
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-guided discovery of a luminescent theranostic toolkit for living cancer cells and the imaging behavior effect†
Chun
Wu‡
a,
Ke-Jia
Wu‡
b,
Jin-Biao
Liu‡
 ac,
Wanhe
Wang
a,
Chung-Hang
Leung
*b and
Dik-Lung
Ma
*a
ac,
Wanhe
Wang
a,
Chung-Hang
Leung
*b and
Dik-Lung
Ma
*a
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon, Hong Kong SAR 999077. E-mail: edmondma@hkbu.edu.hk
bInstitute of Chinese Medical Sciences, State Key Laboratory of Quality Research in Chinese Medicine, University of Macau, Taipa, Macau SAR 999078. E-mail: duncanleung@um.edu.mo
cSchool of Metallurgical and Chemical Engineering, Jiangxi University of Science and Technology, Ganzhou, China
First published on 24th September 2020
Abstract
Dual-functional theranostics are powerful tools that can allow for the in-field understanding of cancer pathology, yet their use is held back by the paucity of suitable theranostics for living systems. Moreover, typical in vitro screening conditions for probe molecules do not necessarily generate candidates that can function effectively in the natural in cellulo environment, limiting their follow-up use in living systems. We introduce herein a general strategy for the development of an iridium(III) theranostic by grafting a well-known inhibitor as a “binding unit” onto an iridium(III) complex precursor as a “signaling unit”. To further optimize their emissive properties, we explored the effect of imaging behavior by incorporating different substituents onto the parental “signaling unit”. This design concept was validated by a series of tailored iridium(III) theranostics 2a–2h for the visualization and inhibition of EGFR in living cancer cells. By comprehensively assessing the theranostic potency of 2a–2h in both in vitro and in cellulo contexts, probe 2f containing electron-donating methoxy groups on the “signaling unit” was discovered to be the most promising candidate theranostic with desirable photophysical/chemical properties. Probe 2f selectively bound to EGFR in vitro and in cellulo, enabling it to selectively discriminate living EGFR-overexpressing cancer cells from normal cells that express low levels of EGFR with an “always-on” luminescence signal output. In particular, its long-lived lifetime enabled its luminescence signal to be readily distinguished from the interfering fluorescence of organic dyes by using time-resolved techniques. Complex 2f simultaneously visualized and inhibited EGFR in a dose-dependent manner, leading to a reduction in the phosphorylation of downstream proteins ERK and MEK, and inhibition of the activity of downstream transcription factor AP1. Notably, complex 2f is comparable to the parental EGFR inhibitor 1b, in terms of both inhibitory activity against EGFR and cytotoxicity against EGFR-overexpressing cancer cells. This tailored dual-functional iridium(III) theranostic toolkit provides an alternative strategy for the personalized diagnosis and treatment of cancers.
Introduction
The combination of an imaging agent and a therapeutic drug into a single molecular entity is known as “theranostics”, and has emerged as a powerful technique used for the timely and personalized monitoring of cancer cells.1,2 Theranostic strategies provide remarkable advantages over conventional probes or drugs as they enable the suppression of specific diseases while simultaneously realizing the in situ detection of the therapeutic dynamics, metabolic pathway, biodistribution or delivery pharmacokinetics of the drug.3 Moreover, the elimination of an additional imaging agent or inhibitor can not only largely simplify the administration process, but also significantly reduce the potential of undesired dose mismatch or drug–drug crosstalk between the imaging agent and therapeutic drug.4 However, it has proven difficult to design synthetic theranostic toolkits tailored for specialized purposes, particularly for the monitoring of targeted analytes in living systems.A desirable theranostic molecule for cancer cells needs to possess biological activity as an inhibitor while simultaneously maintaining the photophysical/chemical properties of a probe. One of the most popular strategies for the development of theranostic agents uses nanomaterials to incorporate drug component and probe component.3,5–7 However, these strategies suffer from limited biocompatibility since the nanomaterial-based platforms generally have larger sizes, with their dimensions up to 100 nm.8–12 An additional drawback of nanomaterial-based theranostic platforms is that the drug and probe components are sometimes still technically separate entities that are simply loaded onto the core/shell regions of the nanomaterial.6,13,14 An alternative and simpler strategy for theranostic has been explored, based on using pure organic scaffolds or metal–organic integrated complexes. Compared to nanomaterials, small molecule-based theranostic agents are more compact and simpler in structure, which confers high reproducibility as well as allowing easy modification of molecular structures to achieve the desired photochemical property, selectivity, biocompatibility, therapeutic activity, cellular uptake efficiency and/or drug excretion with reduced side effects.15–20
Fluorescent dyes, while widely used in theranostic agents, suffer from several defects including high photobleaching rate, short-lived lifetime and low signal-to-noise ratios in the highly autofluorescent cellular environment, which extensively limits their in-field utilization in the complicated biological systems.21,22 In contrast, metal–organic small molecule theranostics have opened up new possibilities for the in situ simultaneous visualization and inhibition of cancer cells due to their advantageous photophysical properties (e.g. long-lived phosphorescence lifetime, high quantum yield, large Stokes shift, etc.)23–27 and the easily tunable nature of their photophysical/chemical properties.28 For example, lanthanide-based complexes (i.e. europium(III), terbium(III), ytterbium(III) complexes) are endowed with typical two-photon excitation behaviour and offer facile discrimination of their signal output with emission bands spanning from visible to near-infrared (NIR) ranges, allowing for deep penetration in biological system.29 Meanwhile, transition metal complex-based strategies (i.e. iridium(III) complex) are relatively photostable compared to most organic fluorophores such as commercially available organic dye DAPI (DAPI = 4′,6-diamidino-2-phenylindole), enabling their favoured applications in long-term observation and dynamic monitoring of the targeted analytes in living biological system.30,31 Encouragingly, metal complexes (including iridium(III), ruthenium(III), platinum(IV) and gold(III) complexes) have been developed as for simultaneous imaging and treatment of cancers, suggesting their great potential as theranostic agents.15,32–35
Epidermal growth factor receptor (EGFR) is a well-known protein tyrosine kinase that plays a crucial role in the pathology of various cancers.36,37 The in situ monitoring of the abnormal overexpression of EGFR in living cancer cells can allow for the timely treatment, diagnosis and assessment of the onset, recurrence, tolerance, efficiency and/or toxicity for cancers. However, small molecule-based EGFR-specific theranostics, particularly those based on long-lifetime transition metal complexes capable for dynamic monitoring in living systems, remain poorly explored. Moreover, typical in vitro screening conditions for probe molecules do not necessarily generate candidates that can function effectively in the natural in cellulo environment, limiting their follow-up use in living systems.38,39 Given the paucity of suitable long-lived theranostic toolkits for the specific targeting of EGFR, we intended to validate our design concept by constructing a series of tailored iridium(III) theranostic agents for the simultaneous visualization and inhibition of EGFR in living cancer cells, which were verified using both in vitro and in cellulo screens (Scheme 1).
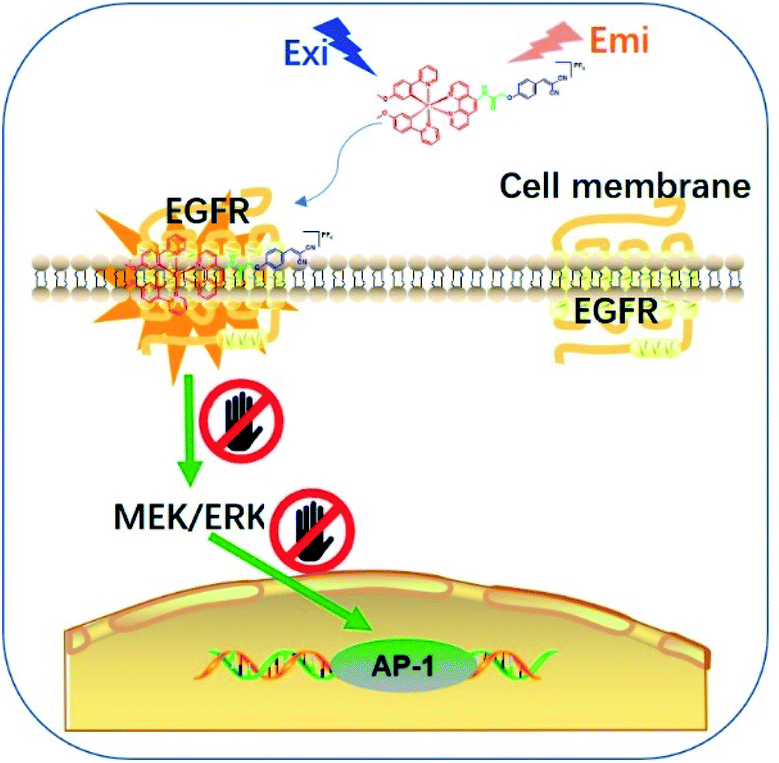 | ||
| Scheme 1 Schematic diagram showing the in situ tracking and suppression of EGFR using a smart and tailored iridium(III) theranostic toolkit. | ||
One key challenge in the design of a transition metal complex-based theranostic agent is being able to acquire biological activity against the target, while retaining desirable photophysical/chemical properties for the imaging of living cells. Poor selectivity of the theranostic towards cancer cells can lead to undesired cytotoxicity against surrounding healthy cells.40 Another challenge that has to be overcome is the fact that many transition metal complexes exhibit strong emission and good stability only in organic solvents, and do not function well in aqueous solutions, such as cellular environment.41 Meanwhile, a red-shifted luminescence is generally the most preferred emission range in order to avoid interference from the autofluorescence, light scattering and/or optical absorption in the cellular environment.42 Moreover, an “always-on” or “switch-on” emission profile is more desirable compared with a “switch-off” detection mode as this increases the accuracy in the discrimination of cancer cells from healthy cells, and/or in the report of the biological events.32,43,44 Finally, the ability of the candidate theranostics to accumulate at the targeted locations (e.g. cytoplasm and/or organelles) in living cells should also be considered, as this could largely limit their use for the monitoring of living cells.45,46
To overcome these problems, this design strategy takes advantage of the tunable photophysical/chemical properties of the transition metal complex precursor 1a (Fig. S1†). A well-known EGFR inhibitor 1b, with high selectivity and inhibition activity against EGFR,47 is introduced to the tailored metal complex. Generally, an assembled metal complex-based theranostic agent consists of a “binding unit” that selectively binds to the targeted analyte with desired biological activity, a “signaling unit” that illuminates the targeted analytes by transducing the sensing event into a dynamic luminescent signal output, and a “linker” that tethers the binding unit and signaling unit into an integrated molecule (Fig. 1). In this study, the “binding unit” is the EGFR-targeting drug moiety 1b, which is expected to confer high selectivity and inhibition activity against EGFR. The final complexes 2a–2h thus all contain an EGFR-targeting moiety 1b grafted onto the N^N ligand of the parent iridium(III) scaffold 1a. To further optimize the emissive behavior of the signaling parent metal complex precursor 1a, we explored the effect of incorporating different substituents such as electron-withdrawing fluorine atoms (2e), electron-donating methyl (2g) or methoxy (2f) groups, different aromatic ring systems (2a–2d) or a sulfur-containing heterocycle (2h). The lead candidate was comprehensively screened by assessment of both in vitro and in cellulo performance. The aim was to obtain a theranostic that retained biological activity while possessing suitable phosphorescence lifetime, quantum yield, Stokes shift, and absorbance and/or emission profiles in living system.
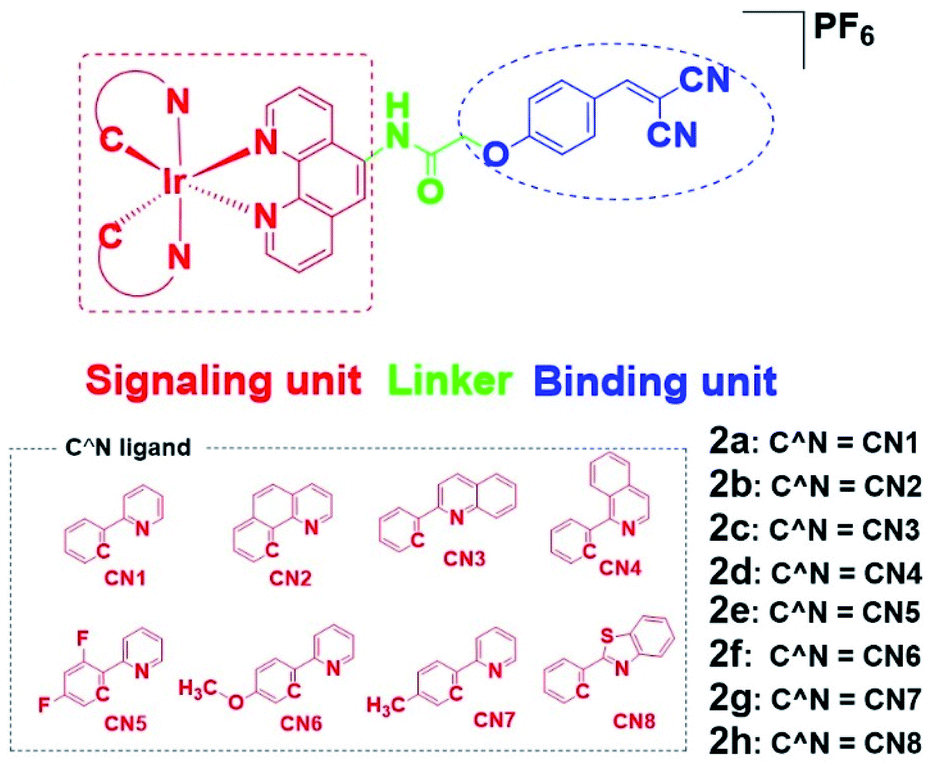 | ||
| Fig. 1 Design principle of EGFR-targeting theranostic agents 2a–2h and chemical structures of the library of auxiliary C^N ligands used for fine-tuning the photo-physical/-chemical property of the parent iridium(III) complex precursor 1a. | ||
Results and discussion
To synthesize the iridium(III) complex-based theranostic molecules, the reference inhibitor 1b was initially attached to the parent complex precursor 1a (Fig. S1†) via two different strategies, either by directly conjugation to the N^N ligand of parent complex 1b (synthesis route 1, Scheme S1†) or a linker tethered parent complex S5 (synthesis routes 2 and 3, Scheme S1†). Unfortunately, synthesis route 1 proceeded poorly owing to the unsatisfactory yield and solubility of the EGFR binding moiety-containing N^N ligand S4. Attempted optimization of the type and amount of the catalyst, alkali, and reaction solvent system failed to produce the desired product. In the next attempt, we switched to an alternative strategy by directly attaching the binding moiety to the parent complex precursor 1a. Complex 1a was first tethered by a linker to obtain an intermediate complex S6. However, complex S6 and 1b showed quite different solubilities in various organic solvents (e.g. ethanol, methanol, acetonitrile and dichloromethane, etc.). Hence, the direct conjugation of the binding moiety 1b to S6 proceeded poorly with many side products either in single solvent or mixed solvent system, causing problems with the subsequent purification process. To address this problem, the conjugation of the targeted 1b moiety was therefore divided into two separate steps, involving an initial substitution of the S1 moiety to give S7, and the subsequent Knoevenagel condensation of propanedinitrile. Encouragingly, this alternative strategy produced much fewer impurities and the yield of the final complex 2a (Fig. 1) could then be further improved by optimization of the reaction temperature, and type and amount of alkali, catalyst and organic solvents involved in the process. The iridium(III) analogues 2b–2h were synthesized in similar fashion via the intermediate parent complex 1a. Complexes 2a–2h were characterized by 1H NMR, 13C NMR, high-resolution mass spectrometry (HRMS) and high performance liquid chromatography (HPLC) techniques (Fig. S2†).We first examined the in vitro inhibition efficiency of iridium(III) complexes 2a–2h against EGFR activity (Fig. 2a), utilizing a commercially available EGFR kinase assay kit. EGFR inhibitor 1b was employed as a reference drug. Excitingly, these complexes displayed 20–50% enhanced EGFR inhibition compared to the drug precursor 1b, which was attributed to the interaction of the drug precursor and the parent iridium(III) complex precursor to form a more biologically active entity. Among these conjugates, 2g bearing methyl groups on its C^N ligands showed the highest inhibitory activity against EGFR, whereas the sulfur heterocycle-containing 2h showed comparatively the weakest activity. These results suggested that the integration of the drug precursor (1b) onto the metal complex core not only retained, but slightly improved the inhibition efficiency against EGFR.
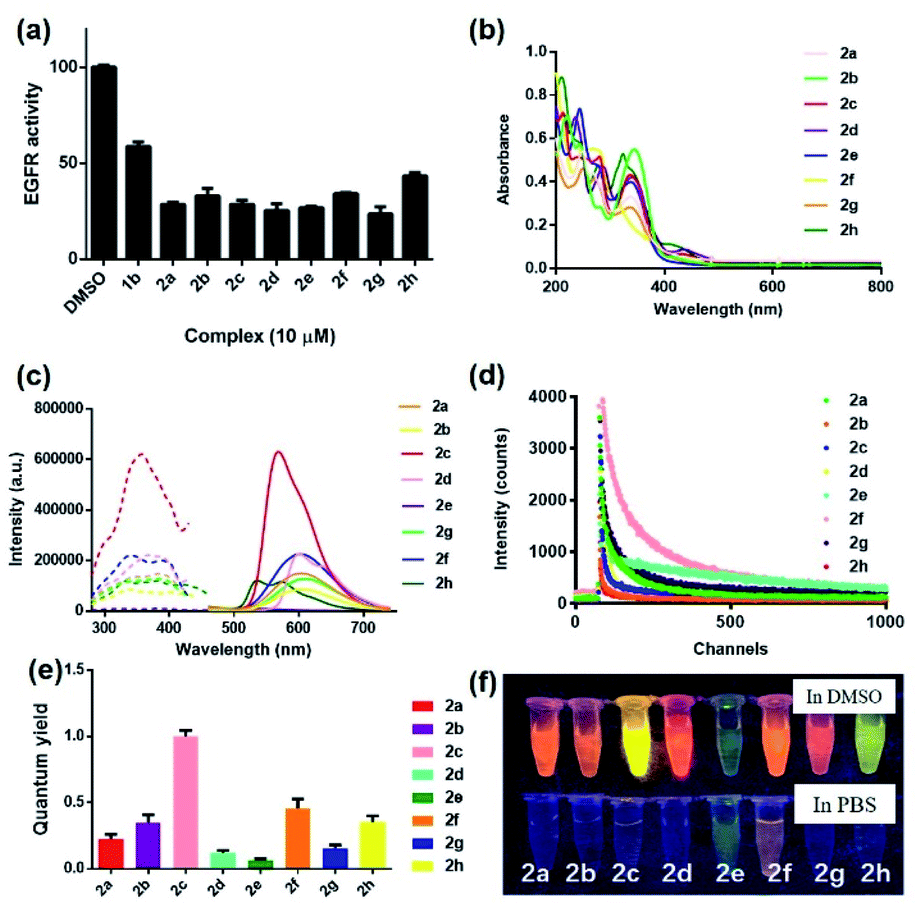 | ||
| Fig. 2 (a) Inhibition activity of complexes 2a–2h and reference EGFR inhibitor 1b (10 μM) against EGFR (10 μg mL−1) as measured by a fluorescence-based kinase assay. The fluorescence intensity was detected at ex/em = 330 nm/450 nm. (b) Absorbance spectra of complexes 2a–2h (5 μM) in ACN. (c) Excitation and emission spectra of complexes 2a–2h (5 μM) in DMSO. (d) Emission decay curves of complexes 2a–2h (5 μM) in PBS buffer containing 0.5% DMSO. (e) Quantum yields of complexes 2a–2h (5 μM). (f) Photographs of complexes 2a–2h (5 μM) observed under UV light (365 nm) in dimethyl sulfoxide (DMSO) and PBS buffer containing 0.5% DMSO, respectively. | ||
In light of this encouraging result, we next studied the intrinsic photophysical properties of 2a–2hin vitro. Complexes 2a–2h showed characteristic absorbance bands of transition metal complexes at 208–346 nm in the UV-Vis spectra (Fig. 2b), which can be assigned to the spin-allowed intraligand (1IL) absorption and metal-to-ligand charge-transfer (1MLCT) transition.48 The maximum emission wavelength of complexes 2a–2h were mainly located in the range of 529–611 nm (Fig. 2c). The presence of electron-withdrawing fluorine atoms (2e) or sulfur-containing heterocyclic ring (2h) also lead to a relatively smaller Stokes shifts (198–205 nm) compared to the other six complexes (211–266 nm). Still, all these complexes had Stokes shifts that are much larger than the typical Stokes shift of fluorescent dyes (around 100 nm), thus making them less susceptible to self-quenching. Notably, complex 2c displayed the strongest luminescence intensity, which might be attributed to subtle variation of the electron density and/or metal-to-ligand-charge-transfer (MLCT) state caused by the joint interplay of the aromatic ring and the π–π conjugation system located in the auxiliary C^N and N^N ligands. Moreover, complexes 2a–2h all displayed characteristic long-lived lifetimes in sub-microsecond regime49,50 (Fig. 2d and Table S1†), which are much longer than the typical nanosecond decay times of organic fluorophores. Complexes 2a–2h exhibited biexponential decay, with the two decay components of each complex showing similar lifetime regimes under the same experimental conditions in PBS buffer containing 0.5% DMSO (Table S1†). The biexponential decay of complexes 2a–2h is possibly attributed to the presence of two distinct conformations.33,51,52 The average lifetime of the decay components of complexes containing electron-withdrawing fluorine atoms (2e), electron-donating methoxy moieties (2f) or sulfur-containing heterocycles (2h) displayed comparatively longer lifetimes of 525, 139 and 112 ns, respectively among the group in the PBS buffer containing 0.5% DMSO (Table S1†). These long lifetimes would enable the emission of complexes 2a–2h to be discriminated from a highly auto-fluorescent background in the complicated cellular environment by use of time-resolved emission spectroscopy (TRES). Additionally, all eight complexes showed comparable (2d, 2e and 2g) (0.057–0.136) or much higher (2a, 2b, 2c, 2f and 2h) (0.202–0.968) quantum yields compared to the widely-used standard Ru(bpy)3 (bpy = 2,2′-bipyridine) (0.062) (Fig. 2e). Notably, the theranostic agents 2a–2h (5 μM) exhibited colourful luminescence upon the irradiation of UV light (365 nm) in dimethyl sulfoxide (DMSO) and PBS buffer containing 0.5% DMSO (Fig. 2f and S3†). Complexes containing either electron-withdrawing groups (2e) or the sulfur-containing heterocyclic ring (2h) on their auxiliary C^N ligands displayed the most blue-shifted emission maxima and appeared blue and green luminescence, respectively, while the other six complexes showed luminescence colors in the yellow or orange region. Overall, these results suggested that the imaging behavior of the iridium(III) theranostic can be fine-tuned by modifying auxiliary functional groups of the parent “signaling unit”.
Inspired by the promising luminescent characteristics of complexes 2a–2h, their practical application as luminescent probes for EGFR imaging in the EGFR-overexpressing A431 human epidermoid carcinoma cell line was next explored (Fig. 3). Living A431 cells were initially incubated with the reference EGFR inhibitor 1b (1 μM) for 3 h as a competing inhibitor, followed by the addition of probes 2a–2h (1 μM) for 2 h at 37 °C. Unexpectedly, probe 2f containing electron-donating methoxy groups on the “signaling unit”, rather than the most luminescent counterpart 2c or the most long-lived counterpart 2d, displayed the most distinguished luminescence change in the presence and absence of the competition EGFR inhibitor 1b over the other seven probes as revealed by the confocal assay. This result suggested that 2f not only enabled the practical detection of EGFR in living A431 cells, but also exhibited better selectivity towards EGFR over the other probes in cellulo. In contrast, probe 2g containing electron-donating methyl groups on the “signaling unit”, exhibited negligible luminescence change in response to the EGFR inhibitor 1b. A likely explanation for the inferior performance of probe 2g compared to probe 2f, even though they both possess electron-donating auxiliary substituents, is that the probes' interactions with the targeted biomolecules can be highly sensitive to differences in hydrophobicity, permeability and binding affinity in living cells.53–56 Moreover, previous studies have suggested that methoxy group played a key role in the interaction between several FDA-approved EGFR inhibitors (e.g. erlotinib, gefitinib, brigatinib and osimertinib) and EGFR.57–60 In summary, although probes 2a–2h all exhibited versatile photophysical characteristics and inhibition activity against EGFR, their sensing performance under the natural in cellulo environment was then considered as the crucial evaluation criterion for use as theranostic agents. Hence, 2f was selected as the final candidate for further investigation due to its superior selectivity for EGFR in living cells.
 | ||
| Fig. 3 Application of complexes 2a–2h as luminescent probes for EGFR imaging in EGFR-overexpressing A431 human epidermoid carcinoma cells. Cells were treated with vehicle control (0.2% DMSO) or complexes 2a–2h (1 μM) containing 0.2% DMSO for 2 h before imaging. The luminescence intensity was detected at ex/em = 405 nm/500–700 nm. Scale bar = 50 μm. | ||
To further support the hypothesis that complex 2f bound to EGFR in cells, the cellular thermal shift assay (CETSA) was used to quantify the extent of drug engagement. Complex 2f significantly stabilized EGFR in A431 cell lysates compared to the DMSO control (Fig. 4a), suggesting that complex 2f engaged EGFR within cells to act as a luminescent probe. We next examined the selectivity of complex 2f for EGFR over common biologically interfering substances, including common amino acids (Arg, His, Asp, Ser, Gln, Cys, Pro, Tyr and Trp), glutathione (GSH), glutathiol (GSSG), homocysteine, homocysteine, metal ions (Mg2+, Fe3+, Cu2+, Al3+, Na+, K+, Ca2+, Zn2+, Fe2+ and Mn2+) and anions (S2−, HCO3−, CO32−, SO32−, Cl−, SO42−, F−, ClO4−, NO3−, HPO42− and H2PO4−) (Fig. S4†). The results indicated that complex 2f was capable of sustaining a robust luminescent signal in the presence of commonly existing biological interfering substances. Stability assays revealed that complex 2f was stable for at least seven days at 298 K, either in a DMSO-d6/D2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution or in an acetonitrile/H2O (9:1) solution as verified by 1H NMR spectroscopy (Fig. S5a†) or UV/Vis spectroscopy (Fig. S5b†), respectively, indicating its suitability for use in further imaging experiments. Overall, these results suggested that complex 2f is a permanent “always-on” EGFR probe with excellent selectivity and stability.
:1) solution or in an acetonitrile/H2O (9:1) solution as verified by 1H NMR spectroscopy (Fig. S5a†) or UV/Vis spectroscopy (Fig. S5b†), respectively, indicating its suitability for use in further imaging experiments. Overall, these results suggested that complex 2f is a permanent “always-on” EGFR probe with excellent selectivity and stability.
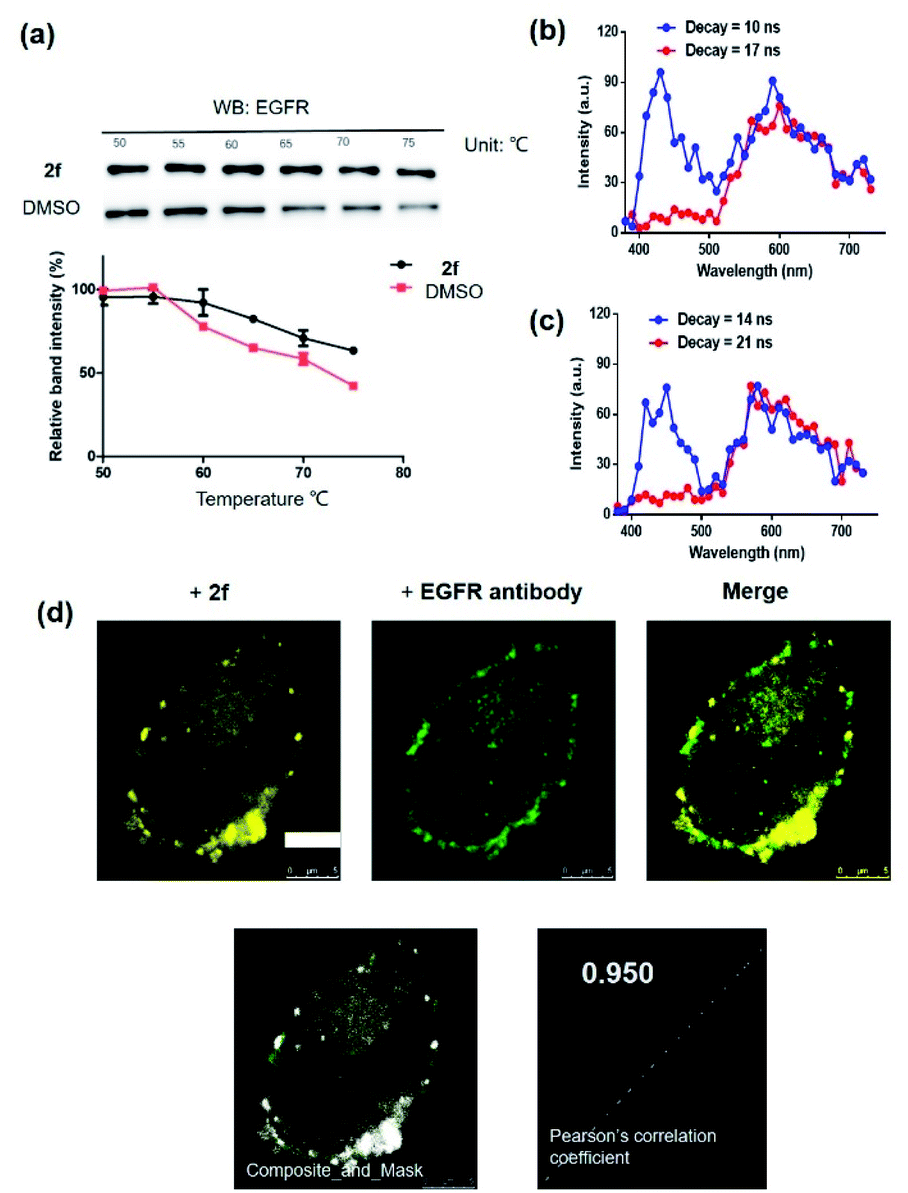 | ||
| Fig. 4 (a) Effects of complex 2f on the thermal stability of EGFR in cell lysates. A431 cells lysates were treated with 2f (1 μM) for 1 h. Time-resolved spectra of complex 2f (10 μM) in the presence of fluorescent organic dyes (b) thioflavin S (THS) and (c) coumarin 460 (Cm460) as two model matrix interferences to simulate the autofluorescence of biological samples in PBS buffer containing 1% DMSO. (d) Co-localization studies of complex 2f (1 μM, 30 min) with EGFR protein in A431 cells. The Pearson's correlation coefficient of complex 2f with EGFR protein was calculated to 0.950. ex/em = 405 nm/500–700 nm. Scale bar = 5 μm. | ||
Compared with traditional organic dye-based probes, the long-lived lifetime of transition metal complex-based probes endows them with the competitive benefit of being able to discriminate their signal output even in a highly autofluorescent background. This hypothesis was verified for 2f by evaluating its emission using TRES on a Horiba fluorescence spectrometer (FL3C-21).61 Two fluorescent organic dyes, coumarin 460 (Cm460) and thioflavin S (THS), were selected as model matrix interferences to simulate the autofluorescence of biological samples with maximum emission at 460 nm and 428 nm, respectively. After 10 ns and 14 ns, which is before the completion of the fluorescence decay of THS or Cm460, respectively, the luminescence of complex 2f was perturbed by the emission peaks of THS at 428 nm (Fig. 4b) and Cm460 at 460 nm (Fig. 4c), leading to reduced accuracy of measurement due to the undesirable interference of the signal output for EGFR. However, after the decay of 17 ns and 21 ns, which is after the completion of the fluorescence decay of THS or Cm460, respectively, the interfering signal from these dyes were almost completely eliminated, while the emission of complex 2f was still retained with considerable intensity. In addition, we recorded the luminescence of complex 2f alone at different decay times, and the spectra was identical to that in the presence of organic dyes after fluorescence decay was completed, further confirming that the observed time-resolved signals were due to complex 2f (Fig. S6†). These results demonstrate the potential use of time-resolved techniques to discriminate the signal of 2f in highly autofluorescent biological samples.
To explore whether the long lifetime of complex 2f could be distinguished from background fluorescence in cellulo, we compared the emission of 2f with a commercially-available cellular dye (4′,6-diamidino-2-phenylindole (DAPI)) in living A431 cells using TRES on a multi-mode microplate reader.31,62 After incubating the cells with both complex 2f and DAPI for 2 h, the signal output from the cells was recorded with the delay time set to 50 μs (Fig. S7a†) and 200 μs (Fig. S7b†), respectively. The results indicated the luminescence of complex 2f could be readily distinguished from potentially interfering fluorescence using TRES. Taken together, complex 2f exhibits a long-lived phosphorescence both in vitro and in cellulo, allowing for its potential for long-lived monitoring of EGFR in living systems via time-gated mode.
Many anticancer drugs have limited ability to discriminate between living normal and tumor cells, which can lead to low efficacy and severe side effects. Given the ability of complex 2f to engage EGFR in vitro, we next investigated whether complex 2f could visualize EGFR in living cancer cells using a co-localization assay (Fig. 4d). Encouragingly, complex 2f (1 μM) showed strong colocalization with EGFR protein in A431 cancer cells as detected via immunofluorescence. A high Pearson's correlation coefficient (Rr = 0.950) between the cell images was detected, suggesting that complex 2f visualizes EGFR through selective binding to EGFR in cells. In order to further verify that complex 2f interacts selectively with the targeted EGFR protein, imaging experiments were repeated in EGFR knockdown A431 cells (Fig. 5a). Notably, a noticeable decrease in the luminescence of 2f was recorded in the EGFR knockdown group compared to untreated cells, suggesting the binding of complex 2f to EGFR is required for the generation of luminescent signal output in A431 cells. Moreover, complex 2f displayed much higher luminescence in A431 cells, which overexpress EGFR, than in normal human hepatocyte LO2 cells that expressed less EGFR (Fig. 5b). This result indicates that 2f has potential for the real-time discrimination of cancer cells that overexpress EGFR compared to normal cells with lower EGFR expression.
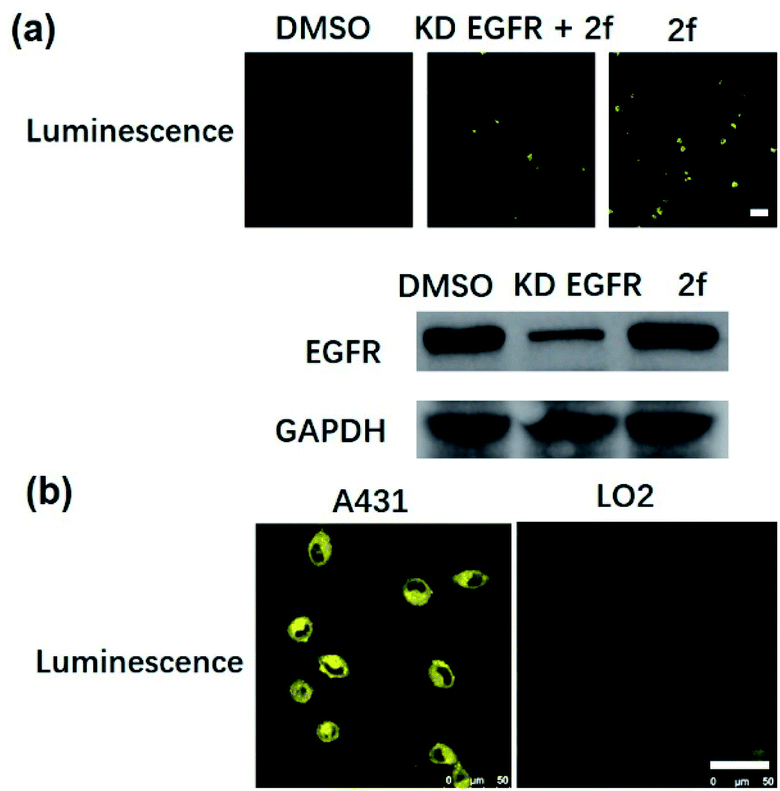 | ||
| Fig. 5 (a) Confocal imaging of EGFR knockdown A431 cells. EGFR knockdown A431 cells were incubated with 2f (1 μM) for 2 h at 37 °C. ex/em = 405 nm/500–700 nm. Scale bar = 50 μm. (b) Confocal imaging of normal LO2 cells and EGFR-overexpressing A431 cells. LO2 cells and A431 cells were incubated with 2f (1 μM) for 2 h at 37 °C. ex/em = 405 nm/500–700 nm. Scale bar = 50 μm. | ||
Given the ability of complex 2f to visualize EGFR in living cancer cells, we next explored the cytotoxicity of complex 2f in either EGFR-overexpressing A431 cancer cells or normal LO2 cells. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT) assay was conducted by using the reference EGFR inhibitor 1b as a positive control. Complex 2f displayed comparable cytotoxicity against EGFR-overexpressing A431 cancer cells (IC50 = 37.40 ± 4.10 μM) compared to the reference EGFR inhibitor 1b (IC50 > 50.0 μM) (Fig. S8a and b†). Many transition metal complexes exhibit inherent toxicity due to the metal center, may lead to reduced biocompatibility and undesired side effects due to the damage to healthy cells or tissues, which can limit their application as therapeutic drugs. Therefore, the cytotoxicity of 2f was also investigated against normal LO2 cells. Encouragingly, complex 2f was less toxic to LO2 cells (IC50 > 50.0 μM) (Fig. S8c and d†) compared to cancer cells. Taken together, these results suggested that complex 2f possesses promising biocompatibility for theranostic applications.
Encouraged by the results above, we further studied the inhibitory activity of complex 2f against EGFR in vitro and in living cancer cells. The in vitro biological potency of 2f in suppressing the activity of EGFR was assessed with 1b as a positive control by using a commercial EGFR kinase assay kit. The results showed that both complex 2f and the positive control 1b could reduce EGFR activity in a dose-dependent manner (Fig. 6a and b). Notably, complex 2f inhibited EGFR activity with an EC50 value of 1.18 ± 0.09 μM, making it comparable to the positive control 1b which had an EC50 value of 4.28 ± 0.22 μM. Taken together, these data indicated that 2f could target and suppress the activity of EGFR in vitro.
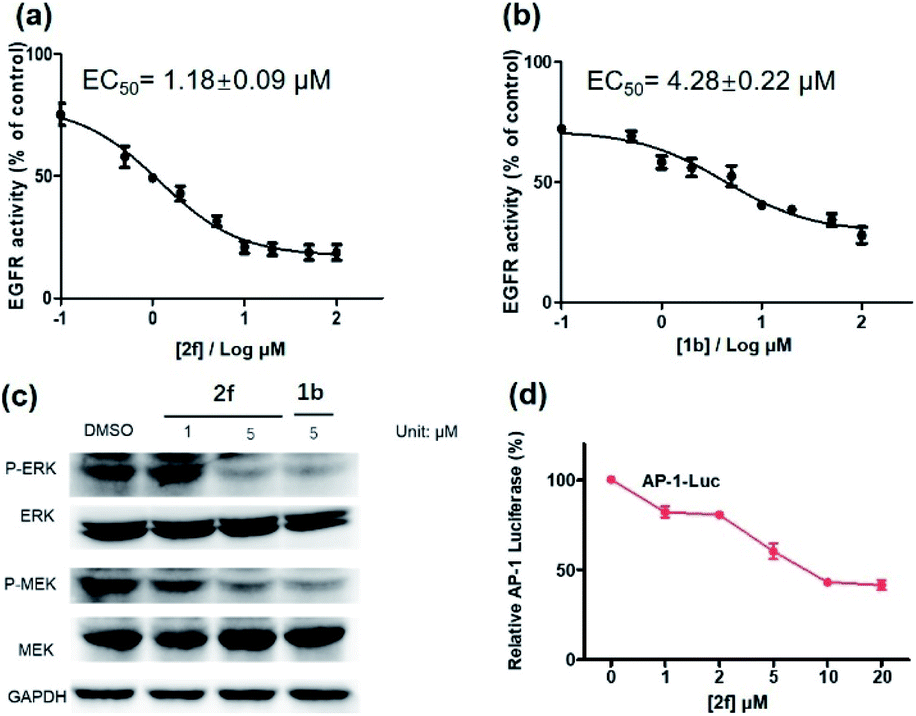 | ||
| Fig. 6 EGFR inhibition activity of complex 2f (a) and the reference inhibitor 1b (b) in EGFR-overexpressing A431 cells. (c) Effects of complex 2f and 1b on P-ERK, ERK, P-MEK and MEK protein expression. A431 cells were treated with 2f for 12 h. The protein levels were revealed by western blot assay. (d) Effects of complex 2f on the transcription activity of downstream protein AP-1. A431 cells were transfected with AP-1 luciferase plasmid for 48 h, and then treated with 2f at different concentration. AP-1 transcription activity was detected. | ||
The activation of EGFR triggers a cascade of phosphorylation events in the MEK/ERK pathway.63 To investigate the ability of complex 2f to inhibit downstream proteins in living A431 cells, the levels of P-ERK and P-MEK in the MEK/ERK pathway was measured by western blotting. Upon pre-incubation with 2f, a significant decrease in the expression levels of P-ERK and P-MEK were recorded compared to the DMSO control, while no significant change on the expression levels of total ERK and MEK proteins were observed (Fig. 6c). The effects of 2f on P-ERK and P-MEK expression were phenocopied by EGFR knockdown using siRNA (Fig. S9†). These results indicate that 2f down-regulates the activation of downstream proteins including P-ERK and P-MEK in living A431 cells, via targeting EGFR. To further explore the ability of complex 2f to block the downstream effects of EGFR in A431 cells, a luciferase reporter assay was performed to evaluate the effect of 2f on the activity of the transcription factor AP-1, a downstream target in the MEK/ERK pathway. Upon the increasing concentration of 2f, the activity of AP-1 was reduced in a dose-dependent manner, suggesting that 2f can decrease AP-1 transcription activity (Fig. 6d). Taken together, these results suggested that 2f could suppress the activity of EGFR in living A431 cells to inhibit downstream pathways.
Conclusions
In summary, using a structure-guided design principle, we have validated our design concept by successfully developing a series of dual-functional iridium(III) complex based theranostic 2a–2h for the simultaneously visualization and inhibition of EGFR in EGFR-overexpressing living cancer cells. A known EGFR inhibitor 1b was grafted to an iridium(III) complex precursor 1a, leading to the endowment of biological activity from the inhibitor while maintaining the desirable photophysical characteristics of the metal complex iridium(III). The properties of the iridium(III) complex-based theranostics were fine-tuned by the modification of auxiliary ligands with electron-withdrawing fluorine atoms (2e), electron-donating methyl (2g) or methoxy (2f) groups, different aromatic ring systems (2a–2d) or a sulfur-containing heterocycle (2h), generating a range of conjugates 2a–2h that all exhibited increased EGFR inhibition activity compared to the known inhibitor 1b while retaining desirable photophysical properties including long-lived lifetime, good stability, high quantum yield, and large Stokes shift. Unexpectedly, probe 2f containing electron-donating methoxy groups on the “signaling unit”, rather than the most luminescent counterpart 2c or the most long-lived counterpart 2d, was the most promising candidate after consideration of both in vitro and in cellulo performance. The candidate theranostic 2f selectively bound to EGFR both in vitro and in living cells as revealed by a cellular thermal shift assay, a western blotting assay, a knockdown assay and a co-localization assay. The long-lived luminescence of 2f in the sub-microsecond regime enabled its luminescence to be readily distinguished from the interfering fluorescence of fluorescent dyes by TRES. Remarkably, complex 2f discriminated living EGFR-overexpressing cancer cells (A431 cells) from normal cells (LO2 cells) that express low level of EGFR with an “always-on” luminescence signal output. Moreover, complex 2f inhibited the activity of EGFR in a dose-dependent manner, decreased the phosphorylation of downstream proteins ERK and MEK, and inhibited the activity of downstream transcription factor AP1. Notably, complex 2f exhibited comparable inhibitory activity against EGFR with an EC50 value of 1.18 ± 0.09 μM compared to the parental EGFR inhibitor 1b (EC50 = 4.28 ± 0.22 μM) and caused a slightly higher cytotoxicity (IC50 = 37.40 ± 4.10 μM) against EGFR-overexpressing A431 cancer cells. Taken together, these results demonstrate the promising potential of complex 2f as an EGFR-selective dual-functional inhibitor and probe (“theranostic”) in targeting living cancer cells. To our knowledge, this is the first small molecule-based iridium(III) theranostic for the simultaneous visualization and inhibition of EGFR in highly autofluorescent living cancer cells.Experimental
Cellular thermal shift assay
Cellular thermal shift assay was performed to monitor the target engagement of 2f in A431 cell lysates. Briefly, cell lysates from 2 × 106 A431 cells were collected, diluted in PBS and separated in the same aliquots. Each aliquot was treated with 2f (10 μM) or DMSO 30 min after incubation at room temperature, the compound-treated lysates were divided into 50 μL in each of PCR tubes and heated individually at different temperatures (Veriti thermal cycler, Applied Biosystems/Life Technologies). The heated lysates were centrifuged and the supernatants were analyzed by SDS-PAGE followed by immunoblotting analysis by probing with the indicated antibody.Immunofluorescence
The cells were initially washed for three times using phosphate-buffered saline (PBS) buffer. Afterwards, 4% PFA was added to the cells for incubation of 30 min, with following triplicate washing by PBS. Next, another 30 min of incubation is required by adding 0.5% Triton X-100 into cells at room temperature, followed by PBS washing for three times. After blocking with 5% BSA for 30 min, the cells were treated by the primary EGFR antibody for overnight incubation at 4 °C and subsequent treatment of the secondary antibody for 3 h at ambient temperature.Western blotting
A431 cells were seeded at a density of 6 × 105 cell in a 6-well plate overnight. Cells were treated with vehicle control, EGFR inhibitor 1b, or complex 2f (in 0.1% DMSO) in 1% FBS medium for 12 h incubation. Cells were thereafter lysed to collect protein for each sample. Western blotting analysis was performed as described.Knockdown assay
A431 cells were seeded in 6-well plate at 80% confluences in DMEM medium for 24 h incubation. EGFR siRNA (5′-GGCACGAGUAACAAGCUCAdTdT-3′ (sense), 5′-UGAGCUUGUUACUC GUGCCdTdT-3′ (antisense)) and Lipo3000 reagent were gently mixed and let for 20 min incubation at ambient temperature. Afterwards, the siRNA/Lipo3000 mixture (500 μL) was added into each well. Cells were next kept in a CO2 incubator for 48 h post-transfection at 37 °C before further experiment.Luciferase reporter assay
The inhibition of AP-1 activity was examined by a luciferase reporter assay (Promega, Madison, WI, USA). Briefly, A431 cells were seeded in a culture dish at a density of 6 × 105 cell for overnight incubation. AP-1-luciferase plasmid was next used to co-transfect the cells using TurboFect Transfection Reagent in a serum-free DMEM medium. The transfected cells were subsequently seeded in a 24-well plate and followed by treatment of 2f at indicated concentrations for 6 h in 1% FBS medium. Passive Lysis Buffer (PLB) (160 μL) was added into the transfected cells for lysis. Next, equal volume of luciferase reporter reagent (LAR) and the cell lysates (50 μL) were added and mixed in a 96-well plate. The transcriptional activity was thus measured by determining the activity of firefly luciferase in SpectraMax M5 microplate reader (Molecular Devices).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Hong Kong Baptist University (FRG2/17–18/003), the Health and Medical Research Fund (HMRF/14150561), the National Natural Science Foundation of China (22077109 and 21775131), the Hong Kong Baptist University Century Club Sponsorship Scheme 2020, the Interdisciplinary Research Matching Scheme (RC–IRMS/16–17/03), Interdisciplinary Research Clusters Matching Scheme (RC–IRCs/17–18/03), Collaborative Research Fund (C5026–16G), SKLEBA and HKBU Strategic Development Fund (SKLP_1920_P02), the Science and Technology Development Fund, Macau SAR (File no. 0072/2018/A2), the University of Macau (MYRG2018–00187–ICMS and MYRG2019–00002–ICMS).Notes and references
- C. J. Adams and T. J. Meade, Chem. Sci., 2020, 11, 2524–2530 RSC.
- T. Lammers, S. Aime, W. E. Hennink, G. Storm and F. Kiessling, Acc. Chem. Res., 2011, 44, 1029–1038 CrossRef CAS.
- K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- J. D. Brown and A. G. Winterstein, J. Clin. Med., 2019, 8, 989 CrossRef CAS.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS.
- J. V. Jokerst and S. S. Gambhir, Acc. Chem. Res., 2011, 44, 1050–1060 CrossRef CAS.
- Y. Xia, W. Li, C. M. Cobley, J. Chen, X. Xia, Q. Zhang, M. Yang, E. C. Cho and P. K. Brown, Acc. Chem. Res., 2011, 44, 914–924 CrossRef CAS.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 24–36 CrossRef.
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2004, 56, 1649–1659 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2014, 115, 327–394 CrossRef.
- R. Liu, C. Hu, Y. Yang, J. Zhang and H. Gao, Acta Pharm. Sin. B, 2019, 9, 410–420 CrossRef.
- C. Chu, M. Su, J. Zhu, D. Li, H. Cheng, X. Chen and G. Liu, Theranostics, 2019, 9, 3134 CrossRef CAS.
- C. Xu, F. Pu, J. Ren and X. Qu, Chem. Commun., 2020, 56, 5295–5298 RSC.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670–6683 RSC.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS.
- E.-J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS.
- Y. Yuan, R. T. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2554 CrossRef CAS.
- R. Weinstain, E. Segal, R. Satchi-Fainaro and D. Shabat, Chem. Commun., 2010, 46, 553–555 RSC.
- G. Nkepang, M. Bio, P. Rajaputra, S. G. Awuah and Y. You, Bioconjugate Chem., 2014, 25, 2175–2188 CrossRef CAS.
- H. Yuan, H. Chong, B. Wang, C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, J. Am. Chem. Soc., 2012, 134, 13184–13187 CrossRef CAS.
- M. Bio, P. Rajaputra, G. Nkepang, S. G. Awuah, A. M. Hossion and Y. You, J. Med. Chem., 2013, 56, 3936–3942 CrossRef CAS.
- T. Zou, C. T. Lum, S. S. Y. Chui and C. M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930–2933 CrossRef CAS.
- C. R. Cardoso, M. r. V. Lima, J. Cheleski, E. J. Peterson, T. Venâncio, N. P. Farrell and R. M. Carlos, J. Med. Chem., 2014, 57, 4906–4915 CrossRef CAS.
- R. R. Ye, Z. F. Ke, C. P. Tan, L. He, L. N. Ji and Z. W. Mao, Chem.–Eur. J., 2013, 19, 10160–10169 CrossRef CAS.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef CAS.
- D.-L. Ma, C. Wu, H. Liu, K.-J. Wu and C.-H. Leung, J. Mater. Chem. B, 2020, 8, 3249–3260 RSC.
- C. Wu, K.-J. Wu, J.-B. Liu, X.-M. Zhou, C.-H. Leung and D.-L. Ma, Chem. Commun., 2019, 55, 6353–6356 RSC.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- M. Yu, Q. Zhao, L. Shi, F. Li, Z. Zhou, H. Yang, T. Yi and C. Huang, Chem. Commun., 2008, 2115–2117 RSC.
- T. C. O'Riordan, A. V. Zhdanov, G. V. Ponomarev and D. B. Papkovsky, Anal. Chem., 2007, 79, 9414–9419 CrossRef.
- C.-N. Ko, G. Li, C.-H. Leung and D.-L. Ma, Coord. Chem. Rev., 2019, 381, 79–103 CrossRef CAS.
- V. Venkatesh, R. Berrocal-Martin, C. J. Wedge, I. Romero-Canelón, C. Sanchez-Cano, J.-I. Song, J. P. C. Coverdale, P. Zhang, G. J. Clarkson, A. Habtemariam, S. W. Magennis, R. J. Deeth and P. J. Sadler, Chem. Sci., 2017, 8, 8271–8278 RSC.
- A. B. Caballero, L. Cardo, S. Claire, J. S. Craig, N. J. Hodges, A. Vladyka, T. Albrecht, L. A. Rochford, Z. Pikramenou and M. J. Hannon, Chem. Sci., 2019, 10, 9244–9256 RSC.
- K. K.-W. Lo, Acc. Chem. Res., 2020, 53, 32–44 CrossRef CAS.
- C. Wang, Z. Wang, T. Zhao, Y. Li, G. Huang, B. D. Sumer and J. Gao, Biomaterials, 2018, 157, 62–75 CrossRef CAS.
- L. G. Meimetis, J. C. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS.
- P. Saraon, J. Snider, Y. Kalaidzidis, L. E. Wybenga-Groot, K. Weiss, A. Rai, N. Radulovich, L. Drecun, N. Vučković, A. Vučetić, V. Wong, B. Thériault, N.-A. Pham, J. H. Park, A. Datti, J. Wang, S. Pathmanathan, F. Aboualizadeh, A. Lyakisheva, Z. Yao, Y. Wang, B. Joseph, A. Aman, M. F. Moran, M. Prakesch, G. Poda, R. Marcellus, D. Uehling, M. Samaržija, M. Jakopović, M.-S. Tsao, F. A. Shepherd, A. Sacher, N. Leighl, A. Akhmanova, R. Al-awar, M. Zerial and I. Stagljar, Nat. Chem. Biol., 2020, 16, 577–586 CrossRef CAS.
- Y. Jia, C. M. Quinn, S. Kwak and R. V. Talanian, Curr. Drug Discovery Technol., 2008, 5, 59–69 CrossRef CAS.
- M. Gao, F. Yu, C. Lv, J. Choo and L. Chen, Chem. Soc. Rev., 2017, 46, 2237–2271 RSC.
- S. Arounaguiri and B. G. Maiya, Inorg. Chem., 1999, 38, 842–843 CrossRef CAS.
- K. Y. Pu, K. Li and B. Liu, Adv. Funct. Mater., 2010, 20, 2770–2777 CrossRef CAS.
- C. Wu, K. Vellaisamy, G. Yang, Z.-Z. Dong, C.-H. Leung, J.-B. Liu and D.-L. Ma, Dalton Trans., 2017, 46, 6677–6682 RSC.
- K. Vellaisamy, G. Li, C.-N. Ko, H.-J. Zhong, S. Fatima, H.-Y. Kwan, C.-Y. Wong, W.-J. Kwong, W. Tan and C.-H. Leung, Chem. Sci., 2018, 9, 1119–1125 RSC.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS.
- W. Wang, C. Yang, S. Lin, K. Vellaisamy, G. Li, W. Tan, C. H. Leung and D. L. Ma, Chem.–Eur. J., 2017, 23, 4929–4935 CrossRef CAS.
- A. Gazit, P. Yaish, C. Gilon and A. Levitzki, J. Med. Chem., 1989, 32, 2344–2352 CrossRef CAS.
- J. S.-Y. Lau, P.-K. Lee, K. H.-K. Tsang, C. H.-C. Ng, Y.-W. Lam, S.-H. Cheng and K. K.-W. Lo, Inorg. Chem., 2008, 48, 708–718 CrossRef.
- S. M. King, S. Claire, R. I. Teixeira, A. N. Dosumu, A. J. Carrod, H. Dehghani, M. J. Hannon, A. D. Ward, R. Bicknell, S. W. Botchway, N. J. Hodges and Z. Pikramenou, J. Am. Chem. Soc., 2018, 140, 10242–10249 CrossRef CAS.
- A. Kazama, Y. Imai, Y. Okayasu, Y. Yamada, J. Yuasa and S. Aoki, Inorg. Chem., 2020, 59, 6905–6922 CrossRef CAS.
- E. C. Glazer, D. Magde and Y. Tor, J. Am. Chem. Soc., 2005, 127, 4190–4192 CrossRef CAS.
- D. Magde, M. D. Magde and E. C. Glazer, Coord. Chem. Rev., 2016, 306, 447–467 CrossRef CAS.
- M. T. Jarlstad Olesen, R. Walther, P. P. Poier, F. Dagnæs-Hansen and A. N. Zelikin, Angew. Chem., Int. Ed., 2020, 132, 7460 CrossRef.
- D. Zhang, Y. Huang, X. You, L. Wang, G. Zhang, S. Gui, Y. Jin and R. Zhao, Angew. Chem., Int. Ed., 2020, 132, 10128 CrossRef.
- C. Mattsson, T. Andreasson, N. Waters and C. Sonesson, J. Med. Chem., 2012, 55, 9735–9750 CrossRef CAS.
- C. Mattsson, T. Andreasson, N. Waters and C. Sonesson, J. Med. Chem., 2013, 56, 4130–4133 CrossRef CAS.
- R. Tjin Tham Sjin, K. Lee, A. O. Walter, A. Dubrovskiy, M. Sheets, T. S. Martin, M. T. Labenski, Z. Zhu, R. Tester, R. Karp, A. Medikonda, P. Chaturvedi, Y. Ren, H. Haringsma, J. Etter, M. Raponi, A. D. Simmons, T. C. Harding, D. Niu, M. Nacht, W. F. Westlin, R. C. Petter, A. Allen and J. Singh, Mol. Cancer Ther., 2014, 13, 1468–1479 CrossRef.
- M. Cichocki, H. Szaefer, V. Krajka-Kuźniak and W. Baer-Dubowska, Mol. Cell. Biochem., 2014, 396, 221–228 CrossRef CAS.
- L. Yang, S. Ying, S. Hu, X. Zhao, M. Li, M. Chen, Y. Zhu, P. Song, L. Zhu, T. Jiang, H. An, N. A. Yousafzai, W. Xu, Z. Zhang, X. Wang, L. Feng and H. Jin, Signal Transduction Targeted Ther., 2019, 4, 25 CrossRef.
- S.-H. I. Ou, J. Cui, A. B. Schrock, M. E. Goldberg, V. W. Zhu, L. Albacker, P. J. Stephens, V. A. Miller and S. M. Ali, Lung Cancer, 2017, 108, 228–231 CrossRef.
- C. Zhang, M. Liu, S. Liu, H. Yang, Q. Zhao, Z. Liu and W. He, Inorg. Chem., 2018, 57, 10625–10632 CrossRef CAS.
- R. I. Dmitriev, H. M. Ropiak, G. V. Ponomarev, D. V. Yashunsky and D. B. Papkovsky, Bioconjugate Chem., 2011, 22, 2507–2518 CrossRef CAS.
- S. Meloche and J. Pouysségur, Oncogene, 2007, 26, 3227–3239 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04576d |
| ‡ The authors contribute equally in this work. |
| This journal is © The Royal Society of Chemistry 2020 |