
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxoiron(V) mediated selective electrochemical oxygenation of unactivated C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C bonds using water as the oxygen source†
C bonds using water as the oxygen source†
Bittu
Chandra
 ,
Hellan
K. M.
,
Santanu
Pattanayak
and
Sayam Sen
Gupta
*
,
Hellan
K. M.
,
Santanu
Pattanayak
and
Sayam Sen
Gupta
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata Mohanpur, West Bengal, India-741246. E-mail: sayam.sengupta@iiserkol.ac.in
First published on 24th September 2020
Abstract
An efficient electrochemical method for the selective oxidation of C–H bonds of unactivated alkanes (BDE ≤97 kcal mol−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of alkenes using a biomimetic iron complex, [(bTAML)FeIII-OH2]−, as the redox mediator in an undivided electrochemical cell with inexpensive carbon and nickel electrodes is reported. The O-atom of water remains the source of O-incorporation in the product formed after oxidation. The products formed upon oxidation of C–H bonds display very high regioselectivity (75
C bonds of alkenes using a biomimetic iron complex, [(bTAML)FeIII-OH2]−, as the redox mediator in an undivided electrochemical cell with inexpensive carbon and nickel electrodes is reported. The O-atom of water remains the source of O-incorporation in the product formed after oxidation. The products formed upon oxidation of C–H bonds display very high regioselectivity (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3°:2° for adamantane) and stereo-retention (RC ∼99% for cyclohexane derivatives). The substrate scope includes natural products such as cedryl acetate and ambroxide. For alkenes, epoxides were obtained as the sole product. Mechanistic studies show the involvement of a high-valent oxoiron(V) species, [(bTAML)FeV(O)]− formed via PCET (overall 2H+/2e−) from [(bTAML)FeIII-OH2]− in CPE at 0.80 V (vs. Ag/AgNO3). Moreover, electrokinetic studies for the oxidation of C–H bonds indicate a second-order reaction with the C–H abstraction by oxoiron(V) being the rate-determining step.
:1, 3°:2° for adamantane) and stereo-retention (RC ∼99% for cyclohexane derivatives). The substrate scope includes natural products such as cedryl acetate and ambroxide. For alkenes, epoxides were obtained as the sole product. Mechanistic studies show the involvement of a high-valent oxoiron(V) species, [(bTAML)FeV(O)]− formed via PCET (overall 2H+/2e−) from [(bTAML)FeIII-OH2]− in CPE at 0.80 V (vs. Ag/AgNO3). Moreover, electrokinetic studies for the oxidation of C–H bonds indicate a second-order reaction with the C–H abstraction by oxoiron(V) being the rate-determining step.
Introduction
Electrochemical oxidation presents an attractive alternative to traditional chemical reagents for large-scale applications, mostly owing to the generation of less toxic waste than that produced by current chemical processes.1–4 However, electrochemical oxidation of unactivated C–H bonds, the most ubiquitous structural motifs in complex natural products, remains a significant challenge. The difficulty in such oxidation is the high redox potentials required to oxidize such moieties by direct electrolysis (frequently above 3.0 V vs. SCE, which is significantly higher than the thermodynamic potential).5,6 A solution to this problem was envisaged by the use of “electron–proton transfer mediator” (EPTM) molecules7 such as N-hydroxyphthalimide (NHPI), quinuclidine, and TEMPO, which have allowed the oxidation of compounds with C–H, including hydrocarbons8,9 and alcohols.3,10,11 In the electrochemical oxidation of compounds with unactivated C–H bonds using an EPTM, the first step involves the H-atom abstraction from the C–H bond by oxidized N-containing tertiary amines (especially quinuclidine) via proton-coupled electron transfer (PCET).8 Subsequently, the radical formed reacts with dioxygen to form an oxygenated product via a radical process, which significantly reduces the regioselectivity of these oxidation reactions.12–15 In Nature, the selectivity in oxidation is achieved by the catalytic generation of high-valent oxometal species, especially high-valent oxoiron intermediates.16–21 These reactive intermediates abstract H-atoms from C–H bonds via PCET and subsequently hydroxylate via a “rebound mechanism, a process that avoids the formation of long-lived radical intermediates that can escape into the bulk”. These high-valent oxometal intermediates are typically formed upon activation of the enzyme-bound metal ion with chemical oxidants such as O2 (together with a biological e−/H+ donor) or with H2O2. Alternatively, high-valent oxometal intermediates are also generated via a PCET process from metal ions ligated to aquo ligands (oxidation of [M − OH2]n+ to [M(O)]n+2), which is postulated to occur in the active site of photosystem II.22,23 Such a PCET process has been used to generate oxometal complexes from their corresponding aqua-bound metal precursors in synthetic complexes of first and second-row transition metals.24–29 For example, RuVI/RuV-oxo species are synthesized electrochemically via PCET from corresponding RuII–OH2 complexes and explored for the oxidation of C–H bonds (e.g. benzyl alcohol, ethylbenzene, and 3° C–H bonds of amine derived substrates).30–32 Recently, a [FeIII-TAML] (TAML = tetra-amido macrocyclic ligand)33 complex has been shown to oxidize activated benzylic C–H bonds via electrochemical formation of a dimeric μ-O-FeIV2 intermediate in a divided cell.34 The advantage of such electrochemical oxidation is that it eliminates the necessity of terminal oxidants like O2, H2O2, or mCPBA. In all these examples, the O-atom of water remains the source of the O-incorporation in the products. Recently, heterogenized iron porphyrins have been used to catalytically oxidize hydrocarbons, including unactivated C–H bonds using molecular O2 at cathodic potentials.35 However, the selective homogeneous electrochemical oxidation of unactivated C–H and CC bonds in organic substrates using water as the O-atom source via the formation of high-valent oxoiron intermediates has been elusive. We have earlier demonstrated that the 5th generation biuret modified TAML, [Et4N]2[Fe-bTAML(Cl)], catalyzes the oxidation of 3° C–H bonds with unprecedented selectivity over 2° C–H bonds using NaOCl or mCPBA as the terminal oxidant.36,37 Epoxidation of CC bonds has also been demonstrated with both NaOCl and O2.38,39 The active intermediate involved in this reaction was identified to be a room temperature stable oxoiron(V) intermediate, [(bTAML)FeV(O)]− [in short, oxoiron(V) or FeV(O)] which has been extensively characterized spectroscopically.36,40 However, this reaction utilizes stoichiometric chemical oxidants, and concurrently produces toxic waste such as m-chlorobenzoic acid at the end of the reaction. Recently, we have demonstrated that this oxoiron(V) intermediate can also be generated electrochemically both in water and in a mixed CH3CN:H2O solvent.41 Since this generation of oxoiron(V) uses only water and electricity, coupling this to the catalytic oxidation of C–H bonds would constitute a clean synthetic process. In this work, we demonstrate the selective electrochemical alkane hydroxylation and epoxidation by employing [(bTAML)FeIII(OH2)]− as the catalyst and water as the oxygen atom source. The substrate included cedryl acetate, a natural product, where selective hydroxylation of an unactivated 3° C–H bond was demonstrated. The electrochemical oxidations were carried out in a simple undivided cell using inexpensive electrodes made from carbon and nickel. To the best of our knowledge, this represents the first example of the use of an iron-complex to catalyze electrochemically selective oxidation of strong C–H bonds (with BDE ≤97 kcal mol−1) and CC bonds using water as the O-atom donor (Scheme 1).
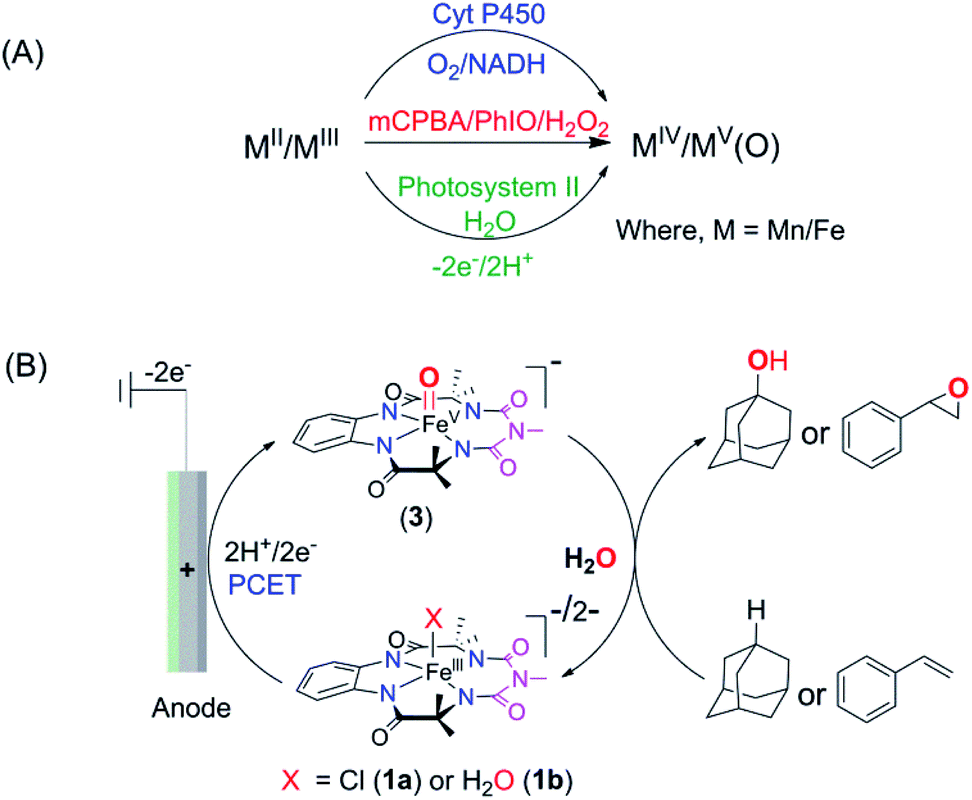 | ||
| Scheme 1 (A) Various approaches of forming high-valent oxometal complexes. (B) Electrochemical approach of forming an oxoiron(V) complex from 1a/1b followed by oxidation of alkanes and alkenes using water as the O-atom source. | ||
Results and discussion
The electrochemical properties of the bTAML complexes 1a and 1b were investigated in 0.1 M Bu4NPF6 acetonitrile solution using glassy carbon (GC) as the working electrode, Pt wire as a counter electrode, and Ag/AgNO3 (0.01 M) as the reference electrode. Complex 1a with an axial chloro (Cl) ligand was converted to its corresponding axially aqua (H2O) complex (1b, PPh4 salt) by a method published before for similar complexes.41 X-ray diffraction quality single crystals of 1b were grown from a concentrated water solution over a week (Fig. S1†). For complex 1a, a reversible one-electron wave at E1/2 = 0.13 V with a near Nernstian peak-to-peak separation of ΔEp = 66 mV was observed by cyclic voltammetry (CV). A second quasi-reversible one-electron wave at E1/2 = 0.72 V with an almost similar peak-to-peak separation of ΔEp = 66 mV was also seen. The peak-to-peak separation of 66 mV of the first and second oxidation process indicates that the processes at 0.18 V and 0.72 V are both 1e− redox processes. We assigned these redox processes to the FeIV/FeIII and FeV/FeIV redox couple, respectively (Fig. 1A). Variation of the scan rates shows that the peak currents (ip) associated with both of the redox events vary linearly with the square root of the scan rates (Fig. S4†) and follow Randles–Sevcik diffusion eqn (1).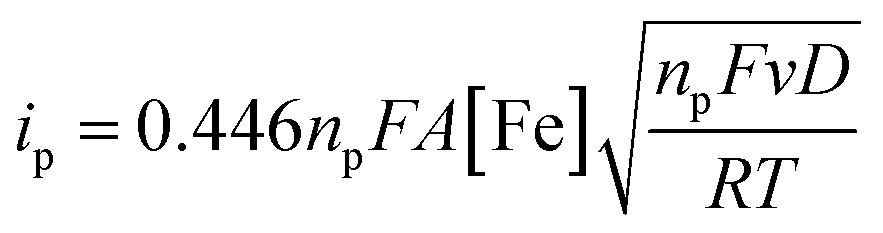 | (1) |
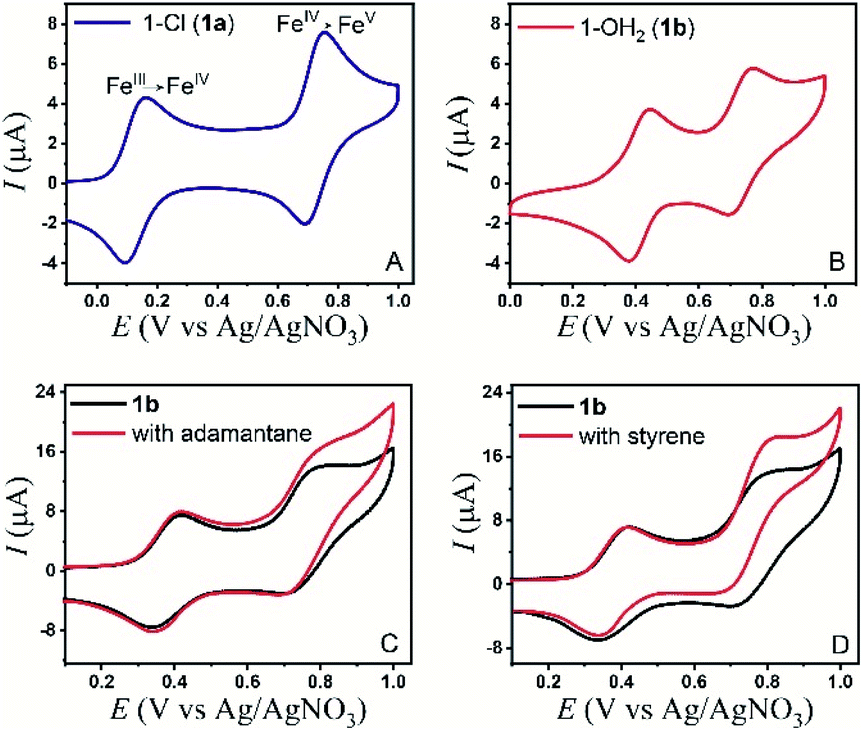 | ||
| Fig. 1 (A) CV of 1a (0.2 mM) in CH3CN, scan rate = 100 mV s−1. (B) CV of 1b (0.2 mM) in CH3CN (95%)–H2O (5%), scan rate = 100 mV s−1. (C) CV of 1b (0.2 mM) with adamantane (30 mM) in CH3CN (90%)–H2O (10%) with a scan rate of 300 mV s−1 and (D) CV of 1b (0.2 mM) with styrene (4 mM) in CH3CN (90%)–H2O (10%) with a scan rate of 300 mV s−1. | ||
In the CV of 1b, which contains an axial H2O ligand instead, the corresponding FeIV/FeIII was shifted to 0.41 V vs. Ag/AgNO3 in CH3CN (95%)–H2O (5%). The peak shift to a higher potential for 1b is reflective of the change in the overall charge of the FeIIIbTAML complex from −2 to −1, upon replacement of the axial anionic Cl ligand with the neutral OH2, in line with similar observations reported earlier.42 Besides, the greater solvation of anionic species due to an increase in the solvent polarity as a result of water addition could also lead to this shift.43 The one-electron redox event at 0.41 V was found to be pH-dependent when CVs and DPVs were recorded at different pH (CH3CN, 80% – 5 mM phosphate buffer, 20%; Fig. S9†). In comparison to that of CH3CN (100%), the FeV/FeIV peak of the catalyst (1a) in CH3CN (90%)–H2O (10%) slightly shifted anodically to an E1/2 of 0.74 V vs. Ag/AgNO3 (Fig. 1B). This change observed might be due to bias voltage change because of the presence of a mixed solvent. However, in contrast to the FeIV/FeIII peak, the FeV/FeIV peak is pH-independent (Fig. S9†). Additionally, the same shifts were observed when the DPV of 1a was performed in CH3CN (90%)–H2O (10%) (Fig. S7†), indicating that the axial chloride ligand was replaced by water under these conditions, as has been reported earlier.41,42 For 1b, the peak currents (ip) also vary linearly with the square root of scan rates (0.01 to 1 V s−1) for both the redox processes (Fig. S5†).
The controlled potential electrolysis (CPE) of a dilute solution (60 μM) of 1b in CH3CN (90%)–H2O (10%) at 0.80 V vs. Ag/AgNO3 for 60 s led to the immediate formation of a light green solution (Fig. S2†). This solution displayed UV-Vis peaks (445 nm and 613 nm), and HR-MS spectra that are characteristic of the well-characterized [(bTAML)FeV(O)]− (3) that had been electrochemically synthesized and reported by our group earlier.41 When the same CPE was carried out at higher concentrations (>0.2 mM of 1b) in CH3CN (90%)–H2O (10%), the formation of a violet species (UV-Vis peaks at 421 nm and 750–1050 nm) characteristic of μ-O-FeIV2 (2) was observed (Fig. S3†).
All these studies indicate that for 1b, the second peak in the CV at E1/2 = 0.74 V can be assigned to a [(bTAML)FeV(O)]−/[(bTAML)FeIV(O)]2− couple (viz. spectroelectrochemistry and pH-independent CV). Since the variation of ip with the square root of the scan rate indicated a diffusion-controlled 1e− redox process, the possibility of an EC mechanism, during which the electro-generated oxoiron(V) underwent very fast comproportionation with bulk 1b to form the dimeric μ-O-FeIV2 complex38 on the CV time scale was excluded. The formation of the μ-O-FeIV2 complex, observed in CPE experiments at higher concentrations of 1b, was a result of electro-generated oxoiron(V) diffusing out of the reaction layer and undergoing a comproportionation reaction with 1b in bulk (Fig. S3†).
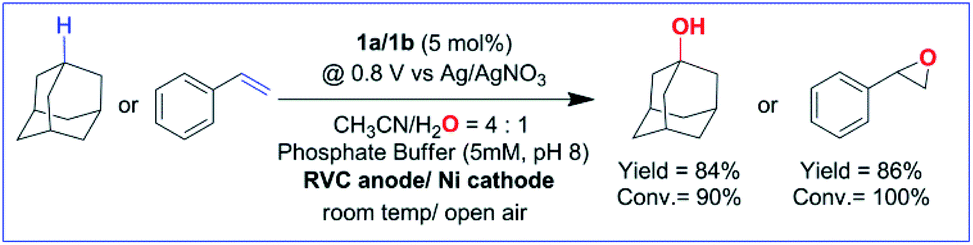
The electrochemical generation of oxoiron(V) at 0.80 V on the CV time scale prompted us to explore the possibility of electrochemical oxidation of substrates containing unactivated C–H and CC bonds. Such substrates, which have C–H bonds with BDE ≤97 kcal mol−1, have been shown to react with the chemically synthesized oxoiron(V) with high selectivity and fast reaction rates.36,37 The CVs of 1b recorded in 0.1 M Bu4NPF6 CH3CN (90%)–H2O (10%) medium in the presence of either styrene or adamantane show an almost unperturbed first oxidation wave at Ep,a = 0.40 V, and a slight increase in current at an Ep,a of 0.80 V (Fig. 1C, D and S10†). The enhancement of the current in the second oxidation wave was indicative of a chemical reaction that was coupled to the oxidation of 1b. Further, the peak currents (ip) did not vary linearly with the square root of scan rates (Fig. S6†). The possibility of the chemical reaction was ascertained by performing CPE at 0.80 V (vs. Ag/AgNO3) in the presence of either adamantane or styrene for 6 hours. GC–MS analysis of the reaction medium showed the formation of styrene oxide when styrene was added during CPE. For adamantane, predominantly 1-adamantanol was observed as the product with ∼3% 2-adamantanone, which indicated a selectivity of 75:1 for 3° C–H bonds over 2° C–H bonds in adamantane. The anodic CV waveform observed in the presence of the strong C–H bond (adamantane) was indicative of low substrate consumption at a high scan rate of 300 mV s−1 (Fig. 1C) as compared to a low scan rate of 50 mV s−1 (Fig. 2C). In both the CV traces, the cathode waveform displays a hybrid peak-plateau shape, where the initial peak shape observed due to depletion of oxoiron(V) at the reaction layer is controlled by a plateau resulting from the slow subsequent catalytic oxidation reaction.44,45 The formation of styrene oxide and 1-adamantanol during the CPE experiments prompted us to perform bulk electrochemical oxidation reactions with a variety of substrates containing C–H bonds with BDE ≤97 kcal mol−1 and CC bonds. All the electrochemical oxidation reactions were performed under constant potential electrolysis (CPE) conditions in an IKA ElectraSyn instrument using a simple 3-electrode undivided electrochemical cell, consisting of a working electrode (RVC), a counter electrode (Ni foam) and a reference electrode (Ag/AgNO3). A reaction mixture of 1a/1b (5 mol%), substrate, and Bu4NPF6 (0.1 M) as the supporting electrolyte in (4:1) CH3CN–H2O was maintained at a constant potential of 0.80 V (vs. Ag/AgNO3) at ambient temperature and pressure. No product formation was observed when the reaction was carried out either in the absence of the catalyst (1a/1b) or water or at constant potential less than 0.80 V (Table S1†). Further, no changes in the yield and the composition of the products were observed for the reaction performed in the presence or absence of O2. Finally, optimization of the reaction conditions showed that the addition of pH 8 phosphate buffer (5 mM) accelerated the catalytic reaction but the yield of the reaction remained unchanged (Table S1†). Phosphate promoted acceleration of reaction rates has been reported by us earlier for the oxidation of C–H bonds using Fe-bTAML and chemical oxidants.37 Since the second redox process at 0.80 V was unaffected by the presence of phosphate buffer, the catalytic reactions were carried out using pH 8 phosphate buffer (5 mM) (Fig. S8† and S9†).
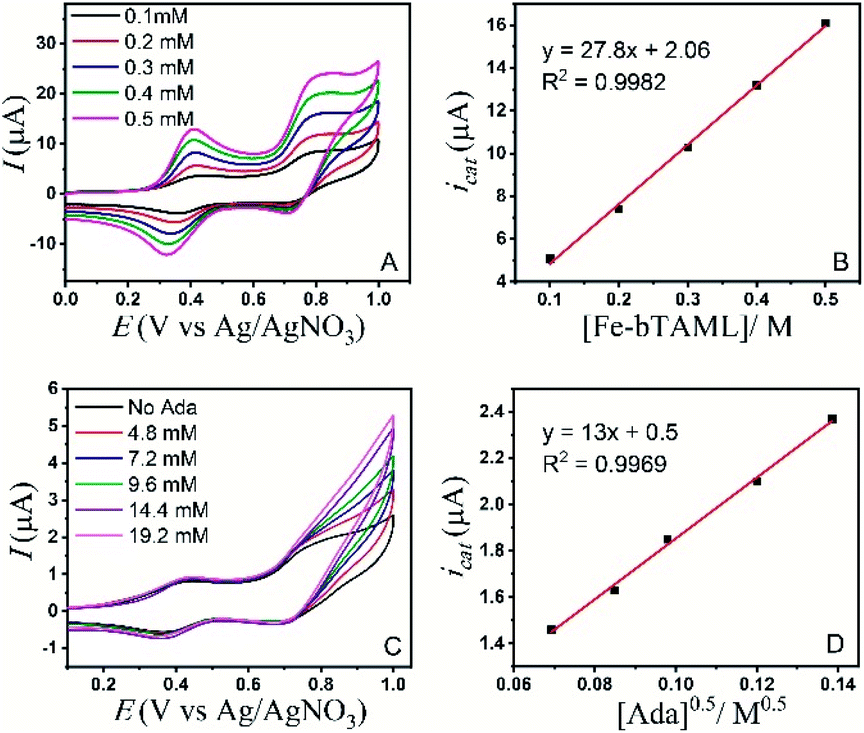 | ||
| Fig. 2 (A) CV of 1b in the presence of adamantane (5 mM) in CH3CN (90%)–H2O (10%) with varying concentration of the catalyst (scan rate = 300 mV s−1). (B) Plot of icatvs. [Fe-bTAML]. (C) CV of 1b (0.08 mM) in CH3CN (90%)–H2O (10%) with varying concentration of adamantane (scan rate = 50 mV s−1). (D) Plot of icatvs. [Ada]0.5. | ||
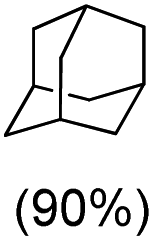
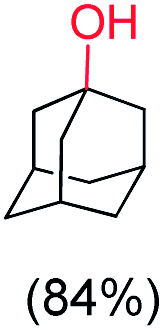
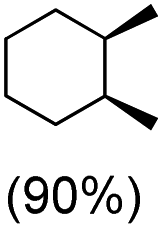
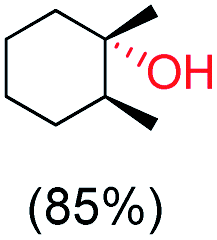
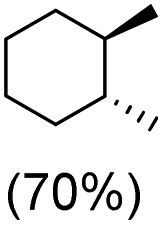
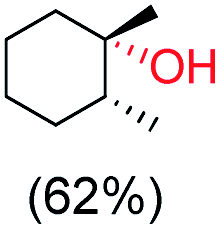
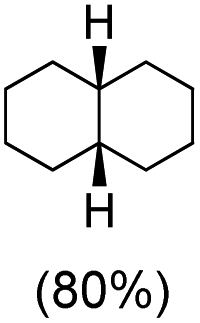
With the optimized reaction conditions, various organic substrates containing 3° C–H bonds with BDE ≤97 kcal mol−1 and activated 2° C–H bonds were explored using this electrochemical method (Table 1 & Fig. S17–S24†). Oxidation at the 3° position occurred preferentially in the presence of the statistically more abundant 2° C–H bond, with excellent conversion, yield, and retention of configuration (RC). For example, oxidation of adamantane, possessing twelve 2° C–H (BDE = 98.5 kcal mol−1)46 and four 3° C–H bonds (BDE = 96.5 kcal mol−1),46 afforded very high yields of 1-adamantanol (∼84%) with very high regioselectivity (i.e., 75:1) of 3° C–H over 2° C–H bonds (Table 1; entry 1). Moreover, a moderate value of faradaic efficiency (∼42%) was observed (Table S2†), which suggests a parallel competitive reaction involving the reaction of electro-generated oxoiron(V) with 1b, resulting in the formation of the μ-O-FeIV2 dimer. cis-1,2-Dimethylcyclohexane (3° C–H BDE = 93.9 kcal mol−1),46trans-1,2-dimethylcyclohexane (3° C–H BDE = 97.4 kcal mol−1),46 and cis-decalin (3° C–H BDE = 93.5 kcal mol−1)46 (Table 1; entries 2, 3 and 4) are the cyclohexane derivatives where the stereochemical orientation of the 3° C–H bonds (axial or equatorial) governs the regioselectivity outcome of the reaction. For cis-dimethylcyclohexane and cis-decalin, mainly 3° hydroxylated products were obtained in 85% and 76% yields, respectively (along with ∼99% RC). On the other hand, electrochemical oxidation of trans-dimethylcyclohexane, afforded moderate yields (62%). Besides, trans-dimethylcyclohexane exhibits compromised regioselectivity, resulting in the formation of both the alcohol and ketone in a ratio (3°:2°) of 97.8:2.2. The difference in reactivity between the cis-isomer (cis-dimethylcyclohexane and cis-decalin) and trans-isomer (trans-dimethylcyclohexane) can be attributed to the strain release in the transition state for the cis-isomers.47 The results obtained in the electrochemical oxidation of these hydrocarbons mirror those obtained in chemical oxidations with complex 1a and mCPBA/NaOCl as the oxidant.36,37 This supports the likely involvement of a high-valent oxoiron intermediate during the electrochemical oxidation reactions with 1a/1b.
| Entry | Substrate (% conversion) | Products (% yield) | Selectivity |
|---|---|---|---|
|
a Reaction conditions: 1a/1b (0.75 mM) and substrate (15 mM) in acetonitrile aqueous phosphate buffer (4:1 v/v, 5 mM, pH ∼8) at a constant potential of 0.80 V (vs. Ag/AgNO3) at room temperature for 10 h. Yields and conversions were estimated by GC–MS.
|
|||
| 1 |
|
|
3°:2° = 75:1 |
| 2 |
|
|
cis:trans = 99:1, 3°:2° = 99:1 |
| 3 |
|
|
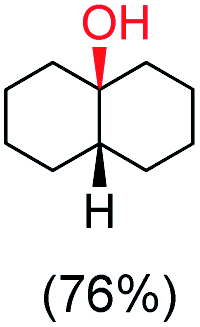
trans:cis = 99:1, 3°:2° = 87.8:22.2 |
| 4 |
|
|
cis:trans = 99:1, 3°:2° = 98.3:1.7 |
| 5 |
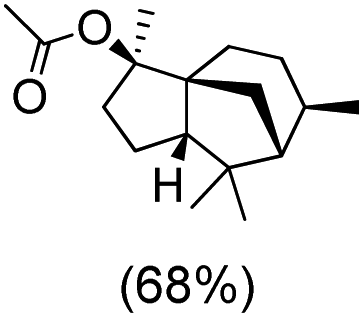
|
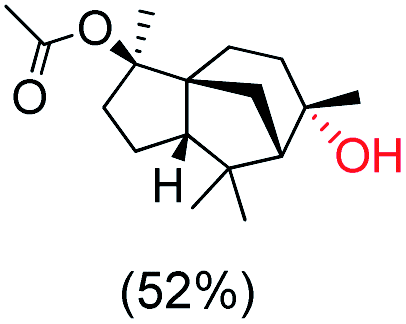
|
Only alcohol |
| 6 |
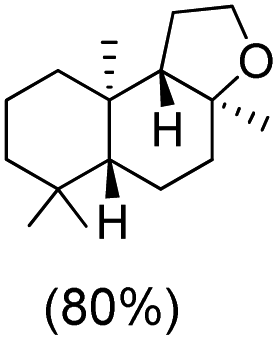
|
|
Only ketone |
| 7 |
|
|
Only ketone |
| 8 |
|
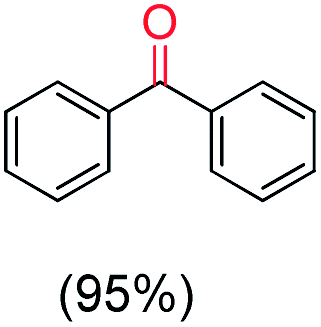
|
Only ketone |
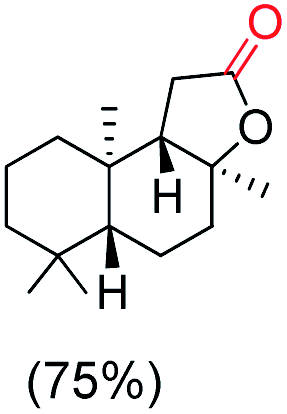
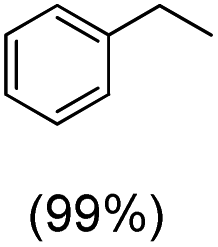
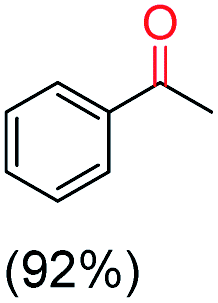
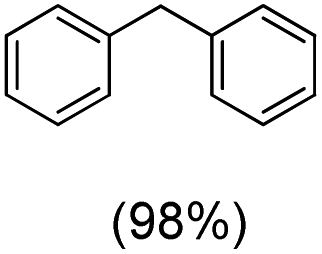
The protocol for oxidation of 3° C–H bonds in simple hydrocarbons was then extended to natural product derivatives. For example, cedryl acetate, a natural product derivative of cedrol, a sesquiterpene alcohol, found in essential oil and used in the chemistry of aroma compounds, having a rigid structure with five 3° C–H bonds, affords a single hydroxylated product in 52% yield (Table 1, entry 5). Ambroxide, a naturally occurring terpenoid used in perfumery, undergoes oxidation at the alpha ethereal C–H bond predominantly among many other electronically and sterically accessible secondary and tertiary C–H bonds. This results in the formation of the ketone product, sclareolide in 75% yield (Table 1, entry 6). The electrochemical oxidation of ambroxide was also attempted at a 1 mmol scale using the same electrochemical setup, and sclareolide was formed in 80% yield (based on conversion). We believe that this methodology can be further scaled up by suitable modifications of the electrodes and the electrochemical cell. Substrates having activated benzylic C–H bonds such as ethylbenzene and diphenylmethane were also explored. Primarily ketone products (acetophenone and benzophenone) were formed with high conversion and yield for ethylbenzene and diphenylmethane, respectively (Table 1, entries 7 and 8). The formation of a ketone as the main product was attributed to the over oxidation of the hydroxylated product, which was first formed during the oxygenation reaction. Over oxidation of alcohols to ketones has been shown by us earlier for the oxidation of cyclohexane (oxidation of cyclohexanol with 3 is ∼350 times faster than cyclohexane oxidation).48
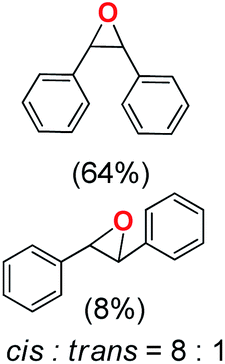
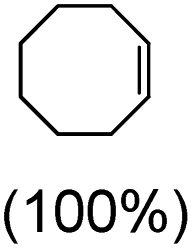
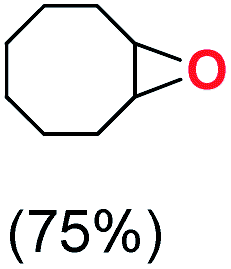
Next, electrochemical oxidation of alkenes was performed at constant potential (0.80 V vs. Ag/AgNO3) using complex 1a/1b as the mediator and water as the oxygen atom source (Table 2 & Fig. S25–S29†). Analysis of the products indicated the predominant formation of alkene oxides in moderate to high yields with only a trace amount of the side-product (mainly aldehydes, ∼5–7% with respect to epoxides). Styrene afforded styrene oxide as the predominant product in 86% yield (Table 2; entry 1). Subsequently, different para-substituted styrene derivatives such as 4-chlorostyrene and 4-methoxystyrene afforded their corresponding epoxides with yields of up to 92% (Table 2; entry 2 and 3). The higher yields obtained for the electron-rich styrene derivative in comparison to the electron-deficient styrene derivative support the likely involvement of an electrophilic high-valent oxoiron intermediate. Oxidation of cis-stilbene, a substrate that contains a sterically constrained double bond resulted in 64% yield (72% conversion) with a cis/trans product ratio of 8:1 (Table 2; entry 4). cis-Cyclooctene was oxidized to cis-cyclooctene oxide in 75% yield (Table 2; entries 5), which also shows the selectivity for the oxidation of the CC bond over the weaker allylic C–H bond.
| Entry | Substrate (% conversion) | Products (% yield) |
|---|---|---|
|
a Reaction conditions: 1a/1b (0.75 mM) and substrate (15 mM) in acetonitrile aqueous phosphate buffer (4:1 v/v, 5 mM, pH ∼8) at a constant potential of 0.80 V (vs. Ag/AgNO3) at room temperature for 4–8 h. Yields and conversions were estimated by GC–MS.
|
||
| 1 |
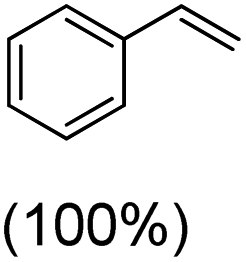
|
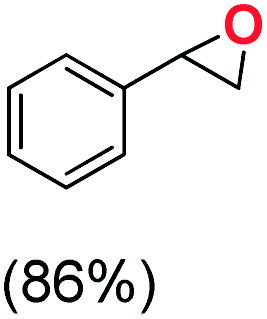
|
| 2 |
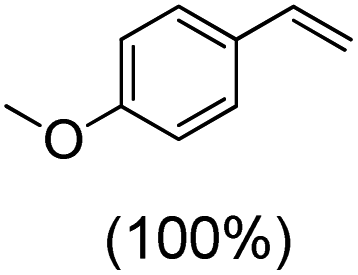
|
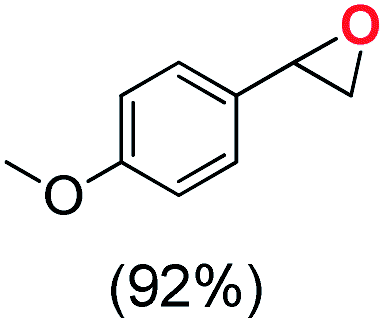
|
| 3 |
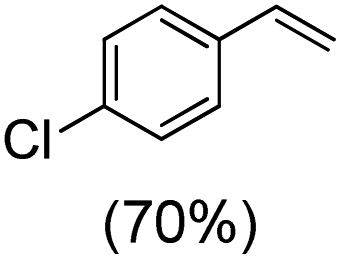
|
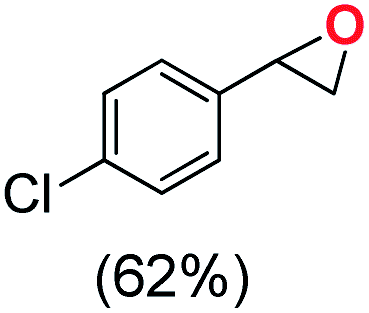
|
| 4 |
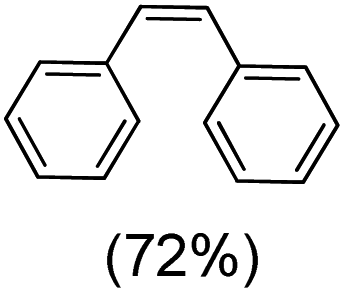
|
|
| 5 |
|
|
Electrochemical epoxidation using iron and manganese complexes and nanoparticles has been reported earlier. For example, Groves and coworkers49 reported the electrochemical generation of a model compound II analog (oxoiron(IV)) from the corresponding porphyrin-FeIII-OH. Although this intermediate was competent in oxidizing styrene derivatives to their epoxides, the selectivity and efficiency were much lower than those of desired model compound I (which is isoelectronic to oxoiron(V)). Collman and coworkers50 and Tanaka and coworkers51 reported electrochemical epoxidation using Mn complexes (porphyrins and salen), but the mechanism involved the electrochemical generation of the chemical oxidant (H2O2 and OCl−), which would subsequently activate the metal complex in a process similar to chemical oxidation. Murray and coworkers52 and recently Dey and coworkers35 showed electrochemical reductive activation of O2 bound to metal porphyrin complexes, which would subsequently form high-valent oxometal intermediates to catalyze oxidation reactions. The only example, which demonstrates the use of water as the O-atom source for epoxidation reactions has been recently reported by Manthiram and coworkers.53 They used manganese oxide nanoparticles to catalyze cyclooctene epoxidation using water as an oxygen source with a faradaic efficiency of ∼30%.
A study of the mechanism of electrochemical oxidation of 3° C–H bonds with BDE ≤97 kcal mol−1 was then attempted with the substrate adamantane. We hypothesized that for the electrochemical reaction, the starting Fe(III)-OH2 (1b) is converted to oxoiron(V), (3) through PCET followed by another electron transfer, which then reacts with adamantane to give rise to the product 1-adamantanol, in turn generating 1b back. The adamantane consumption is small in the reaction layer, and the plateau current is governed by the slow chemical reaction. This reaction can be approximated as proceeding through the ErCi′ mechanism (see ref. 56 for discussion).44,45 To understand the influence of the varying substrate concentration and catalyst concentration on the catalytic current we assume that the maximum catalytic current (icat,max) would change according to eqn (2), where [Ada] is the concentration of the substrate adamantane, and np and ncat are the number of electrons transferred in the absence of the substrate and in the catalytic reaction respectively.
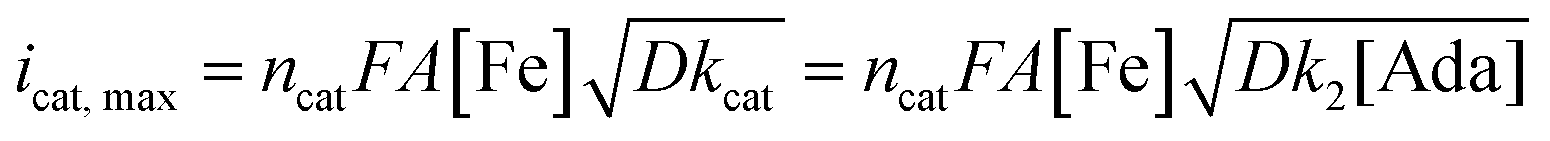 | (2) |
The background-corrected icat increases with increasing concentrations of the catalyst 1b in CH3CN (90%)–H2O (10%) (Fig. 2A) and shows a linear dependence with the catalyst concentration (Fig. 2B). Hence, the catalytic activity is first-order with the concentration of 1b. The catalytic current also varies linearly with the square-root of [Ada], suggesting a first-order kinetic dependence of adamantane (eqn (2), Fig. 2D). When combined, the rate of adamantane oxidation can be expressed as rate = k2 × [Fe] × [Ada]. Similar second-order dependence on the substrate has been found for the electrochemical oxidation of alcohols, which involves high-valent oxoruthenium intermediates.30,31 Finally, the same rate expression was also obtained earlier for hydrocarbon oxidation with chemically synthesized oxoiron(V) under single turnover conditions.36
It's important to note that the CV shape observed for varying catalyst and substrate concentrations is not precisely the S-shaped curves obtained in zone KS (see ref. 57 for discussion), which can be generally achieved at high substrate concentrations or high scan rates.44,45 We were unable to reach a perfect KS zone due to the limited solubility of adamantane in CH3CN (90%)–H2O (10%), and an increasing probability of dimerization at low scan rates. As the CVs show a hybrid peak and plateau at varying substrate concentrations (Fig. 2C & S11†), the plateau current analysis according to eqn (2) to determine kinetic parameters would result in an approximated maximum rate value (k2,max).44 The k2,max value of 0.23 ± 0.02 M−1 s−1 determined from the slope (Fig. 2D and S11†) using eqn (2) was around 6-fold higher than that determined from the reaction of adamantane with chemically generated oxoiron(V) in CH3CN.36 The second-order rate constant (k2,max) was determined from the catalytic response obtained at various substrate concentrations at a fixed scan rate (Fig. 2C), using values of D estimated earlier. The presence of 10% water and higher ionic strength during the electrochemical oxidation could have afforded enhanced rates during the oxidation reactions, as has been reported earlier.54 Moreover, an approximate kinetic isotope effect (KIE) value of 4 was obtained from comparing the catalytic current for adamantane and adamantane-d16 (Fig. S12†). This KIE value is comparable with our previously reported KIE value (∼5) of adamantane during chemical oxidation.55
However, the CVs recorded in the presence of a substrate having C–H bonds with low BDE (xanthene; BDE = 75.5 kcal mol−1) were indicative of a no substrate consumption zone (Fig. S13†).44,45 Catalytic current increased linearly with varying catalyst concentration and with the square-root of the xanthene concentration (Fig. S14 and S15†). Hence, a similar second-order rate for xanthene oxidation can be expressed as rate = k2 × [Fe] × [xanthene]. From the slope of linear variation of icatvs. ν−0.5, the second rate constant (k2,max) for xanthene oxidation was determined to be 182 ± 40 M−1 s−1 (Fig. S16†). This observation does indicate that the rate of the chemical reaction following the electron transfer has an impact on the CV shape.
We proposed a catalytic cycle for the electrochemical oxidation of alkenes and alkanes based on the product analysis, cyclic voltammetry, controlled potential electrolysis, UV-Vis spectroscopy, and previously reported studies. The electrocatalytic cycle begins at the anode (RVC) immediately after applying a constant potential (0.80 V) where complex 1b undergoes an overall 2H+/2e− (PCET) transfer reaction to form oxoiron(V) species, [(bTAML)FeV(O)]− (3). For the hydroxylation reaction, the high-valent oxoiron(V) species (3) hydroxylates the C–H bond in substrates to form the corresponding alcohol by the typical “rebound mechanism” as has been reported earlier.29,36,37 The high regioselectivity observed in adamantane (3°:2° of 75:1) and stereo-retention found for cis-1,2-dimethylcyclohexane (∼99%) show that the alkyl radical formed during the oxidation does not escape the reaction layer to react with O2 present in the solvent to generate unwanted oxygenated products. Such “non-rebound” pathways, if operational, lead to much reduced regioselectivity and stereo-retention. In fact, when catalytic oxidations of adamantane with 1a and NaOCl/mCPBA were carried out in the presence of O2, the regioselectivity of 3°:2° hydroxylation dropped to 30:1 (compared to 75:1, when O2 was rigorously excluded).55 This had indicated that for catalytic oxidation reactions using chemical oxidants, both the “rebound” and “non-rebound” pathways were operational, although oxoiron(V) was shown to be the active intermediate in these reactions. However, during the electrochemical oxidation reactions, the presence or absence of O2 did not affect the selectivity of the reaction, indicating that the radicals formed in the reaction layer at the applied potential exclusively formed the corresponding alcohol via the rebound process. Such a mechanism would regenerate the starting FeIII-OH2 complex (1b), which is expected to be seen in the bulk. However, spectroelectrochemical studies indicate the formation of the μ-O-FeIV2 (2) dimer in bulk during the reaction. Such dimeric species are typically formed by the comproportionation reaction of 3 with 1b. The formation of 2 during the electrochemical catalytic reaction is expected since 3 generated in the anode does not quantitatively react with the substrate due to the inherently slow rates of the reaction. This fact is corroborated in the CV studies of 1b (in the presence of the substrate, Fig. 1C), where the peak due to the reduction of oxoiron(V) to oxoiron(IV) is discernible. Hence a part of 3 generated in the anode diffuses to the bulk solution and is readily converted to the dimeric species μ-O-FeIV2 (2) via a comproportionation reaction with bulk 1b. However, 2 is not stable in the reaction medium and reduces back to the parent 1b. In fact, upon removal of the applied potential during the reaction, spectroelectrochemical studies indicate that the conversion of 2 into the starting FeIII precursor (1b) occurs within minutes. We have reported that the chemically synthesized dimeric μ-O-FeIV2 complex in CH3CN reduces back to the starting FeIII precursor over time, and the rate of reduction increases with an increase in the water concentration. A plausible mechanism for the reduction involves the disproportionation of the dimeric species to corresponding Fe(V)-oxo and Fe(III) complexes in acetonitrile–aqueous phosphate buffer solution. The aqueous phosphate buffer is known to axially bind with the catalyst and can accelerate the disproportionation reaction. Fe(V)-oxo reacts with the substrate and eventually leads to the formation of Fe(III). As there is no applied potential, Fe(V)-oxo is not formed again, and the complete regeneration of Fe(III) is observed. Thus, during the catalytic reaction, the dimeric μ-O-FeIV2 species formed spontaneously converts to FeIII in bulk, which is then re-oxidized in the anode to continue the catalytic cycle. This competitive reaction involving the comproportionation of 3 with 1b and the subsequent self-reduction to the parent FeIII complex is reflected in the low faradaic efficiency of the reaction. On the other hand, the mechanism of the epoxidation reaction probably involves the electrophilic attack of oxoiron(V) onto the alkene followed by a fast ring-closing step to form the epoxide.38 However, additional studies are required to understand the mechanism in more detail.
Finally, there are two things that are important to discuss. First, it is worth comparing the electrochemical oxidation of substrates having 3° C–H bonds with the corresponding chemical oxidation methods using mCPBA as the terminal oxidant. During catalytic chemical oxidation reactions for adamantane with complex 1a, the yield of the product 1-adamantanol obtained was less than 5%.55 Studies have indicated that the excess oxidant and its by-product present in the reaction mixture promoted the demetallation of 1a, resulting in complete loss of its activity. This oxidant-induced demetallation process was completely averted during electrochemical oxidations, which allowed them to be used for reactions with moderate efficiency. The second is the comparison of the prototype Fe-TAML complex with our biuret-substituted Fe-bTAML complex. The prototype Fe-TAML complex has been recently shown to catalyze the electrochemical oxidation of alcohols and substrates containing benzylic C–H bonds, viz. substrates having C–H bonds with BDE less than 90 kcal mol−1.34 In this electrocatalytic approach, the breadth of reactivity demonstrated with the Fe-bTAML complex is not achieved due to the oxidative self-decomposition of the prototype Fe-TAML catalyst under the bulk electrolysis conditions, which limits the catalyst lifetime. In addition, the instability of oxoiron(V) of the prototype Fe-TAML (decomposition at temperatures > −40 ° C)33 leads to its conversion to the corresponding μ-O-FeIV2 dimer, which is a much weaker oxidant. This limits the reactivity to only weak C–H bonds. In contrast, the increased stability of the oxoiron(V) intermediate in our Fe-bTAML (stable at RT) allows this highly reactive intermediate [BDE (FeIVO–H) = 99 kcal mol−1] to catalyze the oxidation of C–H bonds with a BDE of up to 97 kcal mol−1 with very high selectivity.36
Conclusion
In conclusion, we have demonstrated an electrochemical method for selective oxidation reactions of alkanes having sp3 C–H bonds with BDE ≤97 kcal mol−1 using water as the oxygen source. Typically such electrochemical oxidations require very “high overpotential” which can also oxidize other functional groups present in the substrate. However, the use of the FeIII complexes, 1a/1b, allows such oxidations to be achieved at much lower overpotentials and with high selectivity. Spectroelectrochemical analysis shows the involvement of a high-valent oxoiron(V) species, [(bTAML)FeV(O)]−, formed via PCET from the iron(III) complex, [(bTAML)FeIII–OH2]−. Determination of the rate law from cyclic voltammetry experiments and product analysis allows us to conclude that the mechanism of hydroxylation involves abstraction of the H-atom by oxoiron(V) followed by the “rebound” mechanism to form the hydroxylated product. This electrochemical oxidation was performed with a series of substrates including natural products such as cedryl acetate and ambroxide to afford oxygenated products with high conversion and yield. The oxidation of ambroxide was also scaled up to a 1 mmol scale. The electrochemical oxidation was also extended to the epoxidation of alkenes. The main attractiveness of this method includes the use of (i) inexpensive carbon (RVC) and nickel electrodes in a simple undivided cell, (ii) an earth-abundant iron complex as a redox mediator, and (iii) water as the O-atom source, (iv) under an open atmosphere. To the best of our knowledge, our report with 1a/1b demonstrates the first example of homogeneous electrochemical hydroxylation and epoxidation involving the high-valent oxoiron(V) intermediate and using the O-atom of water as the oxygen source.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. G. acknowledges SERB, New Delhi (Grant no EMR/2017/ 003258), and IISER-Kolkata (“Start-up Grant”) for funding. B. Chandra acknowledges UGC-New Delhi for his fellowship.Notes and references
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS
.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS
- K. J. Jiao, Y. K. Xing, Q. L. Yang, H. Qiu and T. S. Mei, Acc. Chem. Res., 2020, 53(2), 300–310 CrossRef CAS
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS
- O. R. Luca, J. L. Gustafson, S. M. Maddox, A. Q. Fenwick and D. C. Smith, Org. Chem. Front., 2015, 2, 823–848 RSC
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC
- Y. Kawamata, M. Yan, Z. Liu, D. H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS
- E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS
- C. Costentin, ACS Catal., 2020, 10, 6716–6725 CrossRef CAS
- H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef CAS
- G. B. Shul’pin, Alkane Funct., 2018, 2, 47–72 Search PubMed
- J. E. Lyons, P. E. Ellis and H. K. Myers, J. Catal., 1995, 155, 59–73 CrossRef CAS
- B. Chandra, P. De and S. Sen Gupta, Chem. Commun., 2020, 56, 8484–8487 RSC
- L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS
- P. R. Ortiz De Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS
- M. K. Coggins, M. T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531–5534 CrossRef CAS
- M. J. Collins, K. Ray and L. Que, Inorg. Chem., 2006, 45, 8009–8011 CrossRef CAS
- W. A. Lee, T. S. Calderwood and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4301–4305 CrossRef CAS
- H. Kotani, T. Suenobu, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 3249–3251 CrossRef CAS
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS
- B. Chandra, K. K. Singh and S. Sen Gupta, Chem. Sci., 2017, 8, 7545–7551 RSC
- A. Paul, J. F. Hull, M. R. Norris, Z. Chen, D. H. Ess, J. J. Concepcion and T. J. Meyer, Inorg. Chem., 2011, 50, 1167–1169 CrossRef CAS
- A. K. Vannucci, Z. Chen, J. J. Concepcion and T. J. Meyer, ACS Catal., 2012, 2, 716–719 CrossRef CAS
- S. G. Robinson, J. B. C. Mack, S. N. Alektiar, J. Du Bois and M. S. Sigman, Org. Lett., 2020, 22(18), 7060–7063 CrossRef CAS
-
(a) T. J. Collins and A. D. Ryabov, Chem. Rev., 2017, 117, 9140–9162 CrossRef CAS
- A. Das, J. E. Nutting and S. S. Stahl, Chem. Sci., 2019, 10, 7542–7548 RSC
- M. Mukherjee and A. Dey, ACS Cent. Sci., 2019, 5, 671–682 CrossRef CAS
- M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. Sen Gupta, J. Am. Chem. Soc., 2014, 136, 9524–9527 CrossRef CAS
- S. Jana, M. Ghosh, M. Ambule and S. Sen Gupta, Org. Lett., 2017, 19, 746–749 CrossRef CAS
- K. K. Singh, M. K. Tiwari, B. B. Dhar, K. Vanka and S. Sen Gupta, Inorg. Chem., 2015, 54, 6112–6121 CrossRef CAS
- K. K. Singh and S. Sen Gupta, Chem. Commun., 2017, 53, 5914–5917 RSC
- S. Pattanayak, A. J. Jasniewski, A. Rana, A. Draksharapu, K. K. Singh, A. Weitz, M. Hendrich, L. Que, A. Dey and S. Sen Gupta, Inorg. Chem., 2017, 56, 6352–6361 CrossRef CAS
- S. Pattanayak, D. R. Chowdhury, B. Garai, K. K. Singh, A. Paul, B. B. Dhar and S. Sen Gupta, Chem. – Eur. J., 2017, 23, 3414–3424 CrossRef CAS
- S. Groni, C. Hureau, R. Guillot, G. Blondine, G. Blain and E. Anxolabéhère-Mallart, Inorg. Chem., 2008, 47, 11783–11797 CrossRef CAS
- A slight change in the CV shape observed in the second redox process in CH3CN–H2O (9:1) could be the result of axial ligation of water molecules to the oxoiron(V) formed. Since chemically synthesized oxoiron(V) undergoes 16O to 18O scrambling upon addition of H2O,18 such change in the coordination environment after formation of oxoiron(V) is possible.
-
(a) E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS
-
(a)
J.-M. Sav'eant, Elements of Molecular and Biomolecular Electrochemistry, John Wiley & Sons, Inc., Hoboken NJ, 2006 CrossRef
-
Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, 2004, vol. 126 Search PubMed
-
(a) K. Chen, A. Eschenmoser and P. S. Baran, Angew. Chem., Int. Ed., 2009, 48, 9705–9708 CrossRef CAS
- M. Ghosh, Y. L. K. Nikhil, B. B. Dhar and S. Sen Gupta, Inorg. Chem., 2015, 54, 11792–11798 CrossRef CAS
- J. T. Groves, Z. Gross and M. K. Stern, Inorg. Chem., 1994, 33, 5065–5072 CrossRef CAS
- H. Nishihara, K. Pressprich, R. W. Murray and J. P. Collman, Inorg. Chem., 1990, 29, 1000–1006 CrossRef CAS
- H. Tanaka, M. Kuroboshi, H. Takeda, H. Kanda and S. Torii, J. Electroanal. Chem., 2001, 507, 75–81 CrossRef CAS
- S. E. Creager, S. A. Raybuck and R. W. Murray, J. Am. Chem. Soc., 1986, 108, 4225–4227 CrossRef CAS
- K. Jin, J. H. Maalouf, N. Lazouski, N. Corbin, D. Yang and K. Manthiram, J. Am. Chem. Soc., 2019, 141, 6413–6418 CrossRef CAS
- K. K. Singh, M. K. Tiwari, M. Ghosh, C. Panda, A. Weitz, M. P. Hendrich, B. B. Dhar, K. Vanka and S. Sen Gupta, Inorg. Chem., 2015, 54, 1535–1542 CrossRef CAS
- M. Ghosh, S. Pattanayak, B. B. Dhar, K. K. Singh, C. Panda and S. Sen Gupta, Inorg. Chem., 2017, 56, 10852–10860 CrossRef CAS
- The “ErCi′ mechanism” associates a reversible electrode electron transfer with a first-order (or pseudo-first-order) follow-up irreversible homogeneous chemical reaction. It is one of the simplest reaction schemes where a heterogeneous Nernstian electron transfer is coupled to an irreversible chemical reaction that takes place in the adjacent solution.44,45.
- A “KS zone” is a zone in a kinetic zone diagram representing the various regimes of competition between diffusion and the follow-up reaction.44,45 In the KS zone of the EC zone diagram for homogeneous catalysis an S-shaped (where the forward and reverse scans trace each other exactly) CV response is expected.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, general instrumentation and additional figures. See DOI: 10.1039/d0sc03616a |
| This journal is © The Royal Society of Chemistry 2020 |