
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-protected gold/silver superatoms: current status and emerging trends
Haru
Hirai
a,
Shun
Ito
a,
Shinjiro
Takano
 a,
Kiichirou
Koyasu
ab and
Tatsuya
Tsukuda
*ab
a,
Kiichirou
Koyasu
ab and
Tatsuya
Tsukuda
*ab
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: tsukuda@chem.s.u-tokyo.ac.jp
bElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Katsura, Kyoto 615-8520, Japan
First published on 21st October 2020
Abstract
Monolayer-protected gold/silver clusters have attracted much interest as nano-scale building units for novel functional materials owing to their nonbulk-like structures and size-specific properties. They can be viewed as ligand-protected superatoms because their magic stabilities and fundamental properties are well explained in the framework of the jellium model. In the last decade, the number of ligand-protected superatoms with atomically-defined structures has been increasing rapidly thanks to the well-established synthesis and structural determination by X-ray crystallography. This perspective summarizes the current status and emerging trends in synthesis and characterization of superatoms. The topics related to synthesis include (1) development of targeted synthesis based on transformation, (2) enhancement of robustness and synthetic yield for practical applications, and (3) development of controlled fusion and assembly of well-defined superatoms to create new properties. New characterization approaches are also introduced such as (1) mass spectrometry and laser spectroscopies in the gas phase, (2) determination of static and dynamic structures, and (3) computational analysis by machine learning. Finally, future challenges and prospects are discussed for further promotion and development of materials science of superatoms.
 Haru Hirai | Haru Hirai received his B.S. degree in 2018 and M.S. degree in 2020 from The University of Tokyo. He is currently a PhD candidate at the University of Tokyo under the supervision of Prof. Tatsuya Tsukuda. His PhD project is the atomically-precise synthesis and functionalization of ligand-protected gold-based clusters. |
 Shun Ito | Shun Ito received his B.S. degree in 2019 from The University of Tokyo. He is currently a PhD candidate at the University of Tokyo under the supervision of Prof. Tatsuya Tsukuda. His PhD project is the characterization of ligand-protected metal clusters in the gas phase. |
 Shinjiro Takano | Shinjiro Takano received his PhD degree in 2016 from The University of Tokyo under the supervision of Prof. Tatsuya Tsukuda. After working as a postdoctoral researcher at the Department of Chemistry, The University of Tokyo for two years, he was promoted to Assistant Professor at the same department in 2018. His research interests are mainly the investigation of the fundamental properties of chemically modified Au clusters through the development of novel atomically-precise synthetic approaches. |
 Kiichirou Koyasu | Kiichirou Koyasu received a PhD degree in 2006 from Keio University under the supervision of Prof. Atsushi Nakajima. After postdoctoral research at the University of Konstanz, Germany and Nagoya University, Japan, he became Research Associate at Keio University in 2009, and then moved to Tohoku University six months later. In 2013 he was promoted to an Associate Professor at The University of Tokyo. His research interests include the exploration and characterization of metal-based clusters in the gas phase. |
 Tatsuya Tsukuda | Tatsuya Tsukuda received a PhD degree in 1994 from The University of Tokyo under the supervision of Prof. Tamotsu Kondow. After postdoctoral research at RIKEN, he was appointed Assistant Professor at The University of Tokyo in 1994, and in 2000 he became an Associate Professor at the Institute for Molecular Science. He was promoted to Professor at the Catalysis Research Center of Hokkaido University in 2007, before moving to The University of Tokyo in 2011. His research interests cover the atomically-precise synthesis and characterization of gold clusters protected by ligands and their catalytic applications. |
1 Introduction
Metal clusters, which are defined as assemblies of several to hundreds of metal atoms, exhibit novel physicochemical properties due to size-specific structures such as nonbulk-like atomic packing and quantized electronic structures.1–5 Because of these unique features, metal clusters have attracted considerable attention as promising building units of new functional materials. Surface protection by organic ligands enables these clusters to be treated as stable chemical entities. Historically, small gold clusters protected by phosphines (PR3) and halides (X−) have been synthesized with atomic precision in the last five decades.6,7 Their geometric structures have been determined by single-crystal X-ray diffraction (SCXRD) and their stability has been theoretically explained in terms of electronic structures. The preparation of thiolate (RS−)-protected Au nanoparticles reported by Brust and Schiffrin in 1994 paved the way for monolayer-protected Au clusters (MPCs) having higher stability than phosphine-protected Au clusters.8 This perspective summarizes the current understanding of the structural properties of MPCs,9,10 followed by recent efforts and new trends in the development of atomically-precise nanochemistry.1.1 Atomically precise synthesis and structural determination
First, the history of the atomically-precise synthesis and structural determination of MPCs is reviewed by highlighting selected benchmark works reported in the last couple of decades. The existence of a series of magic sizes and size-dependent evolution of the electronic structures in MPCs were reported in early works by Whetten and Murray.11–14 Atomically-precise synthesis of MPCs was established not later than 2010 based on the chemical reduction of precursor Au(I)SR complexes followed by size-focusing treatment and fractionation. Mass spectrometric studies by Tsukuda and Jin identified the currently well-known magic clusters [Au25(SR)18]−, Au38(SR)24, and Au144(SR)60.15–17 At present, nearly atomically-precise synthesis up to Au∼2000(SC6H13)∼290 has been achieved by Dass.18 After important theoretical predictions of structural features of MPCs by Landman,12 Garzón,19 and Häkkinen,20 the ground-breaking paper by Kornberg in 2007 revealed the formation of Au(SR)2 and Au2(SR)3 staples on the surface of the Au79 core of Au102(SR)44.21 Kornberg's work also highlighted the importance of the SCXRD method for establishing the correlation between structures and properties. Murray and Jin elucidated that [Au25(SR)18]− and Au38(SR)24 have icosahedral Au13 and bi-icosahedral Au23 cores, respectively, protected by Au(SR)2 and Au2(SR)3 units.22–24 The ability of theoretical calculations to predict structures has also been demonstrated. Grönbeck and Aikens respectively predicted the geometric structures of [Au25(SR)18]− and Au38(SR)24 independently of the SCXRD results.25,26 The structure of Au144(SR)60 predicted by Häkkinen27 was, 10 years later, proved by SCXRD by Wu.28 In the last decade, the library of MPCs with atomically-defined structures has been expanded remarkably. The organic ligands include phosphines, thiolates, alkynyls (RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C−), and N-heterocyclic carbenes (NHC), and the metal elements include Au, Ag, Cu and their alloys with Pd, Pt, Cd, Hg, and Ir. Fig. 1 plots the number of MPCs whose structures have been determined by SCXRD and clearly shows the rapid expansion of the universe of MPCs.
C−), and N-heterocyclic carbenes (NHC), and the metal elements include Au, Ag, Cu and their alloys with Pd, Pt, Cd, Hg, and Ir. Fig. 1 plots the number of MPCs whose structures have been determined by SCXRD and clearly shows the rapid expansion of the universe of MPCs.
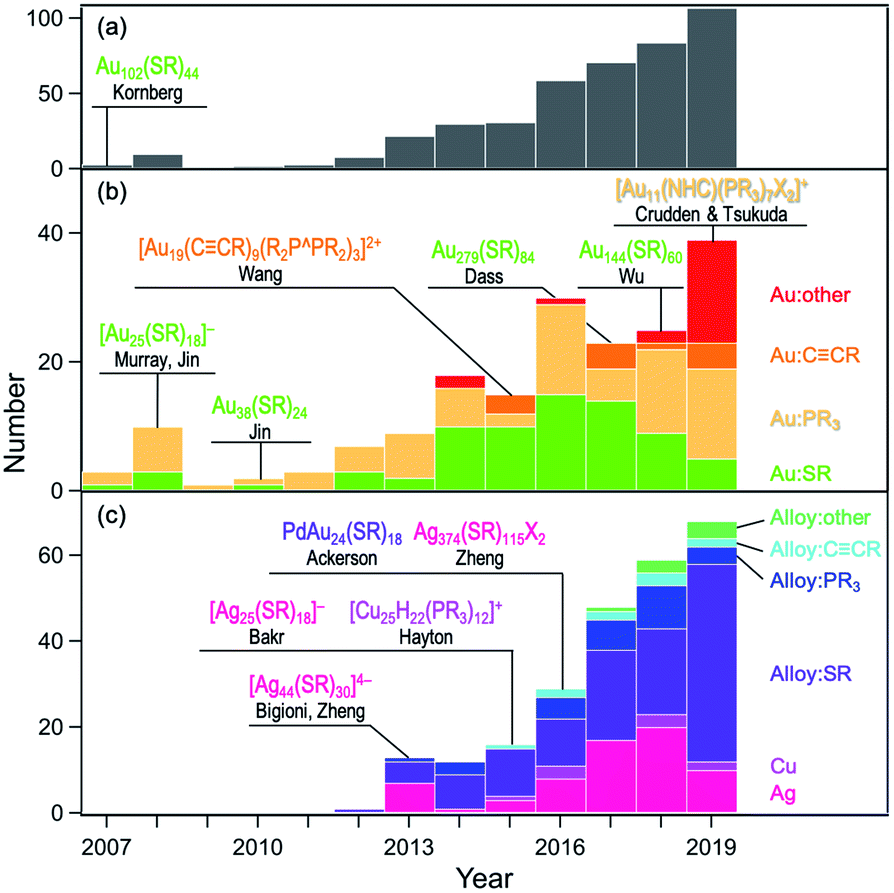 | ||
| Fig. 1 Annual numbers of crystals data of (a) coinage-metal MPCs, (b) Au MPCs, and (c) Ag/Cu/alloy MPCs deposited in the Cambridge Crystallographic Data Centre since 2007, with selected benchmark studies. | ||
1.2 Superatom concept
Since the pioneering works on Al clusters by Castleman, Khanna, and Jena,29–32 the “superatom concept” has been successfully applied to explain the magic stabilities, electronic structures and chemical properties of simple metal clusters in gas phase.4,5,33–36 The jellium model37 qualitatively describes the behavior of valence electrons confined within the clusters of coinage metals (Cu, Ag, Au) having a single valence electron in the ns atomic orbitals (n = 4, 5, 6).38 The spherical jellium model assumes that individual metal atoms delocalize the valence electrons over a uniformly charged spherical volume. Such confinement of the electrons leads to the quantized superatomic orbitals (SOs) labeled 1S, 1P, 1D, 2S, 1F, 2P, 1G, …. The electronic shells of coinage metal clusters are closed when the total number of valence electrons is 2, 8, 18, 20, 34, 40, 58, …. Spherical coinage metal clusters can be viewed as superatoms, given that the electronic shell structures are formed as in the case of conventional atoms. Häkkinen extended this concept to MPCs by introducing a simple counting scheme for the formal number of valence electrons (n*) in MPCs [MxLy]z:39,40| n* = Ax + By − z | (1) |
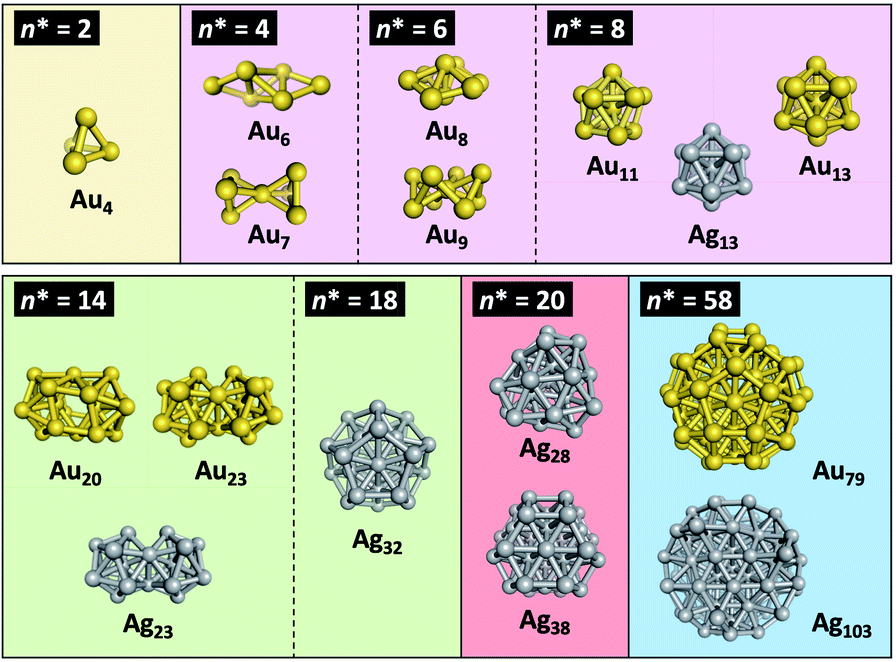 | ||
| Fig. 2 Superatomic Au/Ag cores identified by SCXRD. | ||
The huge impact of structural factors, such as size, shape, and composition, on the electronic structures of superatomic cores can be qualitatively explained within the framework of the spherical and modified jellium model. The origin of important phenomena, such as the closure of electronic shells, evolution of an energy gap between the highest-occupied molecular orbital (HOMO) and the lowest-unoccupied molecular orbital (LUMO), and energy shift of the orbitals upon doping, was qualitatively explained by considering how the volume, shape, and depth of the jellium potential are affected by structural factors. The reduction of the HOMO–LUMO gap with size can be understood by the increase of the volume of the jellium potential and the occupation of SOs in high-energy and dense regions (Fig. 3). When the spherical jellium is deformed, the SOs originally degenerated in energy are split due to the Jahn–Teller effect.42 For example, the energy shift of the SOs upon deformation into oblate and prolate shapes is schematically shown in Fig. 3. Triply degenerated 1P SOs in the spherical jellium are split into two subgroups in the oblate-shaped jellium: 1Pz along the short axis is destabilized compared to 1Px and 1Py. As a result, the oblate superatoms with a closed subshell configuration (1S)2(1P)4 become stable, as can be found in Au8(6e) and Au9(6e) (Fig. 2). In contrast, the prolate superatoms with closed subshell configurations such as (1S)2(1P)2 and (1S)2(1P)6(1D)6 become stable, as can be seen in Au6(4e), Au7(4e), Au20(14e), Au23(14e), and Ag23(14e) (Fig. 2). The effects of doping on the electronic structures of superatoms can be qualitatively predicted by a two-step spherical jellium model,43–45 which is based on the idea that the host and dopant atoms provide different but uniform background potential. When the dopant atom has higher valency than the host atoms, the SOs are stabilized because the effective potential become more attractive, and vice versa (Fig. 3). The energy ordering of SOs can be changed by doping. For example, if the higher-valence dopant is introduced at the center of a spherical superatom, the S and P SOs having density at the center are stabilized more than the D and F SOs having several nodal planes at the center. As a result, the energy ordering of the SOs is changed to 1S, 1P, 2S, 1D, 2P, 1F, … so that the shell closing occurs at n* = 2, 8, 10, 20, 26, 40, …. Although such structural simplification smears out the detailed structural information obtained by SCXRD, these qualitative pictures provide a rational design principle for the development of new functionalized superatoms. However, there are a class of MPCs that fall outside the scope of the superatom concept. For example, the n* values of face-centered cubic (fcc) Au cores of Au8n+4(SR)4n+8 cannot be explained by the simple jellium model,46 but by the grand unified model (GUM).47 In the GUM, the Au cores are viewed as assemblies of smaller superatomic units, such as triangular Au3(2e) and tetrahedral Au4(2e). In this perspective, we will focus only on the MPCs that can be viewed as chemically-modified Au/Ag superatoms.
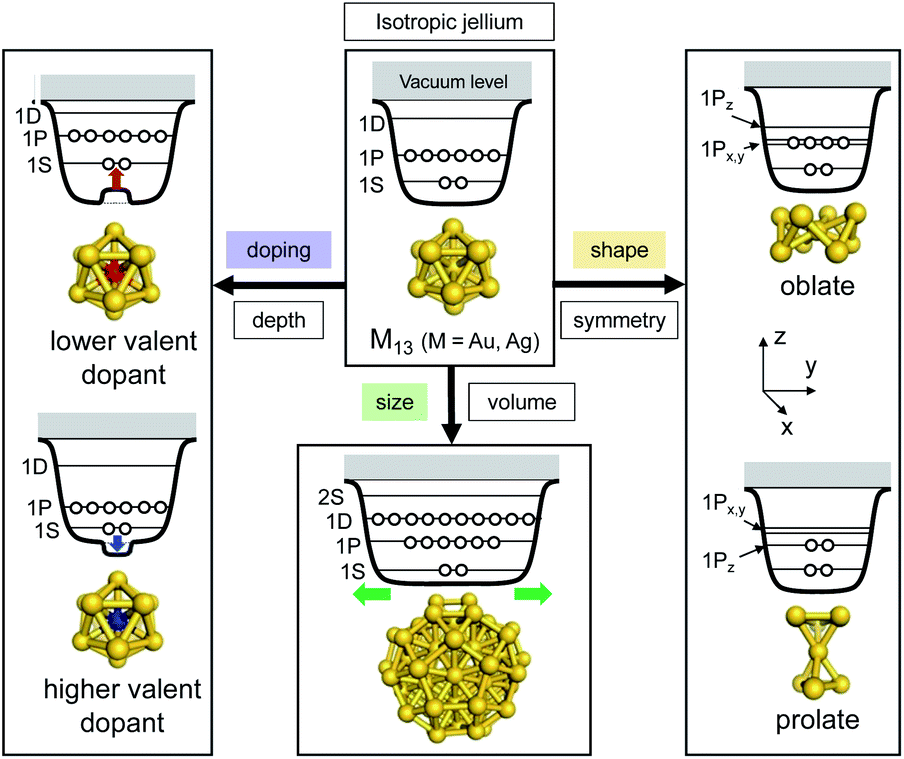 | ||
| Fig. 3 Schematic representation of effects of size, shape and doping on jellium potentials. | ||
1.3 Aim of this perspective
The rapid growth of materials chemistry of chemically-modified superatoms in the last decade has been made possible by the establishment of atomically-precise synthesis and structural determination by SCXRD. Thanks to reproducible synthesis, a wide variety of fundamental properties such as optical and photophysical properties, catalysis, and magnetic properties has been studied by diverse researchers with different backgrounds and interests.48 These studies have demonstrated that chemically-modified superatoms are promising building units of functional materials in the future. In addition, if we could establish a periodic table of the superatoms as artificial elements on a nanoscale, it would create a new paradigm of nanoscience.However, several problems must be overcome for the further development of materials chemistry of superatoms. First, most of the new superatoms have been obtained serendipitously because the formation of superatoms is based on stochastic processes of the aggregation of metal atoms and ligands, and because there is no guarantee that single crystals will be obtained. Thus the synthesis and crystallization of superatoms rely on the carpet-bombing approach of screening the synthetic conditions such as precursor materials, solvents, reducing agents, and reaction temperature. It is necessary to increase the chance of unexpected discoveries and ultimately to establish the targeted synthesis of desired superatoms. Secondly, the stability and scale of the synthesized quantity of superatoms must be increased for future applications. Thirdly, a strategy for creating new properties and functions by taking advantage of cooperative interaction between individual superatoms is needed. In addition to these challenges related to synthesis, new characterization methods which are supplementary to SCXRD are required. For example, the characterization of superatoms isolated in vacuum would provide information of inherent structures and stabilities of the superatoms without the interference from the solvents and counterions. Experimental tools to probe the dynamic aspects of superatoms are also required. This perspective summarizes recent progress toward overcoming the challenges for synthesis and developing new characterization methods.
2 Emerging trends in synthesis
2.1 Targeted synthesis by transformation
This section explains the transformations of a preformed atomically-precise superatom into another superatom with different size, composition, and structure. Synthesis through such transformation has advantages in terms of selectivity and targetability over the conventional methods based on stochastic aggregation of metal atoms and ligands. In addition, such transformations may produce novel products that cannot be obtained by conventional methods. In the following, the transformation processes are categorized into two types: addition and exchange reactions.CR)18]2− upon the addition of a stoichiometric amount of Au(I)–CCR oligomers (Fig. 4d).53
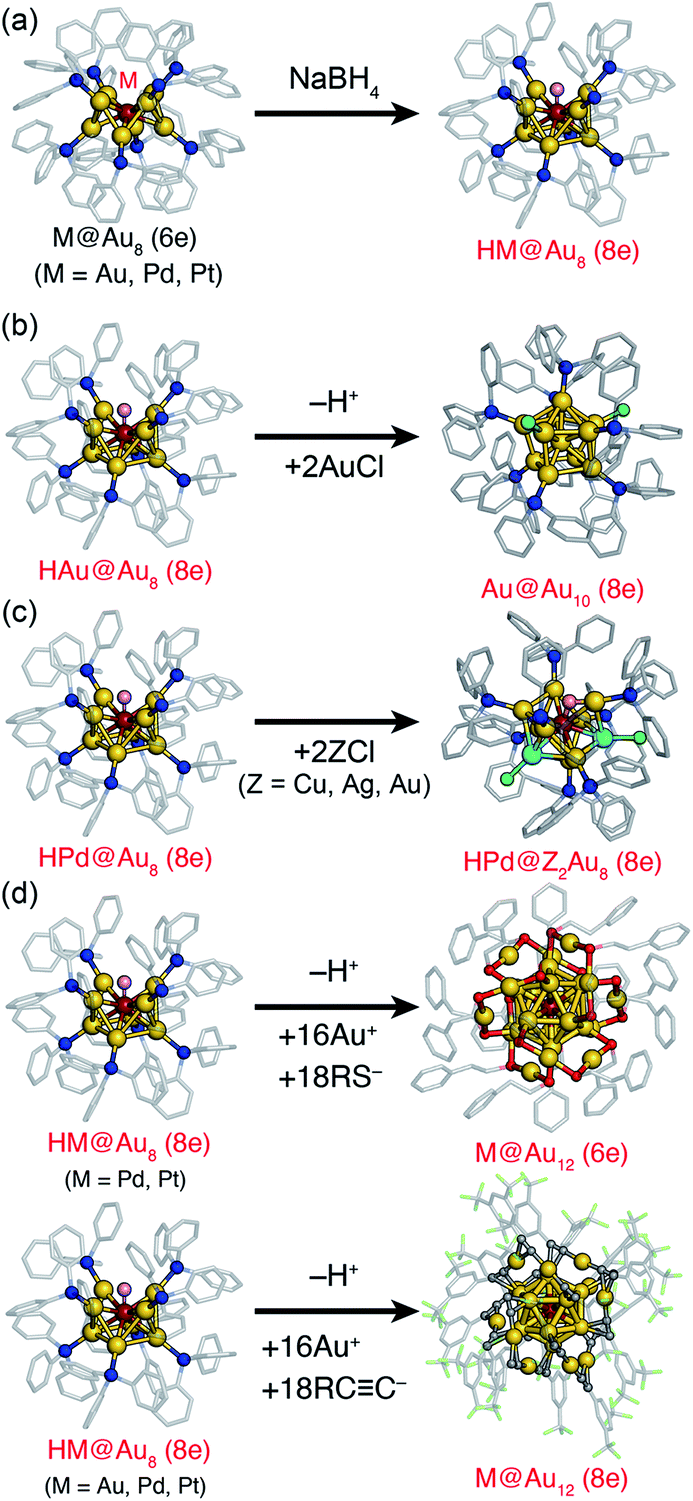 | ||
| Fig. 4 (a) Hydride doping to M@Au8(6e) (M = Au, Pd, Pt) to form HM@Au8(8e). (b) Growth, (c) doping, (d) chemical modification of HM@Au8(8e). Color code: yellow = Au; wine red = Pd or Pt; cyan = Cu, Ag, or Au; pink = H; red = S; green = Cl; blue = P; gray = C. Organic residues are depicted as sticks. | ||
Larger superatoms can be obtained by the controlled growth of “seed” superatoms. Xie achieved selective growth of [Au25(p-MBA)18]− (p-MBA = 4-SC6H4COOH) to Au38(p-MBA)24 and further to [Au44(p-MBA)26]2− by kinetically controlled reduction of Au(I)-p-MBA oligomers by carbon monoxide (CO) in the presence of [Au25(p-MBA)18]− (Fig. 5).54 Two growth mechanisms operate in slow reduction with CO; (1) the LaMer-like growth pathway, in which particles monotonically get larger with the addition of newly reduced Au(I)-p-MBA oligomers and (2) the aggregative growth pathway, in which two particle agglomerate to form a larger particle.
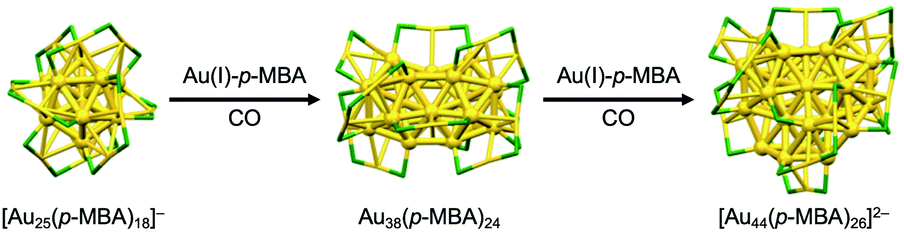 | ||
| Fig. 5 Controlled growth of thiolate-protected Au superatoms. Color code: yellow = Au; green = S. Organic residues are omitted for simplicity. | ||
Ligand exchange of preformed superatoms is a versatile strategy to tune their properties while retaining the core structures. For example, the thermal stabilities of Au11(8e) and Au13(8e) are enhanced by replacing phosphine with NHC65 and thiolate with selenolate,66,67 respectively. In contrast, when introducing thiolates having significantly different steric hinderance, the size and/or atomic packing of the superatomic cores can be modulated. Table 1 shows examples of the ligand-exchange-induced size/structure transformation (LEIST)68–81 observed for thiolate-protected Au/Ag superatoms. The fact that the products depend on the thiolates used indicates that interligand interaction due to steric hinderance of the tail groups of the ligands plays a crucial role. Reversible LEIST was observed as listed in Table 2. Au20(8e) superatomic cores of Au28(TBBT)20 (TBBT = 4-(CH3)3CC6H4S) and Au28(c-HT)20 (c-HT = c-C6H11S) exhibit different catalysis for CO oxidation due to differences in geometric structures.79 ESI-MS analysis by Bakr demonstrated that conversion from [Ag25(2,4-DMBT)18]− to [Ag44(4-FTP)30]4− (4-FTP = 4-FC6H4S) proceeded via dimerization, while reverse conversion proceeded by dissociative disproportionation to form [Ag25(2,4-DMBT)18]− and smaller Ag(I)-SR fragments.80 These examples illustrated that LEIST allows us to access new clusters that cannot be synthesized by the direct reduction method, although it is difficult to predict what type of transformation will be induced by ligand exchange.
| Precursors | Products | Ref. | ||
|---|---|---|---|---|
| Compositiona | Core | Compositiona | Core | |
| a PET = PhC2H4S; TBBT = 4-(CH3)3CC6H4S; 2,5-DCBT = 2,5-Cl2C6H3S; 2,4-DMBT = 2,4-(CH3)2C6H3S; 1,3-BDT = 1,3-C6H4S2; 2,4-DCBT = 2,4-Cl2C6H3S; 4-FTP = 4-FC6H4S; 4-CTP = 4-ClC6H4S. b Unknown. | ||||
| [Au25(PET)18]− | Au13(8e) | Au20(TBBT)16 | Au7(4e) | 70 |
| Au28(TBBT)20 | Au14(8e) | 71 | ||
| Au38(PET)24 | Au23(14e) | Au36(TBBT)24 | Au28(12e) | 72 |
| Au30(StBu)18 | Au22(12e) | 73 | ||
| Au144(PET)60 | Au114(84e) | Au133(TBBT)52 | Au107(81e) | 74 and 75 |
| Au99(SPh)42 | —b | 76 | ||
| [Ag59(2,5-DCBT)32]3− | —b | [Ag25(2,4-DMBT)18]− | Ag13(8e) | 77 |
| [Ag29(1,3-BDT)12(PPh3)4]3− | Ag13(8e) | 77 | ||
| [Ag44(SR)30]4−, SR = 2,4-DCBT/4-FTP/4-CTP | Ag32(18e) | 77 | ||
| Compositiona | Core | Compositiona | Core | Ref. |
|---|---|---|---|---|
| a TBBT = 4-(CH3)3CC6H4S; cHT = c-C6H11S; 2,4-DMBT = 2,4-(CH3)2C6H3S; 4-FTP = 4-FC6H4S; SAdm = C10H16S. | ||||
| Au30(StBu)18 | Au20(12e) | Au36(SR)24, SR = SPh/TBBT | Au24(12e) | 78 |
| Au28(TBBT)20 | Au20(8e) | Au28(cHT)20 | Au20(8e) | 79 |
| [Ag25(2,4-DMBT)18]− | Ag13(8e) | [Ag44(4-FTP)30]4− | Ag32(18e) | 80 |
| [PtAg28(SAdm)18(PPh3)4]2+ | PtAg12(8e) | [PtAg28(cHT)18(PPh3)4]2+ | PtAg12(8e) | 81 |
2.2 Improving robustness
In principle, superatoms are stabilized against aggregation by protection with the organic ligands. However, in practice, most of the superatoms lack long-term stability under ambient conditions and decompose under harsh conditions. Therefore, improvement of the robustness is an essential requirement for practical applications.One of the promising methods for protecting Au superatoms recently found is to use N-heterocyclic carbenes (NHCs) which are known to form highly robust self-assembled monolayers on Au(111) surfaces.82–84 The first NHC-protected Au superatom reported was Au11(8e) in [Au11(NHCiPr)(TPP)7Cl2]+ (NHCiPr = C7H4N2(CH(CH3)2)2) synthesized by exchanging a phosphine ligand of [Au11(TPP)8Cl2]+ with NHCiPr.65 A single NHCiPr ligand selectively replaced the TPP ligand located between two Cl ligands to yield [Au11(NHCiPr)(TPP)7Cl2]+ (Fig. 6a). [Au11(NHCiPr)(TPP)7Cl2]+ showed much higher thermal stability than [Au11(TPP)8Cl2]+ and acted as an electrocatalyst for CO2 reduction. The high stability was ascribed to a strong Au–C bonding based on the collision-induced dissociation (CID) MS and theoretical calculation.65,85 Following the above report, Au superatoms protected only by other NHCs and halogens have been reported.86–88 For example, [Au13(NHCBn)9Cl3]2+ (NHCBn = C7H4N2(CH2Ph)2) (Fig. 6b)86 and [Au13(di-NHC)5Br2]3+ (di-NHC = PhCH2-C7H4N2-(CH2)3-C7H4N2-CH2Ph) (Fig. 6c)88 with an icosahedral Au13(8e) core were synthesized. The latter was robust at temperatures (−70 °C and 100 °C) and under various redox and pH conditions (pH = 1–11) and survived for <6 min even when treated with aqua regia. SCXRD analysis suggested that the ligand layers of NHCs rigidified the Au13(8e) core by the CH–π and π–π interaction between the adjacent NHC ligands. As a result, [Au13(NHCNp)9Cl3]2+ (NHCNp = C7H4N2(CH2(2-C10H7))2) and [Au13(di-NHC)5Br2]3+ exhibit visible photoluminescence with quantum yields as high as 16 and 15%, respectively.86,88
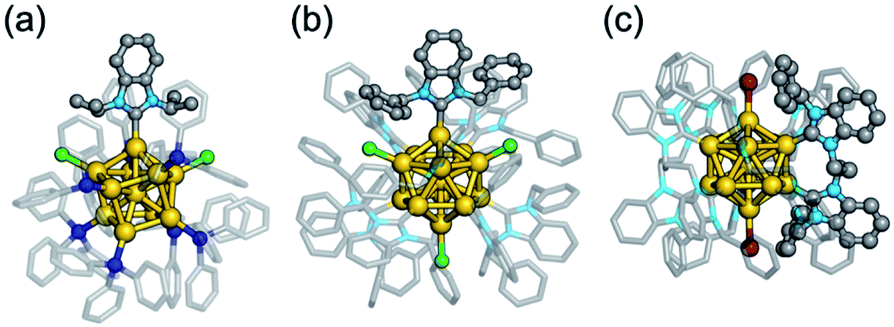 | ||
| Fig. 6 Structures of ultrarobust Au superatoms protected by N-heterocyclic carbenes. (a) [Au11(NHCiPr)(TPP)7Cl2]+, (b) [Au13(NHCBn)9Cl3]2+, (c) [Au13(di-NHC)5Br2]3+. Color code: yellow = Au; gray = C; sky blue = N; blue = P; light green = Cl; dark red = Br. Organic residues are depicted as sticks. | ||
2.3 Scaling up yields
Another challenge for synthesis is to scale up the yield. Typical yields of lab-scale synthesis are on the order of a few milligrams owing to the stochastic nature of the cluster formation and to the loss during rigorous purification. Such low yield limits the characterization methods that can be applied and the scope of practical applications.The most impressive examples are >100 g synthesis of [Ag44(p-MBA)30]4− (Fig. 7a) and >10 g synthesis of [Ag44(3,4-DFBT)30]4− (3,4-DFBT = 3,4-F2C6H3S) having a common Ag32(18e) superatomic core.89,90 The reason for the large-scale synthesis of [Ag44(SR)30]4− is the high yield (>95%) due to extraordinarily high stability: the HOMO–LUMO gap is as large as 0.78 eV and the coordinating solvents as well as the ligands are involved in the protection. Paste-base synthesis in the solid state was also reported as a greener (less wasteful) method.91 Hitherto-known bimetallic clusters [MAu8(TPP)8](NO3)2 (M = Pd, Pt) were selectively synthesized on a large scale (0.2–1 g) by co-reduction of Au(NO3)(TPP) and M(TPP)4 in a one-pot reaction (Fig. 7b).50,53 Tsukuda successfully obtained MAu24(SR)18 on a large scale (50–200 mg) by using [MAu8(TPP)8](NO3)2 as starting materials of conversion (see Section 2.1.1).53 Zheng reported gram-scale synthesis of [Ag28Cu12(2,4-DCBT)24]4− (2,4-DCBT = 2,4-Cl2C6H3S) with the Ag28(20e) superatomic core (Fig. 7c).92 Zhu synthesized [Cu25H22(4-FPP)12]+ (4-FPP = P(4-FC6H4)3) in gram-scale via a high-yield (83%) route under aerobic conditions (Fig. 7d).93 The yield is much higher than that of [Cu25H22(TPP)12]+ under inert conditions (ca. 150 mg, 23%) because 4-FPP is more electron-withdrawing than TPP. The large-scale syntheses outlined above are based on high selectivity due to the intrinsic stability of the products and careful control of the reaction kinetics. However, it is not trivial to scale up the yield while retaining the selectivity by simply increasing the concentration and/or volume of the precursor solutions in the batch synthesis. A possible solution is to use fluidic systems as demonstrated in the synthesis of organic,94 inorganic,95 and nano-materials.96 In contrast to the conventional batch mixing, microfluidic system allows to scale up the quantity of the MPCs while retaining their quality (size distribution) because it provides efficient and homogeneous mixing conditions of solutions regardless of the total volumes of the solutions. Microfluidic synthesis of superatoms will be one of the interesting challenges for synthesis in the future.
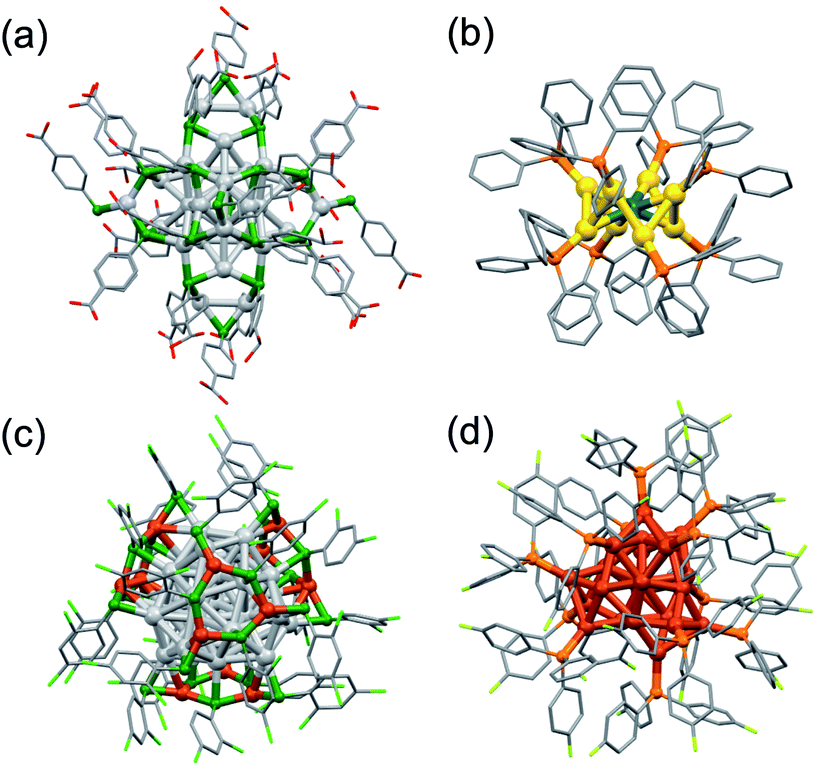 | ||
| Fig. 7 Structures of clusters synthesized on a large scale: (a) [Ag44(p-MBA)30]4−, (b) [PdAu8(TPP)8]2+, (c) [Ag28Cu12(2,4-DCBT)24]4−, (d) [Cu25H22(4-FPP)12]+. Color code: yellow = Au; light gray = Ag; brown = Cu; dark blue = Pd; green = S; orange = P; gray = C; red = O; light green = Cl; light yellow = F. Organic residues are depicted as sticks. | ||
2.4 Bonding and assembly of superatoms
An increasing number of new molecules and materials have been created on the basis of systematic understanding of fundamental properties and bonding interaction of individual elements. Thus, it is an interesting challenge to create a hierarchical world on a nanoscale using superatoms as artificial elements, similar to conventional molecular chemistry. This section examines recent progress in the development of quasi-molecules of superatoms (superatomic molecules) and higher assemblies of superatoms.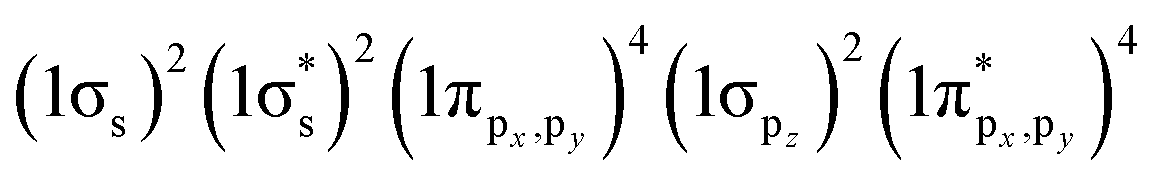 ,97 indicating that the formal bond order between the Au11 and Au13 units is one. Namely, the bonding scheme in Au20(14e) and Au23(14e) is similar to that of the F2 molecule. Similarly, bi-icosahedral Ag23(14e) and Au25(14e) cores correspond to the facet-shared dimer of Ag13(7e) and vertex-shared dimer of Au13(7e), respectively.98,99 Bi-, tri-, and penta-icosahedral superatoms Au25(16e), Au37(24e), and Au60(40e) are constructed by closed-shell Au13(8e) and correspond to a dimer, trimer, and pentamer of the rare gas atoms, respectively.87,100–105 Bimetallic superatomic molecules Pt2Ag33(16e) and Pt3Ag44(22e) contain icosahedral units of PtAg12(8e): their bonding schemes are analogues to those of dimers of rare gas atoms and tri-halogen anions, respectively.106 The Au22(22e) core in [Au22(Ph2P(CH2)8PPh2)6]0 corresponds to a homodimer of Au11(11e)107 which are bonded by a 1D1D triple bond, similarly to transition metal binuclear complexes.108
,97 indicating that the formal bond order between the Au11 and Au13 units is one. Namely, the bonding scheme in Au20(14e) and Au23(14e) is similar to that of the F2 molecule. Similarly, bi-icosahedral Ag23(14e) and Au25(14e) cores correspond to the facet-shared dimer of Ag13(7e) and vertex-shared dimer of Au13(7e), respectively.98,99 Bi-, tri-, and penta-icosahedral superatoms Au25(16e), Au37(24e), and Au60(40e) are constructed by closed-shell Au13(8e) and correspond to a dimer, trimer, and pentamer of the rare gas atoms, respectively.87,100–105 Bimetallic superatomic molecules Pt2Ag33(16e) and Pt3Ag44(22e) contain icosahedral units of PtAg12(8e): their bonding schemes are analogues to those of dimers of rare gas atoms and tri-halogen anions, respectively.106 The Au22(22e) core in [Au22(Ph2P(CH2)8PPh2)6]0 corresponds to a homodimer of Au11(11e)107 which are bonded by a 1D1D triple bond, similarly to transition metal binuclear complexes.108
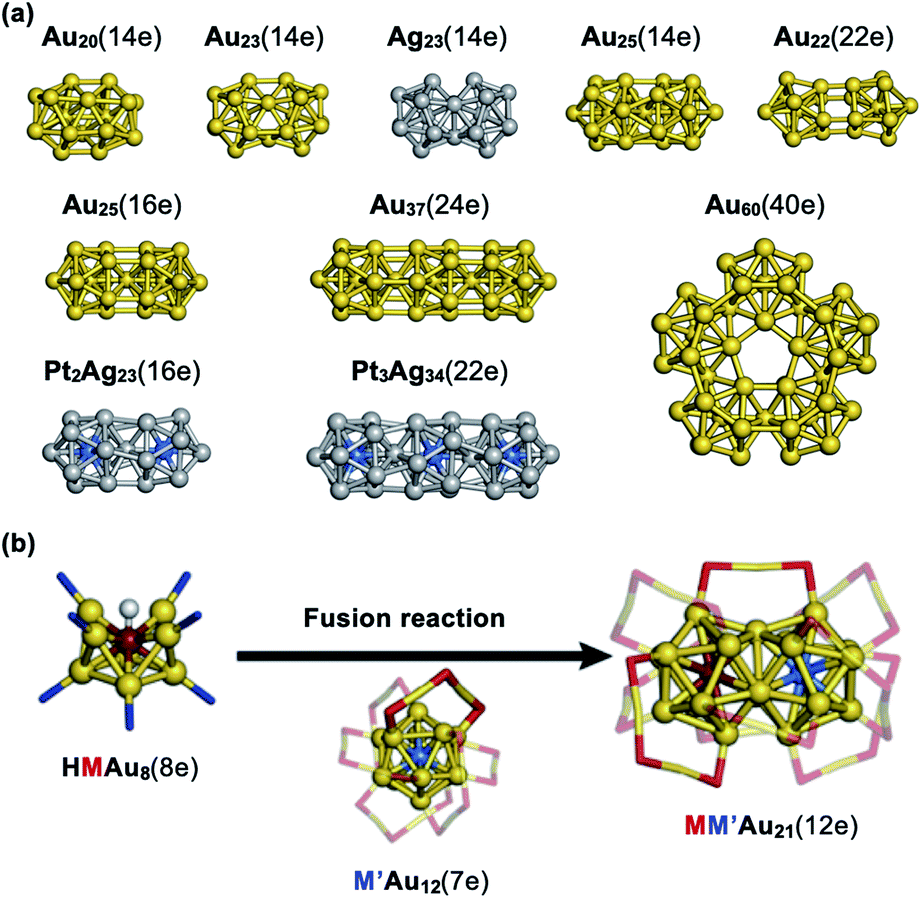 | ||
| Fig. 8 (a) Examples of homo-superatomic molecules. Color code: yellow = Au; light gray = Ag; light blue = Pt. (b) Targeted synthesis of superatomic molecules by fusion. Color code: yellow = Au; wine red = Pd or Pt; blue = P; red = S; white = H. Au–S oligomers and organic residues are omitted for simplicity. | ||
These examples imply that a class of quasi-molecules can be developed by bonding various superatomic units through various bonding schemes. However, their synthesis relies on serendipity; targeted synthesis using well-defined superatoms as precursors is highly demanded for a deeper understanding of the bonding theory of superatoms. Recently, Maran reported the formation of Au38(SR)24 by dimerization of [Au25(SR)18]0 (R = C3H7, C4H9, C8H17, C2H2Ph). This reaction is formally described as:109
| Au13(7e) + Au13(7e) → Au23(14e) + (Au+)3 | (2) |
The faster reaction with smaller thiolates suggests that the overlap of superatomic orbitals is crucial. Takano and Tsukuda synthesized MM′Au21(12e) (M, M′ = Pd, Pt) by the reaction between hydride-doped [HMAu8(TPP)8]+ and monoanionic [M′Au24(PET)18]− (Fig. 8b).110 SCXRD revealed that MM′Au21(12e) corresponds to homo- and hetero-dimers of M@Au12(6e) and M′@Au12(6e). This is the first example of targeted synthesis of hetero-dimers of superatoms via fusion reactions of the corresponding precursors. A rare example of hetero-superatomic molecules can be found in [Au20(H)3(TPP)14]3+; the Au20(14e) core corresponds to a heterodimer of Au9(6e) and Au11(8e) connected via two triangular faces.111
1D assemblies have attracted interest as nanoscale electrical conductors. Maran reported the formation of linear polymers of [Au25(SnBu)18]0 in the crystal connected by intercluster aurophilic interaction (Fig. 9a).113 The polymer of the paramagnetic Au13(7e) units was silent in electron paramagnetic resonance (EPR) measurement because unpaired electrons of neighboring clusters coupled with each other. This electron pairing indicates that the conduction band is fully occupied while the valence band is empty. Theoretical calculation predicted that the polymer of [Au25(SnBu)18]0 has a band gap of ∼0.12 eV at 0 K and shows semiconducting behavior at room temperature. (AuAg)34(CCAdm)20 (Adm = C10H16) also formed a 1D polymer linked via Ag–Au–Ag bonds (Fig. 9b).114 The conductivity along the polymer chain (1.49 × 10−5 S m−1) was 1800 times higher than that along the interchain direction. The conductivity of the (AuAg)34 polymer is 1–3 orders of magnitude higher than that of a 1D polymer of Au15(8e) linked by weak inter-ligand interactions.115 The comparison suggests that direct connection by metal–metal bonds promotes the electron transfer.
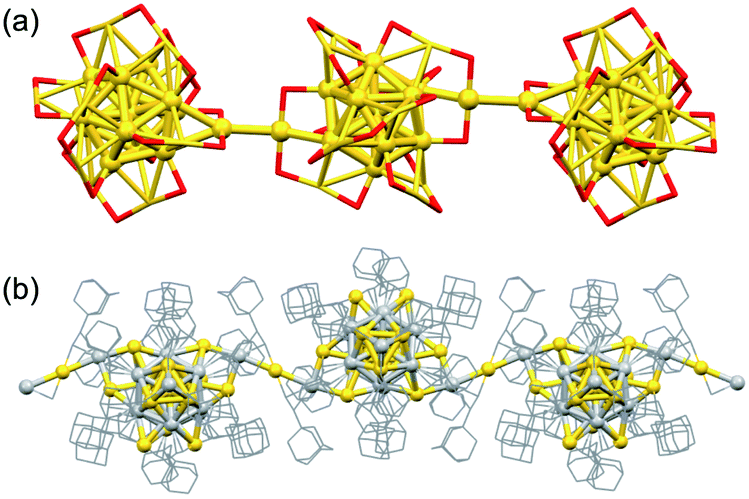 | ||
| Fig. 9 SCXRD structures of polymers of (a) [Au25(SnBu)18]0 and (b) (AuAg)34(CCAdm)20. Color code: yellow = Au; light gray = Ag; red = S; gray = C. Au–S oligomers and organic residues are depicted as sticks. | ||
A super-structure of Ag superatoms with higher dimension has been reported. Zang assembled Ag14(DT-o-C)6 (DT-o-C = B10H10C2S2) having Ag6(2e) superatomic cores using bidentate pyridyl ligands: the assembly forms 1–3D structures depending on the length of linkers (Fig. 10).116 The 3D assembly showed extraordinarily high thermal stability and temperature-dependent photoluminescence, which is different from that of monomer. Recently, a 3D assembly of [AuAg21(SAdm)12]3+ was produced via Ag–F–Ag bonds with SbF6− anions.117 The assembly was enantiopure because of the chiral arrangement of the ligand layer and exhibited circularly polarized luminescence. Red emission was observed in the presence of protic solvents such as methanol, ethanol, and water, but turned off after evaporation of the solvents. There is great scope for developing new functionalized materials by controlling the assembling behaviors of superatoms (e.g., the distance, symmetry, dimensionality and degree of electronic coupling) by appropriately designing the surface modification.
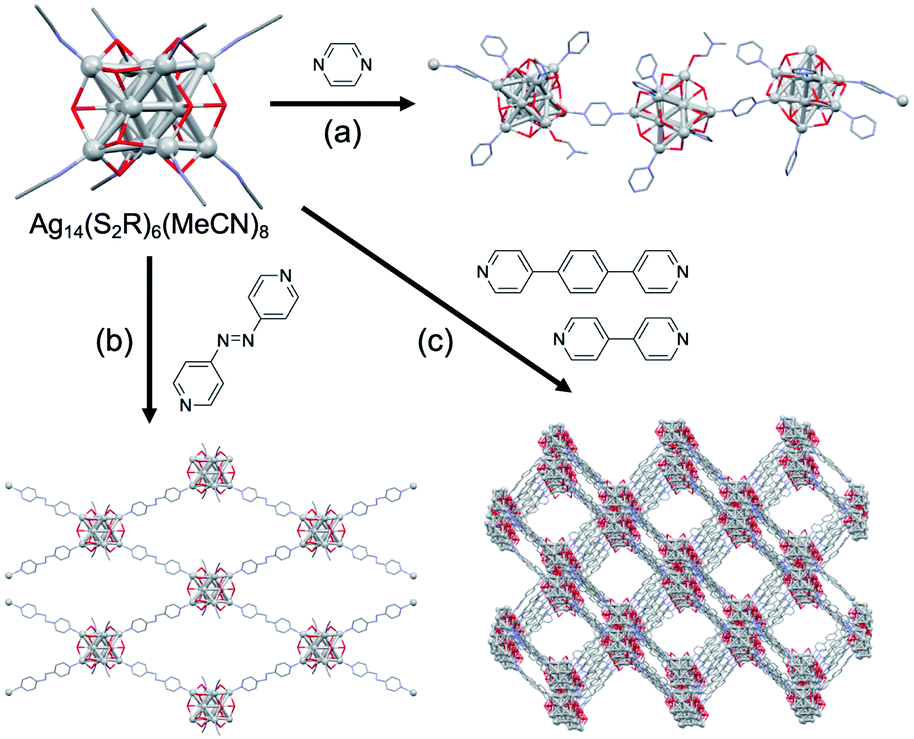 | ||
| Fig. 10 (a) 1D, (b) 2D, and (c) 3D assembly of Ag6(2e) superatoms. Color code: light gray = Ag; red = S; blue = N; gray = C. Organic residues are depicted as sticks. | ||
3 Emerging trends in characterization
Ligand-protected Au/Ag superatoms have been characterized by various experimental methods conventionally used in chemical analysis. The methods include SCXRD, mass spectrometry (MS), X-ray photoelectron spectroscopy (XPS), nuclear magnetic resonance (NMR), pulse voltammetry, X-ray absorption spectroscopy (XAS), aberration-corrected (scanning) transmission electron microscopy (AC(S)TEM), ultraviolet-visible (UV-visible) optical spectroscopy, Fourier-transform infrared spectroscopy (FT IR), photoluminescence (PL) spectroscopy, and circular-dichroism (CD) spectroscopy. In this section, we focus on emerging trends in experimental characterization and theoretical approach for structural prediction.3.1 Gas phase methods
Characterization of ligand-protected superatoms isolated in the gas phase provides intrinsic information of superatoms by eliminating perturbations from the surrounding environments (e.g. solvent, counterion). Especially when SCXRD is available, correlations between electronic and geometric structures can be established. Electrospray ionization (ESI) is a typical interface to introduce the chemically synthesized superatoms in vacuum in the intact form. After mass selection, the superatoms can be characterized by a variety of gas-phase methods such as collision-induced dissociation (CID) MS, ion mobility (IM) MS, photoelectron spectroscopy (PES), photodissociation (PD) MS, and surface-induced dissociation (SID) MS. We herein limit ourselves to MS, CID MS, and laser spectroscopy. The operating principles of other methods and their application to ligand-protected Au/Ag superatoms can be found in recent publications.118–122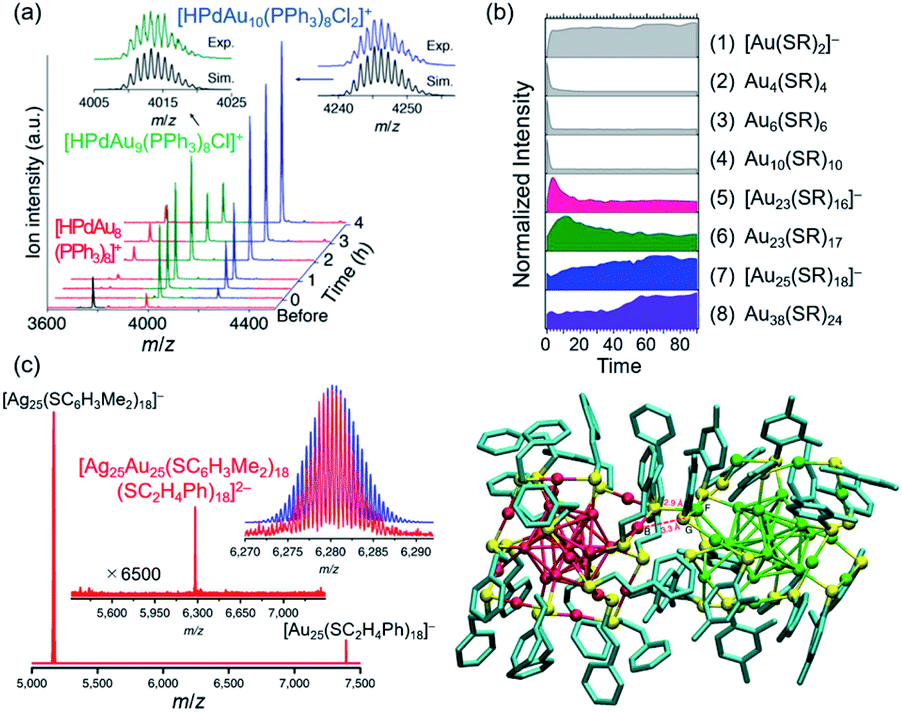 | ||
| Fig. 11 (a) Time-resolved ESI mass spectra of sequential growth of [HPdAu8(TPP)8]+ to [HPdAu10(TPP)8Cl2]+. (b) Time-dependent ESI MS intensity of Au-SR complexes (SR = p-MBA) (1–4) and clusters (5–8) during the synthesis of Au25(SR)18: (1) [Au(SR)2]−, (2) Au4(SR)4, (3) Au6(SR)6, (4) Au10(SR)10, (5) [Au23(SR)16]−, (6) Au23(SR)17, (7) [Au25(SR)18]−, (8) Au38(SR)24. (c) ESI mass spectrum of the mixture of [Au25(PET)18]− and [Ag25(2,4-DMBT)18]− and DFT-optimized structure of [Ag25Au25(2,4-DMBT)18(PET)18]2−. Color code: red = Au; green = Ag; yellow = S; blue = C. Adapted with permission from ref. 50, 124 and 63. Copyright 2018 American Chemistry Society, copyright 2018 American Chemistry Society, and copyright 2016 Nature Publishing Group. | ||
CR)18]2− (M = Pd, Pt; R = 3,5-(CF3)2C6H3) having an icosahedral M@Au12(8e) superatomic core mainly afforded [MAu24(CCR)18−2n]2− (n = 1–6) having (8 + 2n) electrons via sequential reductive elimination of 1,3-diynes RCC–CCR (Fig. 12).126 Theoretical calculations on a model system [MAu24(CCCF3)16]2− predicted that the increased electrons are not accommodated in a 1D superatomic orbital distributed over the cluster, but are localized at the Au2(CCCF3)1 sites formed from the original Au2(CCCF3)3 motif by desorption of CF3CC–CCCF3. Thus, [MAu24(CCR)18−2n]2− can be viewed as novel assemblies of superatoms with 8e and 2e (Fig. 12). The desorption step of 1,3-diynes continued to n = 6, leading to the formation of M@Au12[Au2(CCR)1]6. These results demonstrate the ability of CID MS to explore novel superatoms that cannot be synthesized by the conventional thermal processes. These partially naked Au superatoms produced via hyperthermal CID processes can be tested as new catalysts if they are collected by a proper method such as deposition on solid or powder surface.
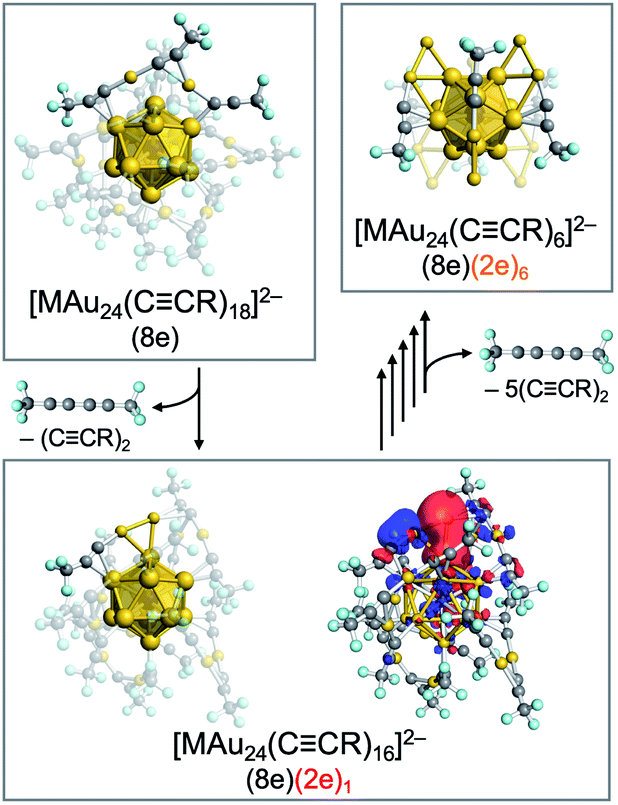 | ||
| Fig. 12 Scheme of the formation of superatomic assemblies via CID of 1,3-diyne from [MAu24(CCR)18]2−. Color code: yellow = Au; gray = C; light blue = F. | ||
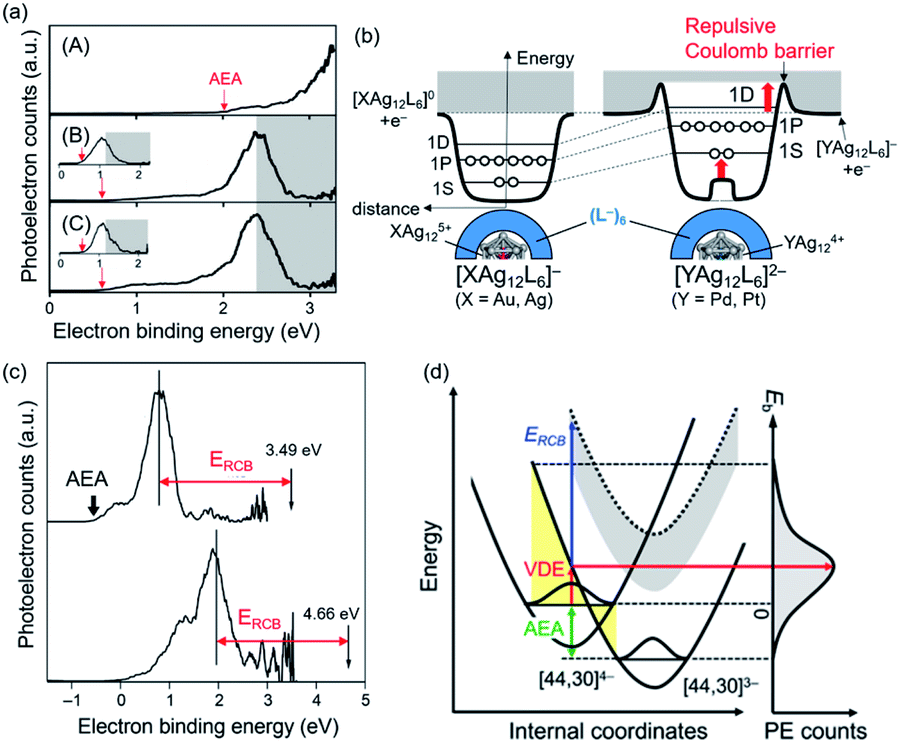 | ||
| Fig. 13 (a) PE spectra of (A) [Ag25(2,4-DMBT)18]−, (B) [PtAg24(2,4-DMBT)18]2−, (C) [PdAg24(2,4-DMBT)18]2− recorded at 355 nm. Insets in panels (B) and (C) are PE spectra recorded at 532 nm. Gray shaded areas in panels (B) and (C) are spectral cutoff regions due to an RCB. (b) Schematic illustration of potentials of (X@Ag12)5+(L−)6 (X = Ag, Au) and (Y@Ag12)4+(L−)6 (Y = Pd, Pt), where L denotes the Ag2(2,4-DMBT)3 staple unit. d is the distance from an electron and superatoms. (c) PE spectra of [Ag44(3,4-DFBT)30]4− recorded at 355 nm (top) and 266 nm (bottom). (d) Potential energy surfaces of [Ag44(3,4-DFBT)30]4− and [Ag44(3,4-DFBT)30]3− explaining the peak A in panel (c). The yellow hatch indicates the Franck–Condon region. The dotted curve is obtained by upshifting the [Ag44(3,4-DFBT)30]3− curve by ERCB. Adapted with permission from ref. 129 and 130. Copyright 2019 Wiley-VCH and copyright 2020 American Chemistry Society. | ||
Johnson established high-resolution UV-visible absorption spectroscopy at cryogenic temperature in the gas phase.132–134 Condensation of inert gases (He, N2) as a tag can freeze the internal motion of the superatoms without noticeable influence on the stable structures. Absorption of UV-visible laser by the tagged superatoms inevitably (with 100% quantum yield) leads to the release of the tags. As a result, the action spectrum of the depletion of the intensity of tagged superatoms corresponds to optical absorption spectra of the ligand-protected superatoms at extremely low temperature. The UV-visible spectrum of [Au9(TPP)8]3+ exhibits much sharper profiles than that in solution phase thanks to the suppression of thermal broadening (Fig. 14a).132,133 The spectra at cryogenic temperature can be compared directly with the theoretical spectra (Fig. 14b).135 It was suggested that hydride and halide affect the electronic structure of [Au9(TPP)8]3+ in similar ways.134
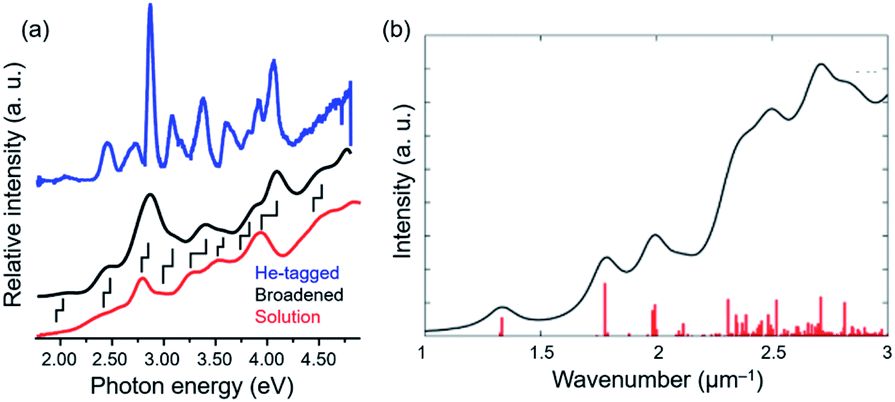 | ||
| Fig. 14 (a) UV-visible absorption spectra of He-tagged [Au9(TPP)8]3+ (blue), [Au9(TPP)8]3+(NO3)3 in methanol (red). The black trace is a numerically broadened spectrum of the blue trace. (b) Simulated absorption spectrum of [Au9(TPP)8]3+. Adapted with permission from ref. 133 and 135. Copyright 2020 American Chemistry Society and copyright 2016 American Chemistry Society. | ||
3.2 Condensed phase methods
SCXRD analysis provides the most straightforward and precise structural information of ligand-protected superatoms in the solid state. However, several challenges in structural characterization remain, such as identification of the position of doped metal atoms with similar electron densities, structural determination of superatoms that cannot be crystallized, and monitoring the fluctuating system. This section summarizes new approaches to obtain structure information complementary to SCXRD by advanced use of techniques such as XAS, AC(S)TEM, NMR, and PL spectroscopy.Ackerson and Billinge demonstrated by high-energy XRD that Au144(SR)60 (R = C4H9, C6H13, C12H25, 4-C6H4COOH) has a distinct isomer with an fcc core,141 in addition to that with an icosahedral core identified by SCXRD.28 However, it was not clear whether they are interconverted with each other or coexist independently. Tsukuda monitored the spatiotemporal change of individual particles of Au144(PET)60 by ACTEM and found reversible isomerization between icosahedral and fcc cores without decomposition.142 Tsukuda and Yamazoe revealed the hierarchy of bond stiffness of Au–Au bonds in Au25(PET)18, Au38(PET)24, and Au144(PET)60 by temperature-dependent extended X-ray absorption fine structure (EXAFS) analysis.143 It was shown that surface Au–Au bonds of the Au cores are more flexible than those in bulk metals while those distributed along the radial direction are stiffer.
Zhang examined the interaction of solvent and superatoms,144,145 which is often overlooked due to the difficulty of characterization in solution. A slight change of geometric and electronic structure of [Ag44(p-MBA)30]4− in solution was monitored by XAS.145 The contraction of the Ag32 core and the reduced electron densities of the S atoms suggested that dimethyl sulfoxide (DMSO) and N,N-dimethylformamide (DMF) directly interact with the Ag32(18e) core (Fig. 15a). These results were supported by quantum mechanics/molecular mechanics (QM/MM) calculations.145 Mitsui conducted quantitative analysis of the association-dissociation equilibria of TPP in [Ag29(BDT)12(TPP)4]3− based on PL measurement.146 The relative abundance of [Ag29(BDT)12(TPP)x]3− (x = 0–4) as a function of the concentration of TPP added in solution ([TPP]) was determined (Fig. 15b). This optical approach provides a nondestructive method for evaluating the concentrations of superatoms under dynamic equilibrium such as ligand-exchange and metal-exchange reactions.
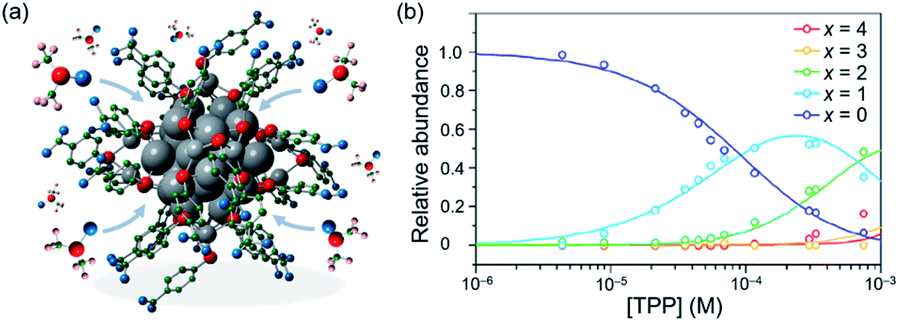 | ||
| Fig. 15 (a) Scheme of interaction between Na4[Ag44(p-MBA)30] and DMSO examined by XAS and QM/MM calculation. (b) Plots of relative abundance of Ag29(BDT)12(TPP)x (x = 0−4) in DMF solution as a function of the concentration of TPP. Color code: gray = Ag; red = S; blue = O; green = C, pink = H. Adapted with permission from ref. 145 and 146. Copyright 2020 American Chemistry Society. | ||
3.3 Computational methods
Initial geometries for structure prediction by DFT calculation are usually constructed based on the intuition of individual researchers; this approach lacks the completeness, efficiency, and subjectivity. Recently, Kärkkäinen and Häkkinen developed an algorithm based on machine learning to predict the structures of ligand-protected superatoms.147 After setting the coordinates of metal atoms, a list of possible positions of interface atoms (P or S) is constructed based on the training set consisting of SCXRD data and computational model structures. Then, complete interfacial structures (∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 models) are created under a restriction based on simple rules of chemistry related to coordination, atomic distances, and local conformations. Finally, model structures are ranked by a criterion called a combined structural error and optimized to the best model structure after adding the organic part of the ligand layer using molecular mechanics or molecular dynamics. In all studied cases, the best-ranked structures reproduced the metal–ligand interfaces of the crystal structures of a variety of Au/Ag superatoms (Fig. 16a). This method also reproduced the steric effect of ligands in Au44(TBBT)26 and Au44(2,4-DMBT)28 (Fig. 16b).
000 models) are created under a restriction based on simple rules of chemistry related to coordination, atomic distances, and local conformations. Finally, model structures are ranked by a criterion called a combined structural error and optimized to the best model structure after adding the organic part of the ligand layer using molecular mechanics or molecular dynamics. In all studied cases, the best-ranked structures reproduced the metal–ligand interfaces of the crystal structures of a variety of Au/Ag superatoms (Fig. 16a). This method also reproduced the steric effect of ligands in Au44(TBBT)26 and Au44(2,4-DMBT)28 (Fig. 16b).
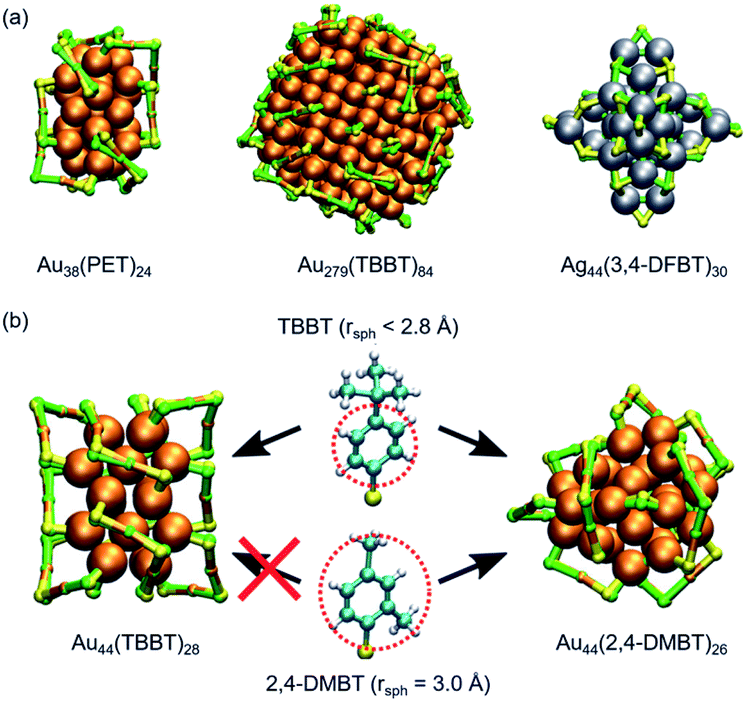 | ||
| Fig. 16 (a) Comparison of the predicted ligand–metal interface structures (green) and those resolved by SCXRD (yellow). (b) The effect of the size of ligands on the structures. Color code: orange = Au; gray = Ag, green and yellow = S; light blue = C; white = H. Adapted from ref. 147 with permission from Nature Publishing Group, copyright 2019. | ||
Optimal conditions for the synthesis and crystallization have also been researched based on the experience of individual researchers. Wang demonstrated that deep learning can accelerate the search for synthesis conditions:148 the conditions for synthesizing Au25(SR)18 reported in the literature were successfully predicted from a database containing only 54 results. Application of this approach to other issues requires the establishment of a reliable database which includes both successful and unsuccessful results. Since published papers include only successful cases, joint research between theoretical and experimental chemists is indispensable to expand this area.
4. Summary and prospects
As briefly summarized in the introduction, atomically-precise synthesis and structural determination by X-ray crystallography have deepened our understanding of the correlation between structures and properties of ligand-protected Au/Ag superatoms. The superatom concept provides a unified picture of how the electronic structures are modified or affected by the size, shape and doping. In spite of great progress in the last two decades, there still remain challenges in the synthesis and characterization of the ligand-protected superatoms. Major issues and possible solutions of the synthesis and characterization of Au/Ag superatoms are summarized below.4.1 Synthesis
Major challenges in the conventional synthesis of superatoms are poor predictability and low yield of the products arising from the stochastic nature of the formation processes. A promising approach includes efficient and selective transformations of well-defined superatoms, such as hydride-mediated growth and doping, seed-mediated growth, and ligand exchange. These transformations will not only achieve the synthesis of desired superatoms on a large (>gram) scale, but also increase the chance of serendipitous discoveries of new superatoms. Another challenge is improvement of robustness for practical application. Recent examples show that N-heterocyclic carbenes are promising candidates for the protecting reagent of Au/Ag superatoms. Large scale synthesis of robust Au/Ag superatoms is a key for the progress of materials science of superatoms as in the case of the development of carbon materials after large-scale production of C60 which was originally discovered as magic species by MS.149 Synthesis of superatomic molecules and ordered assemblies of superatoms will continue to be a challenge for the creation of novel properties and functions by taking advantage of controlled interaction between individual superatoms.4.2 Characterization
Ligand-protected Au/Ag superatoms have been characterized in depth by various methods conventionally used in chemical analysis. X-ray crystallography is without doubt the most direct and powerful method for determining the atomic structures. However, other experimental methods need to be developed to detect transient species that plays an important role in transformation reactions and to gain intrinsic structural information of the superatoms in an isolated environment. ESI MS makes it possible to detect key species in solution. Laser photoelectron spectroscopy probes the superatomic electronic structures and elucidates the effect of doping and total charge on the electronic structures. Information about the stability and structural motifs will be obtained by applying other gas-phase methods such as collision-induced dissociation MS, ion mobility MS, photodissociation MS and surface-induced dissociation MS. Aberration corrected transmission electron microscopy and X-ray absorption spectroscopy give both static and dynamic structural information of superatoms complementary to SCXRD. Theoretical prediction based on machine learning will not only become a powerful tool for the structural exploration of superatoms, but also release scientists from the tedious screening of synthesis conditions.As shown in Fig. 3, superatoms are attractive and promising elements having multi-parameters (size, shape, composition) for the control and tuning of their properties. Some future prospects to maximize the potentials of superatoms are described below.
Chemical synthesis of Au20(20e) with a tetrahedral pyramid structure (Fig. 17a), which has been the representative magic Au cluster since its discovery,152 is a long-pursued target. Wang and Wang independently synthesized Au20(16e) using a tetradentate phosphine ligand (P(C2H4PPh2)3).153,154 However, SCXRD analysis showed that the Au20(16e) core does not have a pyramidal motif, but is composed of an icosahedral Au13 and a helical Au7 motif. Formation of an Au41 with a twisted pyramid structure using bulky arenethiols155 suggests that bulky ligands are possible candidates to stabilize a tetrahedral Au20(20e).
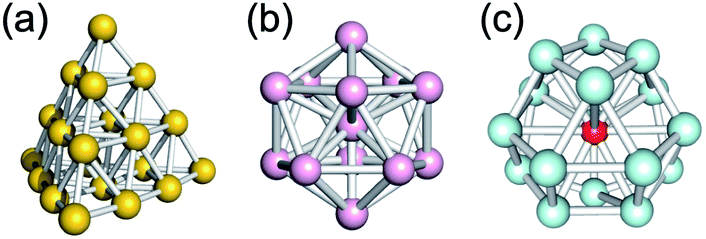 | ||
| Fig. 17 Proposed structures of superatoms generated in the gas phase: (a) Au20, (b) Al13−, and (c) (TM)1@Si16. Color code: yellow = Au; pink = Al, light blue = Si; red = TM. | ||
The ultimate goal, from our perspective, is to create a new paradigm on a nanoscale based on superatoms, which is located in a different hierarchy from that based on atoms and molecules. The efforts described herein will help to establish the fundamentals of superatoms, such as a periodic table, bonding theories, chemical reaction theories of superatoms, and eventually, the materials chemistry of superatoms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by a Grant-in-Aid for Scientific Research (JP17H01182, JP20H00370) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT).Notes and references
- W. de Heer, Rev. Mod. Phys., 1993, 65, 611–676 CrossRef CAS.
- D. M. P. Mingos, T. Slee and L. Zhenyang, Chem. Rev., 1990, 90, 383–402 CrossRef CAS.
- P. Pyykkö, Chem. Soc. Rev., 2008, 37, 1967–1997 RSC.
- Z. Luo, A. W. Castleman Jr and S. N. Khanna, Chem. Rev., 2016, 116, 14456–14492 CrossRef CAS.
- P. Jena and Q. Sun, Chem. Rev., 2018, 118, 5755–5870 CrossRef CAS.
- M. McPatrlin, R. Mason and L. Malatesta, J. Chem. Soc. D, 1969, 334 Search PubMed.
- K. Konishi, Struct. Bonding, 2014, 161, 49–86 CrossRef.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- C. L. Cleveland, U. Landman, T. G. Schaaff, M. N. Shafigullin, P. W. Stephens and R. L. Whetten, Phys. Rev. Lett., 1997, 79, 1873 CrossRef CAS.
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS.
- N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu and T. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610 CrossRef CAS.
- H. Qian and R. Jin, Nano Lett., 2009, 9, 4083–4087 CrossRef CAS.
- S. Vergara, U. Santiago, C. Kumara, D. Alducin, R. L. Whetten, M. J. Yacamán, A. Dass and A. Ponce, J. Phys. Chem. C, 2018, 122, 26733–26738 CrossRef CAS.
- I. L. Garzón, C. Rovira, K. Michaelian, M. R. Beltran, P. Ordejon, J. Junquera, D. Sanchez-Portal, E. Artacho and J. M. Soler, Phys. Rev. Lett., 2000, 85, 5250 CrossRef.
- H. Häkkinen, M. Walter and H. Grönbeck, J. Phys. Chem. B, 2006, 110, 9927–9931 CrossRef.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- H. Quan, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Häkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS.
- O. Lopez-Acevedo, J. Akola, R. L. Whetten, H. Grönbeck and H. Häkkinen, J. Phys. Chem. C, 2009, 113, 5035–5038 CrossRef CAS.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS.
- R. E. Leuchtner, A. C. Harms and A. W. Castleman Jr, J. Chem. Phys., 1989, 91, 2753–2754 CrossRef CAS.
- S. N. Khanna and P. Jena, Phys. Rev. Lett., 1992, 69, 1664–1667 CrossRef CAS.
- S. N. Khanna and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705–13716 CrossRef CAS.
- D. E. Bergeron, A. W. Castleman Jr, T. Morisato and S. N. Khanna, Science, 2004, 304, 84–87 CrossRef CAS.
- A. W. Castleman Jr and S. N. Khanna, J. Phys. Chem. C, 2009, 113, 2664–2675 CrossRef.
- Z. Luo and A. W. Castleman Jr, Acc. Chem. Res., 2014, 47, 2931–2940 CrossRef CAS.
- D. A. Tomalia and S. N. Khanna, Chem. Rev., 2016, 116, 2705–2774 CrossRef CAS.
- A. C. Reber and S. N. Khanna, Acc. Chem. Res., 2017, 50, 255–263 CrossRef CAS.
- W. D. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Y. Chou and M. L. Cohen, Phys. Rev. Lett., 1984, 52, 2141–2143 CrossRef CAS.
- K. J. Taylor, C. L. Pettiette-Hall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 96, 3319–3329 CrossRef CAS.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS.
- H. Häkkinen, Chem. Soc. Rev., 2008, 37, 1847–1859 RSC.
- T. Omoda, S. Takano and T. Tsukuda, Small, 2020 DOI:10.1002/smll.202001439.
- D. M. P. Mingos, Dalton Trans., 2015, 44, 6680–6695 RSC.
- S. B. Zhang, M. L. Cohen and M. Y. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 3455–3458 CrossRef CAS.
- C. Baladrón and J. A. Alonso, Phys. B, 1988, 154, 73–81 CrossRef.
- E. Janssens, S. Neukermans and P. Lievens, Curr. Opin. Solid State Mater. Sci., 2004, 8, 185–193 CrossRef CAS.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS.
- W. W. Xu, X. C. Zeng and Y. Gao, Acc. Chem. Res., 2018, 51, 2739–2747 CrossRef CAS.
- R. Jin, Y. Pei and T. Tsukuda, Acc. Chem. Res., 2019, 52, 1 CrossRef CAS.
- S. Takano, H. Hirai, S. Muramatsu and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 8380–8383 CrossRef CAS.
- S. Takano, H. Hirai, S. Muramatsu and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 12314–12317 CrossRef CAS.
- S. Takano, S. Hasegawa, M. Suyama and T. Tsukuda, Acc. Chem. Res., 2018, 51, 3074–3083 CrossRef CAS.
- H. Hirai, S. Takano and T. Tsukuda, ACS Omega, 2019, 4, 7070–7075 CrossRef CAS.
- S. Takano, S. Ito and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 15994–16002 CrossRef CAS.
- Q. Yao, X. Yuan, V. Fung, Y. Yu, D. T. Leong, D.-e. Jiang and J. Xie, Nat. Commun., 2017, 8, 927 CrossRef.
- Z. Gan, N. Xia and A. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS.
- S. Wang, Q. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2018, 51, 2784–2794 CrossRef CAS.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2015, 128, 934–938 CrossRef.
- K. Zheng, V. Fung, X. Yuan, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2019, 141, 18977–18983 CrossRef CAS.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Nararajan, A. Som and T. Pradeep, Acc. Chem. Res., 2017, 50, 1988–1996 CrossRef CAS.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan and T. Pradeep, Nat. Commun., 2016, 7, 13447 CrossRef CAS.
- M. Suyama, S. Takano, T. Nakamura and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 14048–14051 CrossRef CAS.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garret, M. Nambo, J. H. Hortoon, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS.
- X. Meng, Q. Xu, S. Wang and M. Zhu, Nanoscale, 2012, 4, 4161–4165 RSC.
- W. Kurashige, M. Yamaguchi, K. Nobusada and Y. Negishi, J. Phys. Chem. Lett., 2012, 3, 2649–2652 CrossRef CAS.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- C. Zeng, C. Liu, Y. Chen, N. Rosi and R. Jin, J. Am. Chem. Soc., 2014, 136, 11922–11925 CrossRef CAS.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS.
- C. Zeng, H. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon, R. N. Barnett, R. L. Whetten, U. Landman and R. Jin, Angew. Chem., Int. Ed., 2012, 51, 13114–13118 CrossRef CAS.
- M. Rambukwella, L. Sementa, A. Fortunelli and A. Dass, J. Phys. Chem. C, 2017, 121, 14929–14935 CrossRef CAS.
- C. Zeng, Y. Chen, K. Kirschbaum, K. Appavoo, M. Y. Sfeir and R. Jin, Sci. Adv., 2015, 1, e1500045 CrossRef.
- P. R. Nimmala, S. Theivendran, G. Barcaro, L. Sementa, C. Kumara, V. R. Jupally, E. Apra, M. Stener, A. Fortunelli and A. Dass, J. Phys. Chem. Lett., 2015, 6, 2134–2139 CrossRef CAS.
- P. R. Nimmala and A. Dass, J. Am. Chem. Soc., 2014, 136, 17016–17023 CrossRef CAS.
- E. Khatun, A. Ghosh, D. Ghosh, P. Chakraborty, A. Nag, B. Mondal, S. Chennu and T. Pradeep, Nanoscale, 2017, 9, 8240–8248 RSC.
- A. Dass, T. C. Jones, S. Teivendran, L. Sementa and A. Fortunelli, J. Phys. Chem. C, 2017, 121, 14914–14919 CrossRef CAS.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-e. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS.
- M. S. Bootharaju, C. P. Joshi, M. Alhilaly and O. M. Bakr, Chem. Mater., 2016, 28, 3292–3297 CrossRef CAS.
- X. Kang, L. Hang, W. Liu, L. Xiong, Y. Pei, Z. Sun, S. Wang, S. Wei and M. Zhu, Chem. Sci., 2019, 10, 8685–8693 RSC.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey, T. Seki, E. C. Keste, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef CAS.
- A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS.
- Q. Tang and D.-e. Jiang, Chem. Mater., 2017, 29, 6908–6915 CrossRef CAS.
- M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun, K. G. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS.
- H. Shen, G. Deng, S. Kaapa, T. Tan, Y.-Z. Han, S. Maola, S.-C. Lin, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2019, 58, 17731–17735 CrossRef CAS.
- H. Shen, S. Xiang, Z. Xu, C. Liu, X. Li, C. Sun, S. Lin, B. K. Teo and N. Zheng, Nano Res., 2020, 13, 1908–1911 CrossRef CAS.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. N. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Maola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef.
- B. Bhattarai, I. Chakraborty, B. E. Conn, A. Atnagulov, T. Pradeep and T. P. Bigioni, J. Phys. Chem. C, 2017, 121, 10964–10970 CrossRef CAS.
- J. Yan, H. Su, H. Yang, C. Hu, S. Maola, S. Lin, B. K. Teo, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2016, 138, 12751–12754 CrossRef CAS.
- A. Chen, X. Kang, S. Jin, W. Du, S. Wang and M. Zhu, J. Phys. Chem. Lett., 2019, 10, 6124–6128 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- J. Zhang, C. Gong, X. Zeng and J. Xie, Coord. Chem. Rev., 2016, 324, 39–53 CrossRef CAS.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Chem. Rev., 2017, 117, 1125–11238 CrossRef.
- L. Cheng, C. Ren, X. Zhang and J. Yang, Nanoscale, 2013, 5, 1475–1478 RSC.
- J. Nishigaki, S. Yamazoe, S. Kohara, A. Fujiwara, W. Kurashige, Y. Negishi and T. Tsukuda, Chem. Commun., 2014, 50, 839–841 RSC.
- X. Zou, S. Jin, W. Du, Y. Li, S. Wang, P. Li and M. Zhu, Nanoscale, 2017, 9, 16800–16805 RSC.
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS.
- H. Qian, W. Eckenhoff, M. Bie, T. Pintauer and R. Jin, Inorg. Chem., 2011, 50, 10735–10739 CrossRef CAS.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. L. Rosi, R. R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS.
- Y. Song, F. Fu, J. Zhang, J. Chai, X. Kang, P. Li, S. Li, H. Zhou and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8430–8434 CrossRef CAS.
- Y. Song, S. Jin, X. Kang, J. Xiang, H. Deng, H. Yu and M. Zhu, Chem. Mater., 2016, 28, 2609–2617 CrossRef CAS.
- K. Zheng, J. Zhang, D. Zhao, Y. Yang, Z. Li and G. Li, Nano Res., 2019, 12, 501–507 CrossRef CAS.
- T.-H. Chiu, J.-H. Liao, F. Gam, I. Chantrenne, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2019, 141, 12957–12961 CrossRef CAS.
- J. Chen, Q.-F. Zhang, T. A. Bonaccorso, P. G. Williard and L.-S. Wang, J. Am. Chem. Soc., 2014, 136, 92–95 CrossRef CAS.
- A. Muñoz-Castro, Phys. Chem. Chem. Phys., 2020, 22, 1422–1426 RSC.
- T. Dianese, S. Antonello, S. Bogialli, W. Fei, A. Venzo and F. Maran, ACS Nano, 2018, 12, 7057–7066 CrossRef.
- E. Ito, S. Takano, T. Nakamura and T. Tsukuda, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202010342.
- S.-F. Yuan, J.-J. Li, Z.-J. Guan, Z. Lei and Q.-M. Wang, Chem. Commun., 2020, 56, 7037–7040 RSC.
- A. Ebina, S. Hossain, S. Ozaki, S. Kato, T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 1105 CrossRef CAS.
- M. D. Nardi, S. Antonello, D.-e. Jiang, F. Pan, K. Rissanen, M. Ruzzi, A. Venzo, A. Zoleo and F. Maran, ACS Nano, 2014, 8, 8505–8512 CrossRef.
- P. Yuan, R. Zhang, E. Selenius, P. Ruan, Y. Yao, Z. Zhou, S. Maola, H. Häkkinen, B. K. Teo, Y. Cao and N. Zheng, Nat. Commun., 2020, 11, 2229 CrossRef CAS.
- Q. Li, J. C. Russell, T.-Y. Luo, X. Roy, N. L. Rosi, Y. Zhu and R. Jin, Nat. Commun., 2018, 9, 3871 CrossRef.
- Z.-Y. Wang, M.-Q. Wang, Y.-L. Li, P. Luo, T.-T. Jia, R.-W. Huang, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2018, 140, 1069–1076 CrossRef CAS.
- S. Chen, W. Du, C. Qin, D. Liu, L. Tang, Y. Liu, S. Wang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 7542–7547 CrossRef CAS.
- G. E. Johnson and J. Laskin, Analyst, 2016, 141, 3573–3589 RSC.
- K. Hirata, R. Tomihara, K. Kim, K. Koyasu and T. Tsukuda, Phys. Chem. Chem. Phys., 2019, 21, 17463–17474 RSC.
- P. Chakraborty and T. Pradeep, NPG Asia Mater., 2019, 11, 48 CrossRef.
- T. Chen, Q. Yao, R. R. Nasaruddin and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 11967–11977 CrossRef CAS.
- K. Koyasu, K. Hirata and T. Tsukuda, in Physical Chemistry of Cold Gas-Phase Functional Molecules and Clusters, ed. T. Ebata and M. Fujii, Springer, 2019, pp. 223–253 Search PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS.
- T. Chen, V. Fung, Q. Yao, Z. Luo, D. E. Jiang and J. Xie, J. Am. Chem. Soc., 2018, 140, 11370–11377 CrossRef CAS.
- A. Ghosh, M. Bodiuzzaman, A. Nag, M. Jash, A. Baksi and T. Pradeep, ACS Nano, 2017, 11, 11145–11151 CrossRef CAS.
- S. Ito, K. Koyasu, S. Takano and T. Tsukuda, J. Phys. Chem. C, 2020, 124, 19119–19125 CrossRef CAS.
- K. Hirata, K. Yamashita, S. Muramatsu, S. Takano, K. Ohshimo, T. Azuma, R. Nakanishi, T. Nagata, S. Yamazoe, K. Koyasu and T. Tsukuda, Nanoscale, 2017, 9, 13409–13412 RSC.
- K. Hirata, K. Kim, K. Nakamura, H. Kitazawa, S. Hayashi, K. Koyasu and T. Tsukuda, J. Phys. Chem. C, 2019, 123, 13174–13179 CAS.
- K. Kim, K. Hirata, K. Nakamura, H. Kitazawa, S. Hayashi, K. Koyasu and T. Tsukuda, Angew. Chem., Int. Ed., 2019, 58, 11637–11641 CrossRef CAS.
- Y. Tasaka, K. Nakamura, S. Malola, K. Hirata, K. Kim, K. Koyasu, H. Häkkinen and T. Tsukuda, J. Phys. Chem. Lett., 2020, 11, 3069–3074 CrossRef CAS.
- A. P. Veenstra, L. Monzel, A. Baksi, J. Czekner, S. Lebedkin, E. K. Schneider, T. Pradeep, A. N. Unterreiner and M. M. Kappes, J. Phys. Chem. Lett., 2020, 11, 2675–2681 CrossRef CAS.
- A. Cirri, H. Morales Hernández, C. Kmiotek and C. J. Johnson, Angew. Chem., Int. Ed., 2019, 58, 13818–13822 CrossRef CAS.
- A. Cirri, H. M. Hernández and C. J. Johnson, J. Phys. Chem. A, 2020, 124, 1467–1479 CrossRef CAS.
- A. Cirri, H. M. Hernández and C. J. Johnson, Chem. Commun., 2020, 56, 1283–1285 RSC.
- N. V Karimova and C. M. Aikens, J. Phys. Chem. A, 2016, 120, 9625–9635 CrossRef.
- S. Yamazoe and T. Tsukuda, Bull. Chem. Soc. Jpn., 2019, 92, 193–204 CrossRef CAS.
- Z. W. Wang, O. Toikkanen, B. M. Quinn and R. E. Palmer, Small, 2011, 7, 1542–1545 CrossRef CAS.
- D. Bahena, N. Bhattarai, U. Santiago, A. Tlahuice, A. Ponce, S. B. H. Bach, B. Yoon, R. L. Whetten, U. Landman and M. José-Yacamán, J. Phys. Chem. Lett., 2013, 4, 975–981 CrossRef CAS.
- M. Azubel, J. Koivisto, S. Malola, D. Bushnell, G. L. Hura, A. L. Koh, H. Tsunoyama, T. Tsukuda, M. Pettersson, H. Häkkinen and R. D. Kornberg, Science, 2014, 345, 909–912 CrossRef CAS.
- M. Azubel, A. L. Koh, K. Koyasu, T. Tsukuda and R. D. Kornberg, ACS Nano, 2017, 11, 11866–11871 CrossRef CAS.
- K. M. Ø. Jensen, P. Juhas, M. A. Tofanelli, C. L. Heinecke, G. Vaughan, C. J. Ackerson and S. J. L. Billinge, Nat. Commun., 2016, 7, 11859 CrossRef CAS.
- R. Takahata, S. Yamazoe, Y. Maehara, K. Yamazaki, S. Takano, W. Kurashige, Y. Negishi, K. Gohara and T. Tsukuda, J. Phys. Chem. C, 2020, 124, 6907–6912 CrossRef CAS.
- S. Yamazoe, S. Takano, W. Kurashige, T. Yokoyama, K. Nitta, Y. Negishi and T. Tsukuda, Nat. Commun., 2016, 7, 10414 CrossRef CAS.
- D. M. Chevrier, L. Raich, C. Rovira, A. Das, Z. Luo, Q. Yao, A. Chatt, J. Xie, R. Jin, J. Akola and P. Zhang, J. Am. Chem. Soc., 2018, 140, 15430–15436 CrossRef CAS.
- D. M. Chevrier, B. E. Conn, B. Li, D. E. Jiang, T. P. Bigioni, A. Chatt and P. Zhang, ACS Nano, 2020, 14, 8433–8441 CrossRef CAS.
- Y. Niihori, N. Takahashi and M. Mitsui, J. Phys. Chem. C, 2020, 124, 5880–5886 CrossRef CAS.
- S. Malola, P. Nieminen, A. Pihlajamäki, J. Hämäläinen, T. Kärkkäinen and H. Häkkinen, Nat. Commun., 2019, 10, 3973 CrossRef.
- J. Li, T. Chen, K. Lim, L. Chen, S. A. Khan, J. Xie and X. Wang, Adv. Intell. Syst., 2019, 1, 1900029 CrossRef.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smally, Nature, 1985, 318, 162–163 CrossRef CAS.
- Z. Wu, G. Hu, D.-e. Jiang, D. R. Mullins, Q.-F. Zhang, L. F. Allard Jr, L.-S. Wang and S. H. Overbury, Nano Lett., 2016, 16, 6560–6567 CrossRef CAS.
- G. Hu, Z. Wu and D.-e. Jiang, J. Mater. Chem. A, 2018, 6, 7532–7537 RSC.
- J. Li, H.-J. Zhai and L.-S. Wang, Science, 2003, 299, 864–867 CrossRef CAS.
- X.-K. Wan, S.-F. Yuan, Z.-W. Lin and Q.-M. Wang, Angew. Chem., Int. Ed., 2014, 53, 2923–2926 CrossRef CAS.
- J. Chen, Q.-F. Zhang, P. G. Williard and L.-S. Wang, Inorg. Chem., 2014, 53, 3932–3934 CrossRef CAS.
- J. Nishigaki, R. Tsunoyama, H. Tsunoyama, N. Ichikuni, S. Yamazoe, Y. Negishi, M. Ito, T. Matsuo, K. Tamao and T. Tsukuda, J. Am. Chem. Soc., 2012, 134, 14295–14297 CrossRef CAS.
- H. Schnöckel, Chem. Rev., 2010, 110, 4125–4163 CrossRef.
- K. Koyasu, M. Akutsu, M. Mitsui and A. Nakajima, J. Am. Chem. Soc., 2005, 127, 4998–4999 CrossRef CAS.
- T. Kambe, N. Haruta, T. Imaoka and K. Yamamoto, Nat. Commun., 2017, 8, 2046 CrossRef.
- H. Tsunoyama, H. Akatsuka, M. Shibuta, T. Iwasa, Y. Mizuhata, N. Tokitoh and A. Nakajima, J. Phys. Chem. C, 2017, 121, 20507–20516 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |