
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light triggered templated ligation on surface using furan-modified PNAs†
Alex
Manicardi‡
 *,
Enrico
Cadoni‡
and
Annemieke
Madder
*
*,
Enrico
Cadoni‡
and
Annemieke
Madder
*
Organic and Biomimetic Chemistry Research Group, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281-S4, 9000 Gent, Belgium. E-mail: Alex.Manicardi@Ugent.be; Annemieke.Madder@Ugent.be
First published on 9th October 2020
Abstract
Oligonucleotide-templated reactions are frequently exploited for target detection in biosensors and for the construction of DNA-based materials and probes in nanotechnology. However, the translation of the specifically used template chemistry from solution to surfaces, with the final aim of achieving highly selective high-throughput systems, has been difficult to reach and therefore, poorly explored. Here, we show the first example of a visible light-triggered templated ligation on a surface, employing furan-modified peptide nucleic acids (PNAs). Tailored photo-oxidation of the pro-reactive furan moiety is ensured by the simultaneous introduction of a weak photosensitizer as well as a nucleophilic moiety in the reacting PNA strand. This allows one to ensure a localized production of singlet oxygen for furan activation, which is not affected by probe dilution or reducing conditions. Simple white light irradiation in combination with target-induced proximity between reactive functionalities upon recognition of a short 22mer DNA or RNA sequence that functions as a template, allows sensitive detection of nucleic acid targets in a 96 well plate format.
Introduction
The hybridization between two oligonucleotide strands is a highly specific molecular recognition event, exploited in nature for the transmission and translation of genetic information. This widespread mechanism has frequently been explored for biosensors capable of detecting the presence of target nucleic acid sequences.1 These targets, including DNA and various types of RNAs of biological relevance, hybridize with a capture probe, while a second reporter probe can provide a detectable signal.2,3 However, the use of capture or reporter probes based on natural oligonucleotides for biosensor applications may exhibit drawbacks (e.g. low complex stability and low selectivity among similar sequences), especially when it comes to small target sequences that show high structural homology, such as micro RNAs.4Using non-natural synthetic probes may help overcome these major drawbacks. Peptide nucleic acids (PNAs) are artificial nucleic acid mimics, in which the negatively charged phosphate backbone is replaced by a neutral, poly-amidic backbone.5 Due to the lower electrostatic repulsion between the negative DNA or RNA strands and the uncharged PNA, complex stability and sequence selectivity are higher compared to natural duplexes, with stabilities that are less affected by variations in experimental conditions (i.e. ionic strength, solvent polarity, presence of chaotropic agents).6 On the other hand, destabilization of the complex upon introduction of a single mismatch in the complementary sequence is greater in PNA:D(R)NA duplexes than compared to naturally occurring complexes. Their chemical flexibility together with the unique stability towards nucleases, proteases and chemical degradation,7 render PNA an ideal candidate for detection purposes and enable the creation of devices with a longer shelf-life by the exploitation of various chemical approaches.8–10
With regards to sensing platforms, oligonucleotide templated reactions are becoming widely employed in actual detection strategies.11–17 In this context, the target of interest serves as a template to bring two reactive strands in close proximity to each other. Reaction between the two reactive moieties, located on the two strands, can lead to the formation of a covalent linkage (templated ligation, Fig. 1A) or one of the two strands can trigger the modification of a functional group on the other strand and induce a change in its properties (Fig. 1B).11 In both cases, the effective concentration of reacting probes increases significantly thanks to the hybridization with the target and, as a result, the reaction can occur at low concentrations of target and detection probes.
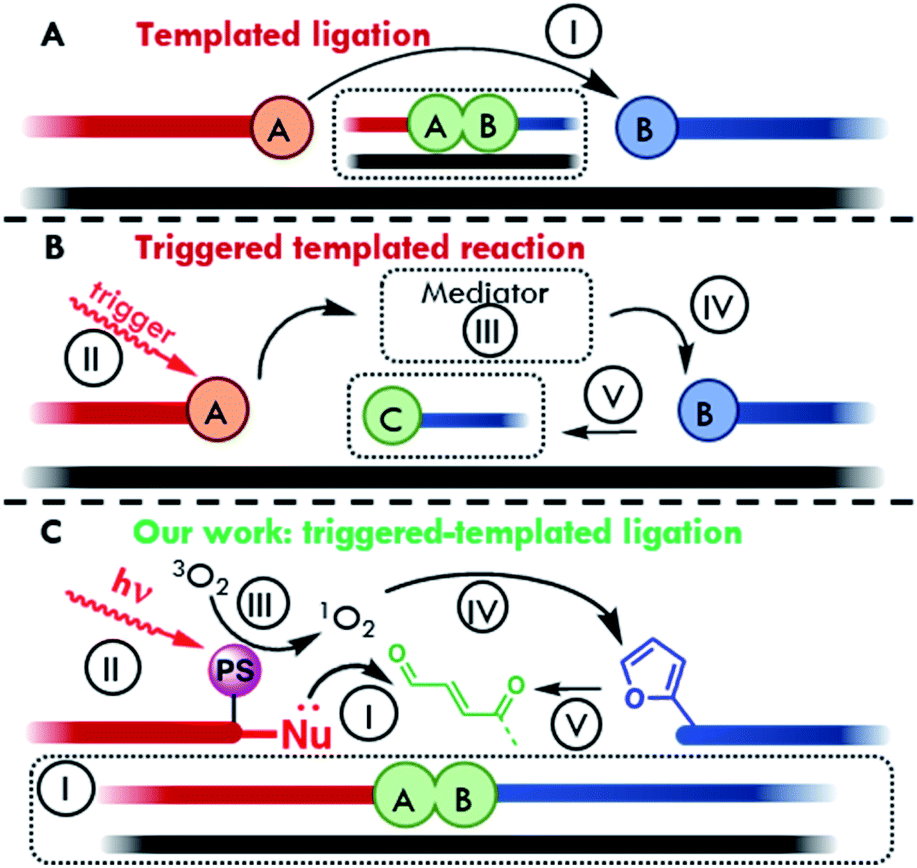 | ||
| Fig. 1 Schematic representation of the main oligonucleotide-templated reaction typologies. (A) Direct templated ligation. (B) Triggered templated reaction. (C) Our light-triggered templated ligation. Visible light irradiation of a photosensitizer allows the excitation of molecular oxygen to reactive singlet oxygen. 1O2 then acts as mediator for furan oxidation. The generated keto-enal further reacts with a nucleophile for the formation of a ligation product, using a target oligonucleotide strand (black line) as template to ensure proximity. (I) Ligation reaction; (II) activation of the antenna system (e.g. light); (III) activation of a mediator; (IV) chemical activation of the pro-reactive moiety; (V) change of properties. | ||
The field of oligonucleotide-templated reactions features many applications of PNA systems, as evidenced by the large number of templated reactions and ligations reported in literature (i.e., carrier release,18–20in cellulo and in vivo nucleic acid imaging,17,21 developments of lateral flow devices for RNA detection,22,23 fluorogenic reactions,10,12,24–27 catalytic and autocatalytic systems,14,28–32 detection and targeting of oligonucleotides involved in genetic-based diseases,15,33,34 artificial aminoacyl transferases and peptide ligation35–39). Oligonucleotide templated reactions, in a wider perspective, have also been explored in the field of nanotechnology. Since the development of the first example of DNA origami back in 2006,40 the nanoscale folding of DNA was used to deliver a growing number of devices and nanomaterials.41,42 The nature-inspired concept of templating has been further employed to program the synthesis of many DNA-decorated rationally designed and oriented nanostructures, engineered thanks to the specific DNA base-pair motif.43
In most cases, when templated ligations are performed, one of the main problems resides in the hydrolysis and degradation of the reactive moieties, thus requiring special precautions in order to maintain the integrity of the system. In this context, the exploitation of an external stimulus can be a valid alternative to unmask the reactive function on demand and enables an increased shelf-life of the probes, avoiding collateral reactions, and obtaining additional spatiotemporal control over the system reactivity.14,44–48
Commonly used activation triggers include chemical, electrochemical and electromagnetic stimuli. In the first case, the presence of a chemical entity is needed in order to activate or reveal the reacting unity. In the second scenario, a variation of the electrochemical properties of the solution provides the necessary input to trigger the system (e.g. a variation of electrolyte concentration or pH). In the last case, the trigger consists of the irradiation of the pro-reactive unit with light of specific wavelengths, capable of exciting the system and generating a reactive center.49,50 In this context, the exploitation of visible light-triggerable systems offers several advantages: the wavelength can be modulated in order to achieve biocompatibility, and its use is universally considered as eco-sustainable.51 Nevertheless, translation of such triggered oligonucleotide-based reactions from solution to surfaces has been poorly explored to date, despite their recognized advantages, such as the production of user-friendly and low-cost devices for DNA and RNA detection, along with increased sensitivity compared to the same detection performed in solution, and the construction of new nanomaterials directly conjugated and engineered on surfaces.
In our lab, a furan oxidation-based methodology, for nucleic acid interstrand crosslinking was developed.52 This strategy was then expanded towards PNA probes for DNA capturing.53 Furan oxidation was initially triggered by using N-bromosuccinimide, but was later accomplished in a visible light-triggered manner by exploiting in situ singlet oxygen production via photosensitizer (PS) induced activation.54–56 This methodology was subsequently applied as a means to immobilize DNA on surfaces,57 and later on extended to the context of peptide labelling and peptide–peptide ligation, exploiting in the latter cases the chemo-selective reactivity of the activated furan moiety towards α-effect nucleophiles.58,59
In this work the first example of a light-triggered templated ligation exploiting a stable but pro-reactive furan moiety which can be oxidized to its reactive keto-enal form by light induced singlet oxygen (1O2) generation (Fig. 1C)60 is shown. By modification of a nucleophile-containing reporter probe with a mild photosensitizer, a pinpoint production of the highly reactive oxygen species was achieved, preventing oxidative damage to any attached payload. Translation of this white light-triggered PNA–PNA oligonucleotide-templated ligation from solution to plastic surface was achieved to allow detection of short 22mer D(R)NA sequences in a 96-well plate format. All steps involved in the construction of the final device were optimized following a bottom-up approach, starting from the optimization of triggered ligation in a PNA:PNA duplex geometry using a free photosensitizer in solution. This was then used as a foundation to develop a templated version of the reaction. Subsequent insertion of the photosensitizer in one of the two strands enabled the creation of a fully integrated system. Finally, to illustrate the feasibility of the translation of the developed technology to other systems, preliminary results on the implementation of this approach onto glass surfaces are also shown.
Results and discussion
The stepwise bottom-up strategy to enable an on-surface templated ligation reaction is illustrated in Fig. 2A in a retrosynthetic fashion. Following careful design of the required PNA probes, the envisaged reaction between an activated furan and a nucleophilic entity was first tested in a PNA:PNA duplex format by employing a photosensitizer in solution (Fig. 2A, I). Upon fine tuning of the conditions, the ligation was then tested in a templated format in the presence of a target nucleic acid sequence (Fig. 2A, II). In a next step, the inclusion of the required photosensitizer onto one of the reacting strands was carried out (Fig. 2A, III). This finally allowed the system translation from solution to a surface and the development of an ELISA-like protocol for detection of nucleic acid sequences in a 96 well plate format.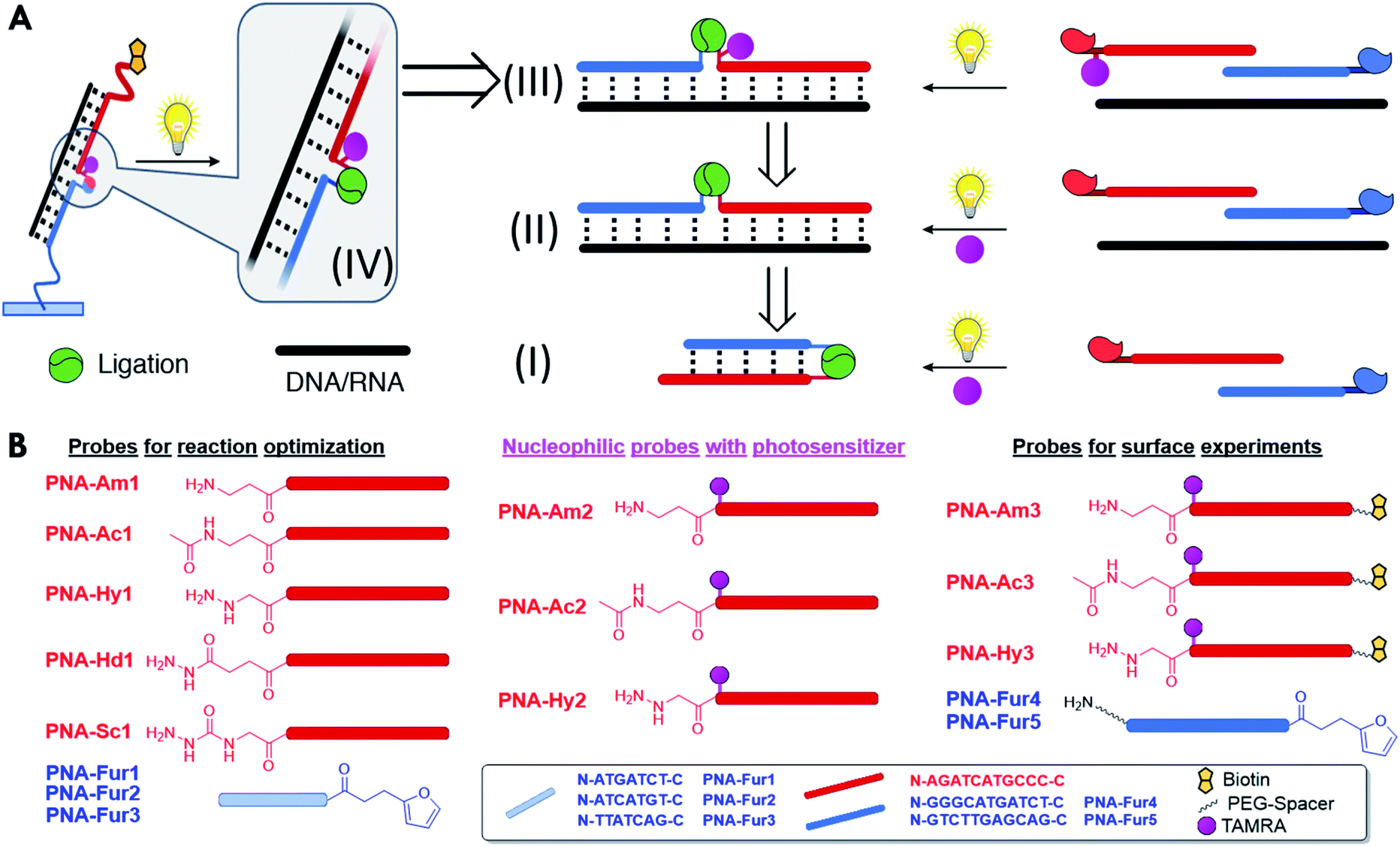 | ||
| Fig. 2 (A) Graphical retrosynthetic representation of the stepwise optimization of the nucleic acid D(R)NA-templated PNA–PNA ligation system from solution to surfaces. Translation of the ligation chemistry to surface (IV) is achieved after optimization of the templated ligation in the fully integrated system, where the photosensitizer is installed in one of the strands (III). Optimization of the templated ligation was first carried out using a classical approach with the photosensitizer in solution (II). A PNA:PNA ligation format was first studied in order to understand the system reactivity and optimize the general experimental conditions (I). (B) PNA design and sequences of the probes used in this study. | ||
As a first step towards the construction of the envisaged system, the potential of light-induced furan-oxidation for efficient PNA–PNA ligation in solution was evaluated. Starting from the results previously obtained in peptide labelling and ligation,58,59 the ligation of two complementary PNA probes was explored. In situ activation of the furan ring to its highly reactive keto-enal form was followed by reaction with a nucleophilic entity (i.e. amine, hydrazine, hydrazide, or semi-carbazide, see Fig. 2B for schematic structures and Fig. S1–S6† for detailed chemical structures) present on the complementary strand with concomitant PNA–PNA ligation in solution (see Fig. 2A, (I)). Visible light irradiation of a photosensitizer present in solution results in the generation of singlet oxygen which, in turn, activates the furan moiety, and leads to stable ligation products through reaction with various nucleophiles. While hydrazines enable the formation of pyridazinium adducts, nucleophiles with a single nucleophilic nitrogen (such as amines, hydrazides, or semicarbazides) lead to the generation of pyrrolidinone species as previously demonstrated.58,59
Preliminary ligation experiments performed on 11mer PNA systems demonstrated the feasibility of the approach, and, at the same time, the fact that HPLC-based techniques are not well suited to distinguish between true ligation products and very stable dsPNA:PNA complexes (data not shown).
For this reason, preliminary optimizations directed towards the identification of suitable experimental conditions (PNA design, photosensitizer, and pH) and analytical techniques were performed (see ESI for extended discussion†).
PNA–PNA ligation (I)
Ligation experiments were performed using 7mer furan PNAs in a PBS buffer at pH 7.4, with Rhodamine B (RhoB) as the photosensitizer. Newly developed urea and sodium dodecyl sulphate polyacrylamide gel electrophoretic conditions (USDS-PAGE) were used to monitor the product formation. The ligation reaction selectivity was tested using fully matched PNA-Fur1, mismatched PNA-Fur2, and scrambled PNA-Fur3 probes. As shown in the USDS-PAGE experiments reported in Fig. 3A, ligation products (L) are only formed for the fully complementary PNA-Fur1. In contrast, the use of mismatched or scrambled sequences only leads to the appearance of a new band that we attributed to the oxidized furan probe (O, as supported by the absence of other high MW species in HPLC-MS, Fig. S25 and S26†) with a mobility lower than the original ssPNA but higher than the ligation product. HPLC-UV analysis further validates these reaction outcomes (Fig. 3B, S16–S23†).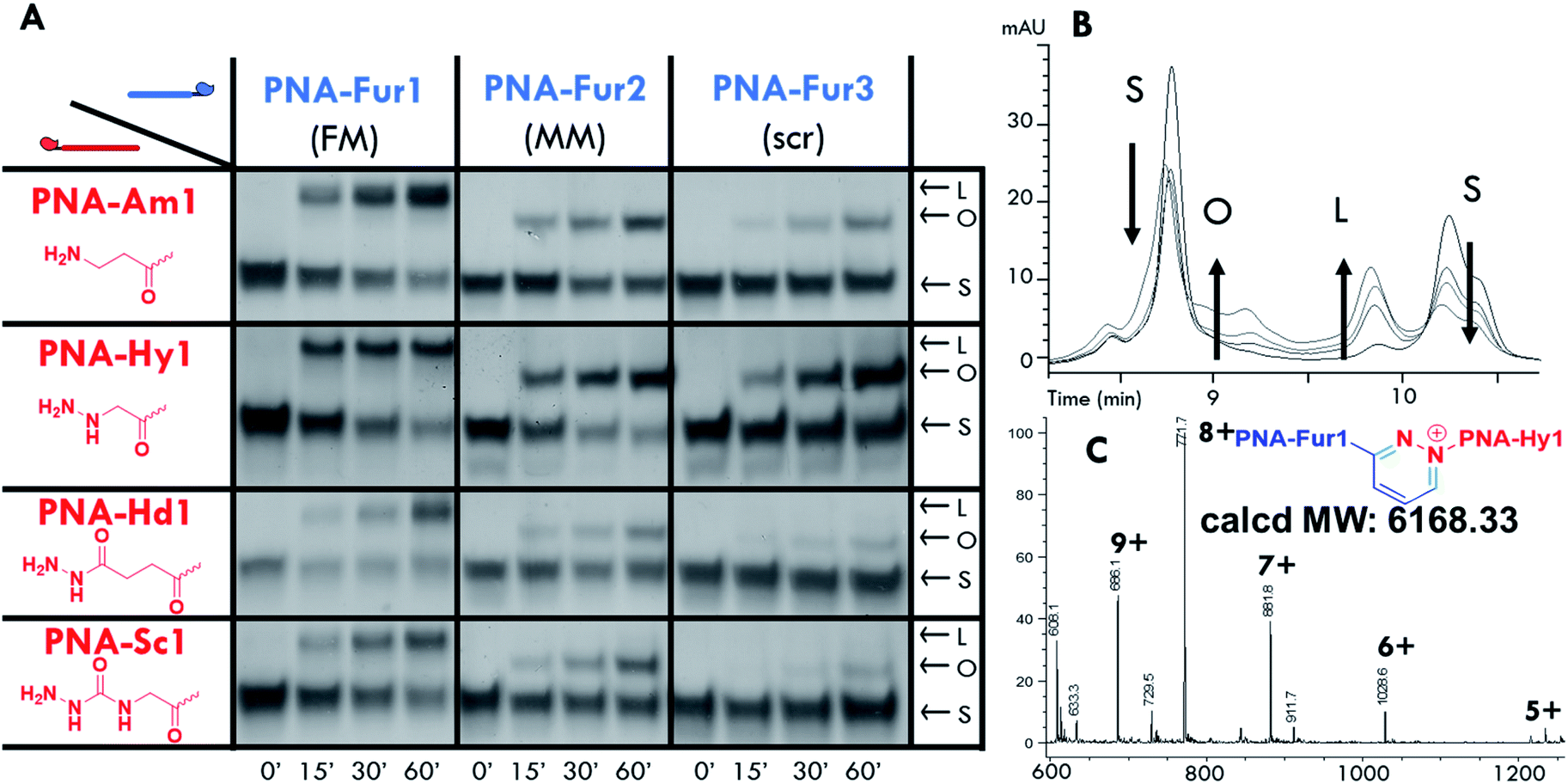 | ||
| Fig. 3 PNA:PNA ligation experiments performed with RhoB in solution (PBS pH 7.4, 25 °C). (A) USDS-PAGE of PNA–PNA ligation (probe and RhoB concentration: 5 μM). (B) Stacked HPLC-UV traces of the ligation experiments between PNA-Hy1 and PNA-Fur1. (C) ESI-MS spectra of the product peak (Rt 9.5 minutes): calcd MW 6168.33: 1234.05 [M + 5H]5+, 1028.6 [M + 6H]6+, 881.8 [M + 7H]7+, 771.7 [M + 8H]8+, 686.1 [M + 9H]9+. Migration bands and chromatographic traces are marked for clarity: starting materials (S), oxidized furan probe (O) and ligation product (L). Reaction mixtures were analyzed after light irradiation for 0′, 15′, 30′, and 60′ for each panel. | ||
Product formation, identity and selectivity were further confirmed by means of HPLC-MS analysis (see Fig. 3C and S24†). The molecular weight of the formed products is in accordance with the calculated masses, based on the different mechanisms of ligation reported earlier.59 From the HPLC-MS chromatograms, signals connected to collateral oxidative damage of the nucleobases have not been detected but signals relative to the degradation of hydrazide and semicarbazide functional groups in PNA-Hd1 and PNA-Sc1 respectively, were found to occur during the irradiation step (data not shown). This degradation seems to occur at the expense of the starting materials, as these by-products were also found when performing the experiments with mismatched and scrambled sequences, where products were not formed. We therefore excluded these probes from further optimization steps and focused on the amine (PNA-Am1) and hydrazine (PNA-Hy1) nucleophilic probes.
From PNA–PNA to templated ligation (II)
Next, we tested the possibility to induce ligation between the furan-modified PNA-Fur3 strand and a PNA strand containing either an amine (PNA-Am1) or hydrazine (PNA-Hy1) functional group, using a DNA or RNA templating strand. Two 14mer DNA and RNA template systems, showing 7 base matches with each probe were employed to evaluate this approach (Fig. 4). Longer sequences (18mers, fully complementary to both target PNA strands) were also tested, but due to the very high Tm of the complex formed with the ligation product, thus preventing its denaturation under USDS-PAGE or HPLC conditions (data not shown), straightforward characterization of covalent bond formation between the two PNA probes was hampered. Furan activation was, as before, achieved by employing RhoB as the photosensitizer in solution. Sequence selectivity of the system was evaluated by means of the mismatched target sequences DNA-2 and RNA-2, containing two mismatches on the portion pairing with the furan-probe.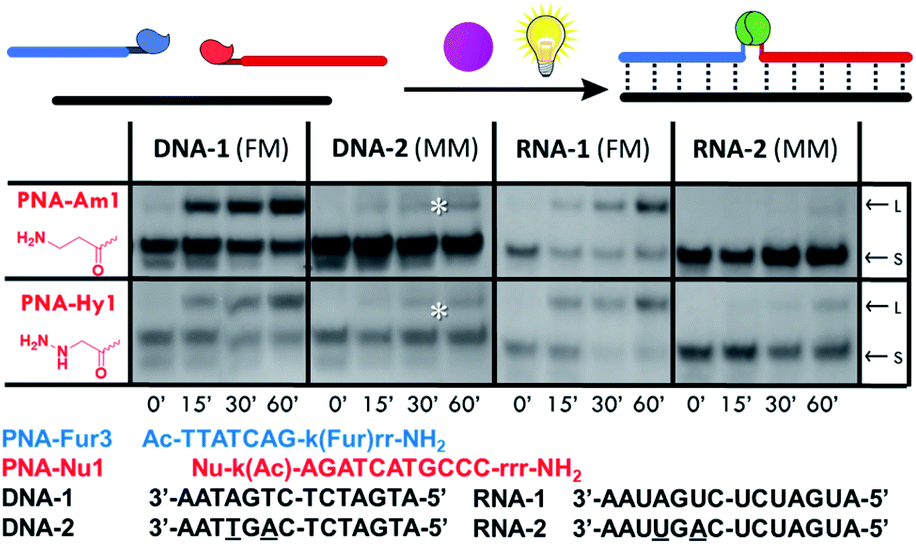 | ||
| Fig. 4 Templated PNA–PNA ligation experiments performed with RhoB in solution (PBS pH 7.4, 25 °C), using PNA-Fur3 (probe and RhoB concentration: 5 μM). Starting material (S) and ligation product (L) migration bands are indicated for clarity. Experiments after 0′, 15′, 30′, and 60′ light irradiation are reported in each panel. *Band corresponding to the formation of the oxidized furan probe. | ||
As shown in Fig. 4, both nucleophilic probes PNA-Am1 and PNA-Hy1 are able to form the desired ligation product in presence of the fully matched target sequences DNA-1 and RNA-1. Sequence selectivity was confirmed by the total absence of ligation when using DNA-2 as the template, while a small peak corresponding to the ligation product was found in the HPLC chromatograms when RNA-2 was used. When lowering strand concentrations to 1 μM, no ligation products could be observed anymore for the RNA-2 case, thus the observation of the product band at the higher 5 μM concentration was ascribed to the higher stability of PNA:RNA complexes even in the presence of mismatches. The results were further confirmed by HPLC-UV and HPLC-MS analyses (see also Fig. S16–S20†).
Towards a fully integrated system: photosensitizer introduction in PNA (from II to III).
With the aim to obtain a fully self-sustained system while reducing the total singlet oxygen content and avoiding collateral damage to the probes or eventual attached payloads, the photosensitizer was inserted into one of the two PNA strands. It was earlier reported that the introduction of a photosensitizer on a furan-containing probe can lead to uncontrolled activation during purification steps, resulting in probe degradation.56 Therefore, it was decided to install the photosensitizer moiety on the nucleophile-containing probe by means of attachment to a neighboring lysine side chain. With this new set-up the only required external trigger is visible light.RhoB coupling to the PNA probe can be achieved in various ways, which in turn affect the spectroscopic properties of the photosensitizer. Therefore, we first evaluated the effect of the type of modification on singlet oxygen generation by synthesizing different rhodamine derivatives (structures of all tested photosensitizers are reported in ESI,† the final selected ones are included in Fig. 5A). Their efficiency of singlet oxygen production was tested following the degradation of an anthracene derivative (9,10-anthracenediyl-bis(methylene)dimalonic acid, ABDA).61 From these experiments (see ESI, Fig. S22, Tables S7 and S8†) 5(6)-carboxytetramethylrhodamine (5(6)-TAMRA) appeared to be the most suitable photosensitizer for the synthesis of the third generation of nucleophilic probes.
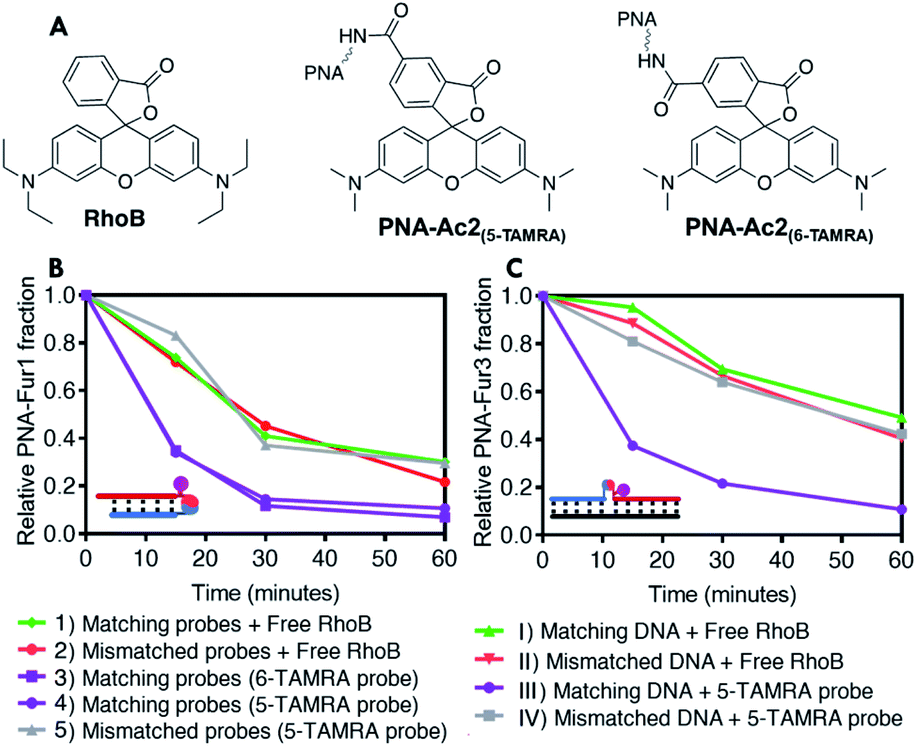 | ||
| Fig. 5 Structure of the studied PSs and furan oxidation experiments performed with a 5 μM probe and photosensitizer concentration using capped nucleophiles, in PBS pH 7.4, at 25 °C. (A) Structure of RhoB and TAMRA derivative studied. (B) PNA–PNA setup: comparison of the furan oxidation profiles between experiments with free PS in solution and probes with covalently attached TAMRA; (1) oxidation of matching furan probe using RhoB in solution (PNA-Fur1 and PNA-Ac1); (2) oxidation of mismatched furan probe using RhoB in solution (PNA-Fur2 and PNA-Ac1); (3) oxidation of matching furan probe using 6-TAMRA-modified PNA (PNA-Fur1 and PNA-Ac2(6-TAMRA)); (4) oxidation of matching furan probe using 5-TAMRA-modified PNA (PNA-Fur1 and PNA-Ac2(5-TAMRA)); (5) oxidation of mismatched furan probe using 5-TAMRA-modified PNA (PNA-Fur2 and PNA-Ac2(5-TAMRA)). (C) DNA Templated setup: comparison of PNA-Fur3 oxidation profiles using PS in solution versus covalently attached PS; (I) oxidation of furan probe in matched template using RhoB in solution (PNA-Ac1 and DNA-1); (II) oxidation of furan probe in mismatched template using RhoB in solution (PNA-Ac1 and DNA-2); (III) oxidation of matched template using 5-TAMRA-modified PNA (PNA-Ac2(5-TAMRA) and DNA-1); (IV) oxidation of mismatched template using 5-TAMRA-modified PNA (PNA-Ac2(5-TAMRA) and DNA-2). | ||
To better understand the influence of the introduction of the photosensitizer in the PNA probe bearing the nucleophilic function, the efficiency of furan activation was first evaluated by assessing the oxidation of fully matching furan-containing probes (PNA-Fur1 and PNA-Fur3 for PNA:PNA and templated ligation experiments respectively) in presence of RhoB and the acetylated probe PNA-Ac1 or with the use of the photosensitizer-containing acetylated PNA-Ac2 (Fig. 5B and C). As the nucleophile in these sequences is rendered inactive by capping, no ligation can take place but the oxidation efficiency can be derived from the speed by which the furan moiety is oxidized, as determined by the disappearance of the furan-containing strand derived from quantitative HPLC analysis. Interestingly, when using the free photosensitizer in solution, the disappearance rate of the furan containing probes, as a result of furan oxidation, drastically drops in the template geometry as compared to the PNA:PNA geometry. This effect can be partially attributed to the reduced singlet oxygen generation capacity of RhoB when it is interacting with DNA.62–64 Given the absence of negative charges in the PNA:PNA case, the positively charged RhoB shows less tendency to interact, thus fully retaining its single oxygen generation capacity. A similar reduction of RhoB-induced singlet oxygen production can be observed when performing the ABDA tests in presence of DNA (see ESI, Table S8†). Moreover, additional shielding or intercalation related effects may also play a role in slowing down furan oxidation to a larger extent in the PNA2:DNA complex than in the PNA:PNA one. When the photosensitizer is covalently attached to a probe that forms a perfectly matched PNA:PNA complex, even faster furan oxidation is obtained as compared to when RhoB is free in solution (Fig. 5B, purple traces versus red trace). Upon photosensitizer attachment to the probe in the template geometry, the furan oxidation rate also increases to be almost as fast as in the PNA:PNA case (dark purple trace in Fig. 5C). No differences in oxidation profiles were observed between the two different TAMRA regioisomers. Reducing the probe concentration from 5 μM to 1 μM clearly results in a reduction of the oxidation rate in the experiments performed with free RhoB in solution, while no significant differences were found when the photosensitizer was installed into the PNA probes forming fully matched PNA2:D(R)NA complexes (see ESI, Fig. S35†). Furthermore, in all cases high glutathione concentrations do not influence furan oxidation profiles (see ESI, Fig. S36†), hinting towards the possibility to perform this reaction in more complex environments such as cell lysates or inside cells.
In ligation experiments, product formation is faster when using PNA-Am2 and PNA-Hy2 probes in presence of the fully matched PNA-Fur1, while oxidation of the mismatched PNA-Fur2 is slower as compared with experiments where RhoB is free in solution (as shown in Fig. 6 for PNA-Am2, and ESI,† for PNA-Hy2). Unfortunately, as a consequence of the insertion of the (hydrophobic) photosensitizer moiety and the resulting increased lipophilicity of the system, when testing the template setup with DNA-1 or RNA-1 (FM sequences) we noticed after light irradiation that the solution became colorless with the formation of a pink precipitate. This precipitation is not observed when the mismatched DNA-2 and RNA-2 were employed. This can be considered as evidence of product formation, as only when the corresponding ligation product is formed, precipitation of the resulting ligated PNA–PNA:D(R)NA-1 complex occurs. This was further confirmed by the absence of the corresponding signals (PNAs and oligos) in HPLC traces (data not shown).
 | ||
| Fig. 6 USDS-PAGE of PNA–PNA ligation experiments performed in PBS pH 7.4, at 25 °C using a PNA:PNA geometry at 5 μM probe concentration (A) and relative intensity of the ligation product band (B). Starting material (S), oxidized furan probe (O) and ligation product (L) migration bands are pointed in the right columns. Experiments after 0′, 5′, 10′, 15′, 20′, and 30′ light irradiation are reported in each panel; band intensities are expressed as relative intensity of the product band as compared to the sum of starting materials and product band intensities of each lane. | ||
Taken together, the data discussed above show the beneficial effect of incorporating the photosensitizer into the system rather than adding it in solution. As a matter of fact, in such a case, activation of the furan sequence followed by ligation, both in the PNA:PNA as well as the PNA2:D(R)NA geometry, requires shorter irradiation times thus reducing the collateral oxidative damage to unrelated sequences. This effect is even more pronounced at lower concentrations.
Light-triggered templated ligation on surface (IV)
The formation of an insoluble ligation product (vide supra) is not a problem when one of the two probes is linked to a surface, provided that the reporter group, required for product quantification, is still functional. In order to exclude a different ligation outcome induced by an altered irradiation geometry upon transferring reactions from Eppendorf tubes to a different setup, preliminary solution ligation experiments were performed in a 96 well plate format. No significant differences in reaction outcome were observed, as shown in Fig. S41 and S42.† Next, our light-triggered template reaction was transferred to the final surface ligation setup. To ensure complex formation at the solution–surface interface it was decided to increase the length of the furan probes from 7mers to 11mers and from 14mers to 22mers for the oligonucleotide templates. The PNA probes were suitably modified in order to allow surface functionalization and the final detection of the ligation product (Fig. 2B). In particular, furan containing probes were equipped with extra PEG spacers (2-(2-aminoethoxy)ethoxy)acetyl spacer, AEEA or O-spacer presenting a free amino function at the N-terminus for surface immobilization. The nucleophile containing PNA-Am3, PNA-Hy3 and acetylated PNA-Ac3 were modified at their C-terminus with a biotin reporter attached through a (PEG)4 spacer. Detection of the final PNA–PNA ligation product is achieved via biotin complexation with a Neutravidin-Horseradish peroxidase conjugate (NAv-HRP) and measurement of the peroxidase activity by oxidation of tetramethylbenzidine (TMB), a common detection methodology in ELISA (schematic representation of the surface functionalization work flow and detection scheme is included in Fig. 7A).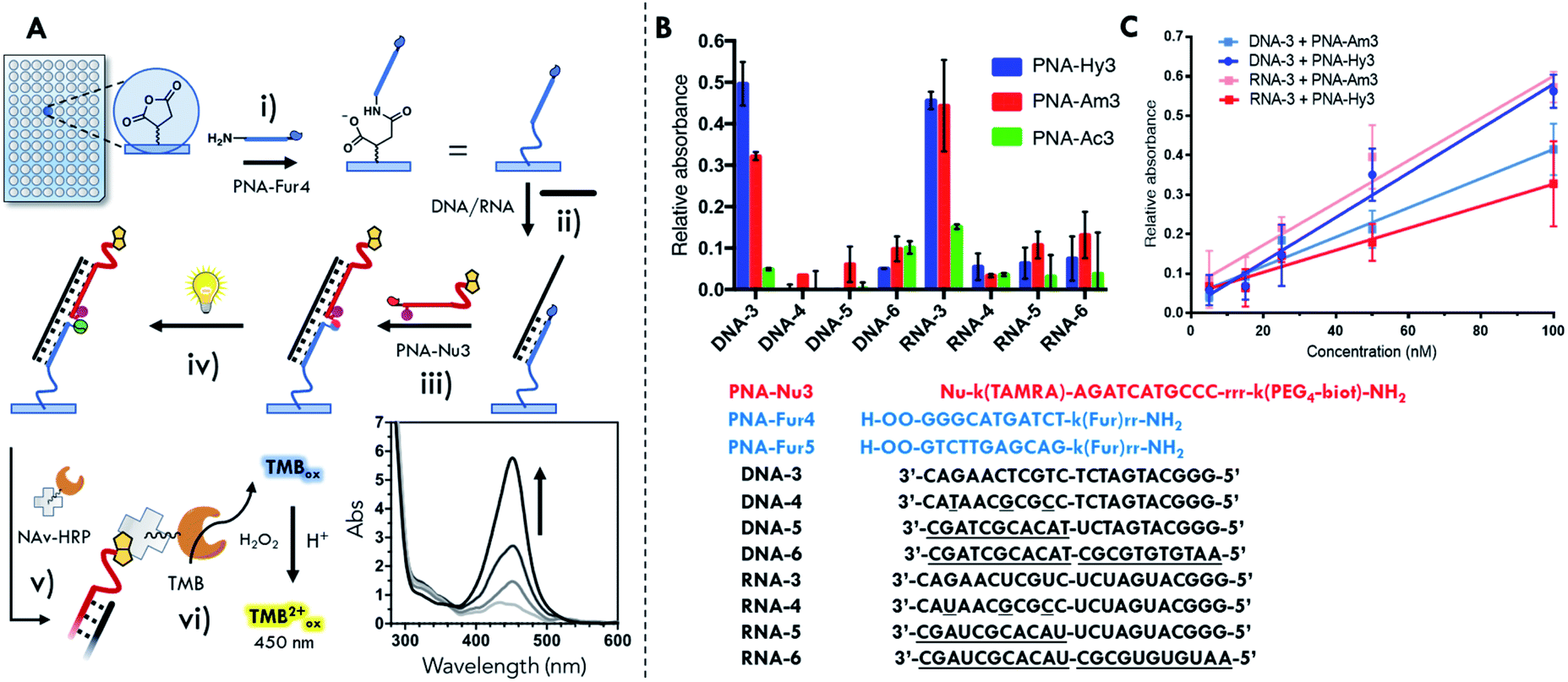 | ||
| Fig. 7 Surface template ligation with PNA-Fur5 functionalized 96-well plates. (A) Schematic workflow of the detection system: (i) immobilization of the furan-containing probes on the surface; (ii) hybridization of the target; (iii) hybridization of the nucleophile-containing probe and formation of the PNA2:D(R)NA complex; (iv) light irradiation and formation of the PNA–PNA:D(R)NA complex; (v) incubation with NAv-HRP; (vi) addition of TMB/H2O2 substrate and UV-Vis read-out after reaction stop with acid solution. (B) Sequence selectivity of PNA-Hy3, PNA-Am3 and PNA-Ac3 in presence of fully matched (DNA-3 and RNA-3) and various mismatched sequences (DNA-4/5/6 and RNA-4/5/6) (oligonucleotide concentration: 200 nM). (C) TMBox signal generation in presence of different concentrations of DNA-3 and RNA-3 template. O: (2-(2-aminoethoxy)ethoxy)acetyl spacer; r: arginine; k: lysine. | ||
Biotin oxidation to the sulfoxide form has recently been reported for a biotin-4-fluorescein dye, as a consequence of light exposure65 and can potentially occur in the current set-up during the photoactivation step. In order to avoid a negative influence of the light irradiation step on the final detection of ligated products, the effect of light irradiation on the biotinylated probes in solution was evaluated. No significant alteration of the oxidation state of biotin was found under our experimental conditions as compared to the use of an external photosensitizer (see Fig. S43†), suggesting that the here developed methodology can be applied to more biologically relevant and sensitive payloads.
Surface ligation tests were first performed using the PNA:PNA geometry in order to optimize the surface functionalization step and light irradiation geometry. Surface immobilized PNA-Am3 was used as a positive control, while surface quenching with ethanolamine was used as a negative (blank) control. After the surface immobilization of PNA-Fur4 and incubation with the complementary nucleophile-PNAs, it was clear that, as a consequence of the extraordinary high stability of these long PNA:PNA duplexes, it was not possible to distinguish between a true ligation product and the hybridized PNA:PNA duplex. In all cases, the generated TMBox signal was as intense as that obtained in the positive control well, where the surface was directly functionalized with biotin-containing PNA-Am3 and light irradiation was not performed. This further confirms that the furan activation step under impulse of a limited amount of locally generated singlet oxygen from the appended TAMRA does not induce significant biotin oxidation, thus allowing the quantification of the attached probes via NAv-HRP TMB oxidation. When the scrambled PNA-Fur5 was immobilized and subsequently incubated with nucleophile-PNAs the TMBox signal turned out as low as the background obtained in the negative control well (data not shown), thus confirming sequence selectivity.
Moving to the template geometry, given the lower stability and higher susceptibility of PNA:DNA complexes to solution composition as compared to PNA:PNA, it should be possible to identify suitably stringent conditions that allow to distinguish between a covalent ligation product and the non-covalent PNA2:DNA duplex. The melting temperature of the complex of each of the PNA probes with target DNA was estimated66 to be 63 °C (as compared to the extrapolated 95.6 °C for PNA:PNA duplex), thus indicating that a harsher washing protocol should allow efficient removal of non-covalently attached probes from the surface. As it is evident from Fig. S44,† increasing the strength of the washing procedure results in a significant decrease of the signal generated with all probes, but only in presence of PNA-Ac3, where ligation is not possible and so the TMBox signal decreases to negative control levels. The harsh denaturing washing conditions required for efficient removal of the non-ligated probe can be associated to an unexpected extra stabilization of the PNA2:DNA complex. This extra stabilization may be connected to the coaxial base stacking hybridization, a typical π–π stacking interaction that occurs between bases on the two sides of a nick, as earlier reported for DNA systems.67
Next, sequence selectivity and concentration dependence of the system were tested. From the data reported in Fig. 7B showing the relative absorbance generated by the oxidized TMB (relative absorbance at 450 nm as compared to the positive well signal), it can be derived that discrimination between fully matched sequences (DNA-3, RNA-3) and mismatched (DNA-4, RNA-4) or scrambled (DNA-5/6, RNA-5/6) sequences is possible. As mentioned earlier, the higher stability of the PNA2:RNA complexes also resulted here in an increased difficulty in complex denaturation and non-ligated probe removal during the washing cycle, as can be noted by the higher residual signal generated with the non-nucleophilic PNA-Ac3.
From the results obtained with DNA-3, when comparing to the residual signal obtained with the acetylated PNA-Ac3, the yield of the surface ligation can be evaluated. The signal obtained from reaction with the hydrazine containing probe PNA-Hy3 is around 50% of that generated by the biotin probe directly attached to the surface. In the amino-containing PNA-Am3 the relative signal is around 30% (see Fig. 7B). The resulting limit of detection (LOD) values obtained here ranged from 15 to 20 nM, depending on nucleophilic probe and the target (see Table S9†). The lowest LOD obtained, enables the detection of about 2000 copies of oligonucleotide per cell (for 3000 μm3, typical HeLa cells). Even though a more thorough investigation is required to define the requirements to analyze real life samples, the obtained LODs open the possibility to apply this methodology to detect a variety of micro RNAs of interest in cells (e.g. miR-29, miR-21, miR-17, miR-16a and miR-15a).68
From the results previously obtained in solution, the reactivity of the system seemed to follow the nucleophilicity of the probe, with hydrazine probes showing higher performance as compared to amine probes. Moving to surface experiments, this behavior is not uniformly maintained, yet seems more complex. The response factor of the hydrazine probe PNA-Hy3 is higher in presence of the DNA-3 template in comparison with RNA-3, while PNA-Am3 exhibits opposite behavior (Fig. 7C). The different behavior of the two probes in the reaction templated by the two classes of target nucleic acids can be attributed to the combination of two different factors: different helical structures and different geometrical requirements in the formation of the piperazine or pyrrolidinone products. The small differences resulting from these two factors in the context of reduced mobility, i.e. the current surface–solution interface, can thus lead to the observed different reaction outcome (see ESI, Fig. S46 and Table S9†).
Finally, we decided to translate this methodology to another type of surface in order to demonstrate the wider applicability of the concept and show the possibility to exploit the TAMRA moiety as both photosensitizer and reporter. Preliminary results obtained on microarrays glass surfaces (reported in Fig. S47†) show the formation of a templated ligation signal on a glass surface. Although the protocols (i.e. spotting, light irradiation, washing steps) still require careful optimization, a similar behavior in terms of PNA:PNA complex stability and sequence selectivity as reported for the 96-well format is observed. These results, even if preliminary, underline the potential of the developed technology and open the door to application of the principles illustrated here in the broader context of different miniaturized rapid test platforms.
Conclusions
The possibility to control the ligation of oligonucleotide strands by means of a light signal has been exploited before by others in a series of applications. In most cases, this type of ligations are reversible and require highly energetic UV radiation,48,69–71 or complex scenarios for intensifying the generated signals.45 On the other hand, the possibility to perform templated reactions at the surface interface was barely explored over the past years.72–74 We here report, for the first time, the possibility to perform the ligation on a surface, templated by short 22mer oligonucleotides, exploiting the in situ activation of a stable pro-reactive moiety using a simple white light lamp. The proposed system, applied to a classical 96-well plate detection platform, was designed to not require delicate enzymatic steps and only involves functional groups with long shelf-lives. The fully integrated approach shown in this work is based on the incorporation of a photosensitizer into a nucleophile-containing PNA probe. Full comprehension of the system reactivity, initially studied by means of a PNA:PNA duplex, was necessary to understand the reactivity of the probes in order to select the best performing nucleophiles and test the ligation in presence of a weak photosensitizer. The activation of the furan and the ligation in a template geometry was then performed, confirming the selectivity of the system towards an external recognition element (DNA and RNA template). Finally, it was shown that activation of the pro-reactive moiety upon the introduction of the weak photosensitizer in the probe was maintained even at low concentration of probes. All these optimization steps enabled the translation of the reaction to an efficient ELISA-like surface format. The performance of the system is not affected by PS dilution, and the local production of the required reactive oxygen species is kept to a minimum in order to achieve efficient activation while avoiding collateral oxidative damage. This, together with the tolerance of the system to work under reducing conditions (cf. glutathione tolerance), opens the way towards applications of this methodology in more complex media or the introduction of more biologically relevant cargos. Finally, the translation of the developed methodology to a different type of surface was shown. The proof-of-concept experiment on microarray glass surfaces represents the first step toward the realization of a genuine miniaturized system, for the development of high throughput devices for multiplex analysis of short nucleotide sequences, such as miR. Moreover, it also opens up new perspectives for the future development of nanoscale construction platforms.Experimental procedures
Ligation experiments in solution
In a typical experiment a 100 μM working solution of PS was freshly prepared from a 1 mM stock solution. In a 1.5 mL Eppendorf vial, 300 μL of buffered solution (PBS, pH 7.4) containing probes at 5 μM concentration was equilibrated for 10 minutes at 25 °C, before the addition of the PS at 5 μM final concentration. The lamp (100 W halogen lamp LE.5210 Euromex EK-1 illuminator, equipped with Euromex LE.5214 dual arm light conductor) was placed on top of the Eppendorf vial for the entire duration of the experiment. 50 μL aliquots were taken at different time intervals and left to react overnight at 25 °C. Results were analyzed via USDS-PAGE and HPLC-UV. For the experiments where PS-containing probes are used, the solutions containing all the probes were equilibrated in the dark before irradiation.The intensity of each lamp was set before starting the irradiation, using a TES 1335 luxmeter equipped with a custom fitting for the lamp bulbs in order to maintain the light intensity constant during all the experiments.
USDS-PAGE
15% polyacrylamide gels (5% crosslinker, 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acrylamide/bisacrylamide) were prepared in 50 mM tris-acetate buffer pH 7.6 containing 7 M urea and 0.1% SDS. The temperature of the gel was stabilized with a Julabo F12 at 25 °C. The power supply used for gel electrophoresis was a consort EV202 and a voltage of 200 V for 0.75 mm thickness or 100 V for 1.0 mm thickness was used to run the gels (15 minutes pre-run). 2 μL of sample solutions were mixed with 3 μL formamide and 5 μL loading buffer (100 mM tris-acetate pH 7.6, 7 M urea, 20% formamide, 2% SDS). From this mixture, 5–8 μL were loaded on the gel. Gels were stained with Pierce Silver Stain (Thermo Fisher Scientific).
:1 acrylamide/bisacrylamide) were prepared in 50 mM tris-acetate buffer pH 7.6 containing 7 M urea and 0.1% SDS. The temperature of the gel was stabilized with a Julabo F12 at 25 °C. The power supply used for gel electrophoresis was a consort EV202 and a voltage of 200 V for 0.75 mm thickness or 100 V for 1.0 mm thickness was used to run the gels (15 minutes pre-run). 2 μL of sample solutions were mixed with 3 μL formamide and 5 μL loading buffer (100 mM tris-acetate pH 7.6, 7 M urea, 20% formamide, 2% SDS). From this mixture, 5–8 μL were loaded on the gel. Gels were stained with Pierce Silver Stain (Thermo Fisher Scientific).
Singlet oxygen production quantification
In a typical experiment a 100 μM working solution of PS was freshly prepared from a 1 mM stock solution. Additionally, a 200 μM ABDA working solution was freshly prepared from a 500 μM stock solution. 300 μL of an air saturated buffered solution containing 100 μM ABDA and 5 μM of PS were added to a 1.5 mL Eppendorf vial. The lamp is placed on top of the Eppendorf and the sample irradiated for the entire duration of the experiment. 20 μL aliquots were taken at different time intervals and protected from light. Absorbance at 380 nm was recorded via UV-Vis spectrophotometry.Ligation experiments on surface
Oligonucleotides and PNA solutions were freshly prepared in PBS (pH 7.4) and supplemented with 0.001% SDS (PBS–S) from 10 μM stock solutions in mQ. Surfaces where pre-wetted for 30 minutes with a 0.001% SDS solution. Then, 50 μL of oligonucleotide solution and 50 μL of 1 μM PNA solution were allowed to equilibrate in the well for 1 h at 40 °C before light irradiation (100 nM final concentration of oligonucleotides unless otherwise specified in ESI†). The strip was placed on top of the light setup and irradiated for 1 h. The strip was then left to react overnight followed by washing and quantification of the attached biotin, using 100 μL of 20 ng mL−1 Pierce High Sensitivity NeutrAvidin-HRPconjugate (Thermo Scientific) and 1-step Ultra TMB-ELISA (Thermo Scientific) as reagent solution. Final readout of the oxidized TMB was performed after the addition of 2 M H2SO4, by recording the absorption at 450 nm after 5 min. For detailed information on the washing protocols used, please refer to the section in the ESI.†Abbreviations
| 1O2 | Singlet oxygen |
| AEEA/O | 2-((2-Aminoethoxy)ethoxy)acetyl spacer |
| ABDA | 9,10-Anthracenediyl-bis(methylene)dimalonic acid |
| HRP | Horseradish peroxidase |
| k | Lysine |
| NAv | Neutravidin |
| PAGE | Polyacrylamide gel electrophoresis |
| PBS | Phosphate buffer saline |
| PNA:PNA | Double stranded PNA complex |
| PNA–PNA | Product of PNA ligation |
| PNA2:D(R)NA | Complex between two PNA strands and DNA or RNA |
| PNA–PNA:D(R)NA | Duplex between PNA–PNA ligation product and DNA or RNA |
| PS | Photosensitizer |
| RhoB | Rhodamine B |
| R | Arginine |
| SDS | Sodium dodecyl sulphate |
| TAMRA | Carboxytetramethylrhodamine |
| TMB | Tetramethylbenzidine |
| TMBox | Oxidized tetramethylbenzidine |
| USDS-PAGE | Urea-sodium dodecyl sulphate polyacrylamide gel electrophoresis |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Silvia Farina for her support in USDS-PAGE optimization and melting temperature experiments. The help provided by Dr Margarida Carvalho with respect to the ELISA system discussions and by Jos Van den Begin for technical support is gratefully acknowledged. We would also like to thank Prof Björn Menten and Geert De Vos for providing the access to the microarray slide reader. Akshay Hattiangadi and Dr Joshua Holloway are acknowledged for final English language checking and editing. This work was supported by the FWO and the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 665501 and by IOF-UGent project F2019/IOF-ConcepTT/188 (Al. M.). Furthermore, this project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no 721613 (E. C.).References
- A. Sassolas, B. D. Leca-Bouvier and L. J. Blum, Chem. Rev., 2008, 108, 109–139 CrossRef CAS.
- A. P. De Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS.
- N. Agrawal, P. V. N. Dasaradhi, A. Mohmmed, P. Malhotra, R. K. Bhatnagar and S. K. Mukherjee, Microbiol. Mol. Biol. Rev., 2003, 67, 657–685 CrossRef CAS.
- P. Nielsen, M. Egholm, R. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS.
- P. E. Nielsen, Mol. Biotechnol., 2004, 26, 233–248 CrossRef CAS.
- V. V. Demidov, V. N. Potaman, M. D. Frank-Kamenetskil, M. Egholm, O. Buchard, S. H. Sönnichsen and P. E. Nlelsen, Biochem. Pharmacol., 1994, 48, 1310–1313 CrossRef CAS.
- H. Shi, F. Yang, W. Li, W. Zhao, K. Nie, B. Dong and Z. Liu, Biosens. Bioelectron., 2015, 66, 481–489 CrossRef CAS.
- Z. Sun, H. Zhang, B. Cai, Z. Zhang, L. Huang and G.-J. Zhang, Biosens. Bioelectron., 2015, 74, 329–334 CrossRef.
- G. A. D. D. Metcalf, A. Shibakawa, H. Patel, A. Sita-Lumsden, A. Zivi, N. Rama, C. L. Bevan and S. Ladame, Anal. Chem., 2016, 88, 8091–8098 CrossRef CAS.
- M. Di Pisa and O. Seitz, ChemMedChem, 2017, 12, 872–882 CrossRef CAS.
- W. A. Velema and E. T. Kool, J. Am. Chem. Soc., 2017, 139, 5405–5411 CrossRef CAS.
- S. Barluenga and N. Winssinger, Acc. Chem. Res., 2015, 48, 1319–1331 CrossRef CAS.
- M. Anzola and N. Winssinger, Chem. - Eur. J., 2019, 25, 334–342 CrossRef CAS.
- A. Kern and O. Seitz, Chem. Sci., 2015, 6, 724–728 RSC.
- Y. Li, P. Zhao, M. Zhang, X. Zhao and X. Li, J. Am. Chem. Soc., 2013, 135, 17727–17730 CrossRef CAS.
- L. Holtzer, I. Oleinich, M. Anzola, E. Lindberg, K. K. Sadhu, M. Gonzalez-Gaitan and N. Winssinger, ACS Cent. Sci., 2016, 2, 394–400 CrossRef CAS.
- K. Gorska, A. Manicardi, S. Barluenga and N. Winssinger, Chem. Commun., 2011, 47, 4364–4366 RSC.
- E. Lindberg, S. Angerani, M. Anzola and N. Winssinger, Nat. Commun., 2018, 9, 1–9 CrossRef CAS.
- K. T. Kim, S. Angerani, D. Chang and N. Winssinger, J. Am. Chem. Soc., 2019, 141, 16288–16295 CrossRef CAS.
- Z. Pianowski, K. Gorska, L. Oswald, C. a. Merten and N. Winssinger, J. Am. Chem. Soc., 2009, 131, 6492–6497 CrossRef CAS.
- J. Sayers, R. J. Payne and N. Winssinger, Chem. Sci., 2018, 9, 896–903 RSC.
- S. Pavagada, R. B. Channon, J. Y. H. Chang, S. H. Kim, D. MacIntyre, P. R. Bennett, V. Terzidou and S. Ladame, Chem. Commun., 2019, 55, 12451–12454 RSC.
- O. Zavoiura, U. Resch-Genger and O. Seitz, Bioconjugate Chem., 2018, 29, 1690–1702 CrossRef CAS.
- D. Al Sulaiman, J. Y. H. Chang and S. Ladame, Angew. Chem. Int. Ed., 2017, 56, 5247–5251 CrossRef CAS.
- H. Wu, B. T. Cisneros, C. M. Cole and N. K. Devaraj, J. Am. Chem. Soc., 2014, 136, 17942–17945 CrossRef CAS.
- K. T. Kim, D. Chang and N. Winssinger, Helv. Chim. Acta, 2018, 101, 1–7 CrossRef.
- A. Singhal and P. E. Nielsen, Org. Biomol. Chem., 2014, 12, 6901–6907 RSC.
- J. Michaelis, A. Roloff and O. Seitz, Org. Biomol. Chem., 2014, 12, 2821–2833 RSC.
- T. A. Plöger and G. von Kiedrowski, Org. Biomol. Chem., 2014, 12, 6908–6914 RSC.
- D. Chang, K. T. Kim, E. Lindberg and N. Winssinger, Bioconjugate Chem., 2018, 29, 158–163 CrossRef CAS.
- D. Chang, E. Lindberg and N. Winssinger, J. Am. Chem. Soc., 2017, 139, 1444–1447 CrossRef CAS.
- E. Oh, Y. Liu, M. V. Sonar, D. E. Merry and E. Wickstrom, Bioconjugate Chem., 2018, 29, 1276–1282 CrossRef CAS.
- R. Bahal, A. Manna, W. C. Hsieh, S. A. Thadke, G. Sureshkumar and D. H. Ly, ChemBioChem, 2018, 19, 674–678 CrossRef CAS.
- A. Singhal, V. Bagnacani, R. Corradini and P. E. Nielsen, ACS Chem. Biol., 2014, 9, 2612–2620 CrossRef CAS.
- T. N. Grossmann and O. Seitz, Chem. - Eur. J., 2009, 15, 6723–6730 CrossRef CAS.
- S. Middel, C. H. Panse, S. Nawratil and U. Diederichsen, ChemBioChem, 2017, 18, 2328–2332 CrossRef CAS.
- A. Erben, T. N. Grossmann and O. Seitz, Angew. Chem. Int. Ed., 2011, 50, 2828–2832 CrossRef CAS.
- O. Vázquez and O. Seitz, Chem. Sci., 2014, 5, 2850–2854 RSC.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2018, 3, 17068 CrossRef CAS.
- A. Heuer-Jungemann and T. Liedl, Trends Chem., 2019, 1, 799–814 CrossRef.
- A. Samanta and I. L. Medintz, Nanoscale, 2016, 8, 9037–9095 RSC.
- K. K. Sadhu and N. Winssinger, Chem. - Eur. J., 2013, 19, 8182–8189 CrossRef CAS.
- D. Mendez-Gonzalez, S. Lahtinen, M. Laurenti, E. López-Cabarcos, J. Rubio-Retama and T. Soukka, Anal. Chem., 2018, 90, 13385–13392 CrossRef CAS.
- R. K. O'Reilly, A. J. Turberfield and T. R. Wilks, Acc. Chem. Res., 2017, 50, 2496–2509 CrossRef.
- A. F. De Fazio, A. H. El-Sagheer, J. S. Kahn, I. Nandhakumar, M. R. Burton, T. Brown, O. L. Muskens, O. Gang and A. G. Kanaras, ACS Nano, 2019, 13, 5771–5777 CrossRef CAS.
- P. K. Harimech, S. R. Gerrard, A. H. El-Sagheer, T. Brown and A. G. Kanaras, J. Am. Chem. Soc., 2015, 137, 9242–9245 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- A. Zhang, K. Jung, A. Li, J. Liu and C. Boyer, Prog. Polym. Sci., 2019, 99, 101164 CrossRef CAS.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem. Int. Ed., 2012, 51, 8446–8476 CrossRef CAS.
- M. O. De Beeck and A. Madder, J. Am. Chem. Soc., 2011, 133, 796–807 CrossRef.
- A. Manicardi, E. Gyssels, R. Corradini and A. Madder, Chem. Commun., 2016, 52, 6930–6933 RSC.
- M. Op de Beeck and A. Madder, J. Am. Chem. Soc., 2012, 134, 10737–10740 CrossRef CAS.
- N. De Laet, E. M. Llamas and A. Madder, ChemPhotoChem, 2018, 2, 575–579 CrossRef CAS.
- N. De Laet and A. Madder, J. Photochem. Photobiol., A, 2016, 318, 64–70 CrossRef CAS.
- C. Véliz Montes, H. Memczak, E. Gyssels, T. Torres, A. Madder and R. J. Schneider, Langmuir, 2017, 33, 1197–1201 CrossRef.
- E. Antonatou, K. Hoogewijs, D. Kalaitzakis, A. Baudot, G. Vassilikogiannakis, A. Madder and N. His, Chem. - Eur. J., 2016, 22, 8457–8461 CrossRef CAS.
- E. Antonatou, Y. Verleysen and A. Madder, Org. Biomol. Chem., 2017, 15, 8140–8144 RSC.
- T. Montagnon, D. Kalaitzakis, M. Triantafyllakis, M. Stratakis and G. Vassilikogiannakis, Chem. Commun., 2014, 50, 15480–15498 RSC.
- N. A. Kuznetsova, N. S. Gretsova, O. A. Yuzhakova, V. M. Negrimovskii, O. L. Kaliya and E. A. Luk’yanets, Russ. J. Gen. Chem., 2001, 71, 36–41 CrossRef CAS.
- A. Al Masum, M. Chakraborty, P. Pandya, U. C. Halder, M. M. Islam and S. Mukhopadhyay, J. Phys. Chem. B, 2014, 118, 13151–13161 CrossRef.
- M. M. Islam, M. Chakraborty, P. Pandya, A. Al Masum, N. Gupta and S. Mukhopadhyay, Dyes Pigm., 2013, 99, 412–422 CrossRef CAS.
- A. Al Masum, M. Chakraborty, S. Ghosh, D. Laha, P. Karmakar, M. M. Islam and S. Mukhopadhyay, J. Photochem. Photobiol., B, 2016, 164, 369–379 CrossRef CAS.
- R. A. Haack, K. M. Swift, Q. Ruan, R. J. Himmelsbach and S. Y. Tetin, Anal. Biochem., 2017, 531, 78–82 CrossRef CAS.
- U. Giesen, W. Kleider, C. Berding, A. Geiger, H. Orum and P. E. Nielsen, Nucleic Acids Res., 1998, 26, 5004–5006 CrossRef CAS.
- V. A. Vasiliskov, D. V. Prokopenko and A. D. Mirzabekov, Nucleic Acids Res., 2001, 29, 2303–2313 CrossRef CAS.
- Y. Song, D. Kilburn, J. H. Song, Y. Cheng, C. T. Saeui, D. G. Cheung, C. M. Croce, K. J. Yarema, S. J. Meltzer, K. J. Liu and T.-H. Wang, PLoS One, 2017, 12, e0180988 CrossRef.
- J. Liu and J. S. Taylor, Nucleic Acids Res., 1998, 26, 3300–3304 CrossRef CAS.
- Y. Yoshimura and K. Fujimoto, Org. Lett., 2008, 10, 3227–3230 CrossRef CAS.
- H. Kashida, T. Doi, T. Sakakibara, T. Hayashi and H. Asanuma, J. Am. Chem. Soc., 2013, 135, 7960–7966 CrossRef CAS.
- S. Pavagada, R. B. Channon, J. Y. H. H. Chang, S. H. Kim, D. MacIntyre, P. R. Bennett, V. Terzidou and S. Ladame, Chem. Commun., 2019, 55, 12451–12454 RSC.
- G. R. Abel, B. H. Cao, J. E. Hein and T. Ye, Chem. Commun., 2014, 50, 8131–8133 RSC.
- T. Ueno and T. Funatsu, PLoS One, 2014, 9, e90920 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed procedures for monomer synthesis, optimization of PAGE conditions for PNA, evaluation of PNA:PNA stability, preliminary results on light-triggered ligation, singlet oxygen quantum yield calculation, detailed procedures for surface ligation protocols, light irradiation set-up overview, raw data, characterization data of probes and small molecules. See DOI: 10.1039/d0sc04875e |
| ‡ Al. M. and E. C. contributed equally to the work. Al. M. conceived the present idea and planned the experiments. Al. M. and E. C. performed the experiments and processed the data. Al. M. and An. M. wrote the manuscript with support from E. C. An. M. supervised the project. All authors discussed the results and commented on the manuscript. |
| This journal is © The Royal Society of Chemistry 2020 |