
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light driven generation and alkyne insertion reactions of stable bis-cyclometalated Pt(IV) hydrides†
Dionisio
Poveda
a,
Ángela
Vivancos
 a,
Delia
Bautista
b and
Pablo
González-Herrero
*a
a,
Delia
Bautista
b and
Pablo
González-Herrero
*a
aDepartamento de Química Inorgánica, Facultad de Química, Universidad de Murcia, Campus de Espinardo, 19, 30100 Murcia, Spain. E-mail: pgh@um.es
bÁrea Científica y Técnica de Investigación, Universidad de Murcia, Campus de Espinardo, 21, 30100 Murcia, Spain
First published on 13th October 2020
Abstract
Hydride complexes resulting from the oxidative addition of C–H bonds are intermediates in hydrocarbon activation and functionalization reactions. The discovery of metal systems that enable their direct formation through photoexcitation with visible light could lead to advantageous synthetic methodologies. In this study, easily accessible dimers [Pt2(μ-Cl)2(C^N)2] (C^N = cyclometalated 2-arylpyridine) are demonstrated as a very convenient source of Pt(C^N) subunits, which promote photooxidative C–H addition reactions with different 2-arylpyridines (N′^C′H) upon irradiation with blue light. The resulting [PtH(Cl)(C^N)(C′^N′)] complexes are the first isolable Pt(IV) hydrides arising from a cyclometalation reaction. A transcyclometalation process involving three photochemical steps is elucidated, which occurs when the C^N ligand is a monocyclometalated 2,6-diarylpyridine, and a detailed analysis of the photoreactivity associated with the Pt(C^N) moiety is provided. Alkyne insertions into the Pt–H bond of a photogenerated Pt(IV) hydride are also reported as a demonstration of the ability of this class of compounds to undergo subsequent organometallic reactions.
Introduction
Transition metal hydride complexes are key intermediates in numerous catalytic transformations and have been the subject of essential studies on their properties, bonding and reactivity that continue to date.1–10 The focus on Pt(IV) hydrides has been particularly intense, mostly in connection with the development of catalytic hydrocarbon activation and functionalization reactions and their mechanistic investigation.11–13 The oxidative addition of C–H bonds to Pt(II) species is the crucial step at the core of these processes, but isolation and characterization of the resulting organometallic Pt(IV) hydrides are rare14,15 and there are no isolated complexes of this kind arising from cyclometalation reactions.16 In addition, studies on the reactivity of Pt(IV) hydrides are mainly limited to reductive elimination16 and only in one instance has the insertion of unsaturated molecules into a Pt(IV)–H bond been investigated.17Visible light promoted transformations involving the activation of C–H bonds are being intensively researched because of the inherent benefits of using readily available substrates and a harmless and sustainable energy source, with most of the methodologies making use of photoredox catalysts that engage in bimolecular single-electron transfer (SET).18–24 Recent research from ours and other laboratories has demonstrated an unprecedented photooxidative C–H addition process that takes place upon visible-light irradiation of Pt(II) complexes of the type cis-N,N-[Pt(R)(C^N)(N′^C′H)], where C^N is a cyclometalated ligand of the 2-arylpyridine type, N′^C′H is an N-coordinated, nonmetalated one, and R can be a methyl or pentafluorophenyl ligand, leading to the cyclometalation of the N′^C′H ligand to give unstable bis-cyclometalated Pt(IV) hydrides [PtH(R)(C^N)(C′^N′)] (Scheme 1).25,26 When R = Me, these hydrides undergo the reductive elimination of methane to give bis-cyclometalated Pt(II) complexes [Pt(C^N)(C′^N′)], whereas pentafluorophenyl derivatives revert to the starting Pt(II) complexes in the dark.25 The C–H addition event in these systems is triggered by a triplet excited state involving a charge transfer from the metal to the coordinated N′^C′H ligand (3ML′CT). This state becomes thermally populated from the lowest triplet excited state, which arises from a photoinduced ligand-centered/metal-to-ligand charge-transfer transition involving the C^N ligand (3LC/MLCT). Therefore, the Pt(C^N) subunit is the key component allowing the formation of a relatively long-lived excited state from which the reactive state can be accessed. A distinctive feature of this process is that a single metal system performs the functions of light harvesting and C–H bond cleavage, the latter occurring through an inner-sphere mechanism.
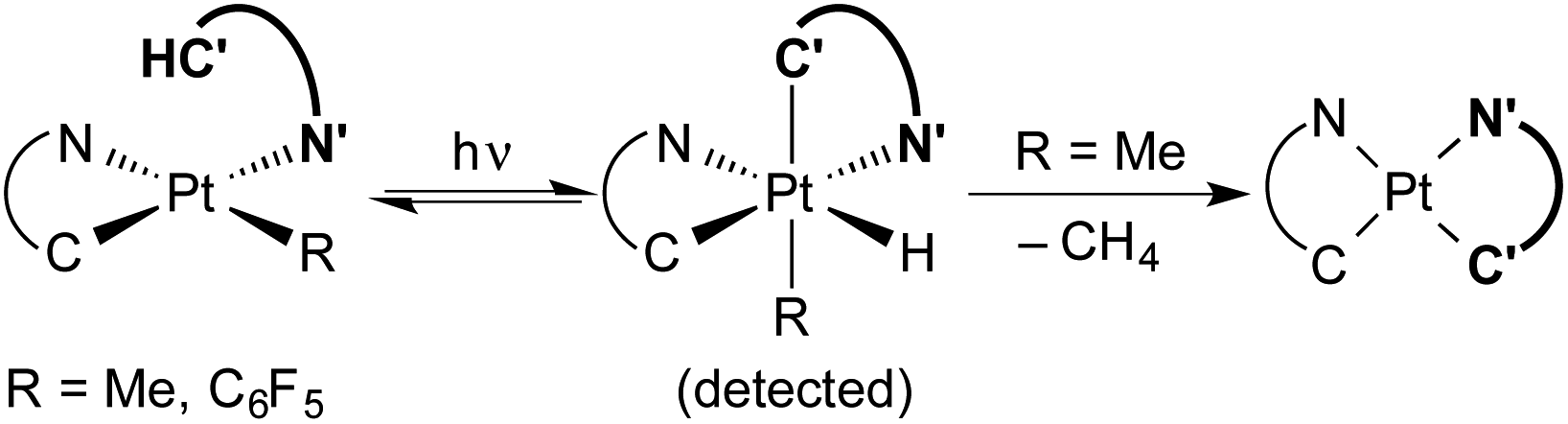 | ||
| Scheme 1 Photooxidative C–H addition reactions of methyl and pentafluorophenyl Pt(II) complexes. C^N = cyclometalated 2-arylpyridine. N′^C′H = N-coordinated 2-arylpyridine. | ||
The exploitation of the photooxidative C–H addition for the development of arene functionalization reactions is highly desirable because it occurs under very mild conditions. However, the synthesis of the Pt(II) precursors employed so far presents some disadvantages, since it requires several steps, the use of organolithium or -magnesium reagents to introduce the methyl or pentafluorophenyl ligands and the handling of unstable intermediates. In addition, the precarious stability of the produced hydrides is an important limitation for further fundamental studies. In this context, it is remarkable that a photooxidative C–H addition has been recently demonstrated as the key step in the borylation of 2-arylpyridines catalyzed by a Rh(I) carbene complex.27
In search for alternative and easily accessible Pt-based platforms for the photooxidative C–H addition reaction, we turned our attention to dichlorobridged dimers [Pt2(μ-Cl)2(C^N)2] (1), which could function as the simplest and most convenient source of the Pt(C^N) subunit. We have recently shown that these dimers establish bridge-splitting equilibria with monomeric complexes cis-N,N-[PtCl(C^N)(N′^C′H)] (cis-N,N-2) upon treatment with different 2-arylpyridines (N′^C′H) in solution at room temperature.28 In the present study, we explore the photoreactivity of these solutions, which has led to the isolation of bis-cyclometalated Pt(IV) hydrides, the discovery of a photochemical transcyclometalation process and a complete elucidation of the involved photochemical and thermal steps. In addition, the reactivity of a bis-cyclometalated Pt(IV) hydride toward alkynes is reported as an initial assessment of the ability of this class of complexes to take part in subsequent organometallic reactions.
Results and discussion
Photogeneration of bis-cyclometalated Pt(IV) hydrides
We initially examined the photoreactivity of three complexes cis-N,N-2 bearing the same 2-arylpyridine as the cyclometalated and N-coordinated ligand, namely 2-(p-tolyl)-, 2-phenyl- or 2-(2,4-difluorophenyl)pyridine (cis-N,N-2aa, 2bb or 2cc, respectively), which were generated in situ from the corresponding dimers 1a–c in acetone (Scheme 2). Irradiation with blue light (λmax = 454 nm) at room temperature for 24 h under rigorous exclusion of oxygen led to very pale yellow solutions that contained the Pt(IV) hydrides [PtH(Cl)(tpy)2] (3aa), [PtH(Cl)(ppy)2] (3bb) or [PtH(Cl)(dfppy)2] (3cc) as the major product. Complexes 3aa and 3bb were isolated as colorless solids in 74 or 71% yield, respectively. Their identity was established by 1H NMR, which showed a hydride signal as a singlet at −17.9 ppm with Pt satellites (JHPt = 1323 Hz) and two sets of resonances corresponding to inequivalent cyclometalated ligands. The orthogonal arrangement of these ligands is demonstrated by a distinctive aromatic resonance at 6.48 (3aa) or 6.64 (3bb) ppm flanked by platinum satellites, arising from the proton ortho to the metalated carbon of one of the ligands, which is considerably shielded by the ring current of the aryl group of the other one. The structure of 3aa was confirmed by single-crystal X-ray diffraction (Fig. 1), which showed the expected ligand arrangement resulting from a cis C–H oxidative addition. The Pt–H bond distance of 1.63(2) Å is close to the mean value of 1.554 Å found for Pt(IV) hydride complexes in the Cambridge Structural Database.29 Derivative 3cc was identified by NMR in solution, but the attempts to isolate it resulted in the precipitation of the poorly soluble Pt(II) complex [Pt(dfppy)2] (4cc) in a large proportion, and therefore it could not be obtained in pure form. This fact indicated the existence of an equilibrium between the Pt(IV) hydrides 3 and the Pt(II) complexes 4 involving reductive elimination/oxidative addition of HCl in solution, which was further supported by 1H NMR monitoring (see below). In accord with this, the bis-cyclometalated Pt(II) complexes 4 were obtained upon treatment of acetone solutions of the in situ generated hydrides with a base (K2CO3 or Et3N) in 71–74% yields with respect to the starting dimers 1. This method represents the shortest and most convenient way to synthesize this kind of Pt(II) complexes.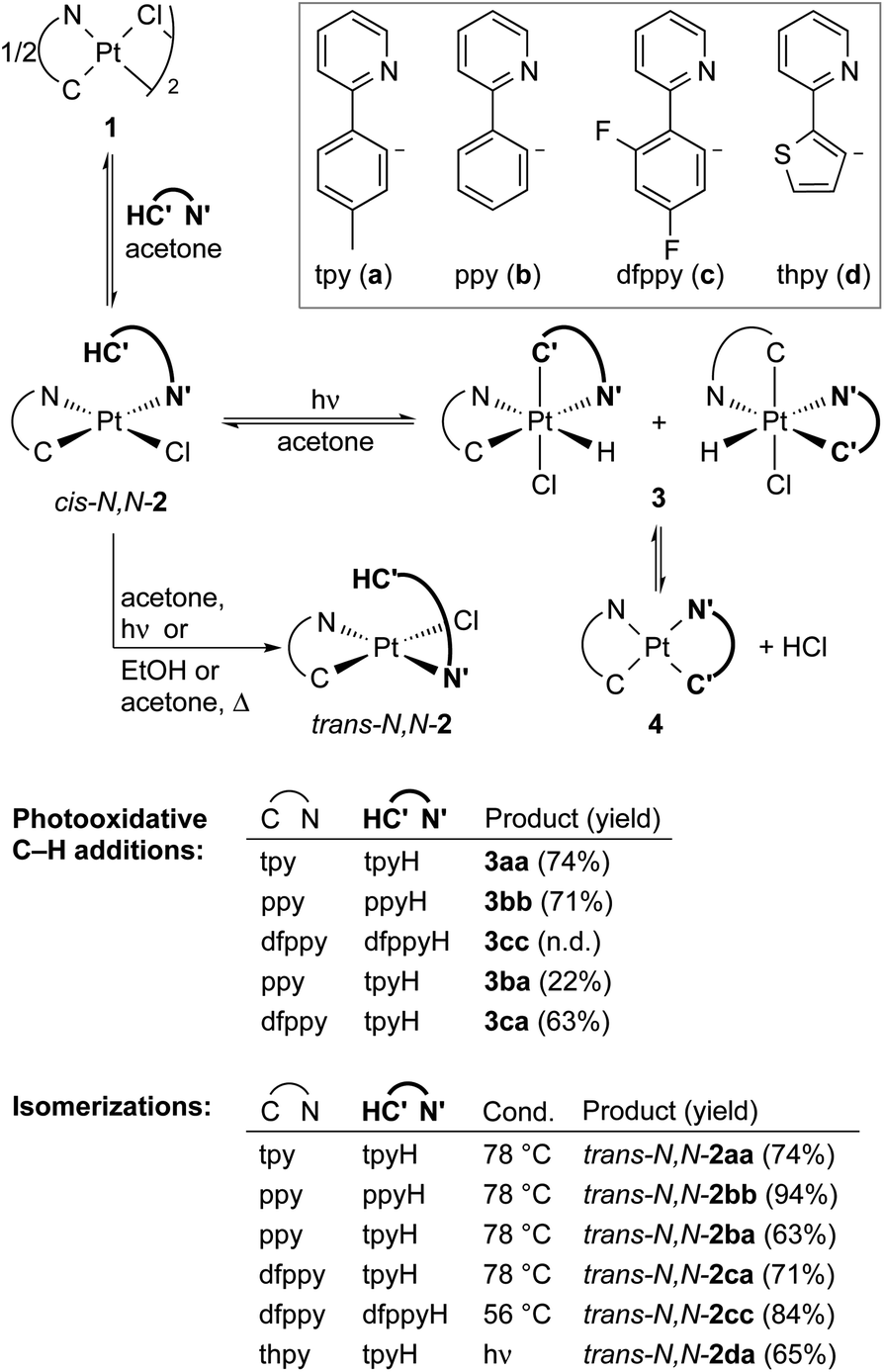 | ||
| Scheme 2 Photochemical and thermal reactions of cis-N,N-2 complexes. | ||
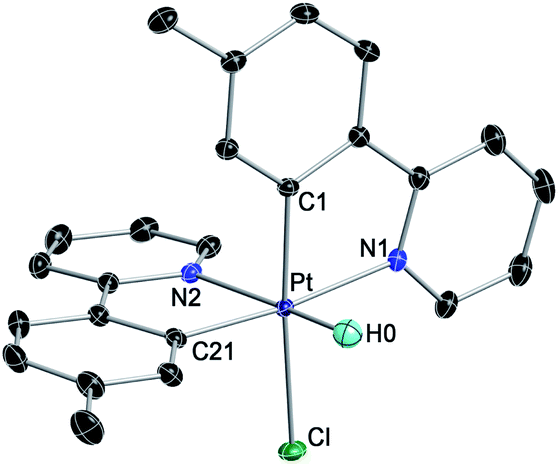 | ||
| Fig. 1 Crystal structure of 3aa·Me2CO (thermal ellipsoids at 50% probability). Hydrogen atoms (except for the hydride) and crystallization solvent are omitted. | ||
To further explore the scope for the photochemical generation of Pt(IV) hydrides, we tested the irradiation of complexes cis-N,N-2 bearing two different 2-arylpyridines. The irradiation of in situ generated solutions of cis-N,N-[PtCl(C^N)(tpyH)], with C^N = ppy (cis-N,N-2ba) or dfppy (cis-N,N-2ca), afforded the corresponding hydrides 3ba or 3ca. Even though a single isomer was expected from the cyclometalation of the tpyH ligand, complexes 3ba and 3ca were obtained as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of two structural isomers (Scheme 2), which implies a rapid isomerization process. The 1H NMR spectra show two shielded aromatic resonances with satellites, each one arising from a different cyclometalated ligand; in the case of 3ca, two hydride resonances are also distinguishable. Isolated yields were appreciably lower than those of 3aa–cc, particularly for 3ba, in part because of the difficulties encountered in precipitating them, associated with the higher solubilities of the isomer mixtures. In fact, the corresponding heteroleptic bis-cyclometalated Pt(II) complexes 4ba and 4ca were obtained in yields above 60% with respect to the starting dimers 1 upon treatment of the generated hydride mixtures with K2CO3.
:1 mixtures of two structural isomers (Scheme 2), which implies a rapid isomerization process. The 1H NMR spectra show two shielded aromatic resonances with satellites, each one arising from a different cyclometalated ligand; in the case of 3ca, two hydride resonances are also distinguishable. Isolated yields were appreciably lower than those of 3aa–cc, particularly for 3ba, in part because of the difficulties encountered in precipitating them, associated with the higher solubilities of the isomer mixtures. In fact, the corresponding heteroleptic bis-cyclometalated Pt(II) complexes 4ba and 4ca were obtained in yields above 60% with respect to the starting dimers 1 upon treatment of the generated hydride mixtures with K2CO3.
In contrast to the previous cis-N,N-2 complexes, irradiation of an acetone solution of cis-N,N-[PtCl(thpy)(tpyH)] [cis-N,N-2da; thpy = cyclometalated 2-(2-thienyl)pyridine] led to a clean isomerization to trans-N,N-2da within less than 3 h (Scheme 2 and Fig. S36†). This compound did not undergo any further transformation upon continued photoexcitation. The isomerization of complexes of the type cis-N,N-2 to their trans-N,N counterparts has not been directly addressed in the literature. However, the fact that the reported bridge-splitting reactions of dimers 1 with 2-arylpyridines at high temperatures give trans-N,N-2 complexes exclusively indicates that these isomers are thermodynamically more stable.30,31 Consistent with this, complexes trans-N,N-2aa, 2bb, 2cc, 2ba and 2ca were obtained upon heating of the corresponding cis-N,N precursors in EtOH or acetone (Scheme 2).
The photochemical generation of 3aa, 3ba and 3ca was monitored by 1H NMR in acetone-d6 to gain additional understanding of the involved processes (Fig. S37–S39†). The hydrides are already detected after 1 h and the two structural isomers of 3ba and 3ca appear simultaneously. Small amounts of trans-N,N-2aa, 2ba or 2ca and the corresponding bis-cyclometalated Pt(II) complex 4 emerge alongside the hydride(s), but remain at low relative proportions (max. 12%). The formation of trans-N,N-2 may indicate that a small fraction of the photoexcited cis-N,N-2 molecules undergo an isomerization process instead of the photooxidative C–H addition. The presence of complexes 4 clearly points to a HCl reductive elimination/oxidative addition equilibrium,16 which explains the presence of the two structural isomers of 3ba and 3ca because the oxidative addition of HCl is not selective. These experiments also showed that the proportion of complexes trans-N,N-2aa and 2ba decreased at the end of the monitored time (21 h), suggesting that they could exhibit photochemical reactivity. A separate experiment showed that trans-N,N-2aa undergoes photodissociation of the coordinated tpyH ligand to give dimer 1a, finally producing 3aavia cis-N,N-2aa (see the ESI for details†).
Additionally, the evolution of pure 3aa was monitored by 1H NMR in CD2Cl2 over a period of 24 h at room temperature in the dark, revealing that the complex is fairly stable under an inert atmosphere. It slowly reverts to cis-N,N-2aa, but it is still the major species after 8 h (Fig. S40†). Complexes trans-N,N-2aa and 4aa form in much smaller proportions. Since cis-N,N-2aa does not isomerize to trans-N,N-2aa at room temperature in the dark,28 the formation of the latter may result from a C–H reductive elimination involving the tpy ligand with the N atom trans to the hydride, which is supported by the behaviour of the analogous hydride complex bearing 2,6-di(p-tolyl)pyridine (see below).
Photochemical transcyclometalation
The photogeneration of Pt(IV) hydrides bearing monocyclometalated 2,6-di(p-tolyl)pyridine (dtpyH) was also attempted. Irradiation of a solution of cis-N,N-[PtCl(dtpyH)(tpyH)] (cis-N,N-2ea) in acetone, generated in situ from the dimer 1e and excess tpyH, produced complex 3aa in 79% isolated yield after 24 h, resulting from the cyclometalation of two tpyH ligands with loss of dtpyH2 (Scheme 3). Therefore, this reaction involves a transcyclometalation32–36 process, leading to the substitution of the dtpyH ligand by tpy. In contrast, the irradiation of an acetone solution of cis-N,N-[PtCl(dtpyH)(dfppyH)] (cis-N,N-2ec) led to the precipitation of the Pt(II) complex [PtCl(dtpyH2-κC)(dfppy)] (5ec, 63% yield; Scheme 3). The X-ray diffraction analysis of this compound (Fig. 2) showed the presence of a C-bound 2,6-di(p-tolyl)pyridinium ligand trans to the N atom of a cyclometalated dfppy. There is a N–H⋯Pt interaction (2.237 Å), for which there are precedents in complexes resulting from the intramolecular deprotonation of Pt(IV) hydrides by metalated aryl ligands bearing an amine functionality.17,37,38 The NH proton appears as a relatively broad resonance at 17.10 ppm with Pt satellites (JHPt ∼ 76 Hz) in the 1H NMR spectrum (CD2Cl2). Complex 5ec must originate from [PtH(Cl)(dtpyH)(dfppy)] (3ec) via decoordination of the pyridine group of the dtpyH ligand, which can then act as a base toward the hydride. This is probably favoured by the steric repulsion between the nonmetalated p-tolyl group and the pyridine ring of the dfppy ligand, as evidenced by the structure of [PtCl2(dtpyH)(tpy)], which shows an elongated Pt–N bond distance.28 The reaction of 5ec with K2CO3 afforded the bis-cyclometalated Pt(II) complex 4ec, i.e., the same reactivity expected from the hydride 3ec. Monitoring of a CD2Cl2 solution of pure 5ec by 1H NMR in the dark showed the gradual formation of 3ec (Fig. S42†), giving rise to a singlet resonance at −20.43 ppm with satellites (JHPt = 1426 Hz). Reasonably, these complexes establish an equilibrium in solution which shifts toward 5ec in acetone because of its low solubility in this solvent. This experiment also showed the formation of small amounts of the new Pt(II) complex trans-N,N-[PtCl(dtpyH2-κN)(dfppy)] (trans-N,N-6ec), arising from a C–H reductive elimination involving the metalated p-tolyl moiety in 3ec. Complex trans-N,N-6ec could be isolated in low yield from an in situ generated solution of 3ecvia irradiation of cis-N,N-2ec under more diluted conditions and its crystal structure was solved by X-ray diffraction (Fig. 2). Reported metal complexes with N-coordinated 2,6-diarylpyridine ligands are extremely rare;39,40 some of them have been found to form via reversible hydrogenolysis of the monocyclometalated ligand, although they have not been isolated.41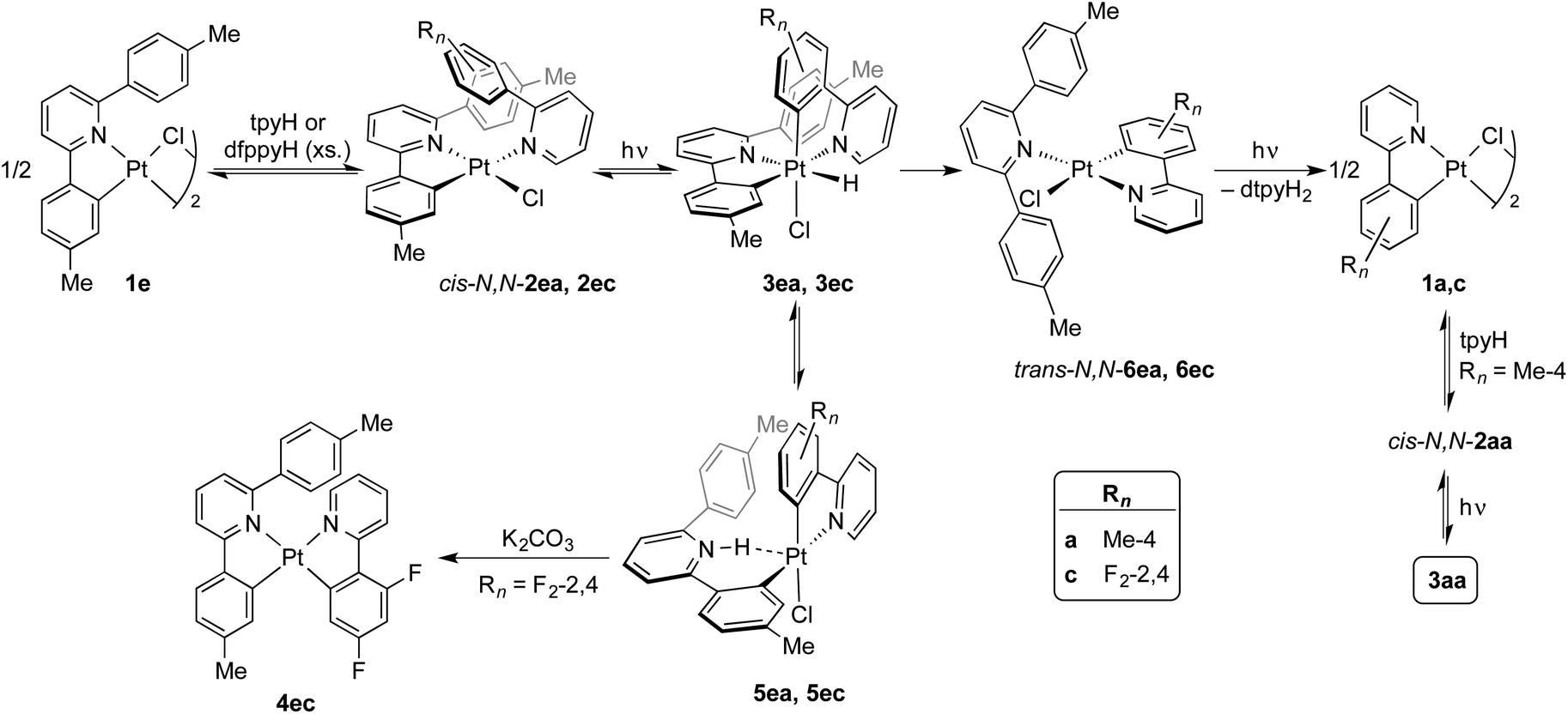 | ||
| Scheme 3 Reactions and intermediates leading to photochemical transcyclometalation. | ||
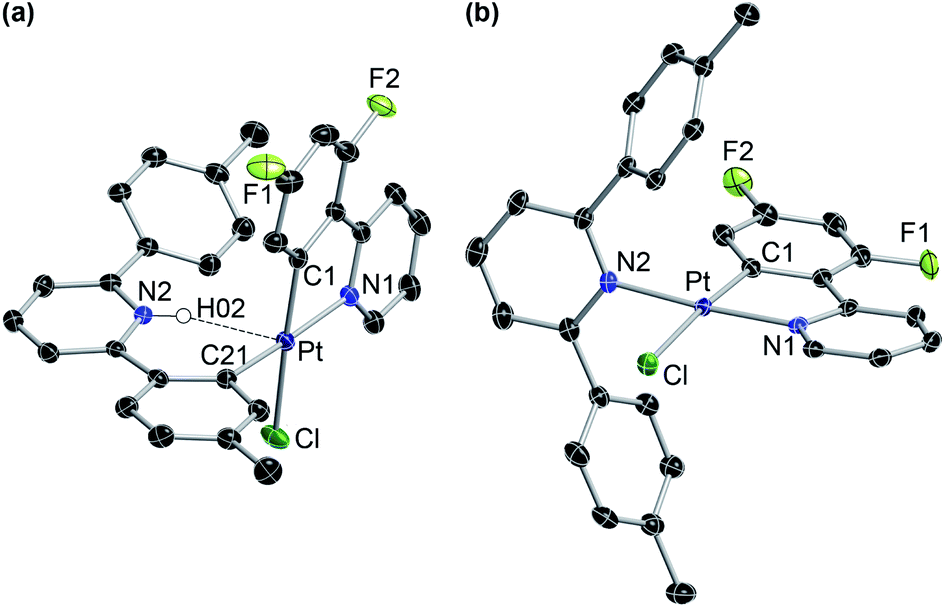 | ||
| Fig. 2 Crystal structures of complexes 5ec (a) and trans-N,N-6ec·CH2Cl2 (b). Solvent molecules and hydrogen atoms are omitted, except for the NH moiety in 5ec. | ||
The identification of complexes 3ec, 5ec and trans-N,N-6ec helped us to find the analogous intermediates in the transcyclometalation reaction leading to 3aa. Monitoring of the irradiation of an acetone-d6 solution of cis-N,N-2ea by 1H NMR (Fig. S43†) demonstrated the formation of the hydride [PtH(Cl)(dtpyH)(tpy)] (3ea) as the major product during the initial stages (ca. 3 h), which is in equilibrium with small amounts of [PtCl(dtpyH2-κC)(tpy)] (5ea). Upon continued irradiation, complexes 3ea and 5ea gradually disappear as trans-N,N-6ea, cis-N,N-2aa and 3aa emerge. The C–H reductive elimination involving the metalated p-tolyl group in 3ea and the substitution of the resulting N-coordinated dtpyH2 ligand in trans-N,N-6ea for a tpyH ligand are thus key to the transcyclometalation reaction. The latter could take place via photodissociation of the dtpyH2 ligand to give dimer 1a, as observed for trans-N,N-2aa, which would then react with tpyH to give cis-N,N-2aa. Continued photoexcitation would then produce 3aa. Indirect support for the photodissociation of dtpyH2 from trans-N,N-6ea was obtained by irradiating a solution of trans-N,N-6ec in acetone-d6, which resulted in the decoordination of the dtpyH2 ligand and the formation of a precipitate of dimer 1c (Fig. S44†).
Excited states of complexes cis-N,N-2aa and 2da
To get an understanding of their contrasting photochemical reactivity, the lowest-energy excited states of derivatives cis-N,N-2aa and 2da were examined by means of electronic absorption spectroscopy and density functional theory (DFT) and time-dependent DFT (TDDFT) calculations at the B3LYP/(6-31G**+LANL2DZ) level considering solvent effects (acetone). The absorption spectrum of cis-N,N-2aa in acetone solution at 298 K shows two low-intensity, broad bands with maxima at ca. 360 and 400 nm, the latter tailing to ca. 450 nm, whereas complex cis-N,N-2da gives rise to an additional maximum at 422 nm (Fig. S48 and S52†). These characteristics are compatible with mixed ligand-centered/metal-to-ligand charge-transfer (LC/MLCT) transitions involving the C^N ligand (tpy or thpy), which are typically observed for cyclometalated Pt(II) complexes and are expected to have a lower energy for cis-N,N-2da because of the lower π–π* transition energy of the thpy ligand relative to tpy.42,43 The TDDFT calculations (Tables S16 and S19†) predict two LC/MLCT transitions involving the C^N ligand as the lowest singlet excitations in both cases. Therefore, irradiation with blue light must mainly populate the lowest 1LC/MLCT state.The observed photoreactivity is expected to involve the lowest triplet excited states because the strong spin–orbit coupling induced by the Pt atom promotes fast intersystem crossing to the triplet manifold. Vibrational relaxation should then lead to the lowest triplet, from which either deactivation to the ground state or thermal population of higher-lying, reactive excited states can occur. Fig. 3(a) shows an energy diagram of the four lowest vertical triplet excitations from TDDFT calculations (see the ESI for details†). The lowest one (T1) corresponds to a LC/MLCT transition involving the cyclometalated ligand (tpy or thpy), lying lower in energy for cis-N,N-2da. A similar LC/MLCT transition incorporating some halide-to-ligand charge transfer character [XLCT or p(Cl) → π*(C^N)] is the second triplet excitation (T2) for cis-N,N-2aa or the third one (T3) for cis-N,N-2da. There is also a low-lying metal-centered excitation (MC), which is lower in energy for cis-N,N-2da (T2) than for cis-N,N-2aa (T3). In both cases, the T4 excitation is a ML′CT transition involving the coordinated tpyH ligand (L′) with a very similar energy in both complexes.
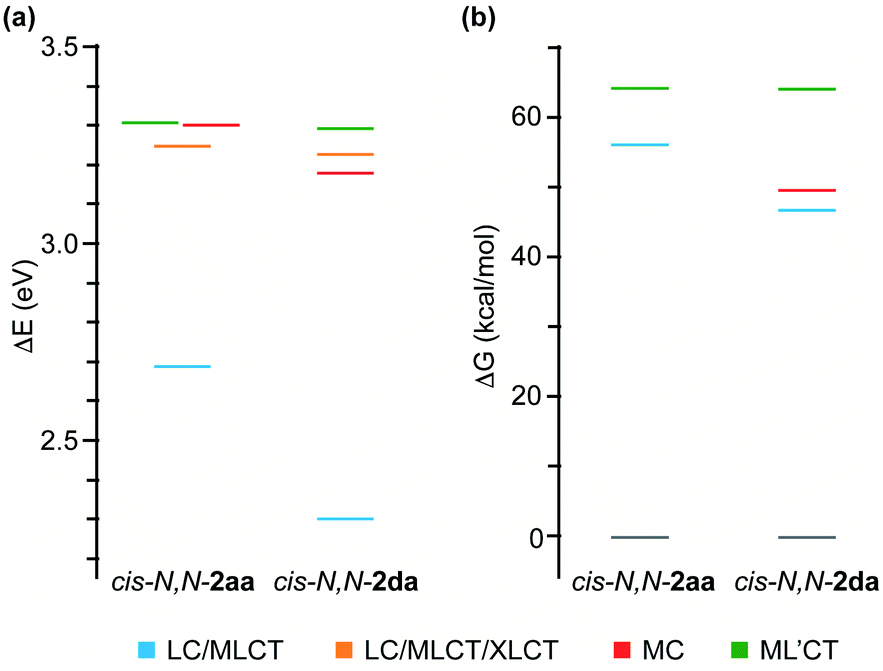 | ||
| Fig. 3 Energy diagrams showing the lowest vertical triplet excitations at the ground-state geometries of cis-N,N-2aa and 2da (a) and free energies of the optimized triplet excited states relative to the respective ground states (b). | ||
The geometries of the 3LC/MLCT and 3ML′CT states of both complexes (T1 and T4, respectively) and the 3MC state of cis-N,N-2da (T2) could be successfully optimized. Their free energies relative to the ground state are represented in Fig. 3(b). The spin density distributions (Fig. 4) agree in all cases with the topology of the expected electronic promotions on the basis of the TDDFT assignments. 3LC/MLCT states similar to T1 in cyclometalated Pt(II) complexes are usually long-lived and non-reactive.42,44,45 The T4 state is postulated as the reactive excited state leading to the photooxidative C–H addition in cis-N,N-2aa because it involves a partial reduction of the tpyH ligand by the metal. Its population could take place from the T1 state, which lies 8.1 kcal mol−1 below. Its relaxed structure reveals noticeable changes in the bond distances (Table S21†) and angles within the coordinated tpyH ligand, including a nearly coplanar arrangement of the p-tolyl and pyridine rings (Fig. S54†), consistent with an electronic promotion to an extended π* orbital.
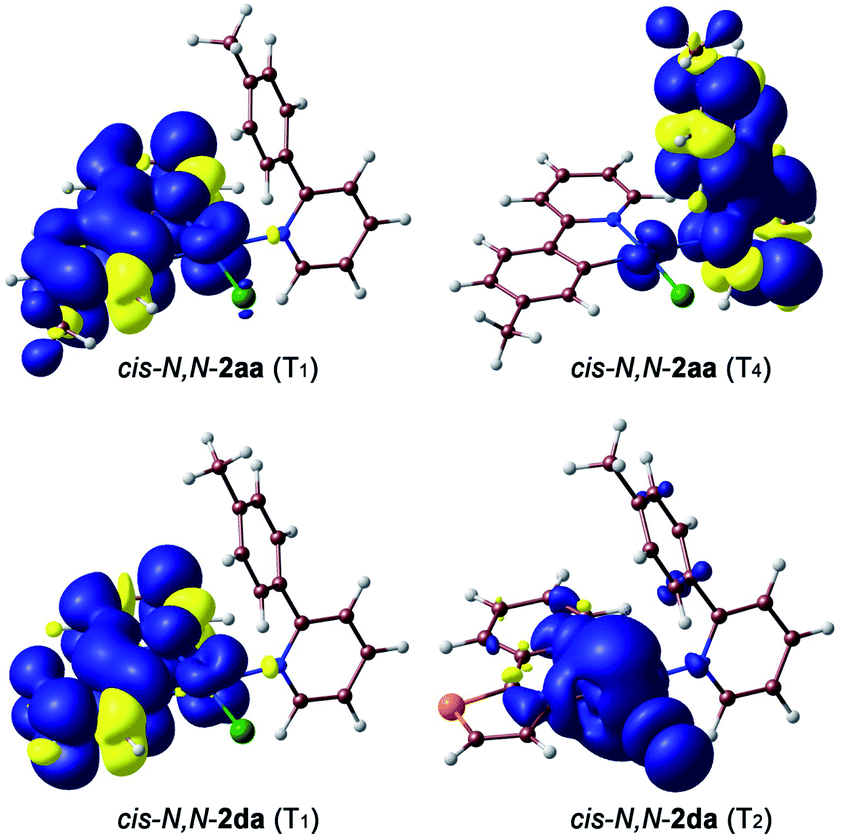 | ||
| Fig. 4 Spin density distributions (0.001 e bohr−3) for the optimized T1 and T4 states of cis-N,N-2aa and T1 and T2 of cis-N,N-2da. | ||
The free energy difference between the T1 and T4 states in complex cis-N,N-2da is considerably higher (17.4 kcal mol−1) than in cis-N,N-2aa, meaning that thermal population of T4 from T1 should be much more difficult. Instead, it is the T2 state that apparently gets easily populated. This state involves an electronic promotion to the metal dx2–y2 orbital, having antibonding character along the Pt–ligand σ-bonds, and its relaxed structure reveals a significant elongation of the N–Pt–Cl axis (Fig. S55 and Table S22†). Hence, its population could be the starting point of the isomerization to trans-N,N-2davia σ-bond labilization and ligand dissociation. From this analysis, it is clear that the photooxidative C–H addition requires that the reactive 3ML′CT state be relatively close in energy to the lowest 3LC/MLCT state and therefore should be more favorable when the involved 2-arylpyridine ligands have similar π–π* transition energies. In addition, the population of low-lying dissociative 3MC states may become competitive in this kind of complexes.
Alkyne insertion reactions
To probe the utility of the photogenerated hydrides for subsequent organometallic reactions, the reactivity of 3aa toward alkynes was investigated. There is only one previous study on the reactions of Pt(IV) hydrides with alkynes,17 which employed complexes of the type [PtH(X)(C^N)2] [C^N = cyclometalated 1-(dimethylamino)naphthalene; X = Cl−, Br− or CF3COO−], but only the reaction of the trifluoroacetate derivative with the highly reactive dimethylacetylene dicarboxylate (DMAD) succeeded to give an alkenyl Pt(IV) complex, for which a Z stereochemistry was proposed. Treatment of an in situ generated solution of 3aa in acetone with DMAD at room temperature afforded the bis-cyclometalated chloro(alkenyl) complex [PtCl(tpy)2(C(CO2Me)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Me)] (7; Scheme 4) in high yield, resulting from the hydroplatination (i.e., insertion into the Pt–H bond) of the triple bond. The reaction was completed within less than 2 h. The 1H NMR spectrum in CD2Cl2 showed that only one of the two possible configurations of the alkenyl ligand is obtained. The alkenylic proton gives a relatively broad resonance at 6.17 ppm with Pt satellites, but the JHPt constant of ca. 72 Hz is not conclusive enough to assign the stereochemistry. An X-ray diffraction analysis revealed an E configuration (Fig. 5) of the alkenyl and also corroborated that the ligand arrangement in 7 is identical to that found for the parent complex 3aa, except for the replacement of the hydride by an alkenyl ligand. The less reactive internal alkynes diphenylacetylene, 1-phenyl-1-propyne or methyl phenylpropiolate did not react with 3aa under the same conditions. Relatively fast reactions did take place with terminal alkynes bearing an inductively withdrawing group to give complexes [PtCl(tpy)2(CHCHR)], with R = Ph (8a), COOMe (8b) or C5H4N-3 (8c) (Scheme 4), whereas tert-butylacetylene proved unreactive. The 1H NMR spectra of the crude reaction mixtures showed a single hydroplatination product, which demonstrates that the reactions are regio- and stereoselective. The two alkenylic protons display JHH values of 11–12 Hz, consistent with cis-vicinal protons, whereas the JHPt values are very different (ca. 30 or 110 Hz), indicating that one of them is geminal with respect to the Pt atom and the other one trans-vicinal. Therefore, the Pt atom binds to the terminal carbon of the alkyne and the resulting alkenyl has a Z configuration, which was corroborated by X-ray diffraction analyses (Fig. 5, S6 and S7†).
CHCO2Me)] (7; Scheme 4) in high yield, resulting from the hydroplatination (i.e., insertion into the Pt–H bond) of the triple bond. The reaction was completed within less than 2 h. The 1H NMR spectrum in CD2Cl2 showed that only one of the two possible configurations of the alkenyl ligand is obtained. The alkenylic proton gives a relatively broad resonance at 6.17 ppm with Pt satellites, but the JHPt constant of ca. 72 Hz is not conclusive enough to assign the stereochemistry. An X-ray diffraction analysis revealed an E configuration (Fig. 5) of the alkenyl and also corroborated that the ligand arrangement in 7 is identical to that found for the parent complex 3aa, except for the replacement of the hydride by an alkenyl ligand. The less reactive internal alkynes diphenylacetylene, 1-phenyl-1-propyne or methyl phenylpropiolate did not react with 3aa under the same conditions. Relatively fast reactions did take place with terminal alkynes bearing an inductively withdrawing group to give complexes [PtCl(tpy)2(CHCHR)], with R = Ph (8a), COOMe (8b) or C5H4N-3 (8c) (Scheme 4), whereas tert-butylacetylene proved unreactive. The 1H NMR spectra of the crude reaction mixtures showed a single hydroplatination product, which demonstrates that the reactions are regio- and stereoselective. The two alkenylic protons display JHH values of 11–12 Hz, consistent with cis-vicinal protons, whereas the JHPt values are very different (ca. 30 or 110 Hz), indicating that one of them is geminal with respect to the Pt atom and the other one trans-vicinal. Therefore, the Pt atom binds to the terminal carbon of the alkyne and the resulting alkenyl has a Z configuration, which was corroborated by X-ray diffraction analyses (Fig. 5, S6 and S7†).
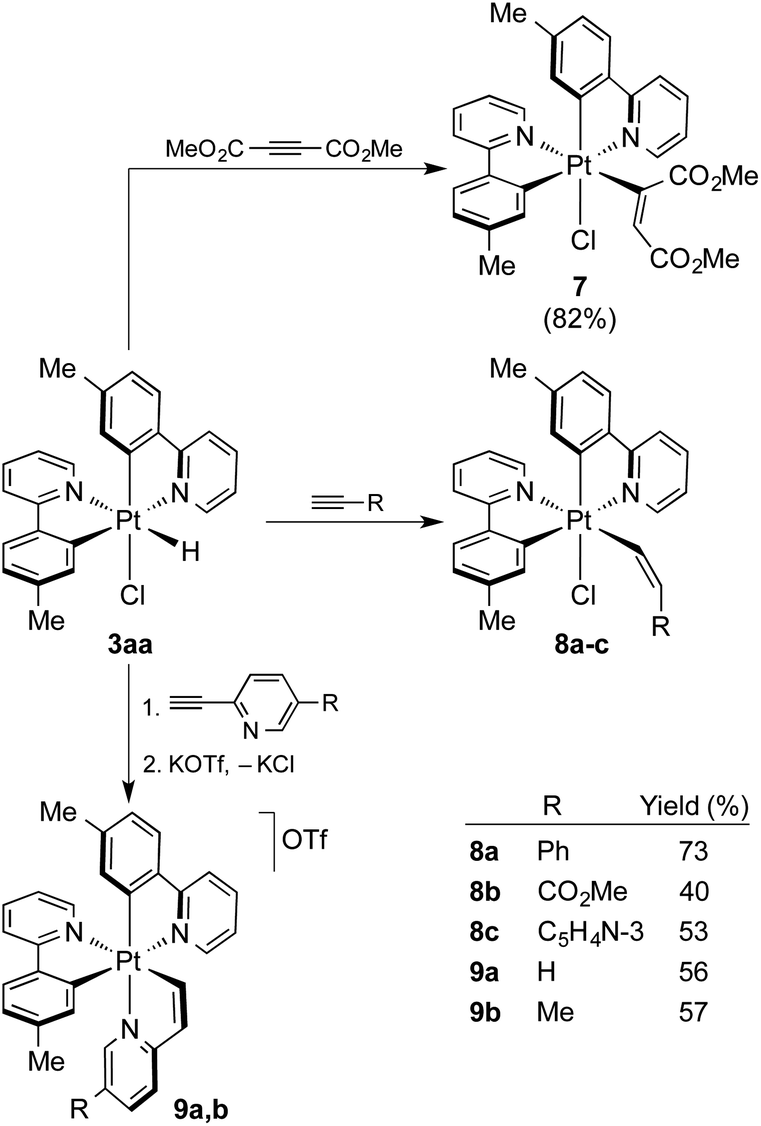 | ||
| Scheme 4 Alkyne insertion reactions into the Pt–H bond of 3aa. Yields are given with respect to precursor 1a. | ||
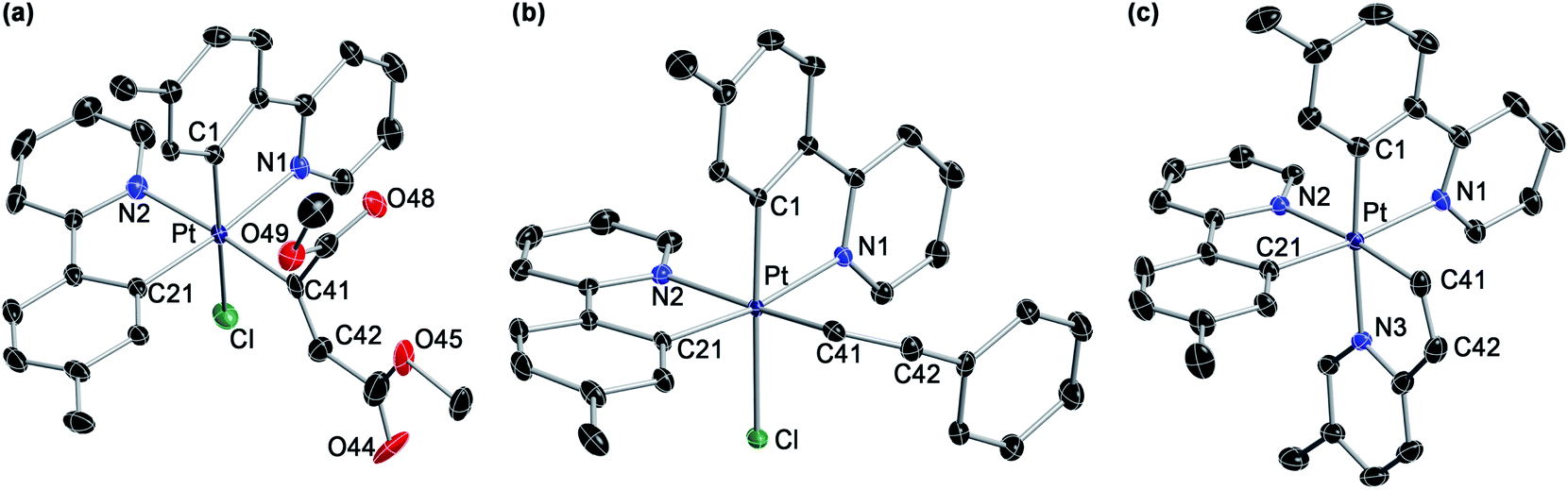 | ||
| Fig. 5 Crystal structures of complexes 7 (a), 8a·CH2Cl2 (b) and 9b-BPh4·CHCl3 (c) (thermal ellipsoids at 50% probability). Hydrogen atoms, crystallization solvent and the anion of 9b-BPh4 are omitted. | ||
The reactions with 2-ethynylpyridine and 2-ethynyl-5-methylpyridine with 3aa led to cationic tris-chelate complexes (9a or 9b, respectively), formally resulting from the hydroplatination of the triple bond to give a Z-configured alkenyl and subsequent displacement of the chloride ligand by the pyridine moiety. These complexes were isolated as the triflate salts by performing the reactions in the presence of KOTf. To get single crystals suitable for an X-ray diffraction study, the BPh4− salt of 9b (9b-BPh4) was also prepared via anion metathesis. Its crystal structure revealed a fac-N,N,N configuration around the metal (Fig. 5), suggesting that the relative arrangement of the tpy ligands remains unchanged in the course of the reaction and that the pyridyl moiety occupies the chloride position. The synthesis of tris-cyclometalated Pt(IV) complexes with a fac configuration is challenging46–48 and only in one previous instance has a crystal structure been successfully solved.49
The above described results indicate that the insertions of alkynes into the Pt–H bond of 3aa require the presence of at least one electron-withdrawing group, implying an electrophilic behaviour of the alkyne. Since most of the tested internal alkynes were found unreactive, it is also clear that the steric requirements of the alkyne substituents have a detrimental effect. In view of the different stereochemistry found for the insertion of DMAD with respect to terminal alkynes, it is possible that different reaction pathways are followed depending on the nature of the alkyne. A migratory insertion mechanism50–54 seems unlikely because the saturated coordination sphere in 3aa is expected to hinder the coordination of the alkyne. Moreover, this mechanism would preferentially lead to an E stereochemistry, which is inconsistent with the observed outcome of terminal alkyne insertions. The present insertions could involve a H atom or hydride transfer2,4,55 as the initial step, although conclusive evidence for either of these possibilities could not be gathered. Further investigations to elucidate the behaviour of hydrides 3 are underway.
Conclusions
Dichlorobridged dimers of the type [Pt2(μ-Cl)2(C^N)2] have been demonstrated as a suitable source of Pt(C^N) subunits that can promote the cyclometalation of 2-arylpyridines (N′^C′H) via photooxidative C–H addition in intermediates cis-N,N-[PtCl(C^N)(N′^C′H)] upon excitation with visible light. The resulting [PtH(Cl)(C^N)(C′^N′)] complexes are fairly stable and represent the first isolated Pt(IV) hydrides arising from a cyclometalation reaction. Bis-cyclometalated Pt(II) complexes [Pt(C^N)(C′^N′)] can be obtained in good yields from the photogenerated hydrides upon addition of a base, which represents the shortest and most convenient synthetic method for this kind of derivatives. When the C^N ligand in the dichlorobridged precursor is a monocyclometalated 2,6-diarylpyridine, a transcyclometalation process takes place, involving three photochemical steps.The observed photooxidative C–H addition process may compete with photoisomerization depending on the C^N/N′^C′H combination. DFT and TDDFT calculations substantiate that a reactive triplet excited state involving a charge-transfer from the metal to the coordinated N′^C′H ligand (3ML′CT) triggers the C–H addition. This state is thermally accessible from the lowest, C^N-based excited state (3LC/MLCT) when the two states are sufficiently close in energy; otherwise, a low-lying, dissociative metal-centered excited state may become populated, resulting in isomerization.
The present results may provide the basis for the systematic development of novel platforms for the photooxidative C–H addition. In addition, the described alkyne insertion reactions into the Pt–H bond of one of photogenerated Pt(IV) hydrides demonstrate that this kind of complexes are amenable for further organometallic reactions, which might allow their incorporation into catalytic cycles for visible light promoted C–H functionalization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Fundación Séneca (19890/GERM/15) and Ministerio de Ciencia, Innovación y Universidades (PGC2018-100719-BI00) for financial support. D. Poveda and Á. Vivancos thank Fundación Séneca for a predoctoral fellowship (20725/FPI/18) and a Saavedra Fajardo Fellowship (20398/SF/17), respectively.References
- G. S. McGrady and G. Guilera, Chem. Soc. Rev., 2003, 32, 383–392 RSC.
- Y. Hu and J. R. Norton, J. Am. Chem. Soc., 2014, 136, 5938–5948 CrossRef CAS.
- R. N. Perutz and B. Procacci, Chem. Rev., 2016, 116, 8506–8544 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS.
- A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS.
- M. A. Esteruelas, A. M. López and M. Oliván, Chem. Rev., 2016, 116, 8770–8847 CrossRef CAS.
- R. H. Morris, Chem. Rev., 2016, 116, 8588–8654 CrossRef CAS.
- A. Pintus, L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2016, 55, 12321–12324 CrossRef CAS.
- L. Rocchigiani, J. Fernandez-Cestau, I. Chambrier, P. Hrobárik and M. Bochmann, J. Am. Chem. Soc., 2018, 140, 8287–8302 CrossRef CAS.
- A. M. Wright, D. R. Pahls, J. B. Gary, T. Warner, J. Z. Williams, S. M. Spring, K. E. Allen, C. R. Landis, T. R. Cundari and K. I. Goldberg, J. Am. Chem. Soc., 2019, 141, 10830–10843 CrossRef CAS.
- L. Johansson, M. Tilset, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2000, 122, 10846–10855 CrossRef CAS.
- M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471–2526 CrossRef CAS.
- J. A. Labinger, Chem. Rev., 2017, 117, 8483–8496 CrossRef CAS.
- D. D. Wick and K. I. Goldberg, J. Am. Chem. Soc., 1997, 119, 10235–10236 CrossRef CAS.
- M. P. Jensen, D. D. Wick, S. Reinartz, P. S. White, J. L. Templeton and K. I. Goldberg, J. Am. Chem. Soc., 2003, 125, 8614–8624 CrossRef CAS.
- R. J. Puddephatt, Coord. Chem. Rev., 2001, 219–221, 157–185 CrossRef CAS.
- I. C. M. Wehman-Ooyevaar, D. M. Grove, P. de Vaal, A. Dedieu and G. van Koten, Inorg. Chem., 1992, 31, 5484–5493 CrossRef CAS.
- R. Kancherla, K. Muralirajan, A. Sagadevan and M. Rueping, Trends Chem., 2019, 1, 510–523 CrossRef.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS.
- S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200–16203 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284–2294 CrossRef CAS.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS.
- C. Michelin and N. Hoffmann, ACS Catal., 2018, 8, 12046–12055 CrossRef CAS.
- F. Juliá and P. González-Herrero, J. Am. Chem. Soc., 2016, 138, 5276–5282 CrossRef.
- N. Giménez, R. Lara, M. T. Moreno and E. Lalinde, Chem.–Eur. J., 2017, 23, 5758–5771 CrossRef.
- J. Thongpaen, R. Manguin, V. Dorcet, T. Vives, C. Duhayon, M. Mauduit and O. Baslé, Angew. Chem., Int. Ed., 2019, 58, 15244–15248 CrossRef CAS.
- Á. Vivancos, D. Poveda, A. Muñoz, J. Moreno, D. Bautista and P. González-Herrero, Dalton Trans., 2019, 48, 14367–14382 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- D. S. C. Black, G. B. Deacon and G. L. Edwards, Aust. J. Chem., 1994, 47, 217–227 CrossRef CAS.
- N. Godbert, T. Pugliese, I. Aiello, A. Bellusci, A. Crispini and M. Ghedini, Eur. J. Inorg. Chem., 2007, 5105–5111 CrossRef CAS.
- M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS.
- M. Albrecht, P. Dani, M. Lutz, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 2000, 122, 11822–11833 CrossRef CAS.
- P. A. Shaw, G. J. Clarkson and J. P. Rourke, Organometallics, 2016, 35, 3751–3762 CrossRef CAS.
- P. A. Shaw, G. J. Clarkson and J. P. Rourke, Chem. Sci., 2017, 8, 5547–5558 RSC.
- Á. Vivancos, D. Bautista and P. González-Herrero, Chem.–Eur. J., 2019, 25, 6014–6025 CrossRef.
- I. C. M. Wehman-Ooyevaar, D. M. Grove, P. van der Sluis, A. L. Spek and G. van Koten, J. Chem. Soc., Chem. Commun., 1990, 6, 1367 RSC.
- I. C. M. Wehman-Ooyevaar, D. M. Grove, H. Kooijman, P. van der Sluis, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 1992, 114, 9916–9924 CrossRef CAS.
- A. Martín, Ú. Belío, S. Fuertes and V. Sicilia, Eur. J. Inorg. Chem., 2013, 2013, 2231–2247 CrossRef.
- A. Habiyakare and E. A. C. Lücken, Z. Naturforsch., A: Phys. Sci., 1990, 45, 224–228 CAS.
- A. C. Albéniz, G. Schulte and R. H. Crabtree, Organometallics, 1992, 11, 242–249 CrossRef.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS.
- E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5, 879–886 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
- F. Juliá, D. Bautista, J. M. Fernández-Hernández and P. González-Herrero, Chem. Sci., 2014, 5, 1875–1880 RSC.
- F. Juliá, G. Aullón, D. Bautista and P. González-Herrero, Chem.–Eur. J., 2014, 20, 17346–17359 CrossRef.
- F. Juliá and P. González-Herrero, Dalton Trans., 2016, 45, 10599–10608 RSC.
- J. C. López-López, D. Bautista and P. González-Herrero, Chem.–Eur. J., 2020, 26, 11307–11315 CrossRef.
- I. Jourdain, M. Knorr, C. Strohmann, C. Unkelbach, S. Rojo, P. Gómez-Iglesias and F. Villafañe, Organometallics, 2013, 32, 5343–5359 CrossRef CAS.
- M. Bassetti, P. Casellato, M. P. Gamasa, J. Gimeno, C. González-Bernardo and B. Martín-Vaca, Organometallics, 1997, 16, 5470–5477 CrossRef CAS.
- T. Foo and R. G. Bergman, Organometallics, 1992, 11, 1811–1819 CrossRef CAS.
- A. Romero, A. Santos and J. López, J. Organomet. Chem., 1990, 391, 219–223 CrossRef CAS.
- M. R. Torres, A. Vegas, A. Santos and J. Ros, J. Organomet. Chem., 1986, 309, 169–177 CrossRef CAS.
- J. M. O'Connor and S. J. Friese, Organometallics, 2008, 27, 4280–4281 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, 1H and 13C{1H} NMR spectra of new compounds, crystallographic information, computational methods and data. CCDC 2018872 (3aa·Me2CO), 2019593 (5ec), 2018874 (trans-N,N-6ec·CH2Cl2), 2018873 (7), 2018875 (8a), 2018876 (8b·CH2Cl2), 2018877 (8c), 2018878 (9b-BPh4·CHCl3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04879h |
| This journal is © The Royal Society of Chemistry 2020 |