
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregation-induced photocatalytic activity and efficient photocatalytic hydrogen evolution of amphiphilic rhodamines in water†
Hajime
Shigemitsu
 *abc,
Youhei
Tani
a,
Tomoe
Tamemoto
a,
Tadashi
Mori
a,
Xinxi
Li
d,
Yasuko
Osakada
de,
Mamoru
Fujitsuka
d and
Toshiyuki
Kida
*a
*abc,
Youhei
Tani
a,
Tomoe
Tamemoto
a,
Tadashi
Mori
a,
Xinxi
Li
d,
Yasuko
Osakada
de,
Mamoru
Fujitsuka
d and
Toshiyuki
Kida
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita 565-0871, Japan. E-mail: shigemitsu@chem.eng.osaka-u.ac.jp; kida@chem.eng.osaka-u.ac.jp
bFrontier Research Base for Global Young Researchers, Graduate School of Engineering, Osaka University, Suita 565-0871, Japan
cGlobal Center for Medical Engineering and Informatics, Osaka University, Suita 565-0871, Japan
dThe Institute of Scientific and Industrial Research (SANKEN), Osaka University, Mihogaoka 8-1, Ibaraki, Osaka 567-0047, Japan
eInstitute for Advanced Co-creation Studies, Osaka University, 1-1 Yamadagaoka, Suita, Osaka 565-0871, Japan
First published on 7th October 2020
Abstract
The development of photocatalysts is an essential task for clean energy generation and establishing a sustainable society. This paper describes the aggregation-induced photocatalytic activity (AI-PCA) of amphiphilic rhodamines and photocatalytic functions of the supramolecular assemblies. The supramolecular assemblies consisting of amphiphilic rhodamines with octadecyl alkyl chains exhibited significant photocatalytic activity under visible light irradiation in water, while the corresponding monomeric rhodamines did not exhibit photocatalytic activity. The studies on the photocatalytic mechanism by spectroscopic and microscopic analyses clearly demonstrated the AI-PCA of the rhodamines. Moreover, the supramolecular assemblies of the rhodamines exhibited excellent photocatalytic hydrogen evolution rates (up to 5.9 mmol g−1 h−1).
Introduction
Photocatalysts are promising materials for the conversion of solar energy into storable chemical energy and are expected to contribute significantly to clean and renewable energy generation.1 In 1974, Fujishima and Honda reported photocatalytic water-splitting using a titanium dioxide electrode, demonstrating the possibility of artificial photosynthesis.2 Since then, a wide range of photocatalysts, based on inorganic,3 molecular,4 and polymeric5 compounds, have been actively developed. Besides their application in artificial photosynthesis, the redox reactivity of photocatalysts has been utilized for environmental remediation,6 organic synthesis,7 and photodynamic therapy.8 The emergence and development of new photocatalysts have contributed to the progress in artificial photosynthesis and generated new opportunities in the related fields.9Based on these backgrounds, we explored a new class of photocatalysts and focused on supramolecular assemblies. The photophysical properties of supramolecular assemblies are different from those of the constituting monomers because of the interaction between the adjacent molecules.10 Various characteristic aggregation-induced photophysical phenomena, such as aggregation-caused quenching (ACQ),11 aggregation-induced enhanced emission,12 light-harvesting,13 and nonlinear optical phenomena (e.g., photon upconversion14 and singlet fission15) have been intensively studied and applied to solar energy collection,16 molecular sensing,17 and biological applications (e.g., bioimaging,18 optogenetics,19 and phototherapy20). However, aggregation-induced photocatalytic activity (AI-PCA) has never been reported despite the high potential for a novel photocatalytic material. Taking into account previous reports on aggregation-induced triplet excited state generation21 and charge carrier migration22 in self-assembled nanostructures of organic dyes, we considered that various organic dyes may cause AI-PCA. These phenomena cause elongation of the excited state lifetime23 and increasing collision frequency with substrates,24 which are important for the progression of photocatalytic reactions. AI-PCA would lead to expansion of the molecular design of photocatalysts that enables adjustment of absorption wavelength and redox potential. In addition, self-assembled supramolecular photocatalysts (SA-SPCs) possessing AI-PCA are expected to produce unprecedented photocatalytic soft-materials25 (gel, liquid crystal, membrane etc.) taking advantages of the unique properties of supramolecular assemblies26 (e.g. reversibility and stimuli-responsiveness).
Herein, we demonstrate the AI-PCA of amphiphilic rhodamines (Fig. 1a). Rhodamines are very common hydrophilic organic dyes with excellent photophysical properties, such as high light absorption and quantum yield, which can be tuned through chemical modification.27 The two SA-SPCs composed of amphiphilic rhodamines (rhodamine B (RhB) and rhodamine 19 (Rh19) (Fig. 1b)) exhibited photocatalytic activity under visible light irradiation in water, while the monomeric rhodamines did not exhibit photocatalytic activity. In particular, the SA-SPCs exhibited excellent hydrogen evolution rates.
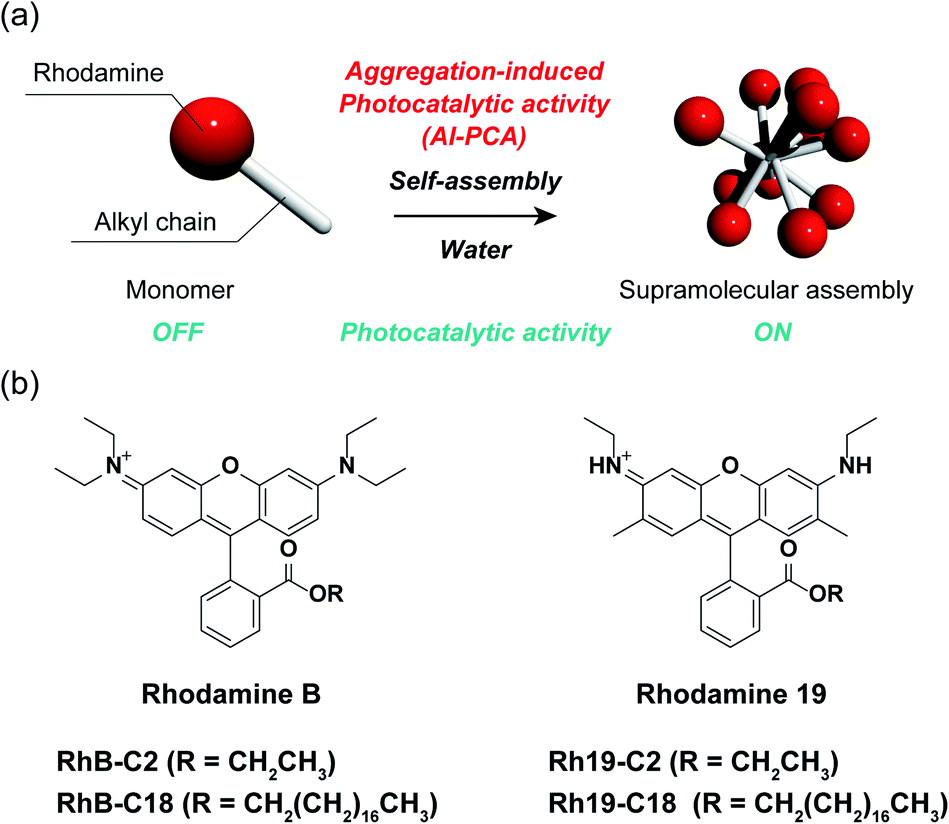 | ||
| Fig. 1 (a) Schematic representation of aggregation-induced photocatalytic activity (AI-PCA). (b) Chemical structures of rhodamine derivatives (left: rhodamine B (RhB), and right: rhodamine 19 (Rh19)). | ||
Results and discussion
Molecular design of the rhodamine derivatives
Four rhodamine derivatives with short and long alkyl chains were used in this study (Fig. 1b). Two common rhodamines with different absorption bands, RhB and Rh19, were selected as hydrophilic organic dyes to examine the concept of AI-PCA. The amphiphilic rhodamines with octadecyl alkyl chains (RhB-C18 and Rh19-C18) were expected to form supramolecular assemblies in water through hydrophobic interaction between the alkyl chains. More hydrophilic rhodamine derivatives with an ethyl ester group (RhB-C2 and Rh19-C2) compared to those with octadecyl alkyl chains were prepared as control compounds to evaluate the effect of self-assembly on the photocatalytic activity.The photophysical properties of monomeric RhB-C2 and RhB-C18 were evaluated from their UV-vis absorption (UV-vis) and photoluminescence (PL) spectra measured in dimethyl sulfoxide (DMSO), which is a good solvent for these compounds (Fig. 2a and b). The absorption spectra of RhB-C2 and RhB-C18 corresponded well with each other (Fig. 2a) and both the compounds exhibited absorption maxima at 566 nm. RhB-C2 and RhB-C18 also exhibited similar PL spectra with an emission peak at λem = 592 nm (Fig. 2b). These results indicate that the electronic states of RhB-C2 and RhB-C18 are quite similar despite the different alkyl chain lengths. We thus conclude that this pair is suitable for evaluating the effect of self-assembly on their photophysical and photocatalytic properties. Further, Rh19-C2 and Rh19-C18 also exhibited similar UV-vis and PL spectra (Fig. S1a and b†) with maxima at λabs = 539 nm and λem = 565 nm, respectively, which indicate that the Rh19-C2/Rh19-C18 pair has similar electronic states regardless of the alkyl chain length.
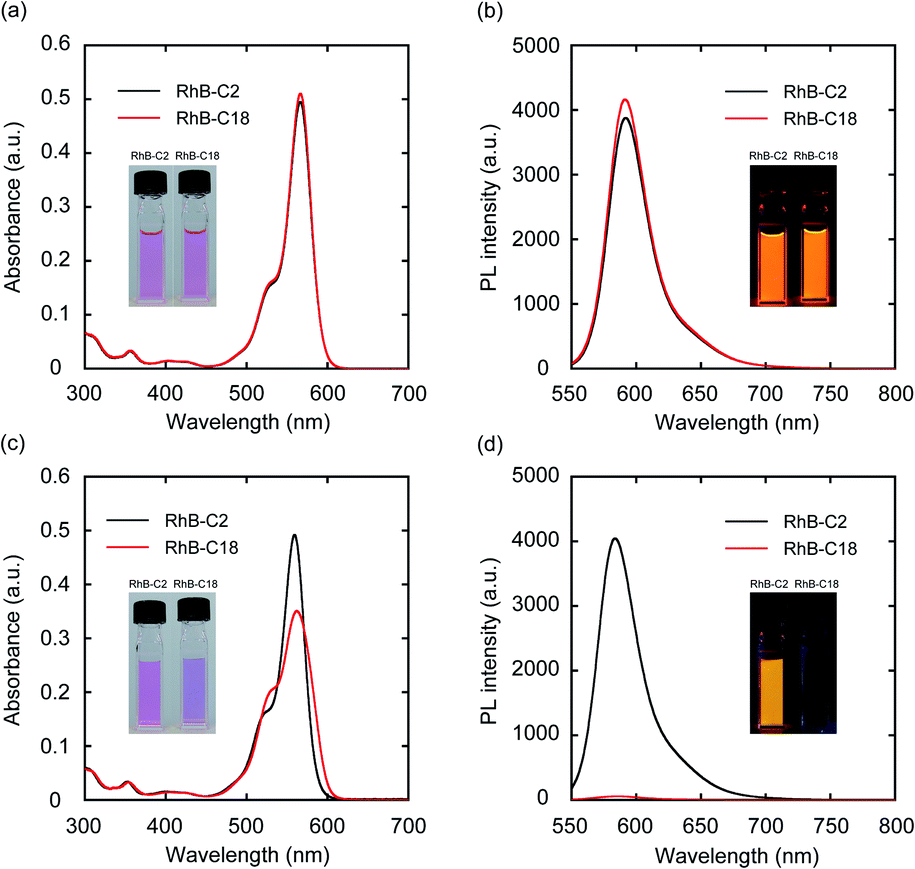 | ||
| Fig. 2 (a, c) UV-vis absorption (UV-vis) and (b, d) photoluminescence (PL) spectra of RhB-C2 and RhB-C18 in DMSO (a, b) and water (c, d). Experimental conditions: [RhB-C2] = [RhB-C18] = 5.0 μM, rt, water, excitation wavelength: 540 nm. Insets: Optical photos of RhB-C2 and RhB-C18 in DMSO and water under (a, c) daylight and (b, d) UV light (365 nm). | ||
Self-assembling properties of the rhodamines in water
To examine the self-assembly properties, the UV-vis and PL spectra of RhB-C2 and RhB-C18 were recorded in water (concentration: 5.0 μM) (Fig. 2c and d). The UV-vis spectrum of RhB-C2 in water is similar to that in DMSO with a slightly red-shifted absorption maximum (λabs: 559 nm (DMSO), 562 nm (water)) (Fig. 2c). In contrast, RhB-C18 exhibited a broad spectrum with split peaks at 530 and 559 nm that can be assigned to the aggregation states of RhB-C18,28 which suggests the formation of a supramolecular assembly of RhB-C18 in water. The PL spectra of RhB-C2 and RhB-C18 were significantly different (Fig. 2d). RhB-C2 exhibited an intense emission in water, whereas RhB-C18 exhibited a very weak emission. This suggests ACQ in RhB-C18.11 Furthermore, the addition of a nonionic surfactant (Triton X-100, 0.3 vol%) to the RhB-C18 aqueous solution drastically increased the PL intensity (Fig. S2b†), which suggests the dissociation of the RhB-C18 supramolecular assembly by Triton X-100. The photophysical properties of the Rh19-C2/Rh19-C18 pair exhibited the same trend as those of the RhB-C2/RhB-C18 pair (Fig. S1c, d and S2c, d†). Rh19-C18 formed supramolecular assemblies, whereas Rh19-C2 did not form supramolecular assemblies in water. With the increasing concentration, the absorbances of the rhodamine derivatives measured in water increased linearly (Fig. S3 and S4†); this indicates that RhB-C2 and Rh19-C2 did not form supramolecular assemblies until 50 μM (Fig. S3†), and the excessive aggregation of RhB-C18 and Rh19-C18 did not occur at least until 100 μM (Fig. S4†).Dynamic light scattering (DLS) measurements and transmission electron microscopy (TEM) observations were performed to confirm the formation of supramolecular assemblies. DLS measurements indicated the presence of supramolecular assemblies of RhB-C18 and Rh19-C18 having average sizes of 200 and 82 nm, respectively (Fig. S5a and b†). TEM revealed the formation of spherical supramolecular assemblies of RhB-C18 and Rh19-C18 (Fig. S5c and d†). The selected area electron diffraction (SAED) patterns obtained by TEM exhibited diffused rings and no diffraction spots, which indicate the amorphous nature of the rhodamine supramolecular assemblies (Fig. S5e and f†).
Photocatalytic activities of the rhodamines in water
Since it became clear that the rhodamine pairs were suitable for evaluation of AI-PCA, we initially examined their photocatalytic activities in water using 1,1′,3,3,3′,3′-hexamethylindotricarbocyanine iodide (HITCI) (Fig. S6a†).29 The rhodamines and HITCI were mixed in water, and the UV-vis spectra were recorded after photoirradiation of the mixture (λ = 560 nm (FWHM: 10 nm, Xe lamp, 300 W)). In the case of RhB-C2, the absorption band at around 735 nm arising from HITCI gradually decreased (Fig. 3a). Considering the self-photooxidation of HITCI upon photoirradiation (Fig. S6b and c†), the photocatalytic activity of RhB-C2 in HITCI oxidation is considered negligible. In contrast, RhB-C18 remarkably oxidized HITCI within a short period of photoirradiation (Fig. 3b and c). In addition, similar to RhB-C2, Rh19-C2 caused slow degradation of HITCI (Fig. S7a†), whereas the supramolecular assemblies of Rh19-C18 rapidly degraded HITCI (Fig. S7b and c†). The rate of HITCI oxidation by the rhodamines was estimated to be 8.2 × 10−3, 3.1 × 10−1, 2.9 × 10−3, and 1.0 × 10−1 min−1 for RhB-C2, RhB-C18, Rh19-C2, and Rh19-C18, respectively, by linear regression fitting (Fig. 3d and S7d†). Compared with RhB-C2 and Rh19-C2, RhB-C18 and Rh19-C18 accelerated HITCI oxidation 39-fold and 34-fold, respectively. Additionally, in the presence of Triton X-100, the rates of HITCI photooxidation by the RhB-C2/RhB-C18 and Rh19-C2/Rh19-C18 pairs were almost the same (Fig. S8†). These results indicate that the formation of supramolecular assemblies of rhodamines enhanced the oxidation of HITCI.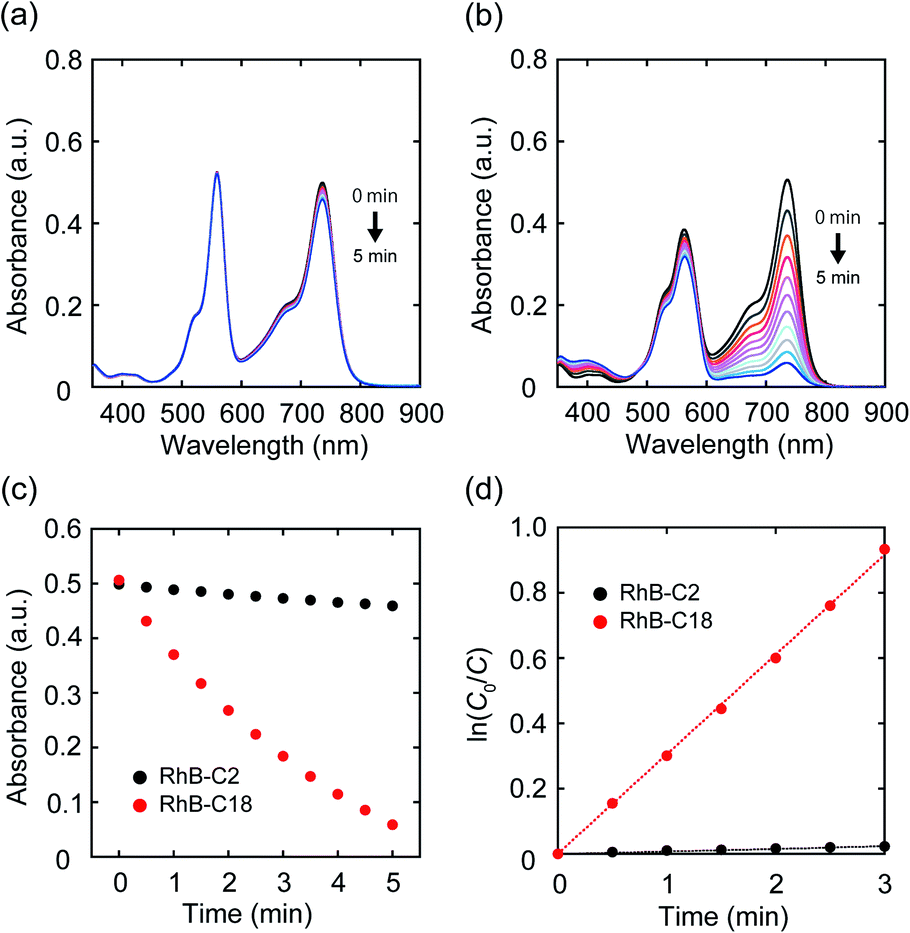 | ||
| Fig. 3 (a, b) UV-vis absorption spectra of the mixture of HITCI with (a) RhB-C2 and (b) RhB-C18 after photoirradiation. (c) Time-course of absorbance at 735 nm after photoirradiation using RhB-C2 (black circle) and RhB-C18 (red circle). (d) Linear regression plots of HITCT absorbance with RhB-C2 (black circle) and RhB-C18 (red circle). Experimental conditions: [RhB-C2] = [RhB-C18] = 5.0 μM, [HITCI] = 2.5 μM solvent: water, rt, irradiation wavelength: 560 nm (FWHM: 10 nm, Xe lamp, 300 W). | ||
Mechanistic study on the photocatalytic activity of the rhodamine SA-SPCs
The photoreaction of SA-SPCs is considered to occur via two types of mechanisms involving electron and/or energy transfer processes.30 Under aerobic conditions, in the energy transfer process, singlet oxygen (1O2) is commonly involved in the photocatalytic reaction, whereas oxygen radicals such as superoxide anion radicals (O2˙−) and hydroxyl radicals (OH˙−) are involved in the electron transfer mechanism (Fig. S9†).31 To explain the mechanism of the photocatalytic reaction, electron spin resonance (ESR) spectroscopy was performed using 4-hydroxy-2,2,6,6-tetramethylpiperidine (4-OH-TEMP)32 and 5,5-dimethyl-1-pyrroline N-oxide (DMPO)33 as spin trap reagents to detect 1O2 and oxygen radicals (e.g. O2˙−, OH˙−), respectively (Fig. S10a and b†). The ESR experimental conditions were first determined using Rose Bengal (Fig. S10c†) as a standard.34 The ESR spectra of RhB-C2 and RhB-C18 exhibited a characteristic 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet corresponding to the TEMPOL radical (Fig. 4a). RhB generates a triplet state despite the low quantum yield of intersystem crossing (quantum yield (ΦT): 0.006).35 Therefore, it is reasonable that RhB-C2 exhibited an ESR signal for the TEMPOL radical. No significant differences were observed in the signal intensities of the TEMPOL radicals of RhB-C2 and RhB-C18, which implies that the rate of energy transfer to oxygen did not drastically change after self-assembly. These results suggest that the photocatalytic reaction of RhB-C18 does not occur through the energy transfer mechanism. Subsequently, we evaluated the generation of oxygen radical species via the electron transfer mechanism. The ESR spectrum for RhB-C2 in DMPO did not exhibit a clear signal for a DMPO adduct (Fig. 4b), whereas the ESR spectrum of RhB-C18 exhibited a signal for a DMPO hydroxyl radical adduct (DMPO-OH). Since the superoxide anion (O2˙−) is unstable in aqueous media, it reacts with protons immediately upon addition to an aqueous medium (Fig. S9a†) and does not react with DMPO to form DMPO-OOH. Hence, no DMPO-OOH peak appeared in the ESR spectrum. However, we confirmed the generation of a hydroxyl radical (OH˙−) that was produced by the chain reaction starting from O2˙− ions through electron transfer in water (Fig. 4b, c and S9a†). The experimental results showed that RhB-C18 exhibits photocatalytic activity mainly through an electron transfer mechanism. Further, the ESR spectra of the Rh19-C2/Rh19-C18 pair were similar to those of the RhB-C2/RhB-C18 pair (Fig. S11†).
:1:1 triplet corresponding to the TEMPOL radical (Fig. 4a). RhB generates a triplet state despite the low quantum yield of intersystem crossing (quantum yield (ΦT): 0.006).35 Therefore, it is reasonable that RhB-C2 exhibited an ESR signal for the TEMPOL radical. No significant differences were observed in the signal intensities of the TEMPOL radicals of RhB-C2 and RhB-C18, which implies that the rate of energy transfer to oxygen did not drastically change after self-assembly. These results suggest that the photocatalytic reaction of RhB-C18 does not occur through the energy transfer mechanism. Subsequently, we evaluated the generation of oxygen radical species via the electron transfer mechanism. The ESR spectrum for RhB-C2 in DMPO did not exhibit a clear signal for a DMPO adduct (Fig. 4b), whereas the ESR spectrum of RhB-C18 exhibited a signal for a DMPO hydroxyl radical adduct (DMPO-OH). Since the superoxide anion (O2˙−) is unstable in aqueous media, it reacts with protons immediately upon addition to an aqueous medium (Fig. S9a†) and does not react with DMPO to form DMPO-OOH. Hence, no DMPO-OOH peak appeared in the ESR spectrum. However, we confirmed the generation of a hydroxyl radical (OH˙−) that was produced by the chain reaction starting from O2˙− ions through electron transfer in water (Fig. 4b, c and S9a†). The experimental results showed that RhB-C18 exhibits photocatalytic activity mainly through an electron transfer mechanism. Further, the ESR spectra of the Rh19-C2/Rh19-C18 pair were similar to those of the RhB-C2/RhB-C18 pair (Fig. S11†).
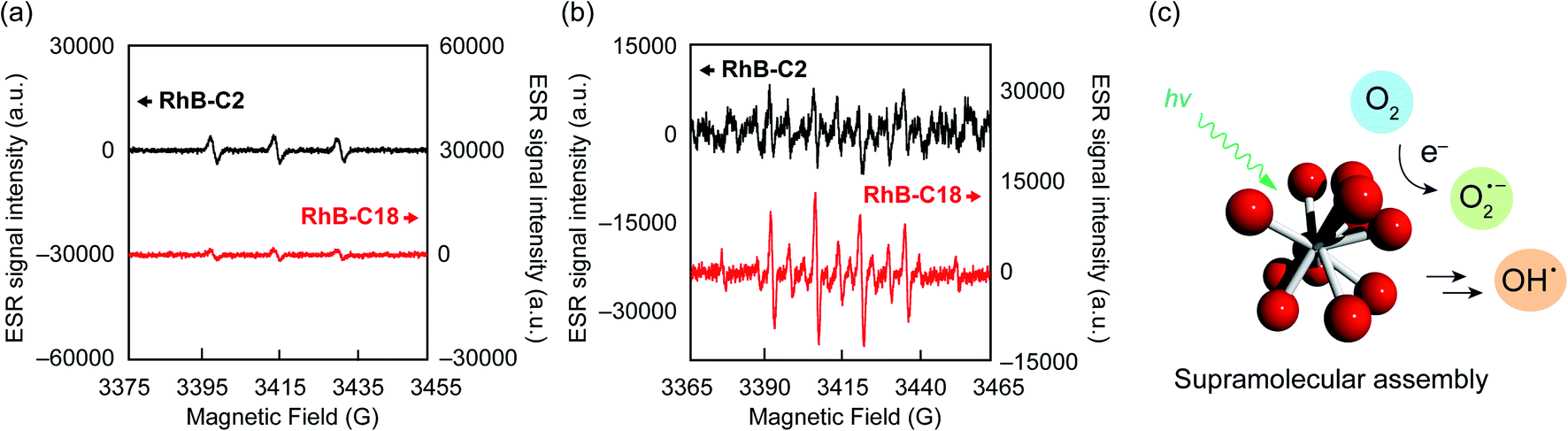 | ||
| Fig. 4 ESR signals of (a) 4-OH-TEMP and (b) DMPO adducts observed for RhB-C2 and RhB-C18 after light irradiation. (c) Schematic representation of generation of a hydroxyl radical through electron transfer and chain reactions. Experimental conditions: [RhB-C2] = [RhB-C18] = 50 μM, [4-OH-TEMP] = [DMPO] = 100 mM, solvent: water, rt, irradiation wavelength: 560 nm (FWHM: 10 nm, Xe lamp, 300 W, 3 min). | ||
Photocatalytic hydrogen evolution using the rhodamine SA-SPCs
Photocatalytic hydrogen evolution is one of the important reactions toward realizing artificial photosynthesis.2 The hydrogen evolution abilities of various photocatalysts have been actively examined to date. The electron transfer mechanism of the SA-SPC encouraged us to investigate the photocatalytic hydrogen evolution by the rhodamine SA-SPCs using Pt nanoparticles as a co-catalyst and ascorbic acid (Asc) as a sacrificial reagent (Fig. 5a). The HOMO levels of the rhodamine derivatives (Table S1,†RhB-C2: −5.4 eV, RhB-C18: −5.5 eV Rh19-C2: −5.4 eV, and Rh19-C18: −5.5 eV) determined by cyclic voltammetry or square wave voltammetry (Fig. S12 and S13†) were sufficiently lower than that of Asc (−4.6 eV),36 while the LUMO levels of the rhodamine derivatives (Table S1,†RhB-C2: −3.2 eV, RhB-C18: −3.5 eV Rh19-C2: −3.1 eV, and Rh19-C18: −3.4 eV) were enough for the proton reduction reaction. The addition of Asc and H2PtCl6 as a precursor of Pt nanoparticles hardly affected the size of SA-SPCs (Fig. S14a and b†).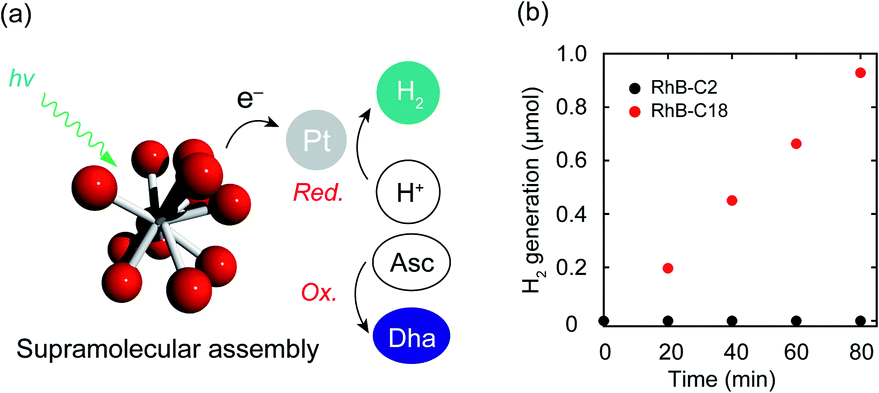 | ||
| Fig. 5 (a) Schematic representation of photocatalytic hydrogen evolution by the SA-SPC composed of rhodamine derivatives. (b) Time-course of hydrogen evolution by RhB-C18 and RhB-C18 under visible light. Experimental conditions: [rhodamines] = 50 μM, [Pt] = 100 μM, [Asc] = 500 mM, solvent: water, rt, irradiation light wavelength: >360 nm. Asc: ascorbic acid, DHA: dehydroascorbic acid. | ||
The time-courses of photocatalytic hydrogen evolution by RhB-C2 and RhB-C18 are shown in Fig. 5b. After light irradiation (λ > 360 nm, 300 W (Xe lamp)), Pt nanoparticles were formed (Fig. S14c–f†) and RhB-C18 exhibited hydrogen generation, while RhB-C2 did not display hydrogen evolution. The RhB-C18 SA-SPC functioned for 80 min without any decrease in the photocatalytic activity, and the average hydrogen evolution rate (HER) was determined to be 3.7 mmol g−1 h−1 (Fig. 5b), which is comparable to that of other excellent organic systems such as g-C3N4 (0.67 mmol g−1 h−1)37 and a covalent organic framework (10.1 mmol g−1 h−1).38 80 min after light irradiation, the photocatalytic activities of the SA-SPCs decreased due to the decomposition of rhodamines. Rh19-C18 exhibited photocatalytic hydrogen evolution (HER: 2.9 mmol g−1 h−1), while Rh19-C2 did not (Fig. S15a†). One of the reasons for the high HER would be intermolecular electron migration among the rhodamines.39 The photoirradiation of rhodamines generated intermolecular charge separation states, and the migration between the rhodamines may have facilitated efficient electron transfer to the Pt nanoparticle. The apparent quantum efficiencies of RhB-C18 and Rh19-C18 were 0.059 and 0.039% under these conditions, respectively.
To examine the effects of the SA-SPC concentration on the hydrogen evolution reaction, the SA-SPC concentrations were increased from 50 to 100 μM. The amorphous self-assembled spherical structures were almost unchanged (Fig. S16,† average particle diameter: RhB-C18: 223 nm, Rh19-C18: 122 nm) after the increase. The hydrogen evolution rates decreased from 3.7 to 2.4 mmol g−1 h−1 for RhB-C18 (Fig. S17a†). On the other hand, in the case of Rh19-C18, the hydrogen evolution rate significantly increased from 2.9 to 5.9 mmol g−1 h−1 (Fig. S17b†). These results indicate that the hydrogen evolution rates are significantly affected by the concentration of the SA-SPC. The versatile factors including the size, morphology, surface area, fluidity, and electric state of the SA-SPC, and interactions between the SA-SPC and Pt nanoparticles or ascorbic acid would have a sensitive effect on the hydrogen evolution. A detailed understanding of the changes of RhB-C18 and Rh19-C18 for photocatalytic hydrogen evolution rates is currently difficult. We will study how each factor has effects on photocatalytic activity in future studies.
Conclusions
In summary, we demonstrated the AI-PCA of two rhodamine derivatives. The rhodamine SA-SPCs showed excellent photocatalytic hydrogen evolution rates (up to 5.9 mmol g−1 h−1). ESR spectroscopic analysis revealed that the photocatalytic reaction proceeded via an electron transfer mechanism. We think that the concept of AI-PCA might be applicable to a wide range of photoactive molecules. Further investigations on the effects of organic dyes, morphologies, and the molecular arrangements of supramolecular assemblies on the photocatalytic activity of SA-SPCs, and the detailed mechanism of AI-PCA are currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed at Osaka University and financially supported by a Grant-in-Aid for Young Scientists (JSPS KAKENHI Grant No. JP18K14189), AMED (Grant No. JP19lm0203007 and JP19lm0203014), Takeda Science Foundation, Kowa Life Science Foundation, Senri Life Science Foundation, and Kato Memorial Bioscience Foundation. We appreciate Mr Y. Murakami for technical support for TEM observation. We also thank Dr Y. Morimoto and Dr K. Oohora for technical support for ESR measurement. We acknowledge Prof. Dr H. Uyama and Dr T. Asoh for DLS measurement.Notes and references
- C. Xu, P. R. Anusuyadevi, C. Aymonier, R. Luque and S. Marre, Chem. Soc. Rev., 2019, 48, 3868 RSC
; J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360 RSC
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS
- J. Xing, W. Q. Fang, H. J. Zhao and H. G. Yang, Chem.–Asian J., 2012, 7, 642 CrossRef CAS
-
Visible light photocatalysis in organic chemistry, ed. C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Wiley-VCH, 2018 CrossRef CAS
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS
- H. Honda, A. Ishizaki, R. Soma, K. Hashimoto and A. Fujishima, J. Illum. Eng. Soc., 1998, 27, 42 CrossRef CAS
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC
- M. S. Kodaimati, K. P. McClelland, C. He, S. Lian, Y. Jiang, Z. Zhang and E. A. Weiss, Inorg. Chem., 2018, 57, 3659 CrossRef CAS
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888 CrossRef CAS
-
Photophysics of Aromatic Molecules, ed. J. B. Birks, John Wiley & Sons Ltd, 1970 CrossRef CAS
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC
- D. M. Eisele, C. W. Cone, E. A. Bloemsma, S. M. Vlaming, C. G. F. van der Kwaak, R. J. Silbey, M. G. Bawendi, J. Knoester, J. P. Rabe and D. A. Vanden Bout, Nat. Chem., 2012, 4, 655 CrossRef CAS
- N. Yanai and N. Kimizuka, Chem. Commun., 2016, 52, 5354 RSC
- A. M. Levine, C. Schierl, B. S. Basel, M. Ahmed, B. A. Camargo, D. M. Guldi and A. B. Braunschweig, J. Phys. Chem. C, 2019, 123, 1587 CrossRef CAS
- Y. Zeng, J. Chen, T. Yu, G. Yang and Y. Li, ACS Energy Lett., 2017, 2, 357 CrossRef CAS
- N. Melnychuk and A. S. Klymchenko, J. Am. Chem. Soc., 2018, 140, 10856 CrossRef CAS
- K. Mizusawa, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 13386 CrossRef CAS
- Y. Sasaki, M. Oshikawa, P. Bharmoria, H. Kouno, A. Hayashi-Takagi, M. Sato, I. Ajioka, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2019, 58, 17827 CrossRef CAS
- X. Li, C.-Y. Kim, S. Lee, D. Lee, H.-M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880 CrossRef CAS
- R. Zhang, Y. Duan and B. Liu, Nanoscale, 2019, 11, 19241 RSC
- A. T. Haedler, K. Kreger, A. Issac, B. Wittmann, M. Kivala, N. Hammer, J. Köhler, H.-W. Schmidt and R. Hildner, Nature, 2015, 523, 196 CrossRef CAS
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022 CrossRef CAS
- N. J. Hestand, R. V. Kazantsev, A. S. Weingarten, L. C. Palmer, S. I. Stupp and F. C. Spano, J. Am. Chem. Soc., 2016, 138, 11762 CrossRef CAS
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404 RSC
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410 RSC
- K. Adachi, K. Watanabe and S. Yamazaki, Ind. Eng. Chem. Res., 2014, 53, 13046 CrossRef CAS
- H. Y. Kim, T. G. Bjorklund, S.-H. Lim and C. J. Bardeen, Langmuir, 2003, 19, 3941 CrossRef CAS
- O. Legrini, E. Oliveros and A. M. Braun, Chem. Rev., 1993, 93, 671 CrossRef CAS
- P. Guo, P. Chen, W. Ma and M. J. Liu, Mater. Chem., 2012, 22, 20243 RSC
- Y. A. Lion, E. Gandin and A. V. D. Vorst, Photochem. Photobiol., 1980, 31, 305 CrossRef CAS
- P. Xiao, P. Wang, H. Li, Q. Li, Y. Shi, X.-L. Wu, H. Lin, J. Chen and X. Wang, J. Hazard. Mater., 2018, 345, 123 CrossRef CAS
- J. J. M. Lamberts and D. C. Neckers, Tetrahedron, 1985, 41, 2183 CrossRef CAS
- V. E. Korobov, V. V. Shubin and A. K. Chibisov, Chem. Phys. Lett., 1977, 45, 498 CrossRef CAS
-
Oxidation-Reduction Potentials of Organic Systems, ed. W. M. Clark, Williams & Wilkins Co, 1960 CrossRef CAS
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 55, 1830 CrossRef CAS
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180 CrossRef CAS
- M. F. Mark, M. W. Kryman, M. R. Detty and D. W. McCamant, J. Phys. Chem. C, 2018, 122, 16519 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04285d |
| This journal is © The Royal Society of Chemistry 2020 |