
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA hierarchical assembly strategy for near-infrared photothermal conversion: unconventional heterogeneous metalla[2]catenanes†‡
Ye
Lu
 *,
Dong
Liu
,
Yue-Jian
Lin
and
Guo-Xin
Jin
*
*,
Dong
Liu
,
Yue-Jian
Lin
and
Guo-Xin
Jin
*
State Key Laboratory of Molecular Engineering of Polymers, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Department of Chemistry, Fudan University, 2005 Songhu road, Shanghai, 200438, P. R. China
First published on 21st September 2020
Abstract
Herein, we report a hierarchical assembly strategy for constructing heterogeneous half-sandwich organometallic D–A (D = π-donor, A = π-acceptor) interlocked structures, and their application in near-infrared (NIR) photothermal conversion. Thienothiophene and diketopyrrolopyrrole groups were selected as the D and A units, leading to two homogeneous metalla[2]catenanes with D–D–D–D and A–A–A–A stacks, respectively. By the ordered secondary assembly of homogeneous metalla[2]catenanes, two unprecedented heterogeneous D–A metalla[2]catenanes comprising an unusual mixed D–A–D–D and unconventional D–A–A–A stacks were realized by the combination of multiple noncovalent interactions, as all demonstrated by a detailed X-ray crystallographic study. Benefiting from the mixed D–A stacking modes, NIR absorption of heterogeneous D–A metalla[2]catenanes is significantly enhanced in contrast to homogeneous metalla[2]catenanes. Thanks to the enhanced NIR absorption and the fluorescence quenching effect from half-sandwich organometallic fragments, heterogeneous D–A metalla[2]catenanes displayed high-performance NIR photothermal conversion properties (η = 27.3%).
Introduction
Research into mechanically interlocked molecules, such as catenanes, rotaxanes, and molecular knots, has been gaining momentum in recent decades.1 Electrostatic interactions between electron-rich (π-donor, D) and electron-poor (π-acceptor, A) aromatics are an important driving force in the self-assembly of interlocked structures.2 Conventional wisdom and the current understanding of D–A interactions have thus far led to most of these catenanes being designed and synthesized with the (presumably most favorable) alternating parallel arrangement of π-rich and π-deficient units, providing D–A–D–A stacks in the final structure of organic [2]catenanes.3 However, the advent of dynamic combinatorial libraries, presented by Sanders et al., provided access to a larger variety of organic unusual donor–acceptor [2]catenanes, such as D–A–D–D, A–D–D–A, D–A–A–D or A–A–D–A stacks.4 Metalla[2]catenanes based on coordination-driven self-assembly have long been limited to combinations of identical organometallic macrocycles or rectangles, which lead to symmetrical D–D–D–D or A–A–A–A stacks.5 The high-yield preparation of some unconventional [2]catenanes, e.g. those based on unconventional D–A–A–A stacks, is extremely challenging even beyond the field of coordination-driven self-assembly.Functional organic materials capable of photothermal conversion, i.e. that generate heat from infrared light, have attracted significant interest since the initial demonstrations of their application in a number of fields, especially on photothermal therapy and photothermal/photoacoustic imaging.6 In order to pursue more high-performance photothermal conversion materials, extensive efforts are paid from two aspects: one is to enhance near-infrared (NIR) or infrared absorption through extending the molecular conjugation or linking D and A fragments; the other is to inhibit the radiative transition process by enhancing the quenching effect.7 In fact, heterogeneous D–A metalla[2]catenanes based on half-sandwich organometallic Cp*Rh (Cp* = pentamethyl-cyclopentadienyl) fragments probably could reach these two aims at the same time. D and A units can be linked together by coordination bond of D–A metalla[2]catenanes and lead to alternating parallel arrangement.8 Meantime, Cp*Rh fragments have inherent ability to quench fluorescence.9
Herein, we present a hierarchical self-assembly strategy for constructing heterogeneous organometallic D–A interlocked structures based on Cp*Rh fragments. We selected thienothiophene and diketopyrrolopyrrole groups as the D and A units, leading to two homogeneous metalla[2]catenanes with D–D–D–D and A–A–A–A stacks, respectively. By the ordered secondary assembly of homogeneous metalla[2]catenanes, two unprecedented heterogeneous D–A metalla[2]catenanes comprising unusual mixed D–A–D–D and D–A–A–A stacks were realized by the combination of multiple noncovalent interactions. Benefiting from the mixed D–A stacking modes and strong D–A interaction, heterogeneous D–A metalla[2]catenanes displayed stronger NIR absorption than homogeneous metalla[2]catenanes. We further took advantage of the enhanced NIR absorption to realize NIR photothermal conversion based on heterogeneous D–A metalla[2]catenanes.
Results and discussion
Homogeneous [2]catenanes
Diketopyrrolopyrrole groups, strong A units, have been widely applied in NIR organic optical materials.10 Thus, their incorporation into metallarectangles could potentially lead to strong D–A interactions. Following this logic, D–A heterogeneous metalla[2]catenanes may can be obtained by ordered self-assembly between a metallarectangle based on D units and another metallarectangle based on A units, namely so-called hierarchical self-assembly strategy.11 Hierarchical self-assembly is a multilevel and multistep self-assembly process that involves initial assembly of elementary molecular units into ordered secondary structures by noncovalent interactions, which has been used for the preparation of soft-matter nanoarchitectures and mechanically-interlocked M8L16 container.12 Hence, we initially prepared two types of metallarectangle, one based on D units, and the other based on A units. Thienothiophene groups were selected as strong D units to act as counterparts of the diketopyrrolopyrrole groups, in the form of the donor-containing pyridyl ligand L1 (L1 = 3,6-di(pyridin-4-yl)thieno[3,2-b]thiophene). We also designed another pyridyl ligand based on the diketopyrrolopyrrole A unit, namely L2 (3,6-di(pyridin-4-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione). The geometrical configurations of L1 and L2 are very similar, apart from their electron rich and poor natures, respectively (Fig. 1a).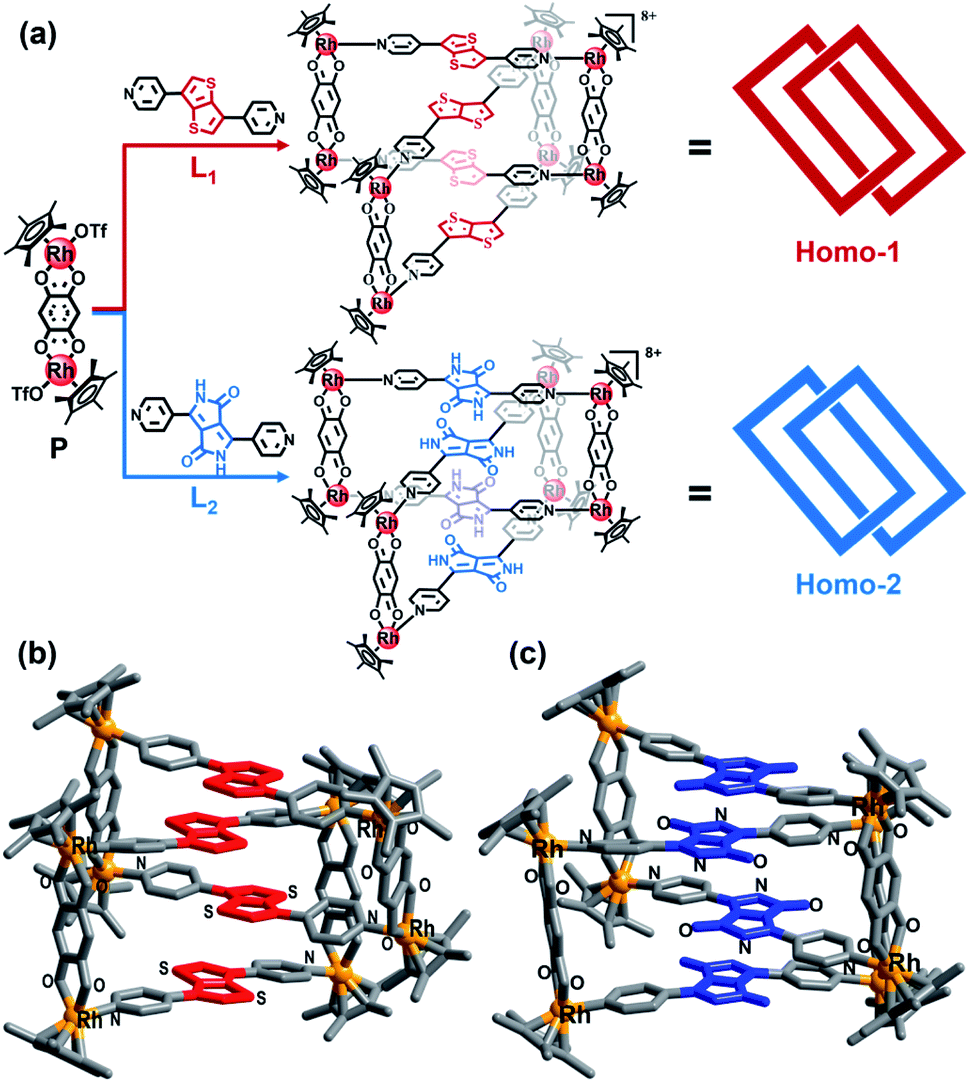 | ||
| Fig. 1 (a) The synthesis of homogeneous metalla[2]catenanes; single-crystal X-ray structures of Homo-1 (b) and Homo-2 (c); D units are shown in red, A units are shown in blue, hydrogen atoms and counter anions are omitted. | ||
The next step was to prepare the individual D and A metallarectangles. We chose the precursor P (based on 2,5-dihydroxy-1,4-benzoquinoneas) as the binuclear precursor in this work as its metal–metal distance is ca. 7.9 Å, close to twice the conventional distance of π–π stacking (7 Å) (Fig. 1a).13 Thanks to the optimized geometry and distance, a homo-[2]-catenanes (Homo-1) was obtained by the reaction of P and pyridyl ligand L1 in methanol, which was confirmed by single-crystal X-ray crystallographic analysis and ESI-MS (Fig. 1b and S28‡). The four planes of the bithienyl solution, groups are nearly parallel to each other and the assembly comprises a D–D–D–D stack (red in Fig. 1b). There is an equilibrium between homo-[2]catenanes Homo-1 and corresponding monomeric rectangle (MR) 1 in methanol solution. Upon dilution, the interlocked Homo-1 transforms to MR 1, which was further confirmed by ESI-MS, 1H, 13C, 1H–1H COSY and DOSY NMR spectroscopy (Scheme S1, Fig. S1–S8 and S29‡). Homo-2 were also obtained by the reaction of P and L2 in methanol solution, which was confirmed by single-crystal X-ray crystallographic analysis and ESI-MS (Fig. 1c and S30‡). The planes of the four electron-deficient units are also nearly parallel to each other, comprising an A–A–A–A stack (blue in Fig. 1c). Similar to Homo-1, the interlocked Homo-2 transforms to corresponding MR 2 upon dilution, which was proven by ESI-MS 1H, 13C, 1H–1H COSY and DOSY NMR spectroscopy (Scheme S1, Fig. S9–S16 and S31‡).
The transformation between homo-[2]-catenanes and MRs also could be observed via absorption spectra, as the strong π–π interaction leads to the narrowing of HOMO–LUMO gaps and remarkable red-shifts in absorption. As shown in Fig. S34,‡ the absorption of MR 1 in methanol solution (0.5 mM) is mainly in the ultraviolet and blue-light region (<500 nm). Upon formation of the interlocked structure (Homo-1), the absorption in the visible light region was significantly enhanced (500–700 nm). This type of red-shift effect caused by π–π interactions can be more clearly observed in the case of Homo-2, the absorption region of which extends to the NIR region (Fig. S35‡).
Heterogeneous [2]catenanes
In an attempt to further enhance the NIR absorption, we attempted to prepare mixed D–A stacking modes by hierarchical self-assembly. According to the conventional understanding of D–A interactions, the most favorable parallel arrangement of π-rich and π-deficient units is the D–A–D–A stacking pattern, containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of π-rich and π-deficient units. Thus, we initially stirred a 1:1 mixture (8.0 mM) of Homo-1 and Homo-2 in an NMR tube with CD3OD for 12 h at room temperature. A range of new signals were observed in the 1H NMR spectrum, indicating the formation of a new compound (Fig. S17 and S18‡). However, the new signals were very crowded and difficult to confidently assign. Moreover, all attempts to grow single crystals were unsuccessful. Four stacking modes potentially exist in such a dimer, including the conventional D–A–D–A, and unconventional D–A–A–D, A–D–D–A and D–D–A–A modes, which cannot be distinguished by NMR alone. However, when the ratio of π-rich and π-deficient units is changed as 3:1 or 1:3, the possible stacking modes would be reduced from four to two. So, we decided to adjust the feed ratio for reducing isomers.
:1 ratio of π-rich and π-deficient units. Thus, we initially stirred a 1:1 mixture (8.0 mM) of Homo-1 and Homo-2 in an NMR tube with CD3OD for 12 h at room temperature. A range of new signals were observed in the 1H NMR spectrum, indicating the formation of a new compound (Fig. S17 and S18‡). However, the new signals were very crowded and difficult to confidently assign. Moreover, all attempts to grow single crystals were unsuccessful. Four stacking modes potentially exist in such a dimer, including the conventional D–A–D–A, and unconventional D–A–A–D, A–D–D–A and D–D–A–A modes, which cannot be distinguished by NMR alone. However, when the ratio of π-rich and π-deficient units is changed as 3:1 or 1:3, the possible stacking modes would be reduced from four to two. So, we decided to adjust the feed ratio for reducing isomers.
We initially changed the feed ratio from 1:1 to 3:1 by stirring a 3:1 mixture (8.0 mM) of Homo-1 and Homo-2 in an NMR tube under the same conditions as described above (Fig. 2a). After 10 min, signals for Homo-1, MR 1, Homo-2 and MR 2 could be clearly observed in the 1H NMR spectrum (Fig. 3, S19 and S20‡). After 12 h, we observed a range of new signals (Fig. 3). Although the new signals were highly complex because of low symmetry, DOSY NMR confirmed that they possessed the same diffusion coefficient (Fig. S21 and S22‡), indicating the formation of a new compound, denoted Hetero-3. According to the relative intensity of the signals of the benzoquinone unit, the proportion of Hetero-3 in 8.0 mM methanol solution exceeded 75%. Thanks to the high proportion of Hetero-3 in solution, we were able to grow single crystals of Hetero-3. As expected, a single-crystal X-ray crystallographic analysis confirmed the hetero-[2]catenanes structure of Hetero-3 and established its unusual D–A–D–D stack (Fig. 2b), which further proved by ESI-MS (Fig. S32‡). Due to its mixed D–A stacking mode, the absorption of Hetero-3 was further shifted to the low-energy region and extended into the NIR region, in marked contrast to the absorption profile of Homo-1 (Fig. S34‡). This observation suggested that the mixed D–A stacking mode in metalla[2]catenanes may be an effective means to improve absorption in the NIR region.
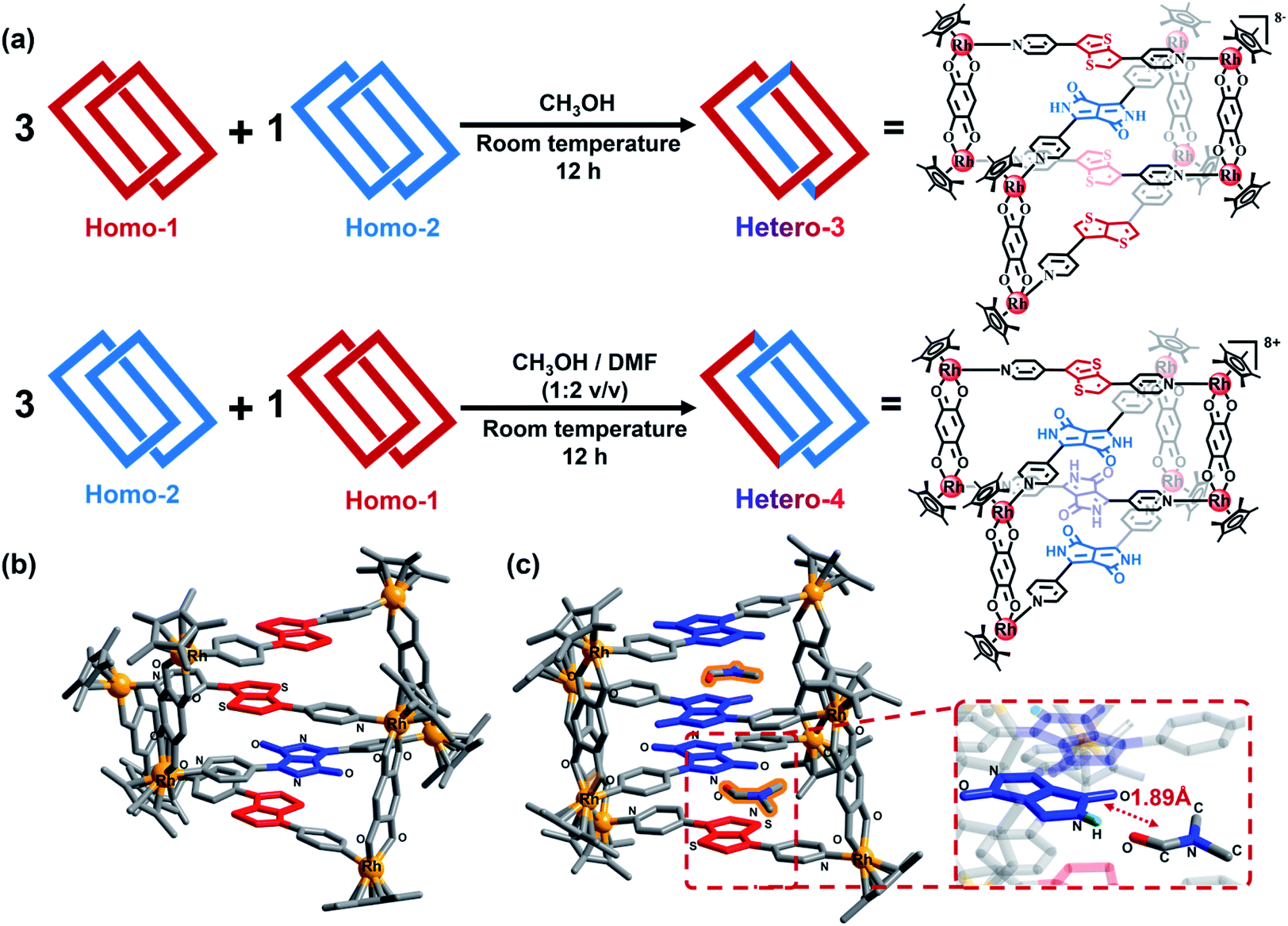 | ||
| Fig. 2 (a) Hierarchical self-assembly for the synthesis of heterogeneous D–A metalla[2]catenanes; single-crystal X-ray structures of Hetero-3 (b) and Hetero-4 (c); the DMF molecule is highlighted with an orange glow, the D-unit is shown in red and the A-unit is shown in blue, the inset is hydrogen bond in Hetero-4, H atoms are shown in green, other hydrogen atoms and counter anions are omitted. | ||
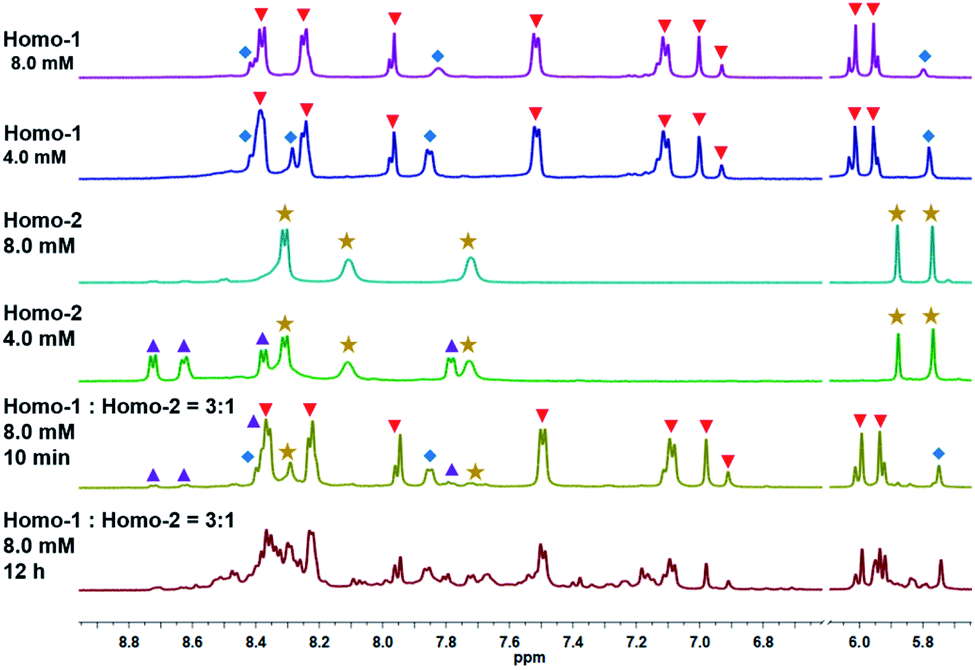 | ||
Fig. 3 Partial 1H NMR spectrum of Homo-1 ( ) + 1 ( ) + 1 ( ) (CD3OD, 8.0 mM), Homo-1 ( ) (CD3OD, 8.0 mM), Homo-1 ( ) + 1 ( ) + 1 ( ) (CD3OD, 4.0 mM), Homo-2 ( ) (CD3OD, 4.0 mM), Homo-2 ( ) + 2 ( ) + 2 ( ) (CD3OD, 8.0 mM), Homo-2 ( ) (CD3OD, 8.0 mM), Homo-2 ( ) + 2 ( ) + 2 ( ) (CD3OD, 4.0 mM), Homo-1 + Homo-2 (the ratio of Homo-1:Homo-2 is 3:1, 10 min) (CD3OD, 8.0 mM) and Homo-1 + Homo-2 (the ratio of Homo-1:Homo-2 is 3:1, 12 h) (CD3OD, 8.0 mM). ) (CD3OD, 4.0 mM), Homo-1 + Homo-2 (the ratio of Homo-1:Homo-2 is 3:1, 10 min) (CD3OD, 8.0 mM) and Homo-1 + Homo-2 (the ratio of Homo-1:Homo-2 is 3:1, 12 h) (CD3OD, 8.0 mM). | ||
Following these results, we attempted to prepare the corresponding hetero-[2]catenanes with a mixed A–D–A–A stack through a similar method. We thus adjusted the feed ratio by using a 1:3 mixture (8.0 mM) of Homo-1 and Homo-2. However, to our disappointment, no obvious change was observed in 1H spectrum even after three days (Fig. S23 and S24‡). Despite this result, we attempted to crystallize this mixture. In our experience, DMF improve the solubility of assemblies, and assist the growth of single crystals during crystallization.13 Hence, similar to the experiments described above, DMF was added to the solution. To our surprise, black block single crystals appeared in the bottom of the tube after two weeks. A single-crystal X-ray crystallographic analysis established the new compound Hetero-4 to also have a hetero-[2]catenanes structure, with a D/A ratio of 1:3, which confirmed by ESI-MS (Fig. S33‡). Moreover, the stacking mode of Hetero-4 was not the expected A–D–A–A arrangement, but an unprecedented D–A–A–A form (Fig. 2c). This assembly can be attributed to the presence of DMF, as obvious hydrogen bonding interactions exist between the N–H groups of the A units and the oxygen atom of DMF, with distances of only ca. 1.89 Å (Fig. 2c). We also observed CH⋯π interactions between the H atoms of the DMF and the pyridyl group of certain pyridyl ligands (Fig. S37‡). The DMF molecules (highlighted in orange in Fig. 2c) act like nails that staple two metallarectangles together and allow formation of the abnormal D–A [2]catenanes. To further confirm the effect of DMF, we gradually added d7-DMF to a solution of the Homo-1/Homo-2 mixture. In 1H NMR spectrum, some signals were found to grow gradually with increasing amounts of d7-DMF (Fig. S25 and S26‡). A DOSY NMR spectrum showed that the growing signals have the same diffusion coefficient (Fig. S27‡). According to the relative intensity of the benzoquinone signal, the proportion of Hetero-4 in an 8.0 mM CD3OD/d7-DMF (1:2 v/v) solution was ca. 80%. Moreover, Hetero-4 displayed stronger NIR absorption (650–750 nm) than Hetero-3 due to its higher proportion of diketopyrrolopyrrole groups (Fig. S35‡).
NIR photothermal conversion
Due to their strong NIR absorption, these D–A metalla[2]catenanes were found to have fascinating photothermal conversion properties. Under 730 nm laser irradiation (0.5 W cm−2) for 250 s, the temperature of Hetero-4 and Hetero-3 sharply increased, reaching temperatures as high as 70 °C and 55 °C, respectively, markedly higher than those of Homo-2 (45 °C) and Homo-1 (37 °C) under the same conditions (Fig. 4a). The NIR-photothermal effect of crystals of Hetero-4 upon exposure to a 730 nm laser can also be clearly observed from Fig. 4c. The cooling curve of Hetero-4 is shown in Fig. S36,‡ from which the photothermal conversion efficiency was calculated (details are shown in ESI‡) to be 27.3%, which is better than the reported organic D–A cocrystal materials.7a This enhanced photothermal temperature and the higher photothermal conversion of heterogeneous metalla[2]catenanes can be attributed to the mixed D–A stacking mode and the fluorescence quenching effect from Cp*Rh fragments. In contrast to cocrystal materials, D–A metalla[2]catenanes still display satisfactory photothermal conversion properties in dissolved solution.7a As shown in Fig. 4b and d, under the same irradiation condition (300 s), the temperature of Hetero-3 in 8.0 mM methanol solution and Hetero-4 in 8.0 mM CH3OH/DMF (1:2 v/v) reach maxima at 49 °C and 58 °C, respectively. As a contrast, physical mixtures of L1 and L2 (L1:L2 = 3:1 or 1:3) in 8.0 mM DMSO solution experience temperature increases of only 2.5 °C and 3 °C under the same conditions (Fig. 4b). This results further confirm D–A metalla[2]catenanes as a promising platform for the development of NIR photothermal conversion materials.
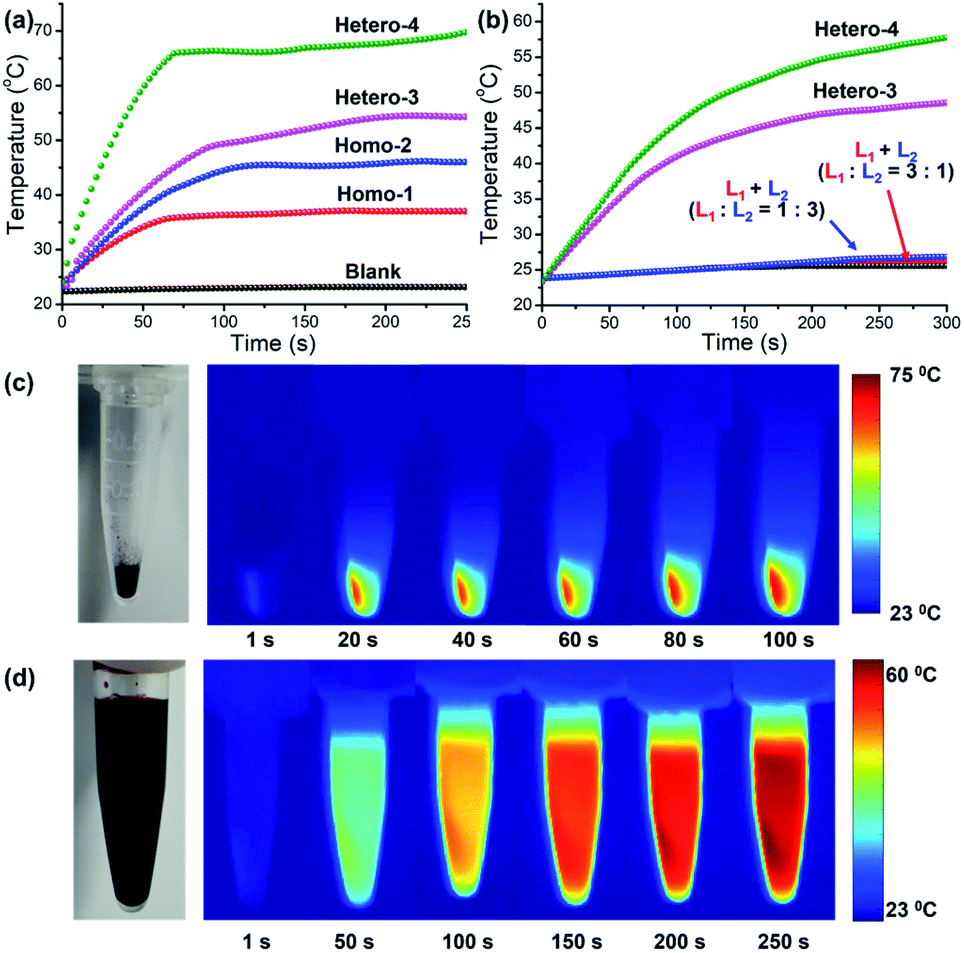 | ||
| Fig. 4 (a) Photothermal conversion behavior of Homo-1, Homo-2, Hetero-3 and Hetero-4 (crystal) under 730 nm laser irradiation; (b) photothermal conversion behavior of Hetero-3 in 8.0 mM methanol solution, Hetero-4 in 8.0 mM CH3OH/DMF (1:2 v/v) and the physical mixture of L1 and L2 (L1:L2 = 3:1 or 1:3) in 8.0 mM DMSO solution under 730 nm laser irradiation; IR thermal images of Hetero-4 crystals (c) and in 8.0 mM CD3OD/d7-DMF (1:2 v/v) (d) under 730 nm laser irradiation. | ||
Conclusions
Hierarchical self-assembly has inherent advantages for the design of heterogeneous interlocked structures. We have employed an hierarchical self-assembly strategy to engineer heterogeneous D–A metalla[2]catenanes, leading to unconventional mixed D–A–D–D and D–A–A–A stacks in metalla[2]catenanes. Benefiting from the mixed D–A stacking modes and the fluorescence quenching effect from Cp*Rh fragments, heterogeneous D–A metalla[2]catenanes displayed high-performance NIR absorption and NIR photothermal conversion properties in crystalline and solution states. These results will not only allow chemists to design and synthesize complex structures that were previously inaccessible, but also provide promising candidates for materials science applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation of China (21531002, 21720102004, 21801045, 22031003) and the Shanghai Science and Technology Committee (19DZ2270100).Notes and references
- (a) R. S. Forgan, J. P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS; (b) G. Gil-Ramirez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef CAS; (c) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC; (d) J. D. Crowley, S. M. Goldup, A. L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC; (e) J. R. J. Maynard and S. M. Goldup, Chem, 2020, 6, 914–1932 CrossRef; (f) W. X. Gao, H. J. Feng, B. B. Guo, Y. Lu and G. X. Jin, Chem. Rev., 2020, 120, 6288–6325 CrossRef CAS; (g) S. L. Huang, T. S. A. Hor and G. X. Jin, Coord. Chem. Rev., 2017, 333, 1–26 CrossRef CAS.
- (a) J. F. Stoddart and H. R. Tseng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4797–4800 CrossRef CAS; (b) P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1396–1399 CrossRef; (c) D. G. Hamilton, J. K. M. Sanders, J. E. Davies, W. Clegg and S. J. Teat, Chem. Commun., 1997, 897–898 RSC; (d) K. Kitajima, T. Ogoshi and T. Yamagishi, Chem. Commun., 2014, 50, 2925–2927 RSC.
- (a) A. C. Fahrenbach, C. J. Bruns, H. Li, A. Trabolsi, A. Coskun and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 482–493 CrossRef CAS; (b) J. M. Lehn, Supramolecular Chemistry. Concepts and Perspectives, Wiley-VCH, Weinheim, Germany, 1995 CrossRef.
- (a) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS; (b) F. B. L. Cougnon, H. Y. Au-Yeung, G. D. Pantos and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3198–3207 CrossRef CAS; (c) H. Y. Au-Yeung, G. D. Pantos and J. K. M. Sanders, J. Am. Chem. Soc., 2009, 131, 16030–16031 CrossRef CAS; (d) H. Y. Au-Yeung, G. D. Pantos and J. K. M. Sanders, Angew. Chem., Int. Ed., 2010, 49, 5331–5334 CrossRef CAS; (e) H. Y. Au-Yeung, G. D. Pantos and J. K. M. Sanders, J. Org. Chem., 2011, 76, 1257–1268 CrossRef CAS.
- (a) M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645–5647 CrossRef CAS; (b) W. L. Shan, Y. J. Lin, F. E. Hahn and G. X. Jin, Angew. Chem., Int. Ed., 2019, 58, 5882–5886 CrossRef CAS; (c) H. Lee, P. Elumalai, N. Singh, H. Kim, S. U. Lee and K. W. Chi, J. Am. Chem. Soc., 2015, 137, 4674–4677 CrossRef CAS; (d) S. A. Boer, R. P. Cox, M. J. Beards, H. X. Wang, W. A. Donald, T. D. M. Bell and D. R. Turner, Chem. Commun., 2019, 55, 663–666 RSC; (e) Y. Lu, D. Liu, Y. J. Lin, Z. H. Li and G. X. Jin, Natl. Sci. Rev., 2020 DOI:10.1093/nsr/nwaa164; (f) M. M. Siddiqui, R. Saha and P. S. Mukherjee, Inorg. Chem., 2019, 58, 4491–4499 CrossRef CAS; (g) Y. W. Zhang, S. Bai, Y. Y. Wang and Y. F. Han, J. Am. Chem. Soc., 2020, 142, 13614–13621 CrossRef CAS; (h) T. Feng, X. Li, Y. Y. An, S. Bai, L. Y. Sun, Y. Li, Y. Y. Wang and Y. F. Han, Angew. Chem., Int. Ed. Engl., 2020, 59, 13516–13520 CrossRef CAS; (i) S. Mukherjee and P. S. Mukherjee, Chem. Commun., 2014, 50, 2239–2248 RSC.
- (a) Y. J. Liu, P. Bhattarai, Z. F. Dai and X. Y. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC; (b) K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012–11042 CrossRef CAS; (c) Y. Y. Li, J. Wang, F. Zhao, B. Bai, G. J. Nie, A. E. Nel and Y. L. Zhao, Natl. Sci. Rev., 2018, 5, 365–388 CrossRef CAS.
- (a) Y. Wang, W. G. Zhu, W. N. Du, X. F. Liu, X. T. Zhang, H. L. Dong and W. P. Hu, Angew. Chem., Int. Ed., 2018, 57, 3963–3967 CrossRef CAS; (b) Q. Song, Y. Jiao, Z. Q. Wang and X. Zhang, Small, 2016, 12, 24–31 CrossRef CAS; (c) A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS.
- (a) Y. Lu, H. N. Zhang and G. X. Jin, Acc. Chem. Res., 2018, 51, 2148–2158 CrossRef CAS; (b) Y. Lu, Y. X. Deng, Y. J. Lin, Y. F. Han, L. H. Weng, Z. H. Li and G. X. Jin, Chem, 2017, 3, 110–121 CrossRef CAS.
- (a) V. Vajpayee, Y. H. Song, T. R. Cook, H. Kim, Y. Lee, P. J. Stang and K. W. Chi, J. Am. Chem. Soc., 2011, 133, 19646–19649 CrossRef CAS; (b) Y. F. Han and G. X. Jin, Acc. Chem. Res., 2014, 47, 3571–3579 CrossRef CAS.
- M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58–77 RSC.
- S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS.
- (a) W. M. Bloch, J. J. Holstein, B. Dittrich, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2018, 57, 5534–5538 CrossRef CAS; (b) X. Z. Yan, T. R. Cook, J. B. Pollock, P. F. Wei, Y. Y. Zhang, Y. H. Yu, F. H. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 4460–4463 CrossRef CAS; (c) R. F. Wen, S. S. Xu, D. L. Zhao, L. X. Yang, X. H. Ma, W. Liu, Y. C. Lee and R. G. Yang, Natl. Sci. Rev., 2018, 5, 878–887 CrossRef CAS.
- Y. Lu, Y. J. Lin, Z. H. Li and G. X. Jin, Chin. J. Chem., 2018, 36, 106–111 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Dr Pierre Dixneuf for his outstanding contribution to organometallic chemistry and catalysis. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1946051 (Homo-1), 1946991 (Homo-2), 1946053 (Hetero-3), 1946054 (Hetero-4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04523c |
| This journal is © The Royal Society of Chemistry 2020 |