DOI:
10.1039/D0SC04174B
(Edge Article)
Chem. Sci., 2020,
11, 12446-12452
A cyclopentadienyl functionalized silylene – a flexible ligand for Si- and C-coordination†
Received
30th July 2020
, Accepted 13th October 2020
First published on 26th October 2020
Abstract
The synthesis of a 1,2,3,4-tetramethylcyclopentadienyl (Cp4) substituted four-membered N-heterocyclic silylene [{PhC(NtBu)2}Si(C5Me4H)] is reported first. Then, selected reactions with transition metal and a calcium precursor are shown. The proton of the Cp4-unit is labile. This results in two different reaction pathways: (1) deprotonation and (2) rearrangement reactions. Deprotonation was achieved by the reaction of [{PhC(NtBu)2}Si(C5Me4H)] with suitable zinc precursors. Rearrangement to [{PhC(NtBu)2}(C5Me4)SiH], featuring a formally tetravalent silicon R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(R′)–H unit, was observed when the proton of the Cp4 ring was shifted from the Cp4-ring to the silylene in the presence of a Lewis acid. This allows for the coordination of the Cp4-ring to a calcium compound. Furthermore, upon reaction with transition metal dimers [MCl(cod)]2 (M = Rh, Ir; cod = 1,5-cyclooctadiene) the proton stays at the Cp4-ring and the silylene reacts as a sigma donor, which breaks the dimeric structure of the precursors.
Si(R′)–H unit, was observed when the proton of the Cp4 ring was shifted from the Cp4-ring to the silylene in the presence of a Lewis acid. This allows for the coordination of the Cp4-ring to a calcium compound. Furthermore, upon reaction with transition metal dimers [MCl(cod)]2 (M = Rh, Ir; cod = 1,5-cyclooctadiene) the proton stays at the Cp4-ring and the silylene reacts as a sigma donor, which breaks the dimeric structure of the precursors.
Introduction
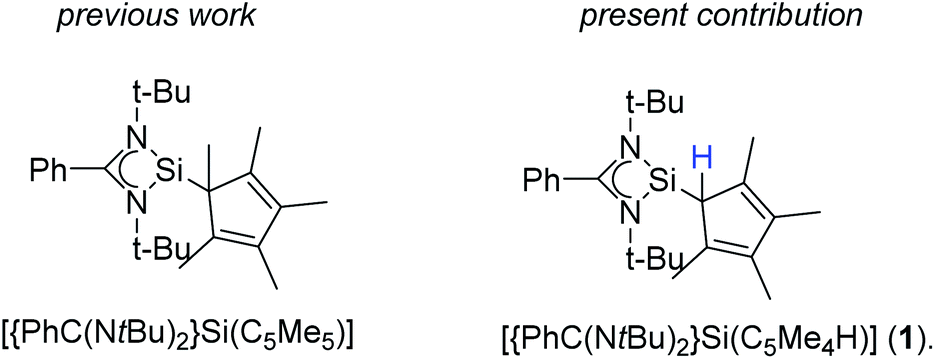
The earliest report of a stable silicon(II) compound (decamethylsilicocene) in 1986 was a landmark achievement in low-valent silicon(II) chemistry, which was only known by matrix-isolations before.1–3 The next significant step towards stable and reactive low-valent main group compounds was achieved in 1991 with the synthesis of N-heterocyclic carbenes.4 This concept was successfully migrated to silicon(II) chemistry with the synthesis of the stable N-heterocyclic silylene (NHSi) in 1994.5 Following this initial milestone, several other NHSi compounds were synthesized and characterized.6,7 Among others, the four-membered NHSi [{PhC(NtBu)2}SiCl] emerged as one of the most valuable silylene reagents reported to date.6,8–19 [{PhC(NtBu)2}SiCl] can easily be derivatized by substitution of the chlorine atom. A wide variety of substituents was applied over the last decade.6,20–25 We recently presented a pentamethylcyclopentadienyl (Cp*) substituted version [{PhC(NtBu)2}Si(C5Me5)] (Scheme 1) and explored its coordination behavior towards transition metals and its reactivity with chalcogens.26–28 In these complexes, we always observed a 1,2-silicon shift of the silylene on the Cp*-ring.
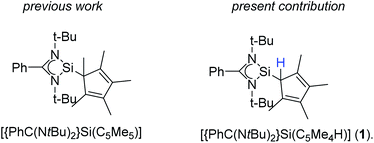 |
| | Scheme 1 Comparison of the previous and herein published silylene. | |
Results and discussion
Synthesis
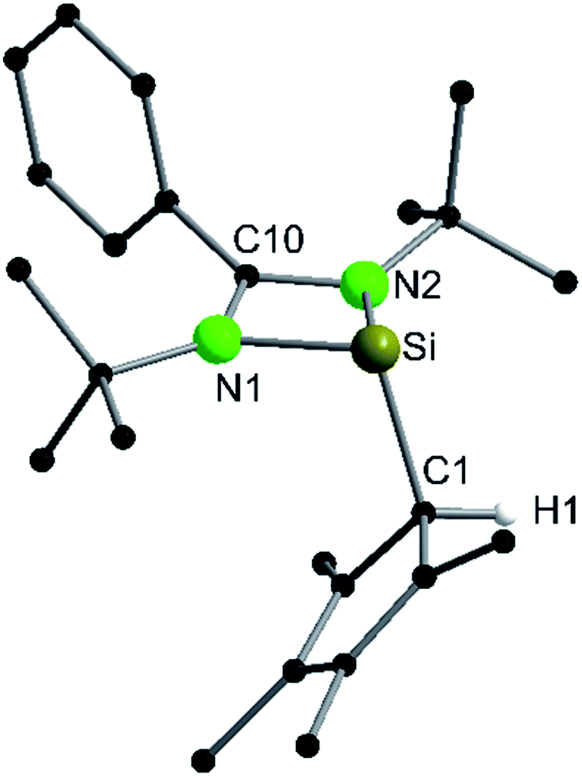
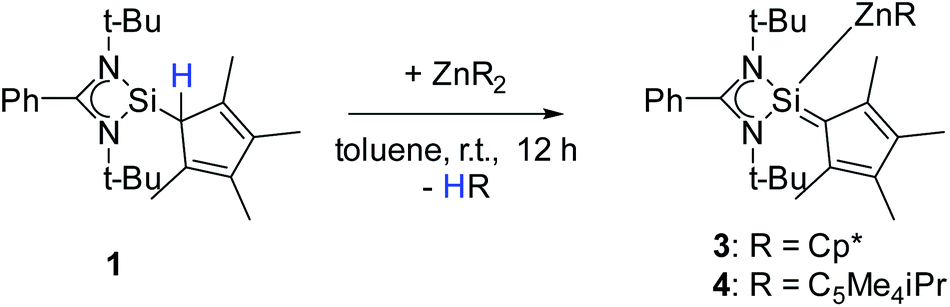
Following our previous results, we were interested in the reaction behaviour of a NHSi with a cyclopentadienyl substituent, which has at least one hydrogen atom at the five membered ring to enable a subsequent deprotonation. 1,2,3,4-Tetramethylcyclopentadienyl (Cp4) emerged as a feasible substituent having one acidic proton at the central ring. Furthermore, due to its relatively high steric demand, the 1,2-silicon shift should be suppressed.26–29 [{PhC(NtBu)2}Si(C5Me4H)] (1) was synthesized in a rational approach from [{PhC(NtBu)2}SiCl] and KCp4 in good yields (89%, Scheme 2). In contrast to its analogue, Cp* substituted compound 1 is soluble in all common but halogen-free solvents. In contrast, halogen-containing solvents cause degradation of the silylene. Compound 1, which was recrystallized from hot n-heptane, crystallizes in the monomeric space group P21/c with one molecule in the asymmetric unit (Fig. 1). As predicted, the silylene binds to the Cp4-ring at the position of the C1 carbon and bears a proton and no methyl group. The Si–C1 bond length (1.972(2) Å) is in the range of literature known carbon–silicon single bonds (1.88–1.98 Å).26,30 The 1H NMR spectrum of 1 shows the expected signals for the [PhC(NtBu)2] amidinate ligand as well as a set of three signals, which correspond to the methyl groups and the proton of the Cp4-ring. The 29Si NMR spectrum shows a strong doublet at 38.0 ppm (2JSi,H = 4.0 Hz) as well as a small doublet at −18.6 (1JSi,H = 234 Hz). The second signal, which we first deemed to be an impurity, was detected in all samples and proved to be an indicator for another silylene species, which was formed during the prolonged 29Si NMR measurements due to a rearrangement reaction. This formation is rather slow at room temperature and can be accelerated at higher temperatures, which could be proven by heating an NMR sample for several days at 80 °C and measuring after different times.
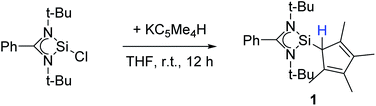 |
| | Scheme 2 Synthesis of [{PhC(NtBu)2}Si(C5Me4H)] (1). | |
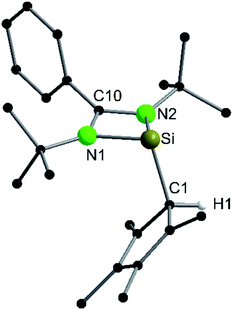 |
| | Fig. 1 Solid state structure of compound 1. Hydrogen atoms are omitted for clarity (except H1). Selected bond lengths [Å] and angles [°]: Si–N1 1.8731(13); Si–N2 1.8732(12); N1–C10 1.342(2); N2–C10 1.339(2); Si–C1 1.972(2); C1–C2 1.492(2); C1–C5 1.499(2); C2–C3 1.352(2); C3–C4 1.462(2); C4–C5 1.353(2), N1–C10–N2 105.50(12); N1–Si–C1 102.71(6); N2–Si–C1 105.45(6); Si–C1–H1 100.3(9); Si–C1–C2 116.86(10); C2–C1–C5 104.07(12); C2–C1–H1 114.4(10); Si–C1–C5 107.29(10); C5–C1–H1 114.2(10). | |
Rearrangement
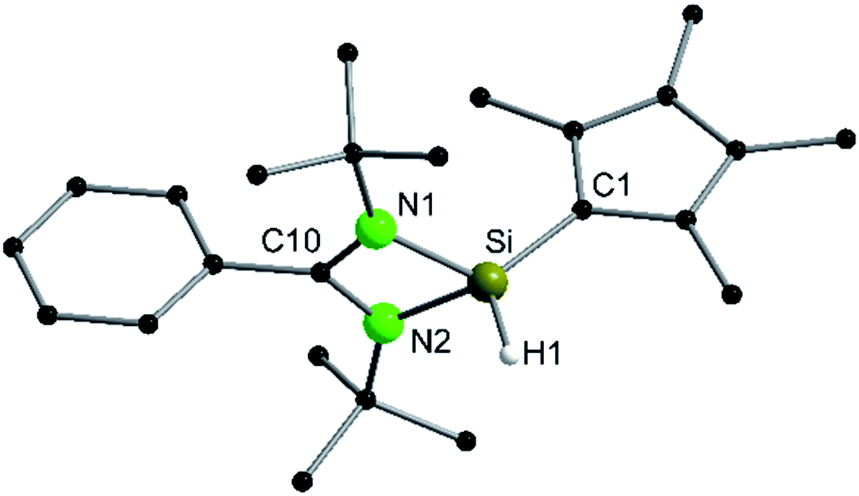
To enforce this rearrangement reaction, compound 1 was treated with [Y{N(SiMe3)2}3] as a weak and well soluble Lewis-acid at room temperature to give the isomer [{PhC(NtBu)2}(C5Me4)SiH] (2) (Scheme 3). Compound 2 crystallizes in the monomeric space group P21/c with one molecule in the asymmetric unit (Fig. 2). Upon rearrangement from 1 to 2 a 1,2-H-shift from the Cp4–carbon ring atom to the silicon atom took place. As a consequence, structural changes in the Cp4–Si bond occur, e.g., the Si–C1 bond is much shorter than that in 1. The observed bond length (1.761(2) Å) is in the range of literature known Si–C double bonds and is, apart from NHC–Si (NHC = N-heterocyclic carbene) interactions, one of only a few known examples of a SiC double bond in which a NHSi is involved.5,11,31–35 Furthermore, the Si–C1 bond is now in-plane with the C–Me bonds of the Cp4-ring and the average C–C bond length within the Cp4-ring is shorter than that in 1. This leads us to the conclusion that Cp4 and the silylene atom form a conjugated system similar to fulvene. The proton H1 of the silicon atom was located and freely refined in the difference Fourier map. The Si–H bond length (1.35(2) Å) is in agreement with those in the literature.24,36 Compared to 1, only minor structural changes are seen in the amidinate backbone of the silylene. In the 1H NMR spectrum of compound 2, the resonance for the proton coordinated to silylene is detected at 5.92 ppm. This is a low field shift of around 3.6 ppm, compared to that in 1. Concomitantly, the resonance for the silylene in the 29Si NMR spectrum is shifted to a higher field (−18.6 ppm) and is observed as a doublet due to 1JSi,H coupling (234 Hz) with the proton. This resonance is in the exact same position as that for the impurity in the NMR spectra for compound 1, leading us to the conclusion that the “impurity” in 1 is in fact compound 2, which formed over a prolonged period of time. The IR spectrum of 2 shows the Si–H valence stretching mode at 2204 cm−1, which is absent in the spectrum of 1.
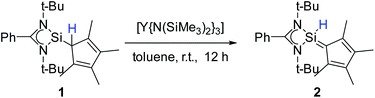 |
| | Scheme 3 Synthesis of [{PhC(NtBu)2}(C5Me4)SiH] (2). | |
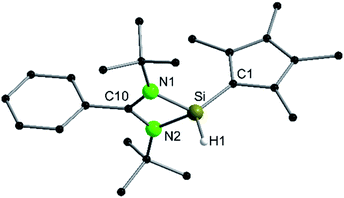 |
| | Fig. 2 Solid state structure of compound 2. Hydrogen atoms are omitted for clarity (except H1). Selected bond lengths [Å] and angles [°]: Si1–H1 1.35(2); Si–N1 1.8219(13); Si–N2 1.8149(13); Si–C1 1.761(2); N1–C10 1.333(2); N2–C10 1.345(2); C1–C2 1.448(2); C1–C5 1.440(2); C2–C3 1.382(2); C3–C4 1.427(2); C4–C5 1.381(2); N1–C10–N2 105.72(12); N1–Si–C1 122.30(6); N2–Si–C1 122.15(6); N1–Si–H1 103.8(7); N2–Si–H1 107.4(7); C1–Si–H1 119.4(7); Si–C1–C2 125.59(11); Si–C1–C5 127.53(11); C2–C1–C5 106.88(12). | |
Interestingly this rearrangement of compound 1 to 2 does not occur in the presence of an excess amount of KCp4, as adding two equivalents of KCp4 to one equivalent of SiCl did not yield compound 2. Furthermore, NMR studies showed that the slow rearrangement of 1 to 2 described before is not accelerated in the presence of excess KCp4. Another interesting point is that while we observed rearrangement from compound 1 to 2 we did not observe the back reaction from 2 to 1. A flame sealed NMR sample of freshly dissolved crystals of compound 2 did not show any traces of compound 1 even after several weeks of continuous heating.
Both 1 and 2 have different possibilities for coordination to metal atoms. Compound 1 is a silylene and thus should coordinate as a soft Lewis-base via the Si atom to metal atoms, while for 2, which formally is a Si(IV) species, a coordination via the Cp4-ring is expected. By deprotonation of either 1 or 2 an anion is formed, which may either act like a substituted cyclopentadienyl ligand or a silyl anion. All these different coordination modes were realized and are now described in the following sections.
Deprotonation
After studying the rearrangement process, we investigated the deprotonation of 1 in the next reaction. After several failed attempts, we finally found 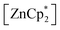 to be a suitable deprotonating agent. Thus,
to be a suitable deprotonating agent. Thus, 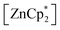 was treated in a straightforward reaction with compound 1 in toluene (Scheme 4), which led to the formation of [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me5)}] (3) as an orange precipitate (starting from compound 2 yields the same result). Upon recrystallization from the hot mother liquor, crystals suitable for X-ray diffraction studies were formed. During the course of the reaction one Cp* ligand was protonated and cleaved off.37 The remaining [ZnCp*]+ fragment was bulky enough to provide steric shielding of the Zn atom.38–40
was treated in a straightforward reaction with compound 1 in toluene (Scheme 4), which led to the formation of [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me5)}] (3) as an orange precipitate (starting from compound 2 yields the same result). Upon recrystallization from the hot mother liquor, crystals suitable for X-ray diffraction studies were formed. During the course of the reaction one Cp* ligand was protonated and cleaved off.37 The remaining [ZnCp*]+ fragment was bulky enough to provide steric shielding of the Zn atom.38–40
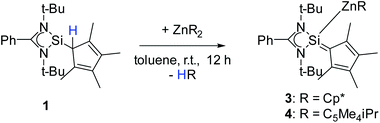 |
| | Scheme 4 Synthesis of [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me5)}] (3) and [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me4iPr)}] (4). | |
Compound 3 crystallizes in the monoclinic space group P21/n with one molecule in the asymmetric unit (Fig. 3). The solid-state structure of compound 3 also shows that the Cp4-ring is arranged as in compound 2 as sila-fulvene but the proton is completely removed from the ligand. The resulting silyl anion is bound to the [ZnCp*] fragment, with a short Zn–Si bond distance of 2.3105(11) Å.26,27,41 Additionally this is one of very few examples of a [ZnR]+-fragment bound directly to a silicon atom.42,43 Due to the rearrangement and deprotonation the coordination of the silyl anion is different from that of typical silylene zinc species, which feature a dative bond between zinc complexes ZnR2 and silylene. The difference in coordination can be attributed to the Cp4-ring, which due to its labile proton acts as non-innocent substituent on the silylene. Since the sila fulvene unit is similar to compound 2, the bond distances and angles are almost equal. The Si–C1 distance is again in the range of the literature known Si–C double bonds.31–33 The [ZnCp*] fragment shows a short distance between Zn and the  of 1.9118(5) Å. In the 29Si NMR spectrum of compound 3, a peak at 6.0 ppm is detected. Compared to that for compound 1 (39.7 ppm) this signal is significantly high field shifted, whereas compared to that for the more similar compound 2 (−18.6 ppm) a downfield shifted is seen.
of 1.9118(5) Å. In the 29Si NMR spectrum of compound 3, a peak at 6.0 ppm is detected. Compared to that for compound 1 (39.7 ppm) this signal is significantly high field shifted, whereas compared to that for the more similar compound 2 (−18.6 ppm) a downfield shifted is seen.
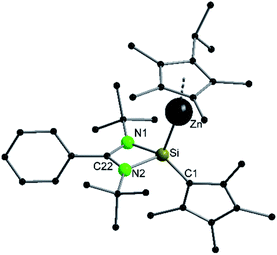 |
| | Fig. 3 Solid state structure of compound 4. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Zn–Si 2.3056(9); Zn–Cpcentr. 1.885(2); Si–N1 1.839(3); Si–N2 1.844(3); Si–C1 1.777(3); N1–C22 1.332(4); N2–C22 1.325(4); C1–C2 1.438(4); C1–C5 1.435(4); C2–C3 1.384(4); C3–C4 1.429(5); C4–C5 1.374(4); N1–C22–N2 106.3(3); N1–Si–Zn 109.99(8); N2–Si–Zn 106.14(9); C1–Si–Zn 122.55(10); N1–Si–C1 117.20(13); N2–Si–C1 118.84(13); Si–C1–C2 125.5(2); Si–C1–C5 128.0(2); C2–C1–C5 106.4(3). | |
Since the bulky substituents on zinc in 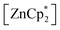 were the key for successful coordination of the silylene, we next used the even more bulky zincocene [Zn(CpiPr4)2] (CpiPr4 = 1-iso-propyl-2,3,4,5-methylcyclopentadienyl) as the precursor (Scheme 4). The reaction of [Zn(CpiPr4)2] with 1 resulted in the product [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me4iPr)}] (4), which was isolated as orange crystals. The product crystallizes in the chiral, monoclinic space group Pc with one molecule in the asymmetric unit (Fig. 4). The chirality is caused by the iso-propyl group on the Cp4iPr-ring, which cannot freely rotate and forms a kind of paddle-wheel geometry. The structure is similar to compound 3 except for the slightly disordered iso-propyl group on the Cp4iPr-ring bound to the zinc atom. The bond length and angles correspond well with those of compound 3 and the NMR spectra show only the additional peaks corresponding to the iso-propyl group.
were the key for successful coordination of the silylene, we next used the even more bulky zincocene [Zn(CpiPr4)2] (CpiPr4 = 1-iso-propyl-2,3,4,5-methylcyclopentadienyl) as the precursor (Scheme 4). The reaction of [Zn(CpiPr4)2] with 1 resulted in the product [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me4iPr)}] (4), which was isolated as orange crystals. The product crystallizes in the chiral, monoclinic space group Pc with one molecule in the asymmetric unit (Fig. 4). The chirality is caused by the iso-propyl group on the Cp4iPr-ring, which cannot freely rotate and forms a kind of paddle-wheel geometry. The structure is similar to compound 3 except for the slightly disordered iso-propyl group on the Cp4iPr-ring bound to the zinc atom. The bond length and angles correspond well with those of compound 3 and the NMR spectra show only the additional peaks corresponding to the iso-propyl group.
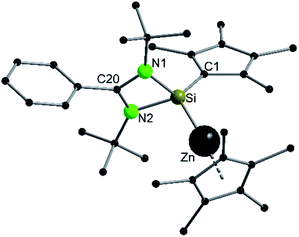 |
| | Fig. 4 Solid state structure of compound 3. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Zn–Si 2.3105(11); Zn–Cpcentr. 1.9118(5); Si–N1 1.835(3); Si–N2 1.843(3); Si–C1 1.779(3); N1–C20 1.346(4); N2–C20 1.329(4); C1–C2 1.447(5); C1–C5 1.442(5); C2–C3 1.389(5); C3–C4 1.421(6); C4–C5 1.387(5); N1–C10–N2 105.8(3); N1–Si–Zn 109.00(10); N2–Si–Zn 107.67(10); C1–Si–Zn 120.62(12); N1–Si–C1 118.1(2); N2–Si–C1 120.17(14); Si–C1–C2 124.6(3); Si–C1–C5 128.6(3); C2–C1–C5 106.7(3). | |
Carbon coordination
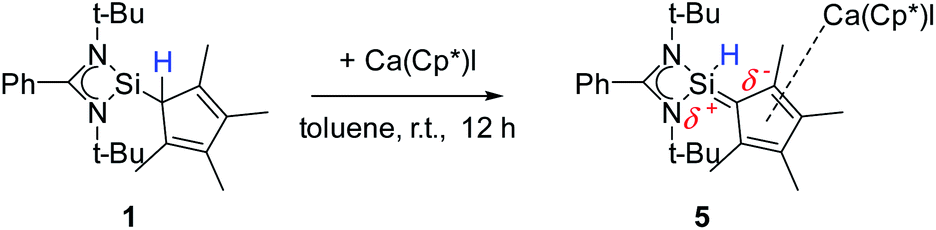
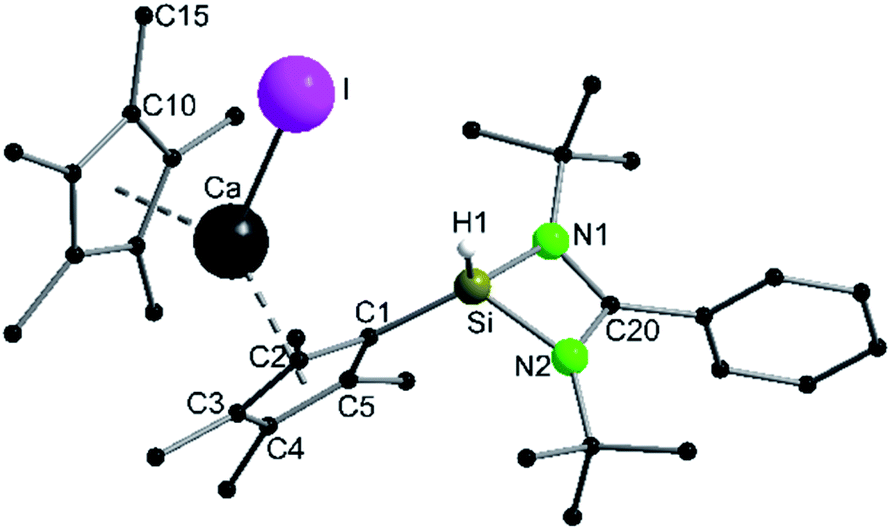
A coordination of compound 2via the carbon atoms of the Cp4-ring was realized in [{PhC(NtBu)2}SiH(C5Me4){Ca(C5Me5)I}] (5) (Scheme 5). When [Ca(C5Me5)I] was stirred together with 1 in toluene overnight a rearrangement of 1 to 2 takes place first, followed by coordination of 2 to the Ca atom (starting from compound 2 yields the same result). As a result, compound 5 was formed as an orange residue, which could be recrystallized from the hot mother liquor to obtain crystals suitable for X-ray diffraction studies.
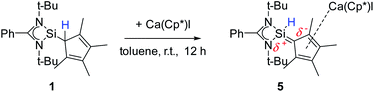 |
| | Scheme 5 Synthesis of [{PhC(NtBu)2}SiH(C5Me4){Ca(C5Me5)I}] (5). | |
Compound 5 crystallizes in the triclinic space-group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule in the asymmetric unit (Fig. 5). The [{PhC(NtBu)2}SiH(C5Me4)] moiety of the structure has nearly the same structural parameters (bonds and angles) as described before in structure 2, 3 and 4. In contrast to the zinc compounds described above, no deprotonation took place during the course of the reaction and the sila fulvene structure of 2 is preserved upon coordination featuring long (C1–C2 1.432(3), C1–C5 1.437(3), and C3–C4 1.426(4) Å) and short (C2–C3 1.388(4) and C4–C5 1.390(3) Å) C–C bonds. Consequently, the calcium atom cannot bind to the silicon atom and a coordination of the Cp4-ring of the sila fulvene is thus observed. Since bond lengths of the C–C double bonds are not influenced upon coordination and the Ca–C1 bond (2.637(3) Å) is the shortest of all Ca–C bonds, we anticipate an electrostatic interaction between the partially negatively charged carbon atom C1 and the calcium atom. π-Coordination of the alkene functions does not seem to play an important role. The calcium atom is coordinated to η5-Cp* and the Cp4-ring as well as to the iodide. Cp* and the Cp4-ring are bent out of plane, enclosing a 143.51(13)° angle. In contrast to many similar structural motifs known in the literature, compound 5 does not dimerize via an iodine bridge.44–48
with one molecule in the asymmetric unit (Fig. 5). The [{PhC(NtBu)2}SiH(C5Me4)] moiety of the structure has nearly the same structural parameters (bonds and angles) as described before in structure 2, 3 and 4. In contrast to the zinc compounds described above, no deprotonation took place during the course of the reaction and the sila fulvene structure of 2 is preserved upon coordination featuring long (C1–C2 1.432(3), C1–C5 1.437(3), and C3–C4 1.426(4) Å) and short (C2–C3 1.388(4) and C4–C5 1.390(3) Å) C–C bonds. Consequently, the calcium atom cannot bind to the silicon atom and a coordination of the Cp4-ring of the sila fulvene is thus observed. Since bond lengths of the C–C double bonds are not influenced upon coordination and the Ca–C1 bond (2.637(3) Å) is the shortest of all Ca–C bonds, we anticipate an electrostatic interaction between the partially negatively charged carbon atom C1 and the calcium atom. π-Coordination of the alkene functions does not seem to play an important role. The calcium atom is coordinated to η5-Cp* and the Cp4-ring as well as to the iodide. Cp* and the Cp4-ring are bent out of plane, enclosing a 143.51(13)° angle. In contrast to many similar structural motifs known in the literature, compound 5 does not dimerize via an iodine bridge.44–48
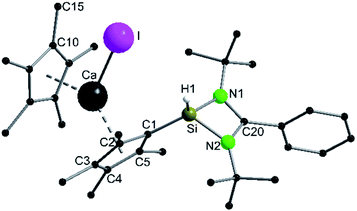 |
| | Fig. 5 Solid state structure of compound 5. Hydrogen atoms are omitted for clarity (except H1). Selected bond lengths [Å] and angles [°]: Ca–I 3.0851(6); 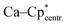 2.368(2); Ca–C1 2.637(3); Ca–C2 2.681(3); Ca–C3 2.780(3); Ca–C4 2.825(3); Ca–C5 2.744(3); Si–N1 1.798(2); Si–N2 1.812(2); Si–C1 1.795(2); Si–H1 1.35(3); N1–C20 1.329(3); N2–C20 1.346(3); C1–C2 1.432(3); C1–C5 1.437(3); C2–C3 1.388(4); C3–C4 1.426(4); C4–C5 1.390(3); Cp*–Ca–Cp4 143.51(13); N2–C20–N1 106.1(2); C1–Si–H1 119.5(12); C1–Si–N1 116.75(11); C1–Si–N2 121.33(10); N1–Si–H1 106.9(13); N2–Si–H1 110.0(12); Si–C1–C2 128.5(2); Si–C1–C5 123.1(2); C2–C1–C5 106.9(2). 2.368(2); Ca–C1 2.637(3); Ca–C2 2.681(3); Ca–C3 2.780(3); Ca–C4 2.825(3); Ca–C5 2.744(3); Si–N1 1.798(2); Si–N2 1.812(2); Si–C1 1.795(2); Si–H1 1.35(3); N1–C20 1.329(3); N2–C20 1.346(3); C1–C2 1.432(3); C1–C5 1.437(3); C2–C3 1.388(4); C3–C4 1.426(4); C4–C5 1.390(3); Cp*–Ca–Cp4 143.51(13); N2–C20–N1 106.1(2); C1–Si–H1 119.5(12); C1–Si–N1 116.75(11); C1–Si–N2 121.33(10); N1–Si–H1 106.9(13); N2–Si–H1 110.0(12); Si–C1–C2 128.5(2); Si–C1–C5 123.1(2); C2–C1–C5 106.9(2). | |
The proton on the silicon atom was detected by NMR and IR spectroscopy. In the 1H NMR spectrum of compound 5, the corresponding signal is seen at 3.27 ppm, which is in contrast to the data of compound 2. In the 29Si NMR spectrum no resonance was observed. The IR spectrum shows the Si–H valence stretching mode at 2205 cm−1.49
Silylene coordination
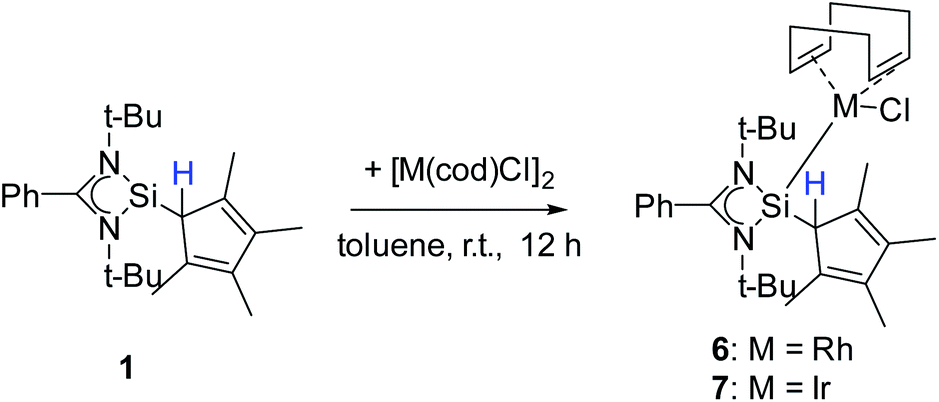
Besides carbon coordination, we were also interested in binding compound 1via the Si atom to transition metals to fully explore the coordination potential of this ligand. Silylenes are known for their ability to bind to, among others, low-valent transition metal complexes.6,20,28,41,50–56 For this purpose, compound 1 was treated with group 9 dimers [RhCl(cod)]2 and [IrCl(cod)]2 (cod = 1,5-cyclooctadiene) to give [{PhC(NtBu)2}(C5Me4H)Si{RhCl(cod)}] (6) and [{PhC(NtBu)2}(C5Me4H)Si{IrCl(cod)}] (7) (Scheme 6). Starting from compound 2 gave the same result.
 |
| | Scheme 6 Synthesis of [{PhC(NtBu)2}(C5Me4H)Si{RhCl(cod)}] (6) and [{PhC(NtBu)2}(C5Me4H)Si{IrCl(cod)}] (7). | |
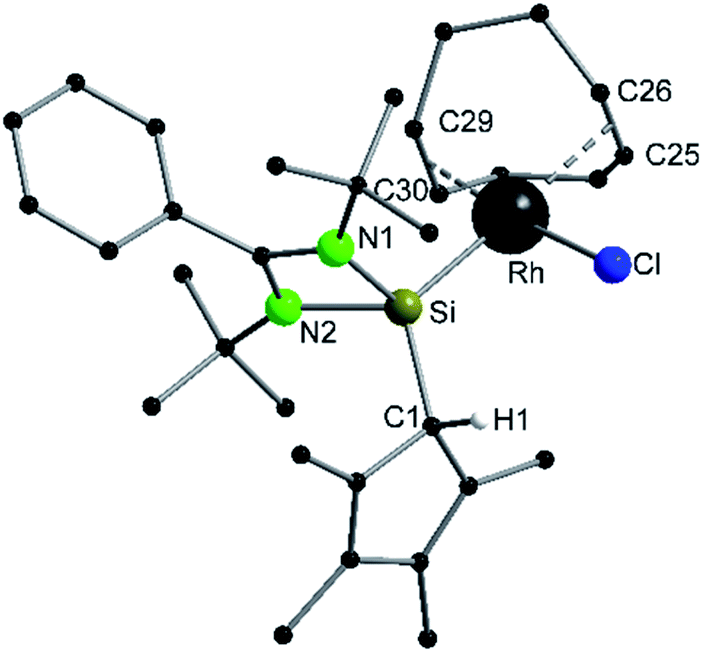
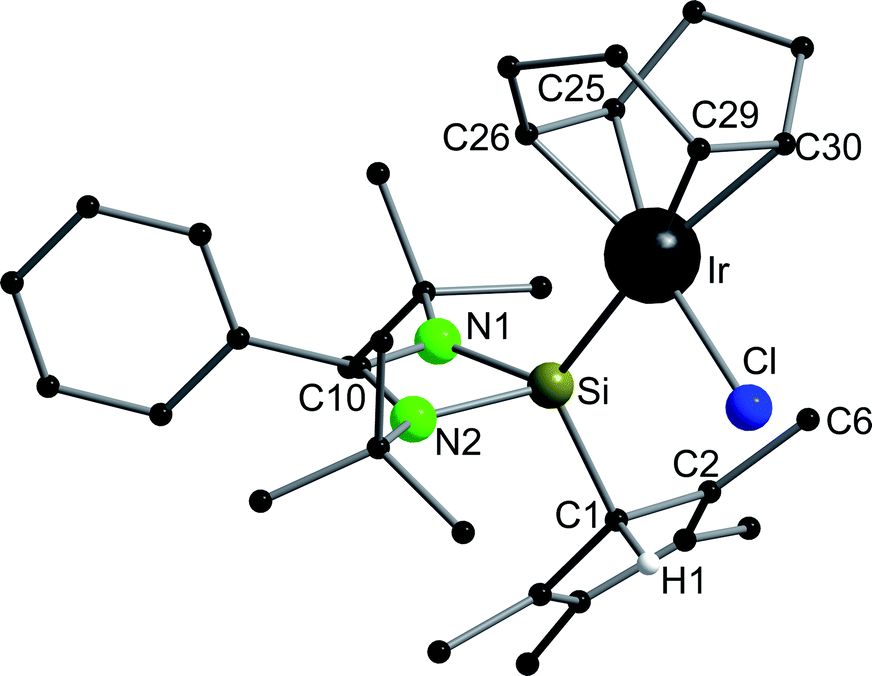
Compound 6 crystallizes in the monoclinic space group P21/n with one molecule in the asymmetric unit (Fig. 6). The [RhCl(cod)] fragment is directly attached to the silylene unit via a Si–Rh bond (Si–Rh 2.2947(5) Å). The Rh atom is coordinated in a square planar arrangement with COD, Cl, and the silylene unit. As expected, the trans influence of the silylene is seen in the elongation of the trans Rh–COD bonds (mean(Rh–Ctrans) = 2.276 Å vs. mean(Rh–Ccis) = 2.101 Å). Compound 1 does not show any transformations such as deprotonation or rearrangement upon coordination. The 29Si spectrum shows a doublet at 42.3 ppm with a coupling constant of 86.4 Hz due to the 1JSi,Rh coupling. Also, the 1H NMR and the IR spectra confirm that the proton is located on the carbon ring. Compound 7 crystallizes in the monoclinic space group C2/c with one molecule in the asymmetric unit (Fig. 7). The structure is isostructural to compound 6. Also, the NMR and IR spectra are similar to those of compound 6.
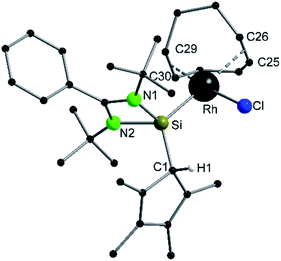 |
| | Fig. 6 Solid state structure of compound 6. Hydrogen atoms are omitted for clarity (except H1). Selected bond lengths [Å] and angles [°]: Rh–Si 2.2947(5); Rh–Cl 2.3785(5); Rh–C25 2.280(2); Rh–C26 2.272(2); Rh–C29 2.094(2); Rh–C30 2.108(2); Si–N1 1.8540(14); Si–N2 1.8443(14); Si–C1 1.911(2); N1–C10 1.336(2); N2–C10 1.339(2); C1–C2 1.514(3); C1–C5 1.501(3); C2–C3 1.343(3); C3–C4 1.468(4); C4–C5 1.339(4); N1–C10–N2 106.92(14); Si–Rh–Cl 87.98(2); C25–Rh–C26 34.68(8); C29–Rh–C30 38.95(7); N1–Si–Rh 112.51(5); N1–Si–C1 108.93(7); N2–Si–Rh 123.17(5); N2–Si–C1 107.78(7); C1–Si–Rh 121.77(6). | |
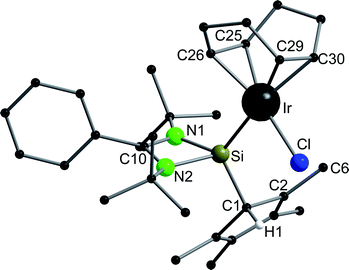 |
| | Fig. 7 Solid state structure of compound 7. Hydrogen atoms are omitted for clarity (except H1). Selected bond lengths [Å] and angles [°]: Ir–Si 2.3423(11); Ir–Cl 2.3552(10); Ir–C25 2.107(3); Ir–C26 2.094(3); Ir–C29 2.211(3); Ir–C30 2.203(3); Si–N1 1.838(3); Si–N2 1.852(3); Si–C1 1.904(3); N1–C10 1.342(4); N2–C10 1.333(4); C1–C2 1.521(5); C1–C5 1.504(5); C2–C3 1.351(5); C3–C4 1.462(6); C4–C5 1.345(5); N1–C10–N2 106.5(3); Si–Ir–Cl 88.12(3); C25–Ir–C26 39.54(13); C29–Rh–C30 36.69(12); N1–Si–Ir 121.68(9); N1–Si–C1 109.23(13); N2–Si–Ir 112.12(9); N2–Si–C1 109.03(14); C1–Si–Ir 121.98(11). | |
The experimental work was supported by theoretical studies. On one hand, the transfer equilibrium between 1 and 2 was investigated. According to quantum chemical DFT calculations, silylene 1 is only 12 kJ mol−1 more stable than silene 2. However, assuming an intramolecular 1,2-H rearrangement this small energy difference is accompanied by a very high energy barrier of 188 kJ mol−1 for 1 and 176 kJ mol−1 for 2. The transition state consists of Si–H or C–H distances of 1.71 and 1.80 Å. These results are in very good agreement with the calculations by Nagase and Kudo57 as well as by H. F. Schaefer III.58 The reaction temperature for the rearrangement 1/2 is expected to be around 400 °C, which has also been demonstrated experimentally for a silaolefin/silandiyl rearrangement.59 According to these calculations, there should be no thermal rearrangement from 1 to 2. This leads us to the conclusion that in solution a more complex process than a simple intramolecular 1,2-H rearrangement takes place.
The experimentally determined 29Si NMR shifts of both isomers are very well in line with the calculated data of 43.5 (1) and −30.7 ppm (2; tetramethylsilane taken as a reference).
Ahlrichs–Heinzmann population analyses based on occupation numbers were carried out to estimate the covalent bonding strengths. The results for the reference molecule silaethen serve as reference values. The shared electron numbers SEN for the C–H and Si–H single bonds as well as the SiC double bond are calculated to be 1.31, 1.33 and 2.13, respectively. The Si–C bond (SEN 1.12) of silylene 1 is in accordance with a single bond, while the one in silene 2 exhibits a bond order of about 1.5 (SEN 1.55).
In order to gain further insight, the binding of ligands 1 and 2 to metal centers was investigated by theoretical methods. The reaction energy of the formation of 5 from Ca(Cp*)I and 2 was determined to be −127 kJ mol−1. After a shift of the negative charge inside the coordinated ligand, the η5 coordination of the C5 unit is predominantly ionic in nature. In contrast, 1 would coordinate only weakly to calcium (−15 kJ mol−1). On the other hand, 1 binds to half an equivalent of [Rh(COD)Cl]2 with a reaction energy of −101 kJ mol−1; for isomer 2 no bonding interaction is found (for details see the ESI†). In both cases examined, the strength of the SiC bonds changes only insignificantly compared to the situation in the respective uncoordinated ligands.
Conclusions
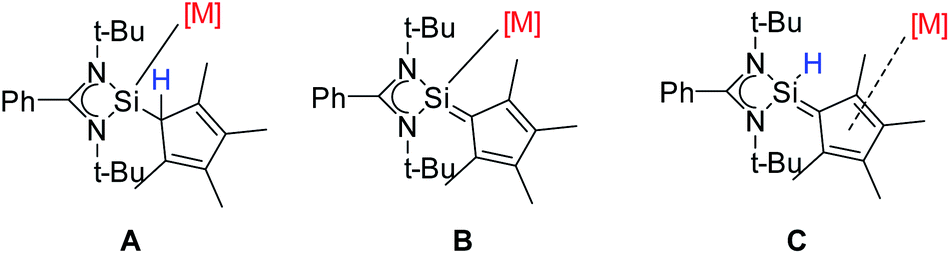
In summary, we have synthesized the newly functionalized four-membered silylene (NHSi) [{PhC(NtBu)2}Si(C5Me4H)] with a Cp4-unit attached to the silylene unit. Due to the labile proton at the Cp4-unit the ligand can be either deprotonated or rearranged to a sila fulvene structure. Both forms were isolated. Depending on the reaction conditions and the metal precursor, the ligand or its derivatives can be coordinated in all three forms: silylene (A), anion (B), and sila fulvene (C) (Scheme 7). This flexibility, which is to the best of our knowledge unique, makes [{PhC(NtBu)2}Si(C5Me4H)] a versatile and flexible (pro)ligand for a wide range of metal complexes.
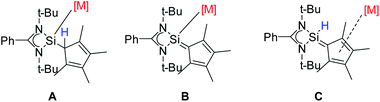 |
| | Scheme 7 Different forms of [{PhC(NtBu)2}Si(C5Me4H)] and its derivatives for coordination: silylene (A), anion (B), and sila fulvene (C). | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The financial support from the German Research Foundation (DFG)-funded transregional collaborative research center SFB/TRR 88 “Cooperative Effects in Homo and Heterometallic Complexes (3MET)” project B3 is gratefully acknowledged. The authors acknowledge the support from the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/467-1 FUGG (JUSTUS cluster).
References
- P. Jutzi, D. Kanne and C. Krüger, Angew. Chem., 1986, 98, 163–164 CrossRef CAS.
- H. Schnöckel, H. J. Göcke and R. Köppe, Z. Anorg. Allg. Chem., 1989, 578, 159–165 CrossRef.
- T. J. Drahnak, J. Michl and R. West, J. Am. Chem. Soc., 1979, 101, 5427–5428 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- B. Blom, M. Stoelzel and M. Driess, Chem.–Eur. J., 2013, 19, 40–62 CrossRef CAS.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS.
- C. W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed. Engl., 2006, 45, 3948–3950 CrossRef CAS.
- M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS.
- M. Okazaki, H. Tobita and H. Ogino, Dalton Trans., 2003, 493–506 RSC.
- N. J. Hill and R. West, J. Organomet. Chem., 2004, 689, 4165–4183 CrossRef CAS.
- M. Scheer, Dalton Trans., 2008, 4372–4386 RSC.
-
B. Marciniec, Hydrosilylation: A Comprehensive Review on Recent Advances, Springer, Dordrecht, 2009 Search PubMed.
- P. Zark, A. Schafer, A. Mitra, D. Haase, W. Saak, R. West and T. Muller, J. Organomet. Chem., 2010, 695, 398–408 CrossRef CAS.
- S. Khan, S. S. Sen, R. Michel, D. Kratzert, H. W. Roesky and D. Stalke, Organometallics, 2011, 30, 2643–2645 CrossRef CAS.
- T. Zell, T. Schaub, K. Radacki and U. Radius, Dalton Trans., 2011, 40, 1852–1854 RSC.
- R. Azhakar, H. W. Roesky, J. J. Holstein, K. Pröpper and B. Dittrich, Organometallics, 2013, 32, 358–361 CrossRef CAS.
- S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS.
- S. S. Sen, S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 3, 659–682 RSC.
- L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef.
- R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Organometallics, 2012, 31, 4588–4592 CrossRef CAS.
- C. W. So, H. W. Roesky, P. M. Gurubasavaraj, R. B. Oswald, M. T. Gamer, P. G. Jones and S. Blaurock, J. Am. Chem. Soc., 2007, 129, 12049–12054 CrossRef CAS.
- S. Khoo, Y. L. Shan, M. C. Yang, Y. Li, M. D. Su and C. W. So, Inorg. Chem., 2018, 57, 5879–5887 CrossRef CAS.
- S. H. Zhang, H. X. Yeong, H. W. Xi, K. H. Lim and C. W. So, Chem.–Eur. J., 2010, 16, 10250–10254 CrossRef CAS.
- Y. L. Shan, B. X. Leong, Y. Li, R. Ganguly and C. W. So, Inorg. Chem., 2017, 56, 1609–1615 CrossRef CAS.
- S. Schafer, R. Koppe, M. T. Gamer and P. W. Roesky, Chem. Commun., 2014, 50, 11401–11403 RSC.
- S. Schafer, R. Koppe and P. W. Roesky, Chem.–Eur. J., 2016, 22, 7127–7133 CrossRef.
- S. Kaufmann, S. Schafer, M. T. Gamer and P. W. Roesky, Dalton Trans., 2017, 46, 8861–8867 RSC.
- N. M. Sergeyev, G. I. Avramenko, A. V. Kisin, V. A. Korenevsky and Y. A. Ustynyuk, J. Organomet. Chem., 1971, 32, 55–77 CrossRef.
- R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Chem. Commun., 2012, 48, 4561–4563 RSC.
- V. W. Day, M. R. Thompson, G. O. Nelson and M. E. Wright, Organometallics, 1983, 2, 494–500 CrossRef CAS.
- T. Iwamoto, N. Ohnishi, N. Akasaka, K. Ohno and S. Ishida, J. Am. Chem. Soc., 2013, 135, 10606–10609 CrossRef CAS.
- S. Spirk, F. Belaj, J. H. Albering and R. Pietschnig, Organometallics, 2010, 29, 2981–2986 CrossRef CAS.
- L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2019, 58, 12007–12010 CrossRef CAS.
-
M. Kaftory, M. Kapon and M. Botoshansky, in The Chemistry of Organic Silicon Compounds, 1998, pp. 181–265, DOI:10.1002/0470857250.ch5.
- A. Jana, D. Leusser, I. Objartel, H. W. Roesky and D. Stalke, Dalton Trans., 2011, 40, 5458–5463 RSC.
- B. Li, C. Wolper, K. Huse and S. Schulz, Chem. Commun., 2020, 56, 8643–8646 RSC.
- S. Mebs, M. A. Chilleck, S. Grabowsky and T. Braun, Chem.–Eur. J., 2012, 18, 11647–11661 CrossRef CAS.
- M. A. Chilleck, T. Braun and B. Braun, Chem.–Eur. J., 2011, 17, 12902–12905 CrossRef CAS.
- T. Li, S. Schulz and P. W. Roesky, Chem. Soc. Rev., 2012, 41, 3759–3771 RSC.
- S. Yadav, E. Sangtani, D. Dhawan, R. G. Gonnade, D. Ghosh and S. S. Sen, Dalton Trans., 2017, 46, 11418–11424 RSC.
- F. M. Mück, J. A. Baus, A. Ulmer, C. Burschka and R. Tacke, Eur. J. Inorg. Chem., 2016, 2016, 1660–1670 CrossRef.
- L. E. Lemmerz, T. P. Spaniol and J. Okuda, Z. Anorg. Allg. Chem., 2016, 642, 1269–1274 CrossRef CAS.
- S. Datta, M. T. Gamer and P. W. Roesky, Dalton Trans., 2008, 2839–2843 RSC.
- M. Arrowsmith, A. Heath, M. S. Hill, P. B. Hitchcock and G. Kociok-Kohn, Organometallics, 2009, 28, 4550–4559 CrossRef CAS.
- S. Yadav, V. S. V. S. N. Swamy, R. Gonnade and S. S. Sen, Chemistryselect, 2016, 1, 1066–1071 CrossRef CAS.
- I. L. Fedushkin, A. A. Skatova, V. A. Chudakova, G. K. Fukin, S. Dechert and H. Schumann, Eur. J. Inorg. Chem., 2003, 2003, 3336–3346 CrossRef.
- I. L. Fedushkin, A. N. Lukoyanov, A. N. Tishkina, G. K. Fukin, K. A. Lyssenko and M. Hummert, Chem.–Eur. J., 2010, 16, 7563–7571 CrossRef CAS.
- R. Anwander, O. Runte, J. Eppinger, G. Gerstberger, E. Herdtweck and M. Spiegler, J. Chem. Soc., Dalton Trans., 1998, 847–858 RSC.
- J. A. Baus, F. M. Muck, H. Schneider and R. Tacke, Chem.–Eur. J., 2017, 23, 296–303 CrossRef CAS.
- E. Neumann and A. Pfaltz, Organometallics, 2005, 24, 2008–2011 CrossRef CAS.
- M. Stoelzel, C. Prasang, B. Blom and M. Driess, Aust. J. Chem., 2013, 66, 1163–1170 CrossRef CAS.
- S. Khoo, H. X. Yeong, Y. Li, R. Ganguly and C. W. So, Inorg. Chem., 2015, 54, 9968–9975 CrossRef CAS.
- N. C. Breit, T. Szilvasi, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958–17968 CrossRef CAS.
- M. Stoelzel, C. Prasang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., Int. Ed. Engl., 2012, 51, 399–403 CrossRef CAS.
- A. Meltzer, S. Inoue, C. Prasang and M. Driess, J. Am. Chem. Soc., 2010, 132, 3038–3046 CrossRef CAS.
- S. Nagase and T. Kudo, J. Chem. Soc., Chem. Commun., 1984, 1392–1394 RSC.
- R. S. Grev, G. E. Scuseria, A. C. Scheiner, H. F. Schaefer and M. S. Gordon, J. Am. Chem. Soc., 1988, 110, 7337–7339 CrossRef CAS.
- A. Sekiguchi and W. Ando, Tetrahedron Lett., 1983, 24, 2791–2794 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR, IR, and MS spectra. XRD data and ORTEP plots. CCDC 2019953–2019959. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04174b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Peter W.
Roesky
and
Peter W.
Roesky
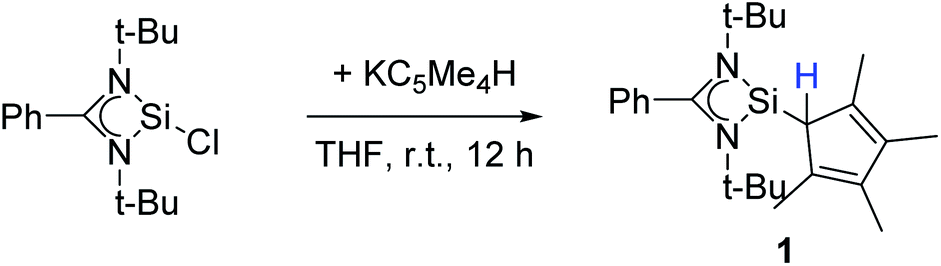
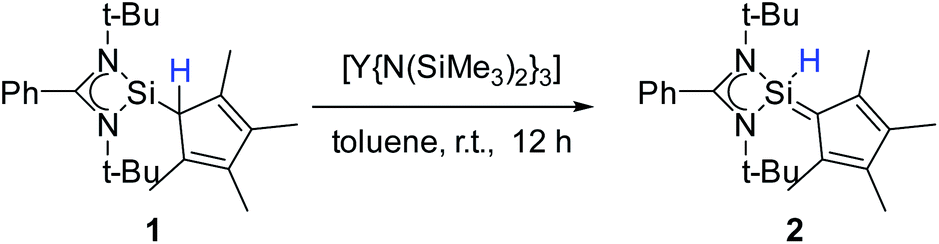
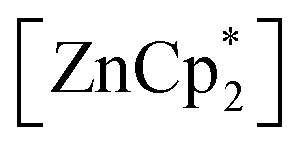 to be a suitable deprotonating agent. Thus,
to be a suitable deprotonating agent. Thus, 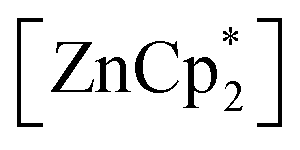 was treated in a straightforward reaction with compound 1 in toluene (Scheme 4), which led to the formation of [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me5)}] (3) as an orange precipitate (starting from compound 2 yields the same result). Upon recrystallization from the hot mother liquor, crystals suitable for X-ray diffraction studies were formed. During the course of the reaction one Cp* ligand was protonated and cleaved off.37 The remaining [ZnCp*]+ fragment was bulky enough to provide steric shielding of the Zn atom.38–40
was treated in a straightforward reaction with compound 1 in toluene (Scheme 4), which led to the formation of [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me5)}] (3) as an orange precipitate (starting from compound 2 yields the same result). Upon recrystallization from the hot mother liquor, crystals suitable for X-ray diffraction studies were formed. During the course of the reaction one Cp* ligand was protonated and cleaved off.37 The remaining [ZnCp*]+ fragment was bulky enough to provide steric shielding of the Zn atom.38–40
 of 1.9118(5) Å. In the 29Si NMR spectrum of compound 3, a peak at 6.0 ppm is detected. Compared to that for compound 1 (39.7 ppm) this signal is significantly high field shifted, whereas compared to that for the more similar compound 2 (−18.6 ppm) a downfield shifted is seen.
of 1.9118(5) Å. In the 29Si NMR spectrum of compound 3, a peak at 6.0 ppm is detected. Compared to that for compound 1 (39.7 ppm) this signal is significantly high field shifted, whereas compared to that for the more similar compound 2 (−18.6 ppm) a downfield shifted is seen.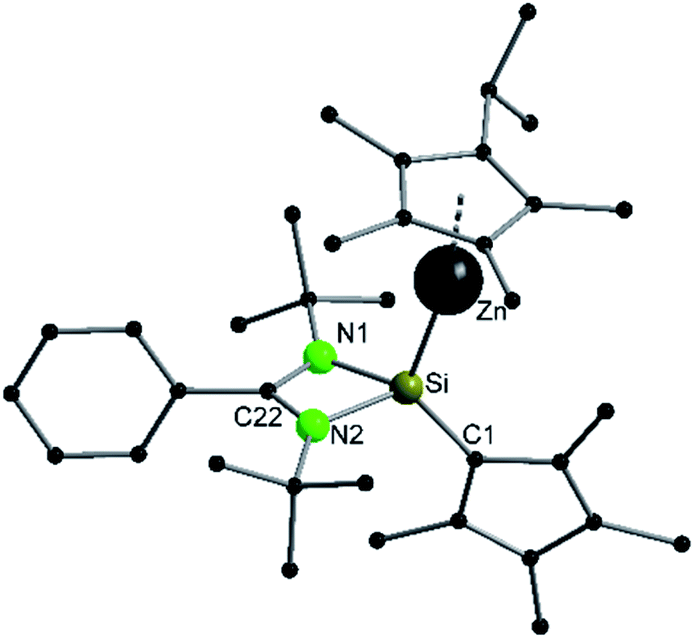
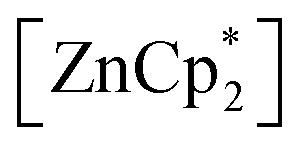 were the key for successful coordination of the silylene, we next used the even more bulky zincocene [Zn(CpiPr4)2] (CpiPr4 = 1-iso-propyl-2,3,4,5-methylcyclopentadienyl) as the precursor (Scheme 4). The reaction of [Zn(CpiPr4)2] with 1 resulted in the product [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me4iPr)}] (4), which was isolated as orange crystals. The product crystallizes in the chiral, monoclinic space group Pc with one molecule in the asymmetric unit (Fig. 4). The chirality is caused by the iso-propyl group on the Cp4iPr-ring, which cannot freely rotate and forms a kind of paddle-wheel geometry. The structure is similar to compound 3 except for the slightly disordered iso-propyl group on the Cp4iPr-ring bound to the zinc atom. The bond length and angles correspond well with those of compound 3 and the NMR spectra show only the additional peaks corresponding to the iso-propyl group.
were the key for successful coordination of the silylene, we next used the even more bulky zincocene [Zn(CpiPr4)2] (CpiPr4 = 1-iso-propyl-2,3,4,5-methylcyclopentadienyl) as the precursor (Scheme 4). The reaction of [Zn(CpiPr4)2] with 1 resulted in the product [{PhC(NtBu)2}(C5Me4)Si{Zn(C5Me4iPr)}] (4), which was isolated as orange crystals. The product crystallizes in the chiral, monoclinic space group Pc with one molecule in the asymmetric unit (Fig. 4). The chirality is caused by the iso-propyl group on the Cp4iPr-ring, which cannot freely rotate and forms a kind of paddle-wheel geometry. The structure is similar to compound 3 except for the slightly disordered iso-propyl group on the Cp4iPr-ring bound to the zinc atom. The bond length and angles correspond well with those of compound 3 and the NMR spectra show only the additional peaks corresponding to the iso-propyl group.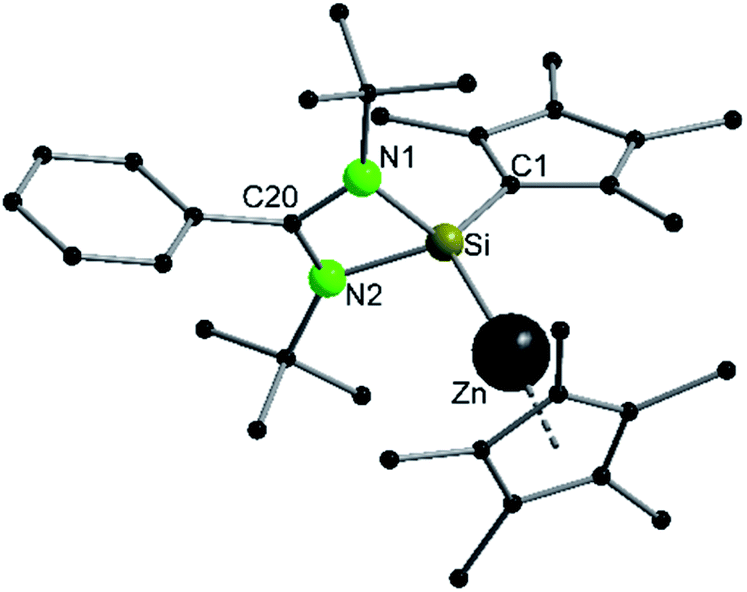
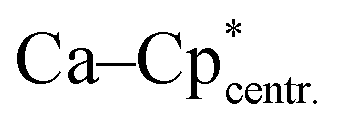 2.368(2); Ca–C1 2.637(3); Ca–C2 2.681(3); Ca–C3 2.780(3); Ca–C4 2.825(3); Ca–C5 2.744(3); Si–N1 1.798(2); Si–N2 1.812(2); Si–C1 1.795(2); Si–H1 1.35(3); N1–C20 1.329(3); N2–C20 1.346(3); C1–C2 1.432(3); C1–C5 1.437(3); C2–C3 1.388(4); C3–C4 1.426(4); C4–C5 1.390(3); Cp*–Ca–Cp4 143.51(13); N2–C20–N1 106.1(2); C1–Si–H1 119.5(12); C1–Si–N1 116.75(11); C1–Si–N2 121.33(10); N1–Si–H1 106.9(13); N2–Si–H1 110.0(12); Si–C1–C2 128.5(2); Si–C1–C5 123.1(2); C2–C1–C5 106.9(2).
2.368(2); Ca–C1 2.637(3); Ca–C2 2.681(3); Ca–C3 2.780(3); Ca–C4 2.825(3); Ca–C5 2.744(3); Si–N1 1.798(2); Si–N2 1.812(2); Si–C1 1.795(2); Si–H1 1.35(3); N1–C20 1.329(3); N2–C20 1.346(3); C1–C2 1.432(3); C1–C5 1.437(3); C2–C3 1.388(4); C3–C4 1.426(4); C4–C5 1.390(3); Cp*–Ca–Cp4 143.51(13); N2–C20–N1 106.1(2); C1–Si–H1 119.5(12); C1–Si–N1 116.75(11); C1–Si–N2 121.33(10); N1–Si–H1 106.9(13); N2–Si–H1 110.0(12); Si–C1–C2 128.5(2); Si–C1–C5 123.1(2); C2–C1–C5 106.9(2).