
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence‘Sacrificial’ supramolecular assembly and pressure-induced polymerization: toward sequence-defined functionalized nanothreads†
Margaret C.
Gerthoffer
 a,
Sikai
Wu
a,
Bo
Chen
ab,
Tao
Wang
c,
Steven
Huss
a,
Shalisa M.
Oburn
a,
Vincent H.
Crespi
acde,
John V.
Badding
acde and
Elizabeth
Elacqua
*ad
a,
Sikai
Wu
a,
Bo
Chen
ab,
Tao
Wang
c,
Steven
Huss
a,
Shalisa M.
Oburn
a,
Vincent H.
Crespi
acde,
John V.
Badding
acde and
Elizabeth
Elacqua
*ad
aDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: elizabeth.elacqua@psu.edu
bBaker Laboratory, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA
cDepartment of Physics, The Pennsylvania State University, University Park, PA 16802, USA
dMaterials Research Institute, The Pennsylvania State University, University Park, PA 16802, USA
eDepartment of Materials Science and Engineering, The Pennsylvania State University, University Park, PA 16802, USA
First published on 3rd September 2020
Abstract
Limited supramolecular strategies have been utilized to synthesize sequence-defined polymers, despite the prominence of noncovalent interactions in materials design. Herein, we illustrate the utility of ‘sacrificial’ aryl-perfluoroaryl supramolecular synthons to synthesize sp3-hybridized nanothreads from sp2-enriched reactants. Our strategy features A–B reactant pairs in the form of a phenol:pentafluorophenol co-crystal that is preorganized for an electronically-biased and sequence-defined polymerization. The polymerization, initiated at 12 GPa, affords an alternating copolymer featuring exogenous –OH functionalities. The external substitution is confirmed through IR spectroscopy. Importantly, the inclusion of the functional unit provides the first experimental glimpse at reaction mechanism: keto–enol tautomerization that can only occur during cycloaddition is observed through IR spectroscopy. Our approach realizes the first example of a functionalized nanothread and attains sequence definition through sacrificial supramolecular preorganization and presents a further approach for de novo design of complex nanothreads.
Introduction
Natural biopolymers (e.g., proteins and DNA) are architected through defined polymer sequences in concert with noncovalent interactions. Inspired by precision sequencing and supramolecular self-assembly in Nature, programming primary structure in synthetic polymers remains a holy grail for organic chemists.1 Methods that utilize differential kinetics of monomer reactivity,2,3 exploit enthalpic or entropic driving forces,4–6 or allow for preorganized monomer sequences to directly transfer to synthetic polymers garner much interest.7–9Despite the prominence of supramolecular organization and self-correction in natural biopolymer synthesis, noncovalent methods to transfer precise sequences directly to products remain underexplored, likely because the need to juxtapose reactants in an eclipsed manner necessitates tactically-precise supramolecular interactions that propagate parallel to the polymerization axis. One emerging method of exerting noncovalent control over molecular reactivity exploits the aryl/perfluoroaryl (Ar/ArF) supramolecular synthon.10,11 Ar/ArF sequences within co-crystals stack molecules in a face-to-face direction, and often are employed to promote a parallel preorganization of reacting π orbitals.11,12 Pioneering work by Grubbs11,13 used the Ar/ArF synthon to mediate covalent bond formation in the solid state. The Ar/ArF quadrupolar synthon has also been observed in the organization of synthetic β-sheet-like polymers17 to invoke chain collapse,18 as well as in co-crystalline and liquid crystalline materials design.10,13,19,20
Ar/ArF synthons10,21 have been used recently to achieve extended carbon solids15 in an effort to control the crystal structure for more uniform synthesis. In particular, a co-crystal containing naphthalene and octafluoronaphthalene was recently polymerized from eclipsed Ar/ArF stacks under pressure (Scheme 1B), resulting in a crystalline material comprising hydrogenated and fluorinated monomeric units.15 The resultant polymer, known as a carbon nanothread, has a one-dimensional poly(adamantane)-like structure featuring an sp3-rich backbone.14 Such sp3-based backbones are theorized to possess the strength of diamond,22 the chemical versatility of graphene, and large insulating band gaps.23 In contrast to simple molecules (e.g., benzene) under pressure, polymerization of this co-crystal produced ordered nanothreads even under fast compression. Despite an electronically-biased preorganization, the pressure needed for reaction was still quite high, ca. 28 GPa.
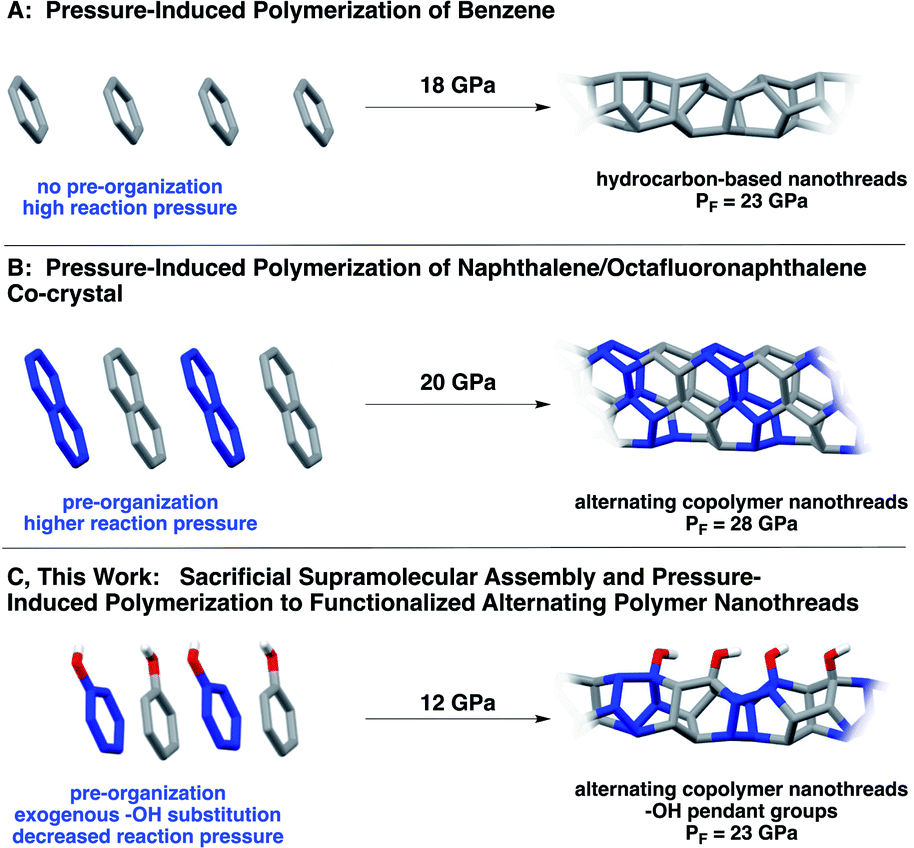 | ||
| Scheme 1 Synthesis of (A) benzene-derived nanothreads14 and (B) Ar/ArF-derived nanothreads.15 Approach (C) uses Ar–OH/ArF–OH co-crystals, wherein a functional group bias is introduced that, along with the sacrificial synthons, effectively decreases the required reaction pressure.16 | ||
Here, we report a functionalized nanothread arising from pressure-induced polymerization of an Ar/ArF co-crystal comprised of phenol and pentafluorophenol (Scheme 1). Our design features a hydroxy substituent, that, in addition to allowing close-packed π–π distances, accesses crystalline nanothreads at markedly lower pressures than those needed for supramolecular designs.15 The lower reaction pressure is attributed to the electronically activating –OH substituent on the ring system, which facilitates participation of the Ar component (phenol) as a diene to promote polymerization with a lower activation barrier. Importantly, compression of Ar–OH/ArF–OH provides an experimental glimpse at reaction mechanism: prominent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC character is observed through IR spectroscopy, suggestive of a keto–enol tautomerization that can only occur after a cycloaddition.
O and CC character is observed through IR spectroscopy, suggestive of a keto–enol tautomerization that can only occur after a cycloaddition.
Our motivation lies in developing precisely sequenced and functionalized nanothread polymers using a supramolecular process. In studies to attain alternating copolymer threads, we realized that ‘sacrificial’ supramolecular synthons would provide a strategic entry point toward A-B-type polymers. Given noncovalent interactions typically template solid-state reactions while remaining intact, we designate our design – the use of sp2-enriched Ar/ArF supramolecular synthons – as sacrificial owing to the solid-state design elements being consumed throughout the duration of the reaction. These synthons additionally guide the establishment of a well-defined sp3-rich polymer backbone.
Results and discussion
Our design required engineering a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 phenol:pentafluorophenol co-crystal to confirm orientation of the Ar/ArF units and exogenous –OH groups prior to compression. Thus, equimolar phenol and pentafluorophenol were crystallized from hexanes through slow cooling. Molecular and crystalline arrangement of the co-crystal was confirmed using single-crystal X-ray diffraction (XRD), in close similarity to a reported deuterated structure.24 As expected, the structure of the co-crystal is dictated by uniaxial π–π stacking quadrupole-type interactions of the Ar/ArF complex (Fig. 1),25 with the functional groups oriented to favor stable hydrogen bonds.21 The alternating arrangement of short π–π stacking distances are preorganized for an alternating copolymerization to occur. The co-crystal molecular structure is further guided by the presence of solely (Ar) O–H⋯O (ArF) and (ArF) O–H⋯O (Ar) hydrogen bonds between electronically opposed aryl groups (Ar/ArF). The hydrogen bonds (dO⋯O = 2.7–2.8 Å) exist entirely between adjacent stacks to form hexameric assemblies (Fig. 1b).
:1 phenol:pentafluorophenol co-crystal to confirm orientation of the Ar/ArF units and exogenous –OH groups prior to compression. Thus, equimolar phenol and pentafluorophenol were crystallized from hexanes through slow cooling. Molecular and crystalline arrangement of the co-crystal was confirmed using single-crystal X-ray diffraction (XRD), in close similarity to a reported deuterated structure.24 As expected, the structure of the co-crystal is dictated by uniaxial π–π stacking quadrupole-type interactions of the Ar/ArF complex (Fig. 1),25 with the functional groups oriented to favor stable hydrogen bonds.21 The alternating arrangement of short π–π stacking distances are preorganized for an alternating copolymerization to occur. The co-crystal molecular structure is further guided by the presence of solely (Ar) O–H⋯O (ArF) and (ArF) O–H⋯O (Ar) hydrogen bonds between electronically opposed aryl groups (Ar/ArF). The hydrogen bonds (dO⋯O = 2.7–2.8 Å) exist entirely between adjacent stacks to form hexameric assemblies (Fig. 1b).
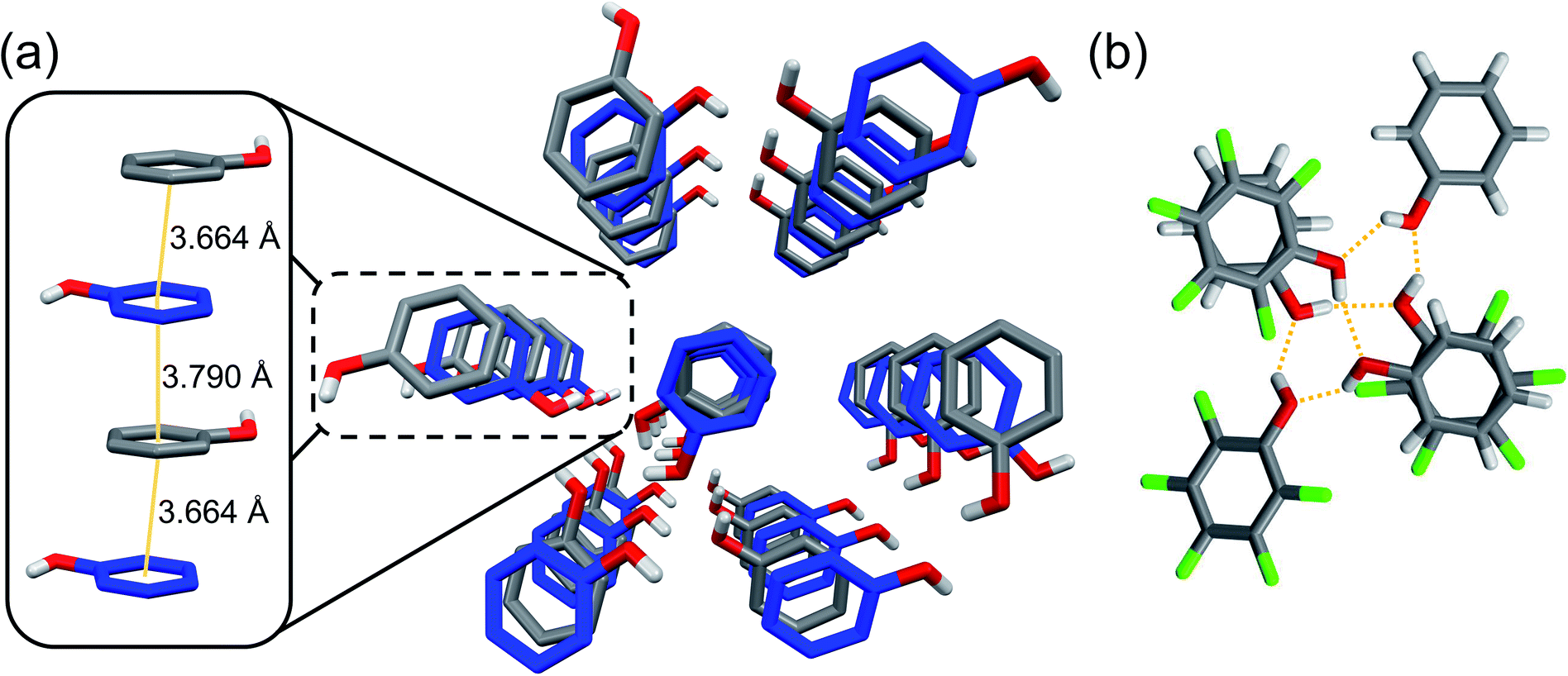 | ||
| Fig. 1 View of (a) Ar–OH (blue)/ArF–OH (grey) co-crystal along the a-axis with the left inset illustrating π–π stacking distance of an Ar/ArF slip-stack along the b-axis, and (b) hydrogen-bonding hexameric unit. | ||
Given high onset pressures ranging from 18–20 GPa for nanothreads derived from benzene and naphthalene/octafluoronaphthalene (Scheme 1), we sought to include an external substituent off of the ring that would realize a lower reaction pressure. We hypothesized that the phenol/pentafluorophenol co-crystal might electronically activate the aryl unit, thus favorably lowering the activation energies required for cycloaddition. The proposed co-crystal affords, in principle, 36 isomers of the [4 + 2] cycloaddition dimer, for which we analyzed the respective energies and activation barriers (see ESI†).26 With phenol acting as the diene, the average activation barrier is 59 kcal mol−1. When pentafluorophenol acts as the diene, the calculated activation barrier increases to 61 kcal mol−1. The determined activation barrier for our Ar/ArF co-crystal is significantly lower than that for benzene-based threads (68 kcal mol−1), suggestive of a lessened onset pressure. Interestingly, we found that the four lowest energy isomers suggest that the C–C bonds within a dimer are meta and para to the hydroxyl group of the ArF unit. These positions are the most likely to exhibit electron-rich character through resonance and are abundant in the precursor crystal as probable reaction sites.
Upon loading the co-crystal in a diamond anvil cell, the powder sample was slowly compressed and analyzed using in situ Raman microscopy (Fig. 2). At 5.9 GPa, we observed the appearance of peaks at 3160 cm−1, characteristic of sp3-hybridized C–H stretching and/or a phase-induced new sp2-hybridized C–H mode. At 5.9 GPa, we additionally noted a decrease in the ring breathing mode at 1007 cm−1, most likely owing to the formation of pressure-induced metastable adducts such as oligomeric threads present below 15 GPa.
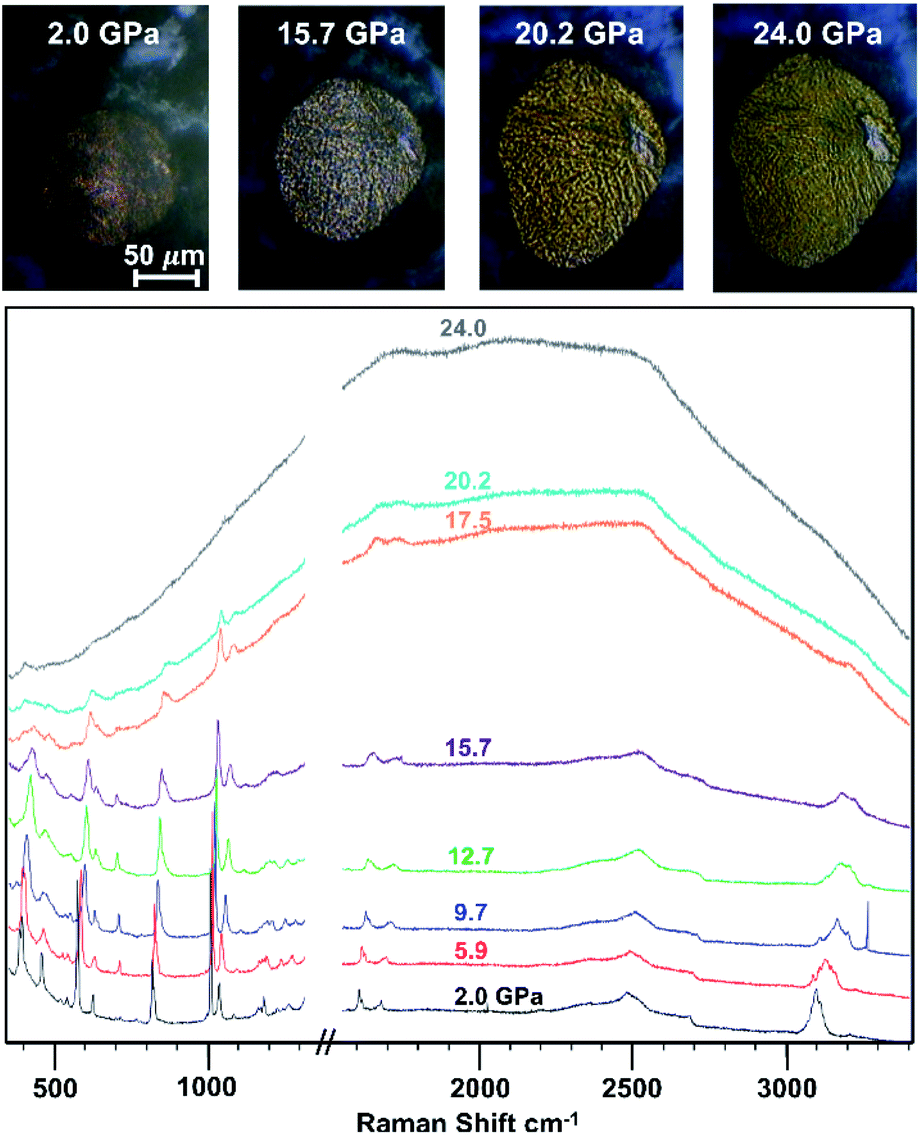 | ||
| Fig. 2
In situ Raman spectral overlay depicting 1:1 phenol:pentafluorophenol co-crystal throughout compression. The top images are taken with the Raman microscope during compression. | ||
Between 15.7 GPa and 17.5 GPa, further reaction occurred, as the sample began to appear yellow-orange, the photoluminescent (PL) background increased, and the gasket deformed (Fig. 2). In contrast, none of these signature characteristics were observed in compression attempts of the individual components (Fig. S2†). Instead, compression yielded mainly amorphous character, with higher reaction pressures being required to observe significant changes. In the co-crystal, an increase in PL background, change in C–H stretching modes, and decrease in ring breathing modes all occurred below 18 GPa. These combined observations confirmed that the lowered onset pressure in comparison to both benzene and the naphthalene:octafluoronaphthalene co-crystal can be attributed to the –OH functional groups favorably providing an electronic bias upon reaction.
The final product was analyzed through X-ray diffraction. The observed six-fold diffraction pattern (Fig. 3) suggests that nanothreads were formed with quasi-hexagonal packing. The diffraction pattern indicated d-spacings of 6.72 Å for one Friedel pair and 6.75 Å for two others.27 We attribute the distinct d-spacings to different orientations of the exogeneous hydroxyl groups around the thread axis. Since the width and subsequent packing of the Ar–OH/ArF–OH co-crystal thread is likely determined by the ArF–OH ring, the small difference in d-spacings is consistent with the ArF ring being approximately circular due to similar C–F and C–OH bond lengths; this supposition is examined further in the simulations of packing described below.
 | ||
| Fig. 3 (Left) Hexagonal six-fold X-ray diffraction pattern obtained for the functionalized nanothread; and (right) 50× images within a stainless-steel gasket, illustrating crystallinity along with (bottom) a polarized light micrograph using a 530 nm waveplate. | ||
A diffraction simulation of the co-crystal at ambient pressure along the Ar/ArF stacks illustrates a similar pattern in that two Friedel pairs have similar d-spacings, here 5.83 Å, with an additional d-spacing of 5.78 Å for the other. The increased d-spacing in the product suggests polymerization from an sp2 supramolecular-bound co-crystal composed of molecular rings to an sp3-rich architecture,16 necessitating an expansion due to rigid covalent constraints. In addition, the functionalized and crystalline nanothread product exhibits birefringence when viewed under polarized light (Fig. 3), suggestive of optical anisotropy.
In designing simulations of how functionalized nanothreads may pack, the combinatorial complexity of kinetic pathways (i.e., several possible [4 + 2] cycloaddition products) motivated a strategy of extracting general lessons from illustrative chiral and achiral thread categories. We therefore investigated the packings of nanothreads based on two archetypes: chiral polytwistane and achiral (3,0)-type28 backbone, both adapted to the alternating Ar–OH/ArF–OH-derived stack. These structures were packed into crystals and relaxed; d-spacings were then calculated (see ESI†). For both types, many isomers exist, reflecting different positions of the –OH groups for polytwistane, as well as eclipsed, gauche, and conserved hexameric hydrogen-bonding pattern arrangements for the (3,0)-type threads (Fig. 4).
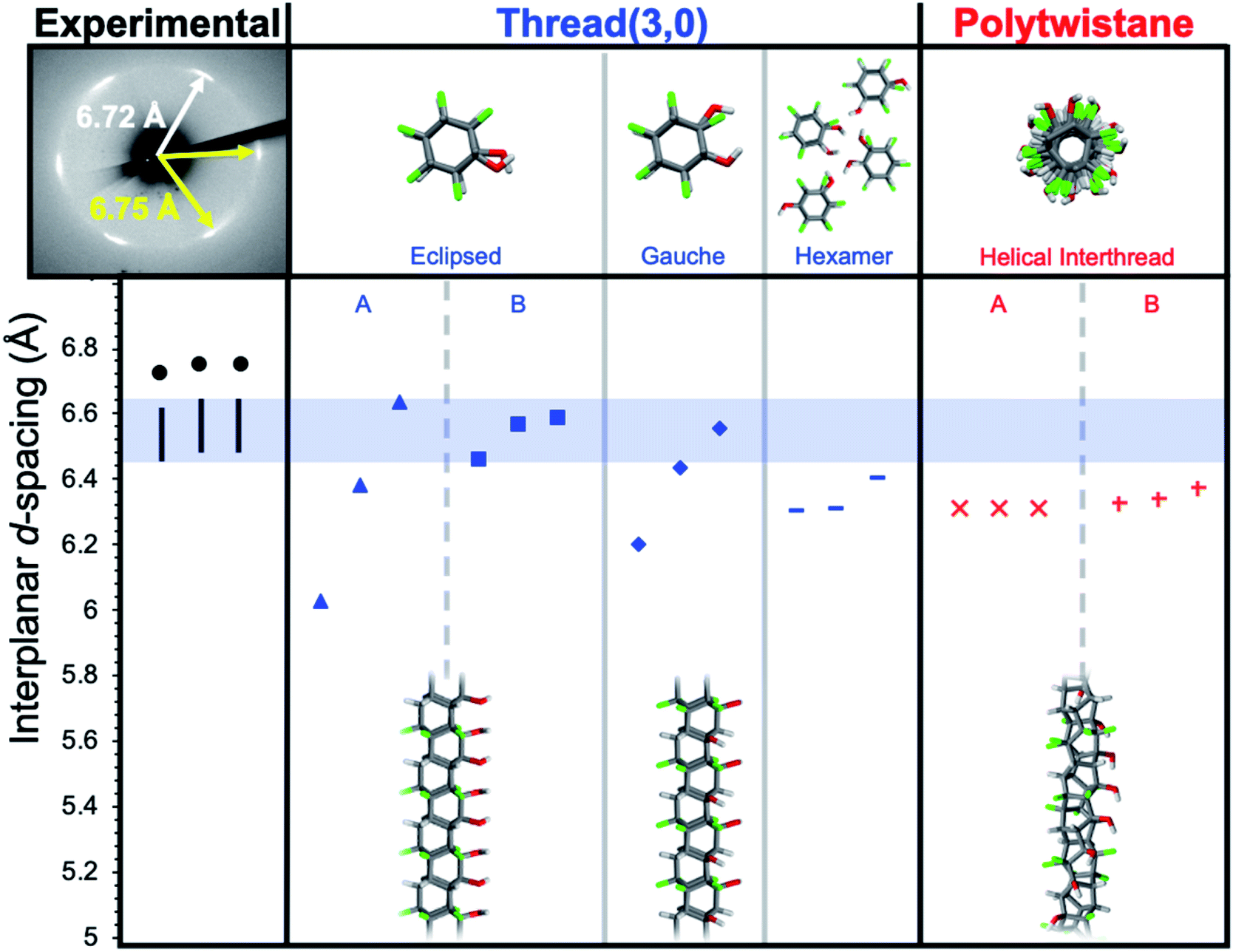 | ||
| Fig. 4 Comparison of interplanar d-spacings for simulated and experimental X-ray diffraction patterns along the co-crystal a-axis. Simulations account for 1–3% subtraction for thermal expansion. | ||
Before addressing the detailed results, we note that the ∼1.1 Å increase in d-spacing from benzene-derived nanothreads to the Ar–OH/ArF–OH-derived threads (i.e., from 5.6 Å to 6.7 Å) corresponds to an increase of about 1.3 Å in the interthread center-to-center separation. The change from C–H to C–F corresponds to a 0.24 Å increase in bond length and a 0.37 Å increase in the van der Waals radius of an F-atom compared to H. Doubling this ∼0.6 Å expansion yields a center-to-center dilation of 1.2 Å, as close to the observed 1.3 Å as would be expected for a rough empirical estimation. A chiral structure, such as the helical polytwistane, cannot easily assume a consistently anisotropic interthread hydrogen-bonding pattern, thus, the simulated d-spacings are closely grouped in magnitude.
Achiral structures such as thread (3,0), in contrast, can naturally assume either near-isotropic (e.g., eclipsed B) or anisotropic (e.g., eclipsed A, gauche) interthread bonding geometries. The experimental observation of a near-hexagonal symmetry with consistent anisotropy in d-spacings rules out an achiral structure with anisotropic inter-thread bonding such as eclipsed A or gauche (absent sufficient structural defects). The information available at present is not definitive enough to decide a specific thread backbone and substituent pattern therein.
Given the successful compression, we sought to understand more about the final nanothread molecular structure and elucidate any mechainstic details that might result from the –OH units being incorporated. We, thus, studied the IR spectroscopic features of the nanothread product in comparison to the starting co-crystal. Prior to compression, the experimental IR spectrum featured prominent modes (Table S5†), including a sharp and strong signal at 3418 cm−1 indicative of the O–H⋯O hydrogen bonding, and sp2 C–H stretching around 3200 cm−1 (Fig. 5, black trace). These modes were in agreement with a simulation of the co-crystal at ambient pressure (Fig. S4†).
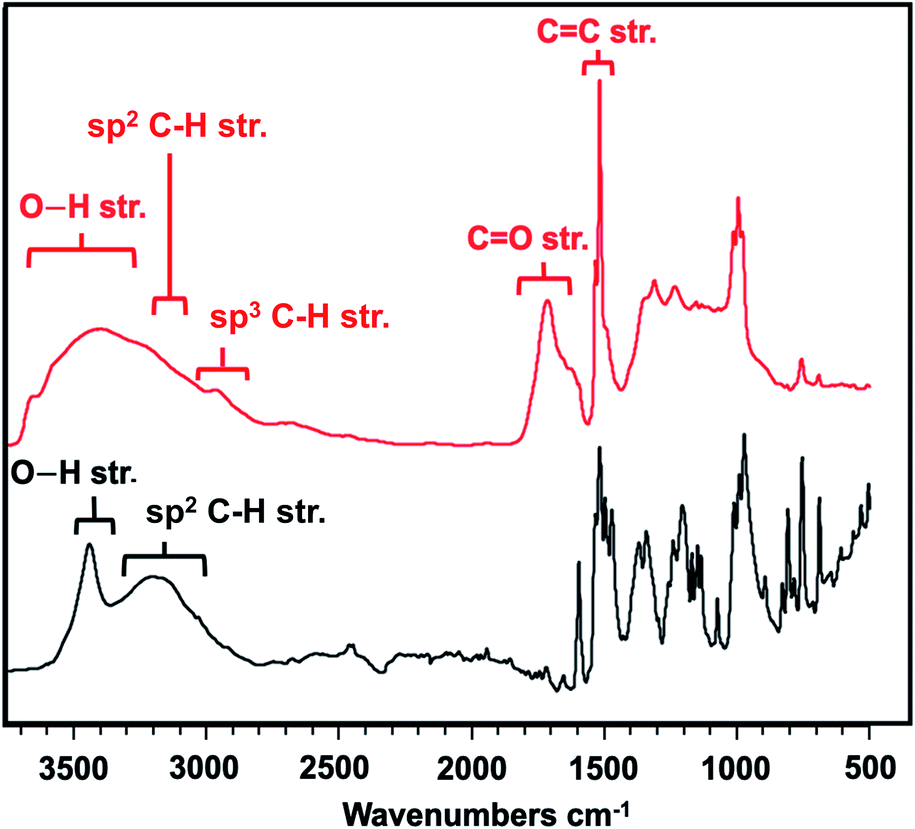 | ||
| Fig. 5 Comparison IR spectra of precursor co-crystal at ambient pressure (black) and final nanothread (red). | ||
In contrast, the IR spectrum of the final nanothread product (Fig. 5) highlighted a larger degree of hydrogen bonding, evidenced by the broadened O–H stretching between 3200–3700 cm−1. The nanothread product also featured sp3 C–H and sp2 C–H stretching centered around 2800 cm−1. Additional modes corresponding to both C–F and C–H bending and stretching in the region of 1000–1400 cm−1 were observed, as expected. In addition, the IR spectrum features a significant CO signal centered around 1700 cm−1, as well as a sharp CC stretching peak.
Whereas the CC signal could result from unreacted co-crystal, the two combined vibrational modes are more likely the result of rapid tautomerization of isolated enolic intermediates to their more prominent keto forms (Scheme 2). Each successful keto–enol tautomerization could, thus, turn an electronically-activated ‘hot’ diene into a deactivated, α,β-unsaturated ketone (akin to a dienophile) or an isolated alkene within the thread structure. Evidence of such a structural detail can also be observed in the IR spectrum. Signals around 1620 cm−1 are commonly observed in 1,3-diketone species wherein an enolic form predominates at one position,29,30 giving rise to a cyclic intramolecular hydrogen-bonding vibration. The strong shoulder on the CO stretching at 1621 cm−1 could suggest the analogous diketone pattern that can arise in proximal rings upon cycloaddition to favor the formation of an enolic H-bond donor within the thread backbone (Scheme 2). Given these signatures of tautomerization are only plausible after an addition reaction that retains some degree of sp2 character, the IR spectral features – combined with the lowering of reaction pressure through installation of a functional group able to induce polarization – suggest that a [4 + 2] Diels–Alder-like cycloaddition is a key step within initiation/propagation events leading to nanothread formation.
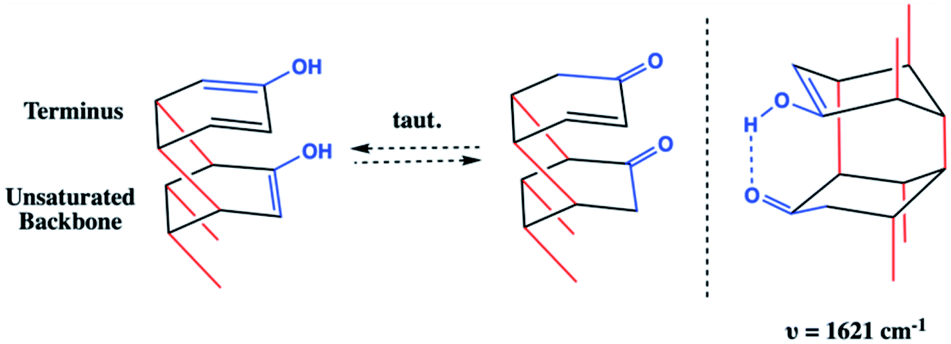 | ||
| Scheme 2 Schematic depicting representative cycloadducts capable of (left) facilitating keto–enol tautomerization and (right) intramolecular enol-assisted hydrogen-bonding. H- and F-atoms have been removed for clarity. | ||
Conclusions
In conclusion, we demonstrate that a functional Ar/ArF-based co-crystal undergoes an electronically-activated pressure-induced polymerization and results in a well-defined crystalline nanothread polymer, as evidenced by a hexagonal X-ray diffraction pattern. Our investigations have highlighted three salient points: (i) the thread-forming polymerization is tolerant of exogenous functional groups, such as –OH; (ii) the combination of ‘sacrificial’ supramolecular assembly and intact hydrogen bonding leads to a lower activation barrier for the reactants; and (iii) isolation of the product highlights key features: the presence of CO in concert with intense CC signals in the IR spectrum suggests tautomerization of –OH groups after a [4 + 2] cycloaddition as a mechanistic lynchpin in nanothread formation. The combined results suggest that the polymerization is influenced by electronic substituents which act to reduce energetic barriers for initiation of the cycloaddition-based polymerization, thus lowering the required reaction pressure.31 Future work will look to pinpoint the effect of electronic bias on polymerization to gain further insights into mechanism, functional group tolerance, and precise theoretical structure elucidation to provide the intuition for designs amenable to post-polymerization functionalization.32
Experimental section
Synthesis of functionalized nanothreads
Compression of the Ar/ArF co-crystal was conducted in a symmetric diamond anvil cell (DAC) with a 400 μm culet while monitoring the pressure-induced polymerization using in situ Raman spectroscopy. The solid co-crystal was cooled in the fridge, ground using a mortar and pestle, and placed on the lower diamond within a stainless-steel gasket. A ruby chip was added to the sample chamber to monitor the pressure through fluorescence. The DAC was closed to form a powder sample by applying 2.0 GPa of pressure. A typical compression proceeded over the course of 16 hours to a maximum pressure of 23 GPa. The compression rate slowed upon the incidence of a photoluminescent (PL) background that indicated polymerization occurring at around 11 GPa to 2–3 GPa h−1. The compression rate was slowed further above 17 GPa to less than 1 GPa h−1. Decompression progressed over the course of 16–18 hours with similar rates on the descent.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Center for Nanothread Chemistry, a National Science Foundation (NSF) Center for Chemical Innovation (CHE-1832471). We acknowledge Prof. Roald Hoffmann for helpful insights and discussions throughout the project and construction of this manuscript. We sincerely thank the HPCAT beamline staff (particularly Dr Changyong Park, Dr Dmitry Popov, and Dr Jesse Smith) at the Advanced Photon Source in Argonne National Laboratory for their guidance at 16-BM-D. We thank Hemant Yennawar for his assistance in collection of the co-crystal X-ray diffraction. The Materials Characterization Laboratory in the Materials Research Institute at the Pennsylvania State University was used to collect Infrared, as well as XPS with the assistance of Jeffery Shallenberger. We acknowledge the computing resource provided by the Extreme Science and Engineering Discovery Environment (XSEDE) Comet cluster at the San Diego Supercomputer Center through allocation CHE180059.References
- J.-F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- S. Pfeifer and J.-F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS.
- D. Moatsou, C. F. Hansell and R. K. O'Reilly, Chem. Sci., 2014, 5, 2246–2250 RSC.
- W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2015, 137, 8038–8041 CrossRef CAS.
- B. R. Elling, J. K. Su, J. D. Feist and Y. Xia, Chem, 2019, 5, 2691–2701 CAS.
- J. A. Nowalk, C. Fang, A. L. Short, R. M. Weiss, J. H. Swisher, P. Liu and T. Y. Meyer, J. Am. Chem. Soc., 2019, 141, 5741–5752 CrossRef CAS.
- E. Elacqua and M. Gregor, Angew. Chem., Int. Ed., 2019, 58, 9527–9532 CrossRef CAS.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- J. W. Lauher, F. W. Fowler and N. S. Goroff, Acc. Chem. Res., 2008, 41, 1215–1229 CrossRef CAS.
- M. D. Blanchard, R. P. Hughes, T. E. Concolino and A. L. Rheingold, Chem. Mater., 2000, 12, 1604–1610 CrossRef CAS.
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 15, 3641–3649 CrossRef.
- M. A. Sinnwell, J. Baltrusaitis and L. R. MacGillivray, Cryst. Growth Des., 2015, 15, 538–541 CrossRef CAS.
- M. Weck, A. R. Dunn, K. Matsumoto, G. W. Coates, E. B. Lobkovsky and R. H. Grubbs, Angew. Chem., Int. Ed., 1999, 38, 2741–2745 CrossRef CAS.
- T. C. Fitzgibbons, M. Guthrie, E. S. Xu, V. H. Crespi, S. K. Davidowski, G. D. Cody, N. Alem and J. V. Badding, Nat. Mater., 2015, 14, 43–47 CrossRef CAS.
- M. D. Ward, W. S. Tang, L. Zhu, D. Popov, G. D. Cody and T. A. Strobel, Macromolecules, 2019, 52, 7557–7563 CrossRef CAS.
- In Scheme 1, PF = (lowest) final pressure needed to synthesize crystalline product.
- E. Elacqua, G. T. Geberth, D. A. Vanden Bout and M. Weck, Chem. Sci., 2019, 10, 2144–2152 RSC.
- J. Lu, N. ten Brummelhuis and M. Weck, Chem. Commun., 2014, 50, 6225–6227 RSC.
- C. R. Patrick and G. S. Praser, Nature, 1960, 187, 1021 CrossRef CAS.
- A. Schwarzer and E. Weber, Cryst. Growth Des., 2014, 14, 2335–2342 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Primagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- R. E. Roman, K. Kwan and S. W. Cranford, Nano Lett., 2015, 3, 1585 CrossRef.
- H. Huang, L. Zhu, M. D. Ward, T. Wang, B. Chen, B. L. Chaloux, Q. Wang, A. Biswas, J. L. Gray, B. Kuei, G. D. Cody, A. Epshteyn, V. H. Crespi, J. V. Badding and T. A. Strobel, J. Am. Chem. Soc., 2020 DOI:10.1021/jacs.9b12352.
- As previously reported in a resolution of a deuterated phenol and pentafluorophenol co-crystal, refinement of the hydroxyl position about the pentafluorophenol ring can prove difficult due to the similar bond lengths of C–F and C–OH of 1.33 Å and similar electron density of F and O. Previous studies with neutron diffraction resolved the hydrogen bonding array to reveal a six-component complex.
- J. E. Bertie and P. Tremaine, J. Chem. Phys., 1973, 58, 854 CrossRef CAS.
- We define the activation barrier to be the necessary energy to initiate formation of an sp3-hybridized carbon, typically through dimer formation, concomitantly exposing a reactive diene.
- A powder CeO2 standard was utilized before diffraction collection and witnessed a FWHM of 0.3 Å. We thus have reasoned that the 6.72 Å and 6.75 Å d-spacings are not symmetrically equivalent, but we cannot rule out that the two recorded 6.75 Å Friedel pairs are or are not symmetrically equivalent to one another by analytical precision.
- E.-S. Xu, P. E. Lammert and V. H. Crespi, Nano Lett., 2015, 15, 5124–5130 CrossRef CAS.
- N. V. Belova, H. Oberhammer, X. Zeng, M. Gerken, H. Willner, R. J. F. Berger, S. A. Hayes and N. W. Mitzel, Phys. Chem. Chem. Phys., 2010, 12, 11445–11453 RSC.
- J. C. Sloop, C. L. Bumgardner, G. Washington, W. D. Loehle, S. S. Sankar and A. B. Lewis, J. Flourine Chem., 2006, 127, 780–786 CrossRef CAS.
- Reduction of the reaction pressure would allow for the use of pressure apparati with larger volume output. Diamond anvil cells (DACs), such as those used in these studies, operate with a sample area eqating to 100 μm, thus affording nanograms of material. The lessened amount of product from DACs precludes conventional analysis including TGA and DSC, as well as post-polymerization functionalization.
- Our current efforts are focused on scalable syntheses (e.g., by using a Paris–Edinburgh press that can afford mg quantities) to further interrogate thermal properties and post-synthetic modification.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details including ambient pressure co-crystal XRD, high pressure experimental setup, compression of individual components, and product characterization details on Raman spectroscopy, IR spectroscopy, XPS, polarized light microscopy, synchrotron XRD, theoretical details including theoretical co-crystal IR spectrum, [4 + 2] barrier calculations, enumeration of thread structures, and calculated XRD on thread structures. CCDC 1980178. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03904g |
| This journal is © The Royal Society of Chemistry 2020 |