
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical oxidative decarboxylation and 1,2-aryl migration towards the synthesis of 1,2-diaryl ethers†
Faxiang
Bu‡
a,
Lijun
Lu‡
a,
Xia
Hu
a,
Shengchun
Wang
a,
Heng
Zhang
 *a and
Aiwen
Lei
*ab
*a and
Aiwen
Lei
*ab
aCollege of Chemistry and Molecular Sciences, The Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, Hubei, P. R. China. E-mail: aiwenlei@whu.edu.cn; hengzhang@whu.edu.cn
bNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, Jiangxi, P. R. China
First published on 2nd September 2020
Abstract
Carboxylic acid compounds are important chemicals and are widely present in various natural products. They are not only nucleophiles, but also radical precursors. Classic transition-metal-catalyzed and photochemical decarboxylation have shown their excellent site selectivity in radical chemistry. However, electrochemical decarboxylation with a long history hasn't got enough attention in recent years. In this work, the electrochemical oxidative decarboxylation and 1,2-aryl migration of 3,3-diarylpropionic acids have been introduced to construct C–O bonds with alcohols. Remarkably, this transformation can proceed smoothly without metal catalysts and external oxidants.
Introduction
1,2-Diaryl compounds are important structures in some pharmaceuticals. For example, dextropropoxyphene is an analgesic in the opioid category, and eslicarbazepine acetate is an anticonvulsant medication.1 Directly using benzyl halides to construct 1,2-diaryl compounds is a difficult process.2 In contrast, 1,2-aryl migration can afford 1,2-diaryl compounds easily.3 The 1,1-diaryl ethyl radical is the key intermediate for achieving 1,2-aryl migration. Previous reports have described some methods for producing 1,1-diaryl ethyl radicals (Scheme 1A). The first method is obtaining radical species by dehalogenation or decarboxylation.4 The second method is adding extra radicals to olefins to produce radical species.5 Intramolecular radical addition is similar to the second method.6 The fourth method is the direct oxidation of the C–H bond to give radical species.7 The addition of radicals to olefins comprises the most common method. But their products have extra functional groups, which influences their further modification. The intramolecular radical addition and C–H oxidation methods are usually limited in terms of the substrate scope, while the decarboxylation process of the first method is relatively simple and efficient.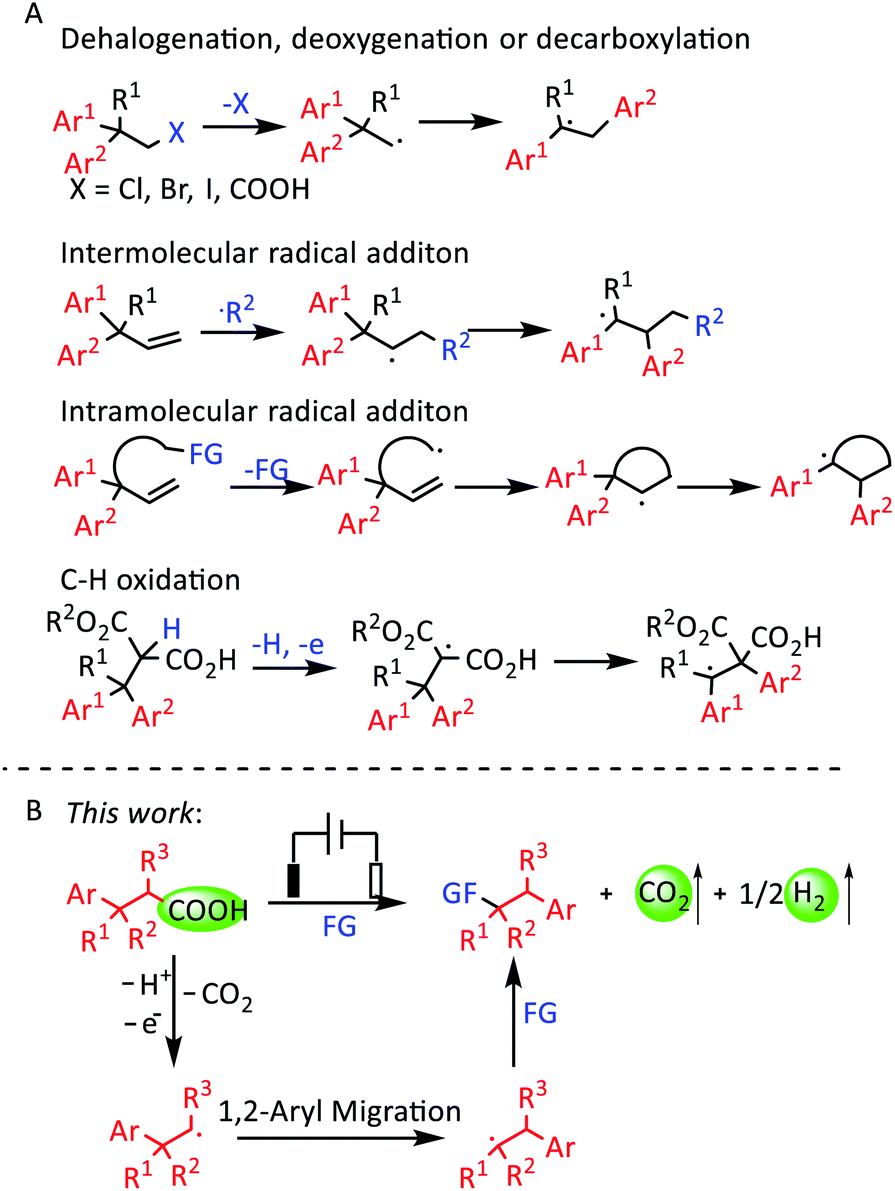 | ||
| Scheme 1 (A) The 1,2-aryl migration of different substrates. (B) Electrochemical oxidative decarboxylation and 1,2-aryl migration reaction. | ||
Carboxylic acids are common precursors of pharmaceuticals, agrochemicals and fine chemicals.8 They are commercially available at low cost and with more structural diversity. Also, they can be prepared through many mature methods.9 Carboxylic acids with the advantages of being non-toxic, stable to air and water, and easy to store and operate are the promising substitutes for organic halides due to the good site selectivity.10 In the past decade, decarboxylative cross-coupling reactions have undergone rapid development in both academia and industry. For example, Macmillan and co-workers made great contributions in photochemical decarboxylative functionalization.11 On the other hand, many breakthroughs have also been achieved in the transition-metal-catalyzed decarboxylative functionalization.12
Recently, electrochemical synthesis has become a hot topic again owing to its mild reaction conditions, which meets the requirement of green and sustainable chemistry.13 Kolbe electrolysis is one of the most famous electrochemical reactions, and involves the anodic oxidation of alkyl carboxylates to afford alkyl radicals.14 Compared with transition-metal-catalyzed and photochemical decarboxylation, electrochemical decarboxylative functionalization can proceed smoothly without transition-metal catalysts, photocatalysts, external oxidants or high temperature. In addition, CO2 and H2 are the only byproducts.15 Due to the above merits, electrochemical decarboxylation is an ideal method to obtain radical species from carboxylic acids. Herein, we report an electrochemical oxidative decarboxylation and 1,2-aryl migration of 3,3-diarylpropionic acids to synthesize a series of 1,2-diaryl ethers (Scheme 1B).
Results and discussion
3,3-Di(2-chlorophenyl)-propionic acid (1a) and MeOH (2a) were selected as model substrates (Table 1). With the combination of carbon cloth as the anode, platinum plate as the cathode, and nBu4NOAc as the electrolyte and base in the mixed solvent of MeOH and HFIP (hexafluoroisopropanol), 80% GC yield of the desired product (3aa) was obtained at 15 mA current and room temperature in 3.5 hours (entry 1). The base was important for this transformation. The yield sharply decreased to 21% with nBu4NBF4 as the electrolyte (entry 2). Replacing nBu4NOAc with other bases, such as KOMe, Li2CO3 or nBu4NOH, gave lower yields (entries 3–5). Using MeOH and DCE (1,2-dichloroethane) as co-solvents, the yield decreased to 70% (entry 6). Reducing the current to 10 mA didn't influence the reaction (entry 7). Notably, only a trace amount of product was detected upon utilizing a platinum plate as the anode (entry 8). When a nickel plate was used as the cathode, the reaction also proceeded smoothly (entry 9). In addition, when the reaction was carried out in air (entry 10) or at 0 °C (entry 11), the yields decreased slightly.|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Standard conditions: carbon cloth anode (15 mm × 15 mm × 0.36 mm), Pt plate cathode (15 mm × 15 mm × 0.3 mm), 1a (0.4 mmol), 2a (12.0 mL), nBu4NOAc (0.6 mmol), HFIP (0.7 mL), constant current, I = 15 mA, 3.5 h, room temperature, N2 atmosphere. Q = 189 C, 4.9 F. b Yields were determined by gas chromatography analysis and calibrated with naphthalene as the internal standard (isolated yield in parentheses). c n Bu4NBF4 (0.4 mmol) and base (0.6 mmol) were used. d MeOH/DCE = 8.0/4.0 mL, no HFIP. | ||
| 1 | None | 80 (75) |
| 2 | n Bu4NBF4 instead of nBu4NOAc | 21 |
| 3c | n Bu4NBF4 and KOMe instead of nBu4NOAc | 57 |
| 4c | n Bu4NBF4 and Li2CO3 instead of nBu4NOAc | 37 |
| 5 | n Bu4NOH instead of nBu4NOAc | 57 |
| 6d | MeOH/DCE as solvent | 70 |
| 7 | I = 10 mA, 5.25 h | 80 |
| 8 | Platinum plate as anode | Trace |
| 9 | Nickel plate as cathode | 74 |
| 10 | In air | 68 |
| 11 | 0 °C | 72 |
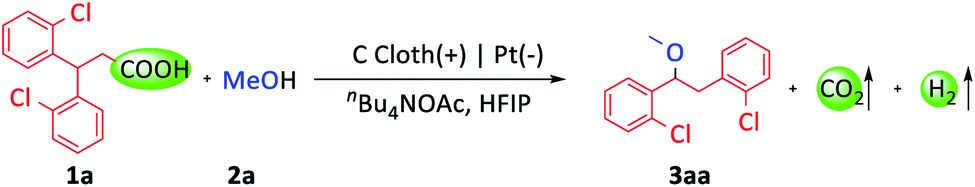
With the optimized conditions in hand, we investigated the substrate scope of this transformation. Firstly, a variety of alcohols were tested (Scheme 2). Primary, secondary and tertiary alcohols could all be tolerated (3aa–3aj, 3ba–3bf). The examples of 3ac and 3bc were noteworthy due to the widespread existence of the trifluoroethoxy group in drug molecules. Then, we focused our attention on the diarylpropionic acids. Diarylpropionic acids with ortho-substituted functional groups, such as methyl, fluorine and bromine, afforded satisfactory yields (3ca–3ea). Substrates with para-substituted electron-donating functional groups, such as methyl (3ga), tert-butyl (3ka) and trifluoromethoxy (3ma), and electron-withdrawing functional groups, such as fluorine (3ha), chlorine (3ia), bromine (3ja) and trifluoromethyl (3la), were all well compatible in this system. Meta-substituted diarylpropionic acids were also transformed into the desired product (3fa). It is worth mentioning that ortho-substituted diarylpropionic acids usually gave higher yields than para or meta substituted ones as they could inhibit the formation of side-products (ESI Schemes S1–S5†). Subsequently, we turned to study the 1,1-diarylpropionic acids with different substituted aryl groups. However, the migration selectivity between different aryl groups was unsatisfactory (3na–3ta, ESI Schemes S6–S8†).
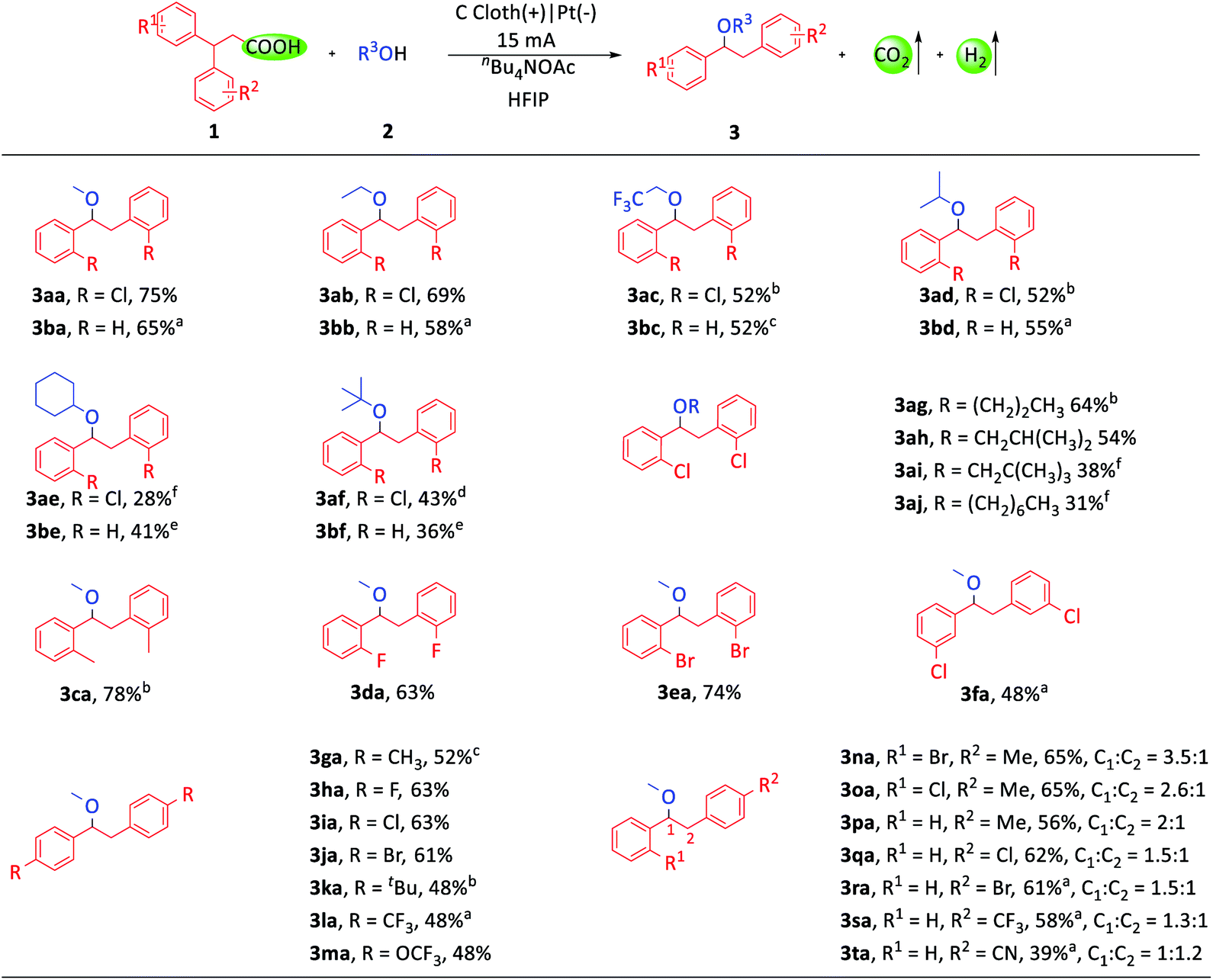 | ||
| Scheme 2 Substrate scope of 3,3-diarylpropionic acids and alcohols. Reaction conditions: 1 (0.4 mmol), 2 (12.0 mL), nBu4NOAc (1.5 equiv. based on 1), HFIP (0.7 mL), room temperature, carbon cloth anode, Pt cathode, constant current, I = 15 mA, 3.5 h. Isolated yields are shown. a2 (8.0 mL), DCE (4.0 mL), no HFIP. bT = 55 °C. c2 (8.0 mL), DCE (4.0 mL), T = 70 °C. dT = 55 °C, I = 5 mA, 10.5 h. e2 (8.0 mL), DCE (4.0 mL), T = 70 °C, I = 5 mA, 10.5 h. f1 (0.2 mmol), 2 (2.0 mL), HFIP (120 μL), T = 55 °C, I = 3.3 mA, 8 h. | ||
Moreover, we explored the applicability of the carboxylic acids with additional α- and β-site substituted groups (Scheme 3). The diarylpropionic acids with an α- or β- methyl group could afford the corresponding products (3ua–3wa). Triphenylpropionic acid gave 76% yield (3xa). Remarkably, this transformation could still proceed when one of the two aryl groups was replaced with a methoxyl or benzyl group (3ya, 3za). The wide substrate compatibility made this protocol promising for application in organic synthesis.
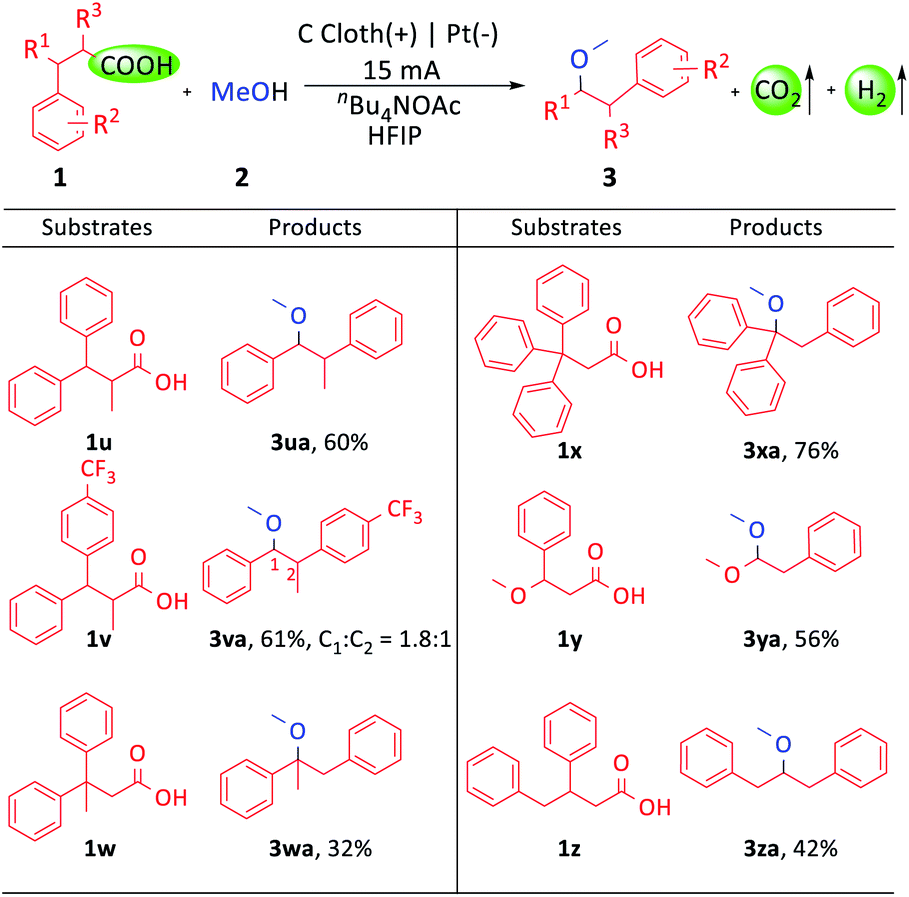 | ||
| Scheme 3 Substrate scope of carboxylic acids. Reaction conditions: 1 (0.4 mmol), 2a (12.0 mL), nBu4NOAc (1.5 equiv. based on 1), HFIP (700 μL), room temperature, constant current, I = 15 mA, 3.5 h. All yields are isolated yields. | ||
To gain further insight into the reaction mechanism, some related experiments were conducted. Firstly, the oxidation potentials of the substrates were measured by using cyclic voltammetry (ESI Fig. S1†). There was no obvious oxidation peak for 3,3-diphenylpropionic acid (1b) alone. However, the oxidation peak could be observed by adding a base into the solution of 3,3-diphenylpropionic acid (1b), which suggested that 3,3-diphenylpropionic carboxylate could be oxidized easily. In addition, 3,3-diphenylpropionic carboxylate and nBu4NOAc have similar oxidation potentials, which might indicate that they could be oxidized simultaneously during the reaction (ESI Scheme S14†).
Furthermore, EPR experiments were performed to investigate the radical intermediate produced by the oxidation of 3,3-diphenylpropionic carboxylate. With the addition of radical spin trapping reagent DMPO (5,5-dimethyl-1-pyrroline-N-oxide), a mixed signal of DMPO trapping carbon radicals (g = 2.0068, AN = 14.30 G, AH = 21.00 G) and hydrogenated DMPO (g = 2.0068, AN = 14.65 G, AH1 = 19.54 G, AH2 = 19.54 G) was identified (Fig. 1A, ESI Fig. S3C†). Meanwhile, the reaction system of the EPR trapping experiment was also tested by high resolution electrospray ionization mass spectrometry (ESI Fig. S3D†). The results demonstrated the possible existence of a carbon radical intermediate (Fig. 1CIII or IV) according to the coincidence of the molecular weight of DMPO trapping products. Moreover, BrCCl3 and O2 were used as radical trapping reagents, respectively. 22% yield of 1,1-diphenyl-2-bromoethane and 16% yield of benzophenone were obtained in the respective experiments, which indicated the possibility of the involvement of the 1,1-diphenylethyl radical (Fig. 1CIII) in the transformation (ESI Schemes S15 and S16†).
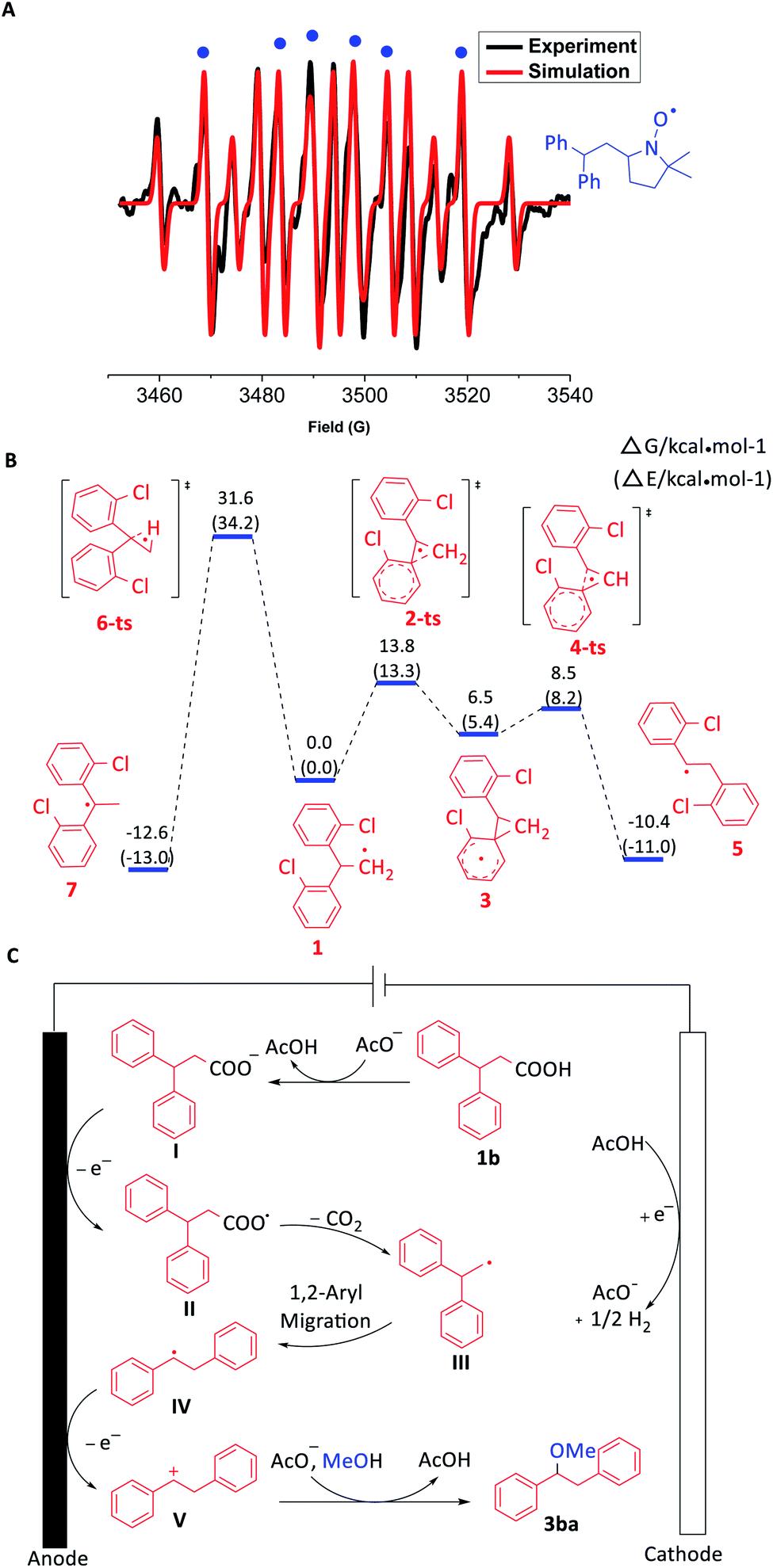 | ||
| Fig. 1 (A) EPR spectrum. (B) Free energy barriers for the 1,2-aryl migration vs. 1,2-H transfer of the radical intermediate. (C) Proposed mechanism. | ||
In order to understand the process of the aryl migration, DFT calculations were conducted (Fig. 1B). According to the calculated free energy barriers, 1,2-aryl migration (13.8 kcal mol−1) of 1,1-diarylethyl carbon radical 1 was much easier than 1,2-H transfer (31.6 kcal mol−1), which matched with the experimental results. Another possibility was that 1,1-diarylethyl carbon radical 1 was oxidized to form a 1,1-diarylethyl carbocation firstly, which further underwent 1,2-aryl migration to give the desired product. However, DFT calculation results indicated that the 1,2-H transfer is more favourable than 1,2-aryl migration for this carbocation intermediate, which didn't meet with experiment results (ESI Fig. S4†).
Based on the above mechanistic experiments and previous reports, a plausible mechanism was proposed (Fig. 1C). Firstly, 3,3-diphenylpropionic acid (1b) is deprotonated in the presence of acetate to produce carboxylate I, which was oxidized at the anode to generate carboxyl radical II. The following decarboxylation can afford the primary carbon radical III. Subsequently, benzyl carbocation V was formed through successive 1,2-aryl migration and anodic oxidation. Finally, the reaction between benzyl carbocation V and methanol can yield the desired product 3ba with the help of acetate. The acetate can be regenerated at the cathode with the liberation of dihydrogen concomitantly.
The product 1-methoxy-1,2-diphenyl ethane (3ba) could be further modified to get useful chemicals. For example, 3ba reacted with azidotrimethylsilane or allyltrimethylsilane to construct a C–N or C–C bond (Scheme 4A, 6a, 6b) by Fe-catalysis.16 In addition, the elimination reaction of 3ba afforded a C–C double bond with the leaving of the methoxy group (6c).17 Notably, the methoxy group could act as the directing group to achieve ortho-C–H olefination of 3ba (6d).18 When 3-phenyl-3-(2-bromophenyl)propionic acid was used as the substrate, two isomers were obtained (Scheme 4B, 5aa, ESI Scheme S13†). When the mixture was employed as a starting material directly in the palladium-catalyzed intramolecular cyclization, the sole product 9,10-dihydro-9-methoxy-phenanthrene was obtained (6f).19 Remarkably, this reaction could proceed smoothly on the gram scale. 1.17 g 3ba was obtained by using 50 mA current without changing the size of the electrodes (Scheme 4C).
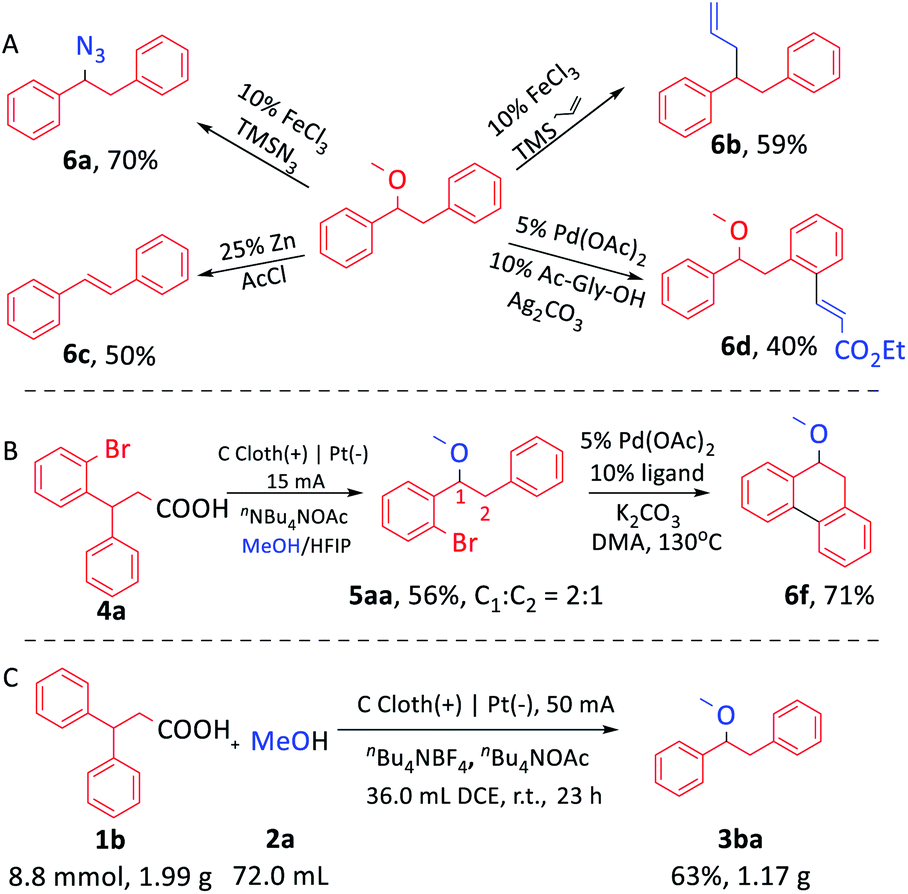 | ||
| Scheme 4 (A) Further functionalization of 1-methoxy-1,2-diphenyl ethane. (B) Derivatization of 5aa. (C) Gram scale electrochemical synthesis of 3ba. | ||
Conclusions
In summary, the strategies of decarboxylation and 1,2-aryl migration have been integrated to achieve electrochemical oxidative cross-coupling between 3,3-diarylpropionic acids and alcohols to yield 1,2-diaryl ethers under mild conditions in one step. The good substrate compatibility and the diversified further functionalization of the target 1,2-diaryl ethers demonstrate the potential application of this protocol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21520102003) and the Hubei Province Natural Science Foundation of China (2017CFA010). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated. The numerical calculations in this paper have been done on the supercomputing system in the Supercomputing Center of Wuhan University. Dedicated to P. H. Dixneuf for his outstanding contribution to organometallic chemistry and catalysis.Notes and references
- (a) W. M. O'Neill, G. W. Hanks, L. White, P. Simpson and K. Wesnes, Eur. J. Clin. Pharmacol., 1995, 48, 447 CrossRef; (b) C. Elger, P. Hala'sz, J. Maia, L. Almeida and P. Soares-da-Silva, Epilepsia, 2009, 50, 454 CrossRef.
- (a) A. H. Stoll, A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 606 CrossRef; (b) M. P. Drapeau, I. Fabre, L. Grimaud, I. Ciofini, T. Ollevier and M. Taillefer, Angew. Chem., Int. Ed., 2015, 54, 10587 CrossRef; (c) G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15, 4398 CrossRef CAS.
- Z. M. Chen, X. M. Zhang and Y. Q. Tu, Chem. Soc. Rev., 2015, 44, 5220 RSC.
- (a) L. Li, P. Cai, Q. Guo and S. Xue, J. Org. Chem., 2008, 73, 3516 CrossRef CAS; (b) N. Tsuji, Y. Kobayashi and Y. Takemoto, Chem. Commun., 2014, 50, 13691 RSC; (c) W. A. Bonner and F. D. Mango, J. Org. Chem., 1963, 29, 430 CrossRef.
- (a) W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480 CrossRef CAS; (b) Z. M. Chen, W. Bai, S. Wang, B. Yang, Y. Tu and F. Zhang, Angew. Chem., Int. Ed., 2013, 52, 9781 CrossRef CAS; (c) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962 CrossRef CAS; (d) A. Bunescu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3132 CrossRef CAS; (e) S. Wang, C. Chuang, J. Lee and S. Liu, Tetrahedron, 1999, 55, 2273 CrossRef CAS.
- W. R. Bowman and J. M. D. Storey, Chem. Soc. Rev., 2007, 36, 1803 RSC.
- Z. Cong, T. Miki, O. Urakawa and H. Nishino, J. Org. Chem., 2009, 74, 3978 CrossRef CAS.
- K. P. C. Vollhardt and N. E. Schore, Organische Chemie, 3. Aufl., Wiley-VCH, Weinheim, 2000, p. 893 Search PubMed.
- L. S. Hegedus and L. Wade, in Compendium of Organic Synthetic Methods, John Wiley & Sons, Hoboken, NJ, 1997, vol. 3, p. 8 Search PubMed.
- (a) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864 CrossRef CAS; (b) B. Song, T. Knauber and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 2954 CrossRef CAS; (c) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS.
- (a) Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83 CrossRef CAS; (b) N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701 CrossRef CAS; (c) L. Chu, J. M. Lipshultz and D. W. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929 CrossRef CAS.
- (a) J. Zhang, J. R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144 CrossRef CAS; (b) K. Takamatsu, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2017, 56, 5353 CrossRef CAS; (c) Q. Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196 CrossRef CAS; (d) P. J. Moon, S. Yin and R. J. Lundgren, J. Am. Chem. Soc., 2016, 138, 13826 CrossRef CAS.
- (a) J. Xiang, et al. , Nature, 2019, 573, 398 CrossRef CAS; (b) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS; (c) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575 CrossRef CAS; (d) R. Hayashi, A. Shimizu and J. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400 CrossRef CAS; (e) P. Xiong, H. H. Xu and H. C. Xu, J. Am. Chem. Soc., 2017, 139, 2956 CrossRef CAS; (f) Y. Qiu, W. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828 CrossRef CAS; (g) X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195 CrossRef CAS.
- (a) H. J. Schaefer, Top. Curr. Chem., 1990, 152, 91 CrossRef CAS; (b) G. N. Wanyoik, O. Onomura, T. Maki and Y. Matsumura, Org. Lett., 2002, 4, 1875 CrossRef; (c) L. B. Rodewald and M. C. Lewis, Tetrahedron, 1971, 27, 5273 CrossRef CAS; (d) R. F. Garwood, Naser-ud-Din, C. J. Scott and B. C. L. Weedon, J. Chem. Soc., Perkin Trans. 1, 1973, 2714 RSC.
- (a) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palmad and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099 RSC; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS; (c) H. Kurihara, T. Fuchigami and T. Tajima, J. Org. Chem., 2008, 73, 6888 CrossRef CAS.
- Y. Sawama, R. Goto, S. Nagata, Y. Shishido, Y. Monguchi and H. Sajiki, Chem.–Eur. J., 2014, 20, 2631 CrossRef CAS.
- S. Bhar and B. Ranu, J. Org. Chem., 1995, 60, 745 CrossRef CAS.
- G. Li, D. Leow, L. Wan and J. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS.
- L. Campeau, M. Parisien, M. Leblanc and K. Fagnou, J. Am. Chem. Soc., 2004, 126, 9186 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03708g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |