DOI:
10.1039/D0SC03744C
(Edge Article)
Chem. Sci., 2020,
11, 11235-11243
Regiocontrolled dimerization of asymmetric diazaheptacene derivatives toward X-shaped porous semiconductors†
Received
8th July 2020
, Accepted 15th September 2020
First published on 16th September 2020
Abstract
Conformationally rigid X-shaped PAHs are attracting interest due to their self-assembly into unique networks and as models to study through-space exciton and charge delocalization in one single molecule. We report here the synthesis of X-shaped PAHs by dimerization of diazaheptacene diimides. The diimide groups are employed to effectively direct the self-assembly into antiparallel dimer aggregates, which assist the compounds to undergo a regiocontrolled [4 + 4] dimerization, leading to an X-shaped conformation bearing electron-poor and -rich subunits. The resulting PAHs are found to pack in 2D layers with large open channels and infinite π⋯π arrays. Furthermore, these highly crystalline porous materials serve as electron-transporting materials in OFETs due to the long-range π-stacked arrays in the layers. This work presents a potentially generalizable strategy, which may provide a unique class of porous semiconductors for organic devices, taking advantage of their open channels.
Introduction
Electroactive, through-space conjugated polycyclic aromatic hydrocarbons (PAHs) bearing aromatic subunits fused to the rigid bridges are garnering interest due to their theoretical importance and potential application in materials science.1 From a theoretical point of view, these rigid bridges limit the conformational orientation of aromatic subunits with respect to bridging moieties that provide an ideal model to investigate through-space intramolecular exciton and charge delocalization between neighboring aromatic subunits in one single molecule, which play a crucial role in charge-carrier transport in organic semiconductors.1a,2 It is also indicated that by reducing conformational flexibility, thus avoiding uncertainties about mechanisms that depend on the relative acene subunit orientation, these compounds have emerged as key platforms to explore the fundamental dynamics of singlet fission (SF). For example, the groups of Campos and Damrauer demonstrated that bridge-linked acene dimers show a faster SF rate with a slower triplet pair recombination than the corresponding dimers with a conjugated linker.3 Importantly, such molecules usually form loosely packed structures and self-assemble into porous films with large interior spaces, which make them useful host molecules for guest inclusion to tune the electronic and optoelectronic properties.1d,4 This makes these compounds appealing active elements in sensory response, organic photovoltaics, and energy storage devices.5
Synthesis of bridge-fused PAHs relies primarily on both divergent (bridge-first) and convergent (subunit-first) strategies.6 The divergent strategy includes synthesizing a rigid bridge, such as aromatic [m.n.o]propellanes, undergoing sequences of reactions to extend π-planes. For example, triptycene and trinaphtho[3.3.3]propellane have been extensively used to prepare extended 3D PAHs by lateral fusion of aromatic rings to the benzene rings.4d,e,7 Recently, we reported a new 3D-symmetric propellane, triperyleno[3.3.3]propellane triimides that possesses three perylene monoimide subunits fused on a propellane, which is compatible with different reactions to readily prepare a quasi-D3h symmetric nanostructure that shows an isotropic charge transport despite the weak intermolecular interactions between the neighboring PAH units.8 The convergent strategy relies on the precision and control afforded by synthetic chemistry to construct aromatic subunits, which can take part in [4 + 2] or [4 + 4] cycloadditions, leading to the formation of the desired targets.9 For example, although acene [4 + 4] cyclodimerization is a well-known decay pathway of acene-based organic devices as a result of decomposition of the active material,10 it has been proposed as a feasible option to prepare rigid X-shaped compounds,9 in particular for 2D polymers.11 A major limitation of the convergent strategy is that the use of large acenes that are not readily available and have a low solubility makes them difficult to manipulate. Therefore, the preparation of X-shaped PAHs remains a considerable synthetic challenge, and it is more difficult for electroactive compounds; consequently their application in organic devices is underexplored.
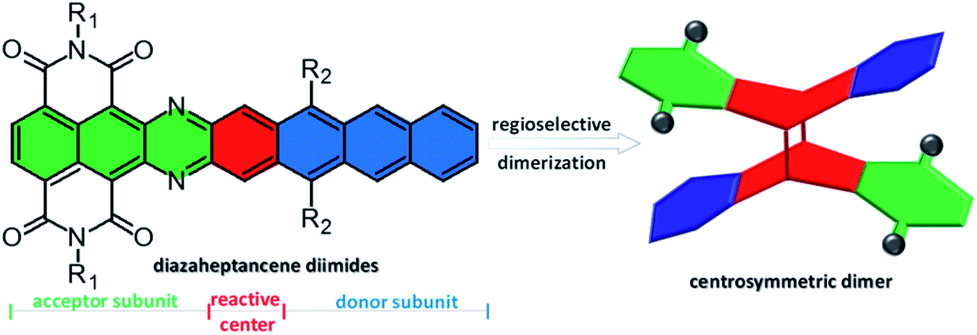
Herein we report, for the first time, conformationally rigid X-shaped PAHs not only bearing electron-poor and -rich subunits but also exhibiting semiconducting behavior, by dimerization of diazaheptacene diimides. The key feature of diazaheptacene diimides is that the imide substituents at only one end of diazaheptacene with the centermost ring (the most reactive ring) are non-functionalized, which not only effectively improves the solubility of heptacene derivatives, but also preserves the reactivity of the centermost rings (Fig. 1). Therefore, these compounds, in principle, should undergo [4 + 4] cycloaddition and lead to the corresponding dimers.12 We also envision that this asymmetric structural pattern with fusion of electron-poor and -rich subunits together, naturally possesses a significant dipole, which might influence the molecular orbital distribution and solid-state packing, thus enabling direct dimerization regioselectively. Additionally, the nitrogen atoms in the structures are expected to stabilize the frontier molecular orbitals and increase the electron affinity.13
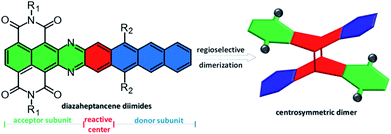 |
| | Fig. 1 The strategy toward centrosymmetric X-shaped PAHs bearing electron-poor and -rich subunits. | |
Results and discussion
Convergent synthesis of X-shaped PAHs
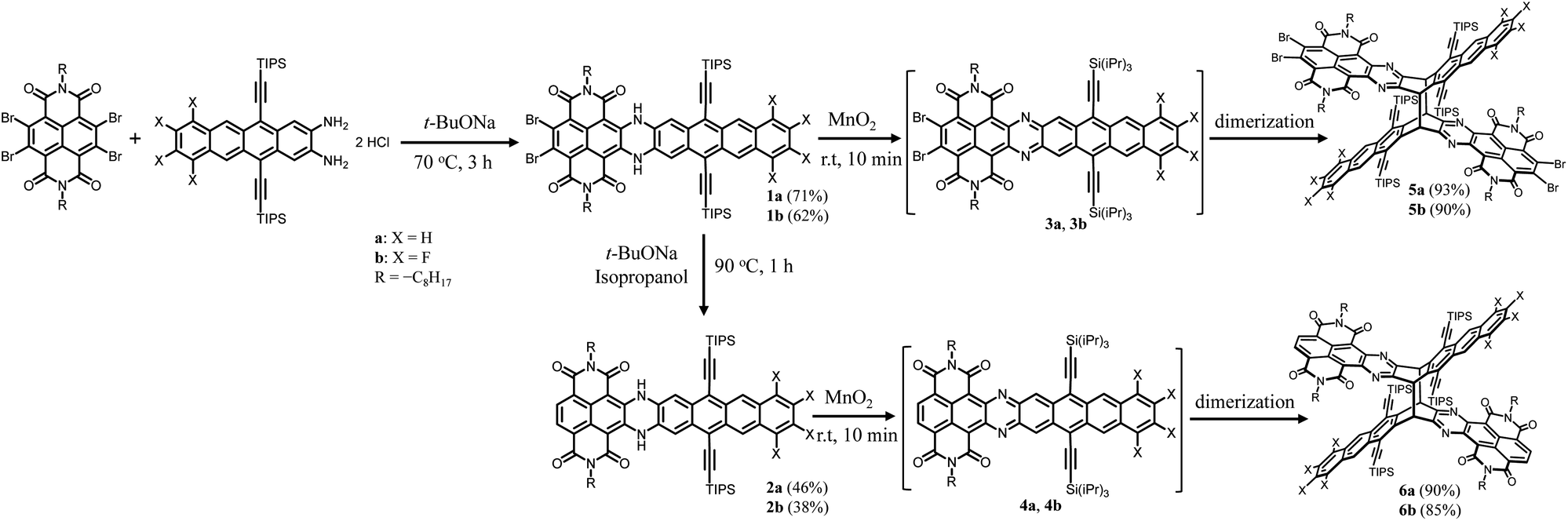
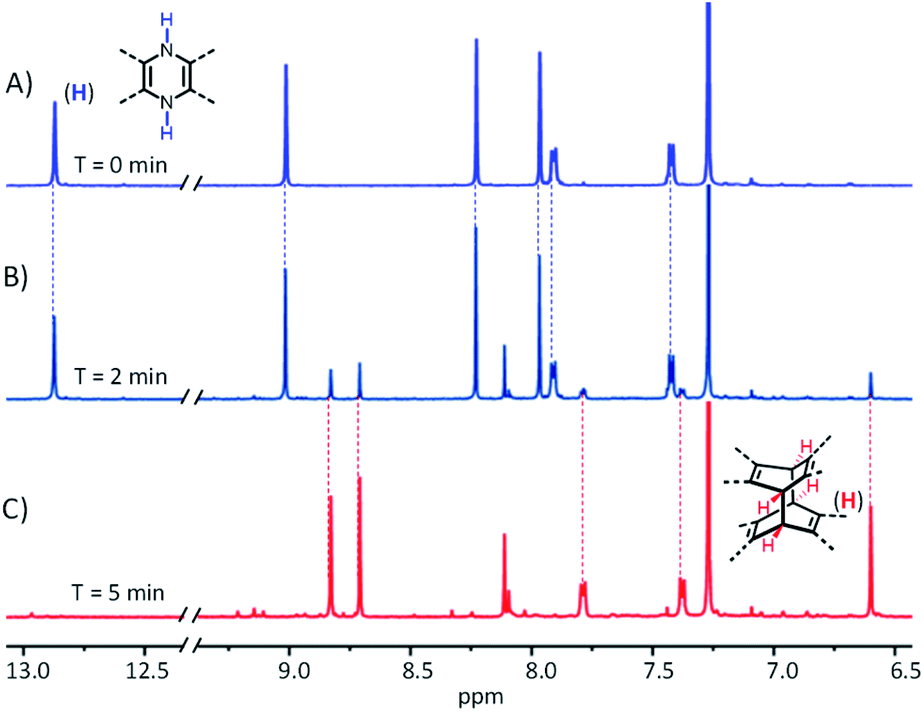
Scheme 1 shows the synthesis of X-shaped compounds. The synthetic route involves the coupling of brominated naphthalenediimides (NDIs) to acene o-diamines via Buchwald–Hartwig amination followed by an oxidation/dimerization cascade reaction to synthesize the dimers. Treatment of tetrabrominated NDI with triisopropylsilyl (TIPS)-ethyl functionalized diaminotetracenes gave the N,N′-dihydro precursors 1a and 1b in 71% and 62% yields, respectively. Upon oxidation of 1a and 1b in the presence of MnO2, we observed a characteristic change in color (from dark blue to yellow) that signified the formation of corresponding dimers 5a and 5b, which was evidenced by the disappearance of the –NH– signal around 13.0 ppm and the appearance of a signal around 6.5 ppm assigned to the bridgehead in the 1H NMR spectrum. A similar oxidation of the precursors 2a and 2b, which were prepared by treatment of 1a and 1b with t-BuONa in the presence of iso-propanol, similarly afforded the dimers 6a and 6b. We anticipated that the N,N′-dihydro precursors were aromatized in situ to produce the corresponding diazaheptacene diimides before dimerization, although they could not even be detected by using 1H NMR (Fig. 2). It is noteworthy that the syntheses of the dimers proceeded in very short time periods with relatively high yields. These dimers are soluble in common solvents and finally confirmed using their X-ray crystal structures.
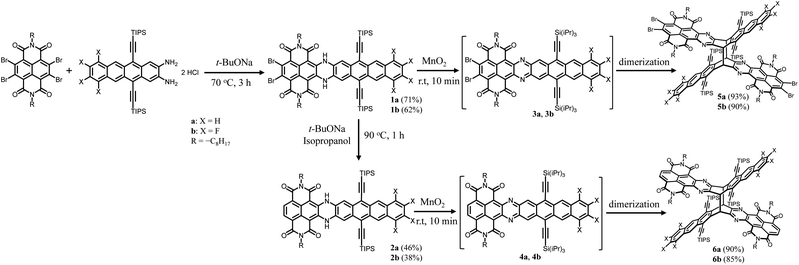 |
| | Scheme 1 Synthetic steps to X-shaped PAHs 5a, 5b, 6a, and 6b. | |
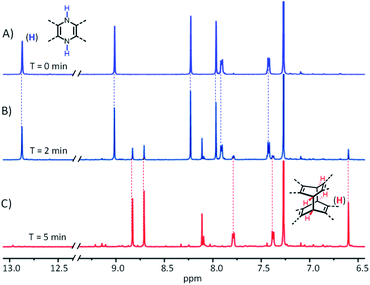 |
| | Fig. 2 Part of the 1H NMR spectra of 2a (3 mM) in CDCl3 by the addition of MnO2 after (A) 0 min, (B) 2 min, and (C) 5 min at 298 K. | |
X-ray crystallography
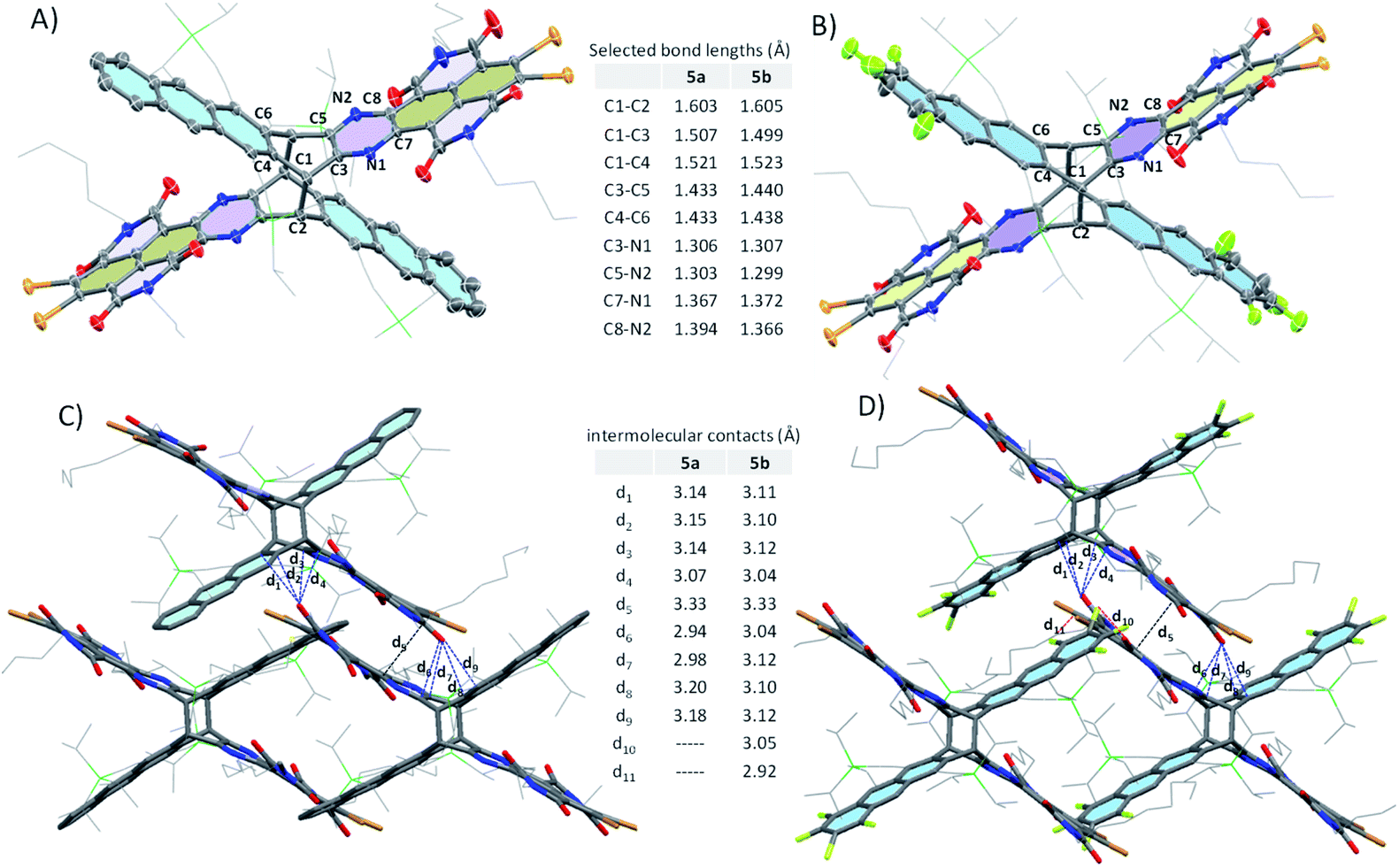
Single crystals of 5a and 5b suitable for X-ray diffraction analysis could be obtained by slow diffusion of methanol into a toluene solution at room temperature.14 As shown in Fig. 3A and B, 5a and 5b adopt an X-shaped conformation, with NDI subunits slightly twisted. The dihedral angles between electron-poor and -rich subunits are ca. 127° and 55°. The bond lengths between the bridgehead carbon atoms (C1–C2) in 5a and 5b are exceptionally long at 1.603 and 1.605 Å, due to the bulky size of the subunits, which provide considerable strain to the C–C bond, whereas the surrounding C–C bond lengths in 5a and 5b (C1–C3 and C1–C4) range between 1.499 and 1.521 Å, which are close to the typical sp3–sp3 C–C bond length. The C3–N1 and C5–N2 bond lengths in 5a and 5b (in the range of 1.299–1.307 Å) are shorter than those of C7–N1 and C8–N2 (in the range of 1.366–1.394 Å), thus confirming the double bond character. In addition, the C3–C5 and C4–C6 lengths are in the range of 1.433–1.440 Å, which fall within the typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond values for polycyclic aromatic hydrocarbons (1.4 ± 0.04 Å).
C bond values for polycyclic aromatic hydrocarbons (1.4 ± 0.04 Å).
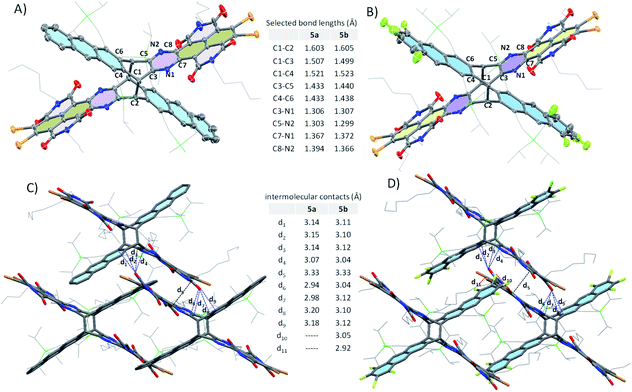 |
| | Fig. 3 Crystal structures with selected bond lengths of 5a (A) and 5b (B); selected packing interactions between adjacent molecules in the crystals of 5a (C) and 5b (D). | |
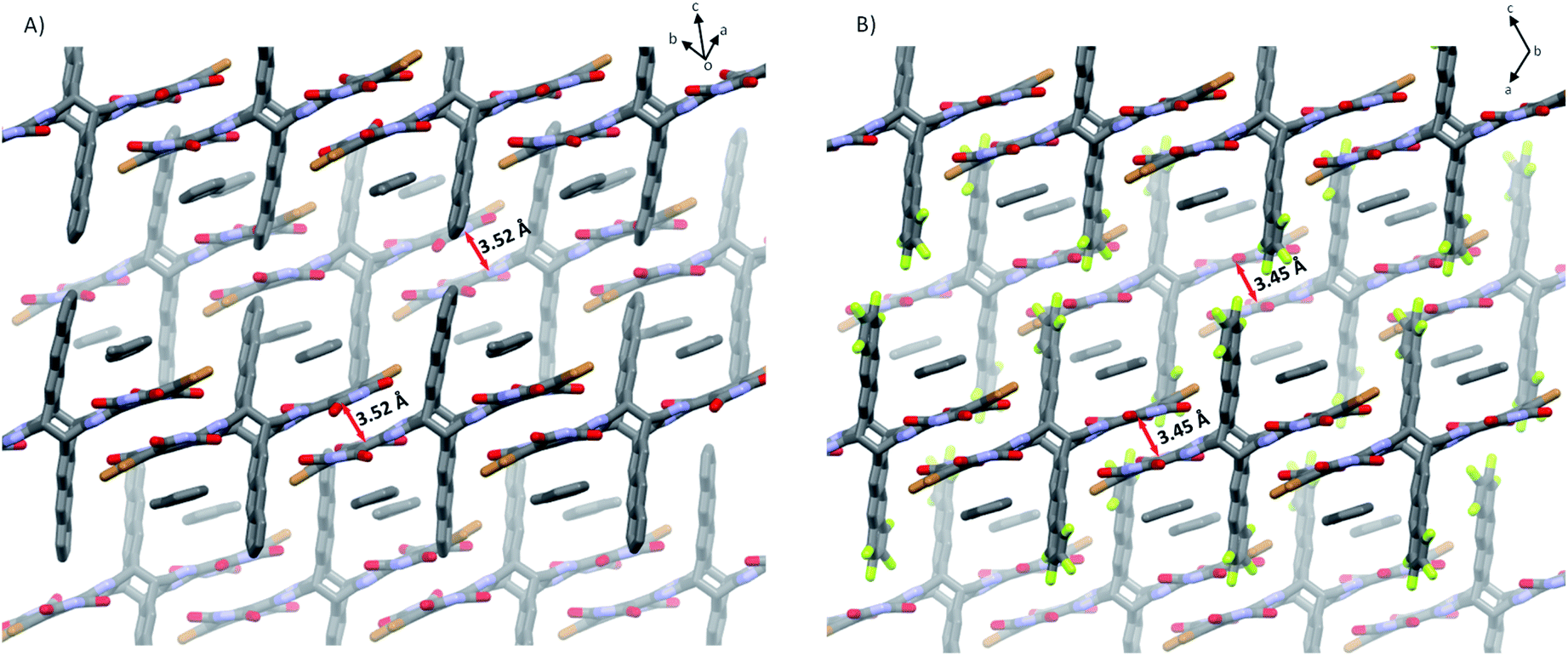
As shown in Fig. 4, 5a and 5b in the crystal pack in two-dimensional (2D) layer structures in the solid state. In an individual layer of 5a, two electron-poor subunits of adjacent molecules adopt a π-stacked dimer motif with a rotation angle of approximately 135° and a π–π distance of 3.5 Å, and, in turn, form infinite arrays with multiple short C⋯O (in the range of 2.94–3.18 Å) and C⋯C (3.33 Å) contacts, thus providing a pathway to enhance the semiconducting behavior in typically insulating X-shaped PAHs. In addition, there are some C–H⋯O interactions between the neighboring layers in 5a. It should be noted that the neighboring layers are offset in a way that defines large rhombic channels, which provide open networks with significant volumes to host guest molecules. As with that of 5a, in an individual layer of 5b, two electron-poor subunits of adjacent molecules also adopt a π-stacked dimer motif with multiple short C⋯O and C⋯C contacts. Therefore, the replacement of H atoms with F atoms does not induce a significant change in the conformation and packing motif. However, the presence of F atoms gives rise to intermolecular O⋯F (3.05 Å) and Br⋯F (2.92 Å) contacts between the molecules in adjacent layers, which reinforce the layers and the channels in the crystals. It is interesting to note that the open channels in 5a and 5b are filled with toluene molecules, which interact with NDI units via multiple intermolecular interactions.
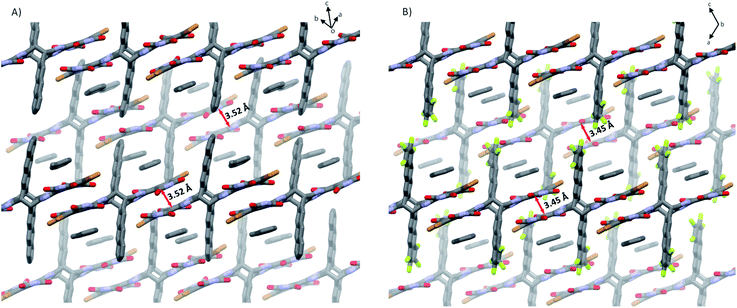 |
| | Fig. 4 Rhombic open channels between two adjacent layers filled with toluene molecules: (A) 5a and (B) 5b. Alkyl chains, TIPS-ethynyl substituents, and hydrogen atoms are omitted for clarity. | |
Theoretical study on dimerization
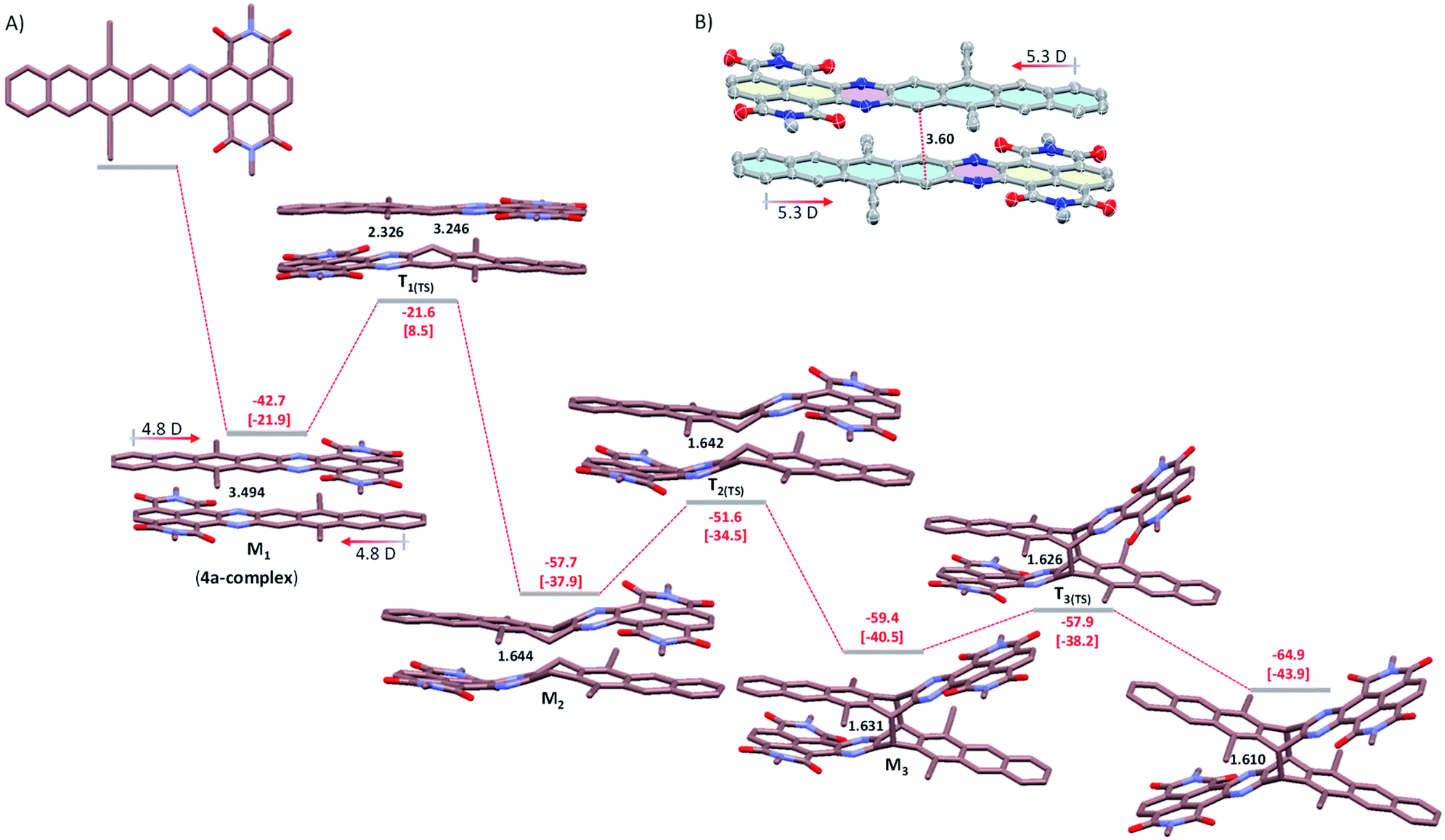
To provide further insight into the mechanism of dimerization, we performed DFT calculations on 6a as a model system at the M06-2X/6-31G(d)+ZPVE level of the theory (Fig. 5A). We based our mechanistic study on previous work by Bendikov et al., in which the multistep dimerization pathway of heptacene was explored computationally.15 The initial step is the exothermic formation of a π-complex of two 4a molecules (the first metastable state, M1), which is stabilized by over 18 kcal mol−1 relative to the π-complex of heptacene calculated at the same level.15 The optimized π-complex structure shows an antiparallel π–π stacking conformation with overlaid electron-poor and -rich subunits, which is attributed to electrostatic interactions arising from the ground state dipole moment (4.8 D for 4a). Although the single crystal of 4a is not obtainable, the X-ray diffraction analysis confirms that 2a with a dipole moment of 5.3 D, crystallizes with electron-poor and -rich subunits stacked face-to-face with a small displacement (Fig. 5B).14 In the calculated π-complex of 4a, the two central benzene rings are parallel and separated by 3.49 Å, which is consistent with the typical π-stacking distances observed in crystalline solids and suitable for a [4 + 4] cyclodimerization.16 Therefore, the calculation results reveal that the antiparallel π–π stacking conformation is indeed energetically more favorable for centrosymmetric dimerization. The π-complex then collapses to form two asymmetric C–C bonds (2.326 and 3.246 Å) and generates the first transition state T1(TS), which is 21.6 kcal mol−1 lower than the reactant (with an activation free energy of 8.5 kcal mol−1). The T1(TS) gives rise to the second metastable state M2 by forming a C–C bond of 1.644 Å, which has strong π–π interactions between electron-poor and -rich subunits. Subsequently, the stepwise breaking of the π–π interactions between electron-poor and -rich subunits on both sides occurs via two transition states T2(TS) (−51.6 kcal mol−1) and T3(TS) (−57.9 kcal mol−1), leading to a more stable product 6a (−64.9 kcal mol−1). Our calculations clearly indicate that the dimerization of 4a may be essentially barrierless relative to the two 4a molecules and the optimized geometry of 6a closely matched the observed single-crystal X-ray structure geometry.
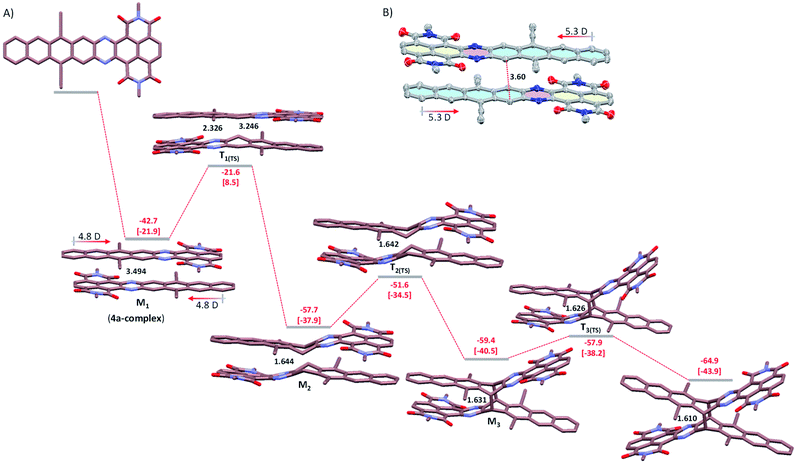 |
| | Fig. 5 (A) Transition state structures and computed activation energies for the dimerization process of 6a. Calculated energies are given in kcal mol−1 and the Gibbs free energies (ΔG, in kcal mol−1) are given in brackets; the newly forming C–C bonds are given in Å; (B) ellipsoid plot of the antiparallel packing π-dimer of 2a with 30% probability of ellipsoids. Alkyl chains and TIPS-ethynyl substituents are replaced by methyl for simplicity. | |
Photo-dissociation of the dimer
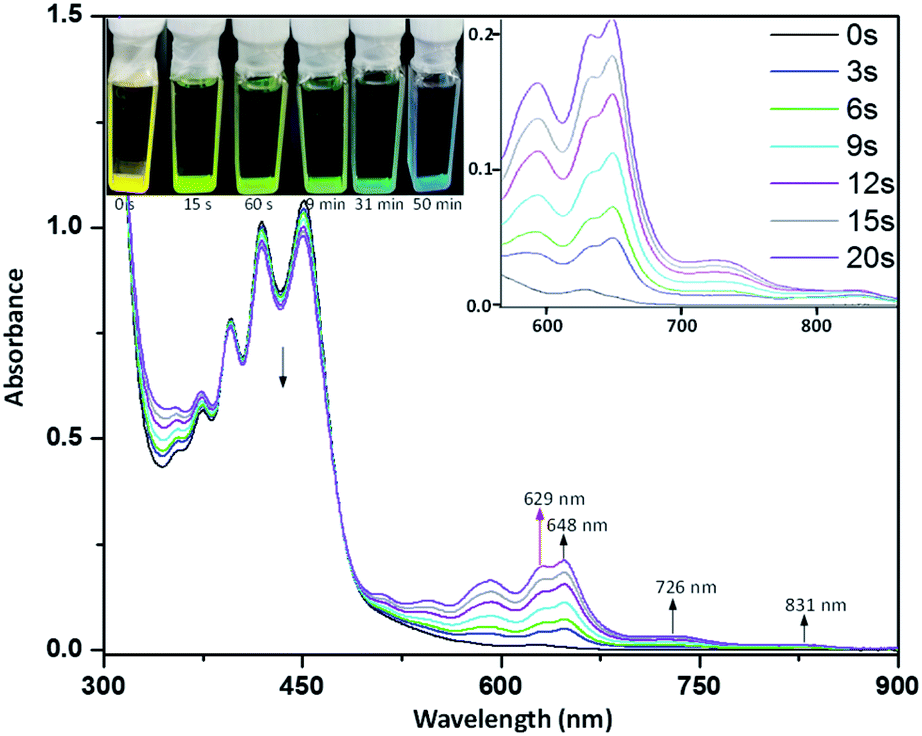
In order to probe the reactive nature of diazaheptacene diimides, we explored the cleavage of X-shaped PAHs into monomers. After many attempts, we found that 5b could be dissociated by photo-irradiation, despite this strategy that is well-known for [4 + 4] cyclodimerization of acenes.9a,17 When a degassed solution of 0.02 mM 5b in toluene (freshly distilled over Na) in a sealed quartz cell was irradiated with a UV lamp (450 nm), a decrease of the absorption bands at 400, 430, and 450 nm was observed (Fig. 6). At the same time, the absorption bands at 594, 629, and 648 nm and two weak broad bands (λmax at 726 and 831 nm, respectively) gradually appeared which indicated that the photo-dissociation of 5b has resulted in a new species. Upon prolonged irradiation, however, the two weak broad bands and the band at 648 nm gradually disappeared (see the ESI for details, Fig. S5†), with a simultaneous increase in the bands at 594 and 629 nm, which are distinct absorption features of 1b, indicating the formation of 1b that is further confirmed by 1H NMR (vide infra). Visually, the spectral changes result in a color change of the solution from yellow to light green to blue. The 1H NMR spectra of 5b in toluene-d8 upon irradiation show a decrease in the bridgehead proton signal along with the appearance of a new signal at 13.4 ppm, characteristic of the –NH– group and the –CH– resonances of the six-membered rings at 8.2 and 9.4 ppm, which are suitable signals for the identification of 1b (see the ESI for details, Fig. S7†). In addition, 1H NMR data showed that 5b can be partially converted to 1b (∼40%) upon irradiation for 4 h and prolonged irradiation of 5b did not significantly increase the conversion yield. 1H NMR spectra also indicate that the photo-dissociation is an extremely clear process, yielding only 1b. A possible explanation involves the photo-dissociation of 5b into 3b that possesses diradical character, and the delocalization of the diradical to the pyrazine ring, results in a new diradical intermediate that might be finally protonated to dihydrodiazaheptacene diimides 1b (see the ESI for details, Fig. S6†), which are usually considered to be stabilized by dihydropyridine ethenamine conjugations.18 It should be noted that, at this stage, we clearly confirmed that the protons involved in the process stem from the residual water in toluene (see the ESI for details, Fig. S8†). The dissociation 5b into 3b is supported by the presence of a band at 648 nm, with two broad bands extending to 900 nm, which is assigned to the HOMO–LUMO transition, according to the TD-DFT calculations (Fig. S4†) that reproduce the optical properties displayed in Fig. 6. Furthermore, the ESR spectroscopy of 5b in toluene upon irradiation shows a signal with a g-value of 2.004 (Fig. S9†), which is characteristic of delocalized singlet diradicaloids.19 Theoretical calculations further predicted that 3b had an open-shell singlet ground state with diradical character (y0 = 0.20) and the spin density is primarily distributed along the zig-zag edge with a significant amount of spin density delocalized on the centermost benzene ring of 3b, leading to the high reactivity of the ring.
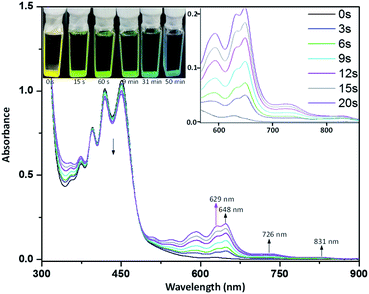 |
| | Fig. 6 Photo-dissociation of 0.02 mM 5b in degassed toluene irradiated with a UV lamp (450 nm) at ambient temperature. Inset: photographs showing the color change of 5b in toluene by increasing irradiation time. | |
Optical and redox properties
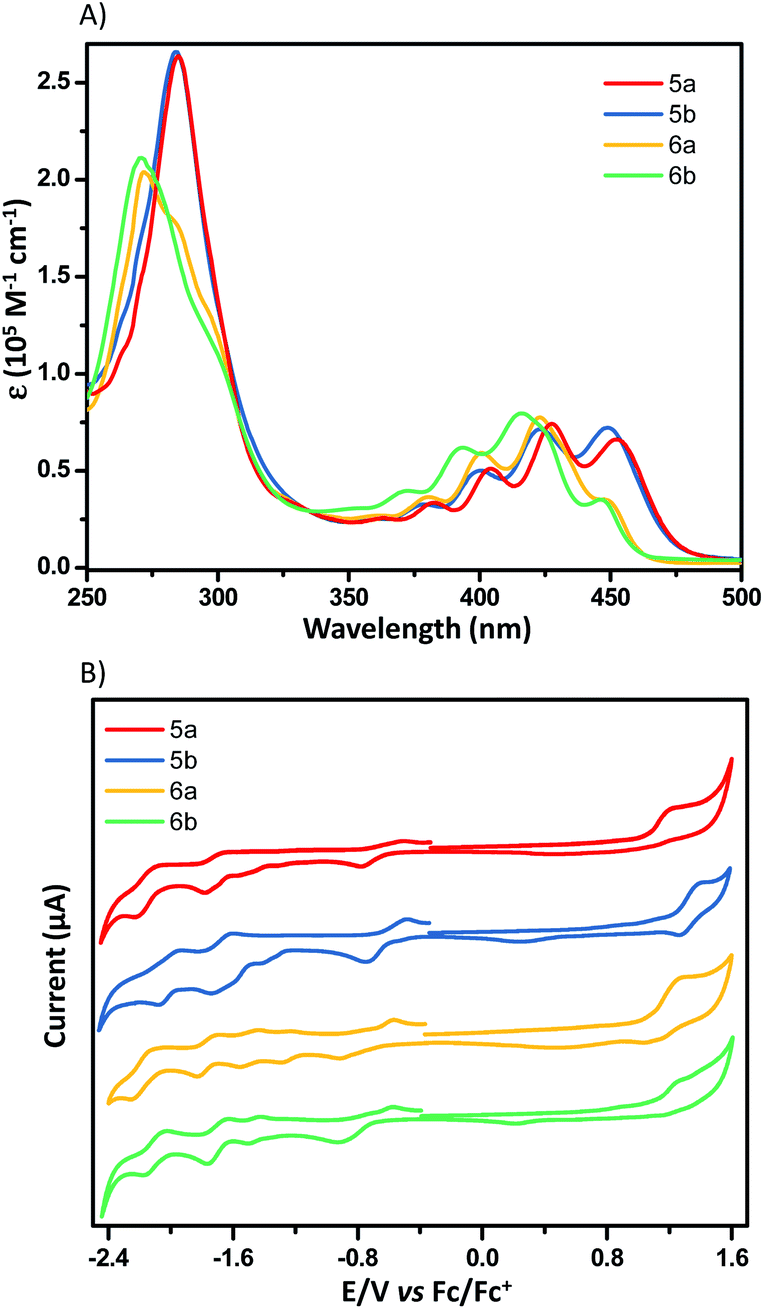
Fig. 7A shows the UV-vis absorption spectra of X-shaped PAHs in chloroform. Apparent from Fig. 7A is that these compounds exhibit very similar electronic transitions, which roughly superimpose the characteristic features of electron-poor and -rich subunits. The absorption around 450 nm is typically from NDI subunits20 and the absorption bands from 370 to 420 nm are largely from acene subunits (Fig. S1†). DFT calculations reveal that the calculated HOMO and LUMO levels of the four compounds are spatially well separated on the electron-rich and -poor subunits, respectively. The electron density distribution of the HOMO is spread over the two electron-rich subunits, while the LUMO is mainly localized on the two electron-poor subunits (Fig. S2†). The excited-state calculations (TD-DFT) show that in these compounds the absorption around 450 nm corresponds to S0 → S8 excitations, which are attributed to a large contribution of the HOMO−3 → LUMO+1 and HOMO−4 → LUMO for 5a (425 nm, fcalc = 0.1450), the HOMO−3 → LUMO and HOMO−2 → LUMO+1 for 5b (421 nm, fcalc = 0.1279), the HOMO−3 → LUMO+1 and HOMO−4 → LUMO for 6a (420 nm, fcalc = 0.3030), the HOMO−3 → LUMO and HOMO−2 → LUMO+1 for 6b (414 nm, fcalc = 0.3796). The strong absorption bands around 420 nm correspond to the S0 → S11 excitations consisting of complicated contributions of multiple transitions (see the ESI† for details). The redox properties of these dimers were examined by cyclic voltammetry (CV; 0.1 M TBAPF6, 100 mV s−1) in dichloromethane, and the onset oxidation–reduction potentials were determined relative to Fc/Fc+ (4.8 eV) (Fig. 7B). All four compounds exhibit one irreversible oxidation peak, arising from the oxidation of the electron-rich subunits. However, the CVs of these compounds show multiple reduction peaks, likely due to the coulombic interactions from the electrons between electron-poor subunits.8,21 According to their onset potentials, the LUMO/HOMO of 5a, 5b, 6a, and 6b are estimated to be −4.17/−5.87, −4.19/−6.04, −4.16/−5.89, and −4.09/−5.95 eV, respectively (relative to Fc/Fc+, 4.8 eV). The differences in potential are consistent with the differences in the respective molecular structures and DFT-computed energy potentials.
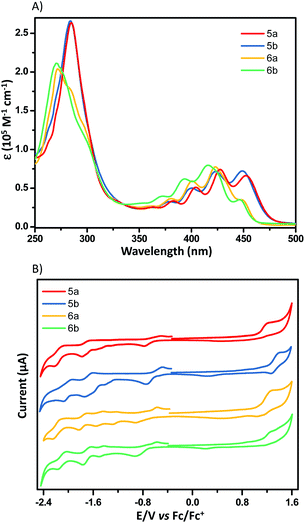 |
| | Fig. 7 (A) UV/Vis absorption of 5a, 5b, 6a, and 6b in chloroform (∼10−5 M) and (B) cyclic voltammograms of 5a, 5b, 6a, and 6b. All CVs were obtained in nitrogen-purged dichloromethane with tetrabutylammonium hexafluorophosphate (TBAPF6, 0.1 M) as the supporting electrolyte with a scan rate of 100 mV s−1. | |
Charge-carrier transport properties
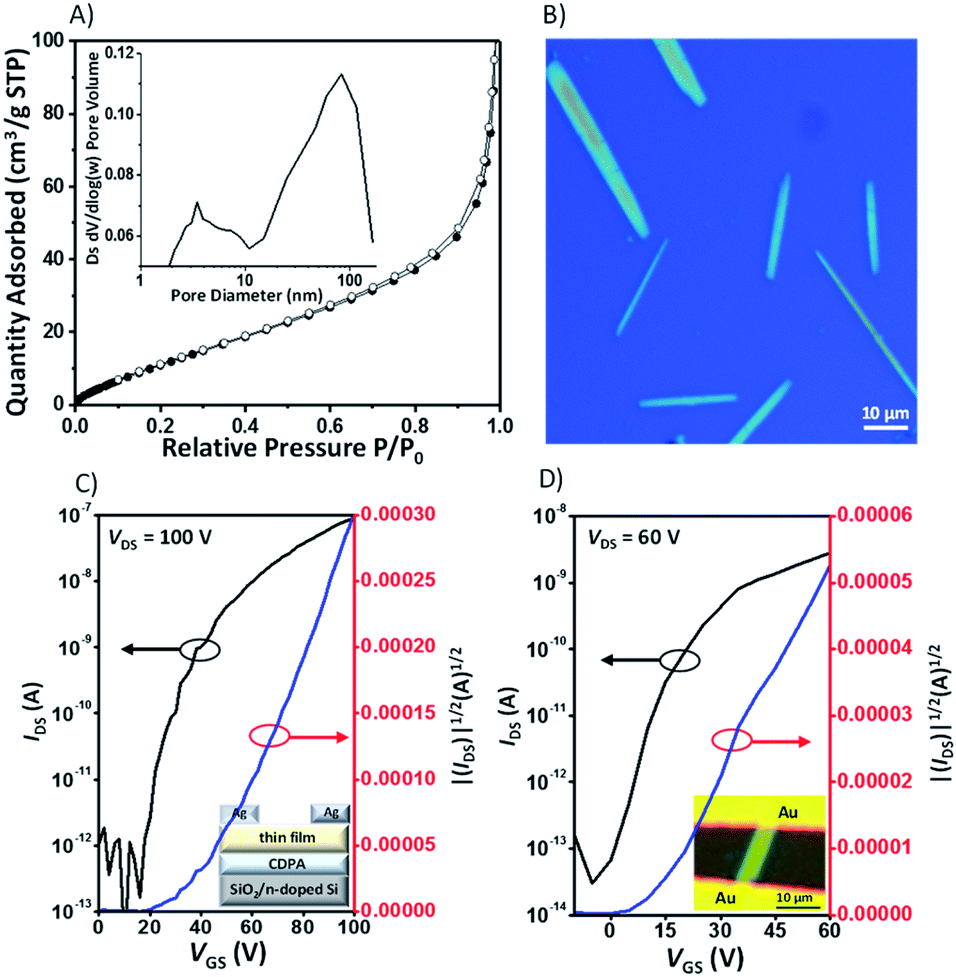
It is well-known that acene dimers have been considered as an insulator and would not be able to participate in the charge transport because the cavities created in the compounds do not provide densely packed structures.22 The Brunauer–Emmett–Teller (BET) surface area of 5a was found to be 60 m2 g−1 according to the nitrogen sorption analysis at 77 K, with the pore size distribution in the range 2.5–7.5 nm, which indicates that it is predominantly mesoporous (Fig. 8A). The low surface area value of 5a is nearly one order of magnitude lower than that of triptycene-based derivatives,4a,23 which indicates that 5a might have a relatively dense packing. Combined with the LUMO levels and the infinite π-stacked electron-poor arrays in the crystals, these X-shaped PAHs are expected, in principle, to be electron-transporting materials. Thin-film organic field effect transistors (OFETs) of these compounds were fabricated in the “bottom-gate top-contact” geometry with SiO2/AlOx as dielectric layers, which were pretreated with a SAM of 12-cyclohexyldodecylphosphonic acid (CDPA).24 The films were prepared by spin-coating a toluene solution onto substrates with Ag as the electrode (see the ESI† for details). X-ray diffraction of the film of 5a exhibits a strong Bragg reflection at 2θ = 5.06° with a d-spacing of 17.37 Å, which could be indexed to [100] according to the crystallographic data for the bulk crystal (Fig. S12†). In contrast, the molecules in the thin film 5b exhibit dissimilar packing to the bulk crystal, indicating a new phase formed in the thin film. The thin films of 6a and 6b also exhibit a strong reflection peak at 2θ = 4.58° and 4.60°, respectively, which indicate the high crystallinity and ordered arrangement of the compounds in the thin films. Unfortunately, without the crystal structure, we are not able to elucidate molecular orientations in the films of 6a or 6b (Fig. S12†). All the thin film OFETs were studied under nitrogen and showed exclusively n-type charge-carrier transport characteristics (5a, Fig. 8C). The device characteristics are summarized in Table 1. For 5a, the devices show an average mobility of (2.9 ± 0.74) × 10−3 cm2 V−1 s−1 and a maximum mobility of 4.4 × 10−3 cm2 V−1 s−1. For 5b, the devices show an average mobility of (3.2 ± 0.53) × 10−3 cm2 V−1 s−1 and a maximum mobility of 3.9 × 10−3 cm2 V−1 s−1. The mobility values of 5a and 5b are nearly one order of magnitude higher than those of their corresponding non-brominated counterparts 6a and 6b. The increase of mobility is due in part to the bromine atoms in 5a and 5b, which enhance the crystallinity in the films, as evident from the AFM images of 5a and 5b that show larger crystalline domains than their non-brominated counterparts 6a and 6b (Fig. S11†), a phenomenon observed in cellular organic semiconductors25 and azaacene-based semiconductors.26
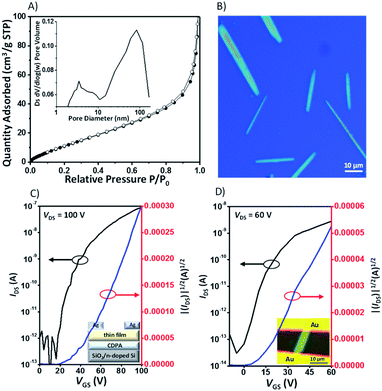 |
| | Fig. 8 (A) Nitrogen adsorption (filled symbols) and desorption isotherms (open symbols) at 77 K for 5a (insert: pore size distribution); (B) optical microscopy image of 5a microcrystals from toluene; the representative transfer curves of OFET devices based on the thin film (C) and single-crystal microbelt (D) of 5a measured under N2. | |
Table 1 Device performances of thin film transistors and single crystal transistors based on X-shaped compounds
|
|
Thin film field-effect transistors |
Single crystal field-effect transistors |
| Substrate |
Electrode |
μ
max
/μaveb (cm2 V−1 s−1) |
I
on/Ioffb |
V
t
(V) |
Substrate |
Electrode |
μ
max
/μavec (cm2 V−1 s−1) |
I
on/Ioffc |
V
t
(V) |
|
Best device.
Average value of fifteen devices.
Average value of ten devices.
|
|
5a
|
CDPA/AlOx/SiO2/Si |
Ag |
4.0 × 10−3/(2.9 ± 0.74) × 10−3 |
106 |
34 |
OTS/SiO2/Si |
Au |
0.023/0.011 ± 0.0087 |
19 |
106 |
|
5b
|
CDPA/AlOx/SiO2/Si |
Ag |
3.9 × 10−3/(3.2 ± 0.53) × 10−3 |
106 |
50 |
OTS/SiO2/Si |
Au |
0.057/0.030 ± 0.0091 |
33 |
104 |
|
6a
|
CDPA/AlOx/SiO2/Si |
Ag |
5.6 × 10−4/(4.6 ± 0.85) × 10−4 |
106 |
48 |
OTS/SiO2/Si |
Au |
0.003/0.002 ± 0.0007 |
11 |
104 |
|
6b
|
CDPA/AlOx/SiO2/Si |
Ag |
3.6 × 10−4/(3.1 ± 0.50) × 10−4 |
104 |
57 |
OTS/SiO2/Si |
Au |
0.005/0.003 ± 0.0008 |
26 |
105 |
Finally, we evaluated the intrinsic charge-carrier transport of the X-shaped semiconductors through single-crystal organic field-effect transistors (SC-OFETs), which were fabricated by a “gold strips” technique.27 The single crystals of the semiconductors were prepared by slow evaporation of the toluene solution, and they have dimensions of several μm width and hundreds of μm length (5a, Fig. 8B). The obtained crystals exhibit sharp Bragg diffraction patterns, which indicate high crystallinity in the microbelts (Fig. S14†). The diffraction pattern of the 5a microbelt is the same as that of the 5a thin film, suggesting the same molecular packing in the microbelts and thin films. For 5b, the microbelt is different in relation to the 5b thin film as it exhibits a reflection peak at 2θ = 4.58°, which could be assigned to the [110] plane, according to its single crystal structure. XRD analysis of single-crystals of 5a and 5b reveals that the electron transport is roughly parallel to the direction of electron-poor subunit aggregation. As a consequence, the stacked electron-poor subunits could bridge and enhance intermolecular charge transport between adjacent molecules, as observed in homoconjugated semiconductors.285a, 5b, 6a, and 6b show electron mobilities of 0.023, 0.057, 0.003, and 0.0047 cm2 V−1 s−1, respectively. These values are 5–10-fold higher than those of the corresponding thin-film transistors. The mobility of 5b is one of the highest values reported for highly crystalline 2D porous semiconductors,2d,29 such as 2D covalent organic frameworks (in the range of 10−6 to 10−3 cm2 V−1 s−1).29 The relatively high mobility is likely due to its dense packing with multiple intermolecular interactions between adjacent layers. To the best of our knowledge, this is the first OFET with conformationally rigid X-shaped PAHs to demonstrate the application of X-shaped PAHs as active materials in organic electronic devices.
Conclusions
We have described the utility of diazaheptacene diimides in which a [4 + 4] cyclodimerization reaction occurs in a quantitative and regioselective manner to synthesize X-shaped semiconductors. In this synthesis, the use of electron-poor and electron-rich units is crucial in directing the dimeric packing motif in the dimerization step, leading to the desired centrosymmetric compounds. The resulting compounds are thoroughly characterized by spectroscopic and redox analysis, DFT calculations, and X-ray diffraction analysis. These molecules pack in 2D layers with large open channels and strong intermolecular interactions, and are able to form active layers in OFETs with an electron mobility of up to 0.057 cm2 V−1 s−1. To the best of our knowledge, this is the first report of the semiconducting behavior in conformationally rigid X-shaped compounds. We believe that the present process would be a useful strategy for design and synthesis of electroactive X-shaped compounds. Further studies for the synthesis of even larger, X-shaped semiconductors and exploration of their application in organic sensors or photodetectors are now in progress.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
L. Z. thanks the National Science Foundation of China (NSFC) (21672020, 21871022), and Beijing Natural Science Foundation (2182049). A. L. thanks the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning. We thank Dr Wei Fan from the National University of Singapore for his helpful discussion.
Notes and references
-
(a) G. P. Bartholomew and G. C. Bazan, Acc. Chem. Res., 2001, 34, 30 Search PubMed;
(b) Z. Hassan, E. Spuling, D. M. Knoll and S. Bräse, Angew. Chem., Int. Ed., 2020, 59, 2156 Search PubMed;
(c) T. M. Swager, Acc. Chem. Res., 2008, 41, 1181 Search PubMed;
(d) A. M. Dilmaç, E. Spuling, A. de Meijere and S. Bräse, Angew. Chem., Int. Ed., 2017, 56, 5684 Search PubMed;
(e) Y. Han, Z. Meng, Y.-X. Ma and C.-F. Chen, Acc. Chem. Res., 2014, 47, 2026 Search PubMed;
(f) D. Kuck, Chem. Rev., 2006, 106, 4885 Search PubMed;
(g) J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301 Search PubMed.
-
(a) J. L. Zafra, A. M. Ontoria, P. M. Burrezo, M. Peña-Alvarez, M. Samoc, J. Szeremeta, F. J. Ramírez, M. D. Lovander, C. J. Droske, T. M. Pappenfus, L. Echegoyen, J. T. L. Navarrete, Z. Martín and J. Casado, J. Am. Chem. Soc., 2017, 139, 3095 Search PubMed;
(b) C.-S. Wang, Y.-C. Wei, K.-H. Chang, P.-T. Chou and T.-T. Wu, Angew. Chem., Int. Ed., 2019, 58, 10158 Search PubMed;
(c) M. R. Talipov, T. S. Navale and R. Rathore, Angew. Chem., Int. Ed., 2015, 54, 14468 Search PubMed;
(d) E. H. Menke, V. Lami, Y. Vaynzof and M. Mastalerz, Chem. Commun., 2016, 52, 1048 Search PubMed.
-
(a) J. D. Cook, T. J. Carey, D. H. Arias, J. C. Johnson and N. H. Damrauer, J. Phys. Chem. A, 2017, 121, 9229 Search PubMed;
(b) E. Kumarasamy, S. N. Sanders, M. J. Y. Tayebjee, A. Asadpoordarvish, T. J. H. Hele, E. G. Fuemmeler, A. B. Pun, L. M. Yablon, J. Z. Low, D. W. Paley, J. C. Dean, B. Choi, G. D. Scholes, M. L. Steigerwald, N. Ananth, D. R. McCamey, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2017, 139, 12488 Search PubMed;
(c) A. T. Gilligan, E. G. Miller, T. Sammakia and N. H. Damrauer, J. Am. Chem. Soc., 2019, 141, 5961 Search PubMed.
-
(a) B. Kohl, F. Rominger and M. Mastalerz, Org. Lett., 2014, 16, 704 Search PubMed;
(b) M. Mastalerz, Chem.–Eur. J., 2012, 18, 10082 Search PubMed;
(c) L. J. Abbott, N. B. McKeown and C. M. Colina, J. Mater. Chem. A, 2013, 1, 11950 Search PubMed;
(d) B. Kohl, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2015, 21, 17308 Search PubMed;
(e) P. Biegger, S. Stolz, S. N. Intorp, Y. Zhang, J. U. Engelhart, F. Rominger, K. I. Hardcastle, U. Lemmer, X. Qian, M. Hamburger and U. H. F. Bunz, J. Org. Chem., 2015, 80, 582 Search PubMed.
-
(a) G. Zhang, V. Lami, F. Rominger, Y. Vaynzof and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 3977 Search PubMed;
(b) D. Meng, H. Fu, B. Fan, J. Zhang, Y. Li, Y. Sun and Z. Wang, Chem.–Asian J., 2017, 12, 1286 Search PubMed;
(c) S. R. Peurifoy, E. Castro, F. Liu, X. Zhu, F. Ng, S. Jockusch, M. L. Steigerwald, L. Echegoyen, C. Nuckolls and T. J. Sisto, J. Am. Chem. Soc., 2018, 140, 9341 Search PubMed;
(d) S. R. Peurifoy, J. C. Russell, T. J. Sisto, Y. Yang, X. Roy and C. Nuckolls, J. Am. Chem. Soc., 2018, 140, 10960 Search PubMed;
(e) Y. Kim, J. Bouffard, S. E. Kooi and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 13726 Search PubMed.
-
(a) T. J. Carey, E. G. Miller, A. T. Gilligan, T. Sammakia and N. H. Damrauer, Org. Lett., 2018, 20, 457 Search PubMed;
(b) T. Hackfort and D. Kuck, Eur. J. Org. Chem., 1999, 2867 Search PubMed;
(c) B. Pei, W. Chan and A. W. M. Lee, Org. Lett., 2011, 13, 1774 Search PubMed;
(d) N. G. White and M. J. MacLachlan, J. Org. Chem., 2015, 80, 8390 Search PubMed;
(e) D. Reinhard, F. Rominger and M. Mastalerz, J. Org. Chem., 2015, 80, 9342 Search PubMed;
(f) C. L. Anderson, J. Liang, S. J. Teat, A. Garzón-Ruiz, D. P. Nenon, A. Navarro and Y. Liu, Chem. Commun., 2020, 56, 4472 Search PubMed.
-
(a) T. Kubo, S. Miyazaki, T. Kodama, M. Aoba, Y. Hirao and H. Kurata, Chem. Commun., 2015, 51, 3801 Search PubMed;
(b) T. Kodama, S. Miyazaki and T. Kubo, ChemPlusChem, 2019, 84, 599 Search PubMed.
-
(a) L. Lv, J. Roberts, C. Xiao, Z. Jia, W. Jiang, G. Zhang, C. Risko and L. Zhang, Chem. Sci., 2019, 10, 4951 Search PubMed;
(b) L. Lv, W. Sun, Z. Jia, G. Zhang, F. Wang, Z. Tan and L. Zhang, Mater. Chem. Front., 2020 10.1039/c9qm00668k.
-
(a) S. Dong, A. Ong and C. Chi, J. Photochem. Photobiol., C, 2019, 38, 27 Search PubMed;
(b) D. Xia, X. Guo, L. Chen, M. Baumgarten, A. Keerthi and K. Müllen, Angew. Chem., Int. Ed., 2016, 55, 941 Search PubMed;
(c) X. Shi, T. Y. Gopalakrishna, Q. Wang and C. Chi, Chem.–Eur. J., 2017, 23, 8525 Search PubMed;
(d) J.-H. Lamm, J. Glatthor, J.-H. Weddeling, A. Mix, J. Chmiel, B. Neumann, H.-G. Stammler and N. W. Mitzel, Org. Biomol. Chem., 2014, 12, 7355 Search PubMed;
(e) T. Geiger, A. Haupt, C. Maichle-Mössmer, C. Schrenk, A. Schnepf and H. F. Bettinger, J. Org. Chem., 2019, 84, 10120 Search PubMed.
-
(a) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 Search PubMed;
(b) B. Purushothaman, S. R. Parkin and J. E. Anthony, Org. Lett., 2010, 12, 2060 Search PubMed.
-
(a) P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774 Search PubMed;
(b) D. J. Murray, D. D. Patterson, P. Payamyar, R. Bhola, W. Song, M. Lackinger, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2015, 137, 3450 Search PubMed;
(c) M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779 Search PubMed.
-
(a) J. U. Engelhart, O. Tverskoy and U. H. F. Bunz, J. Am. Chem. Soc., 2014, 136, 15166 Search PubMed;
(b) J. U. Engelhart, B. D. Lindner, O. Tverskoy, F. Rominger and U. H. F. Bunz, Chem.–Eur. J., 2013, 19, 15089 Search PubMed;
(c) R. Mondal, C. Tönshoff, D. Khon, D. C. Neckers and H. F. Bettinger, J. Am. Chem. Soc., 2009, 131, 14281 Search PubMed;
(d) R. Einholz, T. Fang, R. Berger, P. Grüninger, A. Früh, T. Chassé, R. F. Fink and H. F. Bettinger, J. Am. Chem. Soc., 2017, 139, 4435 Search PubMed.
-
(a) U. H. F. Bunz and J. Freudenberg, Acc. Chem. Res., 2019, 52, 1575 Search PubMed;
(b) U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676 Search PubMed;
(c) Z. Zhang and Q. Zhang, Mater. Chem. Front., 2020 10.1039/c9qm00656g.
- CCDC 2012713 (2a), 2012714 (5a), and 2012715 (5b) contain the supplementary crystallographic data for this paper.†.
- S. S. Zade, N. Zamoshchik, A. R. Reddy, G. Fridman-Marueli, D. Sheberla and M. Bendikov, J. Am. Chem. Soc., 2011, 133, 10803 Search PubMed.
-
(a) M. D. Cohen, Angew. Chem., Int. Ed. Engl., 1975, 14, 386 Search PubMed;
(b) G. M. Schmidt, Pure Appl. Chem., 1971, 27, 647 Search PubMed.
- O. Berg, E. L. Chronister, T. Yamashita, G. W. Scott, R. M. Sweet and J. Calabrese, J. Phys. Chem. A, 1999, 103, 2451 Search PubMed.
- J. I. Wu, C. S. Wannere, Y. Mo, P. V. R. Schleyer and U. H. F. Bunz, J. Org. Chem., 2009, 74, 4343 Search PubMed.
- R. Huang, H. Phan, T. S. Herng, P. Hu, W. Zeng, S. Dong, S. Das, Y. Shen, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 10323 Search PubMed.
- F. Doria, M. di Antonio, M. Benotti, D. Verga and M. Freccero, J. Org. Chem., 2009, 74, 8616 Search PubMed.
-
(a) S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100 Search PubMed;
(b) S. Montanaro, D. G. Congrave, M. K. Etherington and I. A. Wright, J. Mater. Chem. C, 2019, 7, 12886 Search PubMed.
-
(a) P. Coppo and S. G. Yeates, Adv. Mater., 2005, 17, 3001–3005 Search PubMed;
(b) M. Matta, F. Biscarini and F. Zerbetto, J. Phys.
Chem. C, 2016, 120, 13942 Search PubMed.
- R. G. D. Taylor, M. Carta, C. G. Bezzu, J. Walker, K. J. Msayib, B. M. Kariuki and N. B. McKeown, Org. Lett., 2014, 16, 1848 Search PubMed.
- D. Liu, Z. He, Y. Su, Y. Diao, S. C. B. Mannsfeld, Z. Bao, J. Xu and Q. Miao, Adv. Mater., 2014, 26, 7190 Search PubMed.
-
(a) B. Zhang, R. H. Sánchez, Y. Zhong, M. Baill, M. W. Terban, D. Paley, S. J. L. Billinge, F. Ng, M. L. Steigerwald and C. Nuckolls, Nat. Commun., 2018, 9, 1957 Search PubMed;
(b) M. L. Ball, B. Zhang, T. Fu, A. M. Schttman, D. W. Paley, F. Ng, L. Venkataraman, C. Nuckolls and M. Steigerwald, Chem. Sci., 2019, 10, 9339 Search PubMed.
- H. Reiss, L. Ji, J. Han, S. Koser, O. Tverskoy, J. Freudenberg, F. Hinkel, M. Moos, A. Friedrich, I. Krummenacher, C. Lambert, H. Braunschweig, A. Dreuw, T. B. Marder and U. H. F. Bunz, Angew. Chem., Int. Ed., 2018, 57, 9543 Search PubMed.
- Q. Tang, Y. Tong, H. Li, Y. Zhu, Q. Li, W. Hu, Y. Liu and D. Zhu, Adv. Mater., 2008, 20, 1511 Search PubMed.
-
(a) H. Hamada, Y. Itabashi, R. Sahng and E. Nakamura, J. Am. Chem. Soc., 2020, 142, 2059 Search PubMed;
(b) C.-C. Wu, W.-G. Liu, W.-Y. Hung, T.-L. Liu, Y.-T. Lin and H.-W. Lin, Appl. Phys. Lett., 2005, 87, 052103 Search PubMed.
-
(a) C. Li, Y. Wang, Y. Zou, X. Zhang, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 9403 Search PubMed;
(b) J. I. Feldblyum, C. H. McCreery, S. C. Andrews, T. Kurosawa, E. J. G. Santos, V. Duong, L. Fang, A. L. Ayzner and Z. Bao, Chem. Commun., 2015, 51, 13894 Search PubMed;
(c) J. Mahmood, E. K. Lee, S. D. Sohn, N. Park, J. H. Oh, H. J. Shin and J. B. Baek, Nat. Commun., 2015, 6, 6486 Search PubMed;
(d) H. Sahabudeen, H. Qi, B. A. Glatz, D. Tranca, R. Dong, Y. Hou, T. Zhang, C. Kuttner, T. Lehnert, G. Seifert, U. Kaiser, A. Fery, Z. Zheng and X. Feng, Nat. Commun., 2016, 7, 13461 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis and characterization of the compounds; 1H NMR, 13C NMR, and HRMS of the compounds; crystallographic data for 2a, 5a, and 5b; details for photo-dissociation; theoretical calculation details; device fabrication and characterization. CCDC 2012713 (2a), 2012714 (5a), and 2012715 (5b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03744c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c and
Lei
Zhang
c and
Lei
Zhang