DOI:
10.1039/D0SC03858J
(Edge Article)
Chem. Sci., 2020,
11, 9884-9890
Effects of ruthenium hydride species on primary amine synthesis by direct amination of alcohols over a heterogeneous Ru catalyst†
Received
15th July 2020
, Accepted 24th August 2020
First published on 3rd September 2020
Abstract
Heterogeneously catalysed synthesis of primary amines by direct amination of alcohols with ammonia has long been an elusive goal. In contrast to reported Ru-based catalytic systems, we report that Ru–MgO/TiO2 acts as an effective heterogeneous catalyst for the direct amination of a variety of alcohols to primary amines at low temperatures of ca. 100 °C without the introduction of H2 gas. The present system could be applied to a variety of alcohols and provides an efficient synthetic route for 2,5-bis(aminomethyl)furan (BAMF), an attention-getting biomonomer. The high catalytic performance can be rationalized by the reactivity tuning of Ru–H species using MgO. Spectroscopic measurements suggest that MgO enhances the reactivity of hydride species by electron donation from MgO to Ru.
Introduction
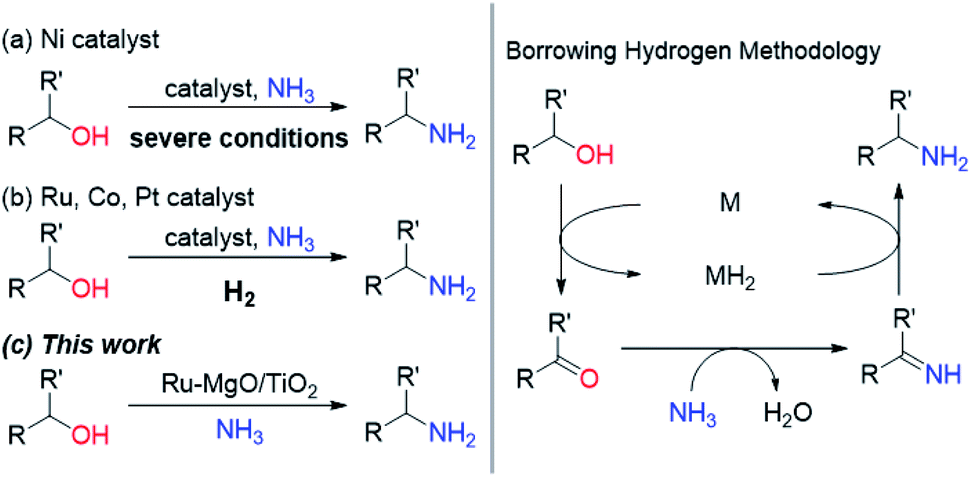
Primary amines are known as versatile building blocks due to their convenient use in different fields of synthetic chemistry and serve as synthons for pharmaceuticals, agricultural chemicals and polymers.1 Thus, a variety of synthetic methods to produce primary amines have been developed including the alkylation of ammonia, and reduction of amides, nitriles and nitro compounds, in addition to special methods (e.g., Staudinger reduction and Hofmann degradation).2 The direct substitution of alcohols with ammonia is considered to be the most rational synthetic protocol for primary amines because of the ready-availability of alcohols, and the low cost of ammonia, and it is attractive in terms of it being environmentally benign. Due to the low leaving-group ability of the hydroxy group, recent attempts have been devoted to the borrowing hydrogen (BH) methodology.3 This system uses a powerful strategy that combines dehydrogenation, transformation of generated unsaturated compounds, and subsequent hydrogenation to form more complex molecules, without the need for tedious separation or isolation processes. Since the pioneering work with a Ru complex bearing a PNP pincer ligand by Gunanathan and Milstein,4 some Ru- and Ir-based homogeneous catalysts have been developed for the selective synthesis of primary amines by the direct amination of alcohols with ammonia.5 In view of the advantages of heterogeneous catalysts, such as ease of recovery, reusability, and stability, there is a substantial need for the development of efficient heterogeneous catalysts that enable selective conversion of alcohols into amines under mild reaction conditions for the environmentally benign production of chemicals. Although some Ni-based heterogeneous catalysts have been developed,6 these systems require severe reaction conditions (Scheme 1 and Table S1†). Ru-, Co- and Pt-based catalysts are also active for the direct amination of alcohols;7–9 however, these catalysts require molecular hydrogen, which is not required in principle. Besides the possible explanation for H2 effects (regeneration of the active metal surface and the removal of coke),7a,b we propose that additional H2 prevents H2-release from the metal hydride species generated through the dehydrogenation of alcohols. Therefore, we have envisioned that electronic tuning of the ruthenium hydride species would result in the development of efficient catalysts for the direct amination of alcohols through a BH strategy. Despite the importance of metal hydride species in many hydrogen-involving reactions, the characterization of metal hydride species is still a challenge for heterogeneous catalysts.10 We have focused on a basic support that can sufficiently stabilize Ru hydride species enough to require hydrogen acceptors for the dehydrogenative oxidation of alcohols catalysed by a supported Ru catalyst.11 In this study, we report a supported Ru catalyst for basic oxide-enabled direct amination of alcohols with ammonia under mild reaction conditions without molecular hydrogen as an accelerator.
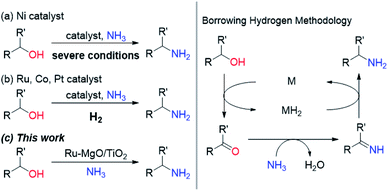 |
| | Scheme 1 Selective synthesis of primary amines by direct amination of alcohols and ammonia over heterogeneous catalysts through borrowing hydrogen methodology. | |
Results and discussion
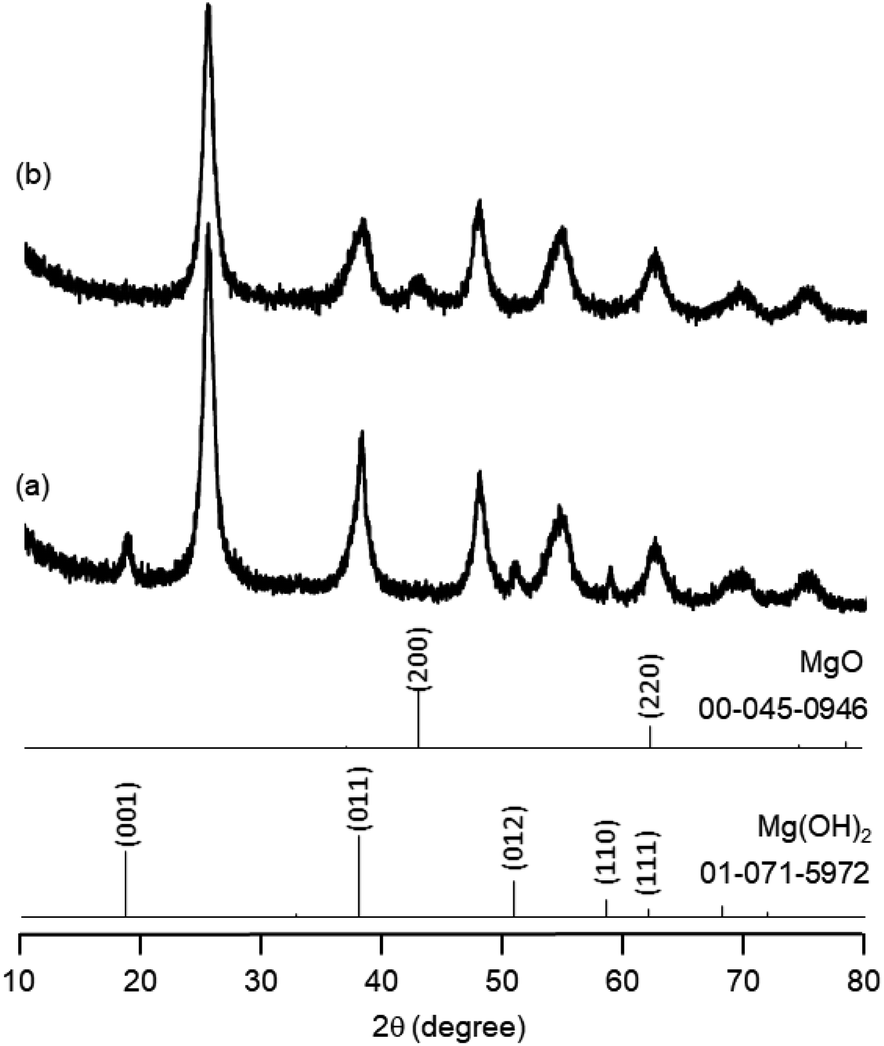
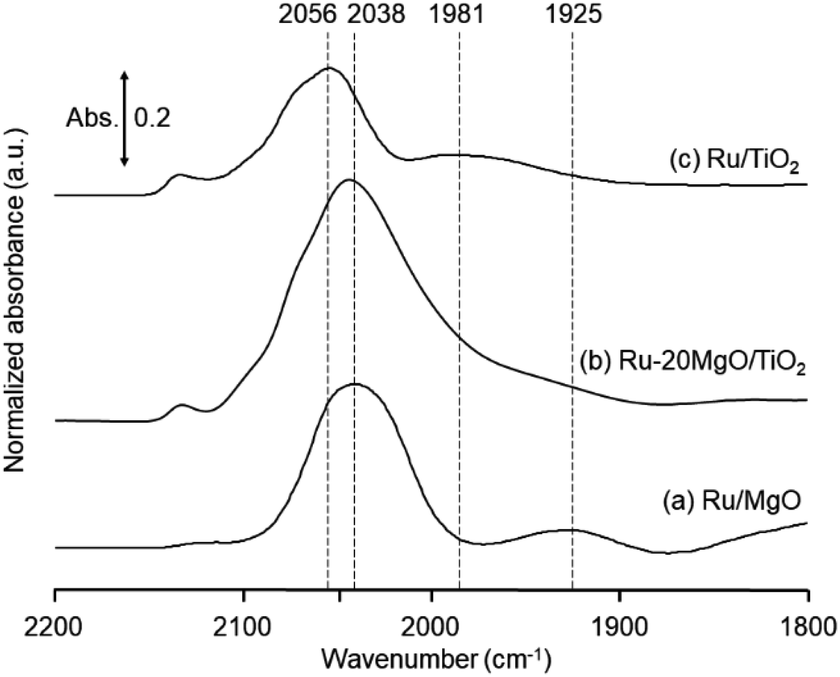
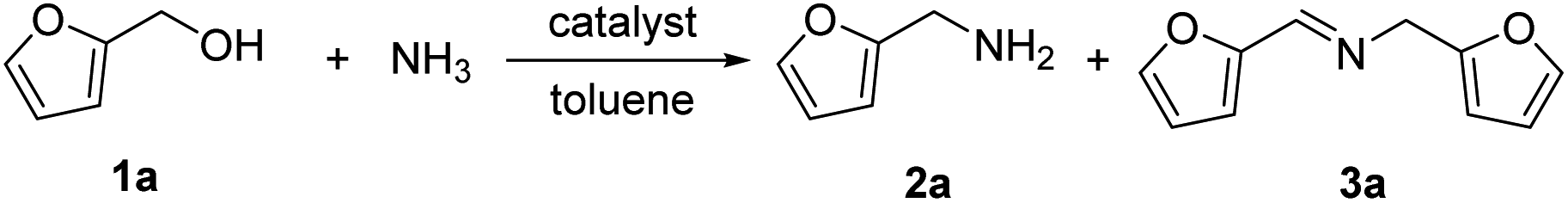
The research began with the direct amination of furfuryl alcohol (1a) using Ru catalysts supported on simple basic metal oxides and hydroxides (Table S2†). Ru/MgO gave the desired primary amine in 77% yield; however, this catalyst could not be reused even after reduction treatment. To achieve more active and stable catalysts, we examined the deposition of Ru and Mg on high-surface-area supports. A Ru and alkaline earth metal co-deposited metal oxide supported catalyst (Ru–xM/support; x = M loading (wt%)) was prepared through the sequential deposition of Ru and an alkaline earth metal on the support by the wet-impregnation method. X-ray diffraction (XRD) analysis was conducted for Ru–20MgO/TiO2 before and after hydrogen reduction (Fig. 1). Mg(OH)2 was observed in addition to the TiO2 (anatase) support before reduction. Reduction treatment of the sample resulted in the formation of MgO. We estimated the average size of MgO nanoparticles on TiO2 from the XRD patterns. The grain size of MgO in Ru–MgO/TiO2 estimated from the (200) diffraction lines using Scherrer's equation was 25 nm. This value is good agreement with the particle size 20–50 nm measured from the field-emission scanning electron microscope (FE-SEM) images of Ru–20MgO/TiO2 (Fig. S1†). Peaks of the Ru species were not observed in the XRD patterns, possibly due to the high dispersion of the Ru species on the support. Transmission electron microscopy-energy dispersive X-ray spectroscopy (TEM-EDS) analysis of Ru–MgO/TiO2 indicated that Ru and Mg were highly dispersed on the TiO2 support (Fig. 2). To obtain information on the environment around Ru particles, the electronic states of Ru particles were examined with CO- adsorption infrared (IR) spectroscopy measurements (Fig. 3). The bands assignable to the stretching vibration of CO adsorbed on Ru can be shifted by the electron-donating character of the Ru particles.12 A broad band (2038 cm−1) with a shoulder (1925 cm−1) was observed in the spectrum for Ru–20MgO/TiO2, and these features were assigned to linearly adsorbed CO on partially oxidized Ru and CO bridged between two metallic Ru atoms, respectively.13 X-ray photoelectron spectroscopy (XPS) measurements for Ru 3d of the catalyst showed that Ru0 coexists with Ru4+ (Fig. S2†), which was consistent with the Fourier transform infrared spectroscopy (FT-IR) results. For Ru/TiO2, both bands appeared at higher wavenumber positions (2056 and 1981 cm−1), which indicated that Ru on Ru–20MgO/TiO2 has higher electron-donating capability than Ru on Ru/TiO2. Fig. 3 also shows that there is no significant difference in the band position between Ru–MgO/TiO2 and Ru/MgO, which suggests that Ru is in contact with MgO rather than TiO2 on Ru–MgO/TiO2, and the electron donation from MgO, a strong electron-donating material, to Ru enhances the electron-donating capability of Ru on Ru–MgO/TiO2. We measured amounts of acid and base sites of Ru–20MgO/TiO2 and Ru/TiO2 by pyridine-adsorbed IR measurements and CO2 temperature-programmed desorption (CO2-TPD) (Table S4†). Ru–20MgO/TiO2 possesses higher amounts of acid sites than Ru/TiO2 probably due to the higher surface area of Ru–MgO/TiO2. Although there was no significant difference in the amounts of base sites between Ru–MgO/TiO2 and Ru/TiO2, the CO2 desorption peaks around 250 °C of Ru/TiO2 shifted to higher temperatures (450–650 °C) in the case of Ru–MgO/TiO2 (Fig. S3†). Thus, the basicity of Ru–MgO/TiO2 would be stronger than that of Ru/TiO2, which would result in the acceleration of the alcohol dehydrogenation step.
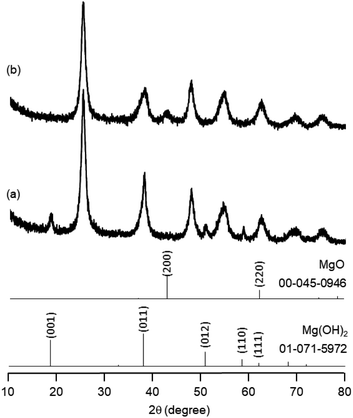 |
| | Fig. 1 XRD patterns for Ru–20MgO/TiO2 before and after reduction. | |
 |
| | Fig. 2 TEM-EDS analysis of Ru–20MgO/TiO2. | |
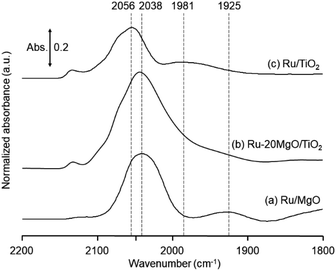 |
| | Fig. 3 Difference IR spectra of CO-adsorbed Ru/MgO, Ru–20MgO/TiO2, and Ru/TiO2. | |
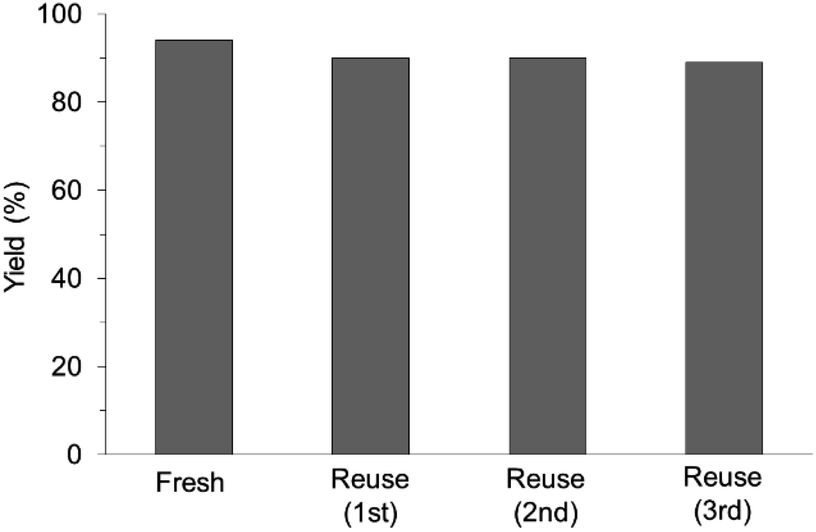
The prepared catalysts were evaluated with respect to the direct amination of 1a with ammonia in toluene at 110 °C for 20 h (Table 1). Ru–20MgO/TiO2 exhibited high catalytic performance to give furfurylamine (2a) in 85% yield with a trace amount of the imine 3a (entry 1). No other side products were observed in the reaction mixture. The turnover frequency (TOF) value is calculated based on the Ru dispersion to be 0.6 h−1. This value is lower than those of reported Ru catalysts (Table S1†) due to the low reaction temperature (110 °C for Ru– MgO/TiO2vs. 150–220 °C for Ru/C, Ru/SiO2, Ru/Al2O3, and RuNPs); however, the reported Ru catalysts showed low selectivity to primary amines in contrast to the present Ru– MgO/TiO2 system. The effect of the support was then examined (entries 4–7). Low yields of 2a were observed for other oxide supports, such as SiO2, Al2O3, ZrO2, and Nb2O5. There was no correspondence between the activity and support features, such as acid/base sites and electron-donating character. Mg loading had a high impact on the catalytic activity of the Ru catalysts. The deposition of lower and higher amounts of MgO resulted in a decrease of the yield of 2a (entries 8 and 9). Other alkaline earth metals were not effective for the direct amination of alcohol (entries 10–12). Ru/TiO2 also exhibited catalytic activity; however, only a 38% yield of 2a was obtained (entry 13). In addition, the result of a physical mixture of Ru/TiO2 and MgO was similar to that of Ru/TiO2 (entry 14), which indicates that direct interaction between Ru and Mg is essential in achieving high activity for the direct amination of alcohols. Ru/Nb2O5 was not effective for the direct amination although it acts as a highly active and selective catalyst for reductive amination (entry 15).14 No reaction was observed when the reaction setup was assembled on a bench outside the glovebox (entry 2). After further optimization studies including solvent screening and the catalyst loading effect (Table S5†), the use of 0.4 g catalyst increased the yield of 2 to 94% (entry 3). Fig. 4 shows the results of the reuse experiment. After base treatment with NaOH and subsequent reduction with H2 at 400 °C, Ru–20MgO/TiO2 could be reused three times without loss of activity.
Table 1 Catalyst screeninga
|
|
| Entry |
Catalyst |
Conv. (%) |
Yield (%) |
|
2a
|
3a
|
|
Reaction conditions: catalyst (0.2 g), 1a (0.5 mmol), pNH3 (0.7 MPa), toluene (5 mL), 110 °C, and 20 h. Conversion and yield were determined by GC analysis. Yield (%) = product (mol)/initial 1a (mol) × 100.
Operated in air.
0.4 g catalyst was used.
0.2 g Ru/TiO2 and 0.04 g MgO were used as the catalyst.
Run at 180 °C.
|
| 1 |
Ru–20MgO/TiO2 |
96 |
85 |
Trace |
| 2b |
Ru–20MgO/TiO2 |
18 |
0 |
0 |
| 3c |
Ru–20MgO/TiO2 |
>99 |
94 |
0 |
| 4 |
Ru–20MgO/SiO2 |
99 |
32 |
0 |
| 5 |
Ru–20MgO/Al2O3 |
99 |
59 |
0 |
| 6 |
Ru–20MgO/ZrO2 |
76 |
24 |
0 |
| 7 |
Ru–20MgO/Nb2O5 |
43 |
17 |
0 |
| 8 |
Ru–10MgO/TiO2 |
82 |
42 |
0 |
| 9 |
Ru–30MgO/TiO2 |
87 |
52 |
0 |
| 10 |
Ru–20CaO/TiO2 |
91 |
44 |
0 |
| 11 |
Ru–20SrO/TiO2 |
79 |
58 |
0 |
| 12 |
Ru–20BaO/TiO2 |
90 |
44 |
0 |
| 13 |
Ru/TiO2 |
72 |
38 |
Trace |
| 14 |
Ru/TiO2 + MgOd |
72 |
35 |
0 |
| 15e |
Ru/Nb2O5 |
46 |
2 |
0 |
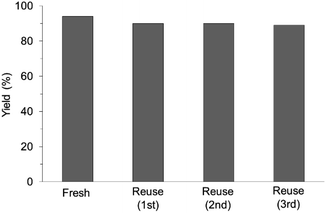 |
| | Fig. 4 Reuse experiments. Reaction conditions: Ru–20MgO/TiO2 (0.4 g), 1a (0.5 mmol), toluene (5 mL), pNH3 (0.7 MPa), 110 °C, and 20 h. | |
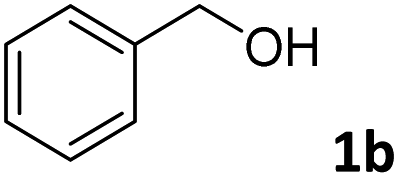
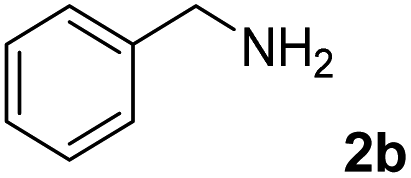
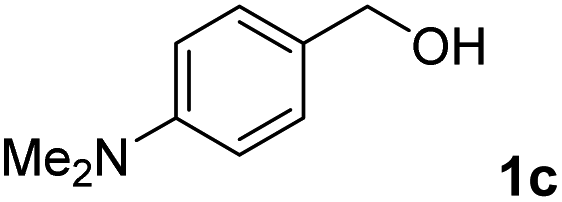
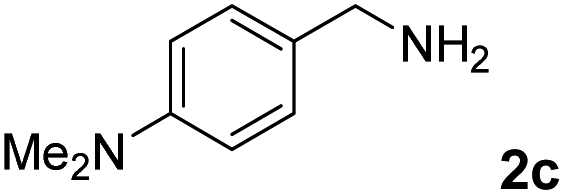
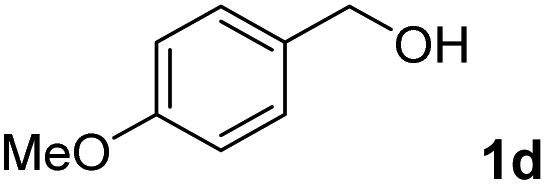
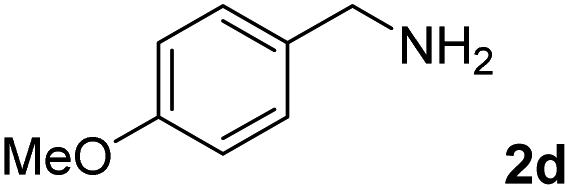
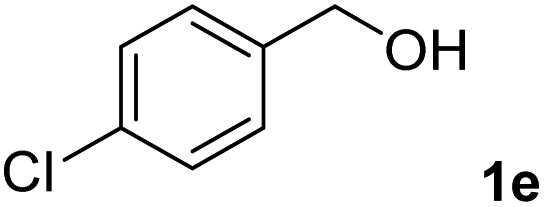
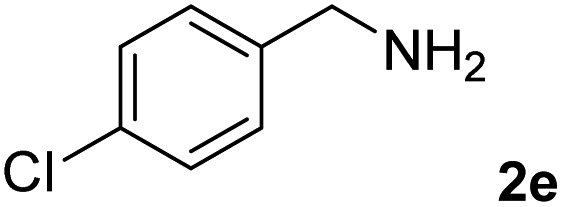
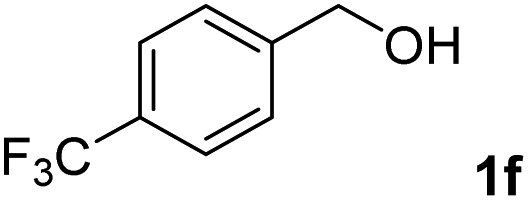
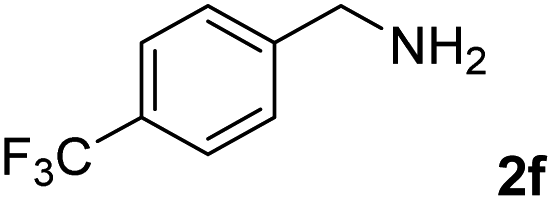
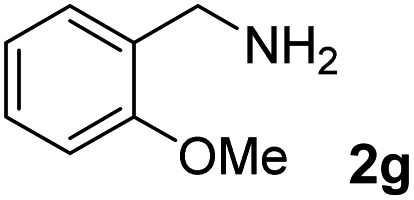
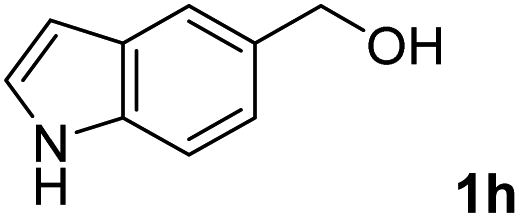
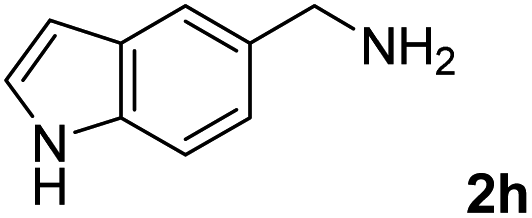
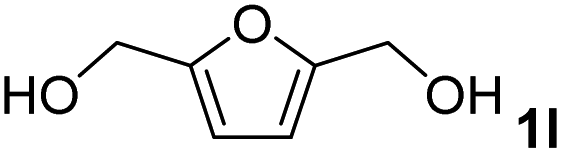
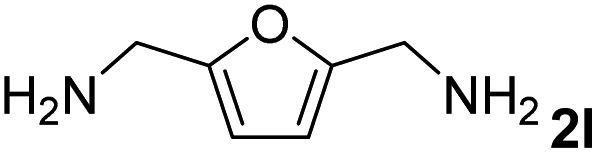
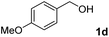
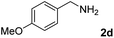
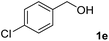
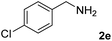
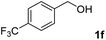
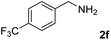
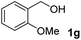
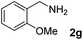
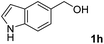
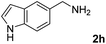
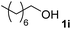
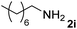
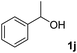
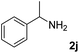
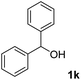
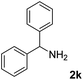
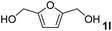
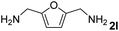
The present catalytic system could be applied to a variety of alcohols (Table 2). Electronic and steric effects were examined using substituted benzyl alcohol. The direct amination proceeded smoothly, regardless of the substituents at the para position, to give the corresponding primary amines in good to excellent yields (entries 2–5). A sterically hindered substrate is also applicable with this catalytic system (entry 6). In addition, the catalytic system has high functional group tolerance. The chloro group and indole skeleton remained intact during the reaction. The present Ru system could be applied to not only benzyl alcohol derivatives, but also to linear alkyl alcohols (entry 8). Secondary alcohols are also good substrates for direct amination with this catalyst system (entries 9 and 10). The product was obtained in moderate yield due to the formation of dehydroxylated products. Finally, the catalytic system was applied to the synthesis of 2,5-bis(aminomethyl)furan (BAMF), which is known as a biomonomer for polyamides and a hardener for epoxy resins.15 When 2,5-bis(hydroxymethyl)furan (BHMF) was used as a substrate, the desired BAMF was obtained in 86% yield (entry 11). Compared with the reported methods for the synthesis of BAMF,16 the present system has superiority in that a high yield of BAMF is produced under mild reaction conditions.
| Entry |
Substrate |
Product |
Yield (%) |
|
Reaction conditions: Ru–20MgO/TiO2 (0.2 g), 1 (0.5 mmol), pNH3 (0.7 MPa), toluene (5 mL), 110 °C, and 20 h.
Run at 100 °C.
Run for 30 h.
|
| 1b |
|
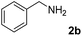
|
91 |
| 2 |
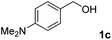
|
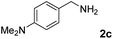
|
82 |
| 3c |
|
|
89 |
| 4 |
|
|
72 |
| 5 |
|
|
76 |
| 6 |
|
|
81 |
| 7 |
|
|
67 |
| 8 |
|
|
78 |
| 9 |
|
|
61 |
| 10 |
|
|
67 |
| 11 |
|
|
86 |
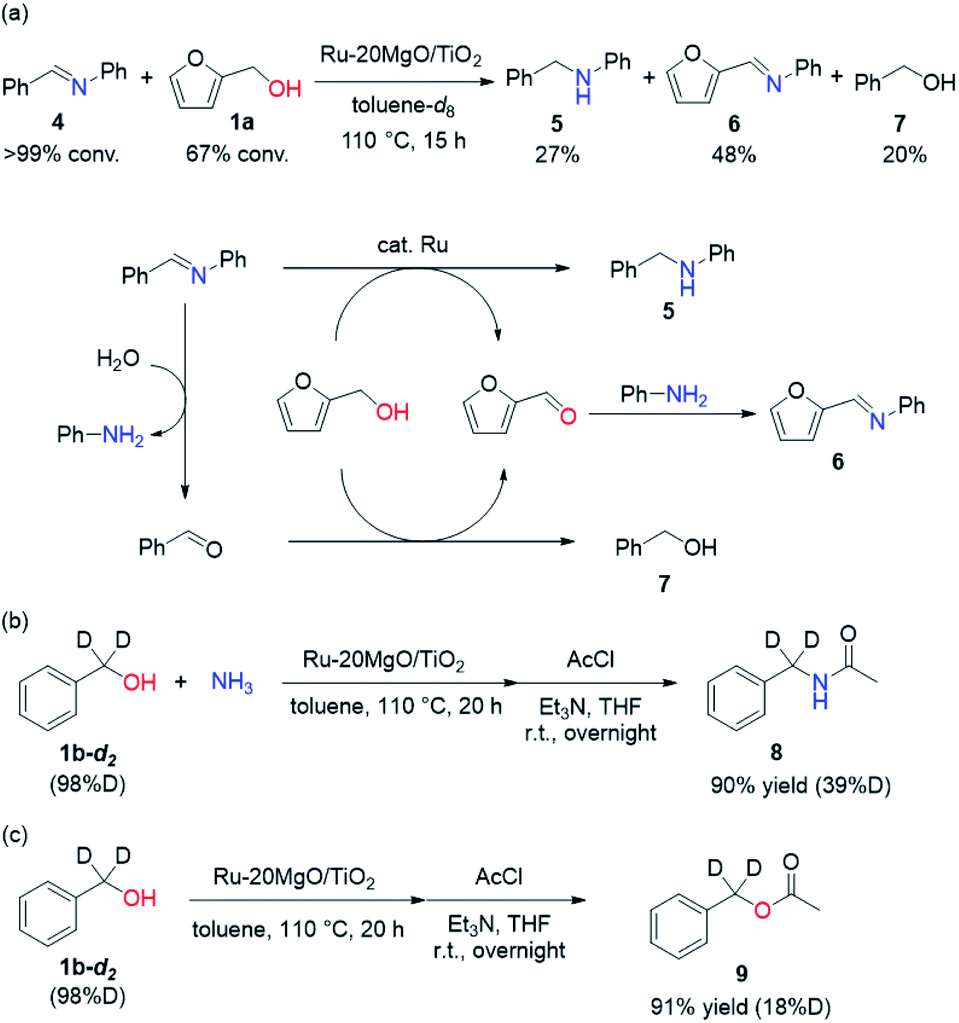
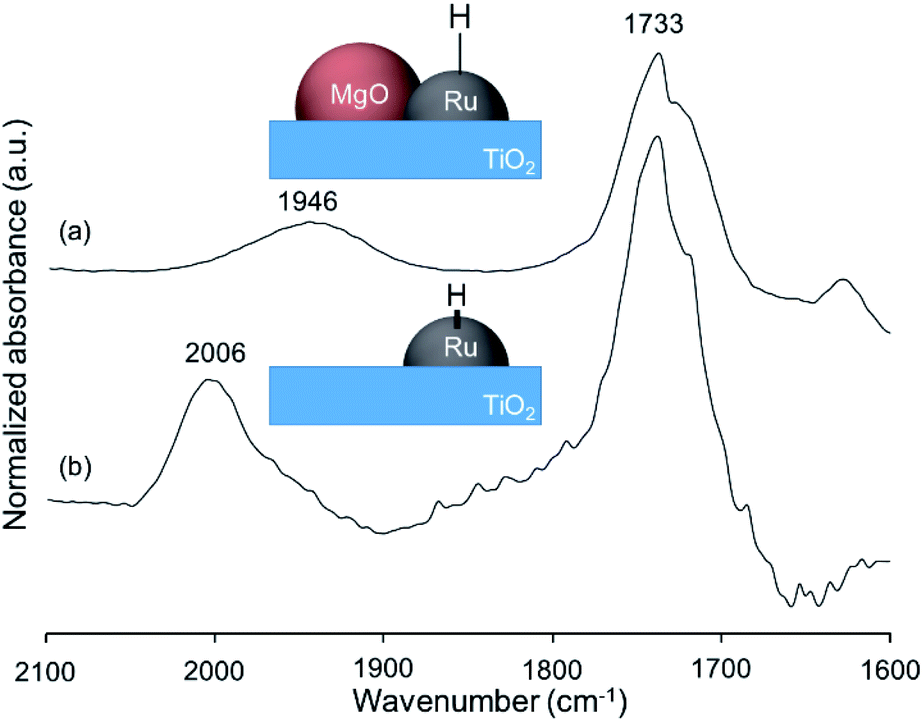
To determine whether the reaction proceeds through the BH strategy or not, we examined the hydrogenation of benzylidene aniline (4) with 1a as a hydride source. As expected, hydrogenated product 5 was obtained in 27% yield along with the formation of furfural derivative 6 in 48% yield (Scheme 2a). 6 was generated through the formation of furfural by the dehydrogenation of 1a and subsequent condensation with aniline derived from the hydrolysis of 4. In addition, deuterium labelling experiments were conducted for the direct amination of benzyl alcohol-α,α-d2 (1b-d2) and subsequent acetylation with acetyl chloride (AcCl) to precisely calculate the deuterium content using 1H nuclear magnetic resonance spectroscopy (NMR) (Scheme 2b). The deuterium content at the benzylic position in 8 was decreased to 39% from 98%. The decrease of the deuterium content suggests that the dehydrogenation of alcohols is reversible under the direct amination conditions. The source of the hydrogen is a hydroxide group or H2O adsorbed on the catalyst surface. The deuterium content was also decreased when 1b-d2 was exposed to the direct amination conditions without ammonia (Scheme 2c). No detection of the dehydrogenated product indicates that Ru–20MgO/TiO2 can prevent H2 release from the Ru hydride species, in contrast to Ru/TiO2 which is known to be active for the dehydrogenation of alcohols under oxidant-free conditions.17 In addition, the H2-TPD analysis was carried out to evaluate the nature of the H species (Fig. S4†). The desorption peak at around 500 °C exclusively appears in the profile of Ru–20MgO/TiO2. This peak is likely assignable to the desorption of spillover hydrogen on the support.18 These results indicate that MgO promotes the hydrogen spillover, contributing to maintenance of H species in the vicinity of Ru particles. The dehydrogenation of alcohols was also confirmed using IR measurements with adsorbed isopropanol. As the temperature increased after the introduction of isopropanol to the IR cell, two bands were observed at 1733 and 1946 cm−1 (Fig. 3 and S6†). The band at 1733 cm−1 is assignable to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of gas phase acetone,19 and that at 1946 cm−1 is due to the Ru–H species on Ru–MgO/TiO2.20 As a complementary result, Ru–D stretching vibration was observed at 1434 cm−1 from measurements using isopropanol-d8 (Fig. S8†). When the hydrogen atom in Ru–H is replaced with a deuterium atom, the wavenumber for the Ru–H stretching vibration is expected to be shifted to 1441 cm−1. The IR spectrum for the measurement using isopropanol-d8 also showed a peak that was assigned to the Ru–H bond, which reflects the H/D exchange by reversible alcohol dehydrogenation. We cannot rule out the possibility that these peaks are assignable to the stretching vibrations of the C–D bond in acetone-d6.21 Comparison of Ru–MgO/TiO2 with Ru/TiO2 revealed that the band due to Ru–H on Ru/TiO2 was at a higher wavenumber position (2006 cm−1) than that on Ru–MgO/TiO2 (Fig. 5). This difference indicates that the Ru–H bond is elongated due to the electron donation from MgO.
O of gas phase acetone,19 and that at 1946 cm−1 is due to the Ru–H species on Ru–MgO/TiO2.20 As a complementary result, Ru–D stretching vibration was observed at 1434 cm−1 from measurements using isopropanol-d8 (Fig. S8†). When the hydrogen atom in Ru–H is replaced with a deuterium atom, the wavenumber for the Ru–H stretching vibration is expected to be shifted to 1441 cm−1. The IR spectrum for the measurement using isopropanol-d8 also showed a peak that was assigned to the Ru–H bond, which reflects the H/D exchange by reversible alcohol dehydrogenation. We cannot rule out the possibility that these peaks are assignable to the stretching vibrations of the C–D bond in acetone-d6.21 Comparison of Ru–MgO/TiO2 with Ru/TiO2 revealed that the band due to Ru–H on Ru/TiO2 was at a higher wavenumber position (2006 cm−1) than that on Ru–MgO/TiO2 (Fig. 5). This difference indicates that the Ru–H bond is elongated due to the electron donation from MgO.
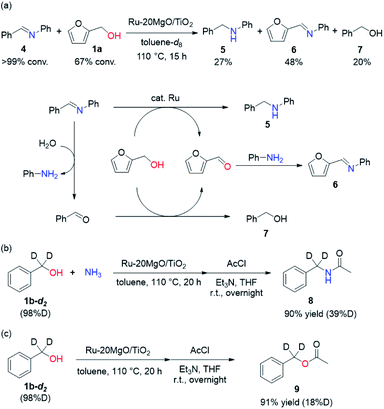 |
| | Scheme 2 Control experiments: (a) hydrogenation of benzylidene aniline (4) using furfuryl alcohol as a hydride source. (b) Direct amination of 1b-d2. (c) Exposure of 1b-d2 to direct amination conditions without ammonia. | |
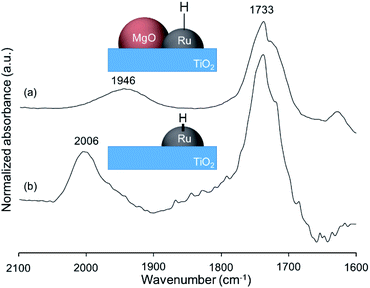 |
| | Fig. 5 Difference IR spectra of isopropanol-adsorbed (a) Ru–20MgO/TiO2 and (b) Ru/TiO2 at 60 °C. | |
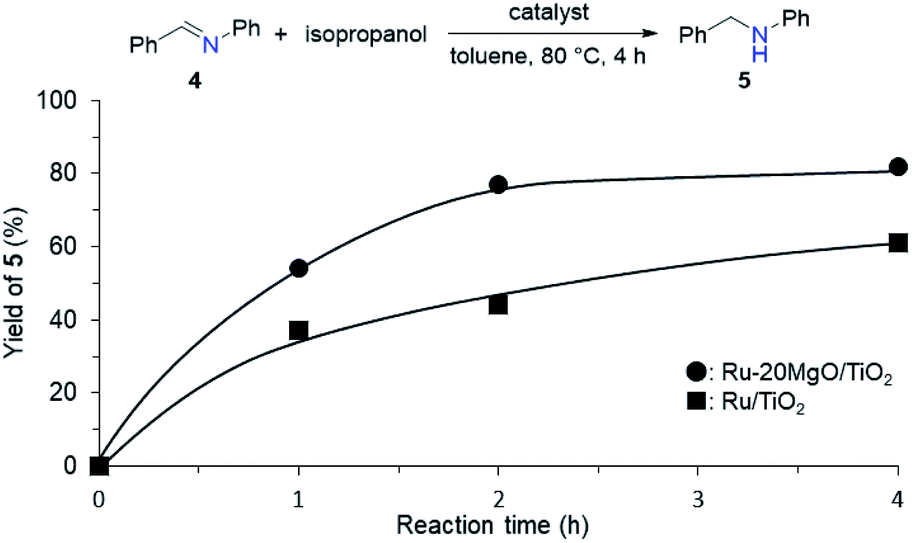
From the elongated Ru–H bond on Ru–MgO/TiO2, hydrogenation ability can be differentiated by the effect of MgO. Therefore, the transfer hydrogenation of 4 was conducted over Ru–MgO/TiO2 and Ru/TiO2 (Fig. 6). When isopropanol is used as a hydride source, the MgO effect on the imine hydrogenation step can be discussed because the dehydrogenation of isopropanol is fast even at 60 °C as observed in IPA-adsorbed IR analysis (Fig. 5). As expected, hydrogenated product 5 was obtained in 81% yield for Ru–MgO/TiO2 and in 60% yield for Ru/TiO2. The higher hydrogenation ability of Ru–MgO/TiO2 can be rationalized by the elongated Ru–H bond, as observed in isopropanol-probed IR measurements, which results in efficient direct amination under mild conditions.
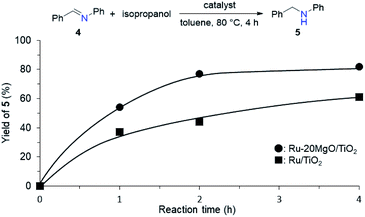 |
| | Fig. 6 Time courses for transfer hydrogenation of 4 over the supported Ru catalyst using isopropanol as a hydride source. Reaction conditions: catalyst (0.05 g), 4 (0.5 mmol), isopropanol (1 mL), toluene (4 mL), and 80 °C. | |
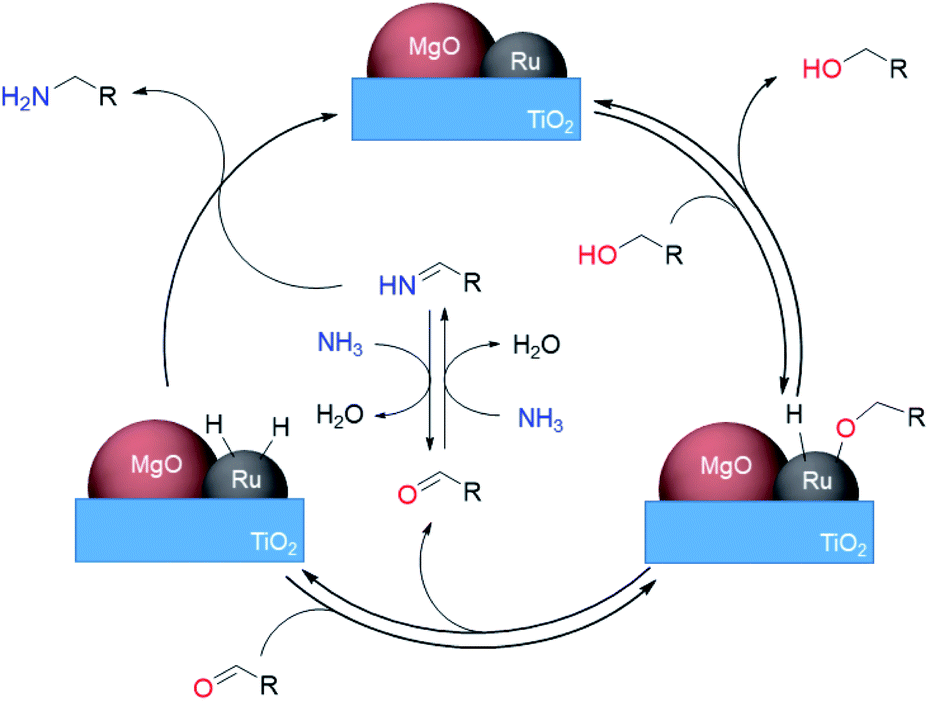
Here we propose a mechanism for this reaction involving the dehydrogenation of alcohols (Scheme 3). Initially, the adsorption of an alcohol on the ruthenium surface forms a deprotonated alkoxide. An aldehyde is generated reversibly formed by β-hydride elimination from the Ru alkoxide. The concomitant Ru–H species, activated by MgO, hydrogenates the NH–imine derived from the aldehyde with ammonia to give the desired primary amines. The high activity of the Ru–H species resulted in high selectivity by the prevention of side reactions of imine intermediates. The lack of H2 requirement could be attributed to the inhibition of Ru–H species decomposition through electronic tuning using MgO.
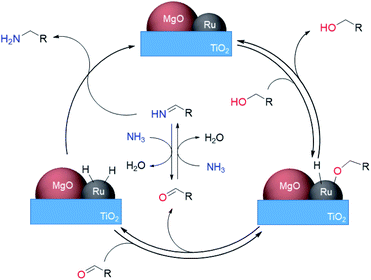 |
| | Scheme 3 Proposed reaction mechanism for direct amination of alcohols over Ru–20MgO/TiO2. | |
Conclusions
We have demonstrated the direct amination of alcohols over Ru–20MgO/TiO2. The direct amination of various alcohols proceeded efficiently to afford the desired primary amines in high yield. The present system is highly advantageous in that it requires no accelerator compared to reported Ru systems, and is reusable without loss of activity after the base treatment and reduction sequence. Direct amination catalysed by Ru–20MgO/TiO2 proceeded through the BH methodology, as evidenced by the formation of ruthenium hydride species from isopropanol. Mechanistic studies revealed that the effect of MgO can be (1) acceleration of the imine hydrogenation step by the electronic tuning of Ru particles, and (2) the anchoring effect that provides large dispersion and small particle size. These results with the metal hydride species are expected to provide a design guide for new heterogeneous catalysts for hydrogen-involving reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Advanced Low Carbon Technology Research and Development Program (ALCA) of the Japan Science and Technology Agency (JST) (JPMJAL1205) and a fund from the Grants-in-Aid for Japan Society for the Promotion of Science (JSPS) Fellows and for Scientific Research from the Ministry of Education, Culture, Science, Sports, and Technology (MEXT) of Japan (18H05251).
Notes and references
-
(a) K. S. Hayes, Appl. Catal. A: Gen., 2001, 221, 187–195 CrossRef CAS;
(b) B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. L. Rotgerink, K. Möbus, D. J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, Appl. Catal. A: Gen., 2005, 280, 17–46 CrossRef CAS;
(c) I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS;
(d) A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS.
-
K. Eller, E. Henkes, R. Rossbacher and H. Höke, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed.
-
(a) A. Corma, J. Navas and M. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS;
(b) K.-i. Shimizu, Catal. Sci. Technol., 2015, 5, 1412–1427 RSC.
- C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS.
-
(a) S. Imm, S. Bähn, L. Neubert, H. Neumann and N. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126–8129 CrossRef CAS;
(b) D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130–8133 CrossRef CAS;
(c) S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Lkasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599–7603 CrossRef CAS;
(d) M. Zhang, S. Imm, S. Bähn, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 11197–11201 CrossRef CAS;
(e) D. Pingen, O. Diebolt and D. Vogt, ChemCatChem, 2013, 5, 2905–2912 CrossRef CAS;
(f) W. Baumann, A. Spannenberg, J. Pfeffer, T. Haas, A. Köckritz, A. Martin and J. Deutsch, Chem.–Eur. J., 2013, 19, 17702–17706 CrossRef CAS;
(g) D. Pingen, M. Lutz and D. Vogt, Organometallics, 2014, 33, 1623–1629 CrossRef CAS;
(h) X. Ye, P. N. Plessow, M. K. Brinks, N. Schelwies, T. Schaub, F. Rominger, R. Paciello, M. Limbach and P. Hofmann, J. Am. Chem. Soc., 2014, 136, 5923–5929 CrossRef CAS;
(i) N. Nakagawa, E. J. Derrah, M. Schlwies, F. Rominger, O. Trapp and T. Schaub, Dalton Trans., 2016, 45, 6856–6865 RSC;
(j) K.-i. Fujita, S. Furukawa, N. Morishima, M. Shimizu and R. Yamaguchi, ChemCatChem, 2018, 10, 1993–1997 CrossRef CAS.
-
(a) K.-i. Shimizu, K. Kon, W. Onodera, H. Yamazaki and J. N. Kondo, ACS Catal., 2013, 3, 112–117 CrossRef CAS;
(b) K.-i. Shimizu, S. Kanno, K. Kon, S. M. A. H. Siddiki, H. Tanaka and Y. Sakata, Catal. Today, 2014, 232, 134–138 CrossRef CAS;
(c) Y. Liu, K. Zhou, H. Shu, H. Liu, J. Lou, D. Guo, Z. Wei and X. Li, Catal. Sci. Technol., 2017, 7, 4129–4135 RSC;
(d) L. Ma, L. Yan, A.-H. Lu and Y. Ding, RSC Adv., 2018, 8, 8152–8163 RSC;
(e) A. Tomer, Z. Yan, A. Ponchel and M. Pera-Titus, J. Catal., 2017, 356, 133–146 CrossRef CAS;
(f) A. Y. K. Leung, K. Hellgardt and K. K. M. Hii, ACS Sustainable Chem. Eng., 2018, 6, 5479–5484 CrossRef CAS;
(g) A. Tomer, B. T. Kusema, J.-F. Paul, C. Przybylki, E. Monflier, M. Pera-Titus and A. Ponchel, J. Catal., 2018, 368, 172–189 CrossRef CAS;
(h) Y. Liu, A. Afanasenko, S. Elangovan, Z. Sun and K. Barta, ACS Sustainable Chem. Eng., 2019, 7, 11267–11274 CrossRef CAS;
(i) K. Zhou, H. Liu, H. Shu, S. Xiao, D. Guo, Y. Liu, Z. Wei and X. Li, ChemCatChem, 2019, 11, 2649–2656 CrossRef CAS;
(j) C. R. Ho, V. Defalque, S. Zheng and A. T. Bell, ACS Catal., 2019, 9, 2931–2939 CrossRef CAS.
-
(a) R. Pfützenreuter and M. Rose, ChemCatChem, 2016, 8, 251–255 CrossRef;
(b) D. Ruiz, A. Aho, T. Saloranta, K. Eränen, J. Wärnå, R. Lino and D. Y. Murzin, Chem. Eng. J., 2017, 307, 739–749 CrossRef CAS;
(c) D. Ruiz, A. Aho, P. Mäki-Arvela, N. Kumar, H. Oliva and D. Y. Murzin, Ind. Eng. Chem. Res., 2017, 56, 12878–12887 CrossRef CAS;
(d) Y. Li, H. Chen, C. Zhang, B. Zhang, T. Liu, Q. Wu, X. Su, W. Lin and F. Zhao, Sci. China Chem., 2017, 60, 920–926 CrossRef CAS;
(e) G. Liang, Y. Zhou, J. Zhao, A. Y. Khodakov and V. V. Ordomsky, ACS Catal., 2018, 8, 11226–11234 CrossRef CAS.
-
(a) A. K. Rausch, E. van Steen and F. Roessner, J. Catal., 2008, 253, 111–118 CrossRef CAS;
(b) J. H. Cho, J. H. Park, T.-S. Chang, G. Seo and C.-H. Shin, Appl. Catal. A; Gen., 2012, 417–418, 313–319 CrossRef CAS;
(c) F. Niu, S. Xie, M. Bahri, O. Ersen, Z. Yan, B. T. Kusema, M. Pera-Titus, A. Y. Khodakov and V. V. Ordomsky, ACS Catal., 2019, 9, 5986–5997 CrossRef CAS.
- T. Tong, W. Guo, X. Liu, Y. Guo, C.-W. Pao, J.-L. Chen, Y. Hu and Y. Wang, J. Catal., 2019, 378, 392–401 CrossRef CAS.
-
(a) C. Copéret, D. P. Estes, K. Larmier and K. Searles, Chem. Rev., 2016, 116, 8463–8505 CrossRef;
(b) F. Polo-Garzon, S. Luo, Y. Cheng, K. L. Page, A. J. Ramirez-Cuesta, P. F. Britt and Z. Wu, ChemSusChem, 2018, 12, 93–103 CrossRef.
-
(a) K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani, K. Jitsukawa and K. Kaneda, Chem. -Eur. J., 2006, 12, 8228–8239 CrossRef CAS;
(b) K. Motokura, D. Nishimura, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 5662–5663 CrossRef CAS.
- G. Delleplane and J. Overend, Spectrochim. Acta, 1966, 22, 593–614 CrossRef.
- S. Y. Chin, C. T. Williams and M. D. Amiridis, J. Phys. Chem. B, 2006, 110, 871–882 CrossRef CAS.
- T. Komanoya, T. Kinemura, Y. Kita, K. Kamata and M. Hara, J. Am. Chem. Soc., 2017, 139, 11493–11499 CrossRef CAS.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS.
-
(a) N.-T. Le, A. Byun, Y. Han, K.-I. Lee and H. Kim, Green Sustainable Chem, 2015, 5, 115–127 CrossRef CAS;
(b) K. Zhou, H. Liu, H. Shu, S. Xiao, D. Guo, Y. Liu, Z. Wei and X. Li, ChemCatChem, 2019, 11, 2649–2656 CrossRef CAS;
(c) H. Yuan, B. T. Kusema, Z. Yan, S. Streiff and F. Shi, RSC Adv., 2019, 9, 38877–38881 RSC.
- Y. Kim, S. Ahn, J. Y. Hwang, D.-H. Ko and K.-Y. Kwon, Catalysts, 2017, 7, 7 CrossRef.
- X. Xue, J. Liu, D. Rao, S. Xu, W. Bing, B. Wang, S. He and M. Wei, Catal. Sci. Technol., 2017, 7, 650–657 RSC.
- M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Langmuir, 2000, 16, 430–436 CrossRef CAS.
- H. D. Kaesz and R. B. Saillant, Chem. Rev., 1972, 72, 231–281 CrossRef CAS.
- K. Aika, A. Ohya, A. Ozaki, Y. Inoue and I. Yasumori, J. Catal., 1985, 92, 305–311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03858j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Midori
Kuwabara
a,
Satoshi
Yamadera
a,
Keigo
Kamata
a,
Midori
Kuwabara
a,
Satoshi
Yamadera
a,
Keigo
Kamata