DOI:
10.1039/D0SC04676K
(Perspective)
Chem. Sci., 2020,
11, 10887-10909
Diverse strategies for transition metal catalyzed distal C(sp3)–H functionalizations
Received
25th August 2020
, Accepted 26th September 2020
First published on 28th September 2020
Abstract
Transition metal catalyzed C(sp3)–H functionalization is a rapidly growing field. Despite severe challenges, distal C–H functionalizations of aliphatic molecules by overriding proximal positions have witnessed tremendous progress. While usage of stoichiometric directing groups played a crucial role, reactions with catalytic transient directing groups or methods without any directing groups are gaining more attention due to their practicality. Various innovative strategies, slowly but steadily, circumvented issues related to remote functionalizations of aliphatic molecules. A systematic compilation has been presented here to provide insights into the recent developments and future challenges in the field. The Present perspective is expected to open up a new dimension and provide an avenue for deep insights into the distal C(sp3)–H functionalizations that could be applied routinely in various pharmaceutical and agrochemical industries.
1. Introduction
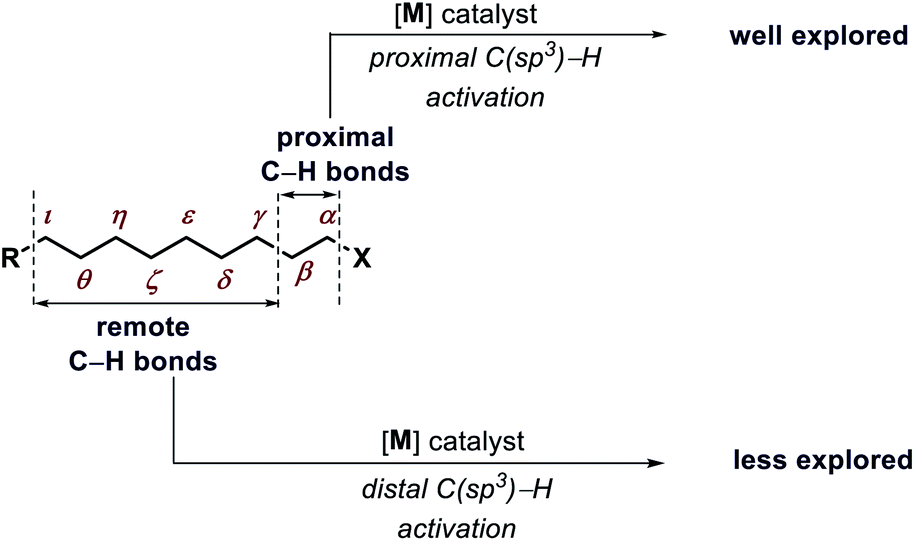
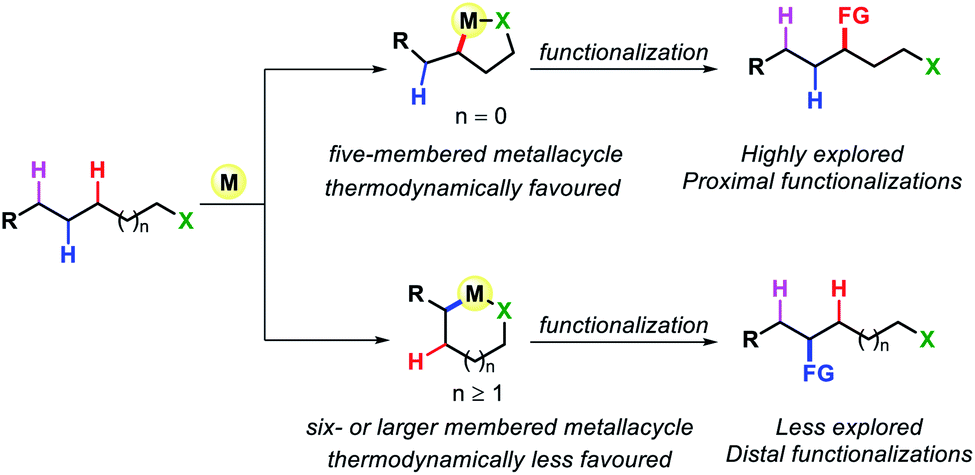
Transformation of chemicals into valuable organic compounds in a green and atom-economic way has long been the core goal of synthetic chemistry. In the last few decades, C–H functionalization has come up as a progressive tool that could be used to serve the purpose in synthetic chemistry. By omitting the need for pre-functionalized substrates, it has become a great tool for synthetic chemists to carry out reactions in a greener way. The key challenges in this field are the inert reactivity of C–H bonds, as well as the selective activation of a specific C–H bond among the abundant number of these bonds in an organic molecule. In the last several decades, sp2 C–H activation has been intensively investigated and found manifold applications in synthetic chemistry. This is evidenced by the excellent accounts and reviews written on this subject.1 In contrast, sp3 C–H activation although, has progressed tremendously but has not been studied as per with the former. The obvious reason for this disparity is the intricacies involved in activating the sp3 C–H bonds. The fluxional nature of aliphatic compounds along with lack of assistance from π groups that could interact with transition metal centers make sp3 C–H bond activation more challenging. Moreover, although proximal C(sp3)–H activation has been well explored in the past decade, distal C(sp3)–H functionalization presents significant challenge (Fig. 1). Reaching out to the distal positions requires intermediacy of larger metallacycles (six-membered or more), which is strenuous because of unfavorable thermodynamic parameters (Fig. 2). To overcome such challenges, researchers in this area are continuously designing and exploring newer catalysts, ligands, and directing groups for functionalization of C(sp3)–H bonds beyond proximity. Various directing group strategies have been employed that could increase the reactivity and selectivity in sp3 C–H functionalization. Along with these, many in situ approaches have been invented for the same.2 Recently the directing group free approaches for sp3 C–H functionalization, where native functional groups present in the molecule (acid, amine, etc.) serve as the chelating group are gaining more attention due to better step and atom economy. Herein, we provide an overview of the transition metal catalyzed sp3 C–H bond activation. However, we would only discuss the distal sp3 C–H functionalization since activation of proximal sp3 C–H bonds (α, β C–H bonds) are well explored and covered in many review articles.3 Asymmetric C–H functionalization is also not part of this review. By detailing the criteria of successful strategies for distal C–H bonds of alkyl chains, the insights and concepts presented are intended to be transferable to other synthetic methodology development and the field of targeted synthesis in general.
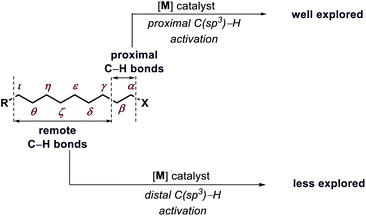 |
| | Fig. 1 Present scenario of C(sp3)–H activation. | |
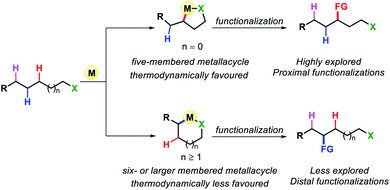 |
| | Fig. 2 Distal C–H functionalization of aliphatic compounds. | |
2. Directing group assisted distal C(sp3)–H functionalizations
The main challenges of C–H functionalization, such as site selectivity among multiple analogous C–H bonds and the inert feature of sp3 C–H bonds can be overcome by introducing a helping entity known as directing groups (DG). Some common functional groups, such as nitrogen, phosphorus, sulfur based functional groups could serve as effective directing groups due to their excellent coordinating ability with transition metals. Through coordination with transition metals, directing groups bring about the proximity effect which can control the inherent steric/electronic properties and eventually command site selective C–H bond activation.
In the past two decades, several directing groups have been invented by different research groups that can carry out selective distal sp3 C–H functionalization of aliphatic substrates. The most common substrates in this category are aliphatic amines, acids, alcohols, thiols, ketones, aldehydes, etc.
Early organometallic findings indicate that five membered cyclometallation is more favored over other ring sizes.4 This has been also proven by numerous examples, such as β-C(sp3)–H functionalization of carboxylic acid or γ-C(sp3)–H functionalization of aliphatic amines. However, this intrinsic preference also brings a tremendous challenge for activation of further distal sp3 C–H bonds as it requires the formation of less preferred six or over six-membered C(sp3)–H cyclometallation.
For the same reason, functionalization of γ-C(sp3)–H bond of aliphatic carboxylic acids or δ-C(sp3)–H bond aliphatic amines present a significant amount of challenge. After tremendous efforts from chemists over the globe, new strategies have been developed to functionalize the distal C–H bonds of aliphatic compounds.
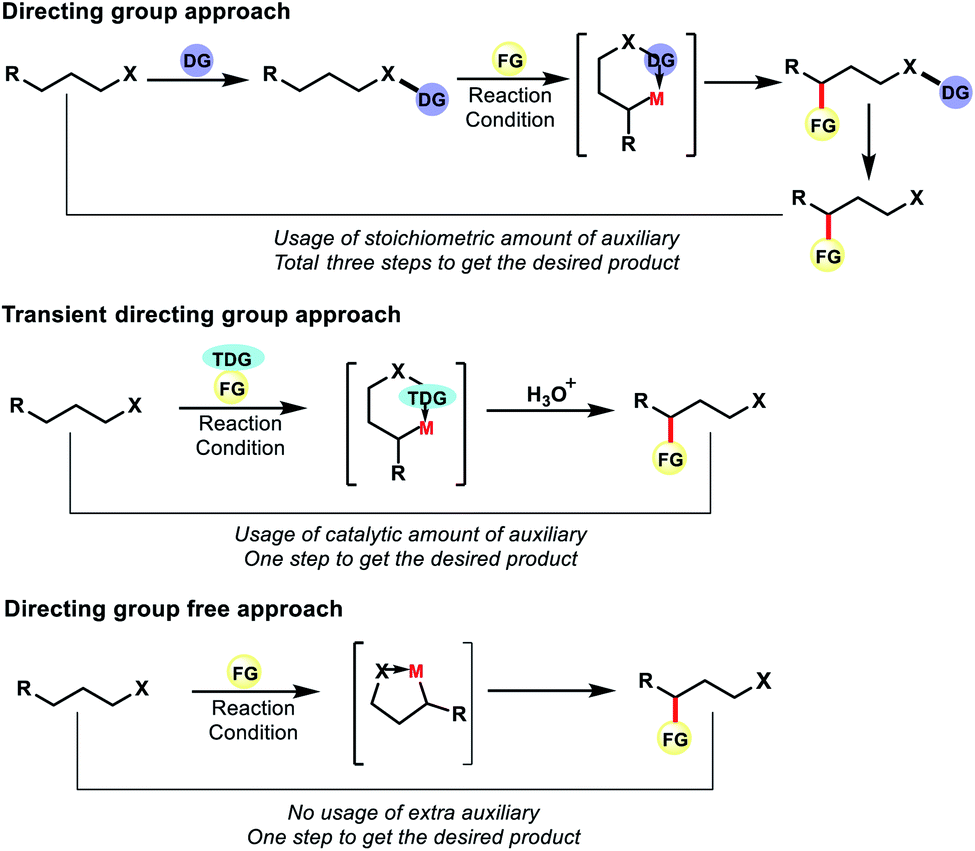
Although directing group approaches have been a great success for remote C–H functionalization of alkyl chains, this approach suffers integral limitations. The necessity of attaching an auxiliary or directing group with the original substrate and then removal of it post functionalization increases the total number of steps in the reaction, leading to unwanted waste products. Installation of the directing group or removal of it often requires harsh conditions. Therefore, such approaches reduce the efficiency of the reactions by decreasing its step as well as the atom economy.
To overcome such drawbacks continuous efforts were made by the chemists. Different tactics have been applied to make distal C–H functionalization more efficient and green. One such tactic is to use a transient directing group which can be generated in situ in the reaction. Unlike the directing group approach, the auxiliary here need not be attached to the substrate covalently. Thus the auxiliary can be used in catalytic amount unlike stoichiometric amount in the former case. The directing group attaches here with the substrate in situ fashion and can be removed via a simple work up and thus does not appear in the product. The usage of reversible transient DGs thus has the potential to reduce the number of synthetic operations through atom economical use of catalytic auxiliaries in a cooperative manner.
However, an ideal approach would be where we do not need to use any auxiliary or directing groups for distal C–H functionalization, be it covalently attached DG or transient DG. The native functional group present in aliphatic compounds can also serve as the directing group for site selective C–H functionalization. Such strategies would make C–H functionalization reactions more efficient and practical for application in organic synthesis (Fig. 3). In recent years such approaches have progressed to a great extent for the functionalization of distal C–H bonds in aliphatic compounds.
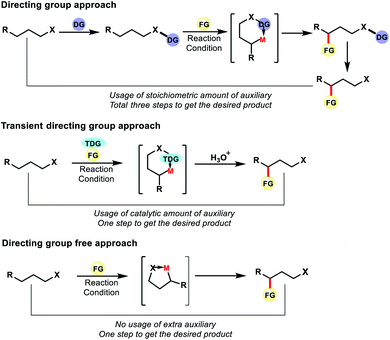 |
| | Fig. 3 Different strategies of distal C–H functionalization. | |
In the following sections, we will discuss the recent advancements on C(sp3)–H functionalization of aliphatic substrates via different strategies such as directing group assisted, transient directing group assisted, and directing group free strategies. The relevant reports are sorted by the classification of different substrate classes.
2.1 Gamma functionalization of aliphatic amines
Aliphatic amines a key functional group in agrochemicals, pharmaceuticals, and materials. Functionalization of such molecules have been the subject of numerous synthetic strategies. In the following sections, various approaches on γ-C(sp3)–H functionalizations have been detailed.
2.1.1. Covalently tethered directing group approach.
2.1.1.1 Arylation.
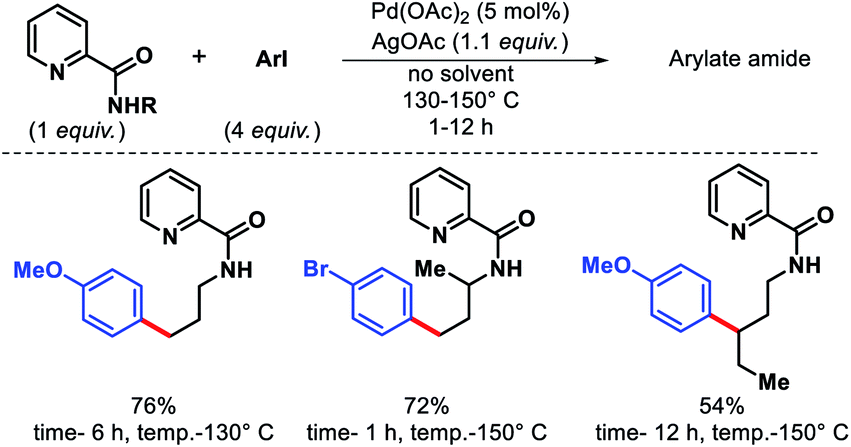
The very initial outcome of γ-C(sp3)–H functionalization was reported by Daugulis and coworkers in 2005.5 γ-C(sp3)–H arylation was achieved with the assistance from 2-picolinic acid auxiliary and palladium catalyst (Scheme 1). This report showed the path for auxiliary assisted distal C(sp3)–H functionalization of aliphatic amines. Both primary and secondary C–H bonds undergo the reaction, though, former one reacts faster which allowed for the selective monoarylation of methyl groups.
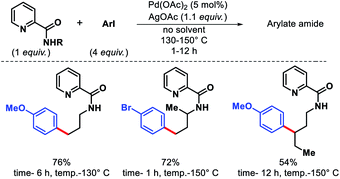 |
| | Scheme 1 Initial report of γ-C(sp3)–H arylation of aliphatic amines. | |
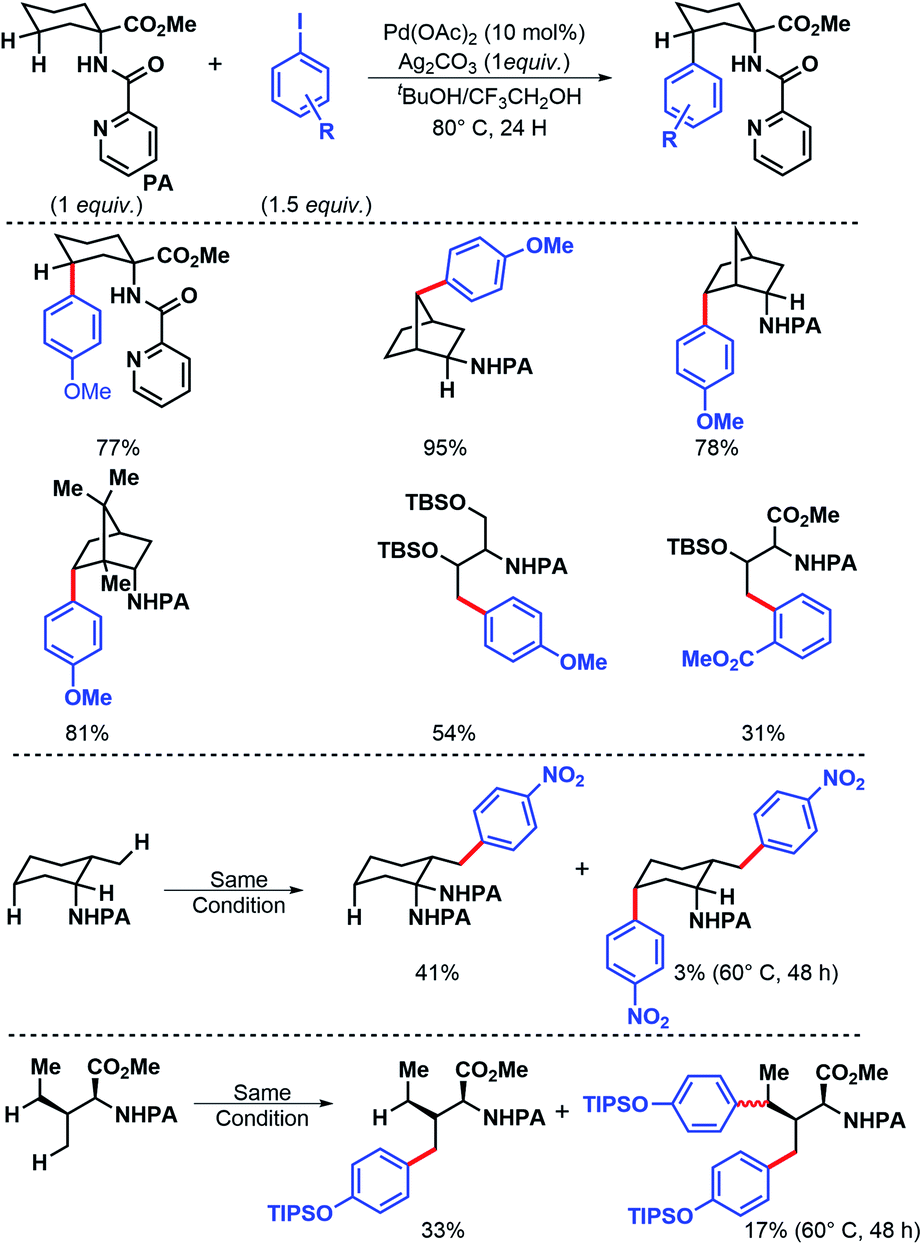
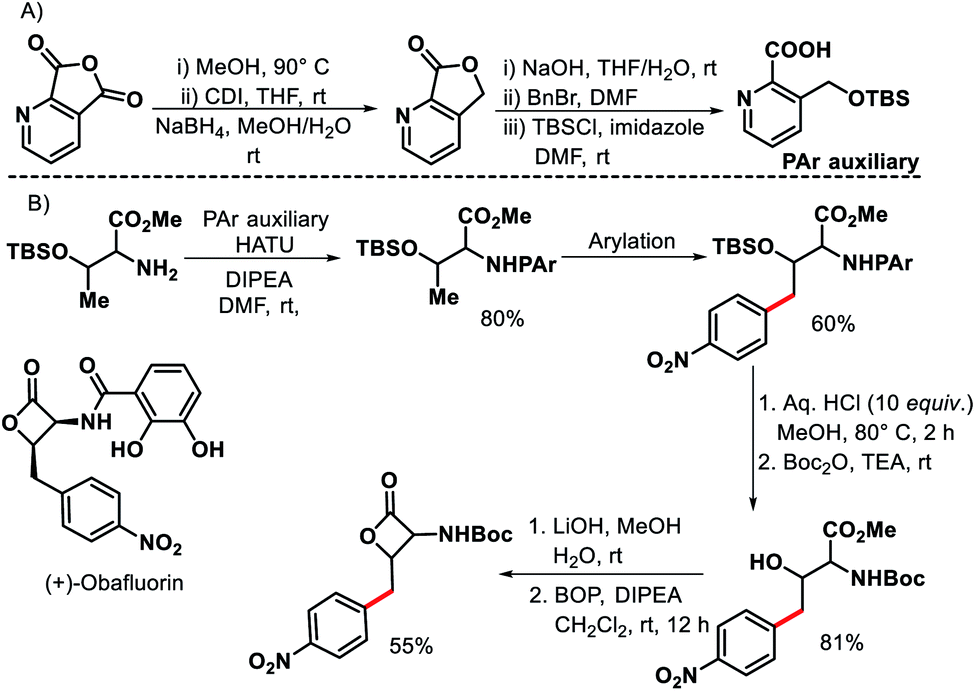
In 2011, Chen and coworkers have come up with a more practical strategy for γ-C(sp3)–H arylation of alkyl amines.6 They showcased a number of substrates such as cyclohexyl amino acid, 2-amino norbornane, bornyl amine, threoninol, and isoleucine derivatives that undergo the reaction with different aryl iodides (Scheme 2A). Substrates with two γ C–H bonds were also subjected to the reaction which resulted in predominant monoarylation of primary C–H bond. A minor bisarylated product was observed after a prolonged reaction at 60 °C. However, no significant amount of gem diarylated product was observed (<3%) in any of the cases presumably because of steric factors (Schemes 2B and C). A more practical directing group which can be removed under mild condition post-functionalization was synthesized from a commercially available anhydride (Scheme 3). Utilizing this easily removable directing group, formal synthesis of a natural product (+)-obafluorin was carried out in starting from a threonine-derived substrate (Scheme 3B).6
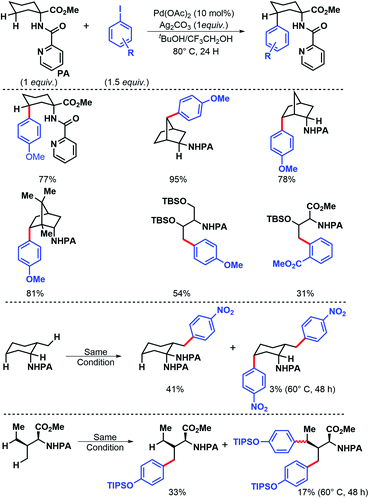 |
| | Scheme 2 Picolinamide (PA) assisted approach for γ-C(sp3)–H arylation. | |
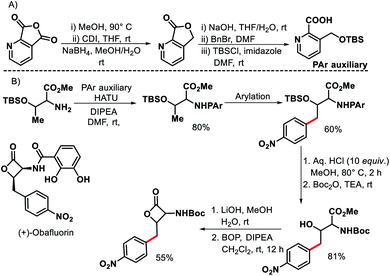 |
| | Scheme 3 (A) Synthesis of PAr auxiliary (B) formal synthesis of natural product (+)-obafluorin. | |
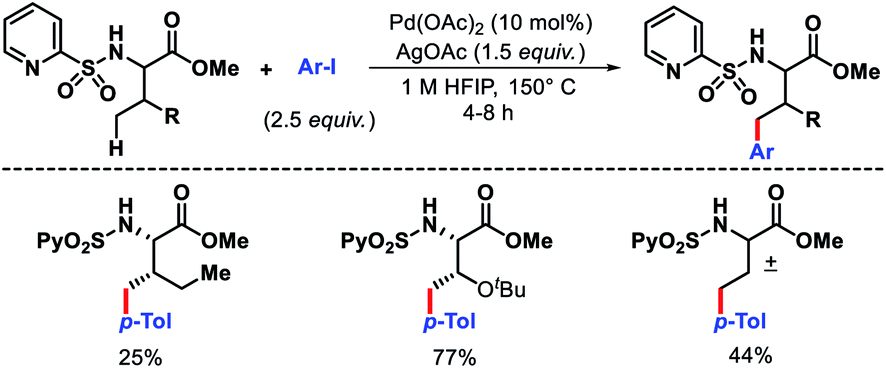
Carretero and colleagues developed N-(2-pyridyl)sulfonyl auxiliary to functionalize the γ-C–H bond of amino acid methyl esters and dipeptides.7 A variety of N-(2-pyridyl)sulfonamide amino acid derivatives, including α-quaternery amino acids, β-amino acids, and dipeptides reacted with aryl iodides to provide γ-arylated products (Scheme 4). A Bimetallic PdII γ-metallated comples was characterized by X-ray that showed the role of the auxiliary in the reaction. The N-(2-pyridyl)sulfonyl template was readily removed by treatment with Zn/NH4Cl. Using the same auxiliary, Carretero group reported a γ-C(sp3)–H carbonylation of α and β amino acids. Mo(CO)6 was utilized as the carbonylating agent in the reaction.8
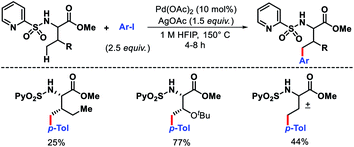 |
| | Scheme 4 γ-Arylation assisted by a N-(2-pyridyl)sulfonyl auxiliary. | |
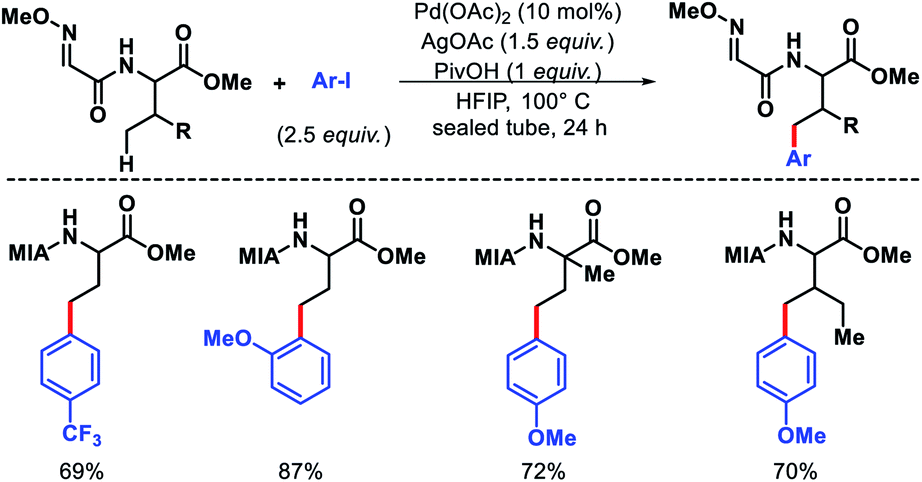
Later, Ma group came up with a 2-methoxyiminoacetyl (MIA) template for promoting γ-C(sp3)–H arylation of amino acids.9 Amino acids such as valine, threonine, and isoleucine derivatives were arylated in moderate to good yields (Scheme 5). Nonetheless, arylation of secondary amides and γ-C(sp3)–H arylation of methylene center were unsuccessful with this approach. Further, the MIA auxilary was transformed into useful moieties.
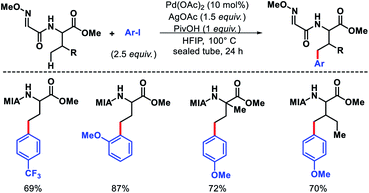 |
| | Scheme 5 2-Methoxyiminoacetyl (MIA) auxiliary for γ-C(sp3)–H arylation. | |
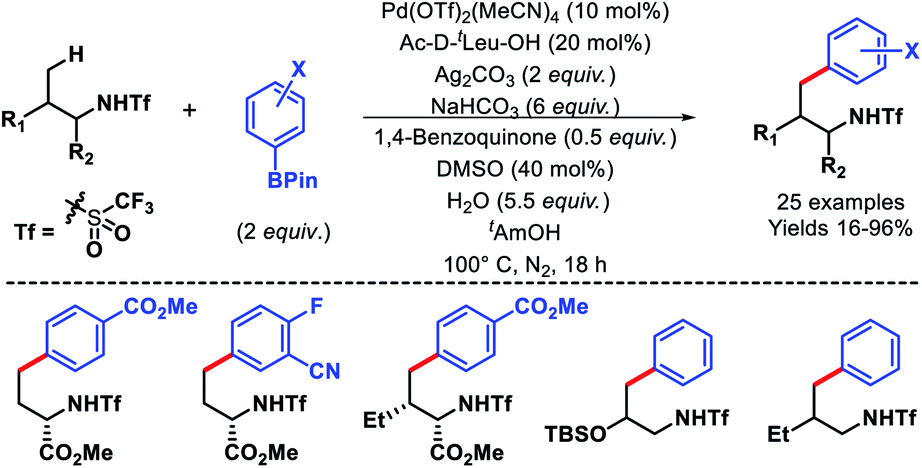
In 2014, Yu and coworkers reported a ligand accelerated γ-C(sp3)–H arylation of aliphatic amines (Scheme 6).10 Triflamide protected amines were used as substrates. It had been hypothesized that the combination of triflamide protecting group that could form an imidate like moiety as a weak coordinating σ donor when deprotonated under a basic condition with a mono-protected amino acid (MPAA) ligand accelerates the reaction. Ac-D-tLeu-OH was found to be the best ligand for the reaction. Various aryl boronic reagents were used as the arylating partner. The scope of amines contained α or β-substituted aliphatic amines. In the absence of the ligand, there was no background reaction observed which shows the crucial role of the ligand in the reaction. Nonetheless, the mechanism of ligand acceleration in the reaction was not stated clearly and need further investigation.
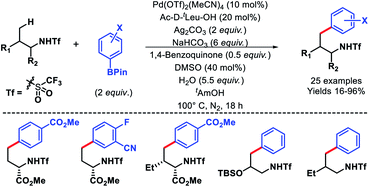 |
| | Scheme 6 Ligand enabled γ-C(sp3)–H arylation of aliphatic amines. | |
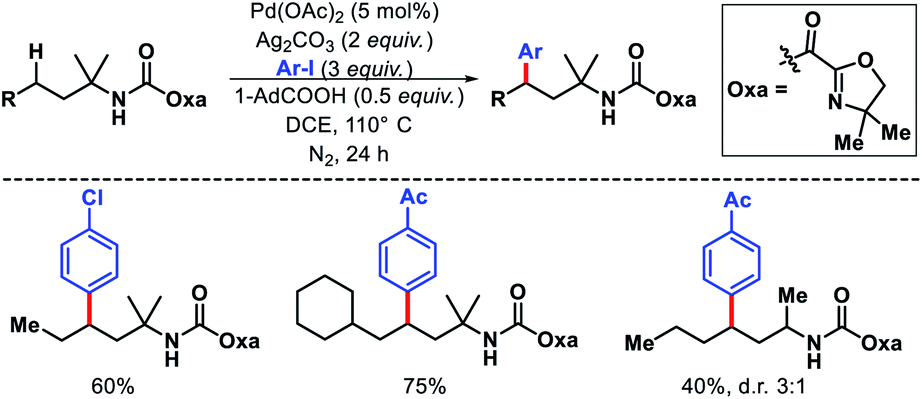
Despite the tremendous progress in γ-methyl γ-C(sp3)–H functionalization of amines, activation of γ-methylene C(sp3)–H bond remained mainly unexplored until Shi and coworkers reported a protocol.11 An oxazoline carboxylate auxiliary was utilized to achieve arylation of γ-methylene C(sp3)–H bonds (Scheme 7). A range of iodoarenes were compatible with the reaction. Several branched and linear alkyl amines containing functional group such as trifluoromethyl, cyano were well tolerated under the reaction condition. A α-quaternery center in the substrate was essential to provide Thorpe–Ingold effect and therefore, better conversion of the substrate. Without it the yield dropped significantly.
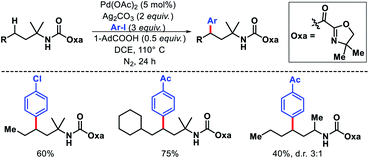 |
| | Scheme 7 γ-Methylene C(sp3)–H arylation of alkyl amines. | |
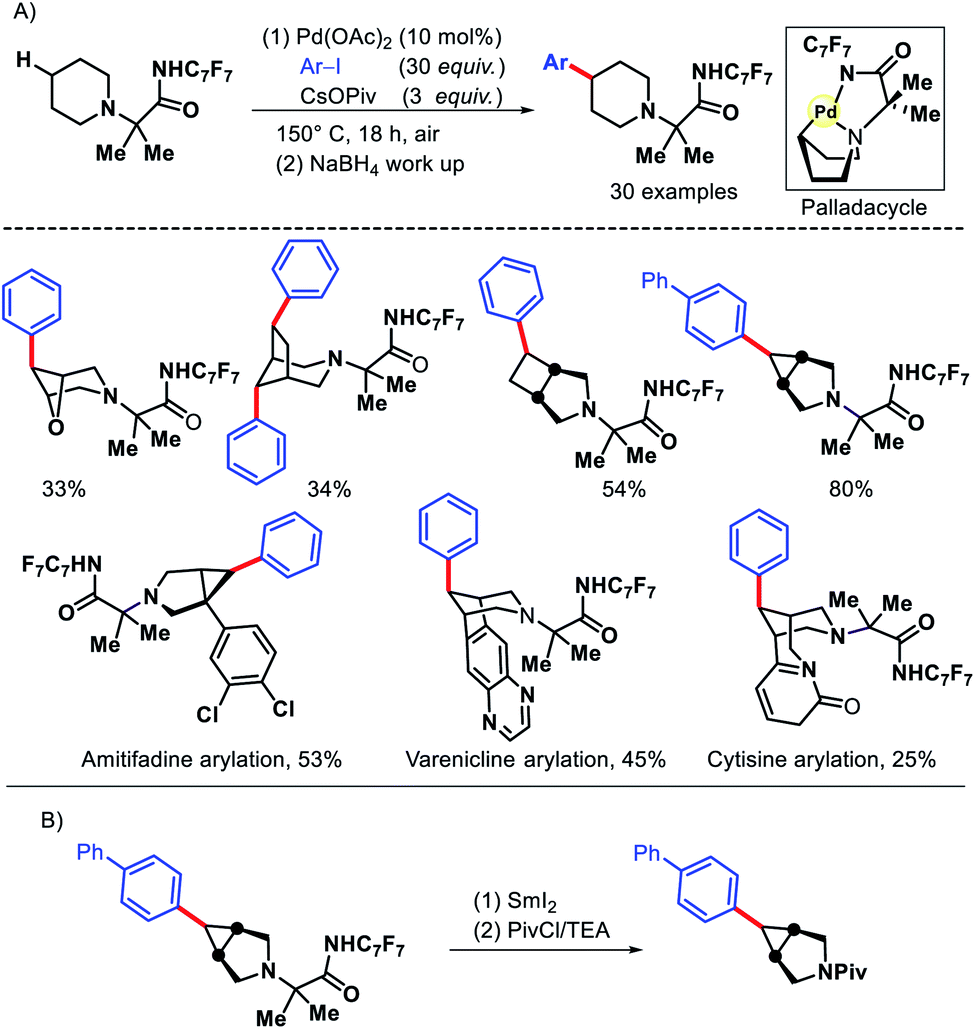
In 2016, Sanford and coworkers reported an elegant approach for remote γ-C(sp3)–H arylation of alicyclic amines.12 Despite the ubiquitous presence of alicyclic amines in pharmaceuticals, remote functionalization of such amines remained a challenge. Most functionalizations are limited to α-C–H bond to nitrogen. Sanford group achieved the γ-C(sp3)–H arylation by covalently attaching a highly electron withdrawing amide (derived from p-CF3-C6F4 aniline) with alicyclic amines to work as a secondary coordinating group. The protocol exhibits a varied scope of aryl iodides and amines including pharmaceutical candidates amitifadine, varenicline, natural product cyticine with moderate to good yields (Scheme 8). The major limitation of the work remains the usage of a large excess (20–30 equiv.) of aryl iodides in most of the cases. Only in the substrate where cyclopropane C–H bond has been activated, 3 equiv. of aryl iodide was enough. The same group later outlined another protocol to transannular γ-C–H arylation of azabicycloalkanes via a second generation Pd catalyst system.13
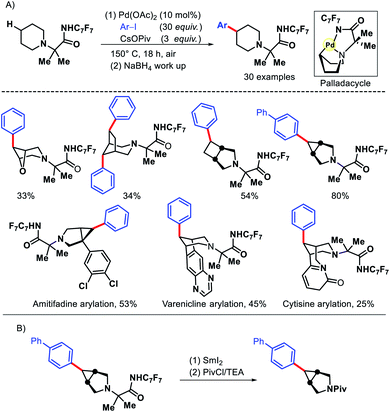 |
| | Scheme 8 (A) γ-C(sp3)–H arylation of alicyclic amines (B) directing group removal from the product. | |
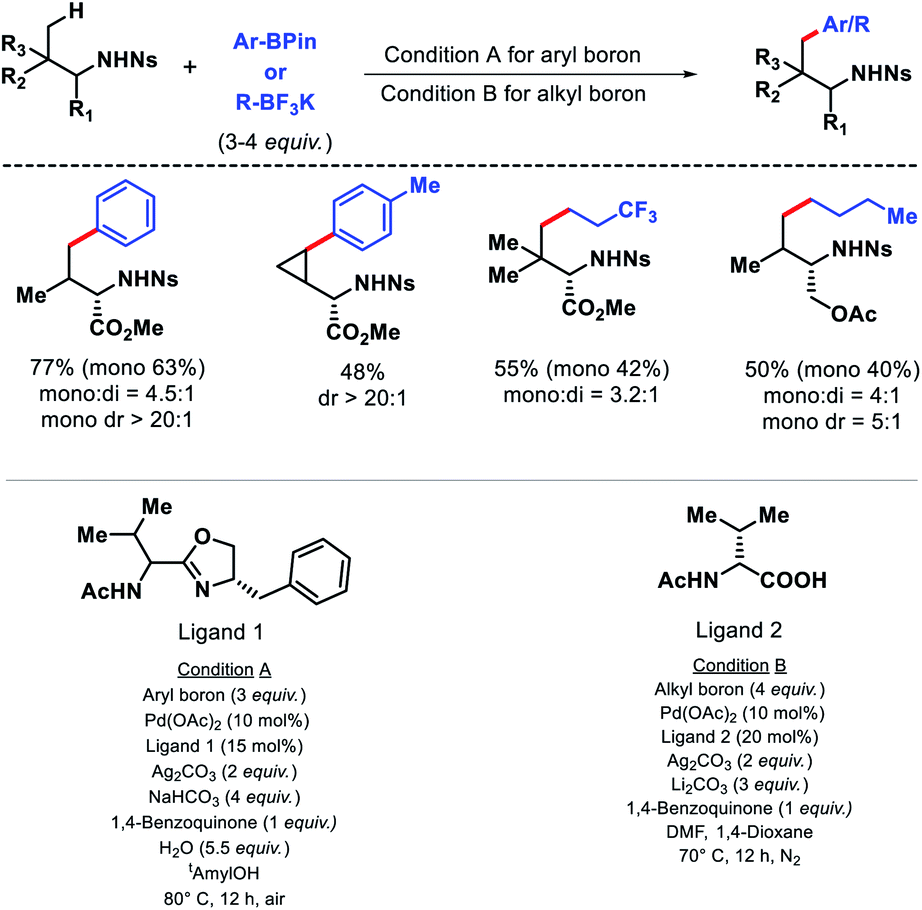
In 2017, Yu and colleagues outlied another γ-C(sp3)–H arylation approach of amines using nosyl (Ns) as the protecting group.14 It is advantageous to utlize nosyl as the directing group due to its ease of removal. Both aryl boron and alkyl boron reagents were used as arylating agent in the reaction in presence of different ligands. An acetyl protected aminomethyl oxazoline (APAO)ligand found to enable arylation with aryl boron reagent, whereas N-acetyl-D-valine ligand promote arylation with alkyl boron reagent. A number of amines and amino acids were arylated at γ-position with various aryl and alkyl boron reagents in good yield and diasterioselectivity (Scheme 9). Gram scale synthesis of γ-arylated amino acids was carried out to demonstrate practacilty of the reaction.
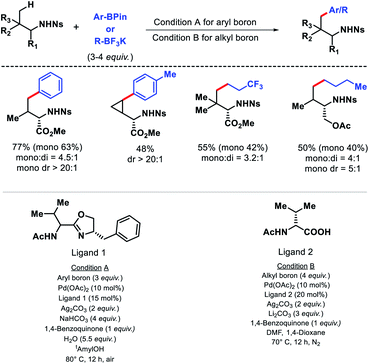 |
| | Scheme 9 γ-C(sp3)–H arylation of nosyl protected amines. | |
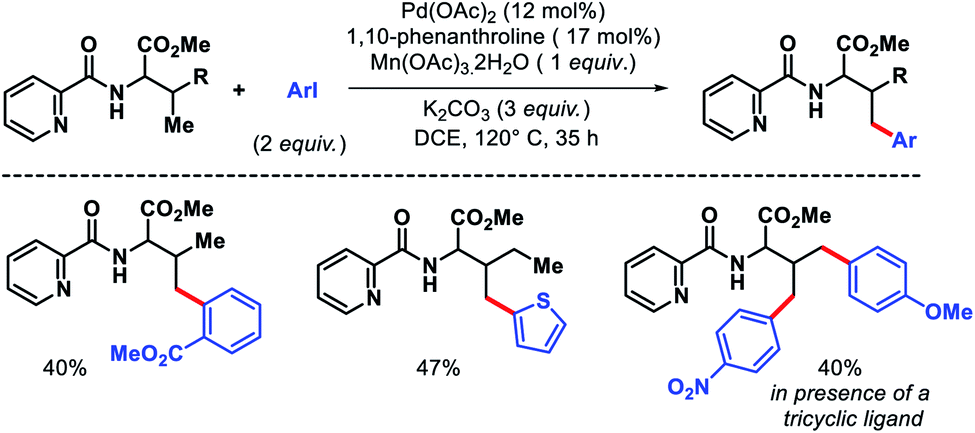
Jana and coworkers have recently reported a 1,10-phenanthroline ligand enabled γ-arylation of amino acids (Scheme 10).15 Along with monoarylation, unsymmetrical diarylation was carried out utilizing a tricyclic ligand. Notably, inexpensive Mn(OAc)3 dihydrate was utilized as oxidant in the reaction instead of expensive silver salts.
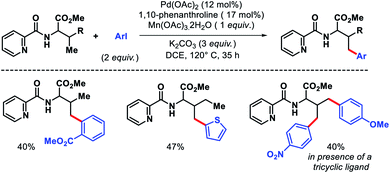 |
| | Scheme 10 Picolinamide directed γ-C(sp3)–H mono and diarylation of amino acids. | |
 |
| | Scheme 11 PA assisted γ-C(sp3)–H alkenylation. | |
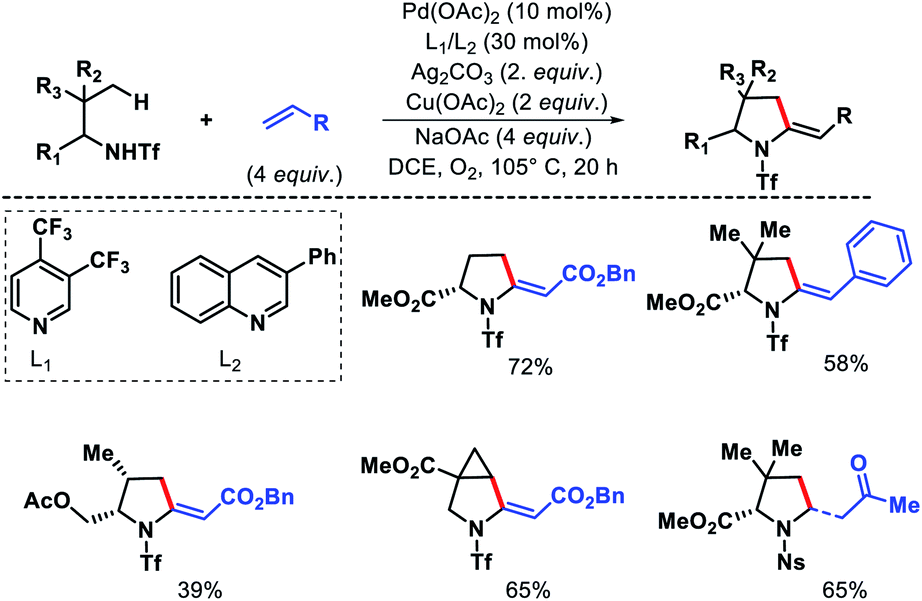
Yu and coworkers later developed a ligand enabled protocol for γ-C(sp3)–H alkenylation of aliphatic amines and α-amino esters to provide pyrrolidines.16 Both electron deficient alkenes and styrenes were utilized as olefin partners using pyridine or quinolone based ligands. Both triflyl (Tf) and nosyl (Ns) groups were used as directing groups with different set of ligands (Scheme 12). More substituted amine substrates showed superior reactivity to less biased amines due to Thorpe–Ingold effect.
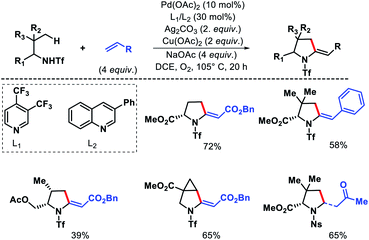 |
| | Scheme 12 γ-C(sp3)–H olefination of alkyl amines to form pyrrolidines. | |
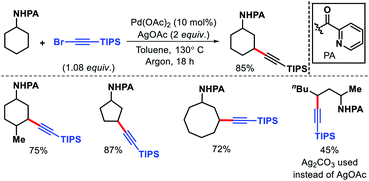 |
| | Scheme 13 PA assisted γ-C(sp3)–H alkenylation. | |
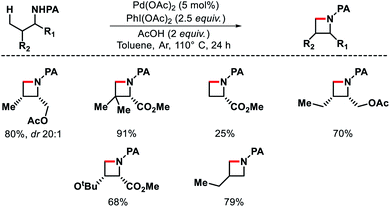 |
| | Scheme 14 PA assisted γ-C(sp3)–H intramolecular amination. | |
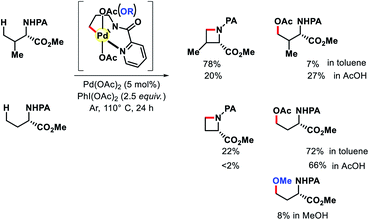 |
| | Scheme 15 Proof of concept for PA directed alkoxylation. | |
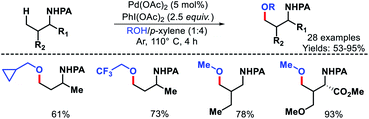 |
| | Scheme 16 Picolinamide assisted γ-C(sp3)–H alkoxylation. | |
 |
| | Scheme 17 Oxalyl amide assisted γ-C(sp3)–H carbonylation. | |
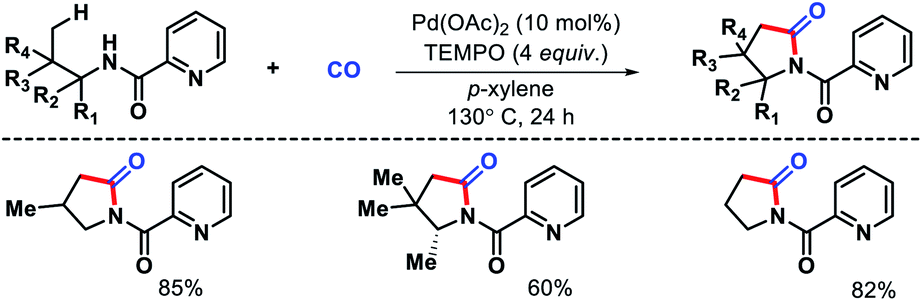
Wang and colleagues also developed a γ-C(sp3)–H carbonylation procedure for aliphatic amines using picolinamide as auxiliary (Scheme 18).22 The reaction took place in presence of TEMPO as the sole oxidant and Pd(OAc)2 as catalyst. Alkyl amines containing substitions at α and β position proceeded smoothly to afford pyrrolidines in good yields. Alkyl amine bearing no sybstitutions was also well compatible with the reaction. The synthetic utility was demonstrated by synthesizing rac-pregabalin.
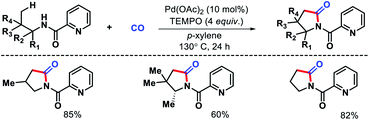 |
| | Scheme 18 Picolinamide directed γ-C(sp3)–H carbonylation. | |
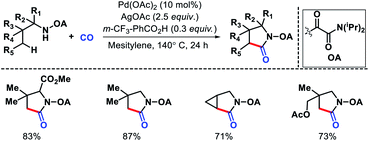
 |
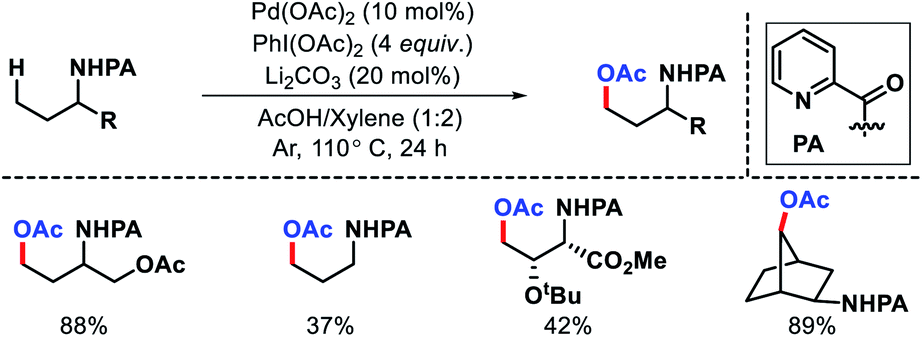
| | Scheme 19 PA directed γ-C(sp3)–H acetoxylation. | |
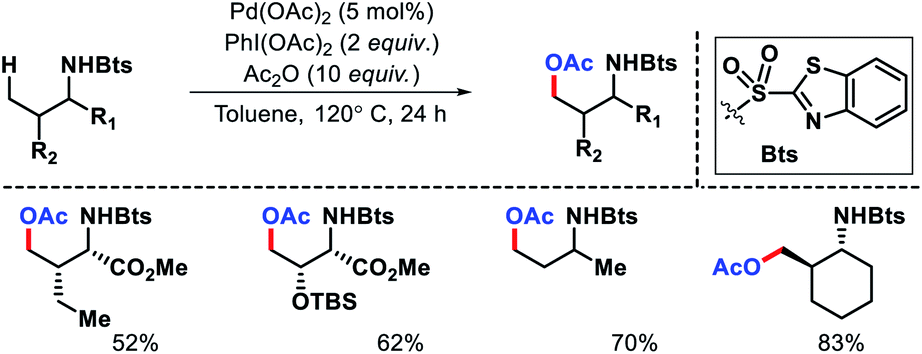
Later other groups came up with different protocol for γ-acetoxylation of aliphatic amines. Xuan and coworkers utilize a benzothiazol-2-sulphonyl as an auxiliary to carry out γ-C(sp3)–H acetoxylation (Scheme 20).24 Similarly, γ-acetoxylation of triflyl protected amines were achieved by Jia et al.25
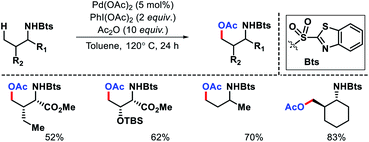 |
| | Scheme 20 Benzothiazol-2-sulphonyl as directing group for g-acetoxylation of alkyl amines. | |
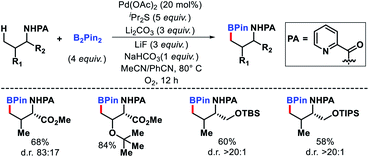 |
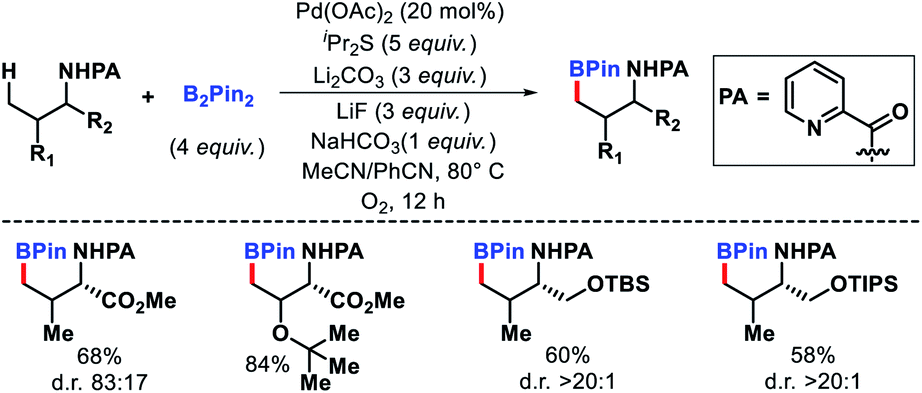
| | Scheme 21 Primary γ-C(sp3)–H borylation of amines. | |
2.1.2. Transient directing group approach.
2.1.2.1. Arylation.
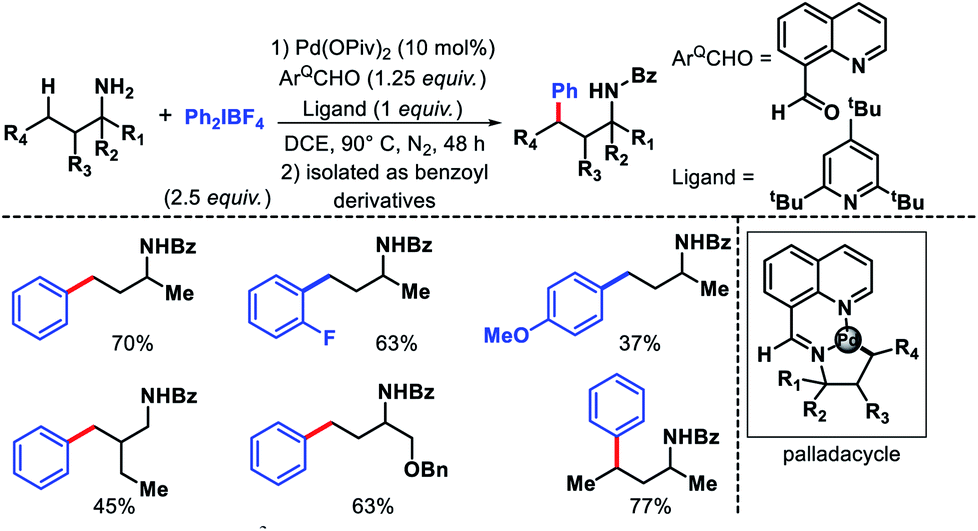
The earliest report on distal γ-C(sp3)–H functionalization via in situ generated directing group was disclosed by Dong and co-workers in 2016.27 8-Formyl quinoline was used as the additive in the reaction which formed imine functionality by reacting with amine substrate in the reaction. This in situ generated imine then directs the activation of distal γ-C(sp3)–H bond. In the absence of 8-formyl quinoline, no desired product formed which showed the key role of imine formation in the reaction. The reaction required highly active aryl iodonium [Ar2IBF4] salts as the aryl source. Aliphatic amine substrates with varied α-substitutions were employed in the reaction. A few examples were also shown for methylene C–H bond arylation. The products are isolated as benzoyl derivatives for the ease of isolation (Scheme 22). Although it was the first report of its kind, it had several limitations such as usage of equivalent amount (125 mol%) additive, the necessity of using glovebox, use of highly active aryl iodonium salts, etc.
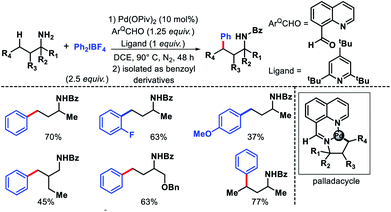 |
| | Scheme 22 γ-C(sp3)–H arylation via in situ generated imine from 8-formyl quinolone. | |
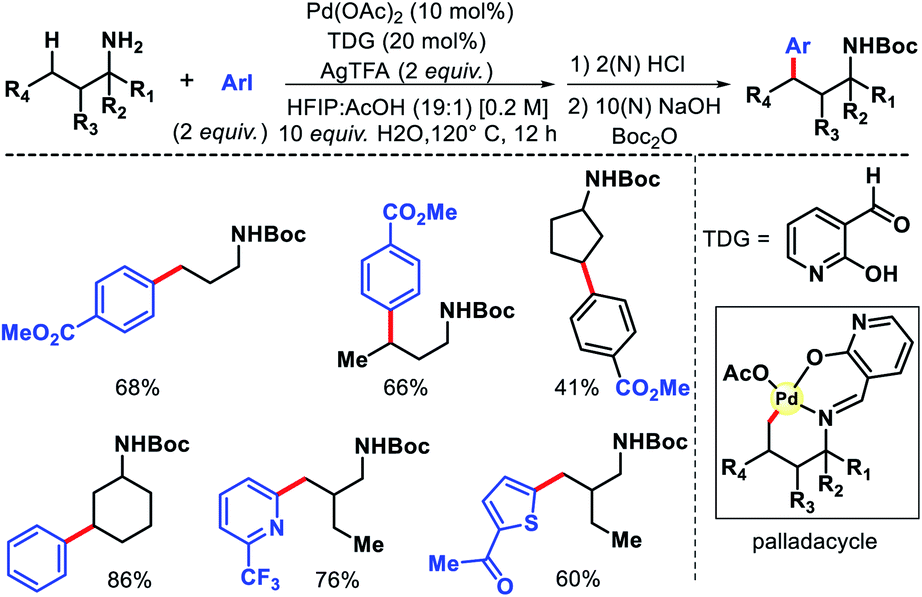
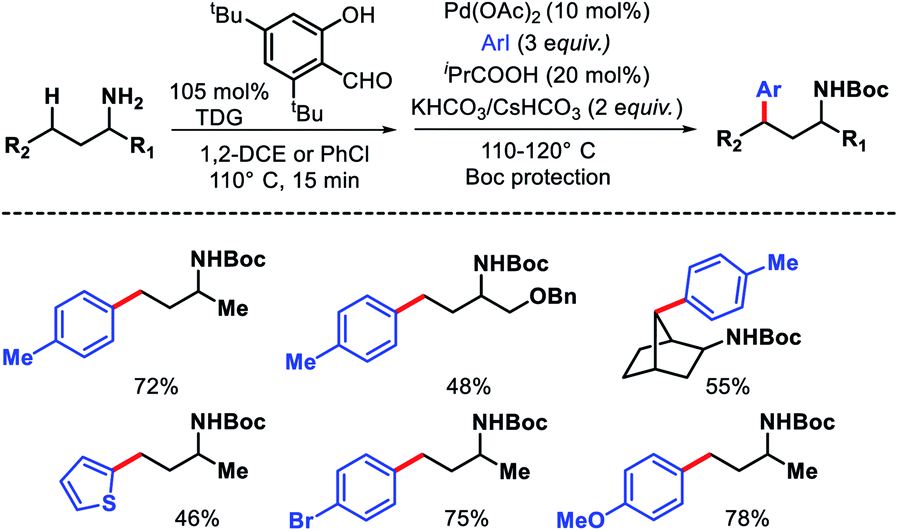
In the same year, Yu group overcame the limitations associated with Dong's work by using a catalytic amount of 2-hydroxy nicotinaldehyde as the additive or transient directing group (TDG) for γ-C(sp3)–H arylation of alkyl amines (Scheme 23).28 Bidentate chelation mode of the imine and hydroxyl moiety from TDG provided a robust reaction with aryl iodides as the coupling partner. A variety of aryl iodide was utilized for the distal functionalization of cyclohexyl amine. Heteroaryl iodides were also found to be compatible under their reaction condition. Notably, unbiased amines such as propyl amine, isobutyl amine, and others without any α-substitutions were well tolerated in this reaction. Further, methylene C–H arylation of cyclic and acyclic amines was also achieved. The products of the reaction were isolated as Boc derivatives of amines (Scheme 23).
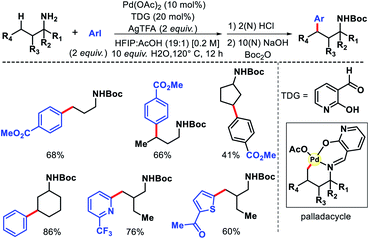 |
| | Scheme 23 2-Hydroxy nicotinaldehyde as TDG for γ-C(sp3)–H arylation. | |
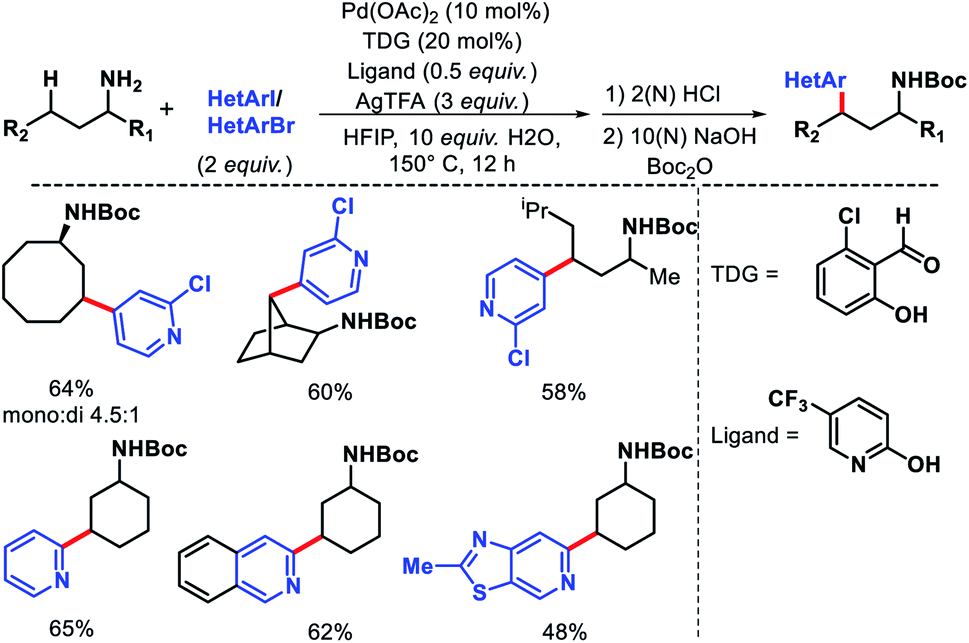
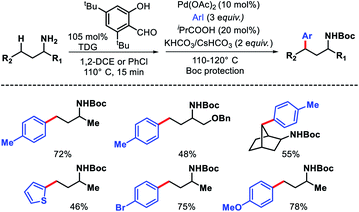
The same group in 2018 came up with another similar approach for in situ imine directed γ-C(sp3)–H arylation of methyl and methylene C–H bonds.29 However, this time the focus was mainly to use heteroaryl iodides and bromides as arylating agents through ligand development. 5-Trifluoromethyl pyridone was found to be the best ligand along with 2-hydroxy-6-chloro-benzaldehyde as TDG. Several heteroaryl iodides and bromides were employed in the reaction at elevated temperature (slightly higher temperature for heteroaryl bromides) (Scheme 24). The scope of alkyl amines contained various cyclic as well as acyclic α-substituted amines. Usage of cheaper heteroaryl bromides (compared to frequently used iodides) is one of the key highlights of this work. Because of their inherently stronger C–Br bond strength, heteroaryl bromides are rarely used as the coupling partner in C–H activation reactions.
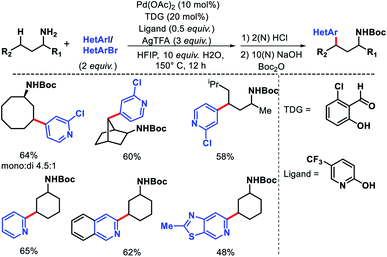 |
| | Scheme 24 γ-C(sp3)–H arylation via cooperation of transient directing group and ligand development. | |
An elegant approach in this area was reported by Ge and coworkers in 2017.30 They used catalytic amount of (20 mol%) glyoxalic acid as the transient directing group (TDG) for γ-C(sp3)–H arylation (Scheme 25). The reaction tolerated a diverse range of aryl iodides along with mostly α-disubstituted alkyl amines as amine substrates (Scheme 25A). α-Disubstitution on the amines was necessary to provide suitable geometrical alignment via Thorpe–Ingold effect. Without α-disubstitution, yields decreased drastically. Methylene C–H arylation was also sluggish via this methodology. One great aspect of this work lies in the isolation of arylated amines. A simple acidic/basic workup was enough to isolate products, no extra column chromatography was required. The five membered cyclopalladated intermediate was also characterized by the X-ray that led to a better insight into the reaction mechanism (Scheme 25B). The synthesis of fingolimod analogues (a drug for treating multiple sclerosis) was carried out using the methodology in three steps.
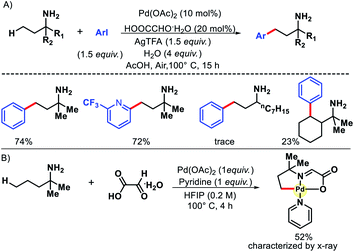 |
| | Scheme 25 (A) Glyoxalic acid as TDG for γ-C(sp3)–H arylation (B) synthesis of cyclopalladated intermediate. | |
3,5-Di-tert-butylsalicylaldehyde was found to be another effective TDG by Murakami and co-workers (Scheme 26).31 Precondensation between amine substrates and TDG was carried out before the addition of other reagents. The TDG was used in an equivalent amount to form imine from aliphatic amines. Various α-substituted alkyl amines and aryl iodides were compatible. Products were isolated as Boc derivatives.
 |
| | Scheme 26 Pd catalyzed γ-C(sp3)–H arylation of alkyl amines using 3,5-Di-tert-butylsalicylaldehyde as TDG. | |
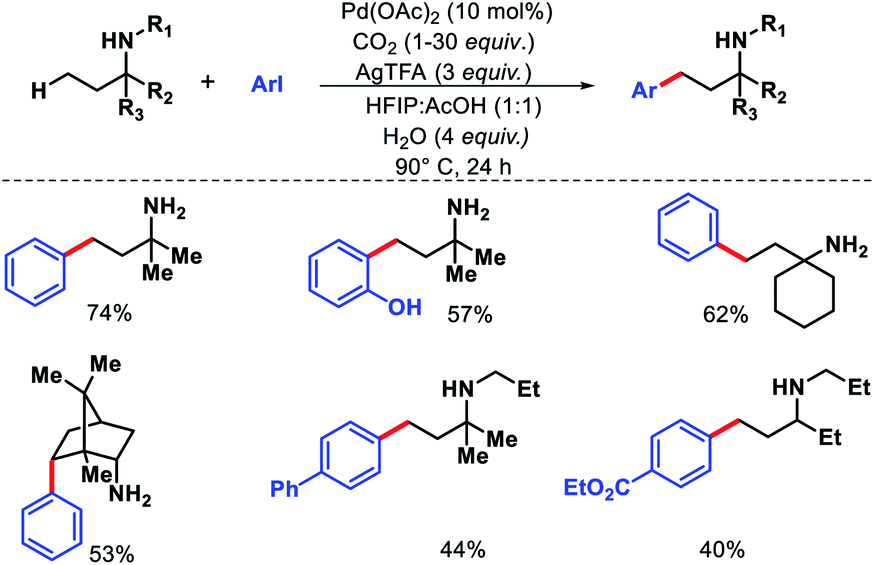
Recently, Young and colleagues utilized CO2 in the form of dry ice as a transient directing group for γ-C–H arylation.32 The in situ generated carbamate can readily coordinate with palladium and chemically more robust than imine moiety. No exogeneous ligand is necessary to carry out the reaction. Both primary and secondary amines were employed as the successful substrates with a range of aryl iodides (Scheme 27). In case of secondary amines an excess functionalization reactions. Amount of CO2 was essential. At lower concentration of CO2 the yield decreased significantly due to oxidation of the amine starting material. Mechanistic experiments revealed that C–H activation is the rate limiting step of the reaction. Usage of CO2 as transient DG instead of traditional imine moiety or other protecting group is a fruitful addition to improve the sustainability of C–H activation.
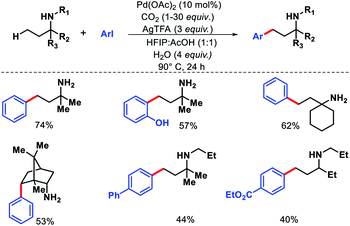 |
| | Scheme 27 CO2 mediated γ-C(sp3)–H arylation of amine substrates. | |
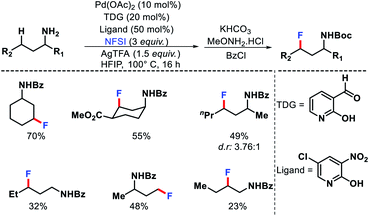 |
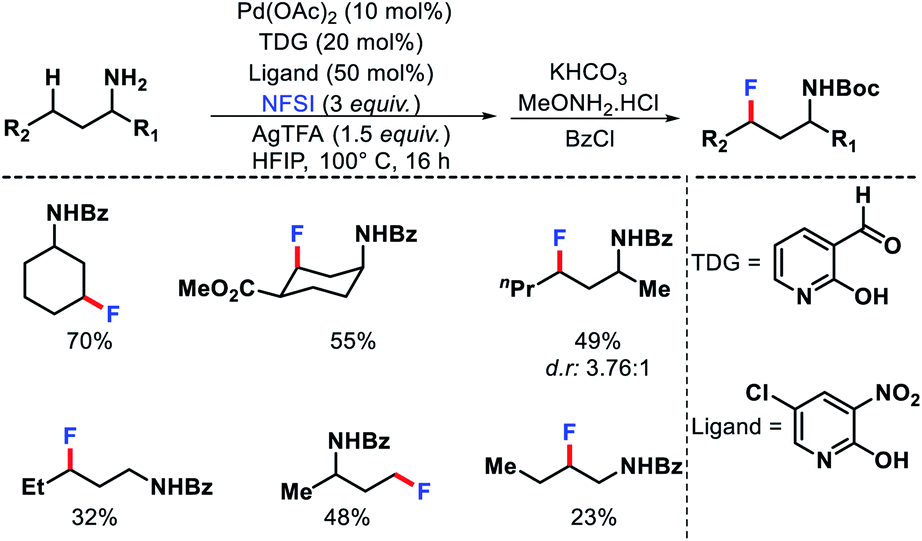
| | Scheme 28 A transient group mediated γ-C(sp3)–H fluorination of aliphatic amines. | |
2.1.3 Directing group free approach.
2.1.3.1. Alkenylation.
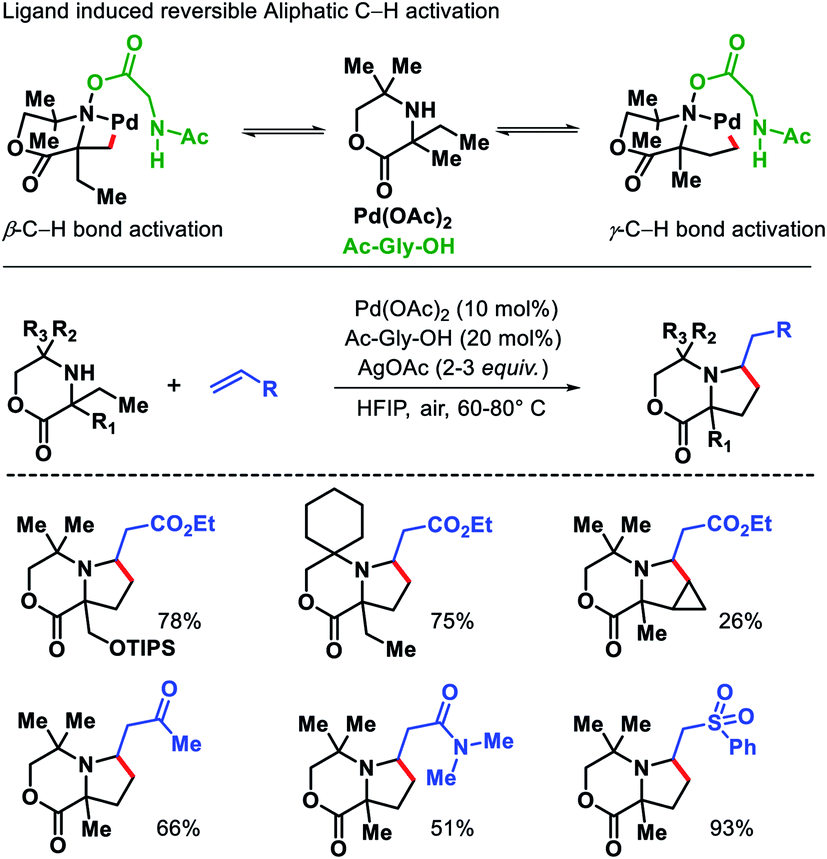
Functionalization of unprotected free amines is extremely challenging due to the rapid formation of bis-amine metal complex of free amines with transition metals, which hinders the formation of C–H activated intermediate. Moreover, reactivity of α-C–H bonds makes it even more challenging to functionalize distal C–H bonds. Instead of these challenges, Gaunt and coworkers reported a γ-C(sp3)–H alkenylation of morpholinone derivatives to form functionalized pyrrolidines.34 The alkene insertion is followed by aza-Michael cyclization to form pyrrolidines (Scheme 29). The sterically specialized substrate used by Gaunt group hinders the formation of bis-amine metal complexes (by inducing steric strain) and favour the formation of required mono-amine metal complex. An amino acid ligand Ac-Gly-OH played a key role in the reaction. By rendering the cyclopalladation step reversible, Ac-Gly-OH was found to favour a 5-membered cyclopalladation pathway instead of four for morpholinone derivatives. Different α,α′-substituted morpholinones were compatible in the reaction along with various activated olefins. Mechanistic investigations revealed that without ligand the C–H bond cleavage is irreversible and occurs after rate determining step. While in the presence of ligand C–H bond cleavage become reversible.
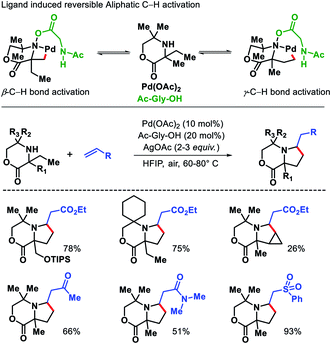 |
| | Scheme 29 Ligand induced γ-C(sp3)–H alkenylation of morpholinones. | |
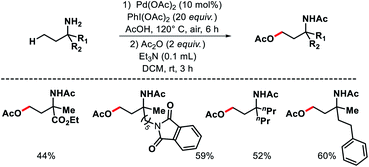 |
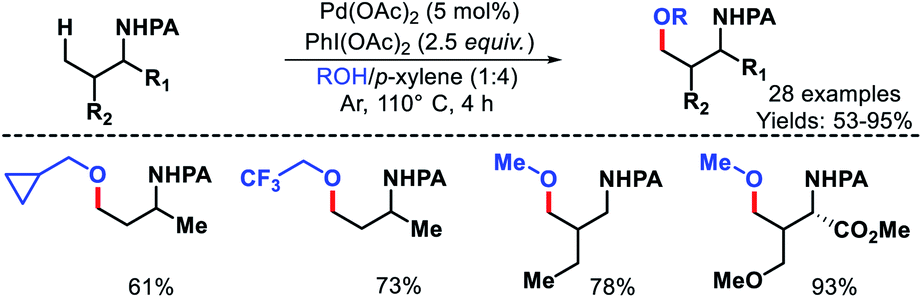
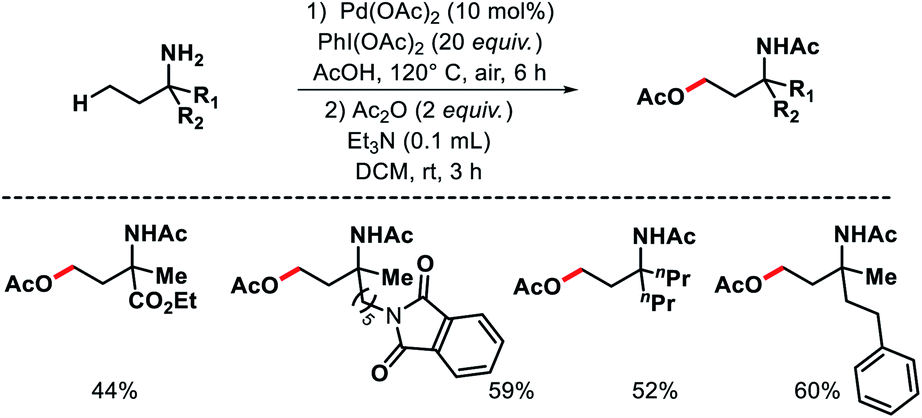
| | Scheme 30 Pd catalyzed γ-C(sp3)–H acetoxylation of free primary amines. | |
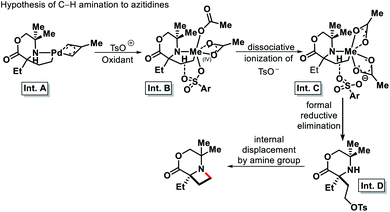 |
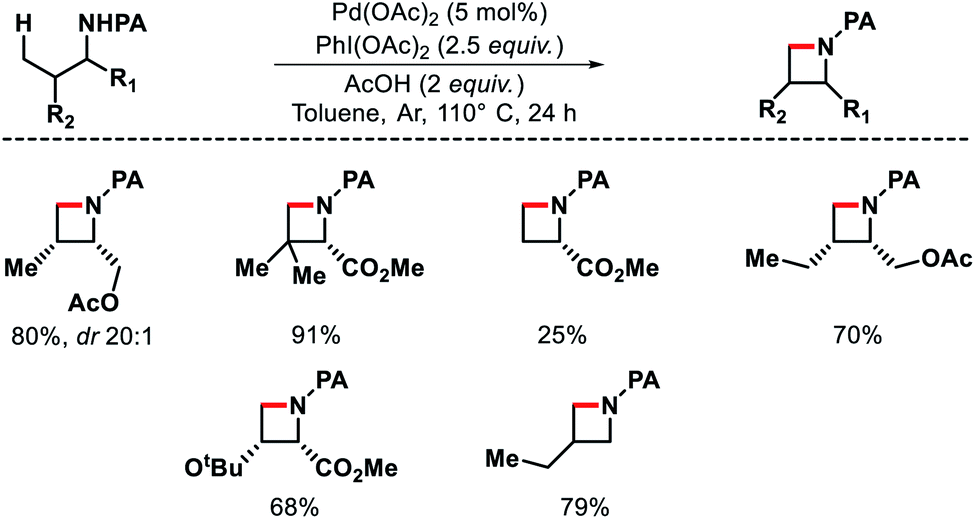
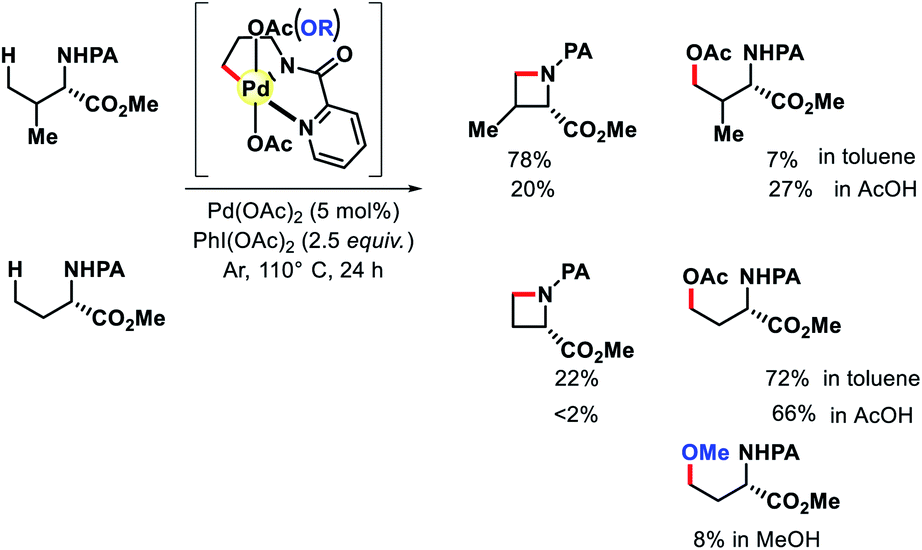
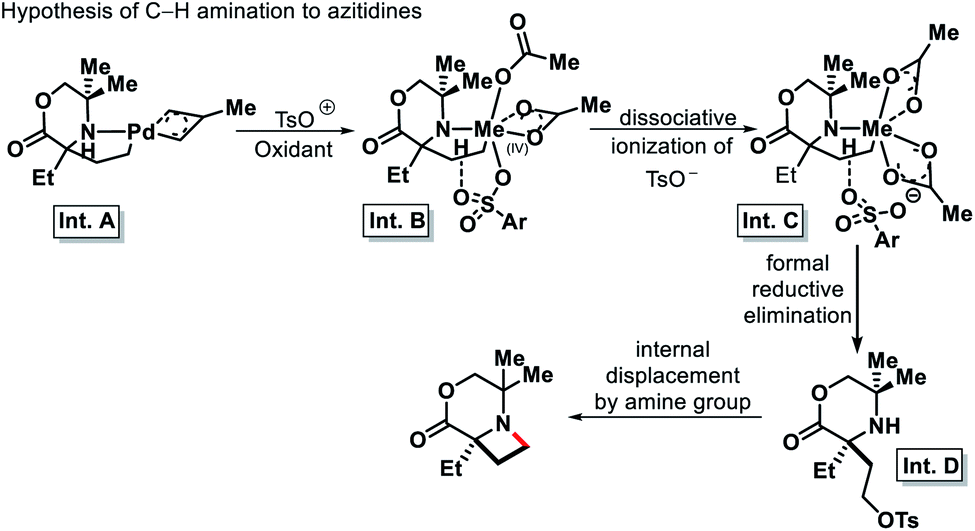
| | Scheme 31 Proposed C–H amination mechanism to form azitidine. | |
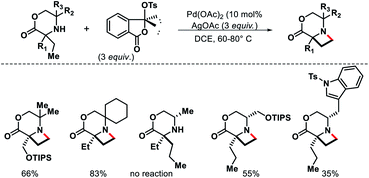 |
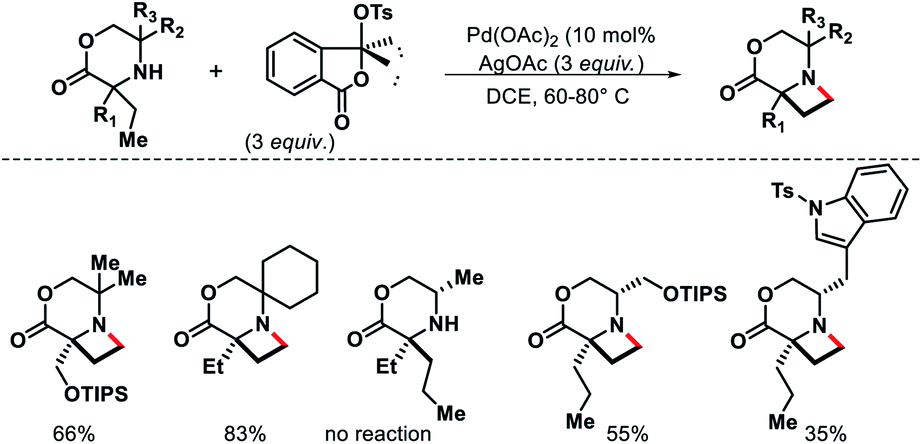
| | Scheme 32 Pd catalyzed intermolecular γ-C(sp3)–H amination of morpholinones. | |
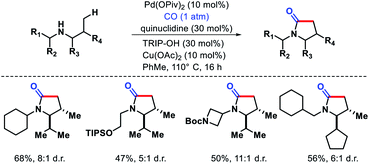 |
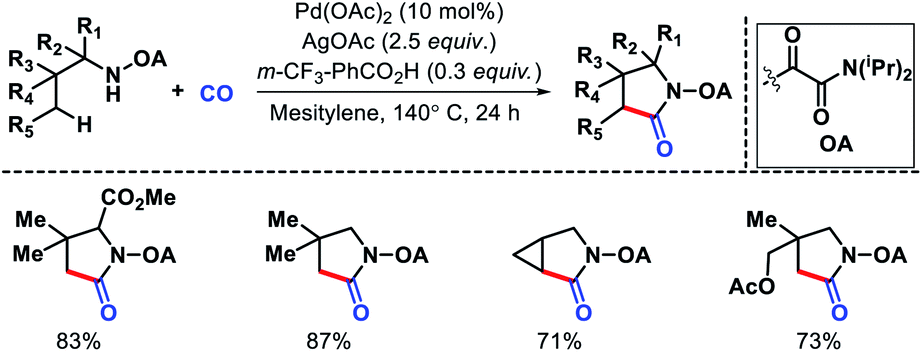
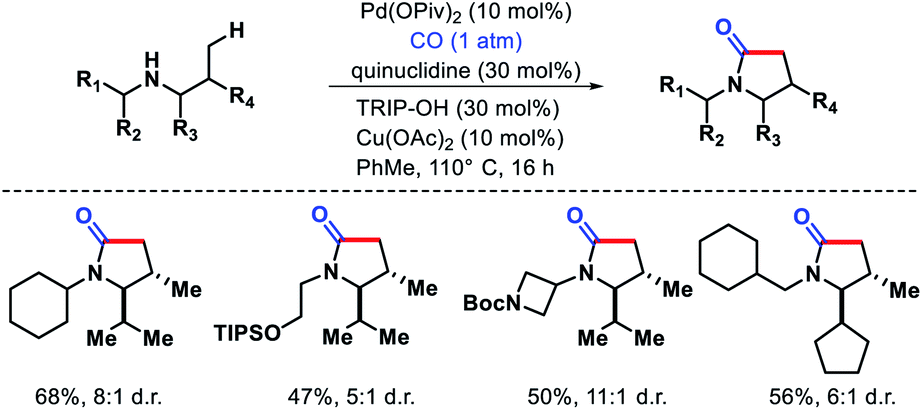
| | Scheme 33 γ-C(sp3)–H carbonylation of secondary alkyl amines to form γ-lactams. | |
2.2 Delta functionalization of aliphatic amines
Functionalization of further distal δ-C(sp3)–H bonds in aliphatic amines present an even greater challenge, as it requires the formation of thermodynamically less favored six-membered metallacycle as an intermediate. For this reason, activation of δ-C(sp3)–H bonds in alkyl amines are still in its infancy with just a few transformations reported till date. Below we will discuss the δ-C(sp3)–H functionalizations of alkyl amines that have been reported using different strategies.
2.2.1 Covalently tethered directing group approach.
2.2.1.1. Arylation.
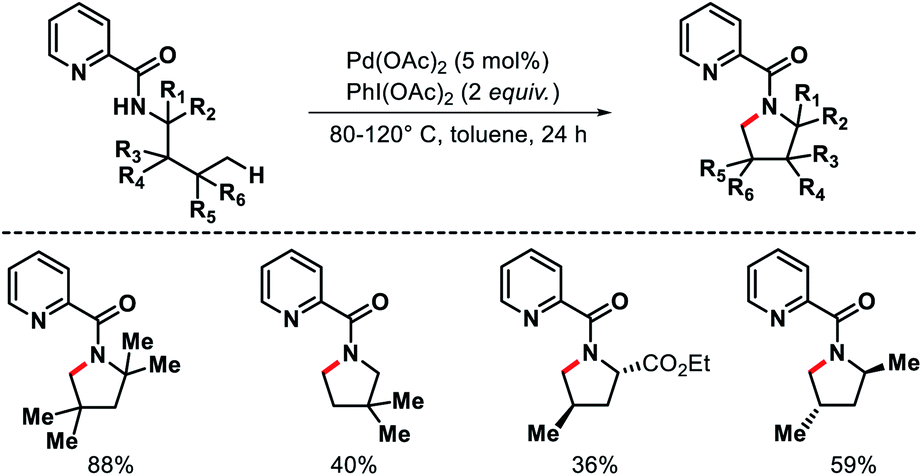
The first report of δ-C(sp3)–H bond activation came from the group of Daugulis in 2012.38 Pyrrolidine derivatives were formed via δ-C–H activation followed by N–H cyclization with the assistance from a covalently attached picolinamide directing group (invented by the same group previously). The reaction required Pd(OAc)2 as a catalyst, PhI(OAc)2 as an oxidant, and toluene as solvent at 80–120 °C (Scheme 34). Only a few examples of δ-C(sp3)–H activation were showcased in the report with mostly δ-C(sp2)–H activation to form indoline derivatives. The aliphatic amines used in the reaction as substrates were mostly biased, where γ-C(sp3)–H bond was either not accessible (γ-quaternary center) or present as γ-methylene center which was more difficult to activate compared to δ-methyl center. Nonetheless, it was the first report for distal δ-C(sp3)–H activation of alkyl amines, which opened a new avenue for further investigation of distal C(sp3)–H functionalization.
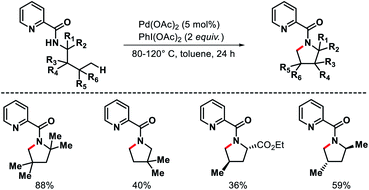 |
| | Scheme 34 Pd catalyzed δ-C(sp3)–H activation of alkyl picolinamides. | |
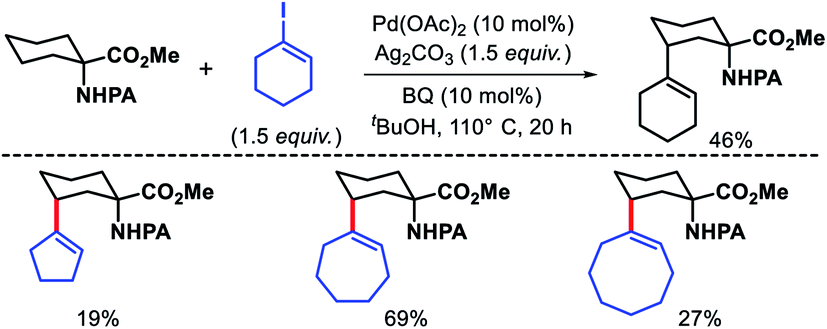
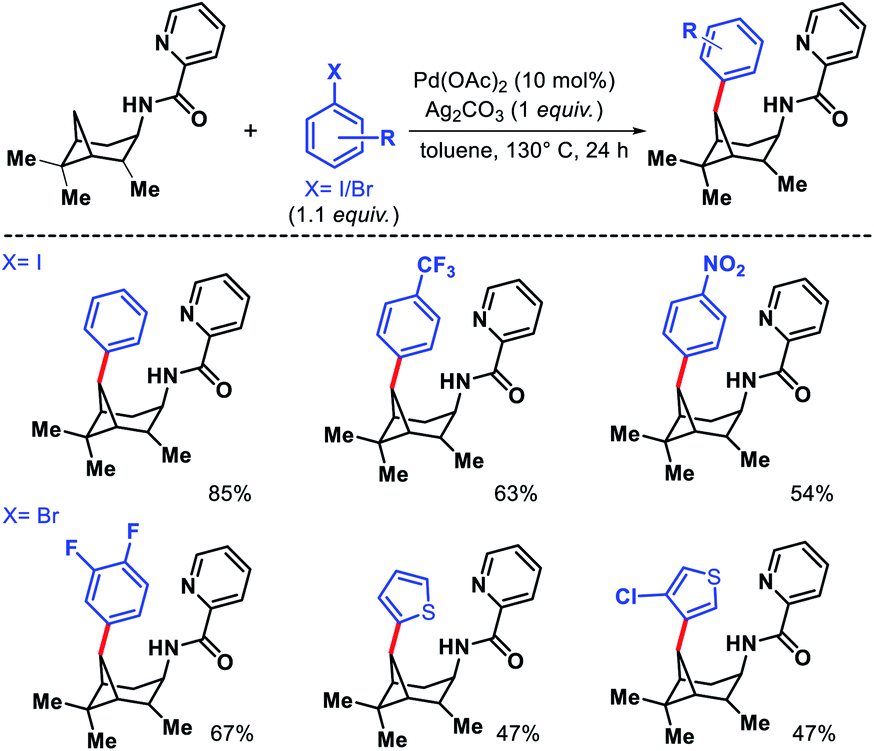
Wang and coworkers outlined δ-C(sp3)–H arylation of 3-pinanamine using the same bi-dentate picolinamide as the directing group (DG).39 The conformational biasness of the substrates makes the γ-C–H bond inaccessible to reach for the directing group and the δ-C–H (methylene center) becomes proximal C–H bond to the DG. For the same strain induced proximity, stereoselective arylation at the δ-methylene center pointing toward the amino group was found. Various aryl iodides as well as aryl bromides were utilized as the arylating partner in the reaction. 2 and 3-bromo thiophenes found to be suitable partners for the reaction but nitrogen containing heterocyclic halides were ineffective (Scheme 35). An easily removable directing group PAr (used by Chen group previously) was employed to get the free amine post arylation.
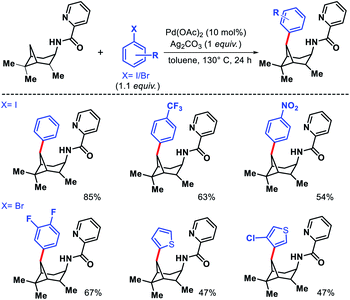 |
| | Scheme 35 Pd catalyzed δ-C(sp3)–H arylation of 3-pinanamine. | |
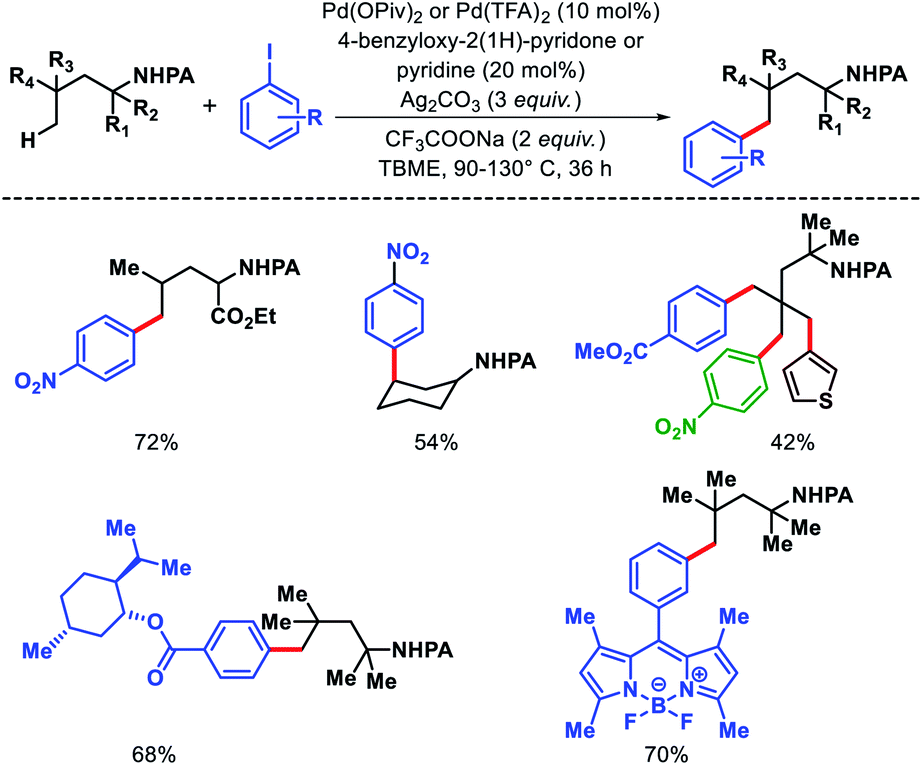
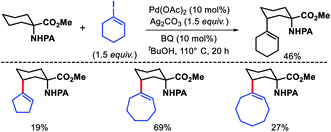
We reported a δ-C(sp3)–H arylation and heteroarylation of amino acids and aliphatic amines aided by picolinamide DG, where the tuning of different ligands played crucial role for different purpose.40 Leucine, homoleucine, bicyclic amines like 3-pinanamine and others were successfully subjected for δ-C–H bond arylation using 4-benzyloxy-2(1H)-pyridone as the ligand. 2,4,4-Trimethylpentane-2-amine was monoarylated at δ position with various aryl and natural products containing complex iodides utilizing pyridine as a ligand (which suppressed diarylation). The same aliphatic amine was triarylated sequentially at δ positions at a slightly higher temperature. A number of heteroaryl iodide was employed as the coupling partner with 1-hydroxy isoquinoline as ligand in the reaction. Notably, iodides derived from fluorogenic BODIPYs were successfully incorporated into δ-C–H bond of aliphatic amines in a ligand free reaction condition (Scheme 36). The mechanistic investigation of the reaction was carried out by experimental and computational studies, which revealed the different mechanisms for different substrates and the role of different ligands.
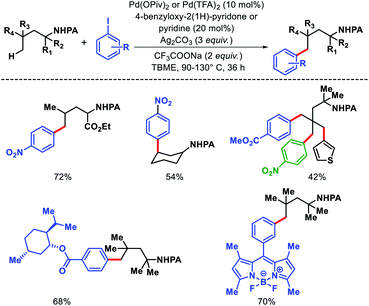 |
| | Scheme 36 Pd catalyzed δ-C(sp3)–H (hetero)arylation of amino acids and aliphatic amines. | |
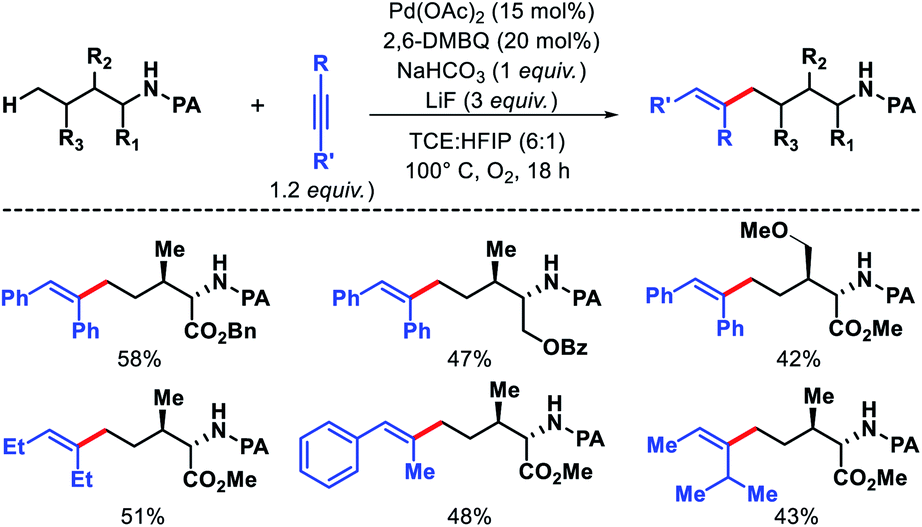
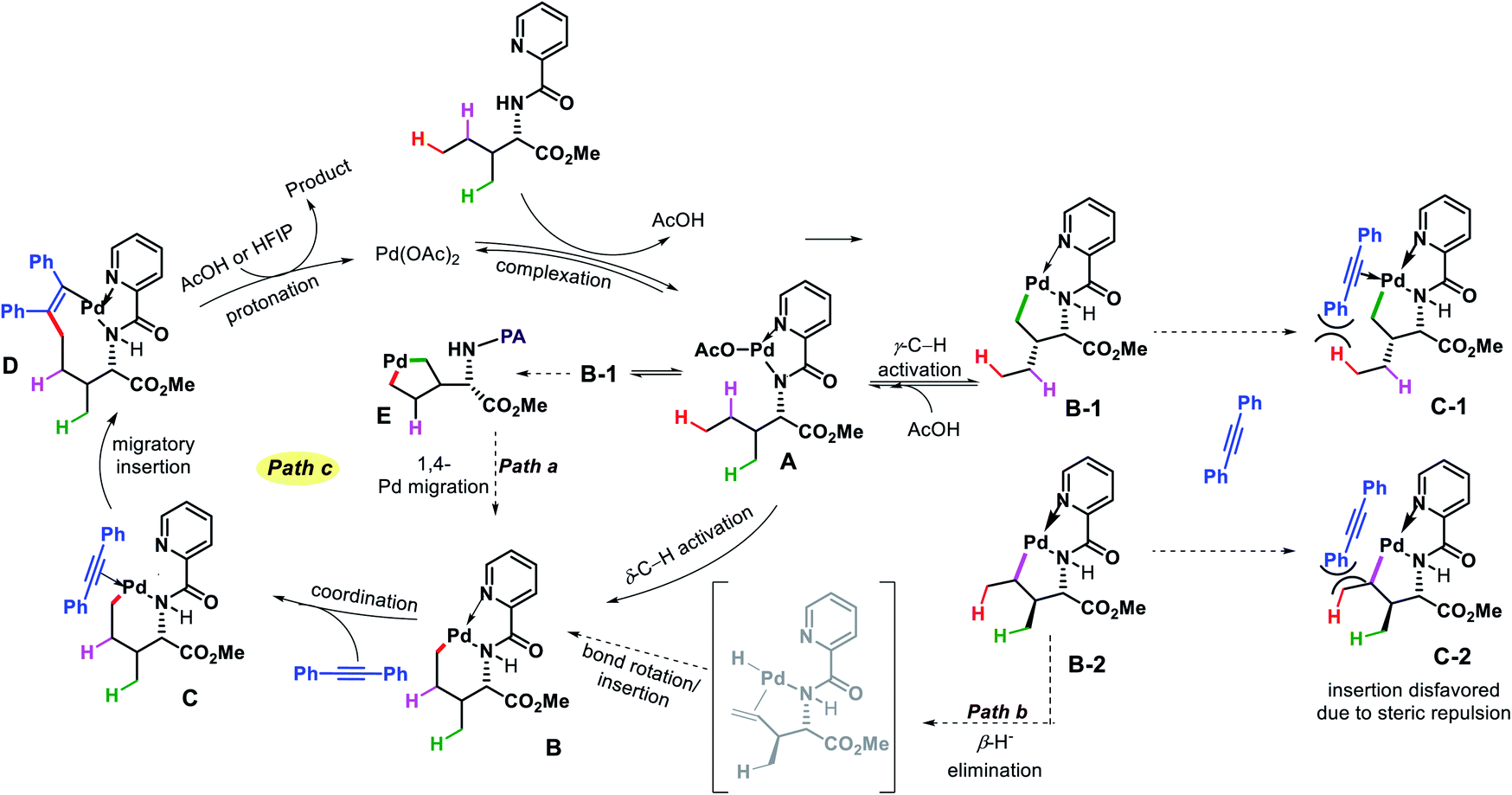
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HFIP (6:1) as solvent, and 1,2-disubstituted alkyne as the alkenylating agent under O2 atmosphere in the reaction. The scope of substrates were pretty narrow with amino acid derivatives such as isoleucine derivatives, norvaline, benzyl, and benzoyl protected amino alcohols were utilized as successful substrates in the reaction. Other aliphatic amines bearing γ-methoxy or δ′-ester were compatible substrates for the reaction. The yields, however, were poor in most of the cases. It is noteworthy that all the substrates used in the reaction were α-substituted and therefore, some biasness is required in the substrates to successfully functionalize the δ-C(sp3)–H bond under this reaction condition. Various symmetrical and non-symmetrical alkynes were well tolerated for this reaction, giving alkenylated products in moderate to good yields. The picolinamide (PA) directing group was removed from the alkenylated product by treatment with PCl5/lutidine, followed by MeOH and aqueous work up. The mechanism was one of the interesting aspects of this reaction that explained why δ-C(sp3)–H alkenylated product formed which goes via a less preferred six membered palladacycle instead of forming γ-C(sp3)–H alkenylated product that required the formation of kinetically favored five membered palladacycle. Based on the deuteration experiment, the γ-C–H bond cleavage was found to be reversible but not the δ-C–H bond cleavage. These results indicate that either δ-C–H bond activation is irreversible or migratory insertion step is faster than the reversal of C–H activation step. No formation of γ-C(sp3)–H product was explained by the authors via Curtin–Hammett principle that, due to steric repulsion it unlikely for palladacycles C-1 and C-2 (Scheme 38) to undergo migratory insertion. While the migratory insertion of C to form an eight membered intermediate D is sterically favored process. Finally, protonation of D lead to the desired product and completes the cycle (Scheme 36, path C). Alternatively, there is a possibility (path A) where reaction could proceed via generation of palladacycle E by 1,4-palladium migration of B-1. A third possibility was also pointed out where β-hydride elimination of B-2, followed by bond rotation/insertion could lead to formation of palladacycle B (path B). However, further mechanistic investigation with deuterated substrate and 1H-NMR study ruled out path A and B.
:HFIP (6:1) as solvent, and 1,2-disubstituted alkyne as the alkenylating agent under O2 atmosphere in the reaction. The scope of substrates were pretty narrow with amino acid derivatives such as isoleucine derivatives, norvaline, benzyl, and benzoyl protected amino alcohols were utilized as successful substrates in the reaction. Other aliphatic amines bearing γ-methoxy or δ′-ester were compatible substrates for the reaction. The yields, however, were poor in most of the cases. It is noteworthy that all the substrates used in the reaction were α-substituted and therefore, some biasness is required in the substrates to successfully functionalize the δ-C(sp3)–H bond under this reaction condition. Various symmetrical and non-symmetrical alkynes were well tolerated for this reaction, giving alkenylated products in moderate to good yields. The picolinamide (PA) directing group was removed from the alkenylated product by treatment with PCl5/lutidine, followed by MeOH and aqueous work up. The mechanism was one of the interesting aspects of this reaction that explained why δ-C(sp3)–H alkenylated product formed which goes via a less preferred six membered palladacycle instead of forming γ-C(sp3)–H alkenylated product that required the formation of kinetically favored five membered palladacycle. Based on the deuteration experiment, the γ-C–H bond cleavage was found to be reversible but not the δ-C–H bond cleavage. These results indicate that either δ-C–H bond activation is irreversible or migratory insertion step is faster than the reversal of C–H activation step. No formation of γ-C(sp3)–H product was explained by the authors via Curtin–Hammett principle that, due to steric repulsion it unlikely for palladacycles C-1 and C-2 (Scheme 38) to undergo migratory insertion. While the migratory insertion of C to form an eight membered intermediate D is sterically favored process. Finally, protonation of D lead to the desired product and completes the cycle (Scheme 36, path C). Alternatively, there is a possibility (path A) where reaction could proceed via generation of palladacycle E by 1,4-palladium migration of B-1. A third possibility was also pointed out where β-hydride elimination of B-2, followed by bond rotation/insertion could lead to formation of palladacycle B (path B). However, further mechanistic investigation with deuterated substrate and 1H-NMR study ruled out path A and B.
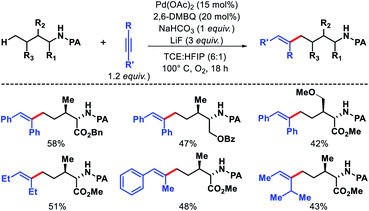 |
| | Scheme 37 Pd catalyzed δ-C(sp3)–H alkenylation of aliphatic amines. | |
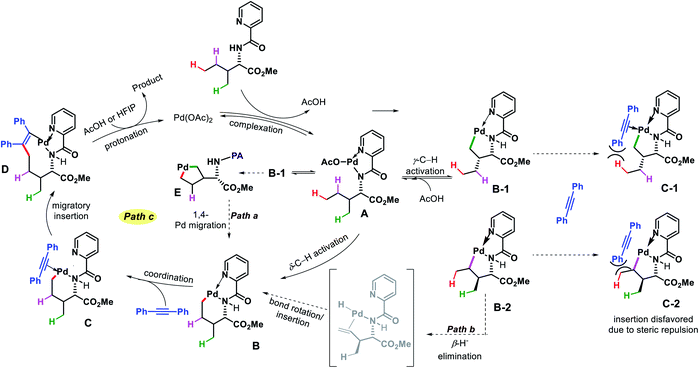 |
| | Scheme 38 Proposed mechanism for selective δ-C(sp3)–H alkenylation. | |
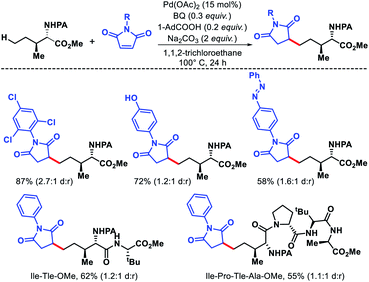 |
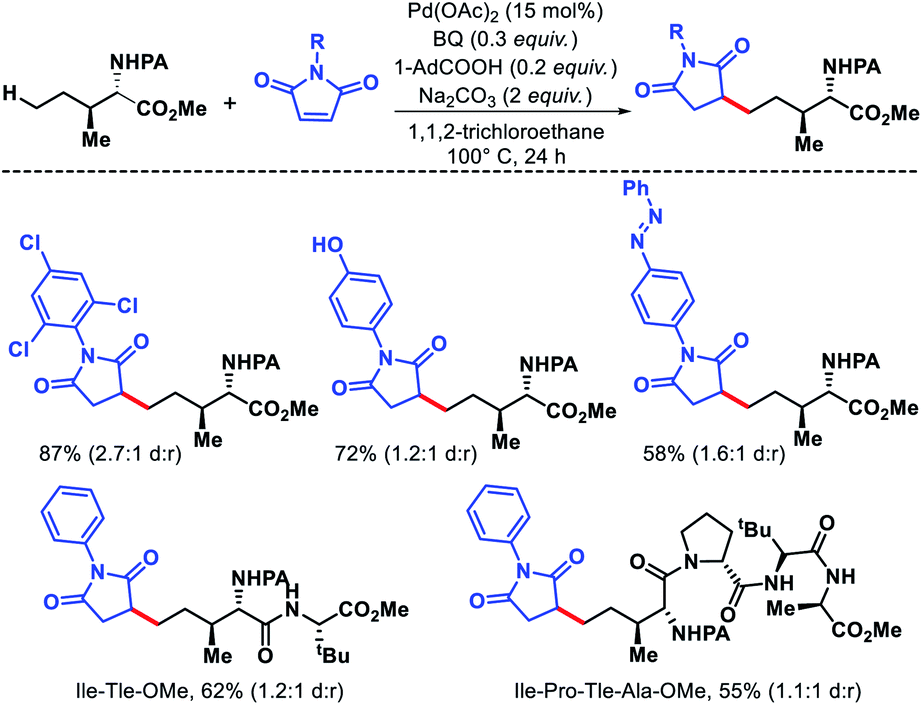
| | Scheme 39 Pd catalyzed δ-C(sp3)–H alkylation of amino acids and peptides. | |
2.2.2. Transient directing group approach.
2.2.2.1. Arylation.
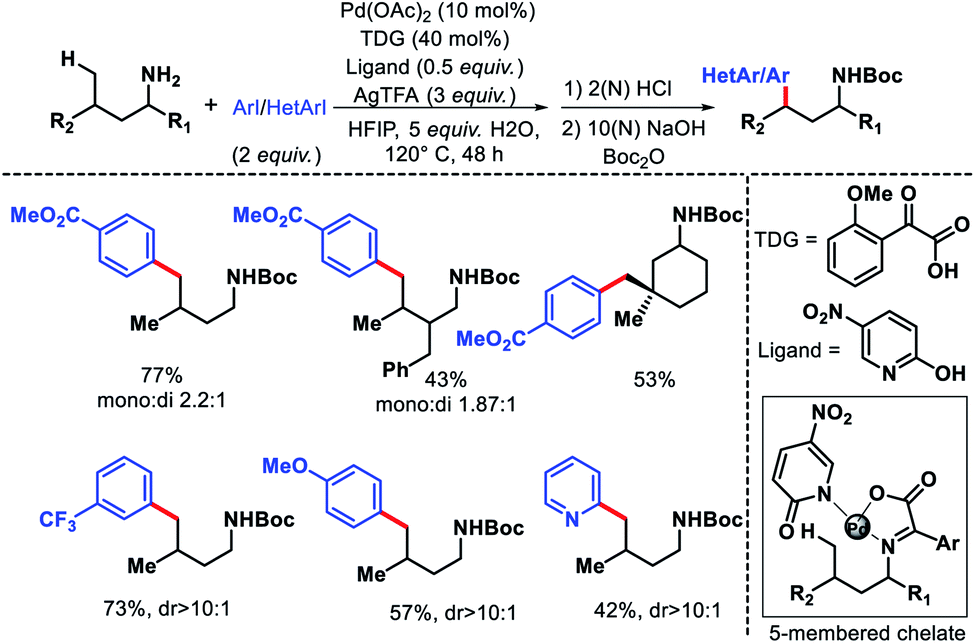
The only report of δ-C(sp3)–H functionalization of alkyl amines via a transient group strategy came from group of Yu in 2018.29 The authors employed glyoxylic acid derivative as the transient directing group (TDG) along with a 5-nitro pyridone as the optimum ligand for δ-C(sp3)–H arylation and heteroarylation of alkyl amines. The TDG coordinate with Pd(II) and substrate to form a five membered chelate and activate the δ-C–H bond. In absence of either TDG or ligand the reaction did not proceed and together a cooperative effort increased the reactivity of the reaction. Although, the exact role the TDG and ligand in increasing the reactivity is not known yet. Further study is required to comprehend their roles. A range of aryl iodides as well as heteroaryl iodides were utilized as the aryl partner in the reaction. The arylated products were isolated as Boc derivative of amines for the ease of isolation. Both cyclic and acyclic primary amines were well compatible in the reaction (Scheme 40). The aliphatic amines employed in this reaction were all γ-substituted (along with α and β-substitutions in some cases), which make the γ-C–H bond less accessible (as it is a methylene center) compared to δ-C–H bond.
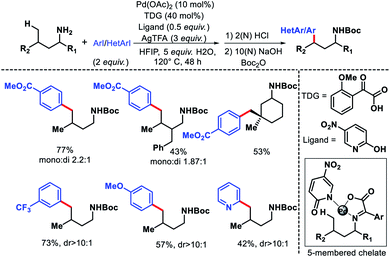 |
| | Scheme 40 δ-C(sp3)–H arylation of aliphatic amines via a cooperative effort of TDG and ligand. | |
2.2.3. Directing group free approach.
2.2.3.1. Arylation.
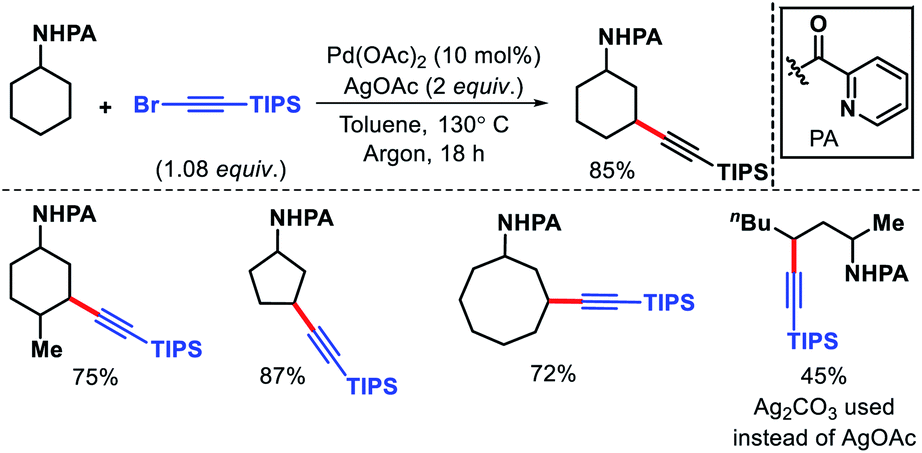
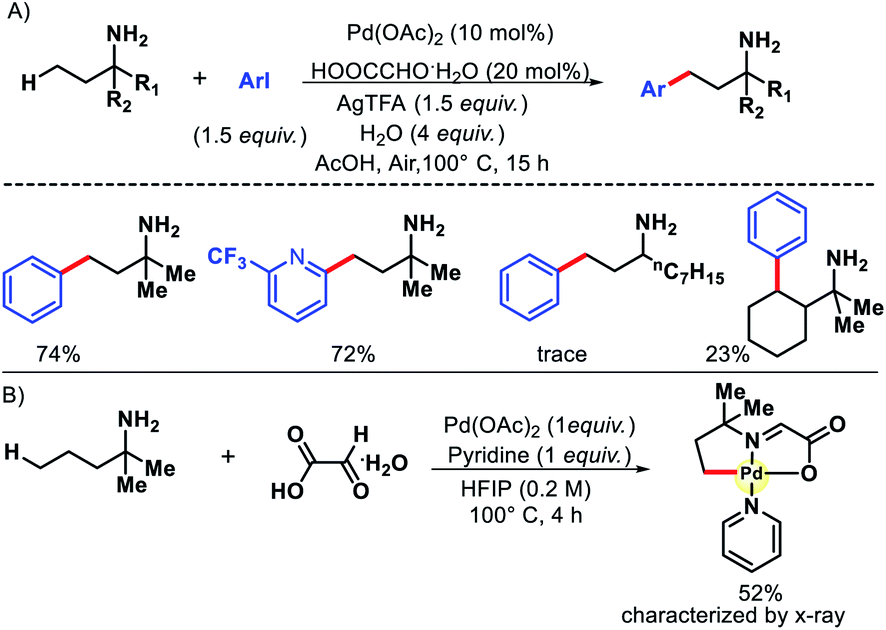
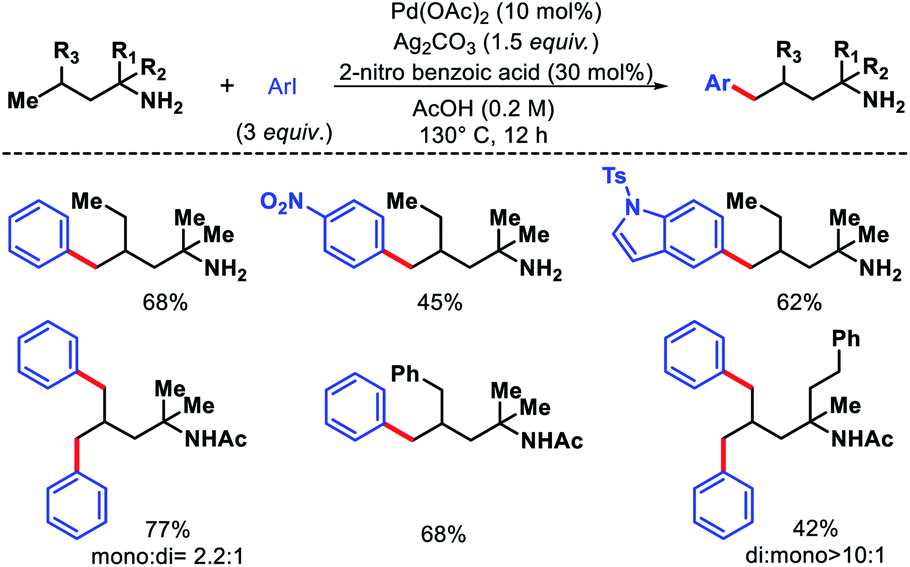
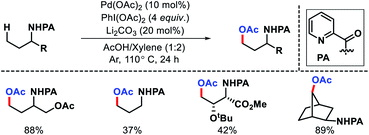
Bannister and colleagues detailed an elegant approach for δ-C(sp3)–H arylation of alkyl amines without using any exogenous directing group.43 A weak acidic solvent AcOH along an acidic additive 2-nitro benzoic acid was crucial for the success of the reaction. The acidic additives aid the dissociation of the unwanted bis-amine complex (of the amine substrates) while not halting coordination to palladium through conversion to ammonium salts. The scope of amine substrates is somewhat limited with α,α-disubstitution essential for providing with Thorpe–Ingold effect. In cases, where two or three δ-C–H bonds are equally accessible, a mixture of mono, di, and tri products formed. An array of aryl iodides and one example of heteroaryl iodide is shown to work well as the aryl partner in this reaction (Scheme 41). Furthermore, control experiments done by the authors showed the crucial role played by the free amino group and acetic acid solvent in the reaction. Overall, it is the first directing group free method for δ-C(sp3)–H functionalization and has opened new possibilities to activate distal C–H bonds of amines in a more efficient manner.
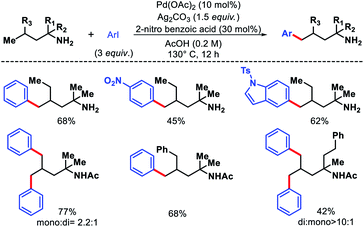 |
| | Scheme 41 Directing group free δ-C(sp3)–H arylation of aliphatic amines. | |
2.3. Gamma functionalization of aliphatic acids
2.3.1. Covalently tethered directing group approach.
2.3.1.1. Arylation.
The first fascinating outcome of γ-C(sp3)–H functionalization was detailed by Corey and coworkers in 2006.44 With the combination of 8-amino quinoline bidentate group and palladium catalysis, γ-C(sp3)–H arylation of amino acids was achieved. Amino acids such as L-valine, L-iso-leucine, and L-tert-leucine derivatives were arylated at the γ-position with good yield. Excellent diastereoselectivity was obtained in the case of L-valine (Scheme 42).
 |
| | Scheme 42 γ-C(sp3)–H arylation of amino acids. | |
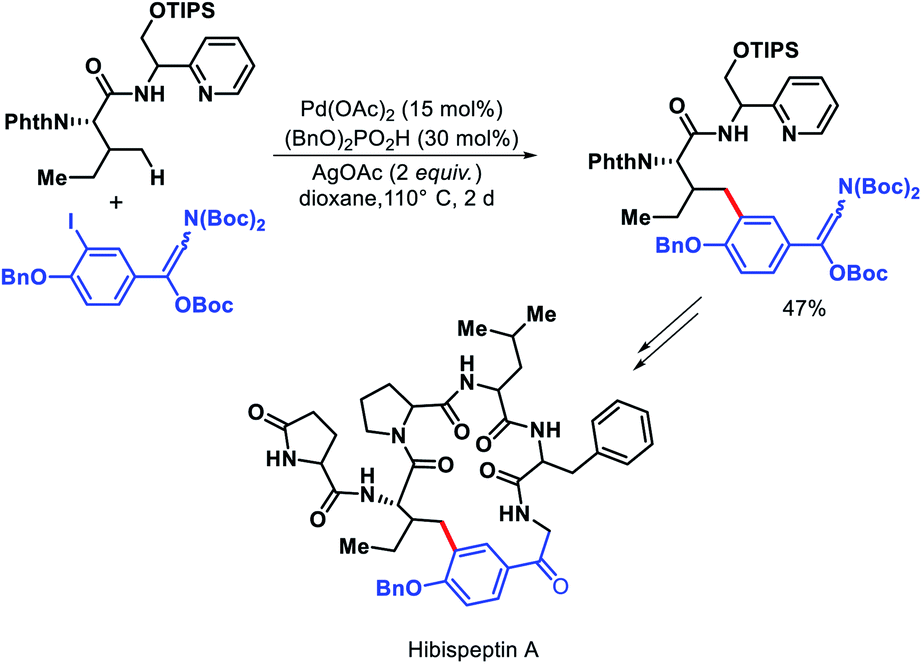
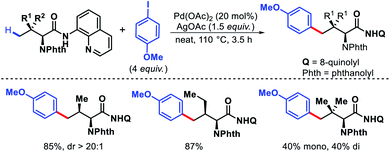
Chen and colleagues explored a number of bidentate directing groups for γ-C(sp3)–H arylation of isoleucine for total synthesis of hibispeptin A (Scheme 43).45 A pyridylmethyl-based directing groups (PR) was found to be the optimum DG which permitted arylation with sterically hindered ortho-substituted aryl iodide and can be removed under mild condition.
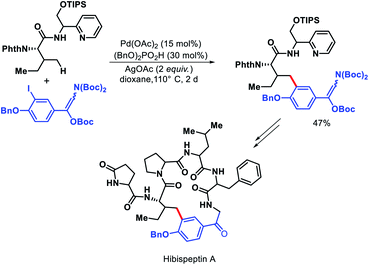 |
| | Scheme 43 Total synthesis of hibispeptin A via γ-C(sp3)–H arylation of isoleucine. | |
Yu group came up with an arylation approach using a highly electron deficient aryl amine directing group and a tricyclic quinoline based ligands.46 Along with simple aliphatic acids containing β-quaternary centers, phthalimide protected amino acids including valine, isoleucine, and tert-leucine were arylated at γ-C–H bond (Scheme 44). Ligands played crucial for enhanced reactivity of the reaction. The directing group was removed either to thioester or carboxylic acid via different procedures.
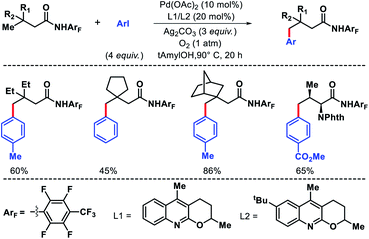 |
| | Scheme 44 Ligand enabled γ-C(sp3)–H arylation of aliphatic acids. | |
We have also contributed in this area. In 2017, we disclosed a protocol for regioselective γ-C(sp3)–H arylation of aliphatic acids using bidentate 8-amino quinoline as a directing group.47 The reaction requires no exogenous ligand to perform which makes it cost-effective for practical applications selective arylation of aliphatic acids and amino acids containing β-quaternary or secondary centers were achieved by employing excess acid derived substrate to inhibit multi arylation by competing for coordination (Scheme 45). A range of aryl iodides with different electronic and steric substitutions were utilized as aryl coupling partner in the reaction. The removal of the directing group performed in presence of excess TsOH and MeOH as solvent.
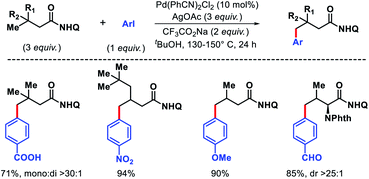 |
| | Scheme 45 A bidentate group assisted γ-C(sp3)–H arylation of aliphatic acids. | |
Later Maiti and coworkers came up with another approach for γ-C(sp3)–H arylation of β-quaternary aliphatic acids.48 Instead of 8-amino quinoline, they developed a new 2-pyridone-based N,O-bidentate directing group that enables mono-selective γ-C(sp3)–H arylation without requiring excess acid substrate (Scheme 46). The usage of aryl iodides were a little limited with electron deficient aryl iodides majorly provided better yields. The removal of the directing group was established through treatment of arylated products with LiOH.
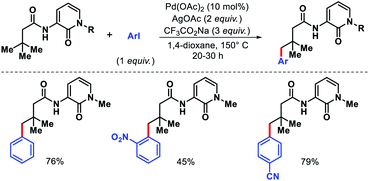 |
| | Scheme 46 Pyridone based directing group for γ-C(sp3)–H arylation of aliphatic acids. | |
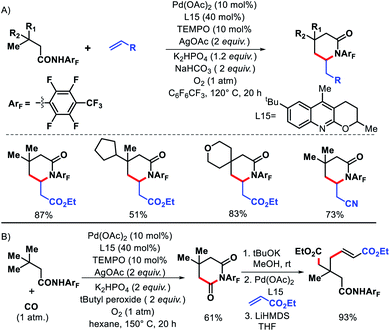 |
| | Scheme 47 (A) γ-C(sp3)–H olefination of aliphatic acids using a combination of highly electron withdrawing amide directing group and tricyclic ligand. (B) Sequential γ-C–H carbonylation/olefination. | |
We also disclosed palladium-catalyzed γ-C(sp3)–H alkenylation of aliphatic acids using 8-amino quinoline as the auxiliary.50 Both acrylates and alkenyl iodides were compatible as coupling partners in the reaction (Scheme 48). 4,4′-Di-tert-butyl-2,2′-bipyridine (dtbpy) was employed as an efficient promoter when acrylates were subjected to reaction conditions. Along with aliphatic acids, α-amino acids such as valine, tert-leucine, and isoleucine were also olefinated at γ-position. A sequential γ-C(sp3)–H arylation/olefination was performed to form highly functionalized aliphatic acid derivative.
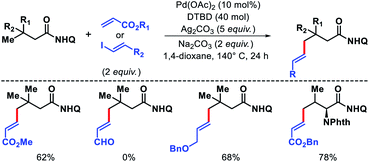 |
| | Scheme 48 8-Amino quinoline (Q) as template for γ-C(sp3)–H olefination of aliphatic and amino acids. | |
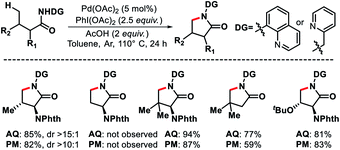 |
| | Scheme 49 Pd catalyzed γ-C(sp3)–H intramolecular amination of aliphatic acid derivatives. | |
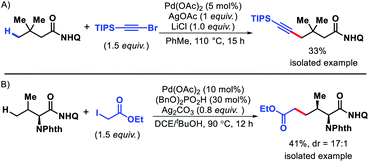 |
| | Scheme 50 Preliminary examples of (A) γ-C(sp3)–H alkynylation and (B) γ-C(sp3)–H alkylation. | |
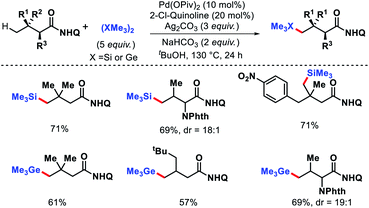 |
| | Scheme 51 Pd catalyzed γ-C(sp3)–H silylation and germanylation of aliphatic acid derivatives. | |
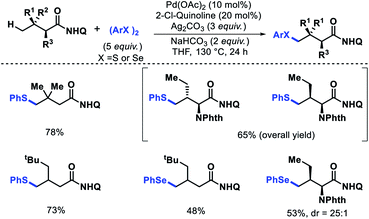 |
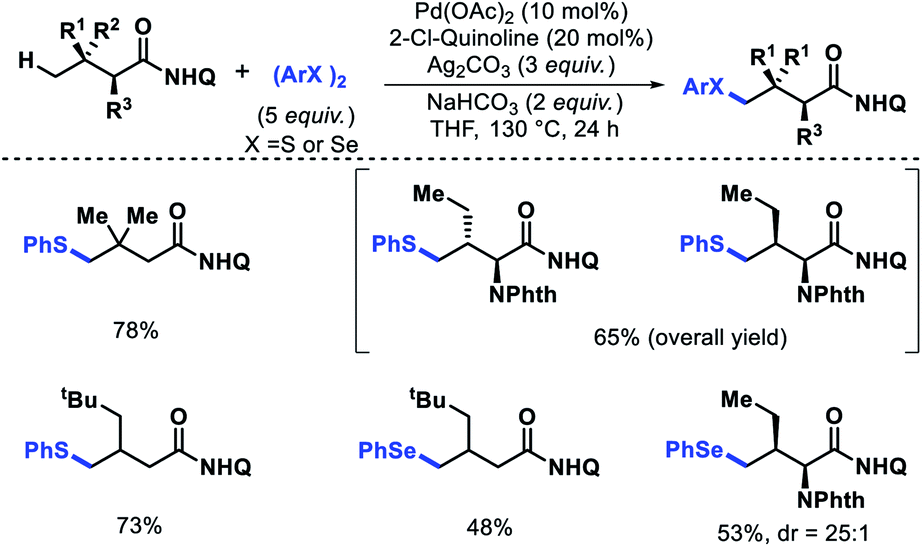
| | Scheme 52 Pd catalyzed γ-C(sp3)–H thioarylation and selenoarylation of aliphatic acid derivatives. | |
Benzothiepinone derivatives were synthesized from thioarylated products in two steps. Most importantly, the protocol enables the synthesis of bioactive chalcogen containing α-amino acids in fewer steps.
2.3.2. Directing group free approach.
2.3.2.1. Arylation.
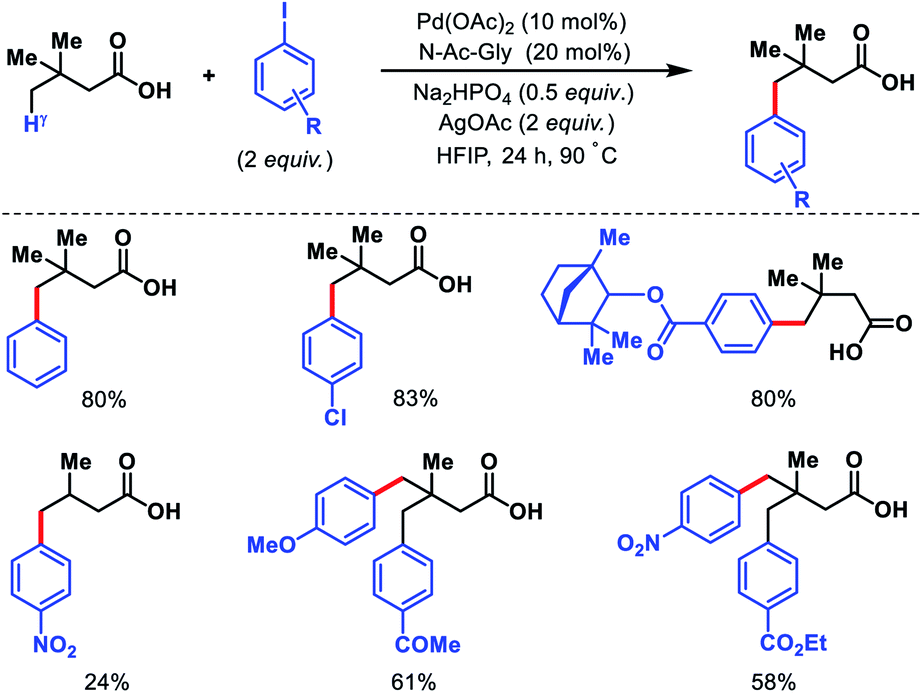
Despite the elegance of above mentioned methods the requirement of install and remove templates reduces the practical usefulness of the reaction. Strategies without directing would be more interesting and practical. Our group has reported the first γ-C(sp3)–H functionalization of free aliphatic carboxylic acids.56 With the aid of a mono-protected amino acid, selective γ-C–H arylation of 3,3-dimethyl butanoic acid detailed. Alkali metal base thought to play a crucial role as it facilitates switching the equilibrium from κ2 to κ1 mode and thus palladium has an opportunity to activate the distal γ-C(sp3)–H bond. Various aryl iodides as well as some bulky natural products containing aryl iodides were utilized as the coupling partner in the reaction (Scheme 53). At a slightly higher temperature, sequential diarylation of the free acid was carried out. C–H activation was found out to be the rate determining step of the reaction via mechanistic experiments.
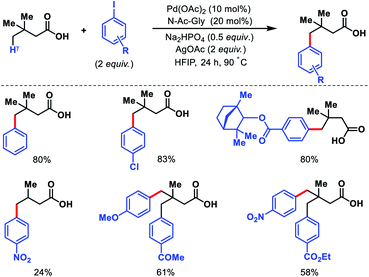 |
| | Scheme 53 Pd catalyzed γ-C(sp3)–H arylation of free aliphatic acids. | |
Other than 3,3-dimethyl butanoic acid, isovaleric acid was found to give γ-C(sp3)–H arylated product, albeit, in poor yield. Nonetheless, butyric acid remained silent under the reaction condition.
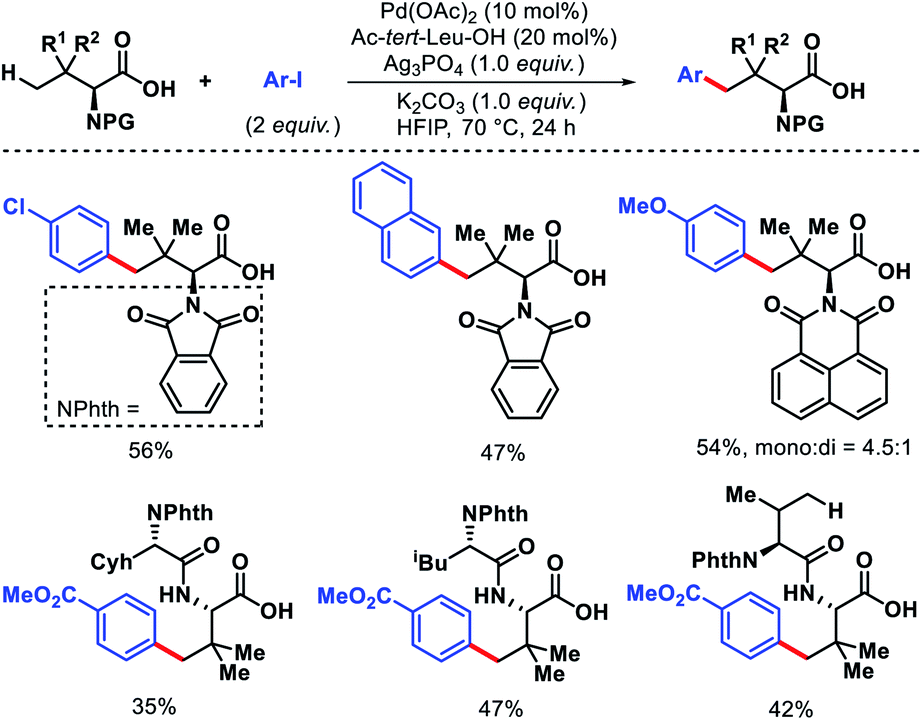
Shi and coworkers have also detailed γ-C(sp3)–H arylation of tert-leucine and its derived peptides without using any exogenous template.57 Acetyl protected tert-leucine has been utilized as ligand in the reaction. Different electronic substitutions on the aryl iodide are well compatible with the reaction. Although, mono and di arylated products formed in most cases but the selectivity of mono:di were moderate to good. Late stage γ-C(sp3)–H arylation of tert-leucine containing di and tri peptides were carried out using directing ability of carboxylate (Scheme 54). It is interesting to note that weakly coordinating carboxylate outcompetes strongly coordinated bidentate DG of the peptide backbone and produced selective γ-C(sp3)–H arylation of tert-leucine residue. Various protecting groups for amine group of tert-leucine were tolerated under the reaction condition. Nonetheless, protecting groups such as Fmoc, Cbz, Boc were incompatible with this protocol.
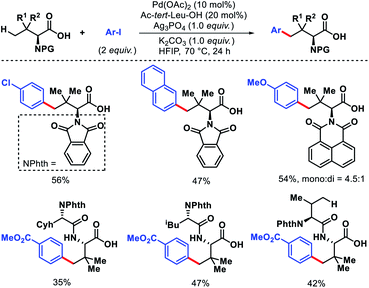 |
| | Scheme 54 Pd catalyzed γ-C(sp3)–H arylation of tert-leucine and its derived peptides. | |
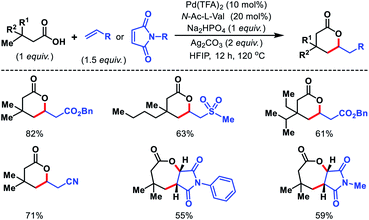 |
| | Scheme 55 Directing group free γ-C(sp3)–H olefination of aliphatic acids. | |
2.4. Gamma functionalization of aliphatic ketones and aldehydes
2.4.1. Arylation.
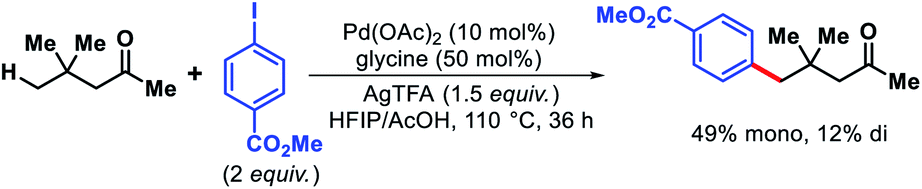
In 2016, Yu and coworkers reported a preliminary example of γ-C(sp3)–H arylation of 4,4-dimethylpentan-2-one to afford 49% monoarylated and 12% diarylated product using glycine as transient directing group (Scheme 56).61
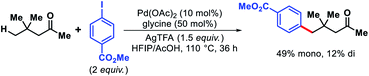 |
| | Scheme 56 Palladium-catalyzed γ-C(sp3)–H arylation of a ketone enabled by transient directing group. | |
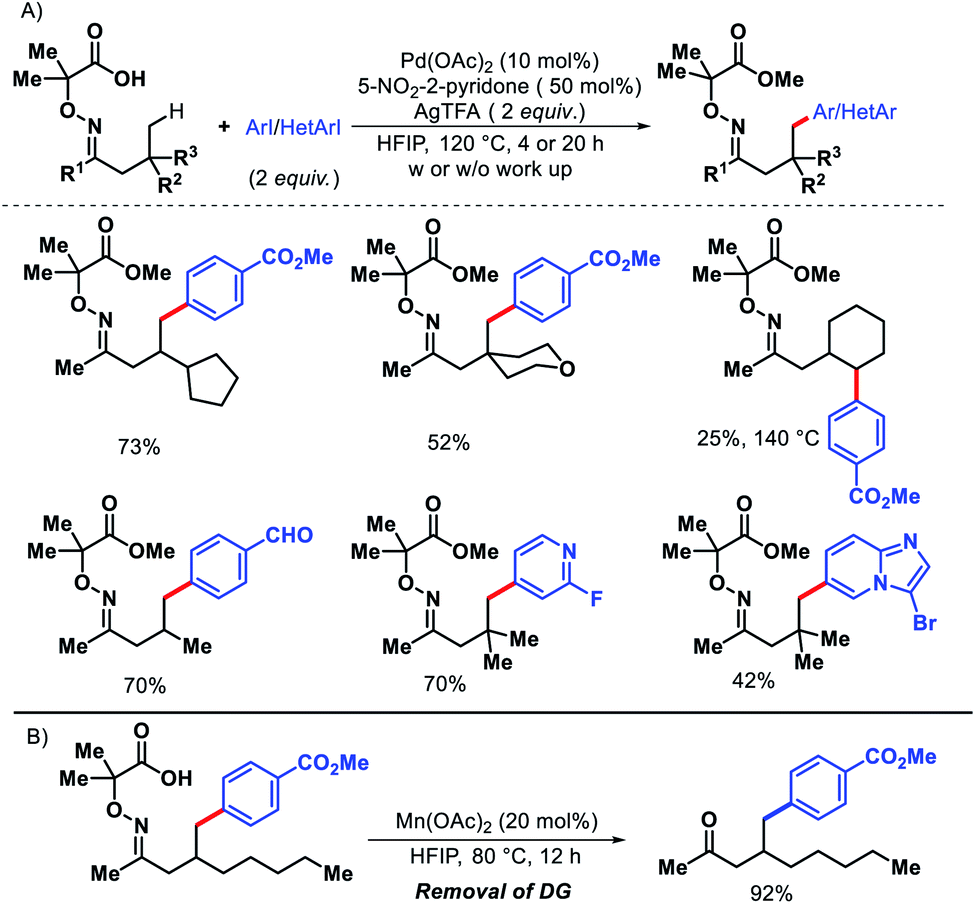
Later in 2018, the same group detailed a directing group assisted approach for γ-C(sp3)–H arylation and heteroarylation of aliphatic ketones.62 An unique template 2,2-dimethyl aminooxyacetic acid was preinstalled in ketones. 5-Trifluoromethyl-2-pyridone found tobe the most effective ligand for increasing yield of the arylated ketones. Aliphatic ketones even without β-quaternary centers were compatible with the reaction. Functional groups in the substrate such as aryl, amino, hydroxyl, ester groups were well tolerated under the reaction. An example of γ-methylene C–H activation was also showcased, albeit in low yield (Scheme 57A). However, diarylated products formed in some cases with prolonged reaction time. Along with aryl groups, various heteroaryl groups were also installed at γ-position of aliphatic ketones. When vinyl iodides were subjected to the reaction, the γ-C(sp3)–H vinylation was also achieved in good yield. Easy removal of the directing group was developed via Mn(II) catalyzed radical based reaction (Scheme 57B).
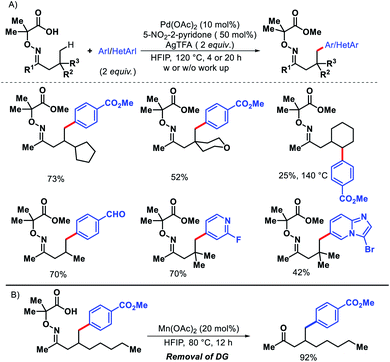 |
| | Scheme 57 (A) Directing group assisted γ-C(sp3)–H arylation and heteroarylation of aliphatic ketones (B) removal of directing group. | |
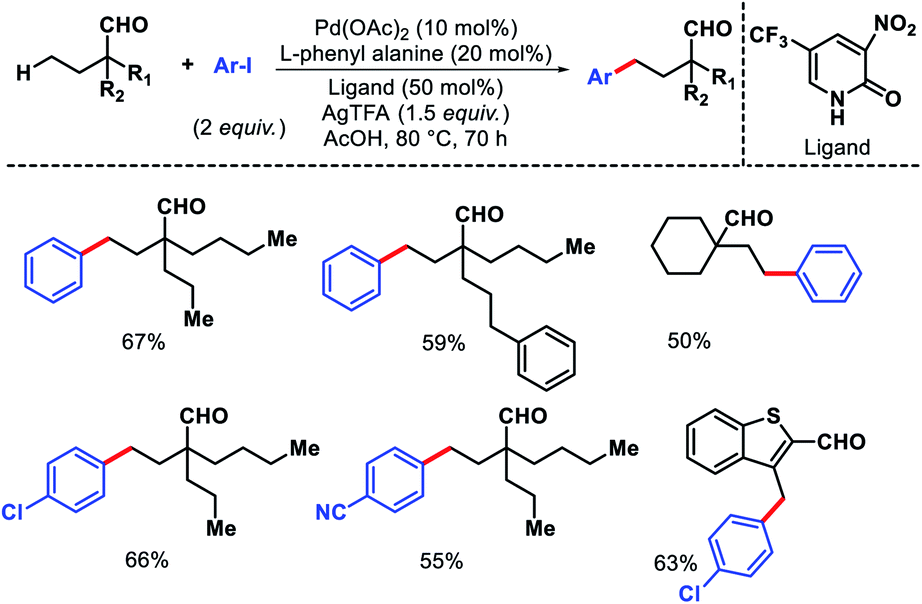
Ge and collaborators reported a Pd(II) catalyzed primary γ-C(sp3)–H arylation of aliphatic aldehydes, which was enabled by the combination of a transient auxiliary (L-phenyl alanine) and an external ligand (3-NO2-5-CF3-2-pyridone).63 A series of α-quaternary aldehydes were arylated at γ-position with diverse iodobenzene derivatives (Scheme 58). Interestingly, heteroaryl aldehydes were also compatible with this protocol. However, when both β and γ-primary C(sp3)–H bonds exist in the substrate, the reaction proceeded preferentially at the β-position. Mechanistic experiments revealed that C–H cleavage is the rate limiting step of the reaction. The authors showed a potential application of the reaction by synthesis of new mechanofluorochromic materials from heteroaryl aldehyde.
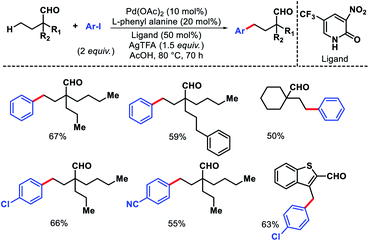 |
| | Scheme 58 Pd catalyzed γ-C(sp3)–H arylation of aliphatic aldehydes. | |
2.4.2. Alkenylation.
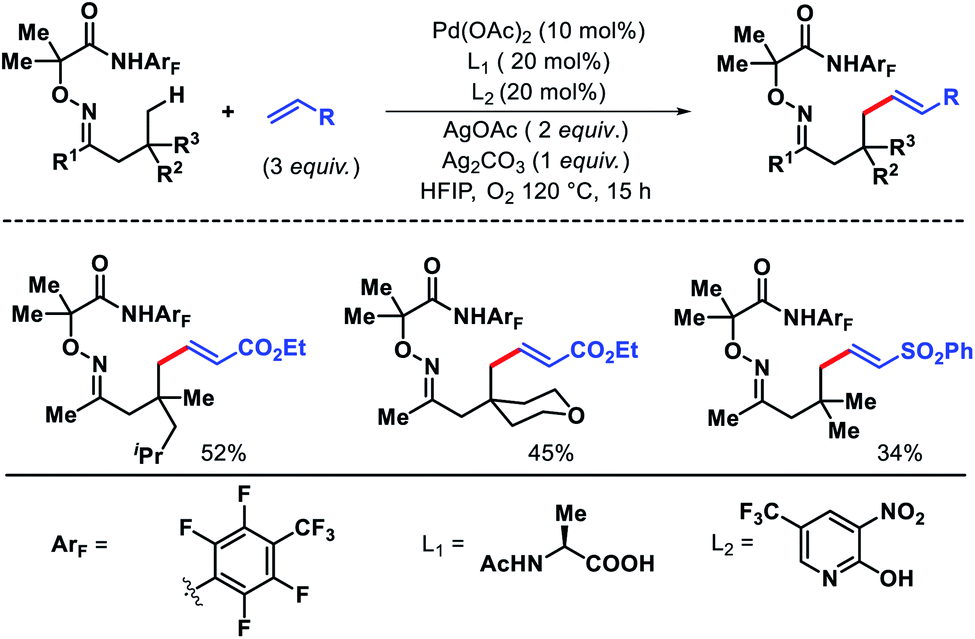
Very recently, Yu group has introduced a new approach for γ-C(sp3)–H olefination of ketones by tuning the directing group and ligands.60 Instead of carboxylic acid (Scheme 53A) they used highly electron deficient amide auxiliary as the template. A combination of mono-protected amino acid and electron withdrawing 2-pyridone ligand was found to be most effective for the reaction. Aliphatic ketones with various β-substituents were well compatible with this protocol. Nonetheless, a β-quaternary center in the ketones was necessary for the reaction to proceed. Activated olefins such as acrylates, complex acrylic acid derivatives, vinyl sulphones, phosphonates were utilized as the olefin partners (Scheme 59). Further, the robustness of the reaction was showcased by scaling it up (2 mmol scale).
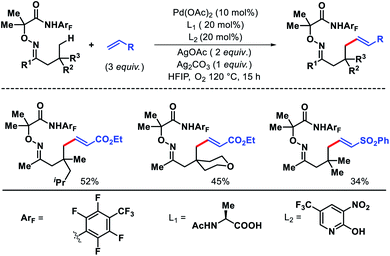 |
| | Scheme 59 Pd catalyzed γ-C(sp3)–H olefination of ketones. | |
2.5. Gamma functionalization of aliphatic alcohols
2.5.1. Acetoxylation.
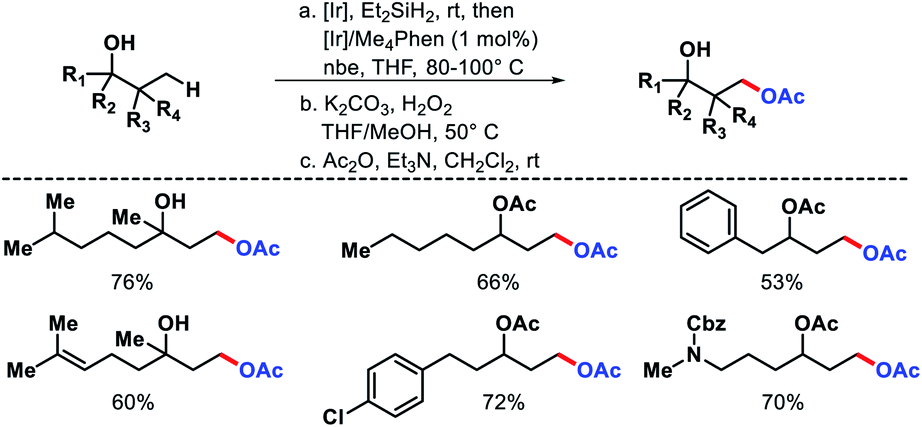
In 2012, Hartwig and coworkers outlined a strategy for distal functionalization of aliphatic alcohols.64 A single dihydridosilane reagent was utilized as the director of the functionalization. It served two purposes (A) attached to oxygen atom of alcohols and formed (hydrido)silyl ether (B) which then directed the γ-C–H bond functionalization. An oxasilolane intermediate was formed due to C–H bond activation which further subjected to Tamao–Fleming oxidation to obtain a 1,3-diol. [Ir(COD)OMe]2 as the catalyst, 3,4,7,8-tetramethyl-1,10-phenanthroline as ligand, and norbornene as hydrogen acceptor employed in the reaction. Both secondary and tertiary alcohols were oxygenated at the γ-position (Scheme 60). Functional groups such as aryl halides, trifluoromethyl, internal double bonds were well compatible with the reaction. Products were isolated as acetyl derivatives for the ease of isolation. It is noteworthy that natural products such as fenchol, camphor, etc. were derivatized via this protocol.
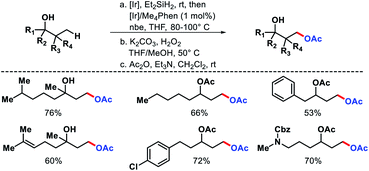 |
| | Scheme 60 Ir catalyzed γ-oxygenation of aliphatic alcohols. | |
2.5.2. Hydroxylation.
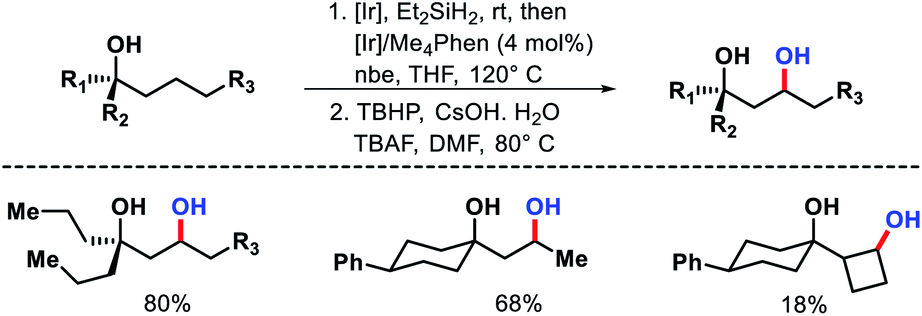
A few years later the same group reported a γ-C(sp3)–H oxygenation procedure for secondary C–H bond.65 The same (hydrido)silyl ether reagent was used for activating the γ-C–H bond of alcohols. The reaction condition for the protocol remained almost the same as in their aforementioned protocol. The temperature was raised to 120 °C and a different condition was applied to oxidize the oxasilolane to 1,3-diol.
A variety of 1,3-diols prepared from tertiary alcohols (Scheme 61). Mechanistic studies suggested C–H cleavage step to be the rate limiting step. The rate of silylation for primary C–H bond was found to be 40–50 times faster than secondary C–H bond.
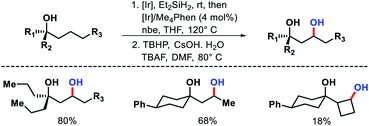 |
| | Scheme 61 γ-C(sp3)–H oxygenation of secondary C–H bonds. | |
2.5.3. Arylation.
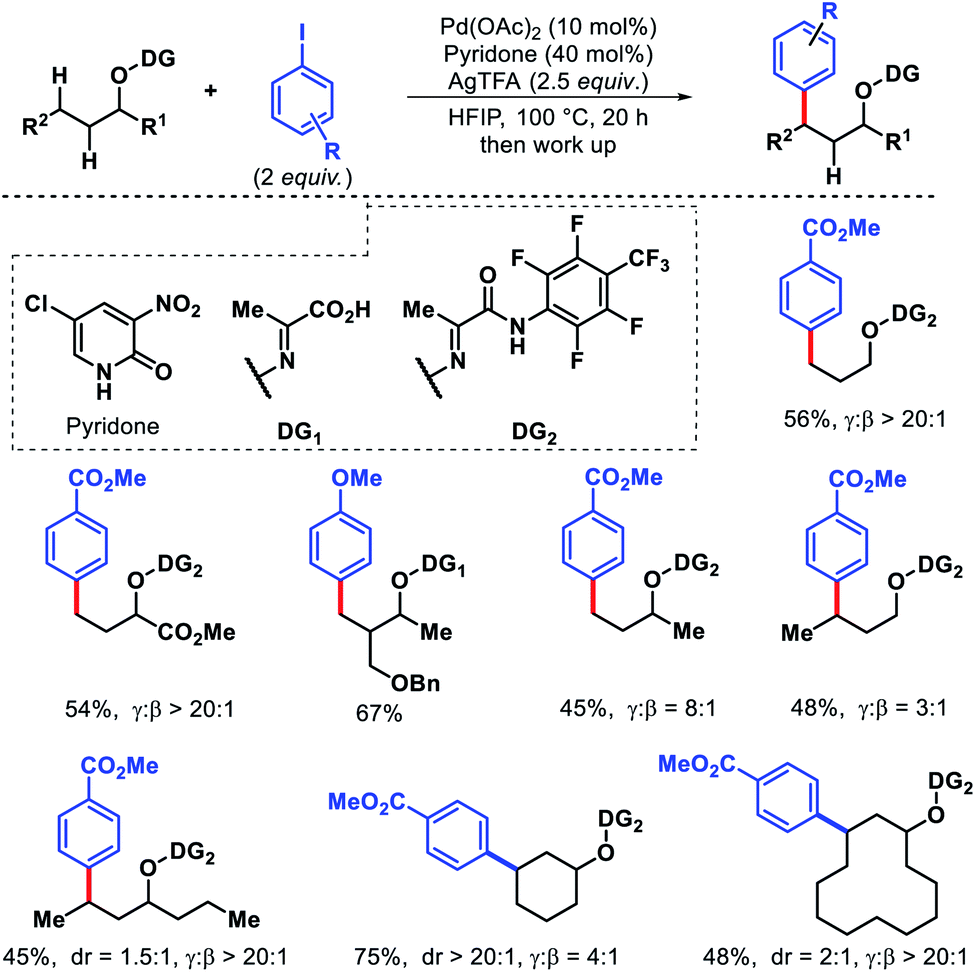
In 2019, Yu and colleagues designed a pyruvic acid-based directing group for remote γ-C(sp3)–H arylation of alcohols.66 3-NO2-5-Cl-2-pyridone ligand was the most effective ligand for this conversion. Various aliphatic alcohols containing β-C(sp3)–H bonds were selectively arylated at γ-position, regardless of whether the γ-C–H bonds were primary or the less reactive secondary. The unconventional reactivity was attributed to the increased ring strain of the bicyclic palladium intermediate. Due to the presence of multiple unsaturated bonds in the directing group, the conventional 5,5-bicyclic palladium intermediate became too high, leaving the 5,6-bicyclic palladium intermediate favorable. The protocol worked with a wide range of functional groups in the alcohol. Various aryl iodides were also compatible with the reaction (Scheme 62).
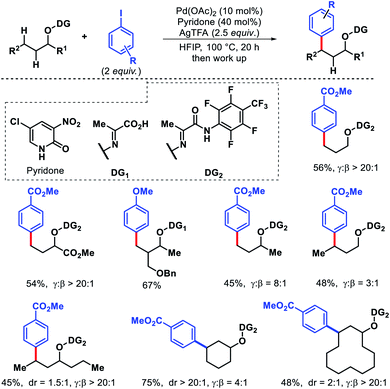 |
| | Scheme 62 Directing group assisted γ-C(sp3)–H arylation of alcohols in presence of β-C(sp3)–H centers. | |
2.5.4. Carbonylation.
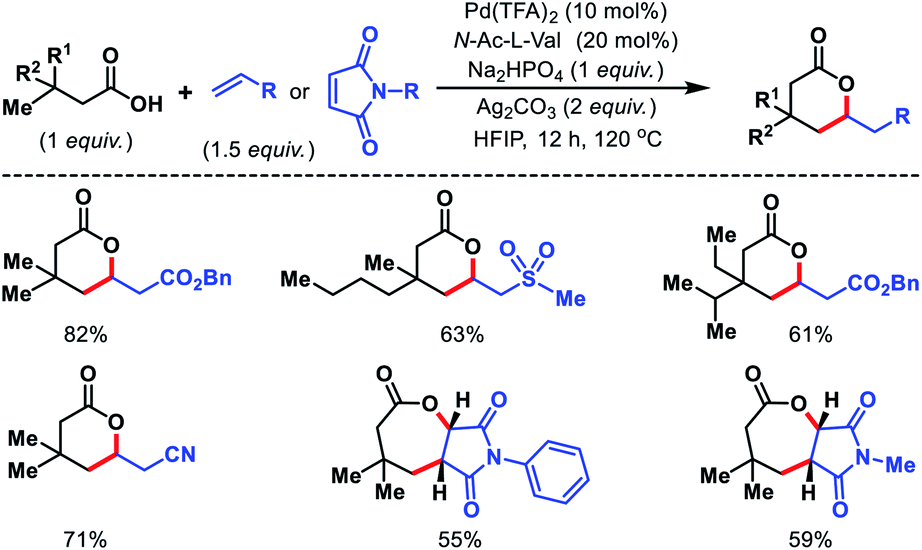
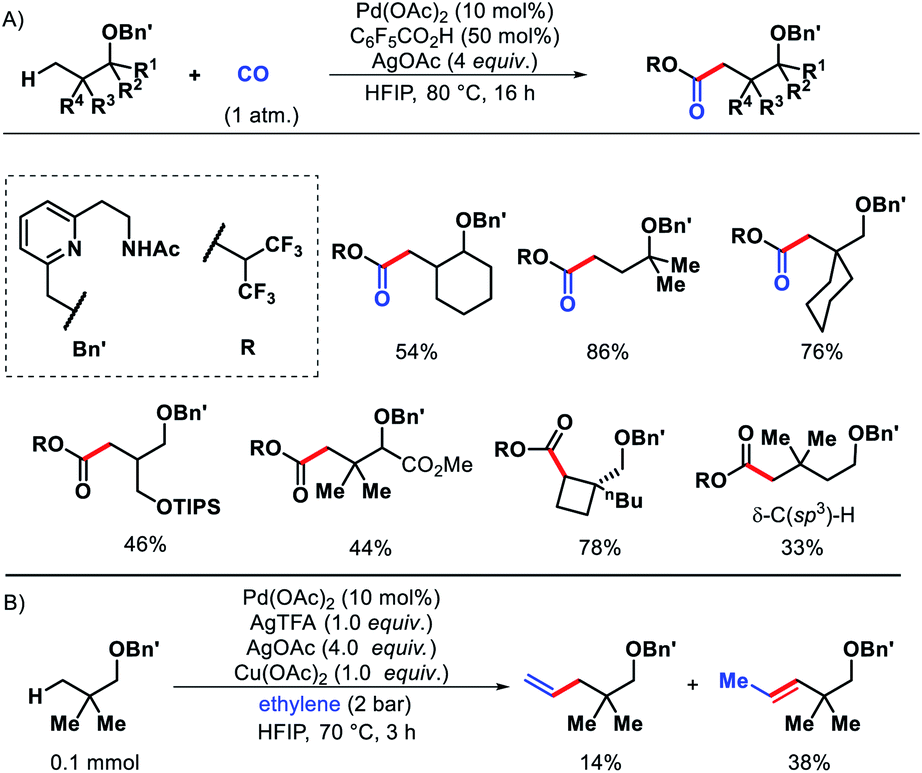
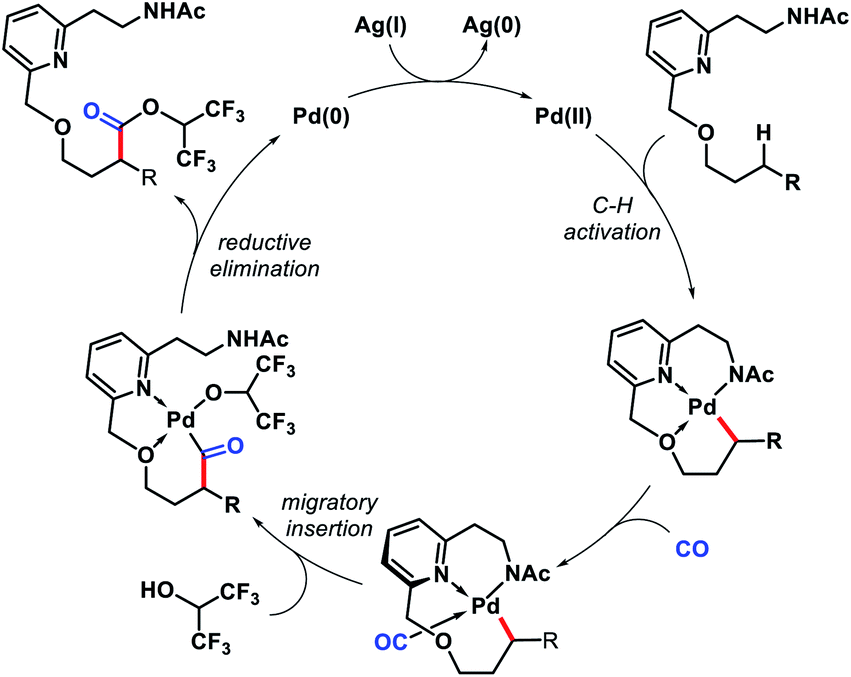
Later, Yu and co-workers designed a tridentate directing group for palladium-catalyzed C(sp3)–H functionalization of alcohols.67 A hemilabile directing group was utilized which exploits chelation of a benzyl ether moiety to direct distal carbonylation. Primary, secondary, and tertiary all kinds of alcohols were carbonylated at γ-position with moderate to high yield in some cases. Methylene γ-C–H bond of cyclopropane or cyclobutane substrates were also converted to esters (Scheme 63A). δ-C(sp3)–H carbonylation of a γ-quaternary center containing substrate was achieved in the same reaction condition. A single example of ethylene insertion into the γ-C–H bond was showcased (Scheme 63B). During the reaction process, the hemilabile ether moiety assists the γ-C(sp3)–H activation and then dissociates to allow other reaction partners to bind onto the C–H cyclopalladation intermediates (Scheme 64).
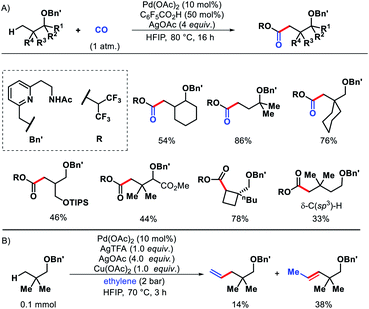 |
| | Scheme 63 (A) A hemilabile directing group promoted γ and δ-C(sp3)–H carbonylation of alcohols (B) ethylene insertion into γ-C–H bond of aliphatic alcohol. | |
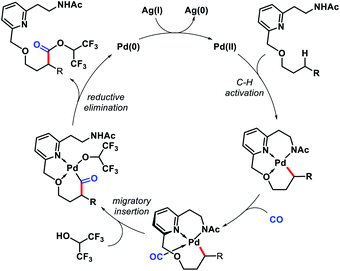 |
| | Scheme 64 Catalytic cycle for γ-C(sp3)–H carbonylation. | |
2.6. Gamma functionalization of aliphatic thiols
2.6.1. Arylation.
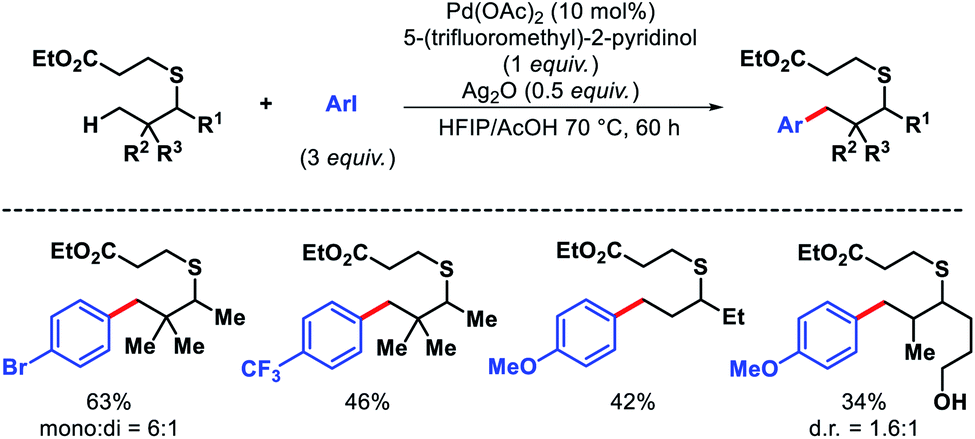
The first distal functionalization of thiols was reported by Dong and coworkers.68 The catalytic poison ability of sulphur along with its high reactivity oxidants and electrophiles make thiols extremely difficult substrates for carrying out C–H functionalization. Dong group masked thiols as thioethers that can be removed easily after functionalization. Aliphatic thiol containing a β-quaternary center was arylated with aryl iodides having different electronic properties. Less rigid thiols gave diminished yields (Scheme 65). Nonetheless, primary thiols and secondary C–H bonds were not reactive under this protocol.
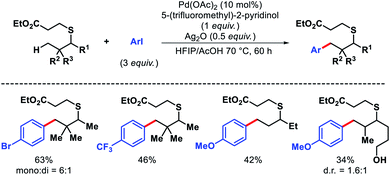 |
| | Scheme 65 Pd catalyzed γ-C(sp3)–H arylation of aliphatic thiol. | |
3. Non-directed C(sp3)–H functionalizations
3.1 Metal catalyzed radical initiated strategies
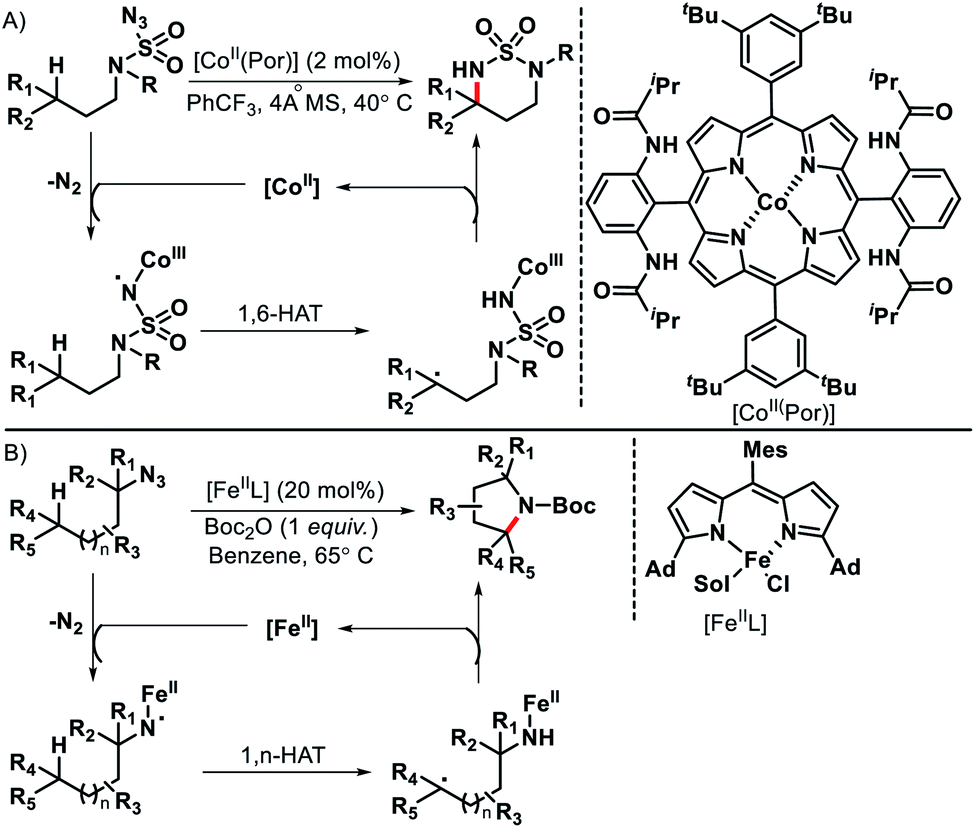
Although radicals are known for their unpredictable behavior, if generated in a controlled manner they can be utilized for selective C–H functionalization. Radical initiated distal C(sp3)–H functionalization has benifitted greatly by the development of transition metal, photoredox, and electrochemical promoted single electron transfer processes. For convenience, we are not going to discuss photoredox or electrochemical promoted radical processes in this review. Separate reviews are available on this topic.69 We would focus mainly on just transition metal catalyzed radical initiated distal C–H functionalizations. Single electron reduction (SET) of organiz azides could lead to metal coordinated N-centered radicals. The formed N-centered radical then undergo 1,n-hydrogen atom transfer to provide remote C(sp3)–H functionalization. Zhang70 and Betley71 group reported Co(II) and Fe(II) catalyzed C(sp3)–H amination to generate azaheterocycles (Schemes 66A and B). The reaction proceeds via (1) single electron reduction of azides with metal catalyst to form metal-imidoradical along with elimination of N2 (2) then intramolecular hydrogen abastraction via a 1,n-hydrogen atom transfer process (3) Finally, radical recombination to provide N-heterocyclic products and release of metal catalysis.
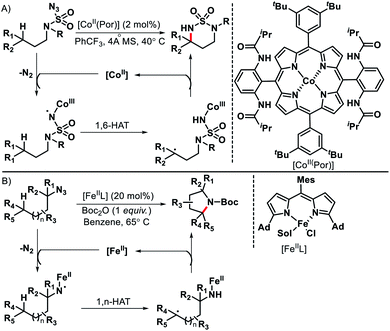 |
| | Scheme 66 (A) Co(II) catalyzed remote C–H amination of organic azides (B) Fe(II) catalyzed intramolecular amination with azides. | |
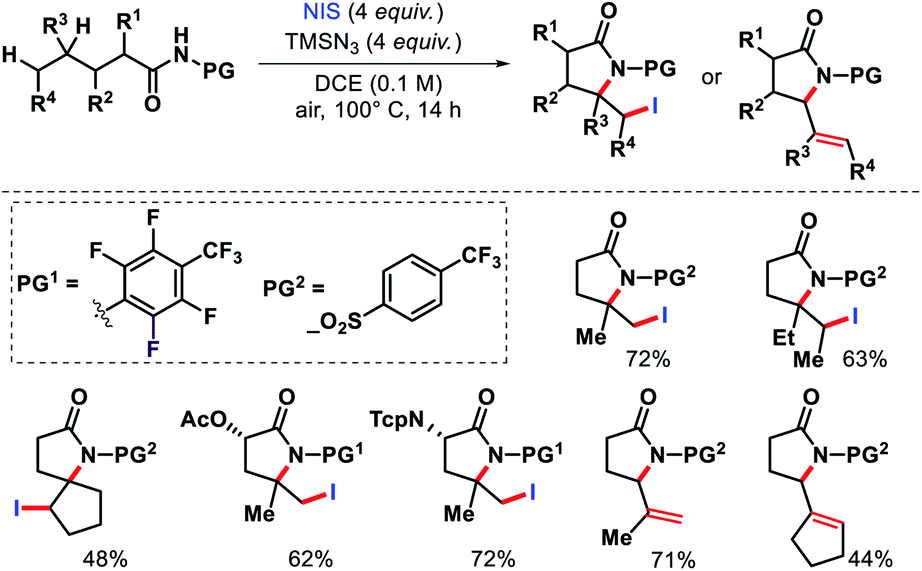
In 2015, Yu and colleagues detailed a selective γ and δ-lactamization to form highly functionalized lactams from aliphatic acids via radical 1,5 and 1,6-H abstraction.72 The highly electron deficient N-heptafluorotolylamine and N-para-trifluoromethylsulphonylamine were used as the protecting groups. A combination of PhI(OAc)2 and I2 facilitated the lactamization process. Further, N-iodosuccinimide and TMSN3 were utilized to intercept the γ-carbon centered radical prior to cyclization. Aliphatic amides that contained secondary or tertiary γ-C–H bond and primary or secondary δ-C–H bonds generated δ-iodo-γ-lactams. But amides with tertiary C–H bond provided δ,ε-dehydrogenated-γ-lactams (Scheme 67).
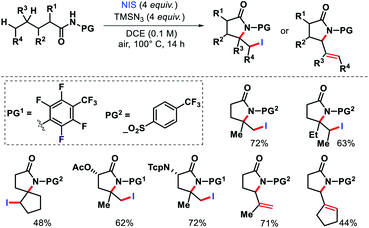 |
| | Scheme 67 γ,δ,ε-C(sp3)–H functionalization via radical 1,5 and 1,6-H abstraction. | |
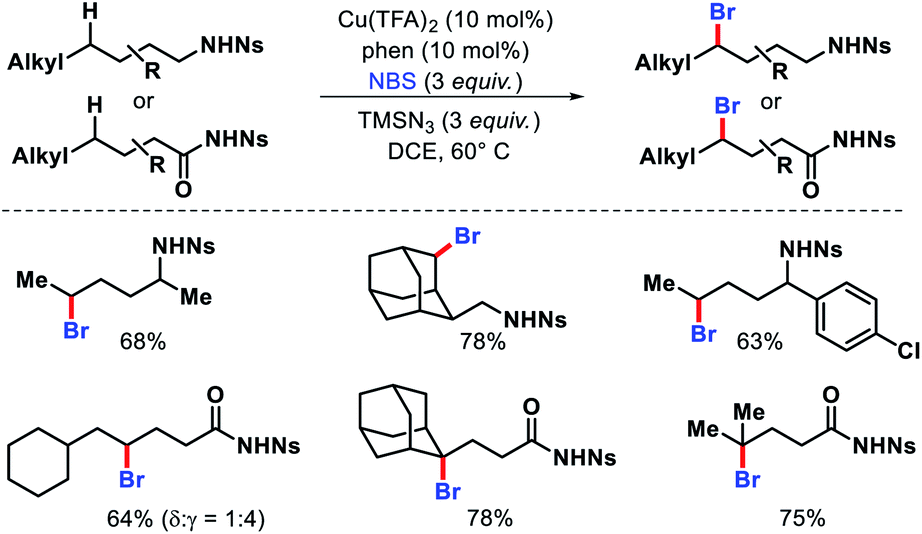
The same group reported distal bromination of nosyl protected aliphatic amines and amides via a radical approach.73 γ-Methylene C(sp3)–H bonds of amides and δ-methylene C(sp3)–H bonds of alkyl amines were brominated using NBS as brominating source (Scheme 68). CuII/phenanthroline complex was utilized as the optimum catalyst in the reaction. Although, 1,5-H abstraction is generally the favoured pathway for the reaction, in some substrates steric effects lead to 1,6-H abstraction pathway for distal radical formation.
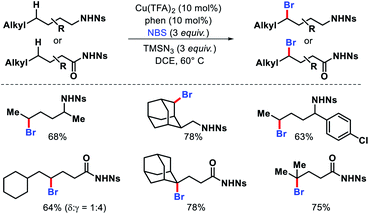 |
| | Scheme 68 Distal bromination of aliphatic amides and amines via a radical strategy. | |
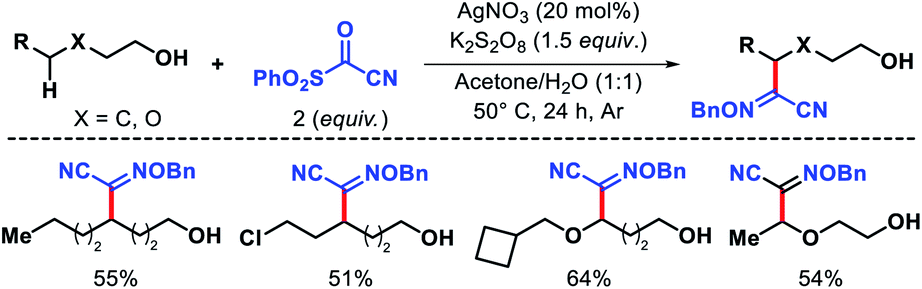
Later Zhu and coworkers developed a similar protocol for arylation of N-fluorocarboxamides and N-fluorosulphonamides.74 In 2018, Jiao group reported an elegant Ag catalyzed protocol for selective δ-functionalization of aliphatic alcohols.75 Generating alkoxy radicals from free alcohol via O–H bond hemolytic cleavage is extremely challenging due to its bond energy. AgNO3 was successfully utilized to generate alkoxy radical along with K2S2O8 as the oxidant. The reaction was conducted in acetone and water as solvents. The reaction undergoes an alkoxy radical mediated 1,5-hydrogen atom abstraction, following which a sulphonyl oxime ether reagent trapped the radical. Various primary and secondary alcohols were afforded δ-functionalized products with moderate to good yields and excellent chemoselectivity (Scheme 69). β-Substituted alcohols were unsuccessful under this reaction condition due to a β-scission process.
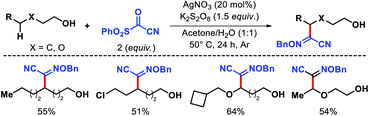 |
| | Scheme 69 Ag catalyzed δ-functionalization of aliphatic alcohols. | |
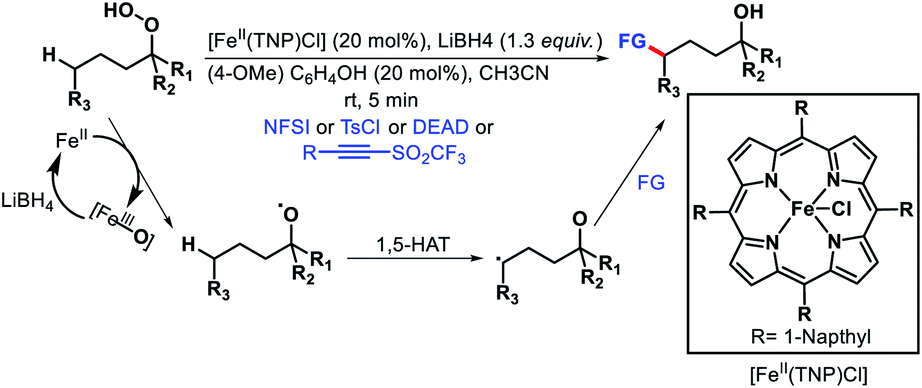
Reduction O–O bond in alkyl hydroperoxides by a low valent metal is another way to generate oxygen radical. Recently, Liu and coworkers developed an Fe(II) catalyzed remote functionalization of inert methylene and methine C–H bonds guided by an alkoxy radical (Scheme 70).76 The protocol enabled site-selective aliphatic fluorination, chlorination, amination, and alkynylation of varied primary, secondary, and tertiary hydroperoxides with excellent functional group tolerance. Fe(II) catalyst mediated one electron reduction affords an alkoxy radical that undergoes a 1,5-HAT process to generate an alkyl radical. Finally, the alkyl radical combines with various radical acceptors to provide the final product. Several other reports for radical promoted distal fluorination of alkyl bonds have been reported very recently.77
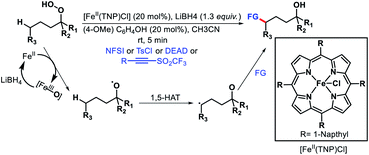 |
| | Scheme 70 Fe(II) catalyzed δ-functionalization of alkyl hydrperoxides. | |
3.2. Catalyst guided C–H functionalization processes
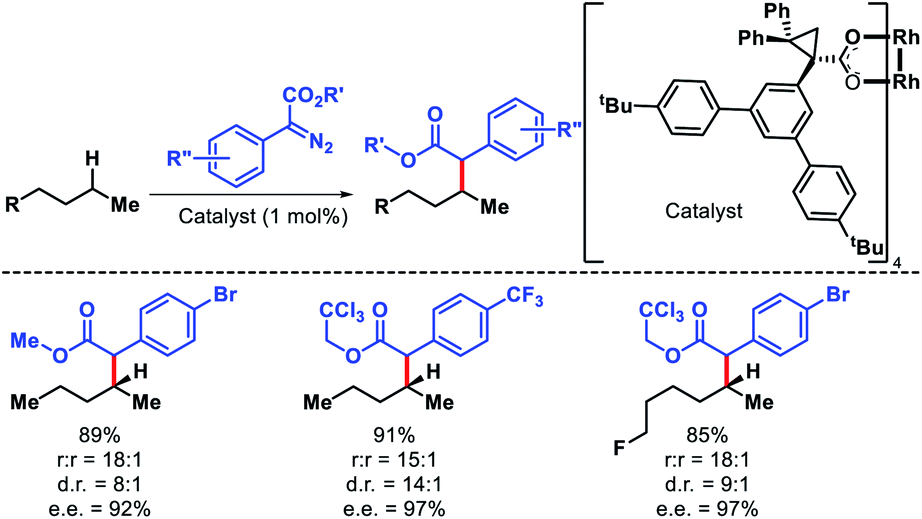
The strategies discussed so far in this perspective have shown a trend that it is easiest to activate a primary C(sp3)–H bonds, followed by secondary and tertiary C–H bonds, which is true sterically. However, electronically a tertiary C–H bond should be easiest to functionalize than secondary or primary. Carbenes are traditionally used for C–H functionalization by various researchers for C–C bond formation. Davies and coworkers took carbene chemistry for C–H functionalization one step forward via a combination of sterically demanding dirhodium catalyst and donor/acceptor carbenes. By altering the nature of the catalyst site selectivity of different C–H bonds were dictated. Dirhodium catalyst with sterically demanding tricyclopropanecarboxylate ligand was employed to favour functionalization at most accessible secondary C–H bond with excellent regioselectivity.78 Simple alkyl chain like n-pentane was functionalized at C2 position with excellent regioselectivity, diastereoselectivity, and enantioselectivity (Scheme 71).
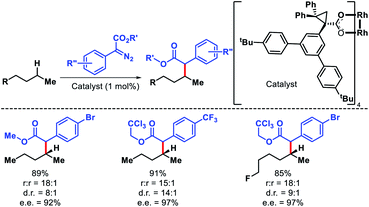 |
| | Scheme 71 A dirhodium catalyst controlled secondary C–H bond functionalization of alkanes and alkyl halides. | |
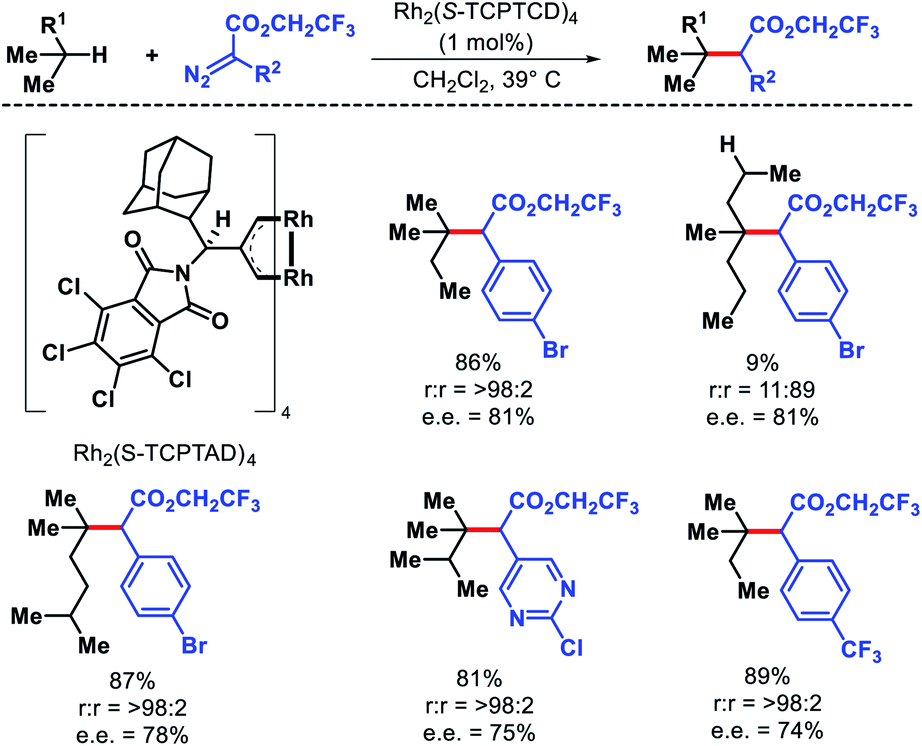
Terminally substituted n-alkyl compounds that contained functional groups like halides, silanes, and ester were compatible with the reaction. Later the same group developed a less sterically demanding dirhodium catalyst Rh2(S-TCPTAD)4 to functionalize least sterically accessible tertiary C–H bonds.79 The regioselectivity for tertiary C–H bonds were excellent except in cases where tertiary center become crowded, secondary C–H functionalization is preferred. Carbene precursors that contained heterocycles such as pyridyl or pyrimidyl heterocycles were also utilized in the reaction (Scheme 72). The protocol was also extended to functionalize tertiary C–H bonds of natural products like cholesteryl acetate, vitamin E acetate, phytyl pivalate, etc. These studies by Davies group showed that it is possible to achieve site selectivity at different C–H bonds within a substrate simply by designing the appropriate catalyst. Just like nature utilizes its enzymes to activate specific C–H bonds, catalysts with defined chiral pockets can control site selectivity, diastereoselectivity, and enantioselectivity of aliphatic substrates. Works of Hu Davies group paved the path for more of such concepts.
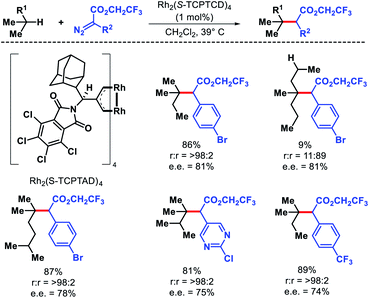 |
| | Scheme 72 Catalyst controlled tertiary C–H bond functionalization of alkanes. | |
4. Conclusion and future outlook
Undoubtedly, transition metal catalyzed distal C–H functionalization has witnessed substantial advancement in recent years. Multifarious strategies were being developed to achieve remote site selectivity of aliphatic molecules. Given the inherent inertness and fluxionally of alkyl C–H bonds, the progress made over the last decade is commendable. The trend of distal C–H activation that started from the usage of strongly coordinating directing group gradually evolved to a point where more efficient and robust approaches are being invented. With the appropriate ligands, weakly coordinating directing groups were also capable of achieving remote C–H activation regioselectively. Transient directing groups were identified to avoid the complications of covalently attached directing groups. Imine functionality was primarily utilized as in situ directing groups in such approaches. In recent times, usage of native functional groups as the director for remote C–H functionalization has come up as a great strategy due to improved atom and step economy. Several such reports are published in recent years. Along with these, metal catalyzed radical approaches such as 1,5 and 1,6-hydrogen atom transfer processes and metal catalyzed carbene transfer strategies have enabled chemists to functionalize distal C–H bonds of almost all kinds of aliphatic molecules including amines, acids, ketones, aldehydes, thiols, alcohols, alkanes, etc. All these varied strategies for distal C–H functionalization are distinct in terms of the reactivity of different C–H bonds and complement each other well. Having said that, more effort is required to circumvent the current limitations associated with this field. Formation of five and six membered metallacycles have enabled to activate distal alkyl C–H bonds like δ-C–H bond of amine and γ-C–H bond of acid. But to functionalize further distal positions one needs to come up with unique strategies. Stabilization of seven and higher membered metallacycles can be one of the ways to achieve further remote C–H functionalization. Development of new directing groups can also pave the path for activating remoter C–H bonds. The area of non-directed C(sp3)–H functionalization has a great prospect in the future since it does not require the formation of particular size metallacycles. Currently, palladium is the most versatile catalyst for distal C–H functionalization. Future efforts must be devoted to replace noble metals like palladium with earth abundant metals. In particular, attempt to decrease catalyst loading, use of inexpensive oxidants must be made to make reactions more economic. Development of distal enantioselective C(sp3)–H functionalization is another exciting area that must be looked closely by researchers of academia and industry. Transition metal catalyzed distal C(sp3)–H functionalizarion has become a powerful tool in the arsenal of synthetic chemists. Further honing is required to make such reactions more economic and practical so that more of such strategies can find its application in targeted molecule synthesis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank SERB India (CRG/2018/003915) and BRNS-India (RD/0118-BRNS000-002) for funding. J. D. thanks CSIR India for financial support.
Notes and references
-
(a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC;
(b) T. Gensch, M. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC;
(c) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord and T. Besset, Chem. Soc. Rev., 2018, 47, 6603 RSC.
-
(a) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS;
(b) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456 RSC.
-
(a) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191 RSC;
(b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS;
(c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS;
(d) J. He, M. Wasa, K. S. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2016, 117, 8754 CrossRef;
(e) Y. Xu and G. Dong, Chem. Sci., 2018, 9, 1424 RSC.
- A. J. Cheney, B. E. Mann, B. L. Shaw and R. M. Slade, J. Chem. Soc. D, 1970, 1176 RSC.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS.
- G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192 CrossRef CAS.
- N. Rodriguez, J. A. Romero-Revilla, M. Ý. Fernandez-Ibanez and J. C. Carretero, Chem. Sci., 2013, 4, 175 RSC.
- E. Hernando, J. Villalva, A. M. Martínez, I. Alonso, N. Rodríguez, R. G. Arrayas and J. C. Carretero, ACS Catal., 2016, 6, 6868 CrossRef CAS.
- M. Fan and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12152 CrossRef CAS.
- K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146 CrossRef CAS.
- P.-X. Ling, S.-L. Fang, X.-S. Yin, K. Chen, B.-Z. Sun and B.-F. Shi, Chem.–Eur. J., 2015, 21, 17503 CrossRef CAS.
- J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220 CrossRef CAS.
- P. J. Cabrela, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2018, 140, 5599 CrossRef.
- Q. Shao, J. He, Q.-F. Wu and J.-Q. Yu, ACS Catal., 2017, 7, 7777 CrossRef CAS.
- S. Das, G. Bairy and R. Jana, Org. Lett., 2018, 20, 2667 CrossRef CAS.
- H. Jiang, J. He, T. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 2055 CrossRef CAS.
- V. G. Landge, A. Parveen, A. Nandakumar and E. Balaraman, Chem. Commun., 2018, 54, 7483 RSC.
- G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS.
- X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Peterson and X. Shi, Chem. Sci., 2013, 4, 3712 RSC.
- S.-Y. Zhang, G. He, Y. Zhao, K. Wright, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2012, 134, 7313 CrossRef CAS.
- C. Wang, L. Zhang, C. Chen, J. Han, Y. Yao and Y. Zhao, Chem. Sci., 2015, 6, 4610 RSC.
- P.-L. Wang, Y. Li, Y. Wu, C. Li, Q. Lan and X.-S. Wang, Org. Lett., 2015, 17, 3698 CrossRef CAS.
- Q. Li, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Adv. Synth. Catal., 2014, 356, 1544 CrossRef CAS.
- Y. Zheng, W. Song, Y. Zhu, B. Wei and L. Xuan, J. Org. Chem., 2018, 83, 2448 CrossRef CAS.
- W. L. Jia and M. A. Fernández-Ibáñez, Chem.–Eur. J., 2018, 44, 6088 CrossRef.
- L.-S. Zhang, G. Chen, X. Wang, Q.-Y. Guo, X.-S. Zhang, F. Pan, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 3899 CrossRef CAS.
- Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554 CrossRef CAS.
- Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084 CrossRef CAS.
- Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884 CrossRef CAS.
- Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26 CrossRef CAS.
- A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073 CrossRef CAS.
- M. Kapoor, D. Liu and M. C. Young, J. Am. Chem. Soc., 2018, 140, 6818 CrossRef CAS.
- Y.-Q. Chen, S. Singh, Y. Wu, Z. Wang, W. Hao, P. Verma, J. X. Qiao, R. B. Sunoj and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 9966 CrossRef CAS.
- C. He and M. J. Gaunt, Chem. Sci., 2017, 8, 3586 RSC.
- K. Chen, D. Wang, Z. W. Li, Z. Liu, F. Pan, Y. F. Zhang and Z. J. Shi, Org. Chem. Front., 2017, 4, 2097 RSC.
- M. Nappi, C. He, W. G. Whitehurst, B. G. N. Chappell and M. J. Gaunt, Angew. Chem., Int. Ed., 2018, 57, 3178 CrossRef CAS.
- Z. M. Png, J. R. Cabrera-Pardo, J. P. Cadahía and M. J. Gaunt, Chem. Sci., 2018, 9, 7628 RSC.
- E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7 CrossRef CAS.
- W. Cui, S. Chen, J.-Q. Wu, X. Zhao, W. Hu and H. Wang, Org. Lett., 2014, 16, 4288 CrossRef CAS.
- S. Guin, P. Dolui, X. Zhang, S. Paul, V. K. Singh, S. Pradhan, H. B. Chandrashekar, S. S. Anjana, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 5633 CrossRef CAS.
- J. W. Xu, Z. Z. Zhang, W. H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750 CrossRef CAS.
- B. B. Zhan, Y. Li, J. W. Xu, X. L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858 CrossRef CAS.
- H. Lin, X. Pan, A. L. Barsamian, T. M. Kamenecka and T. D. Bannister, ACS Catal., 2019, 9, 4887 CrossRef CAS.
- B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS.
- G. He, S.-Y. Zhang, W. A. Nack, R. Pearson, J. Rabb-Lynch and G. Chen, Org. Lett., 2014, 16, 6488 CrossRef CAS.
- S. Li, R.-Y. Zhu, K.-J. Xiao and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 4317 CrossRef CAS.
- A. Dey, S. Pimparkar, A. Deb, S. Guin and D. Maiti, Adv. Synth. Catal., 2017, 359, 1301 CrossRef CAS.
- T. K. Pati, S. Debnath, M. Kundu, U. Khamrai and D. K. Maiti, Org. Lett., 2018, 20, 4062 CrossRef CAS.
- S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267 CrossRef CAS.
- N. Thrimurtulu, S. Khan, S. Maity, C. M. R. Volla and D. Maiti, Chem. Commun., 2017, 53, 12457 RSC.
- G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS.
- Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS.
- K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906 RSC.
- A. Deb, S. Singh, K. Seth, S. Pimparkar, B. Bhaskararao, S. Guin, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 8171 CrossRef CAS.
- S. Guin, S. A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, ACS Catal., 2018, 8, 2664 CrossRef CAS.
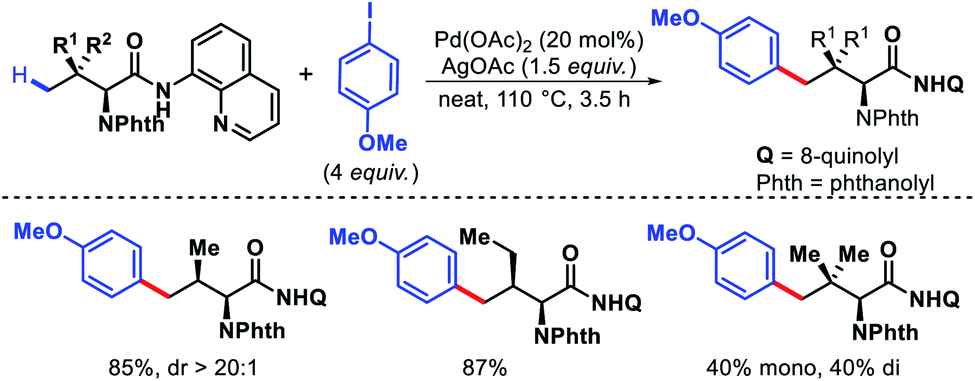
- P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773 CrossRef CAS.
- L. Liu, Y.-H. Liu and B.-F. Shi, Chem. Sci., 2020, 11, 290 RSC.
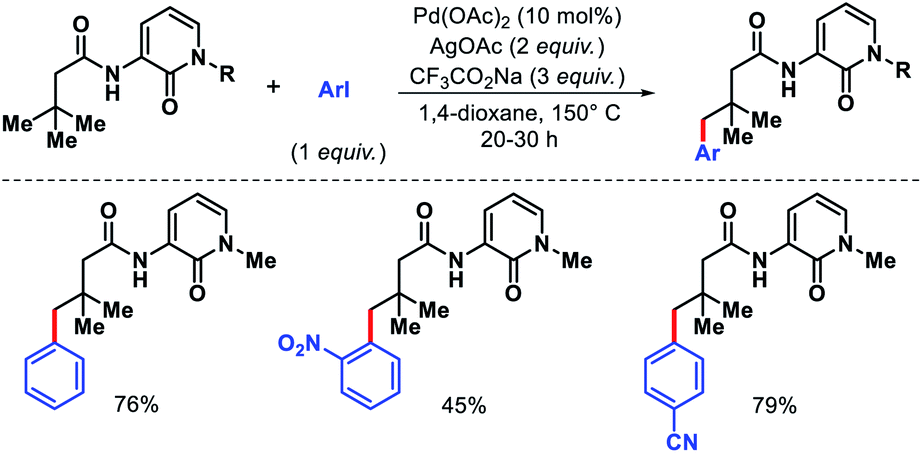
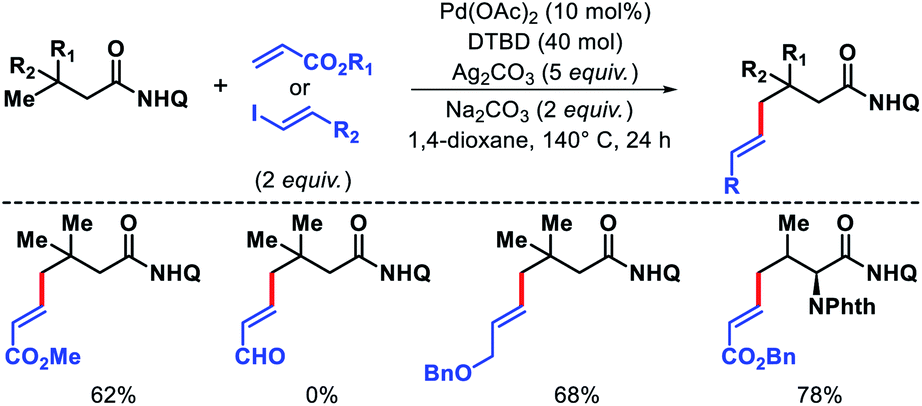
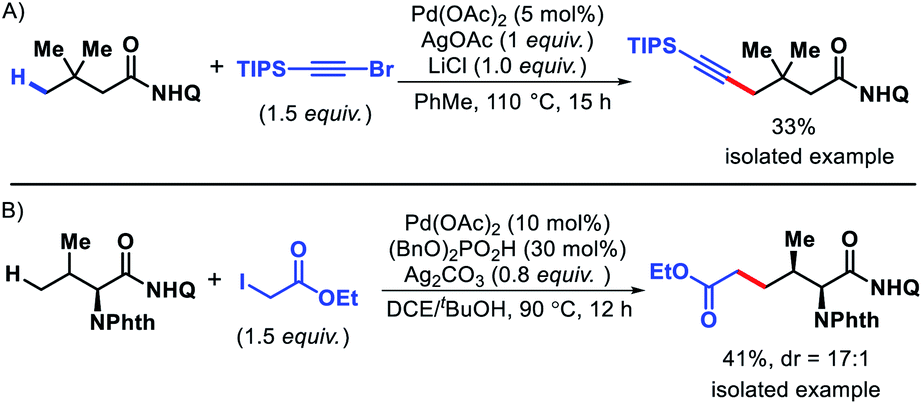
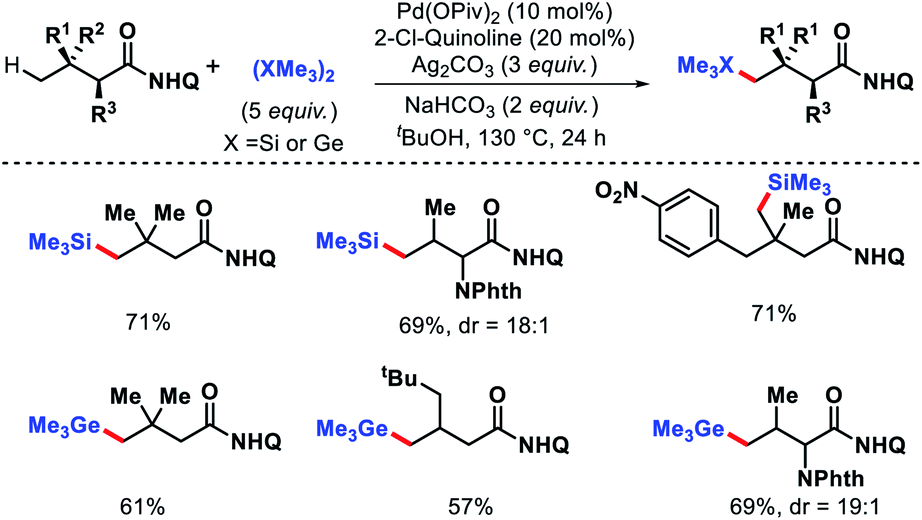
- J. Das, P. Dolui, J. Biswas, W. Ali, H. B. Chandrashekar, G. Prakash and D. Maiti, Chem. Sci., 2020, 11, 9697 RSC.
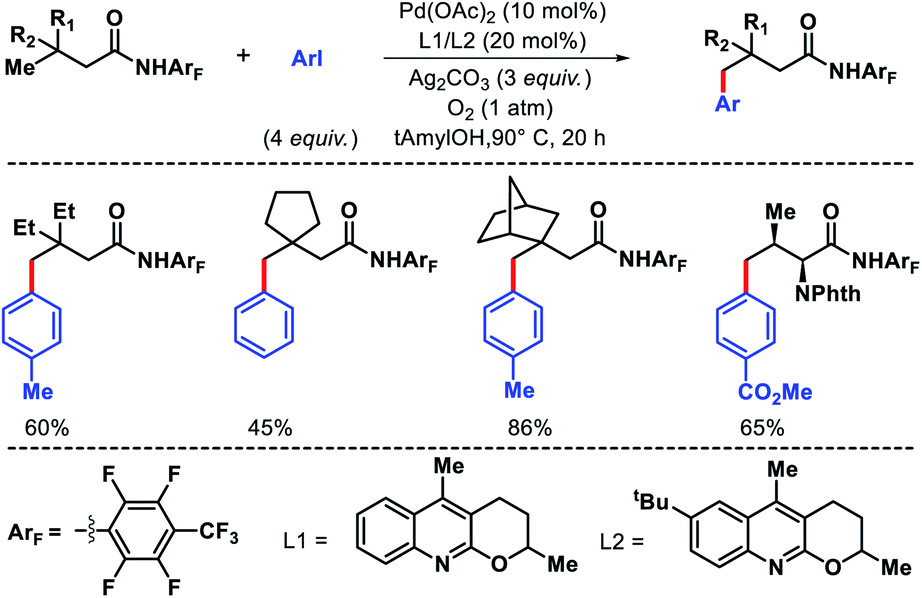
- K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 12848 CrossRef CAS.
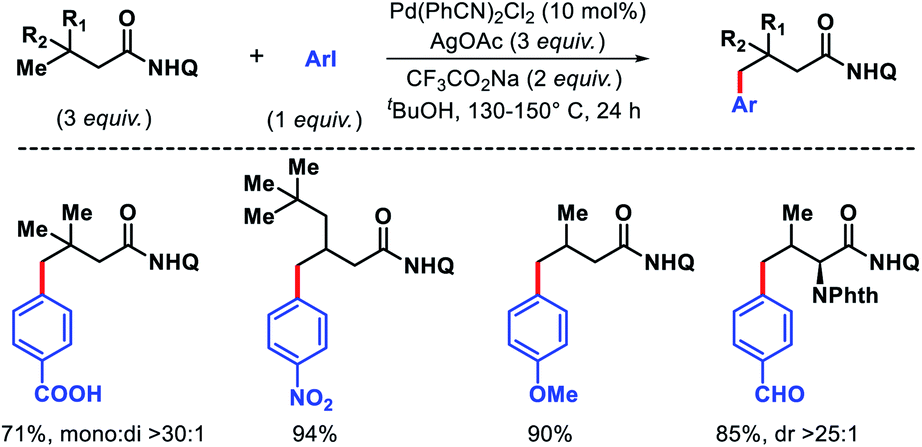
- H. S. Park, Z. Fan, R. Y. Zhu and J. Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 12853 CrossRef CAS.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS.
- R.-Y. Zhu, Z.-Q. Li, H. S. Park, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564 CrossRef CAS.
- B. Li, B. Lawrence, G. Li and H. Ge, Angew. Chem., Int. Ed., 2019, 59, 3078 CrossRef.
- E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70 CrossRef CAS.
- B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 6586 CrossRef CAS.
- G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J.-Q. Yu, Nat. Chem., 2019, 11, 571 CrossRef CAS.
- K. Tanaka, W. R. Ewing and J.-Q. Yu, J. Am. Chem. Soc., 2019, 141, 15494 CrossRef CAS.
- L. Jin, J. Wang and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 12352 CrossRef CAS.
-
(a) N. Goswami and D. Maiti, Isr. J. Chem., 2020, 60, 303 CrossRef CAS;
(b) M. Nechab, S. Mondal and M. P. Bertrand, Chem.–Eur. J., 2014, 20, 16034–16160 CrossRef CAS.
- H. Lu, H. Jiang, L. Wajtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 59, 10192 CrossRef.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS.
- T. Liu, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 5871 CrossRef CAS.
- T. Liu, M. C. Myers and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 306 CrossRef CAS.
- Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288 CrossRef CAS.
- Y. Zhu, K. Huang, J. Pan, X. Qiu, X. Luo, Q. Qin, J. Wei, X. Wen, L. Zhang and N. Jiao, Nat. Commun., 2018, 9, 2625 CrossRef.
- H. Guan, S. Sun, Y. Mao, L. Chen, R. Lu, J. Huang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 11413 CrossRef CAS.
-
(a) E. Nobile, T. Castanheiro and T. Besset, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202009995;
(b) I. Marek, F.-G. Zhang, X.-Q. Wang, Y. Zhou, H.-S. Shi, Z. Feng and J.-A. Ma, Chem.–Eur. J., 2020 DOI:10.1002/chem.202003416.
- K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230 CrossRef CAS.
- K. Liao, T. C. Pickel, V. Boyarskikh, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2017, 551, 609 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Srimanta
Guin
and
Debabrata
Maiti
,
Srimanta
Guin
and
Debabrata
Maiti