
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase transfer of metal cations by induced dynamic carrier agents: biphasic extraction based on dynamic covalent chemistry†
Aline
Chevalier
ab,
Artem
Osypenko
 b,
Jean-Marie
Lehn
*b and
Daniel
Meyer
*a
b,
Jean-Marie
Lehn
*b and
Daniel
Meyer
*a
aInstitut de Chimie Séparative de Marcoule (ICSM), CEA, CNRS, ENSCM, Université de Montpellier, UMR 5257, Bâtiment 426, BP 17171, 30207 Bagnols-sur-Cèze, France. E-mail: daniel.meyer@cea.fr
bLaboratoire de Chimie Supramoléculaire, Institut de Science et d’Ingénierie Supramoléculaires (ISIS), UMR 7006, 8 Allée Gaspard Monge, 67000 Strasbourg, France. E-mail: lehn@unistra.fr
First published on 2nd October 2020
Abstract
In contrast to the classical method where a single molecule is designed to extract metal cations under specific conditions, dynamic covalent chemistry provides an approach based on the implementation of an adaptive dynamic covalent library for inducing the generation of the extractant species. This approach has been applied to the liquid–liquid extraction of copper(II) nitrate based on a dynamic library of acylhydrazones constituents that self-build and distribute through the interface of a biphasic system. The addition of copper(II) cations to this library triggers a modification of its composition and the up-regulation of the ligand molecules driven by coordination to the metal cations. Among these, one species has proven to be sufficiently lipophilic to play the role of carrier agent and its formation by component exchange enables the partial extraction of the copper(II). The study of different pathways to generate the dynamic covalent library demonstrates the complete reversibility and the adaptability of the system. The detailed analytical investigation of the system provides a means to assess the mechanism of the dynamic extraction process.
Introduction
Solving a liquid–liquid metal extraction problem is usually based on a “one problem–one solution” approach. For a given problem such as that raised by treatment of spent nuclear fuel,1 mining of minerals2 or any other metal extraction procedure,3 liquid–liquid extraction offers a molecular system that can be designed, synthesised, characterised and studied under various conditions. Usually, the nature of any study is highly specific and depends upon the composition of the aqueous phase in regard to the metal cations, their counter-anions and the acidity. The properties of such an extraction system for a given aqueous phase depend strongly on the water-immiscible organic solvent and the molecular extractant as a pair.‡ In order to be able to extract a metal cation from an aqueous phase and carry it into an organic phase, the extractant molecule must have three main characteristics: (i) strong and selective complexation of metal cations, (ii) organic phase solubility and (iii) limited aqueous solubility.The properties of an extracting system can be modified in several ways with or without changing the molecular complexation agent itself. The type of modification performed depends upon the relative importance of the different interactions as driving forces for the liquid–liquid extraction of specific metallic species. The easiest modification lies in the reformulation of the extractant–solvent pair.‡ Nevertheless, if the extracting molecule needs to be modified, two approaches may be considered: (i) keeping the metal complexing site unchanged while modifying other molecular features, or (ii) opting for a total modification of the extractant. In the first case, variation of the properties, such as that of lipophilicity, is mainly ruled by the physicochemical features resulting from the interactions with the medium. In the second case, modifying the complexing pattern of the extracting molecule will drastically change the direct metal–ligand interactions with a marked effect on the behaviour of the system.
As the reformulation and molecular structural modifications primarily concern weak interactions between the metal complex and the molecules in the organic phase, a proper understanding of these relationships is essential when considering the liquid–liquid extraction of metal.4 In general, the organic molecular connectivity remains the same during a liquid–liquid extraction of metal cations because the molecules are covalently built, that is to say, “static”, thus preventing any major change in their interactions. In strong contrast, the Dynamic Covalent Chemistry approach (DCC)5 allows for in situ variations in molecular constitution by component exchange and recombination induced in an adaptive fashion by the system itself.
It implements reversible chemical reactions within sets of initial components to generate dynamic constitutional diversity and gives access to Dynamic Covalent Libraries (DCLs). DCC relies on the reversible and continuous formation and cleavage of chemical bonds so as to generate the constituents consisting of all possible combinations obtained by exchange of their components. The resulting distribution can undergo spontaneous modifications in response to a physical stimulus or a chemical effector such as metalloselection,6 photoselection7 or kinetic and thermodynamic changes.8 DCLs thus display adaptive behaviour in response to the environment/medium and the applied agents.
The use of binary liquid mixtures enables the examination of the behaviour of a DCL in a phase separation process.9 The dynamic library adapts to the phase separation and undergoes a constitutional reorganisation generating a distribution of the fittest constituents for each phase. Component exchange across the interface between the separated liquid layers is markedly slowed down and depends on the phase affinities of the different components. Thus, the system may be kinetically blocked in an out-of-equilibrium distribution.10 When the phases are re-united into a single phase, the dynamic library returns to its initial equilibrium state, thus displaying the full reversibility of the system. Phase transfer of multiple metal cations leads to the generation of cation specific distributions.11
In the present work, the structural, kinetic and thermodynamic features of several DCLs have been explored in order to define the parameters and conditions required for achieving suitable constituent formation and distribution as well as dynamic covalent exchanges of component across the interface. The dynamism of the implemented libraries takes place at different level: (i) formation and exchange of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds; (ii) metal/ligand exchange; (iii) phase transfer; (iv) protonation/deprotonation of the ligands.
N bonds; (ii) metal/ligand exchange; (iii) phase transfer; (iv) protonation/deprotonation of the ligands.
Most significantly, the addition of a metal salt to the DCLs triggers the redistribution within as well as between both phases of the components, the ligand constituents and the corresponding complexes depending on the nature of the added chemical effector. The present systems are thus adaptive, responding to a specific metal cation and allowing for its transfer from an aqueous phase to an organic phase. Such a behaviour has been implemented for the extraction of Zn(II) or Cd(II) ions by means of a dynamic combinatorial Schiff-base library.12
The DCL-based approach introduces a new paradigm in the liquid–liquid extraction–separation field because intrinsic chemical modifications occur in the system during the extraction process. Indeed, the interactions and their consequences are not frozen but add dynamic degrees of freedom that offer additional opportunities to study the importance of the strong–weak interactions in metal extraction in biphasic systems. More specifically, the work reported here describes the adaptive behaviour of DCLs based on aldehyde/hydrazide components and their tridentate acylhydrazone ligand constituents in the process of inducing copper(II) phase transfer from an aqueous phase to an organic phase. It represents an exploration of the fundamentals of dynamic covalent chemistry as applied to liquid–liquid extraction. The main goal is the selective transfer of metals from one phase to another in a non-miscible biphasic system.13
Results and discussion
Choice of the DCL components and of experimental extraction conditions
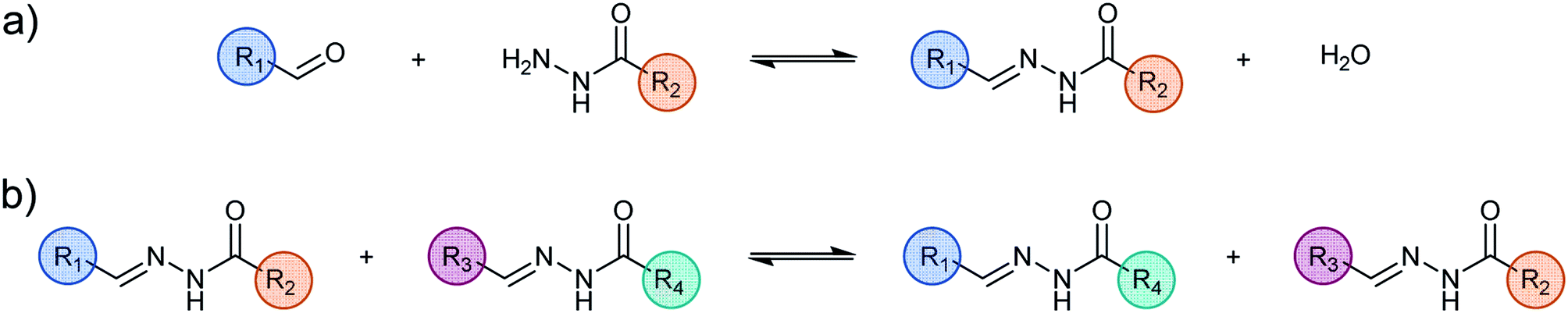 | ||
| Scheme 1 (a) Reaction of formation (b) reaction of exchange of acylhydrazone. | ||
Based on complexation and solubility properties, the presence of a deprotonatable site and hydrophilic–hydrophobic properties, 16 aldehydes and 4 hydrazides were identified as potential components (Fig. S17 in ESI†). The four hydrazides B1–B4 were chosen as they span a range of lipo-hydrophilic balance, the more lipophilic, B2, being a good candidate for extraction into an organic phase (Fig. 1). From a first series of formation tests based on the reactivity of B2 with the 16 aldehydes, only 10 gave the acylhydrazone in the present conditions.
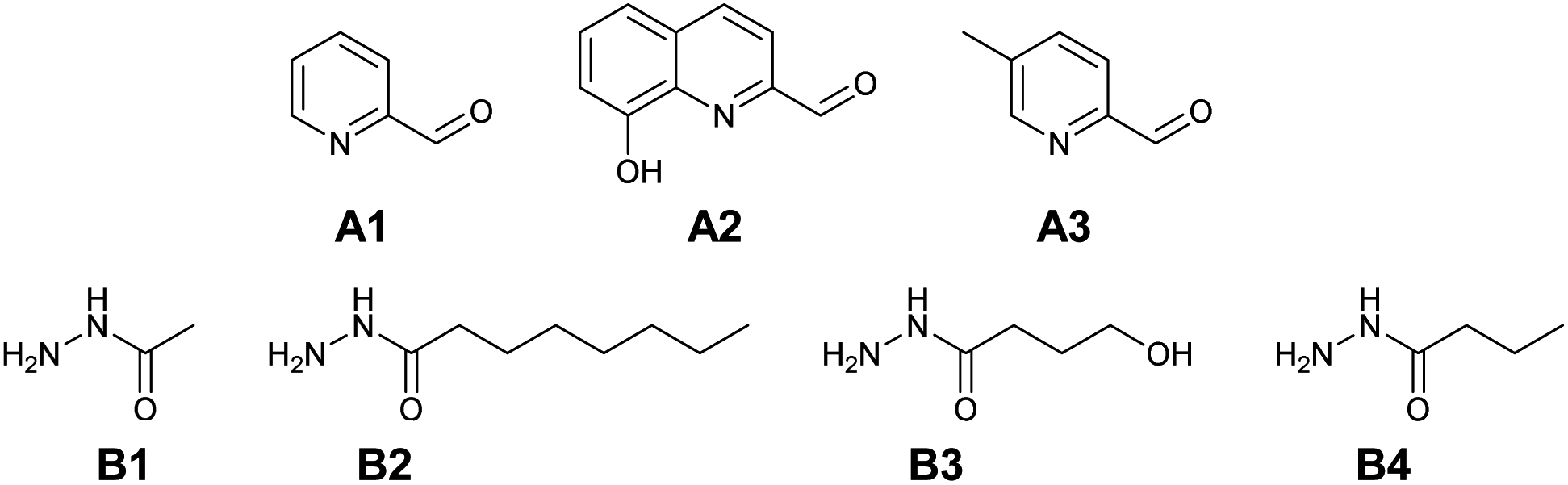 | ||
| Fig. 1 Reference library of aldehyde A1–A3 and hydrazide B1–B4 components. | ||
As the present work is mainly focused on the role played by a labile proton and the hydrophobic–hydrophilic balance, the two aldehydes A1 and A2 were mainly considered. A3 as structural analogue of A1 and B3 as an analogue of B4 were used only in the study of kinetic features. Altogether, the final set of molecules A1–A3 and B1–B4 forms the reference library used here (Fig. 1).
Qualitative screening of the extraction of Cu(II) in water/organic systems was conducted for chloroform, dichloromethane, toluene, dodecane, 1-octanol and nitromethane. All the solvents showed, by direct observation, a similar behaviour towards Cu(II) extraction by the ligands formed by the reference library (Fig. S18 and S19 in ESI†). Chloroform was chosen for further investigations, as it presents balanced solubilisation properties and is easily available in deuterated form for extraction experiments monitored by 1H nuclear magnetic resonance spectroscopy (NMR).
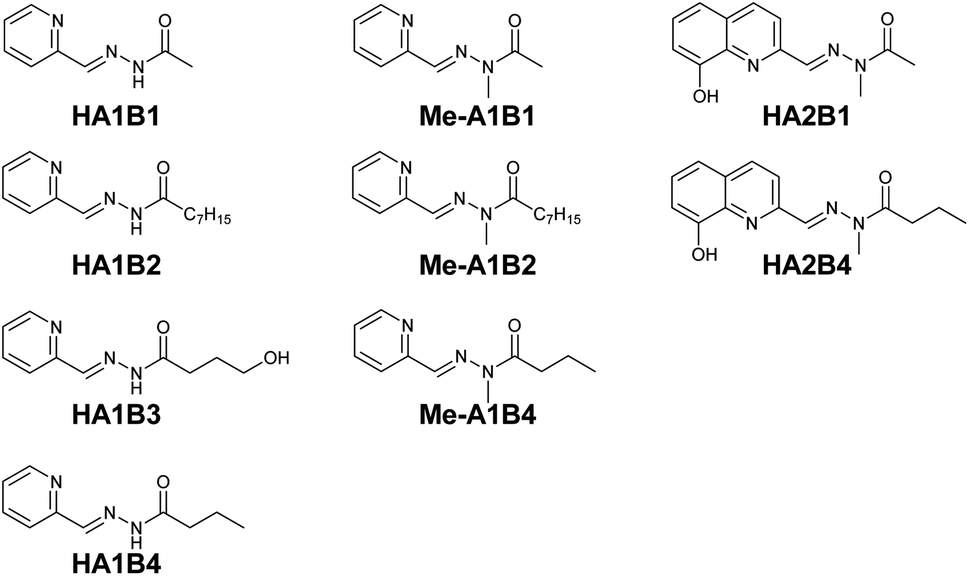 | ||
| Fig. 2 Acylhydrazone ligand constituents studied in the preliminary extraction tests with Cu(NO3)2·3H2O. | ||
The extraction experiments were conducted by introducing the studied ligand constituent into a H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CHCl3 (1:1, v/v) biphasic system and stirring until equilibration at room temperature. Then 0.5 equivalent of Cu(II) was added and the system was stirred for one hour. The electronic absorptions were recorded and exhibited new peaks in the visible region for the N–H acylhydrazones (Fig. S24–S27 in ESI†), whereas the spectra of the corresponding N-methylated derivatives did not show any such peaks. The occurrence of these peaks is consistent with the formation of a complex of the NH acylhydrazone and Cu(II). The coordination involves the carbonyl-O, the imine-N and the pyridine-N atoms.
:CHCl3 (1:1, v/v) biphasic system and stirring until equilibration at room temperature. Then 0.5 equivalent of Cu(II) was added and the system was stirred for one hour. The electronic absorptions were recorded and exhibited new peaks in the visible region for the N–H acylhydrazones (Fig. S24–S27 in ESI†), whereas the spectra of the corresponding N-methylated derivatives did not show any such peaks. The occurrence of these peaks is consistent with the formation of a complex of the NH acylhydrazone and Cu(II). The coordination involves the carbonyl-O, the imine-N and the pyridine-N atoms.
The effect of the chain length was studied using A1 and the hydrazides with different carbon chain lengths: CH3, C7H15 and C3H7 for HA1B1, HA1B2 and HA1B4 respectively. The absorption band of the coordination complex of Cu(II) with HA1B1 was present in the spectrum of the aqueous phase only (Fig. S24 in ESI†). With HA1B4 (propyl chain), the peak was visible in the two phases. Assuming identical molar extinction coefficients in water and chloroform, the distribution of the complex [Cu(II)(HA1B4)2](NO3)2 is 75:25% (aqueous:organic) (Fig. S27 in ESI†). For HA1B2 (heptyl chain), the aqueous phase stayed colourless and the organic phase turned green due to the presence of the complex in the organic phase (Fig. S26 in ESI†).
Behaviour of the DCLs
Before starting the metal extraction study, the behaviour of the acylhydrazones and the exchange processes within the library were characterised experimentally in one- and two-phase systems. The reactions were run at room temperature (23 °C) at 20 mM in CDCl3 and D2O at various pD values and monitored by 1H NMR spectroscopy.For the biphasic studies, the initial distributions of the components and constituents between D2O and CDCl3 were determined at pD = 6 with an error of ±5%.16A2, A3, B2, HA1B2, Me-A1B2 and Me-A1B4 were dissolved in chloroform (water content below 5%). B1, B3, and HA1B3 were dissolved in water (chloroform content below 5%). For the other members of the DCL, the distribution in D2O:CDCl3 was: A1 (5:95); B4 (85:15); HA1B1 (40:60); Me-A1B1 (5:95); HA1B4 (13:87) (Table S1 in ESI†).
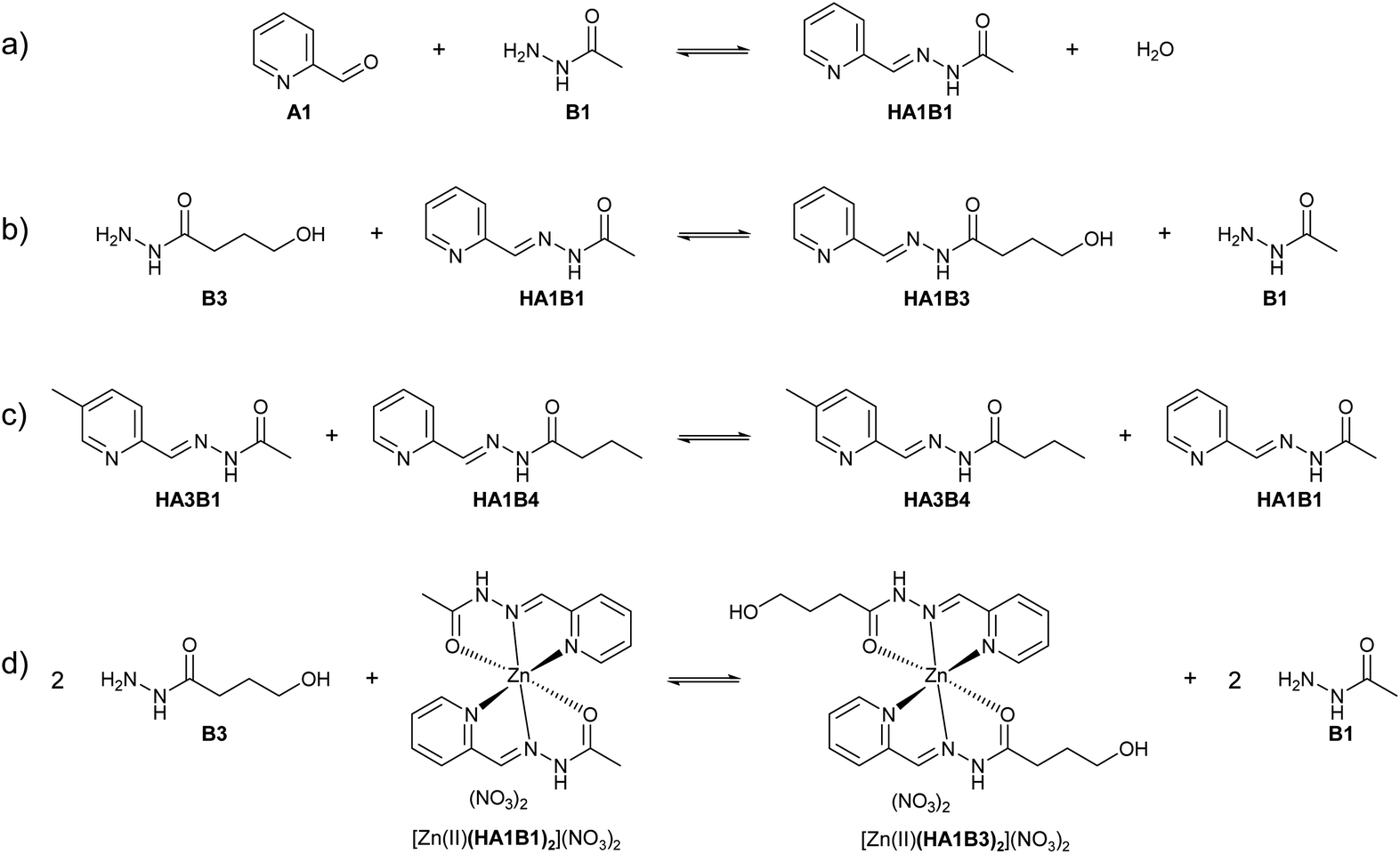 | ||
| Scheme 2 (a) Reaction of formation of acylhydrazone – types of exchange processes: (b) exchange 1 – between a hydrazide and an acylhydrazone – (c) exchange 2 – between two acylhydrazones – (d) exchange 3 – reaction between a hydrazide and the ligands on a Zn(II) complex. | ||
| Reaction | CDCl3 | D2O | ||
|---|---|---|---|---|
| pD = 7 | pD = 5 | pD = 2.5 | ||
| (a) Formation | 3 h 15 | 1 h 45 | <10 min | <10 min |
| (b) Exchange 1 | — | 20 h | 2 h | 40 min |
| (c) Exchange 2 | — | >3 d | >3 d | 24 h |
| (d) Exchange 3 | — | — | 5 h | — |
Like the formation reaction, exchange 1 and exchange 2 are acid catalysed (Table 1b–d; Fig. S29 and S30 in ESI†). The equilibration time of the exchange decreased drastically with pD. In water, the exchange between an acylhydrazone/metal complex and a hydrazide, was made using [Zn(II)(HA1B1)2](NO3)2 and B3. Zn(II) was used instead of Cu(II) as the two metals have similar Lewis acidity and the reaction is monitored by 1H NMR spectroscopy. The exchange reached equilibrium within 5 hours at pD = 5 (Fig. S31 in ESI†). Altogether, the single-phase experiments indicated that the rate of component exchange of free and complexed constituents in acidic condition was suitable for extraction processes (Fig. S28–S31 in ESI†).
:CDCl3 system at different pD values of the aqueous phase. As expected, for both acylhydrazones, the reaction was pD dependent and became faster in acidic conditions (Table 2 and Fig. S32 in ESI†).
| pD | 8 | 6.3 | 2.6 |
|---|---|---|---|
| (a) HA1B1 formation | 100 h | 25 h | <1 h |
| (b) HA1B2 formation | Conversion 40% (100 h) | 100 h | <1 h |
| (c) HA1B1/B2 exchange | >3 weeks | 10 days | 10 h |
| (d) HA1B2/B1 exchange | >3 weeks | >3 weeks | >3 weeks |
| (e) A1/B1/B2 exchange | >3 weeks | Conversion 80% (10 days) | 5 h |
For the formation of HA1B2, solutions of the components A1 and B2 were initially introduced into the chloroform phase where the reaction occurs (Fig. S33 in ESI†). The reaction rate increased with the acidity of the water phase in view of the presence of residual water in the wet organic phase (about 0.87 g water per L)19 (Table 2b).
Comparing the formation reactions at pD = 2.5 shows that acid catalysis greatly increased the reaction rate and there was almost no difference in the formation rates of HA1B1 and HA1B2, both completed within one hour. At pD = 6.3, the formation of HA1B1 is 4 times faster than the one of HA1B2. At pD = 8 HA1B1 formation is complete within 100 h whereas HA1B2 reaches 40% of conversion. These comparisons highlight the importance to perform reactions at low pD (Table 2a and b).
Exchange reactions in biphasic conditions were performed on the HA1B1/B2 and HA1B2/B1 sets. At pD 8 and 6, the exchange reactions were slow and raising the temperature to 333 K (60 °C) after one week showed that the thermodynamic equilibrium had not yet been reached (Fig. S34 and S35 in ESI†). As observed previously, the biphasic exchange reactions were catalysed under acidic conditions (Table 2c and d). After one day at pD = 2.5, HA1B1/B2 exchange reached equilibrium and 90% yield whereas the HA1B2/B1 pair did not react (within the 5% estimated error of determination).
When the 3 components A1, B1, B2 were mixed at pD 6.3 or 8, the equilibration of formation and component exchange took longer than 100 hours and an increase in temperature from 296 to 333 K had a very limited impact (Table 2e). It is assumed that reaction proceeded by the rapid formation of HA1B1 in the aqueous phase followed by its transfer to the chloroform phase where the exchange with B2 occurred (Fig. S36 in ESI†).
Considering the [A1 + B1 + B2] system, acid catalysis led to a reasonable formation/exchange rate and fortunately, the system is also consistent with a transition metal(II) extraction20 at pD 2 to 3.5.
During the study of dynamic reactions in biphasic systems, the determination of the concentrations of each compound in each phase is essential to understand where the reaction takes place and to follow the redistributions of reactants (Fig. S32–S36 in ESI†). A vigorous stirring is necessary as the components/constituents interact only through the interface between the two phases. The stirring of the mixture ensures a large surface area of contact between the two immiscible phases and thus a high rate of molecular transfer between them.
Comparing the mono and biphasic systems, the acylhydrazone formation is significantly slower in the biphasic system than in a single-phase system except at the lowest pD (Tables 1a and 2a). The exchange reaction between HA1B1 and B2 is also faster in the aqueous medium itself but is nevertheless fast enough to reach equilibrium in the biphasic system at a timescale (10 hours) suitable for performing the liquid–liquid extraction.
Complexation of Cu(II) nitrate by acylhydrazones
Efficient extraction of Cu(II) ions by acylhydrazones in a biphasic system is dependent on the complexation of metal cation by the ligand. To this end, the coordination properties of the Cu(II) cation with the acylhydrazone HA1B1 were investigated through single-crystal structure determinations. As the acylhydrazones N–H present a labile proton, three complexes of different charges, 2+, 1+ and neutral, may be formed between a Cu(II) cation and two tridentate HA1B1 ligands depending on the ligand ionization: respectively [Cu(II)(HA1B1)2](NO3)2 (type 1), [Cu(II)(HA1B1)(A1B1)](NO3) (type 2), and [Cu(II)(A1B1)2] (type 3). In view of their potential biological applications, the [Cu(acylhydrazone)n] complexes have been extensively studied and the structures of all three types of complexes have been reported.21Single crystals of [Cu(II)(HA1B1)2](NO3)2 and [Cu(II)(HA1B1)(A1B1)](NO3) were obtained and their molecular structures determined by X-ray crystallography (Fig. 3 and S37, Table S2 in ESI†). For [Cu(II)(A1B1)2] (type 3), we have tested different conditions for crystal growth to obtain suitable crystals but unfortunately only chain polymers of [–Cu(L−)2–CuCl−]n type had been obtained (Fig. S37 and Table S3 in ESI†).
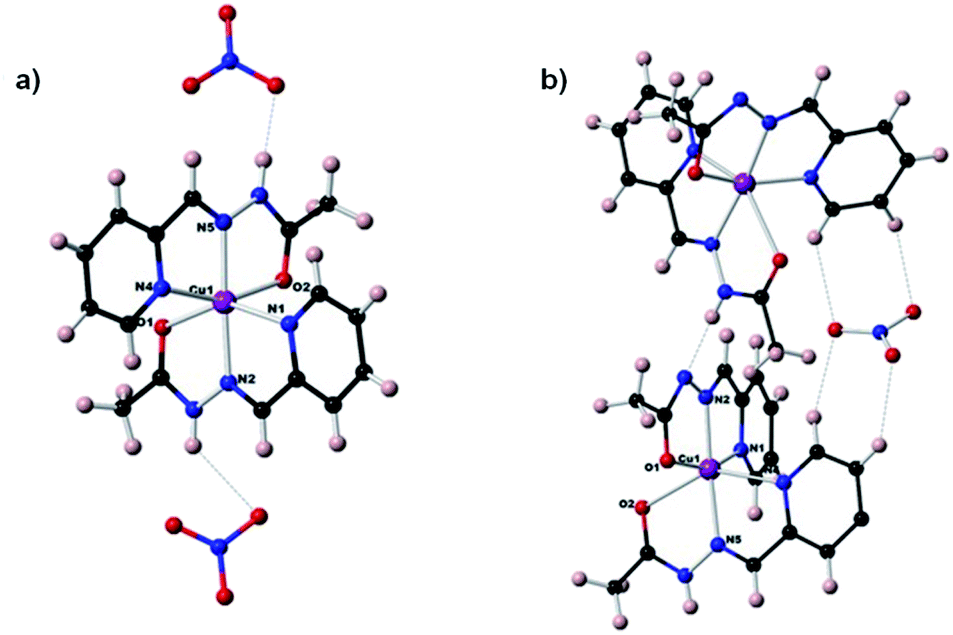 | ||
| Fig. 3 Molecular structures of the complexes (a) [Cu(II)(HA1B1)2](NO3)2 (Type 1) (b) [Cu(II)(HA1B1)(A1B1)](NO3) (Type 2). | ||
The earlier structural studies21 of complexes of types 1 and 2 involved anions other than nitrate but no significant differences from the present are apparent. In both present cases, the nitrate ions are in the second coordination sphere and form hydrogen bonds with the N–H sites of the acylhydrazone ligands, contributing to the overall stabilization of the crystal lattice. Both complexes have a strongly deformed octahedral coordination geometry (Fig. S37 and Table S4 in ESI†). The trans bonds show large deviation from 180°, with the following angles: N4–Cu–O2: 156.5° and N1–Cu–O1: 147° for complex 1; and 156.7° and 145.6° for complex 2. Comparison between (type 1) and (type 2) complexes, especially the shortening of one of the O–Cu bonds from over 2.1 Å to about 2 Å, indicates the deprotonation of one ligand out of two favours the enol-imine form over the keto-amine form of the protonated ligand left. This is in line with a shortening of the OC–N bond and a lengthening of NC–O. This structural feature is strongly dependent on the NH–X hydrogen bond, type 1: X = NO3− and type 2: X = N− site of another complex (Fig. S38 in ESI†).
Extraction properties of components and constituents
The extraction properties of the components A1, B1, B2 and the constituents HA1B1 and HA1B2 were first studied in the presence of copper(II) nitrate in a H2O:CHCl3 (v/v, 1:1) biphasic system by spectrophotometry and by direct Cu analysis using inductively coupled plasma optical emission spectrometry (ICP-OES; Fig. S39 and S40 in ESI†). At pH = 4.4, A1, B1 and HA1B1 did not extract Cu(II) ions, whereas B2 and HA1B2 showed promising properties, extracting respectively 60 ± 2% and 45 ± 1% of Cu(II) into the organic phase (Fig. S39 and S45, Tables S5–S10 in ESI†).
Further liquid–liquid extraction studies on B2 and HA1B2 separately showed that pH (2 to 5) and stirring time (5 to 900 min) had a limited effect on the phase transfer process (Tables S5–S10 in ESI†). When the amount of Cu(II) increased from 0.05 to 0.5 equiv. with respect to 1 equiv. of HA1B2 ligand, the fraction of extracted cation decreased. On the other hand, the fraction extracted by B2 increased between 0.05 to 0.25 equivalent of Cu(II) and then decreased slightly at 0.5 equivalent (Fig. S40 in ESI†).
Dynamic component exchange enabling metal phase transfer
The dynamic behaviour of the DCL of components A1, B1, B2 in the presence of Cu(II) was investigated by different experiments involving 2 or 3 steps. A typical experiment consisted of starting with two initial components, followed by the addition of copper(II) nitrate and thereafter the third component (Fig. 4 and Section 8 of ESI†).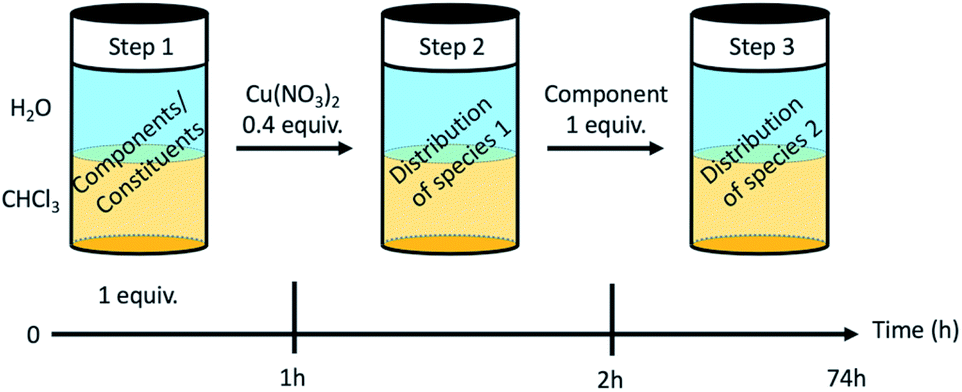 | ||
| Fig. 4 Dynamic extraction procedure. | ||
Four different extraction experiments were performed (Table 3), designed to determine the behaviour of both components and constituents independently. The main issue was the determination of the speciation of the library in each phase, using ICP-OES, NMR spectroscopy (after Cu(II) precipitation) and mass spectrometry to characterise and quantify the system. In order to compare the different experiments, the steps 1 and 2 were conducted over 1 hour (except for extraction III). The final step (the step 2 for extraction III) was left to equilibrate for up to 72 h. In all cases, the pD was around 3 (2.8 to 3.5).
:CDCl3 at pD = 3 and 296 K)
| Extraction | Step 1 | Step 2 | Step 3 | Efficiency (% of Cu in CDCl3) |
|---|---|---|---|---|
| I | HA1B1 | Cu(II) | B2 | 41% |
| II | A1 + B1 | Cu(II) | B2 | 42% |
| III | A1 + B1 + B2 | Cu(II) | — | 43% |
| Reverse | HA1B2 | Cu(II) | B1 | 32% |
Extraction experiment I
:56 (D2O:CDCl3) with an amount of hydrolysis below the detection limit (5%) (Fig. 5a and S60, Tables S12, S13 in ESI†).
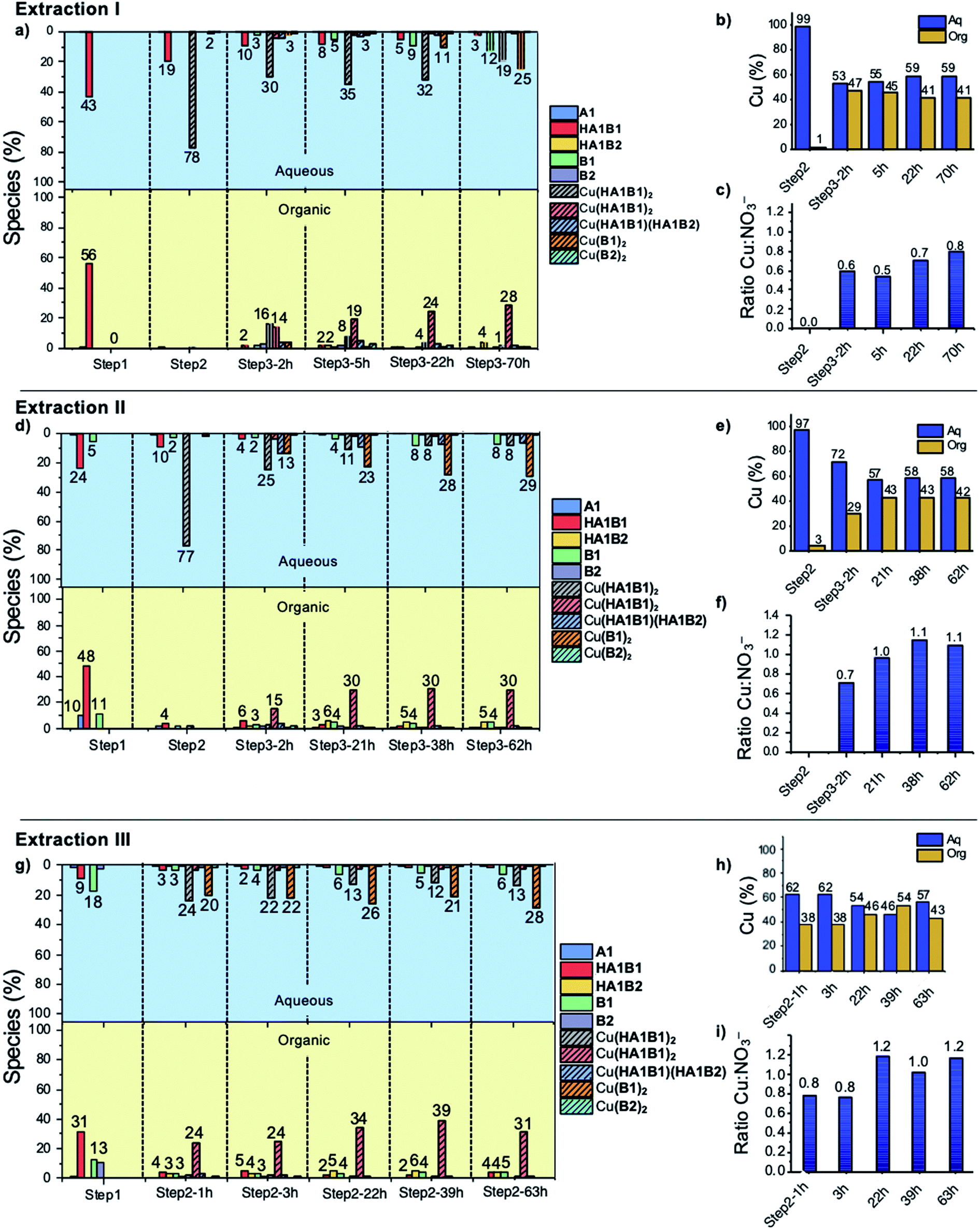 | ||
| Fig. 5 Extraction I (a) distribution (%) of species between the two phases (b) distribution (%) of Cu(II) in each phase (c) molar Cu:NO3 ratio in chloroform phase – extraction II (d) distribution (%) of species between the two phases (e) distribution (%) of Cu(II) in each phase (f) molar Cu:NO3 ratio in chloroform phase – extraction III (g) distribution (%) of species between the two phases (h) distribution (%) of Cu(II) in each phase (i) molar Cu:NO3 ratio in chloroform phase. | ||
:NO3 ratio in chloroform gives access to the number of deprotonated ligands. Five hours after the addition of B2, the ratio was 0.5, meaning that two nitrate anions were extracted with the complex and thus that the two ligands were protonated (HA1B2). At thermodynamic equilibrium after 70 h, the ratio was 0.8, indicating that one of the two ligands had slowly and partially deprotonated (with liberation of the elements of nitric acid transferred into the water phase) leading to the formation of the complex as [Cu(II)(HA1B2)(A1B2)](NO3) (Fig. 5c, Scheme 3).
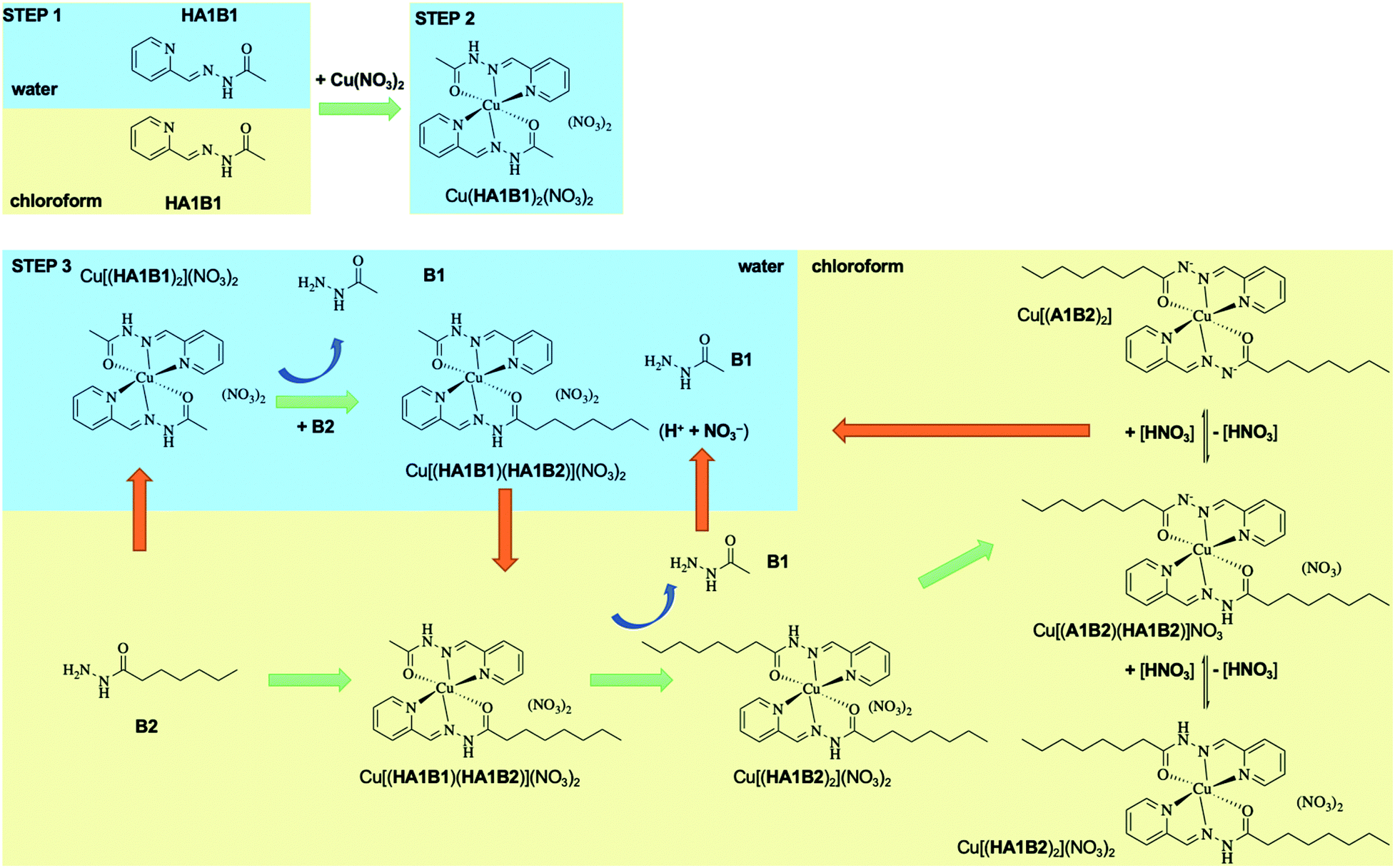 | ||
| Scheme 3 Proposed mechanism of the covalent dynamic Cu(II) extraction from water to chloroform operating through dynamic covalent extracting agents along the sequence of steps followed in the extraction experiment I above. Only the main species are represented. | ||
In the aqueous phase, the total amount of copper(II) was 59% (Fig. 5b). The species containing copper(II) were found to be [Cu(II)(HA1B1)2](NO3)2 (19%), [Cu(II)(B1)2](NO3)2 (25%) and [Cu(II)(HA1B1)(HA1B2)](NO3)2 (2%). The concentration of free B1 in water increased over time up to 12%. Conversely, HA1B1 decreased in the aqueous phase to release free A1 that transferred to the chloroform phase and reacted with B2 to form HA1B2. In these changes resides the operation of the dynamic extraction process.
Taken together, the addition of the hydrophobic component B2 into the organic phase led to a reaction with the hydrophilic complex [Cu(II)(HA1B1)2](NO3)2. The reactions occurs between the two phases and induces the transfer of Cu(II) from the aqueous phase to the organic phase in the form of the [Cu(II)(HA1B2)2](NO3)2 and [Cu(II)(HA1B2)(A1B2)](NO3) complexes. The occurrence of the covalent dynamic B1/B2 component exchange determines the Cu(II) extraction capability of the process.
Extraction experiment II
:2 (aqueous:organic). The same occurred for the remaining free A1 and B1 (Fig. 5d; Tables S14 and S15 in ESI†).
:NO3 ratio was 1:1 which means that the acylhydrazone complex species are on the average monodeprotonated, the most abundant (30%) being [Cu(II)(HA1B2)(A1B2)](NO3) (Fig. 5f; Tables S14, S15, S18, S19 in ESI†). The final state of the water phase composition displays roughly the same histogram signature as for extraction I, which means that even if the intermediate steps (1 and 2) were not completely at equilibrium, the system finally reached thermodynamic equilibrium.
Extraction experiment III
As in previous extractions, the comparison of the histogram signatures shows that the final state of the chloroform phase is very close to that of the other extraction experiments.
Global discussion of the extraction experiments
All three extraction experiments described above led to almost the same distribution of Cu(II) between the aqueous and the organic phases. They converge to a common final histogram signature after different sequences of additions/exchanges leading to a partial extraction of Cu(II).A simplified experiment, starting from HA1B2 and forming [Cu(II)(HA1B2)(A1B2)](NO3) in chloroform after copper(II) nitrate (0.4 equiv.) addition, showed that subsequent addition of B1 (1.0 equiv.) triggered the transfer of Cu(II) from the chloroform to the aqueous phase by forming hydrophilic complexes (Fig. 6; Table S20 in ESI†).
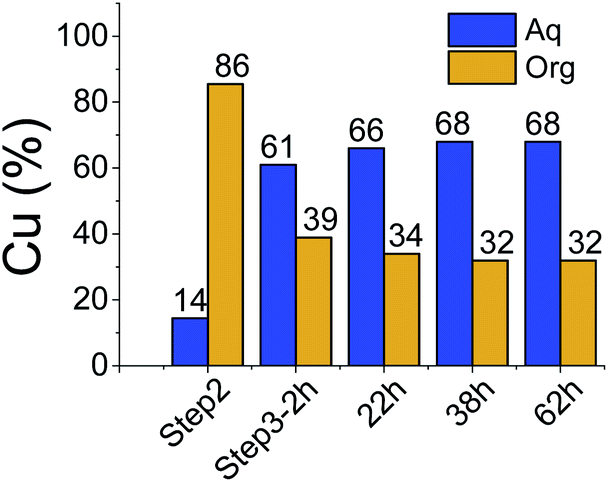 | ||
| Fig. 6 Distribution (%) of Cu(II) in each phase in the reverse experiment. | ||
This experiment demonstrates the complete reversibility of the extraction process.
The present dynamic studies have shown that it is possible to transfer Cu(II) from an aqueous phase to an organic phase or from an organic phase to an aqueous phase using the adaptive properties of a 3-component dynamic covalent library (Scheme 3).
The in-depth analytical study of the species and their behaviour over time provides the data for deriving the pathways and entities of the extraction mechanism for Cu(II) phase transfer (Scheme 3, Fig. S68 in ESI†). The analyses show a complicated evolution of the interactions between Cu(II) and the constituents HA1B1 and HA1B2. To some extent, Cu(II) also interacts with the components B1 and B2.
The main pathway leading to Cu(II) extraction by the present process involving dynamic covalent extracting agents may be considered to occur in 4 steps.
(1) The addition of the Cu(II) nitrate to the constituent HA1B1 in a biphasic D2O:CDCl3 set-up leads to the formation of [Cu(II)(HA1B1)2](NO3)2 in the aqueous phase only.
(2) The lipophilic B2 added thereafter reacts with [Cu(II)(HA1B1)2](NO3)2 through the interface by an acid-catalysed exchange reaction. In the first instance, [Cu(II)(HA1B1)(HA1B2)](NO3)2 is produced and distributed in both phases. B1 is released mainly into the aqueous phase and forms a small amount of [Cu(II)(B1)2](NO3)2 in water.
(3) As a result of the B1/B2 exchange, [Cu(II)(HA1B2)2](NO3)2 is produced and passes preferentially into the organic phase. The small amount of residual B2 produces a little [Cu(II)(B2)2](NO3)2 complex in chloroform.
(4) Finally, in chloroform, the [Cu(II)(HA1B2)2](NO3)2 complex by releasing the elements of nitric acid (that rapidly transfers to the aqueous phase) generates an equilibrium mixture of three complexes in different states of protonation [Cu(II)(HA1B2)2](NO3)2, [Cu(II)(HA1B2)(A1B2)](NO3) and [Cu(II)(A1B2)2]. The overall process is thermodynamically driven by the deprotonation of the ligand through the increase in N–H acidity resulting from Cu(II) complexation (Scheme 3).
Conclusions
The present work achieves the liquid–liquid extraction of copper(II) cations by means of a dynamic covalent library performing adaptation through reversible component exchange. It provides a detailed analysis of the ability of a DCL of acylhydrazones to complex and transfer copper(II) cations from an aqueous phase to an organic chloroform phase by the generation of a DCL of ligands through component exchange across the interface of the two-phase system. The final constituents of the library strongly coordinate the metal ions and the lipophilic acylhydrazone ligand generated enables the metal extraction. The addition of the metal salt to the DCL induces a dynamic covalent reshuffling of the system. The different ways used to obtain the dynamic library lead to the same thermodynamic equilibrium distribution of entities after copper addition showing the reversibility of the process. Finally, studying the reversibility and the adaptability of the system provides some understanding of the mechanism of extraction. However, the analytical investigations show both the novel possibilities offered and the complicated behaviour introduced by the use of DCC in liquid–liquid extraction methodologies. The versatility of the approach paves the way to more complex systems both from a dynamic library point of view and from that of metal ion extraction/separation processes, which may be made selective by suitable choice of the components of the DCL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the ERC (Advanced Research Grant SUPRADAPT 290585), the ANR Grant DYNAFUN as well as the University of Strasbourg Institute for Advanced Study (USIAS) and the Commissariat à l’Énergie Atomique et aux Énergies Alternatives (CEA) for financial support. AC thanks the CEA for a doctoral fellowship, Pr. Jack Harrowfield for discussions, Dr Jean-Louis Schmitt, Cyril Antheaume and Dr Bruno Vincent for helpful discussions of NMR and HRMS data as well as Corinne Bailly, and Dr Lydia Karmazin for the X-ray crystallographic structure determinations. AC thanks Dr Anne Boos, Ilsah El Masoudi and Pascale Ronot for ICP-AES analyses.Notes and references
-
(a) V. Manchanda and P. Pathak, Sep. Purif. Technol., 2004, 35, 85–103 CrossRef CAS
; (b) D. Whittaker, A. Geist, G. Modolo, R. Taylor, M. Sarsfield and A. Wilden, Solvent Extr. Ion Exch., 2018, 36, 223–256 CrossRef CAS
-
(a) T. Lasheen, M. El-Ahmady, H. Hassib and A. Helal, Miner. Process. Extr. Metall. Rev., 2015, 36, 145–173 CrossRef CAS
-
(a) T. Nguyen and M. Lee, Miner. Process. Extr. Metall. Rev., 2019, 40, 278–291 CrossRef CAS
-
(a) R. Poirot, X. Le Goff, O. Diat, D. Bourgeois and D. Meyer, ChemPhysChem, 2016, 2112–2117 CrossRef CAS
-
(a) J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS
-
(a) G. Vantomme, S. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518 CrossRef CAS
-
(a) G. Men and J.-M. Lehn, J. Am. Chem. Soc., 2017, 139, 2474–2483 CrossRef CAS
-
(a) J. Armao and J.-M. Lehn, J. Am. Chem. Soc., 2016, 138, 16809–16814 CrossRef CAS
-
(a) N. Hafezi and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 12861–12868 CrossRef CAS
- J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS
- A. Osypenko, S. Dhers and J.-M. Lehn, J. Am. Chem. Soc., 2019, 141, 12724–12737 CrossRef CAS
-
(a) D. Epstein, s. Choudhary, M. R. Churchill, K. Keil, A. Eliseev and J. Morrow, Inorg. Chem., 2001, 40, 1591–1596 CrossRef CAS
- X.-Q. Chen, Y.-D. Cai, W. Jiang, G. Peng, J.-K. Fang, J.-L. Liu, M.-L. Tong and X. Bao, Inorg. Chem., 2019, 58, 999–1002 CrossRef CAS
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC
-
(a) A. Ekström, J. Wang, J. Bella and D. Campopiano, Org. Biomol. Chem., 2018, 16, 8144–8149 RSC
- J. Wyman and E. Ingalls, J. Am. Chem. Soc., 1938, 60, 1182–1184 CrossRef CAS
-
(a) E. Cordes and W. Jencks, J. Am. Chem. Soc., 1962, 84, 832–837 CrossRef CAS
- D. Kölmel and E. Kool, Chem. Rev., 2017, 117(15), 10358–10376 CrossRef
-
A. L. Horvath and F. W. Getzen, IUPAC Solubility Data Series, 1995, vol. 60 Search PubMed
-
(a)
D. Peppard, Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, 1966, pp. 1–80 Search PubMed
-
(a) R. Patel, Y. P. Singh, Y. Singh, R. Butcher, M. Zeller, R.-K. Singh and O. U-Wang, J. Mol. Struct., 2017, 1136, 157–172 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2014872–2014874. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04098c |
| ‡ Researchers and engineers working in the field of liquid–liquid separation define the solvent as a combination of a diluent, an extractant and sometimes, additional molecules often called co-extractant. In the present context, the term of solvent used corresponds to the diluent. |
| § The notation AaBb designates a deprotonated acylhydrazone group with a site N−. |
| This journal is © The Royal Society of Chemistry 2020 |