and H. X. Wang, Mater. Chem. Front., 2019, 3, 571–578 RSC.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS.
- P. A. Szijj, C. Bahou and V. Chudasama, Drug Discovery Today: Technol., 2018, 30, 27–34 CrossRef.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS.
- L. N. Tumey, M. Charati, T. He, E. Sousa, D. Ma, X. Han, T. Clark, J. Casavant, F. Loganzo, F. Barletta, J. Lucas and E. I. Graziani, Bioconjugate Chem., 2014, 25, 1871–1880 CrossRef CAS.
- M. E. B. Smith, M. B. Caspersen, E. Robinson, M. Morais, A. Maruani, J. P. M. Nunes, K. Nicholls, M. J. Saxton, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2015, 13, 7946–7949 RSC.
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 RSC.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS.
- Y. Cui, Y. Yan, Y. Chen and Z. Wang, Macromol. Chem. Phys., 2013, 214, 470–477 CrossRef CAS.
- J. K. Y. Tan, J. L. Choi, H. Wei, J. G. Schellinger and S. H. Pun, Biomater. Sci., 2015, 3, 112–120 RSC.
- N. Forte, M. Livanos, E. Miranda, M. Morais, X. Yang, V. S. Rajkumar, K. A. Chester, V. Chudasama and J. R. Baker, Bioconjugate Chem., 2018, 29, 486–492 CrossRef CAS.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS.
- J. T. Husband, A. C. Hill and R. K. O'Reilly, Polym. Int., 2019, 68, 1247–1254 CrossRef CAS.
- S. Ramesh, P. Cherkupally, T. Govender, H. G. Kruger, F. Albericio and B. G. De La Torre, Chem. Commun., 2016, 52, 2334–2337 RSC.
- A. Wall, K. Nicholls, M. B. Caspersen, S. Skrivergaard, K. A. Howard, K. Karu, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2019, 17, 7870–7873 RSC.
- Z. Chen, S. D. Boyd, J. S. Calvo, K. W. Murray, G. L. Mejia, C. E. Benjamin, R. P. Welch, D. D. Winkler, G. Meloni, S. D'Arcy and J. J. Gassensmith, Bioconjugate Chem., 2017, 28, 2277–2283 CrossRef CAS.
- F. F. Schumacher, M. Nobles, C. P. Ryan, M. E. B. Smith, A. Tinker, S. Caddick and J. R. Baker, Bioconjugate Chem., 2011, 22, 132–136 CrossRef CAS.
- A. B. Mabire, M. P. Robin, W. Quan, H. Willcock, V. G. Stavros and R. K. O'Reilly, Chem. Commun., 2015, 51, 9733–9736 RSC.
- Y. Xie, J. T. Husband, M. Torrent-Sucarrat, H. Yang, W. Liu and R. K. O'Reilly, Chem. Commun., 2018, 54, 3339–3342 RSC.
- F. Brotzel, C. C. Ying and H. Mayr, J. Org. Chem., 2007, 72, 3679–3688 CrossRef CAS.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS.
- B. Elsadek and F. Kratz, J. Controlled Release, 2012, 157, 4–28 CrossRef CAS.
- R. E. Kontermann, Expert Opin. Biol. Ther., 2016, 16, 903–915 CrossRef CAS.
- D. Sleep, J. Cameron and L. R. Evans, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5526–5534 CrossRef CAS.
- M. T. Larsen, M. Kuhlmann, M. L. Hvam and K. A. Howard, Mol. Cell. Ther., 2016, 4, 1–12 CrossRef.
- K. K. Schelde, K. Nicholls, F. Dagnæs-Hansen, K. Bunting, H. Rawsthorne, B. Andersen, C. J. A. Finnis, M. Williamson, J. Cameron and K. A. Howard, J. Biol. Chem., 2019, 294, 3735–3743 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- X. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS.
- M. F. Debets, J. S. Prins, D. Merkx, S. S. Van Berkel, F. L. Van Delft, J. C. M. Van Hest and F. P. J. T. Rutjes, Org. Biomol. Chem., 2014, 12, 5031–5037 RSC.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 1–20 CrossRef CAS.
- M. B. Soellner, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2006, 128, 8820–8828 CrossRef CAS.
- A. S. Cohen, E. A. Dubikovskaya, J. S. Rush and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 8563–8565 CrossRef CAS.
- M. Fernández, A. Shamsabadi and V. Chudasama, Chem. Commun., 2020, 56, 1125–1128 RSC.
- S. Wang, H. Ma, J. Li, X. Chen, Z. Bao and S. Sun, Talanta, 2006, 70, 518–521 CrossRef CAS.
- L. Ducry and B. Stump, Bioconjugate Chem., 2010, 21, 5–13 CrossRef CAS.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS.
- K. Kurrikoff, M. Gestin and Ü. Langel, Expert Opin. Drug Delivery, 2016, 13, 373–387 CrossRef CAS.
- F. Wang, Y. Wang, X. Zhang, W. Zhang, S. Guo and F. Jin, J. Controlled Release, 2014, 174, 126–136 CrossRef CAS.
- C. Bechara and S. Sagan, FEBS Lett., 2013, 587, 1693–1702 CrossRef CAS.
- J. E. Dixon, G. Osman, G. E. Morris, H. Markides, M. Rotherham, Z. Bayoussef, A. J. El Haj, C. Denning and K. M. Shakesheff, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E291–E299 CrossRef CAS.
- M. Broncel, R. A. Serwa, P. Ciepla, E. Krause, M. J. Dallman, A. I. Magee and E. W. Tate, Angew. Chem., Int. Ed., 2015, 54, 5948–5951 CrossRef CAS.
- E. M. Storck, J. Morales-Sanfrutos, R. A. Serwa, N. Panyain, T. Lanyon-Hogg, T. Tolmachova, L. N. Ventimiglia, J. Martin-Serrano, M. C. Seabra, B. Wojciak-Stothard and E. W. Tate, Nat. Chem., 2019, 11, 552–561 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05128d |
| This journal is © The Royal Society of Chemistry 2020 |