
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMerging C(sp3)–H activation with DNA-encoding†
Zhoulong
Fan‡
 a,
Shuai
Zhao‡
a,
Tao
Liu
a,
Peng-Xiang
Shen
a,
Zi-Ning
Cui
a,
Zhe
Zhuang
a,
Qian
Shao
a,
Jason S.
Chen
b,
Anokha S.
Ratnayake
c,
Mark E.
Flanagan
c,
Dominik K.
Kölmel
c,
David W.
Piotrowski
c,
Paul
Richardson
d and
Jin-Quan
Yu
*a
a,
Shuai
Zhao‡
a,
Tao
Liu
a,
Peng-Xiang
Shen
a,
Zi-Ning
Cui
a,
Zhe
Zhuang
a,
Qian
Shao
a,
Jason S.
Chen
b,
Anokha S.
Ratnayake
c,
Mark E.
Flanagan
c,
Dominik K.
Kölmel
c,
David W.
Piotrowski
c,
Paul
Richardson
d and
Jin-Quan
Yu
*a
aDepartment of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA. E-mail: yu200@scripps.edu
bAutomated Synthesis Facility, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA
cPfizer Medicinal Chemistry, Eastern Point Road, Groton, Connecticut 06340, USA
dPfizer Medicinal Chemistry, 10578 Science Center Drive, San Diego, CA 09121, USA
First published on 7th September 2020
Abstract
DNA-encoded library (DEL) technology has the potential to dramatically expedite hit identification in drug discovery owing to its ability to perform protein affinity selection with millions or billions of molecules in a few experiments. To expand the molecular diversity of DEL, it is critical to develop different types of DNA-encoded transformations that produce billions of molecules with distinct molecular scaffolds. Sequential functionalization of multiple C–H bonds provides a unique avenue for creating diversity and complexity from simple starting materials. However, the use of water as solvent, the presence of DNA, and the extremely low concentration of DNA-encoded coupling partners (0.001 M) have hampered the development of DNA-encoded C(sp3)–H activation reactions. Herein, we report the realization of palladium-catalyzed C(sp3)–H arylation of aliphatic carboxylic acids, amides and ketones with DNA-encoded aryl iodides in water. Notably, the present method enables the use of alternative sets of monofunctional building blocks, providing a linchpin to facilitate further setup for DELs. Furthermore, the C–H arylation chemistry enabled the on-DNA synthesis of structurally-diverse scaffolds containing enriched C(sp3) character, chiral centers, cyclopropane, cyclobutane, and heterocycles.
Introduction
The concept of using DNA sequences to encode each single reagent in every reaction step during the split-pool synthesis was originally proposed by Brenner and Lerner.1 DNA-encoded library (DEL) affinity selection against target proteins can now be performed at the benchtop, with millions or billions of DEL molecules incubated with the immobilized target protein in few experiments.2 The application of DEL technology has led to rapid identification of lead compounds for drug discovery.3 Currently, increasing the hit rate as well as improving the drug-like properties of the lead compounds is a primary concern for constructing superior DELs. In this context, establishing diverse types of organic transformations compatible with DEL technology (water as solvent, presence of DNA, 1 mM concentration of DNA substrate) is of pivotal importance.4 Development of on-DNA reactions faces a number of distinct challenges. DNA backbone degradation could occur under conventional reaction conditions; metal catalysts could be poisoned by oligonucleotides; at least 20% water is required as co-solvent for dissolving DNA-tagged substrates. Over the past decade, advances in DNA-compatible reactions focused on nucleophilic aromatic substitution (SNAr), cross-coupling, cycloaddition, and click chemistry to construct C(sp2)–C(sp2),5 C–N,6 C–O,7 S–X bonds,8 and heterocycles.9 Considering the well-known trends for incorporating C(sp3) carbon centers to build C(sp2)–C(sp3) bond10 and avoiding high molecular mass and lipophilicity, coupling C(sp3)–H bonds of simple aliphatic acids and ketones with DNA-encoded heteroaryls will be highly desirable. The availability of multiple C–H bonds of a wide range of carboxylic acids and ketones offers a unique opportunity to expand the accessible chemical space for DELs.Our recent development of a wide range of transformations of β-C(sp3)–H bonds demonstrates the potential for creating unprecedented diversity from simplicity (Fig. 1A).11 The attractiveness of using C–H arylation of carboxylic acid derived substrates to build DNA-encoded libraries is evident from a recent study where C–H activation reactions were performed in organic solvent and the DNA tags were subsequently attached individually. However, this approach does not allow encoding each single reagent in every reaction step during the split-pool synthesis, thus limiting the number of available building blocks and thereby the size of the library (Fig. 1B).12 In contrast, using C–H activation as a coupling step on DNA would allow much larger libraries to be constructed. Notably, despite the use of a very powerful directing group, C(sp3)–H activation reactions have not been successful in the presence of DNA thus far.13 We envision the development of on-DNA C(sp3)–H activation of different classes of monofunctional building blocks to participate in the key cycle for DEL buildup with enriched C(sp3) character, chiral centers, small rings and heterocycles. The reserved functional group could also be directly employed for further setup in DEL synthesis (Fig. 1C).
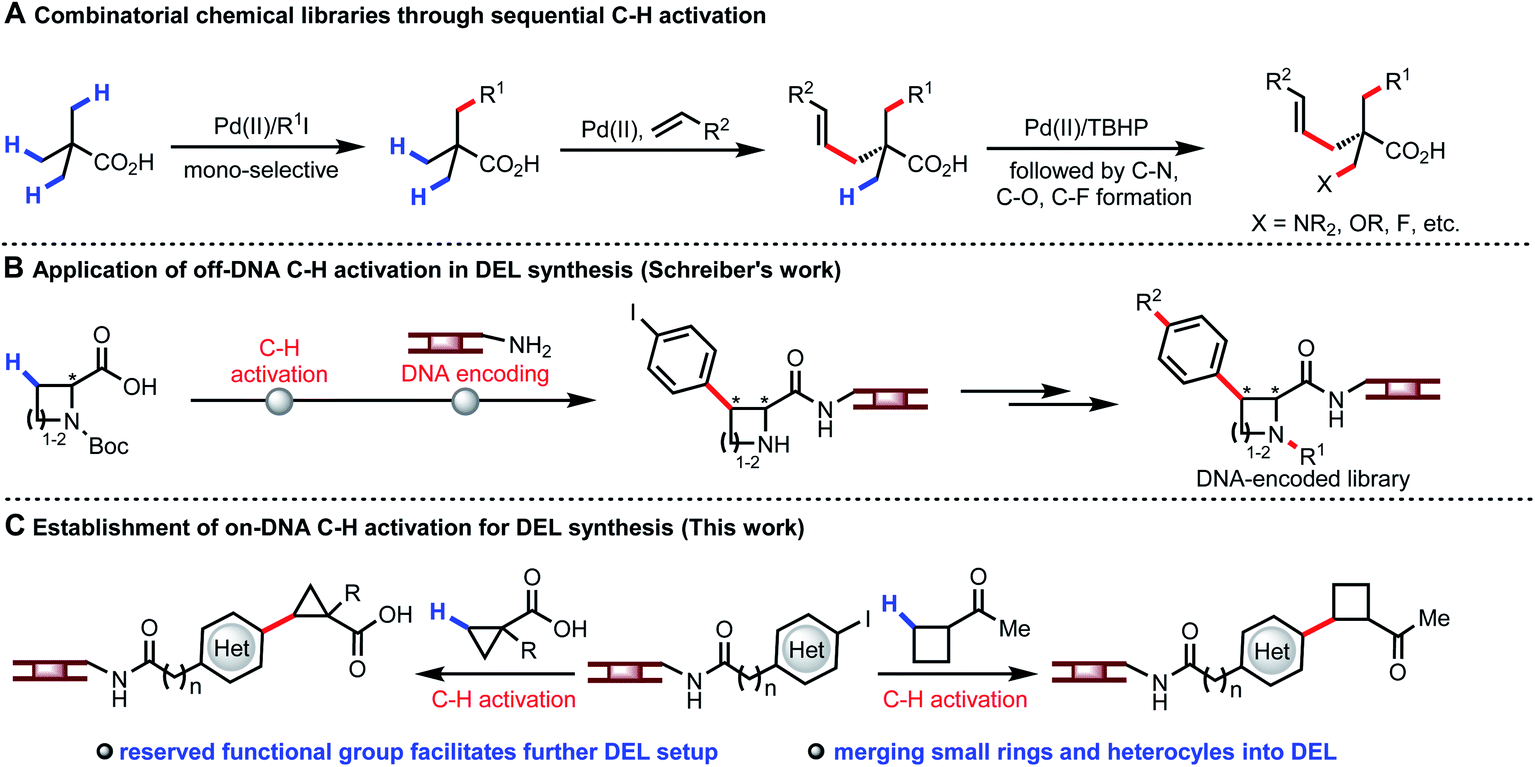 | ||
| Fig. 1 Exploiting diversity of C–H activation in DELs. | ||
Results and discussion
Since carboxylic acids and (hetero)aryl iodides are ubiquitous building blocks for DNA-encoded libraries, we first studied the coupling of a DNA-tethered aryl iodide with a free carboxylic acid through Pd-catalyzed β-C–H arylation. This presented formidable challenges beyond the need to adapt the chemistry to aqueous conditions, notably the limited stability of the DNA towards low pH and heat, the potential for interfering reactivity of the DNA bases, and the high dilution of the DNA-tethered component. Nonetheless, we decided to develop this chemistry around free carboxylic acids. Guided by our previous C–H arylation efforts,14 we screened palladium source, ligand, silver source, base, and co-solvent. The optimized conditions gave product 1 in 78% yield with no di- or tri-arylation of pivalic acid (entry 1, Fig. 2). Palladium, silver, and heat were mandatory (entries 2 to 4). No ligand was required (entry 5), but amino-acid derived ligands improved the yield (see ESI†) and L1 was found to be optimal. The main side reaction, protodeiodination (see S1b), was always present to a small extent due to the use of excess silver salt and palladium. Compound S1b containing no iodide was subjected to the reaction conditions and recovered intact, confirming that coupling occurs at the aryl iodide and not at some undetermined location on the DNA tag. Dithiocarbamate releases DNA from the DNA–palladium complex as a result of its stronger coordination with palladium leading to an insoluble complex. After each reaction, the mixture was incubated with sodium diethyldithiocarbamate trihydrate in order to scavenge palladium. The level of DNA recovery was significantly influenced by the loading of the dithiocarbamate scavenger (see ESI†).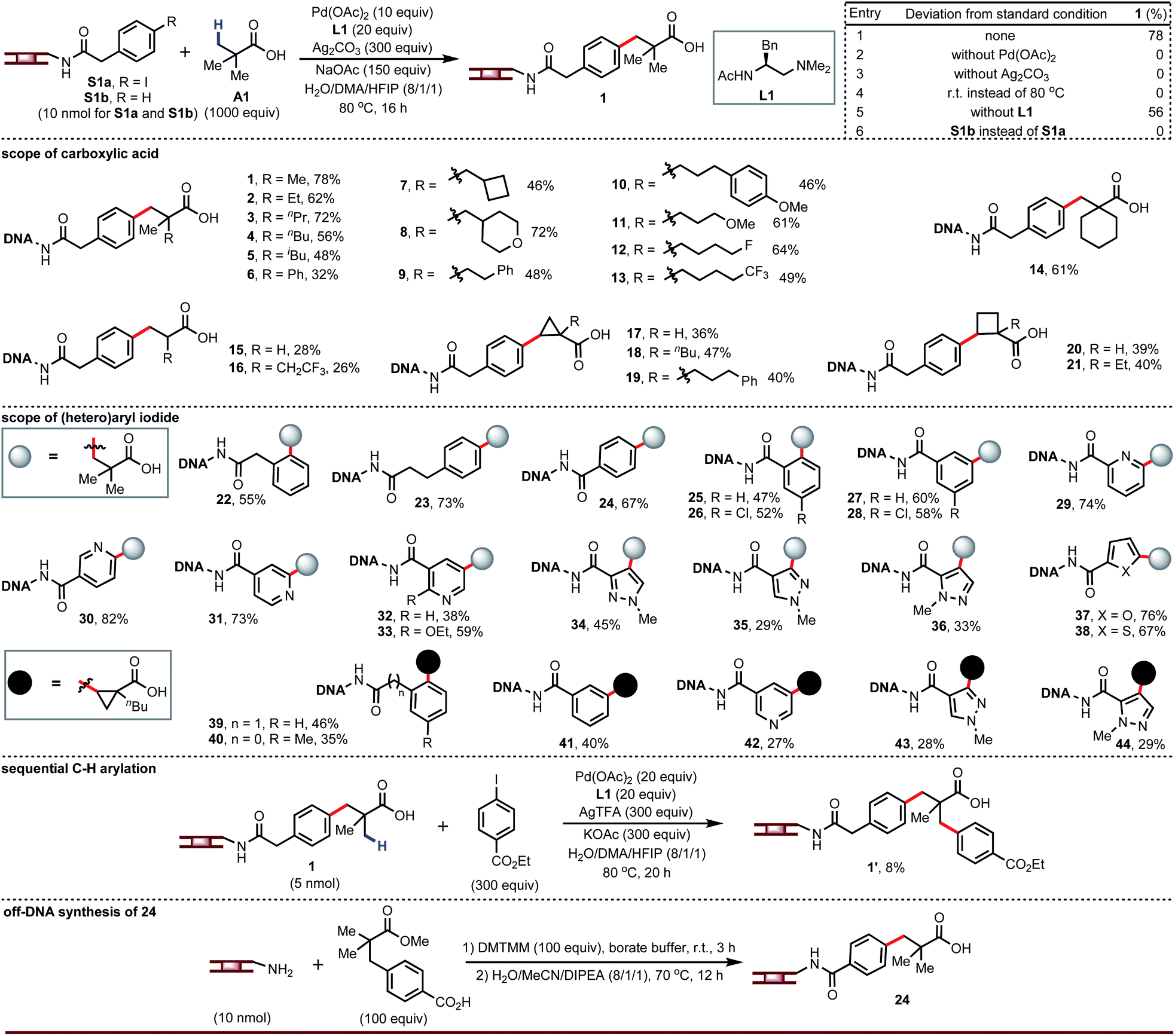 | ||
| Fig. 2 Ligand-promoted on-DNA C–H arylation of free carboxylic acids. Unless otherwise noted, condition of entry 1 was used as the standard condition. For 2: corresponding A, 500 equiv. For 3–14, 18, 19, and 39–44: AgTFA, 300 equiv.; corresponding A, 300 equiv.; 36 h. For 15: Ag3PO4, 200 equiv.; corresponding A, 300 equiv.; 36 h. For 21: corresponding A, 300 equiv.; 36 h. For 16: AgOTf, 300 equiv.; corresponding A, 300 equiv.; 36 h. For 17: H2O/DMA (6/1). | ||
A broad range of carboxylic acids adjacent to quaternary carbon atoms are suitable for this chemistry, including those containing ethers or fluorine (2 to 14). However, acids adjacent to a secondary or tertiary carbon react with 26–28% yields (15 to 16). Importantly, cyclopropane- and cyclobutanecarboxylic acids (desirable as alkene isosteres) are competent coupling partners (17 to 21). The mechanism of the Pd-catalyzed β-C–H arylation is well known to give the cis stereoisomer for cyclic carboxylic acid exclusively. Importantly, these cis products are resistant to epimerization in the presence of strong bases.14b Our LCMS analysis was unable to determine whether chiral ligand L1 gave any absolute stereoinduction. Though we screened different silvers for achieving best yields for all of carboxylic acid substrates (see ESI†), silver trifluoroacetate gave acceptable yields in most of cases and can be used in split-pool synthesis for a DEL buildup.
The aryl iodide substitution pattern is flexible (22 to 28, 39 to 41). Although our previously-published reaction conditions in organic solvent could not couple heteroaryl iodides,14 we were optimistic that conditions that tolerate DNA—the bases of which contain nitrogen heterocycles—should allow coupling of heteroaryl iodides (e.g., pyridines and pyrazoles). This was indeed the case; heteroaryl iodides successfully reacted with carboxylic acids under the same reaction conditions (29 to 38, 42 to 44). Since aryl and heteroaryl iodides react under the same conditions, they can both be present in a split-pool synthesis of a DEL. In addition, the β-C(sp3)–H arylated product attached to DNA can undergo the second carboxylate-directed C(sp3)–H arylation to obtain the DNA-tagged product 1′. Furthermore, we selected product 24 to proceed off-DNA synthesis and demonstrate the reliability of the present on-DNA C–H reaction.
The carboxylic acids in the products from the above chemistry can be further derivatized (vide infra). One interesting possibility is coupling to chiral amino acids in order to enhance the chiral recognition potential of the resultant DNA-encoded libraries. We recognized an opportunity in these cases to develop a ligandless Pd-catalyzed β-C–H arylation. Rather than couple the free carboxylic acid and then amide couple, one could pre-synthesize amides derived from carboxylic acids and amino acids and take advantage of the presence of a bidentate directing group to run a more facile Pd-catalyzed arylation.15 Re-evaluation of the silver source, base, and co-solvent in the absence of ligand led to optimized conditions that afforded compound 45 in 69% yield (entry 1, Fig. 3). Palladium is essential for this transformation (entry 2). Silver salts and bases are not strictly required but have a significant impact on yield (entries 3 to 9). Surprisingly, we found this reaction also worked well at room temperature (entry 10).
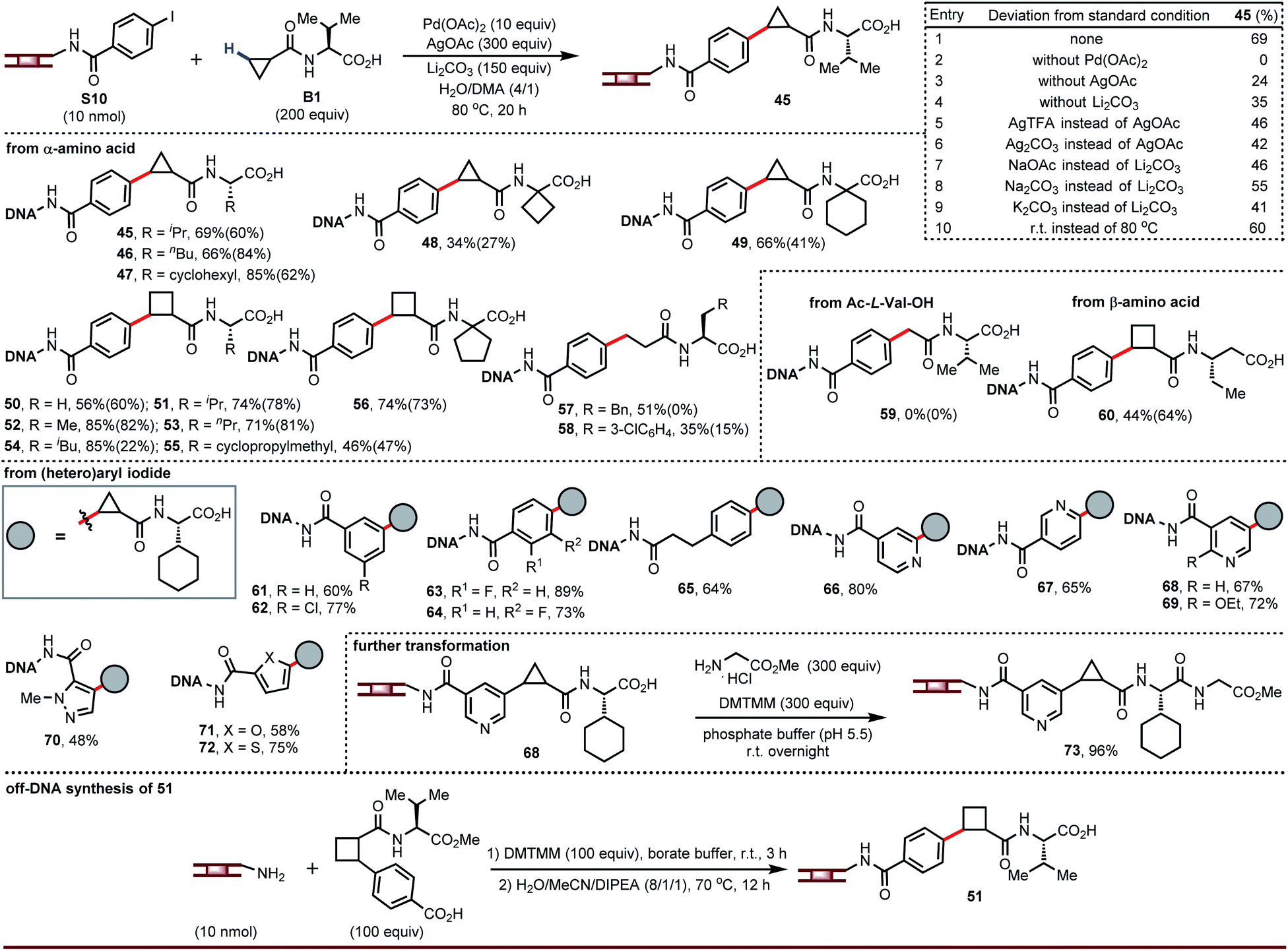 | ||
| Fig. 3 On-DNA C–H arylation of amides. Unless otherwise noted, condition of entry 1 was used as the standard condition. Yields at room temperature are listed in parentheses. DMTMM, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. | ||
Amides derived from cyclopropane- or cyclobutanecarboxylic acid and a broad range of α-amino acids reacted smoothly both heated and at room temperatures (45 to 56). Pd-catalyzed arylation proceeds only at the β-C–H bond from the amide; attempted coupling of Ac-L-Val-OH gave none of arylation product 59 indicating that α-arylation did not occur. Two LC peaks having same mass were due to the generation of diastereomers after the C–H arylation. To demonstrate this, we ran a representative off-DNA reaction to synthesize product 51. As expected, a mixture of diastereomers was observed, although the ratio is slightly lower due to the different reaction temperature. Although we focused on amides derived from α-amino acids and containing cyclopropyl or cyclobutyl rings, the chemistry can be extended to other alkyl carboxylic acids (57 and 58) and to β-amino acids (60). Diverse arene substitution patterns on the DNA-tethered aryl iodide were tolerated (61 to 65). As was the case for β-C–H arylation of carboxylic acids, the DNA-tolerant conditions for amide arylation can also be employed for coupling DNA-tethered heteroaryl iodides such as pyridines and pyrazoles (66 to 72). If desired, the carboxylic acid of the product can be further modified (73).
Having developed DNA-compatible C(sp3)–H arylations for carboxylic acids and amides, we turned our attention to ketones. Ketones are useful monomers for building DELs since they can be further elaborated via reductive amination. Guided by our previous work using aminooxyacetic acids as removable directing groups to recruit palladium to activate the β-C–H bond of ketones,16 we optimized a DNA-tolerant version of this reaction. The optimized reaction gave arylation product 83 in 62% yield (entry 1, Fig. 4). This reaction requires palladium and heat (entries 2 and 3) and is strongly influenced by the silver salt (entries 4–6); ligand and base play a lesser role (entries 7 and 8). We also found L8 could decrease the degradation of DNA and provide clean LC traces. Although a large excess of palladium is often associated with degradation of the DNA tag, this chemistry gives higher yield at 40 equiv. Pd/L (entry 1) than at 30 equiv. (entry 9).
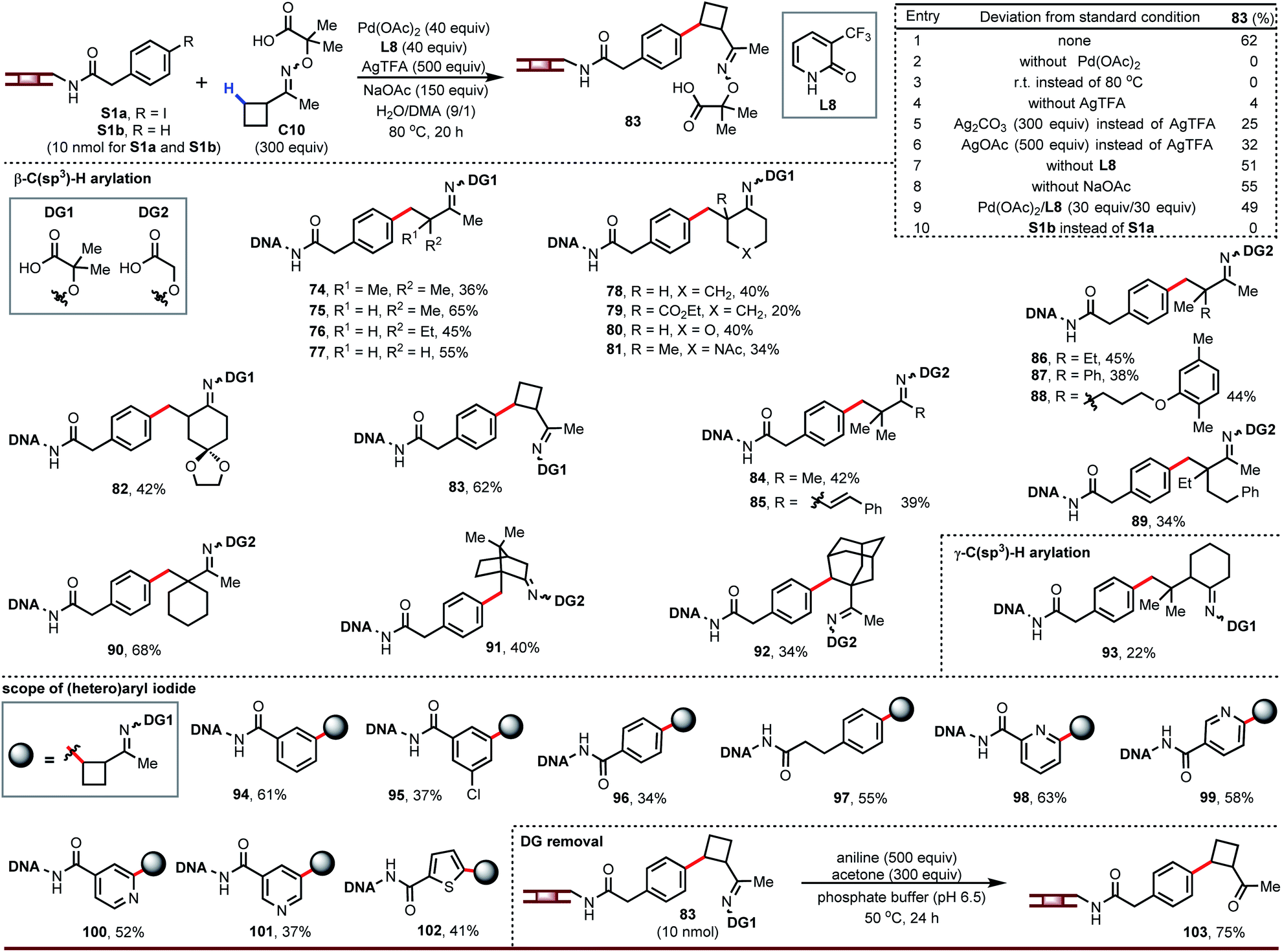 | ||
| Fig. 4 DNA-compatible C–H arylation of ketones. Unless otherwise noted, condition of entry 1 was used as the standard condition. For 89 and 90: Pd(OAc)2, 30 equiv.; L8, 30 equiv.; H2O/DMA (2/1). | ||
The above conditions affect β-C–H arylation in diverse settings, including on acyclic ketone derivatives (74 to 77, 84 to 89), at positions next to simple or complex rings (78 to 82, 90, 91), and on simple or complex rings (83, 92). A broad range of functional groups are tolerated, including esters, ethers, acetals, and amides. Ketone derivatives bearing β-quaternary centers can be γ-arylated (93). The DNA-tethered aryl iodide accepts different substitution patterns (94 to 97) and heteroaryl iodides can react under the same conditions (98 to 102). We also noticed that cis and trans-isomers of directing group were separated in the LC traces.
The ability to convert the oxime ethers back into ketones is critical for implementing this chemistry in a DEL buildup. We discovered that these oxime ethers readily hydrolyze in the presence of aniline and acetone (103), likely through equilibrium transimination with aniline and trapping of the free aminooxyacetic acid with acetone.17
Each of these C(sp3)–H activation reactions, as new disconnections for DEL synthesis, can incorporate unique structural motifs. The combination of multiple C(sp3)–H activations in DEL synthesis can further enhance the diversity. Hence, we embarked on a multi-step synthesis on DNA consisting of β-C–H arylation of a carboxylic acid, amide formation, β-C–H arylation of a masked ketone, and ketone deprotection (Fig. 5A) in order to demonstrate how these C–H activation chemistries can be combined to develop large DELs of diverse, drug-like compounds. A representative analog synthesis is shown in Fig. 5B. Thus, pyridyl iodide S18 and pivalic acid were coupled to form intermediate 31, and amide coupling with p-iodobenzyl amine then set up a second C–H arylation event. Coupling with a masked cyclobutyl ketone gave oxime ether 105. The above is only one of many sequences that one could design by using sequential multiple C–H arylation steps in combination with common DNA-encoded library-building steps such as amide formation or reductive amination. The use of multiple C–H activation reactions connects commonly used building blocks at different carbon centers hence providing complementary diversity of chemical space.
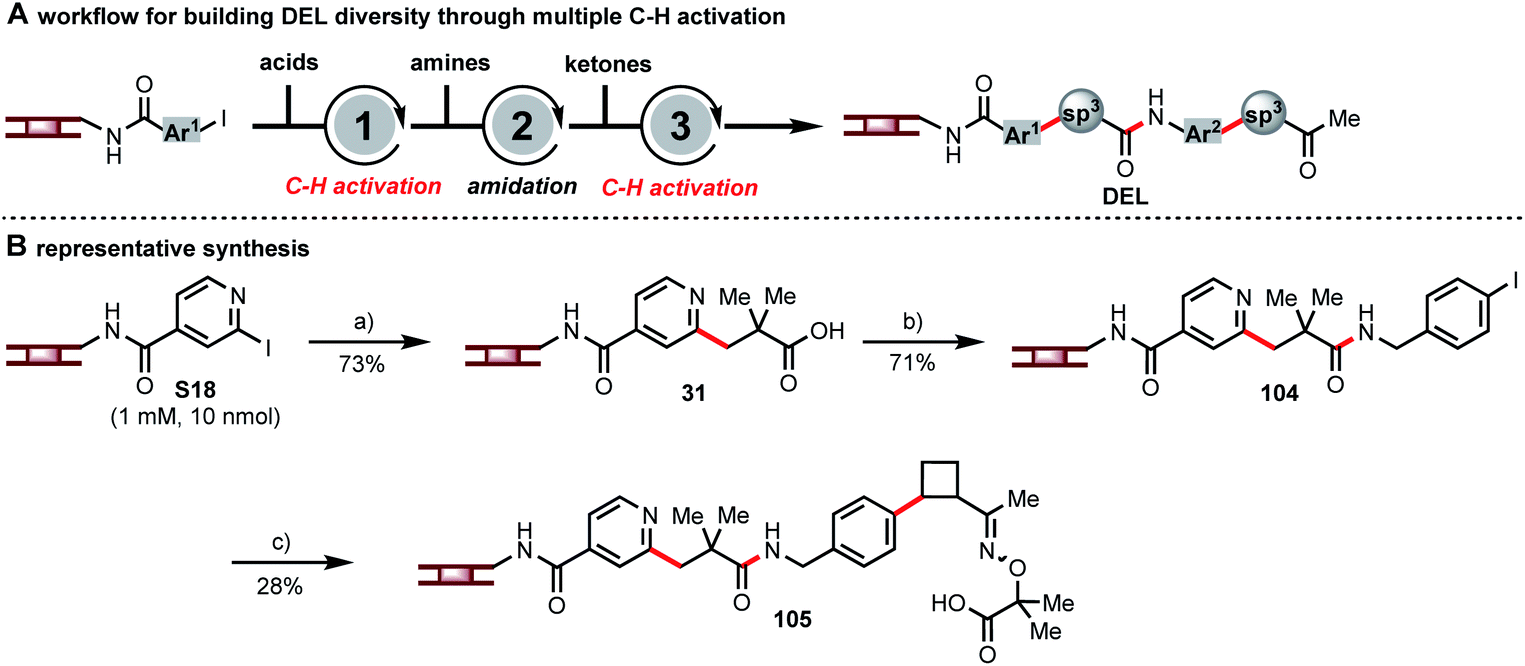 | ||
| Fig. 5 The utility of multiple C–H activation in DEL synthesis. (A) Workflow for building DEL diversity through multiple C–H activation; (B) representative synthesis. Conditions: (a) A1 (1000 equiv.), Pd(OAc)2 (10 equiv.), L1 (20 equiv.), Ag2CO3 (300 equiv.), NaOAc (150 equiv.), H2O/DMA/HFIP (8/1/1), 80 °C, 16 h. (b) 4-Iodobenzylamine (300 equiv.), DMTMM (300 equiv.), phosphate buffer (pH 5.5), r.t. 16 h. (c) C10 (300 equiv.), Pd(OAc)2 (40 equiv.), L8 (40 equiv.), AgTFA (500 equiv.), NaOAc (150 equiv.), H2O/DMA (9/1), 80 °C, 20 h. | ||
In order to evaluate DNA compatibility of the C–H activation chemistry with DEL synthesis, a select set of chemically modified on-DNA analogs (C–H arylation products 1, 45 and 83), along with their starting aryl iodide analog S1a were enzymatically ligated to a 65-mer dsDNA, so that the resulting oligomers were approximately equal in length to a encoding tag of a 3-cycle DEL build. All four ligation reactions proceeded smoothly, indicating that chemistry had no significant impact on encodability. In order to determine the amount of amplifiable DNA remaining after exposure to C–H activation conditions, the ligation products from 1, 45 and 83 were amplified by PCR and compared with that of S1a (untreated control). All three reactions showed satisfactory PCR viability (60–80% amplifiable DNA remaining). Moreover, Sanger sequencing reads also confirmed the integrity of their nucleobase sequence structures (see ESI†).
As purification is an inherently difficult process in split-pool synthesis, the reactivity required for DNA-compatible reactions must be devoid of unidentified byproducts that can complicate analysis. In this case, the main byproducts generated through our reaction platform consists only of starting material or its protodehalogenated derivative. Finally, we were able to obtain all products through our on-DNA C–H arylation platform in moderate and synthetically useful yields; higher than the threshold of 25% deemed practical in DEL synthesis.10f Gratifyingly also, we were able to obtain 60–80% DNA recovery from qPCR experiments, greater than the acceptable 30% threshold deemed practical in these processes.4c Altogether, these promising results further demonstrate the practicality of our DEL-compatible C(sp3)–H activation platform, enabling practitioners to rapidly generate structural complexity and diversity in a modular manner.
Conclusions
In summary, we have developed DNA-compatible C(sp3)–H activation reactions of carboxylic acid, amides, and ketones. The use of universal building blocks and reserved functional group facilitates direct access to next setup for DEL synthesis. These protocols are compatible with the C(sp3)–H bond of small rings and heterocyclic coupling partners that are desirable for improving drug-like properties. These C(sp3)–H activation reactions for DEL synthesis provides a unique tool for constructing chemical diversity containing high C(sp3) character.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge The Scripps Research Institute and Pfizer for their financial support. S. Z. thanks Jiangsu Overseas Research & Training Program for University Prominent Young & Middle-aged Teachers and Presidents. Chris O'Donnell, Russell Dushin, Bruce Lefker, and Joe Abramite are acknowledged for their suggestions during the early phases of this work. Brian M. Paegel is acknowledged for the evaluation of DNA compatibility with aqueous conditions for C–H activation. Brittany Sanchez and Emily Sturgell are acknowledged for their assistance with LC-MS analysis.Notes and references
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381 CrossRef CAS.
- (a) R. A. Goodnow, A Brief History of the Development of Combinatorial Chemistry and the Emerging Need for DNA-Encoded Chemistry, A handbook for DNA-encoded chemistry, Wiley-Blackwell, 2014, pp. 19–43 CrossRef; (b) R. M. Franzini, D. Neri and J. Scheuermann, Acc. Chem. Res., 2014, 47, 1247 CrossRef CAS; (c) G. Zimmermann and D. Neri, Drug Discovery Today, 2016, 21, 1828 CrossRef CAS; (d) R. A. Lerner and S. Brenner, Angew. Chem., Int. Ed., 2017, 56, 1164 CrossRef CAS; (e) D. Neri, ChemBioChem, 2017, 18, 827 CrossRef CAS; (f) B. Shi, Y. Zhou, Y. Huang, J. Zhang and X. Li, Bioorg. Med. Chem. Lett., 2017, 27, 361 CrossRef CAS; (g) A. L. Satz, ACS Med. Chem. Lett., 2018, 9, 408 CrossRef CAS; (h) G. Zhao, Y. Huang, Y. Zhou, Y. Li and X. Li, Expert Opin. Drug Discovery, 2019, 14, 735 CrossRef CAS.
- (a) P. Zhao, Z. Chen, Y. Li, D. Sun, Y. Gao, Y. Huang and X. Li, Angew. Chem., Int. Ed., 2014, 53, 10056 CrossRef CAS; (b) Y. Ding, H. O'Keefe, J. L. DeLorey, D. I. Israel, J. A. Messer, C. H. Chiu, S. R. Skinner, R. E. Matico, M. F. Murray-Thompson, F. Li, M. A. Clark, J. W. Cuozzo, C. Arico-Muendel and B. A. Morgan, ACS Med. Chem. Lett., 2015, 6, 888 CrossRef CAS; (c) C. C. Arico-Muendel, Med. Chem. Commun., 2016, 7, 1898 RSC; (d) S. L. Belyanskaya, Y. Ding, J. F. Callahan, A. L. Lazaar and D. I. Israel, ChemBioChem, 2017, 18, 837 CrossRef CAS; (e) D. Neri and R. A. Lerner, Annu. Rev. Biochem., 2018, 87, 479 CrossRef CAS; (f) L. H. Yuen, S. Dana, Y. Liu, S. I. Bloom, A. G. Thorsell, D. Neri, A. J. Donato, D. Kireev, H. Schüler and R. M. Franzini, J. Am. Chem. Soc., 2019, 141, 5169 CrossRef CAS; (g) B. Cai, D. Kim, S. Akhand, Y. Sun, R. J. Cassell, A. Alpsoy, E. C. Dykhuizen, R. M. Van Rijn, M. K. Wendt and C. J. Krusemark, J. Am. Chem. Soc., 2019, 141, 17057 CrossRef CAS.
- (a) A. L. Satz, J. Cai, Y. Chen, R. Goodnow, F. Gruber, A. Kowalczyk, A. Petersen, G. Naderi-Oboodi, L. Orzechowski and Q. Strebel, Bioconjugate Chem., 2015, 26, 1623 CrossRef CAS; (b) R. M. Franzini and C. Randolph, J. Med. Chem., 2016, 59, 6629 CrossRef CAS; (c) M. L. Malone and B. M. Paegel, ACS Comb. Sci., 2016, 18, 182 CrossRef CAS.
- (a) Y. Ding and M. A. Clark, ACS Comb. Sci., 2015, 17, 1 CrossRef CAS; (b) Y. Ding, J. L. DeLorey and M. A. Clark, Bioconjugate Chem., 2016, 27, 2597 CrossRef CAS; (c) X. Lu, L. Fan, C. B. Phelps, C. P. Davie and C. P. Donahue, Bioconjugate Chem., 2017, 28, 1625 CrossRef CAS.
- (a) X. Lu, S. E. Roberts, G. J. Franklin and C. P. Davie, Med. Chem. Commun., 2017, 8, 1614 RSC; (b) Y. Ruff and F. Berst, Med. Chem. Commun., 2018, 9, 1188 RSC.
- D.-Y. Wang, X. Wen, C.-D. Xiong, J.-N. Zhao, C.-Y. Ding, Q. Meng, H. Zhou, C. Wang, M. Uchiyama, X.-J. Lu and A. Zhang, iScience, 2019, 15, 307 CrossRef CAS.
- (a) N. Probst, R. Lartia, O. Théry, M. Alami, E. Defrancq and S. Messaoudi, Chem.–Eur. J., 2018, 24, 1795 CrossRef CAS; (b) F. Liu, H. Wang, S. Li, G. A. L. Bare, X. Chen, C. Wang, J. E. Moses, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2019, 58, 8029 CrossRef CAS; (c) D. T. Flood, X. Zhang, X. Fu, Z. Zhao, S. Asai, B. B. Sanchez, E. J. Sturgell, J. C. Vantourout, P. Richardson, M. E. Flanagan, D. W. Piotrowski, D. K. Kölmel, J. Wan, M.-H. Tsai, J. S. Chen, P. S. Baran and P. E. Dawson, Angew. Chem., Int. Ed., 2020, 59, 7377 CrossRef CAS.
- (a) H. Li, Z. Sun, W. Wu, X. Wang, M. Zhang, X. Lu, W. Zhong and D. Dai, Org. Lett., 2018, 20, 7186 CrossRef CAS; (b) C. J. Gerry, Z. Yang, M. Stasi and S. L. Schreiber, Org. Lett., 2019, 21, 1325 CrossRef CAS; (c) P. Ma, H. Xu, J. Li, F. Lu, F. Ma, S. Wang, H. Xiong, W. Wang, D. Buratto, F. Zonta, N. Wang, K. Liu, T. Hua, Z.-J. Liu, G. Yang and R. A. Lerner, Angew. Chem., Int. Ed., 2019, 58, 9254 CrossRef CAS; (d) M. K. Škopić, K. Götte, C. Gramse, M. Dieter, S. Pospich, S. Raunser, R. Weberskirch and A. Brunschweiger, J. Am. Chem. Soc., 2019, 141, 10546 CrossRef; (e) M. Potowski, F. Losch, E. Wunnemann, J. K. Dahmen, S. Chines and A. Brunschweiger, Chem. Sci., 2019, 10, 10481 RSC.
- (a) D. K. Kölmel, R. P. Loach, T. Knauber and M. E. Flanagan, ChemMedChem, 2018, 13, 2159 CrossRef; (b) J. Wang, H. Lundberg, S. Asai, P. Martín-Acosta, J. S. Chen, S. Brown, W. Farrell, R. G. Dushin, C. J. O'Donnell, A. S. Ratnayake, P. Richardson, Z. Liu, T. Qin, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6404 CrossRef CAS; (c) D. T. Flood, S. Asai, X. Zhang, J. Wang, L. Yoon, Z. C. Adams, B. C. Dillingham, B. B. Sanchez, J. C. Vantourout, M. E. Flanagan, D. W. Piotrowski, P. Richardson, S. A. Green, R. A. Shenvi, J. S. Chen, P. S. Baran and P. E. Dawson, J. Am. Chem. Soc., 2019, 141, 9998 CrossRef CAS; (d) J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723 CrossRef CAS; (e) S. O. Badir, J. Sim, K. Billings, A. Csakai, X. Zhang, W. Dong and G. A. Molander, Org. Lett., 2020, 22, 1046 CrossRef CAS; (f) Y. Ruff, R. Martinez, X. Pellé, P. Nimsgern, P. Fille, M. Ratnikov and F. Berst, ACS Comb. Sci., 2020, 22, 120 CrossRef CAS.
- (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS; (b) Z. Zhuang and J.-Q. Yu, Nature, 2020, 577, 656 CrossRef CAS.
- C. J. Gerry, M. J. Wawer, P. A. Clemons and S. L. Schreiber, J. Am. Chem. Soc., 2019, 141, 10225 CrossRef CAS.
- X. Wang, H. Sun, J. Liu, D. Dai, M. Zhang, H. Zhou, W. Zhong and X. Lu, Org. Lett., 2018, 20, 4764 CrossRef CAS.
- (a) G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 1506 CrossRef CAS; (b) P.-X. Shen, L. Hu, Q. Shao, K. Hong and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 6545 CrossRef CAS.
- (a) W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940 CrossRef CAS; (b) T. Liu, J. X. Qiao, M. A. Poss and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10924 CrossRef CAS.
- (a) R.-Y. Zhu, L.-Y. Liu, H. S. Park, K. Hong, Y. Wu, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 16080 CrossRef CAS; (b) R.-Y. Zhu, Z.-Q. Li, H. S. Park, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564 CrossRef CAS.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03935g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |