
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRenewable resources for sustainable metallaelectro-catalysed C–H activation
Ramesh C.
Samanta
 a,
Tjark H.
Meyer
ab,
Inke
Siewert
bc and
Lutz
Ackermann
*ab
a,
Tjark H.
Meyer
ab,
Inke
Siewert
bc and
Lutz
Ackermann
*ab
aInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
bWoehler Research Institute for Sustainable Chemistry (WISCh), Georg-August-Universität Göttingen, Tammannstrasse 2, 37077 Göttingen, Germany
cInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany
First published on 31st July 2020
Abstract
The necessity for more sustainable industrial chemical processes has internationally been agreed upon. During the last decade, the scientific community has responded to this urgent need by developing novel sustainable methodologies targeted at molecular transformations that not only produce reduced amounts of byproducts, but also by the use of cleaner and renewable energy sources. A prime example is the electrochemical functionalization of organic molecules, by which toxic and costly chemicals can be replaced by renewable electricity. Unrivalled levels of resource economy can thereby be achieved via the merger of metal-catalyzed C–H activation with electrosynthesis. This perspective aims at highlighting the most relevant advances in metallaelectro-catalysed C–H activations, with a particular focus on the use of green solvents and sustainable wind power and solar energy until June 2020.
 Ramesh C. Samanta | Ramesh C. Samanta has received his B. Sc. in chemistry from Vidyasagar University in 2008. After completion his M. Sc. from the Indian Institute of Technology (IIT), Kanpur in 2010, he moved to Germany for his doctoral study and has received his PhD from the University of Münster under the supervision of Prof. Armido Studer in 2013, working on oxidative N-heterocyclic carbene catalysis. Then, he was working as a Japan Society for the Promotion of Science (JSPS) postdoctoral fellow with Prof. Hisashi Yamamoto in Japan on chiral Brønsted acid catalysis before he moved to the Georg-August-Universität Göttingen (Germany) for his second postdoctoral study. Since 2018, he is working there as an Alexander von Humboldt postdoctoral fellow with Prof. Lutz Ackermann, focusing on electrochemical C–H activation. |
 Tjark H. Meyer | Tjark H. Meyer studied chemistry at the Georg-August-University Göttingen and obtained his MSc in 2016 under the supervision of Prof. L. Ackermann. During his studies he joined the group of Dr Stephen P. Thomas at the University of Edinburgh for a four-month research stay, and later, he performed an industrial placement at Bayer AG, Wuppertal. In 2017, he started his doctoral studies in the group of Prof. L. Ackermann. His current research is focused on synthetic organic electrochemistry, with a major interest on the merger of metal-catalysed C–H activation and electrocatalysis. |
 Inke Siewert | Inke Siewert studied chemistry at the Humboldt University of Berlin (Germany). In 2009 she received her PhD from the same university under the supervision of Prof. Dr C. Limberg. She then moved to the University of Oxford (UK) as a postdoctoral fellow in the group of Prof. Dr S. Aldridge. In 2011 she joined the Georg-August-University Göttingen (Germany) as an independent researcher, since 2013 funded by the Emmy Noether program of the DFG. In 2017 she was appointed as an associate professor. Her research is focused on molecular electrochemistry for the activation of small molecules. |
 Lutz Ackermann | Lutz Ackermann studied chemistry at the Christian-Albrechts-University Kiel and obtained his PhD in 2001 with Alois Fürstner at the MPI für Kohlenforschung in Mülheim/Ruhr. He was a postdoctoral fellow with Robert G. Bergman (UC Berkeley) before initiating his independent research in 2003 at the Ludwig-Maximilians University München, supported within the Emmy Noether Program of the DFG. In 2007, he became full professor at the Georg-August-University Göttingen, where he served as the Dean of Research and Dean of Chemistry as well as the director of the Wöhler Research Institute for Sustainable Chemistry. The development of novel concepts for homogeneous catalysis and their applications to late-stage peptide diversification by bond activation, and electrocatalysis are among his main current research interests. |
1 Introduction
During the last decades, research in academia and agrochemical and pharmaceutical industries has increasingly focused on the development of resource-economical,1 synthetic methodologies, reducing the amount of byproducts while enabling more efficient molecular transformations of decreased environmental impact.2–5 In line with the UN-sustainability goals,6 catalysis7 represents an indispensable tool for process chemists by providing access to unique bond-forming transformations not only with a high atom economy.8 Indeed, the required reaction temperature and the amount of otherwise needed stoichiometric reagents, can be minimized.9,10 As a result, scientists have focused their efforts towards the avoidance of waste product formation and more efficient resource utilization in molecular sciences.11 Therefore, ambitious guidelines have been set in place to define these challenges, such as the twelve-principles of green chemistry,12,13 or circular chemistry.14In this context, C–H activation constitutes one of the most promising strategies for directly forming new chemical bonds and consequently synthesizing novel organic compounds in a sustainable and step efficient fashion,15–22 in line with the principles of green chemistry.23 For example, in stark contrast to classical metal-catalysed cross-coupling reactions,24 the direct conversion of otherwise inert C–H bonds circumvents the laborious prefunctionalisation of synthetic building blocks and avoids by-product generation (Scheme 1).25
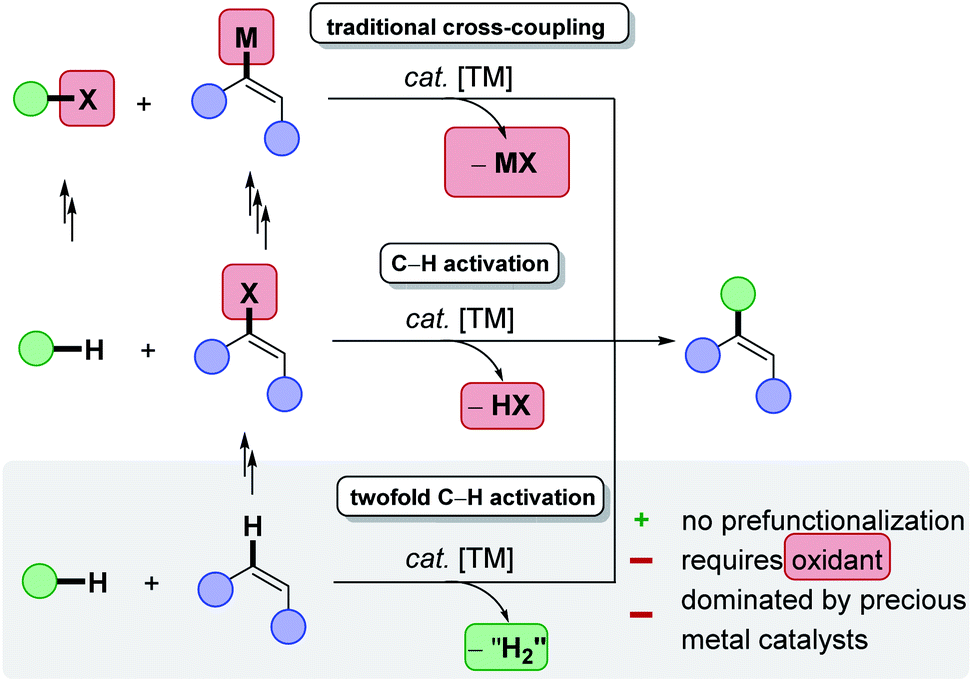 | ||
| Scheme 1 Twofold C–H activation as step and resource economical alternative to cross-coupling reactions. | ||
Although classical C–H activation avoids the prefunctionalisation of one coupling partner, additional steps are required to synthesise often required organic electrophiles, such as organic halides.26 In theory, the most atom- and step-economic approach would be the cross-dehydrogenative coupling (CDC),27 or twofold C–H activation, since formally molecular hydrogen would be generated as the sole byproduct. Despite tremendous successes in the field of twofold C–H activations, the latter predominantly suffers from harsh reaction conditions, such as high reaction temperatures, toxic, halogenated solvents, precious transition metal catalyst, stoichiometric amounts of often ecologically damaging and cost-intensive oxidants, thus compromising the overall sustainability of twofold C–H activations. A remarkable progress has been made by the replacement of the commonly used fossil-based and often toxic solvents, with biomass-derived solvents, thereby considerably reducing the ecological footprint.28,29 Furthermore, recent accomplishments, especially with Earth-abundant base metal catalysts has indicated the great potential towards an overall resource-economic functionalisation of ubiquitous C–H bonds (Scheme 2).30 Despite of these notable advances, one challenge – the requirement of chemical oxidants – remained largely unsolved until recently.31,32
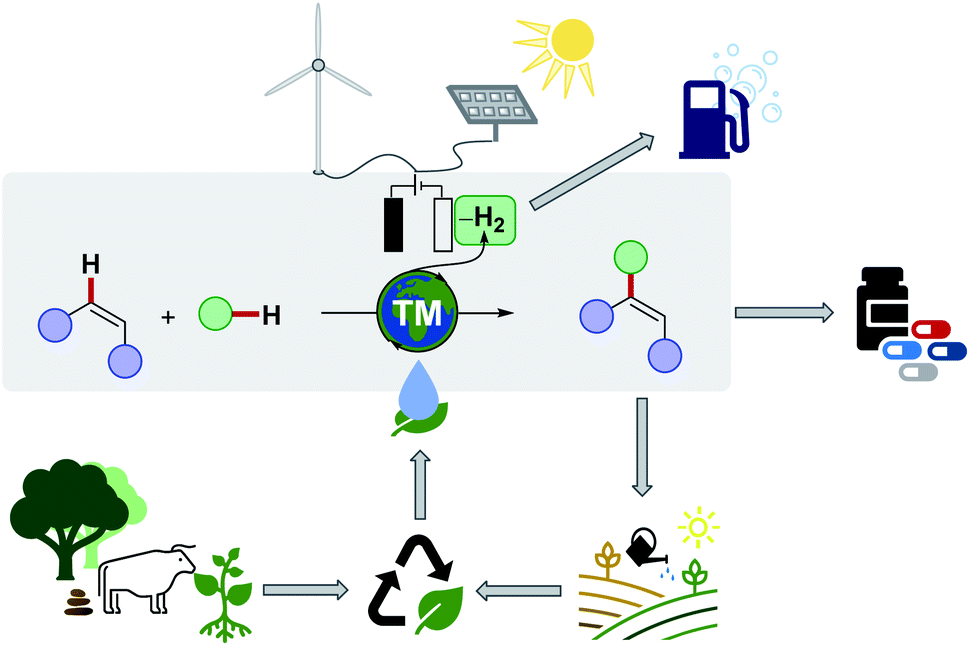 | ||
| Scheme 2 Electrochemical C–H activation in biomass-derived solvents, powered by renewable energy sources, catalysed by Earth-abundant catalyst. | ||
Meanwhile, organic electrochemistry has experienced a renaissance during the recent years and evolved from a underappreciated technique into an attractive concept in academia and industries.33–40 Through the use of electrochemistry it is possible to direct sustainable power, harvested from sunlight, wind, biomass or hydropower, towards the desired chemical transformation drastically improving the overall environmental footprint of the required chemical transformation.41,42 Consequently, scientists enabled to facilitate catalytic reactions through the use of electricity for the assembly of chemical compounds or renewable fuels,43 based on greenhouse gases such as carbon dioxide (CO2).44–46 Furthermore, more complex synthetic tasks can be tackled with the aid of electrosynthesis,47 and recent successes in synthetic organic electrochemistry, have emerged as a strong testament to the versatility of electrochemically promoted organic transformations (Scheme 2).38,48–52
The advantages of the merger of C–H activation with electrochemistry include the replacement of chemical redox reagents and the discovery of new synthetic methodologies such as twofold C–H activations amongst others.53–57 The only by-product formed – molecular hydrogen gas – can further be used as a sustainable energy source58 or utilized in various chemical reduction reactions, ideally by paired electrolysis.59
Within this perspective, key examples highlight the recent developments in metallaelectro-catalysed C–H activation, with a major focus on Earth-abundant base metal catalysis, biomass-derived reaction media, and the combination of sustainable power sources for overall resource economic chemical bond transformations until June 2020.1,31,32,51
2. Merger of electrochemistry and C–H activation
Oxidative transformations commonly employ chemical oxidants and thus generate stoichiometric amounts of by-products, which reduces the atom- and resource-economy of the process at hand.1 As a more sustainable alternative, anodic oxidation can be used with molecular hydrogen as the sole by-product formed through cathodic reduction. Direct anodic oxidation for metal-catalysed redox reactions was early realized in 1987 by Tsuji, for a modification of the Wacker reaction, using anodic oxidation with benzoquinone as a redox mediator.60 In 2007, Amatore and Jutand reported an electrochemically-modified Fujiwara–Moritani alkenylation of acetanilides 1 with alkene 2 (Scheme 3).61 A divided cell setup was used to prevent the deposition of palladium black on the cathode. A catalytic amount of p-benzoquinone served as a shuttle for the electrons between the anode and the metal catalyst to reoxidise the palladium(0) species to the catalytically relevant palladium(II) complex. Indeed, this otherwise powerful approach has an adverse impact on the overall atom economy. Instead, the direct anodic oxidation of the metal catalyst on the electrode surface to achieve a proton-coupled electron transfer (PCET) in C–H activation is more desirable, yet significantly more challenging (vide infra).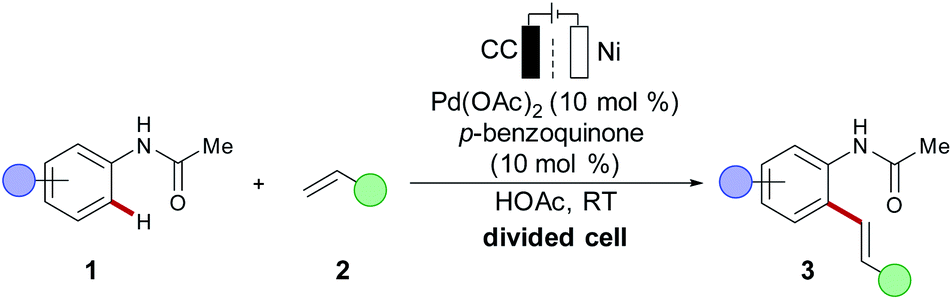 | ||
| Scheme 3 Early work on palladaelectro-catalysed C–H activation. | ||
In this context, Mei disclosed a palladium-catalysed C–H acetoxylation, for which the catalyst was directly oxidized at the electrode surface.62 However, palladium-catalysed electrochemical transformations including halogenations63 and alkylations64 commonly required a more complex divided cell set up. In order to circumvent the problems accompanied by a divided cell setup, the Ackermann group introduces an unprecedented, user-friendly undivided cell arrangement for performing rhoda-65 and ruthena-electro-catalysed66 C–H/C–H activation and C–H/O–H annulation reactions respectively. Subsequently, other 4d and 5d metals including ruthenium,66–70 rhodium71–75 and iridium76–78 were explored for electrochemical C–H activations.
Electrochemical 3d base metal-catalysed oxidative C–H activation
The preference for 3d base metals over precious 4d and 5d metals in organic catalysis is due to several reasons including (a) low costs (b) environmentally-benign nature (c) Earth abundant (d) generally less toxic than 4d and 5d metals, and arguably intellectually most notably (e) mechanistically unique pathways in molecular transformations. In recent years, 3d metals30 have experienced tremendous applications to metal-catalysed C–H activations, featuring the power of copper,79 iron,80,81 manganese,82,83 cobalt84–86 and nickel.87,88 Despite significant advances in this field, the use of renewable electricity to power oxidative and redox-neutral C–H activations remained elusive until 2017.Cobalt-catalysed electrooxidative C–H activation
In 2017, the Ackermann group disclosed the merger of electrochemistry with 3d metal-catalysed C–H activation.89,90 The strategy provided access to the desired C–H oxygenated products 6 at ambient temperature from a variety of amides 4 using primary alcohols 5 with the assistance of a pyridine N-oxide amide (Scheme 4). Inspired by this finding, cobalt-catalysed oxidative C–H transformations were heavily explored with the aid of electricity as a green oxidant. Herein, operationally simple undivided cells were predominantly used by the Ackermann group for numerous C–H transformations, such as C–H amination,91 C–H/N–H annulations with allenes,92,93 alkynes94,95 and bis-alkynes96 and C–H annulation reactions with carbon monoxide 19 or isocyanides 2097 (Scheme 5A(a–e)). Although the initial finding on C–H oxygenation was assisted by pyridine N-oxides 4, later an electro-removable hydrazide 11 proved viable likewise.95 In contrast, the Lei group employed thereafter a rather complex divided cell setup to achieve oxidative cobalt catalysis under electrochemical conditions namely for C–H aminations, alkyne annulations98 and a C–H/N–H carbonylation of quinoline amides 25 (Scheme 5B).99,100 Very recently the same group have developed a [4 + 2] annulation method for the synthesis of sultams 24 using the undivided cell setup (Scheme 5A(f)).101 | ||
| Scheme 4 Merger of electrochemistry with cobalt-catalysed C–H oxygenation. | ||
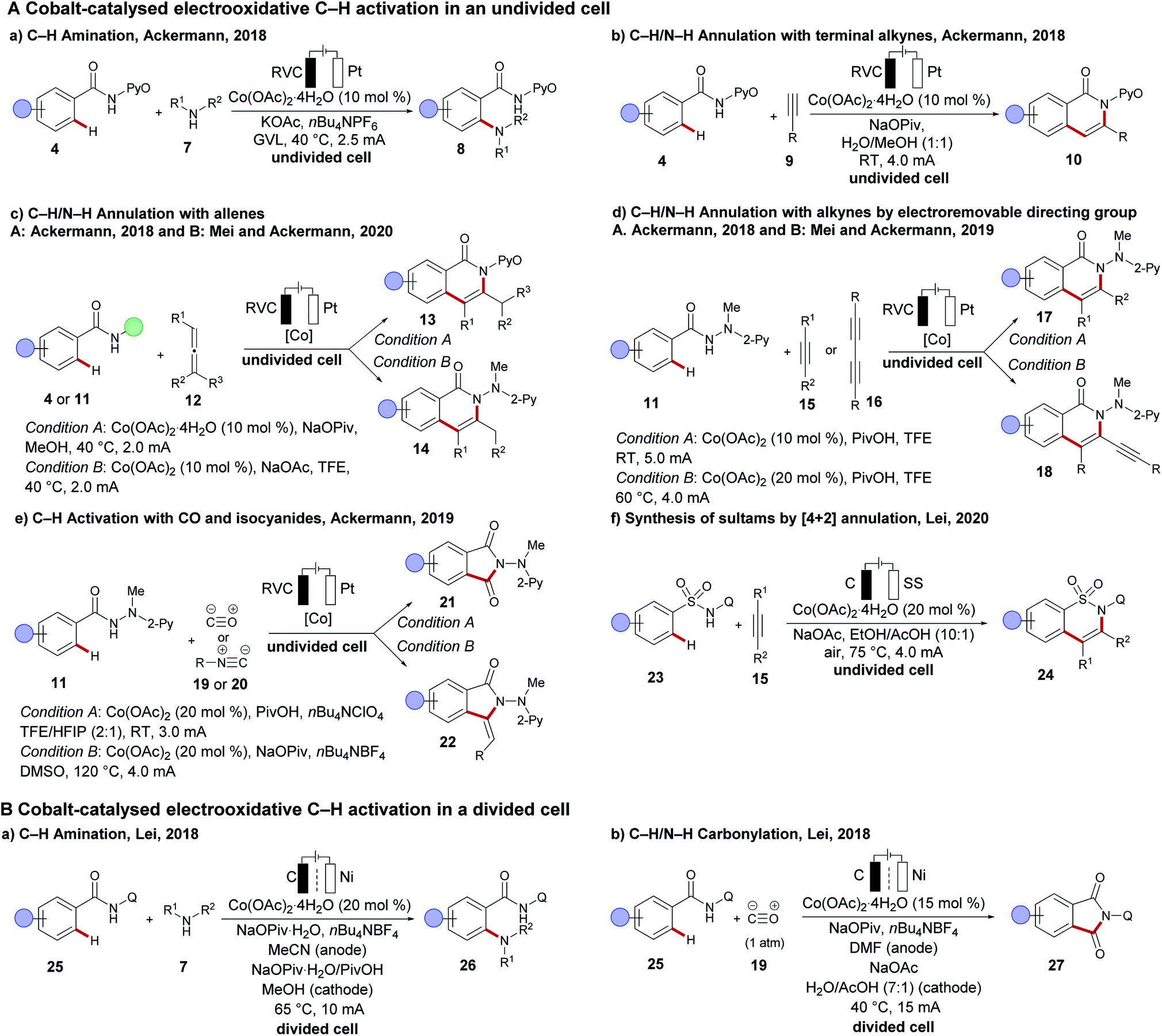 | ||
| Scheme 5 Recent advances on cobaltaelectro-catalysed C–H activation (A) in an undivided cell setup; (B) in a divided electrolysis setup. | ||
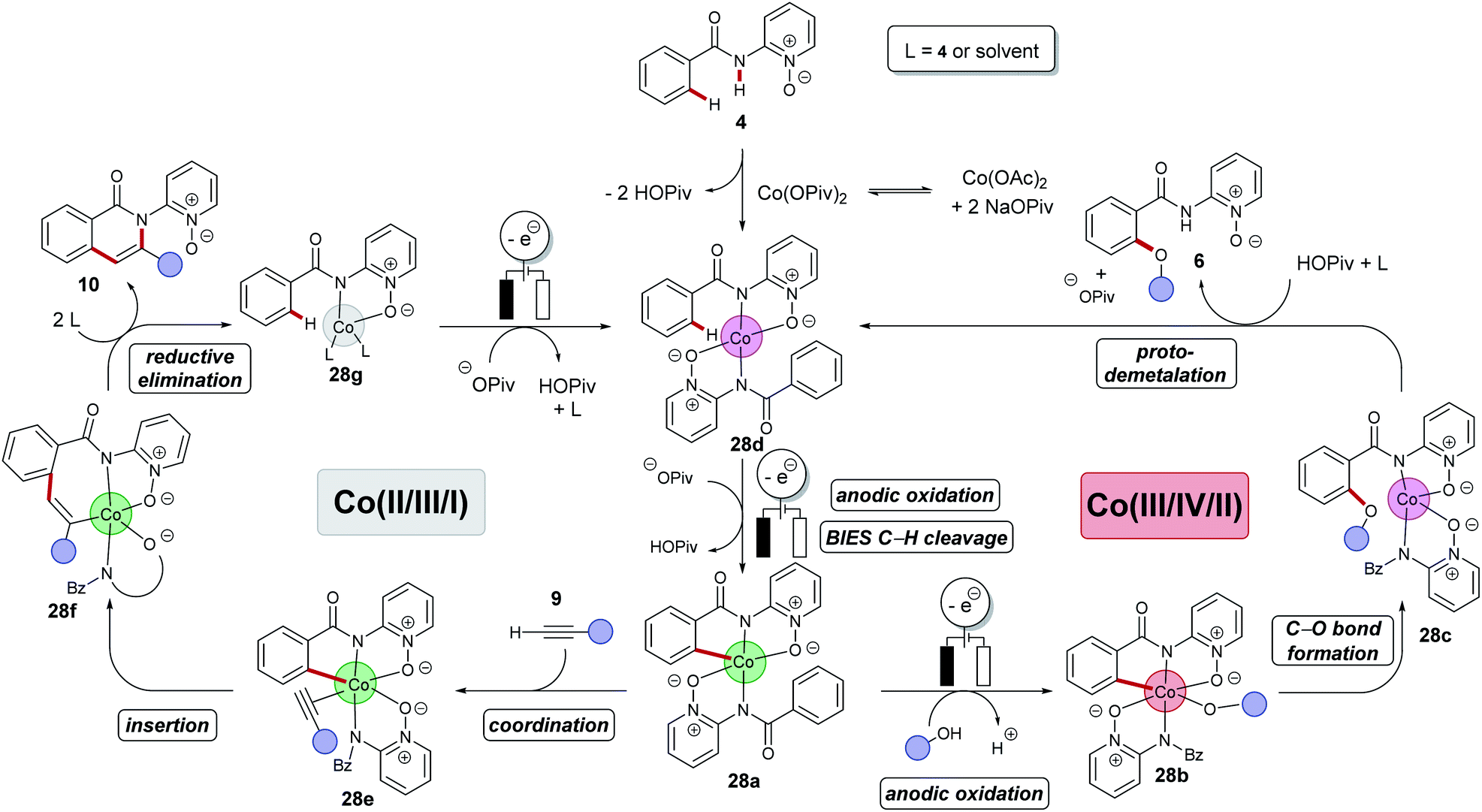
Despite of extensive reports on cobaltaelectro-catalysed transformations, until recently, there was only very little understanding on the exact working mode of these novel transformations. Moreover, these reactions are mechanistically intriguing as they can be performed under exceedingly mild conditions, in stark contrast to reactions performed with chemical oxidants. Rather these reactions were suggested to occur via a cobalt(II/III/I)-catalytic cycle involving a base-assisted internal electrophilic-type substitution (BIES) type C–H activation. However, the role of the electricity presumably only served as a green oxidant to reinitiate the catalytic turnover by anodic oxidation of the reduced cobalt-species. To shed light on the mechanism of the cobaltaelectro-catalysed C–H activation, very recently a detailed mechanistic study was performed by the Ackermann group.53 Here, key cyclometalated cobalt(III) complexes were synthesized with the aid of potentiostatic electrolysis and were subjected to detailed mechanistic studies. A series of stoichiometric reactions were performed with the isolated cobalt(III) complex 28a and the results revealed that in the case of alkoxylation, a second oxidation was required, whereas for alkyne annulation reactions, the product was formed without any further oxidation (Scheme 6). This observation indicated that the alkyne annulation reactions are likely to proceed via a cobalt(II/III/I) manifold, which is in agreement with the previously proposed oxidative cobalt catalysis (Scheme 7, left cycle).53 However, the authors proposed the cobaltaelectro-catalysed C–O forming reaction to proceed via an oxidatively-induced reductive elimination from a high-valent cobalt(IV) complex. Their hypothesis was further supported by means of cyclic voltammetry (CV) (Fig. 1).
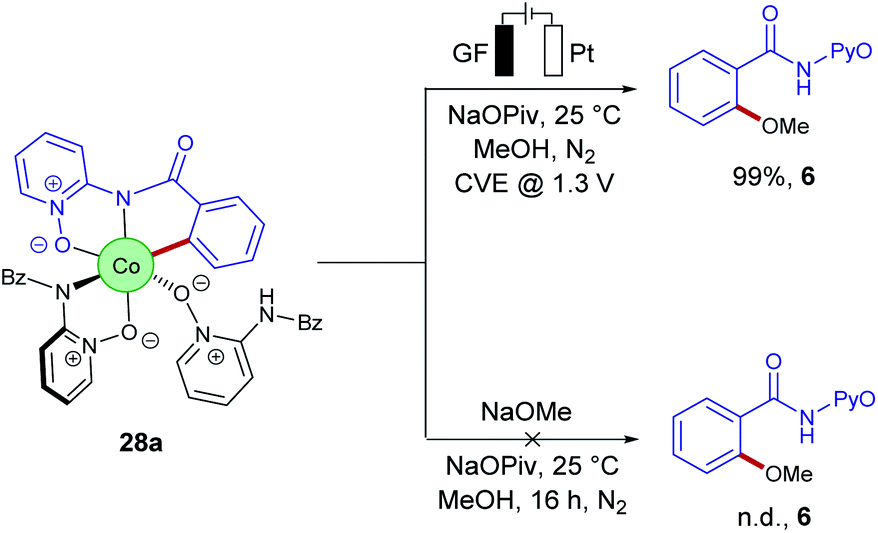 | ||
| Scheme 6 Stoichiometric reactions with cyclometalated [Co(III)] complex 28a. | ||
 | ||
| Scheme 7 Proposed catalytic cycle for cobaltaelectro-catalysed C–H alkoxylations. | ||
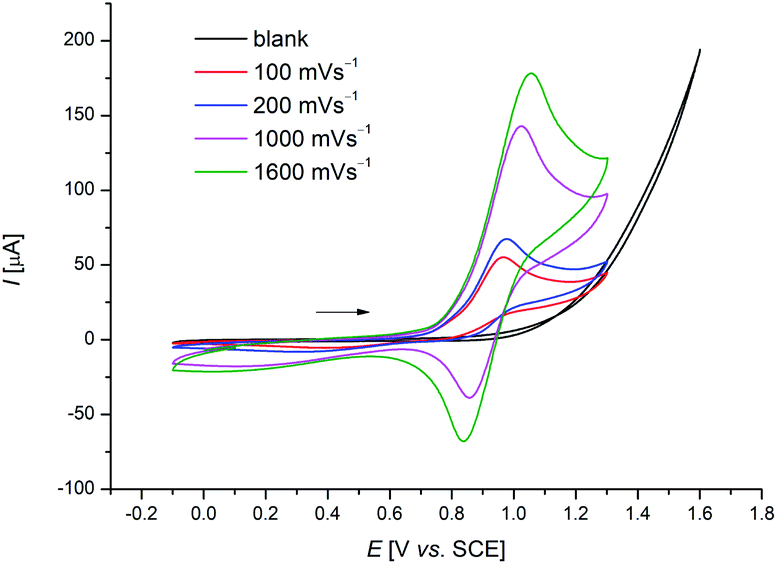 | ||
| Fig. 1 Cyclic voltammograms of cobaltacycles [Co(III)] in MeOH (3.5 mM) at different scan rates. The voltammograms were recorded in 0.1 M [n-Bu4N][PF6] at 273 K. | ||
When the CV-experiments were performed at lower temperatures, a reversible redox event was observed, which could be assigned to the transient generation of high-valent Co(IV) intermediates.
Based on their mechanistic studies a cobalt(III/IV/II) manifold was put forward for the electrocatalytic C–H alkoxylations, which was further supported by DFT calculations for an oxidatively-induced reductive elimination. In this new mechanism, a C–H activation takes place after anodic oxidation to the active cobalt(III) catalyst with a BIES mechanism to generate intermediate 28a (Scheme 7, right cycle). It is noteworthy that this scenario likely proceeds within a concerted PCET, which contrasts with the previously reported mediated anodic reoxidation of palladium complexes (vide supra, Scheme 3). The subsequent anodic oxidation results in a highly reactive cobalt(IV) complex 28b which is followed by a facile reductive elimination leading to cobalt(II) intermediate 28c. A proto-demetalation releases the product and the reduced cobalt(II) species 28d. Finally, anodic oxidation and subsequent C–H activation regenerates the catalytically competent cobalt(III) intermediate 28a.
A cobalt(III/IV/II) catalytic cycle seems likewise plausible for the cobaltaelectro-catalyzed amination, as multiple oxidations appear prior entering the catalytic cycle similar as in the C–O forming reaction.89,91 The potential of the amination reaction is lower than for the latter reaction. However, the estimated bond dissociation energy of R2N–H is lower than of RO–H suggesting that its oxidation (via a bidirectional PCET reaction) appears at lower potential.102
The Ackermann group was further able to use the mechanistically gained insights in the high-valent cobaltaelectro-catalyzed C–H activation to develop an unprecedented twofold C–H/C–H arylation of benzamides 4 (Scheme 8).
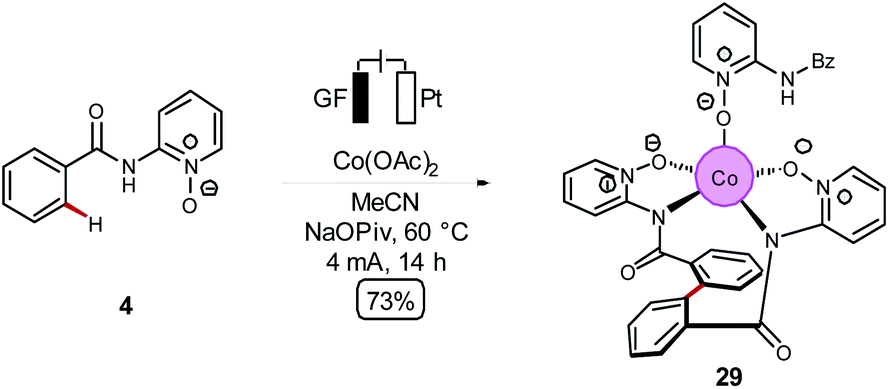 | ||
| Scheme 8 Cobaltaelectro-induced twofold C–H/C–H arylation. | ||
This study, among others56,57 demonstrates how electrochemical manipulation of the oxidation states of the metal catalyst by single-electron transfer (SET) processes, offers the possibility to access new mechanistic pathways that were previously inaccessible.
Nickelaelectro-catalyzed C–H activation
A plethora of C–H activations have been realized by palladium catalysis.17 Nickel catalysts feature advantages in contrast to palladium, including its low costs, Earth-abundant nature, as well as being generally less toxic.103 However, an electrochemical C–H activation104 using nickel catalysis was unprecedented until very recently when Ackermann and coworkers achieved an electrochemical C–H amination of benzamides 25 with cyclic and acyclic secondary amines 7 (Scheme 9).105 C–H amination products 26 were hence obtained by chelation assistance without the necessity of additional chemical oxidants. Cyclic voltammetry studies suggested a possible nickel(IV) intermediate and hence an oxidatively-induced reductive elimination step was proposed to be operative. The most recent notable advancement in electrooxidative nickel catalysis was again made by the Ackermann group. Thus, they disclosed an electrochemical C–H oxygenation of diversely decorated benzamides 30 with a wealth of secondary alcohols 31, which otherwise were not viable with any other chemical oxidant or transition metals, such as palladium, cobalt, copper, among others.54 As to the reaction mechanism, a cyclometalated nickel(III) complex 33 was independently synthesised. As a result of detailed mechanistic studies, an oxidatively-induced reductive elimination step at a nickel(III) complex 33 was suggested experimentally, and computational studies at the PBE0/Def2TZVP level of theory indicated that a non-innocent ligand effect was of relevance for the SET-oxidation.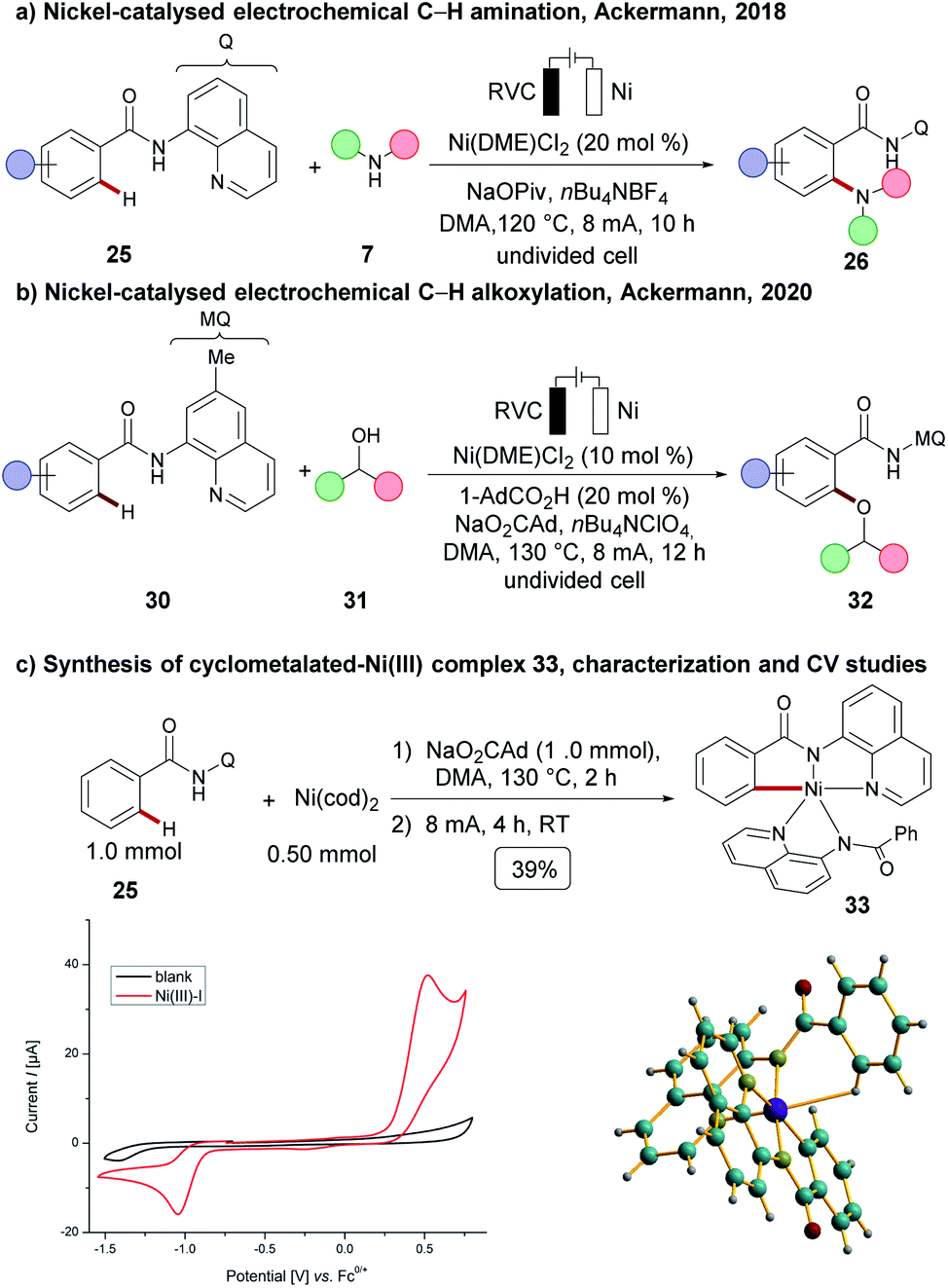 | ||
| Scheme 9 Nickel-catalysed electrooxidative C–H activation. | ||
Copper-catalysed electrooxidative C–H activation
Due to the low costs and high natural abundance of copper, a number of copper-catalysed or copper-mediated C–H activations have been reported.30,79 Furthermore, simple copper salts or complexes have often served as the oxidants in oxidative transformations. Despite these precedents, the merger of electrosynthesis with copper-catalysed C–H activation remained dormant until very recently. Thus, the Mei group reported on the copper-catalysed electrochemical C–H amination of anilides 34, using various cyclic secondary amines 7 as the coupling partner (Scheme 10).106 An undivided cell setup was used to access densely decorated the ortho C–H amination products 35. Here, tetrabutylammonium iodide (TBAI) acted as a redox mediator to shuttle the electrons between the anode and the reduced copper complex. Based on insightful mechanistic studies, a radical mechanism was proposed. The active catalyst features a copper(II) species that is coordinated by anilide 34 and amine 7 to form the copper(II) intermediate 36a, which then undergoes oxidation to copper(III) species 36b by the iodine radical. According to kinetic studies, the formation of the copper(III) species 36b is expected to be the rate-limiting step. A single-electron-transfer (SET) at the copper(III) complex 36b forms the proposed radical copper(II) intermediate 36c. Intramolecular amine transfer and subsequent electron transfer forms the copper(I) intermediate 36d. Finally, the product 35 is released and the resulting copper(I) is oxidized by the iodine mediator to regenerate the active copper(II) catalyst. Thereafter, a redox mediator-free cupraelectro-catalysed C–H transformation was reported by Nicholls for quinoline amides 25. The C–H amination products 26 could only be obtained in moderate yields at elevated temperatures of 60 °C and with higher catalyst loading (20 mol%), albeit with a broad scope, including active pharmaceutical ingredients (APIs), such as antipsychotic Haloperidol 26a, Perospirone 26c and antidepressant Fluoxetine 26b.107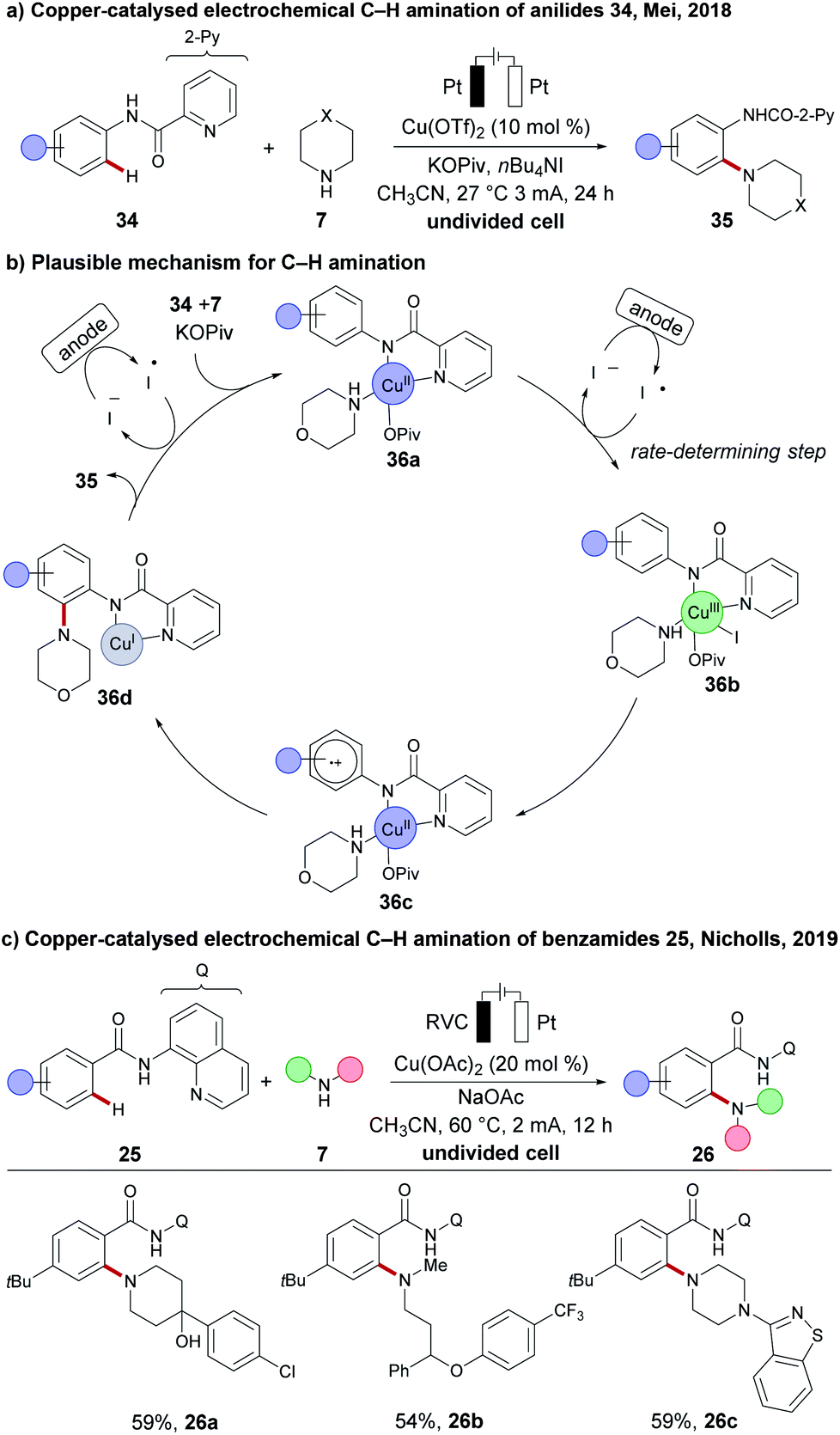 | ||
| Scheme 10 Copper-catalysed electrochemical C–H activation. (a) C–H amination of anilides 34. (b) Mechanistic proposal. (c) C–H amination of quinoline amides 25. | ||
Concurrently, the Ackermann group reported on the first cupraelectro-catalyzed C–C formation by C–H activation on amides 25 with terminal alkynes 9 in a simple undivided cell setup (Scheme 11a).108 Their mechanistic findings for redox mediator-free conditions comprise an electrochemical C–H alkynylation under copper catalysis, followed by a base-mediated cyclization of alkyne 39 to the annulation products 37 (Scheme 11b). Moreover, the strategy was operative up to gram scale without loss of catalytic efficacy.
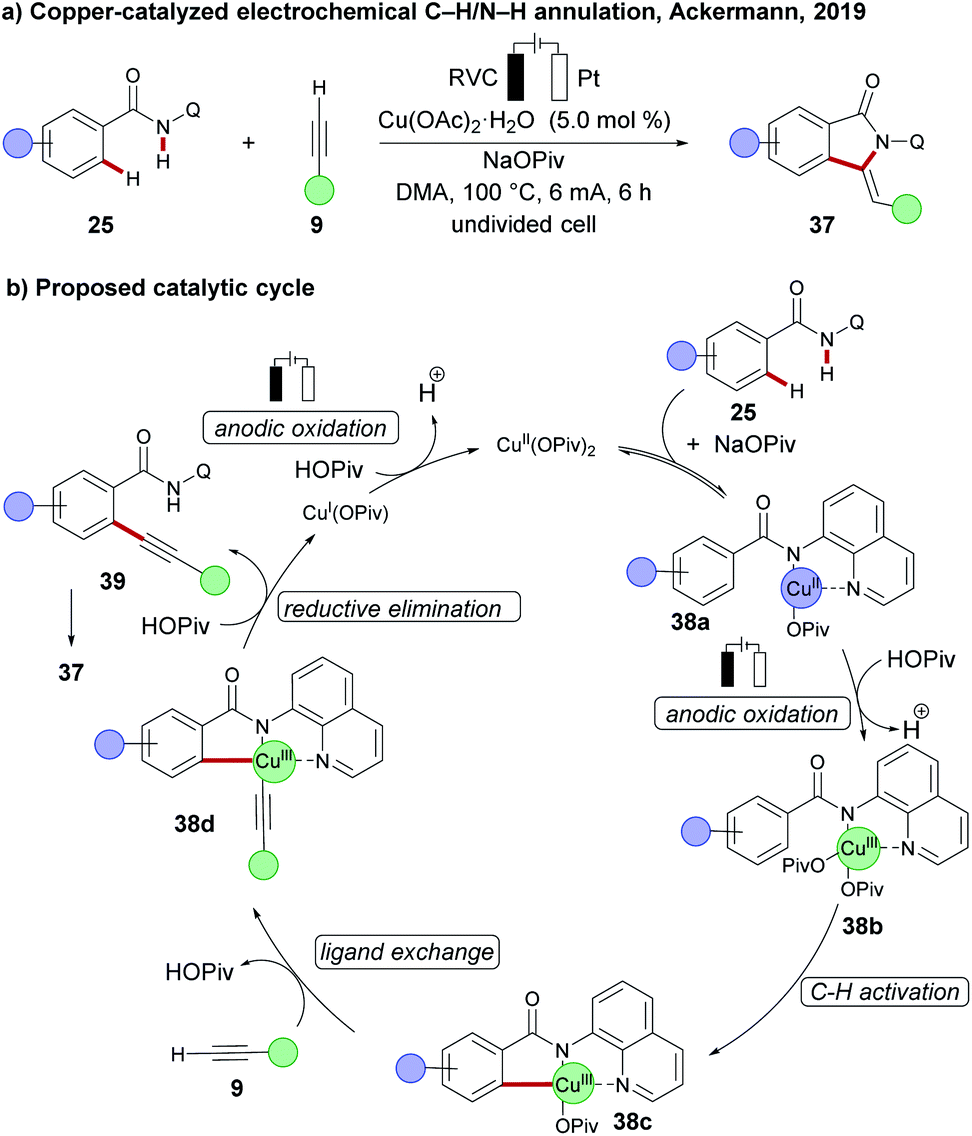 | ||
| Scheme 11 (a) Copper-catalysed electrochemical C–C formation by C–H activation. (b) Proposed mechanism. | ||
Furthermore, electrochemical C–H/C–C cleavage was realized with alkyne carboxylic acids 40 to deliver the annulation products 37 upon decarboxylation in good yields and with high E/Z selectivity (Scheme 12).
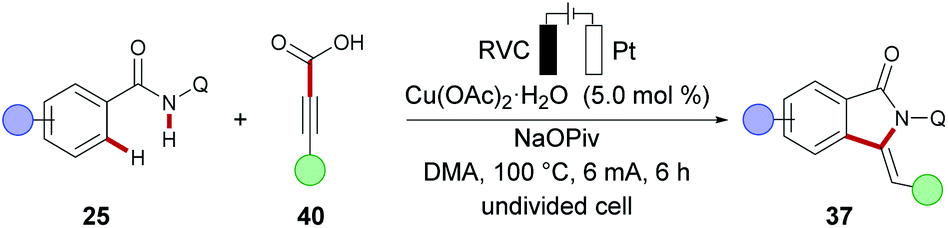 | ||
| Scheme 12 Copper-catalysed C–H/N–H annulation of benzamides via C–C cleavage. | ||
Iron-catalysed electrooxidative C–H activation
Iron being the most abundant transition metal in the earth's crust and having less toxicity compared to other transition metals, can have a remarkable impact on C–H activation. However, the predominant requirement of dichloroisobutane (DCIB) as a sacrificial stoichiometric oxidant made iron-catalysed oxidative C–H activation rather unattractive.30 This longstanding problem in iron catalysis was solved recently by successfully replacing the chemical oxidant DCIB through carefully chosen electrochemical reaction conditions by the Ackermann group. A chelation assisted C–H arylation of TAM-amide 41, using arylating reagent 42 under electrochemical iron catalysis, enabled the formation of arylated products 43 in excellent yield, with a broad scope (Scheme 13a).109 Detailed mechanistic studies proved indicative of an iron(II) complex as the active catalyst. The proposed catalytic cycle involves first a ligand-to-ligand hydrogen transfer (LLHT), followed by transmetalation to generate complex 44c and the key subsequent anodic oxidation to deliver the iron(III) complex 44d, which finally undergoes reductive elimination to deliver the desired product 43 (Scheme 13b).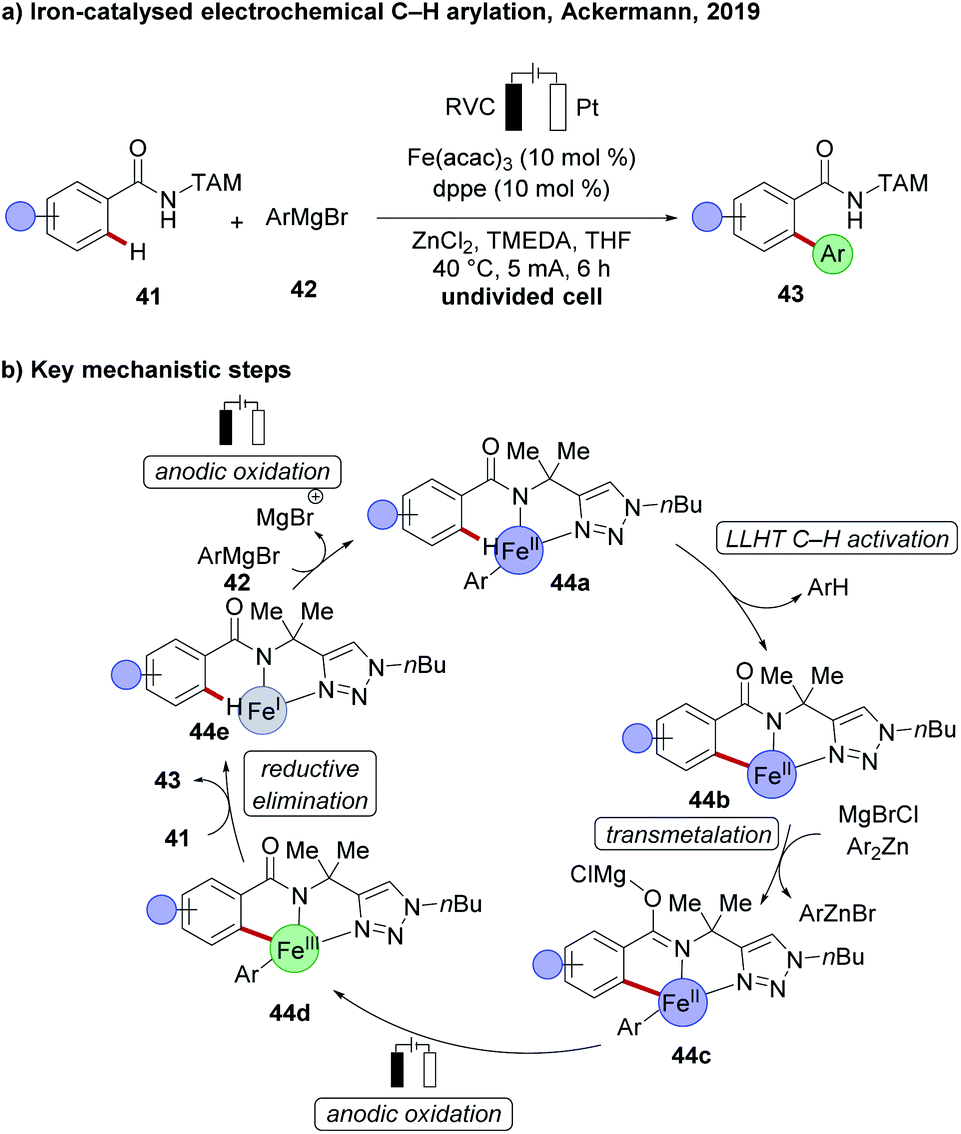 | ||
| Scheme 13 (a) Electrochemical C–H arylation under iron catalysis. (b) Key mechanistic steps. | ||
Manganese-catalysed electrooxidative C–H activation
While the electrochemical C–H activation with 3d metal catalysts has been established in recent years as a green method, an approach for the merger of manganese-catalysed oxidative C–H transformations with electrosynthesis was not achieved until recently. As a proof-of-concept, in the same study Ackermann first reported on electrosynthesis with environmentally-benign manganese catalysis for a C–H arylation of pyridine amide 45 with arylating reagent 42 in 70% yield of 46.109 The reaction proceeded in an undivided cell under zinc additive free conditions, being indicative of the unique potential of manganaelectro-catalyzed C–H activation (Scheme 14).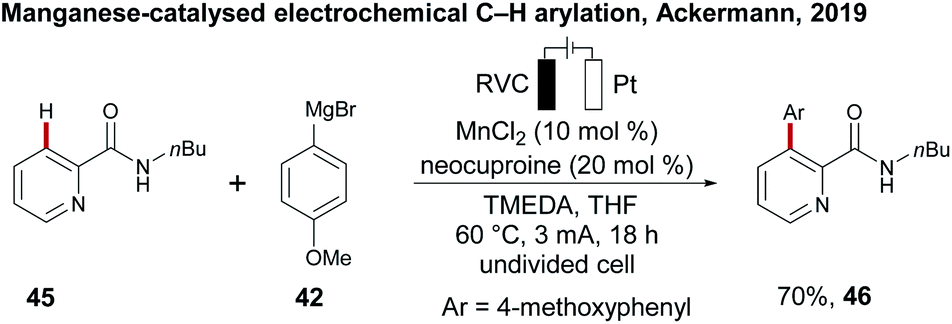 | ||
| Scheme 14 Electrochemical C–H arylation under manganese catalysis. | ||
Electrochemical net redox-neutral C–H activation: nickel-catalysed electrochemical C–H alkylations
In oxidative C–H transformations, electricity serves as an oxidant, and plays a crucial role in terms of lowering the activation energy, which then facilitates the desired transformations. In contrast, net redox-neutral nickel-catalysed C–H activation reactions do not require a stoichiometric oxidant, but were largely limited to harsh reaction conditions. Specifically nickel-catalysed C–H alkylations require strong bases, such as lithium tert-butoxide (LiOtBu) or lithium bis(trimethyl)silylamide (LiHMDS), and high reaction temperatures of 140–160 °C, translating into a significantly reduced substrate scope.87Very recently, the Ackermann group have demonstrated that nickel-catalysed C–H alkylations can be achieved under mild reaction conditions at room temperature using Et3N as a mild base (Scheme 15a).55 Both primary and secondary alkylation proceeded efficiently, using the corresponding alkyl iodides 47 as the alkylating reagents. It is further noteworthy to mention that chain-walking110,111 – commonly observed in nickel-catalysed cross-coupling reactions – was not observed under the optimized reaction conditions.55 The organometallic nature of the transformation was further supported by the independently prepared cyclometalated nickel(III) complex 33, which was found to be catalytically competent under the electrochemical conditions (Scheme 15b).
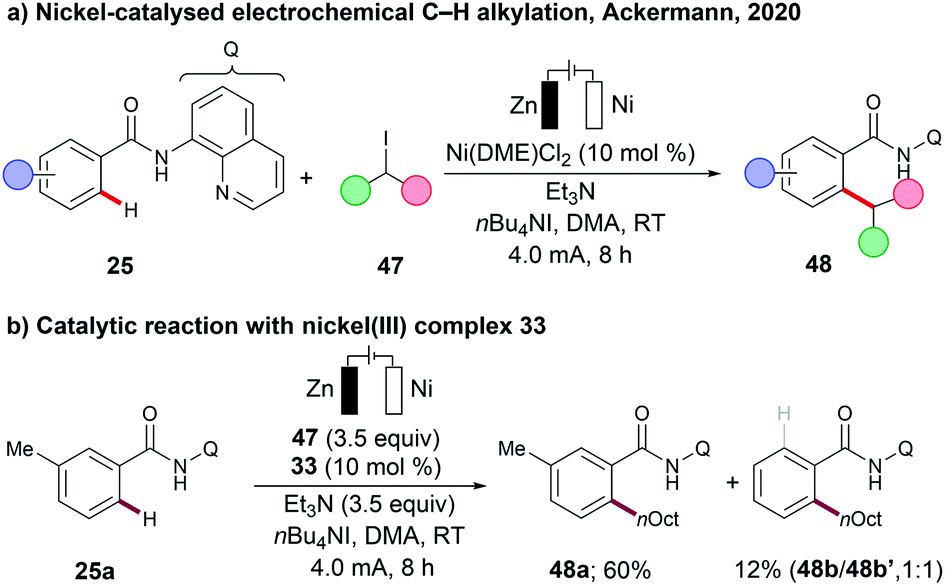 | ||
| Scheme 15 (a) Nickel-catalysed electrochemical C–H alkylation. (b) Catalytic reaction with nickel(III) complex 33. | ||
Based on detailed mechanistic studies,55 the authors proposed a nickel(I/III/III) manifold. A cyclometalated nickel(III) complex 49b was formed which was then reduced at the cathode to produce the nickel(II) intermediate 49c. Following a single electron transfer (SET), and addition of 47, gives the nickel(III) complex 49d. Complex 49d then undergoes cathodic reduction, followed by radical recombination, which generates nickel(III) intermediate 49e. Subsequent reductive elimination, and coordination of substrate 25, releases the C–H alkylation product 48 and regenerates the catalytically competent nickel(I) intermediate 49a (Scheme 16).
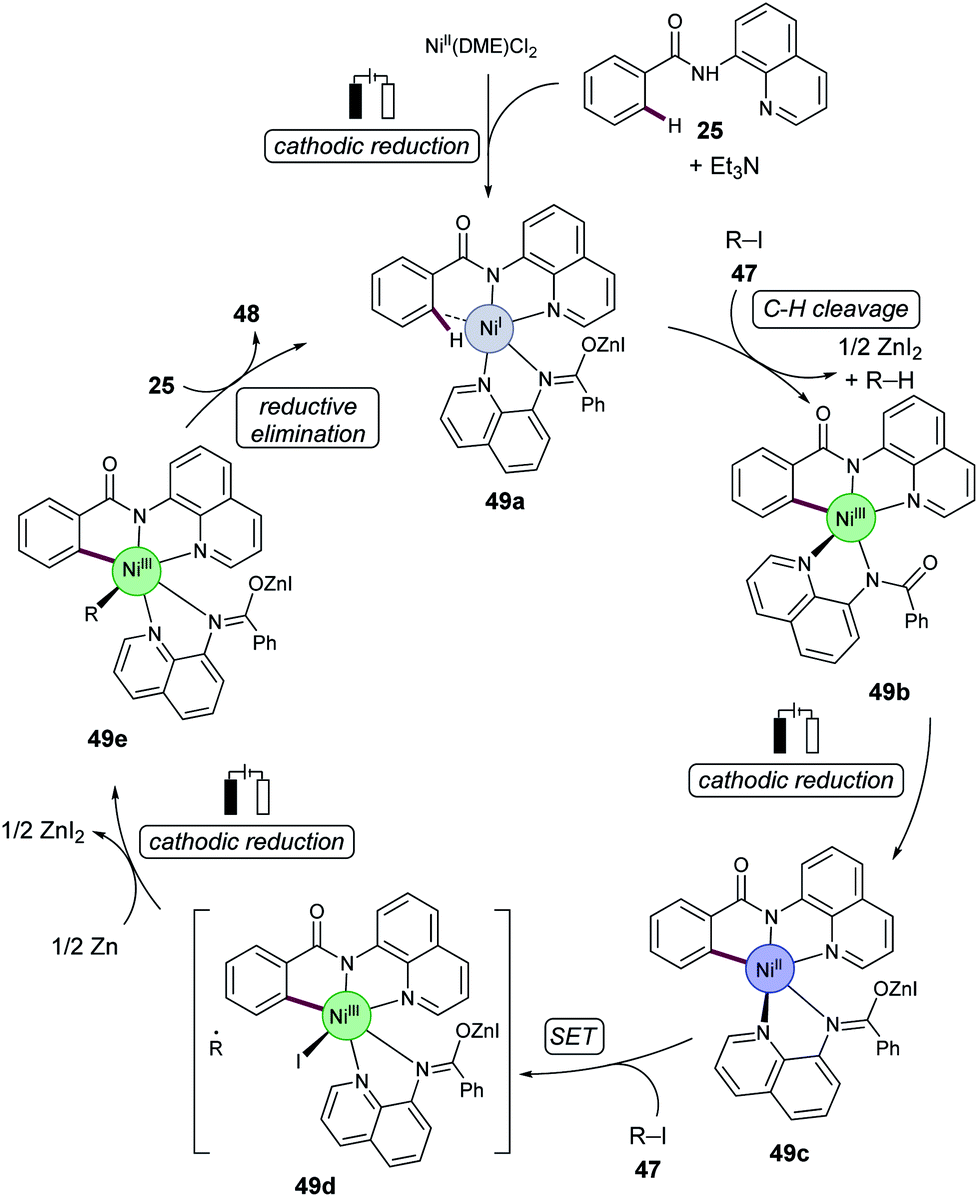 | ||
| Scheme 16 Proposed catalytic cycle for nickelaelectro-catalysed C–H alkylations. | ||
3 Renewable solvents in electrochemical C–H activation
γ-Valerolactone (GVL)
γ-Vialerolactone (GVL) has gained significant attention in recent years as a potential green and renewable reaction medium for C–H activation reactions.29 GVL can be produced from levulinic acid, obtained upon degradation of lignocellulose biomass.112 It is characterised by a low toxicity, it is non-flammable, shows good chemical stability, and its biodegradable nature renders it a suitable alternative for polar aprotic, non-renewable, toxic and often flammable solvents, such as DMF, DMA and NMP. GVL was first used in electrochemistry for C–H activation by the group of Ackermann in a cobalt-catalysed C–H amination reaction (Scheme 17a).91 To overcome the low conductivity, nBu4NPF6 was added as conducting salt. Under the reaction conditions, various aromatic and heteroaromatic amides 4 were aminated, with various cyclic secondary amines 7 to furnish the ortho C–H aminated products 8 with high reaction efficacy. Later, the same group found that GVL again was the optimum solvent for a cobaltaelectro-catalysed C–H oxygenation, using various benzoic acids 50 (Scheme 17b).113 An ortho-substituent to the amide 25 was found to be useful in order to obtain the desired C–H oxygenation products 51 in good yield and high selectivity.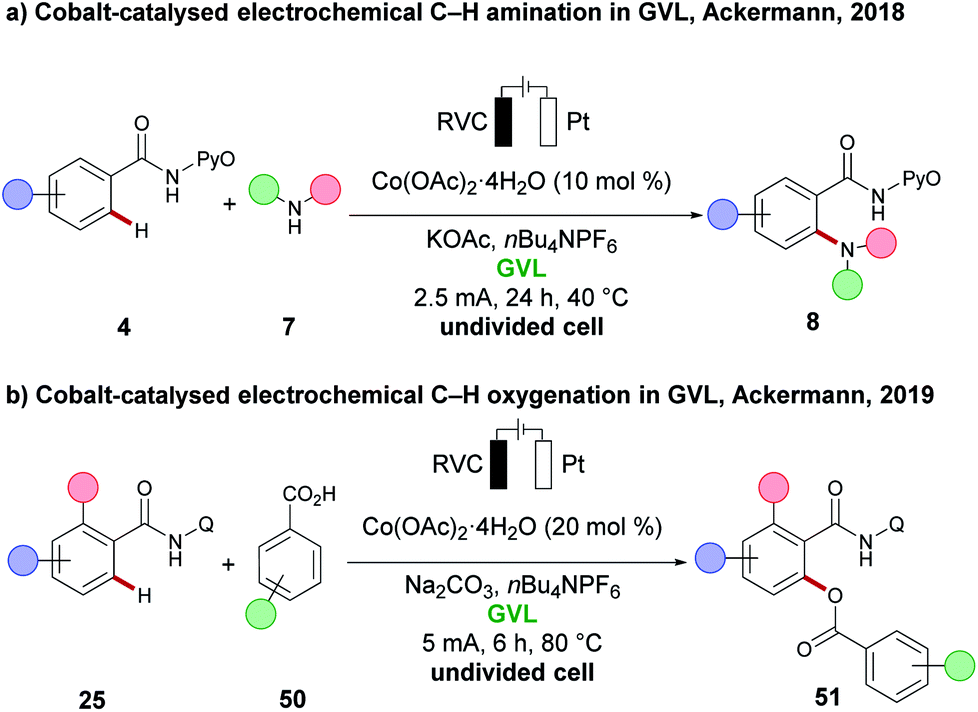 | ||
| Scheme 17 (a) Cobalt-catalysed electrochemical C–H amination in GVL. (b) Cobalt-catalysed electrochemical C–H oxygenation in GVL. | ||
Glycerol
Glycerol is produced as a by-product from biodiesel synthesis and can be obtained at a low cost. Several features, including low toxicity, a high boiling point, and its biocompatibility make it a potential green alternative solvent for numerous transformations.114,115 However, a major disadvantage arises from its high viscosity which results difficulties in mixing and diffusion of reactants inside this reaction medium and must be solved for using glycerol as a green solvent in electrochemistry. Due to its high polarity, it is miscible with water and thereby the viscosity of the overall mixture can be reduced, which now transforms it in a suitable protic solvent.116 Indeed, this strategy was in a proof-of-concept validated by the Ackermann group earlier this year. They could successfully replace non-renewable solvents for electrochemical transformations with identical and even better reaction efficacy using a mixture of glycerol/water as the renewable solvent of choice. Furthermore, their approach did not require electrolytes, which further boosts the catalyst's sustainability. In the first case, they performed a cobalt-catalysed oxidative C–H/N–H annulation of amides 4 with alkynes 9 (Scheme 18a).117 An improved yield of the annulation products 10 was observed in the glycerol/water solvent mixture. They also explored the C–H/N–H annulation reaction of amides 4 with allenes 12 (Scheme 18b), which transformed the substrates in slightly reduced yield to product 13, compared to the previously published solvent system.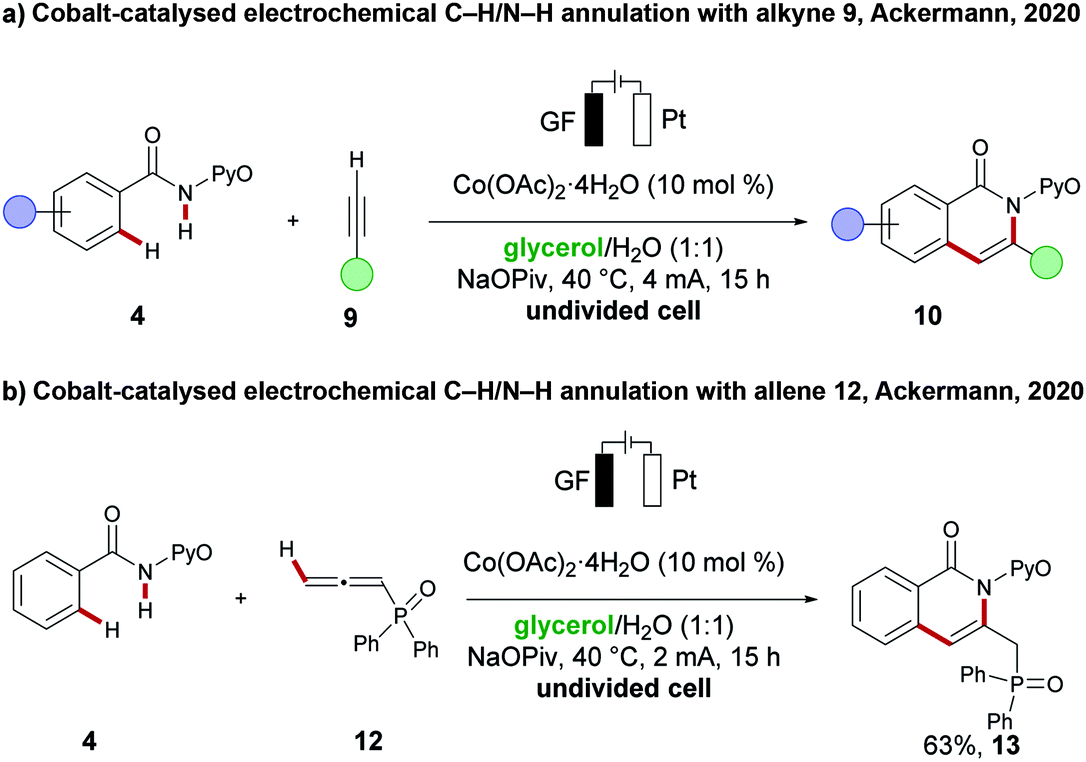 | ||
| Scheme 18 Cobaltaelectro catalysis in glycerol/H2O. (a) Alkyne annulation. (b) Allene annulation. | ||
4 Renewable energy sources in organic electrochemical transformations
In order to meet the increasing demand of global energy consumption, fossil fuels are not adequate and therefore the direct utilization of renewable energies has become a highly demanding field of research.41–43,118,119 Hydrogen production from water will certainly have a high impact, however, this requires the associated oxygen evolution reaction (OER) which is characterized by a high overpotential.120–123 In this regard electrosynthesis has been identified as a powerful tool for direct conversion of electricity to high-value chemicals, rather than producing oxygen with a lower economical footprint.124The direct utilization of sunlight for chemical energy conversion is an active research field.125 Although its impact in organic syntheses has been realized by the development of suitable photocatalysts for energy and electron transfer reactions,126 it is unfortunately often limited to the use of energy-intensive light sources of a specific wave length.127–129 In this context, the direct utilization of solar energy for electrosynthesis was reported by Moeller for oxidative transformations with a crystallin silicon photovoltaic module (Scheme 19a).130,131 In a representative example the direct oxidative cyclization of 52 to the tetrahydrofuran derivative 53 was achieved. A similar photovoltaic device was also used for the catalyst regeneration in an asymmetric dihydroxylation of 54 to form chiral alcohol 55 by means of anodic oxidation (Scheme 19b).130 Recently, the group of Rueping has demonstrated the use of solar energy for the reductive nickel-catalyzed cross-electrophile coupling between halides 56 and 57 to form the chain-walking product 58 (Scheme 19c).111
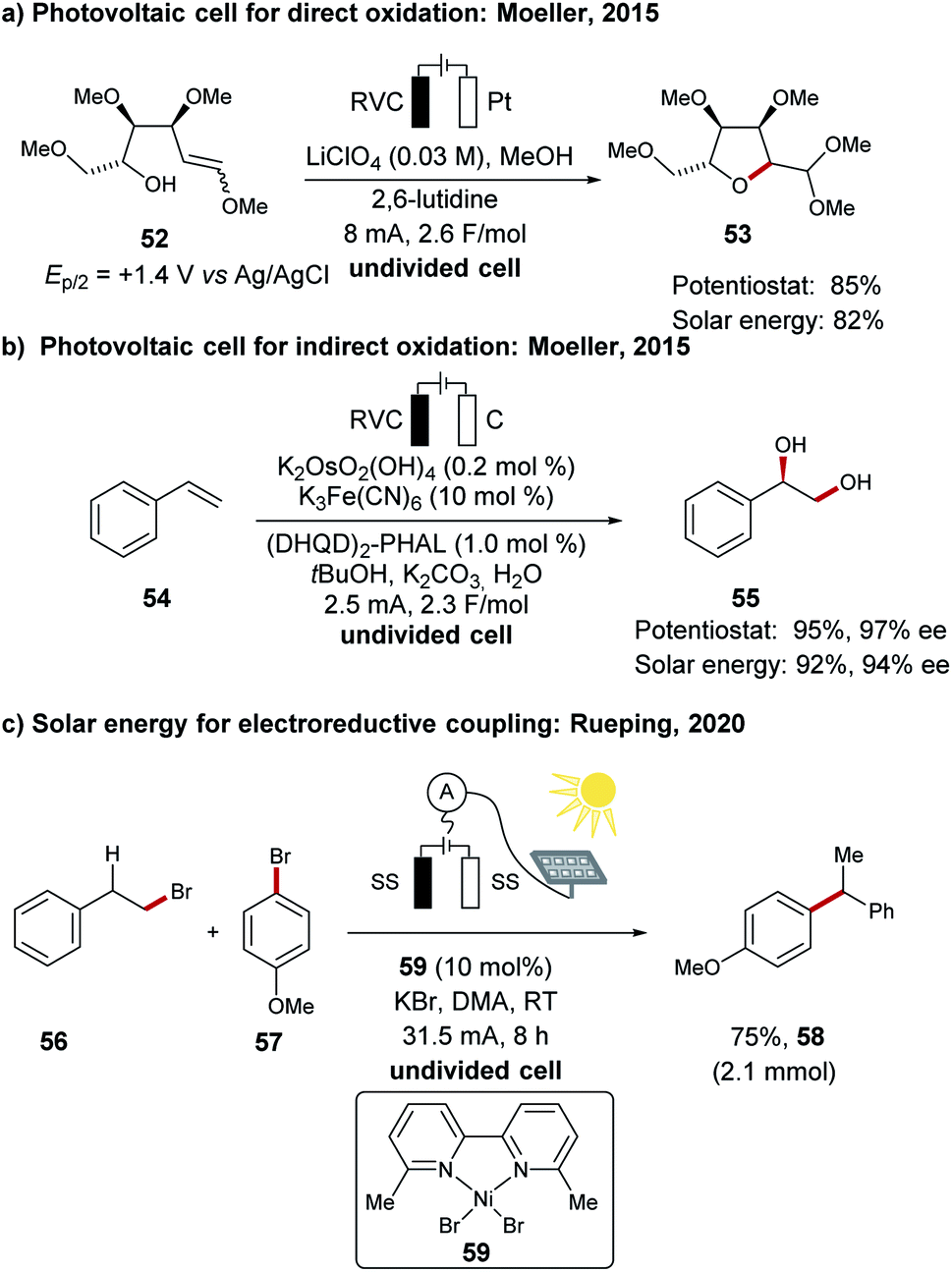 | ||
| Scheme 19 Selected examples for the direct utilization of solar energy in chemical transformations. | ||
To guarantee full resource economy of C–H activations, the merger of electrochemistry with Earth-abundant 3d base metal catalysis have made a notable impact (vide supra). However, in a recent proof-of-concept study, the Ackermann group showed that the direct utilization of renewable energies for oxidative C–H transformations by renewable energy sources, such as solar energy and wind energy, is indeed viable. Here, the cobaltaelectro-catalysed C–H/N–H annulation of amide 4 with alkyne 9 in biomass derived glycerol served as model reaction and was powered by either a commercially available photovoltaic module or a commercially available wind turbine (Scheme 20).117
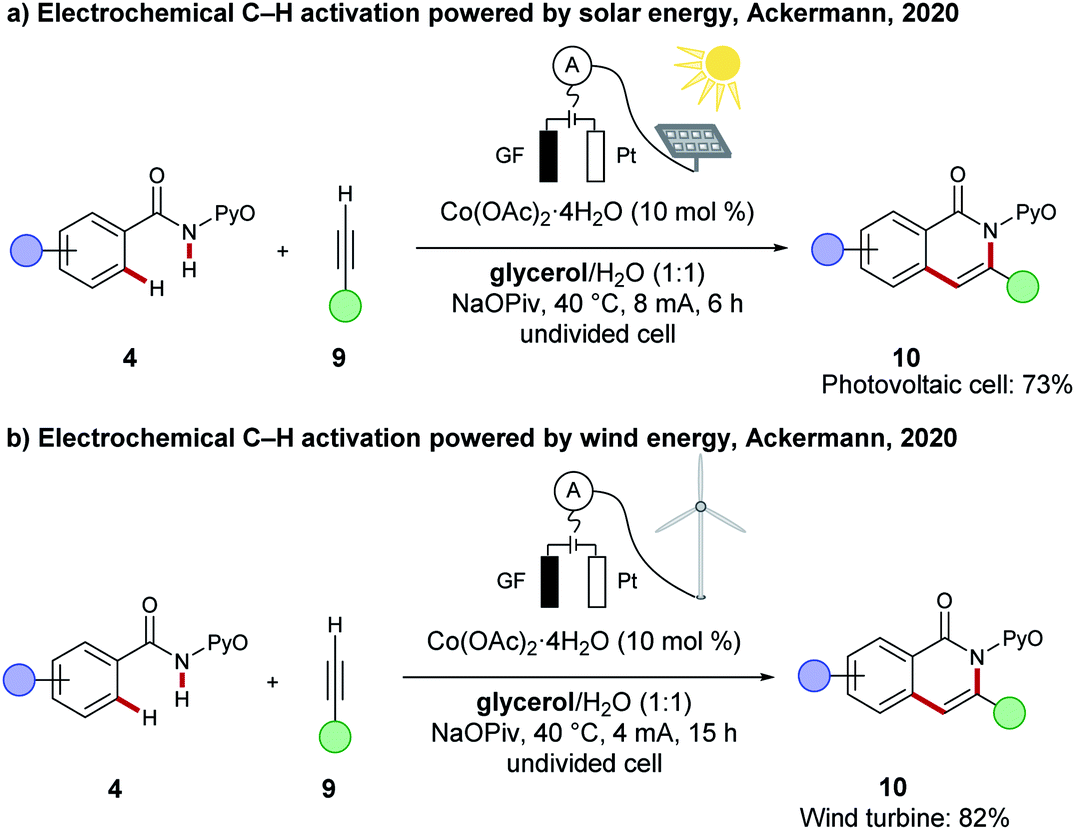 | ||
| Scheme 20 Electrochemical C–H activation powered by renewable electricity. | ||
5 Summary and outlook
Metallaelectro-catalysed C–H activations have significantly improved the sustainability and resource-economy of organic synthesis by the direct utilisation of renewable energy sources, Earth-abundant metal catalyst, and the employment of biomass-derived solvents. Herein, we have discussed, in the first part, cost-effective, less-toxic and mechanistically less explored 3d base metal-catalysed transformations that with the aid of electrochemistry enabled unexplored mechanistic pathways, such as oxidatively-induced transformations can be revealed. In the second part, biomass-derived solvents such as GVL and glycerol have proven to be viable solvents in cobaltaelectro-catalysed C–H transformations, and thus further improved the overall sustainability of the methodology. Polar protic solvents, such as glycerol, serve as an excellent reaction medium for organic electrochemical synthesis as no additional electrolyte is needed. Finally, the direct utilization of renewable energies, such as solar or wind energy, have served as an ideal proof-of-concept for the robustness of the method.Overall, the recent state-of-the-art allows an environmentally-benign strategy for C–H activation with molecular hydrogen being produced as the sole by-product. The proper utilization of the on-site generated hydrogen gas for further useful transformations may lead to complete electron economy by paired electrolysis, which has barely been explored59 and thus enables synthetic strategies in a circular fashion. Paired electrolysis could further be used for a combined strategy in organic CO2 functionalisation and fixation.132,133 However, unsolved challenges remain for the broader implementation of metallaelectro-catalysed C–H activations. For instance, in most of the presented studies, a bidentate directing group was required to stabilise the often high-valent metal-catalyst. Innovative ligand design will therefore be mandatory to overcome this challenge to adjust the reactivity of the 3d metal catalysts. Here precious metal-catalysts show a significantly higher robustness as was presented in recent reports by the group of Ackermann,48 using chiral amino-acids as transient directing groups in asymmetric palladaelectro-catalysed C–H olefinations.134 Thus, axially-chiral biaryls 61 were accessed from the biaryl aldehydes 60via atroposelective C–H alkylations under electrochemical conditions (Scheme 21a). Moreover, the thus-obtained biaryls 61 were readily converted into the diversified axially-chiral biaryl compounds 62, 63 and helicenes 64, 65 (Scheme 21b).
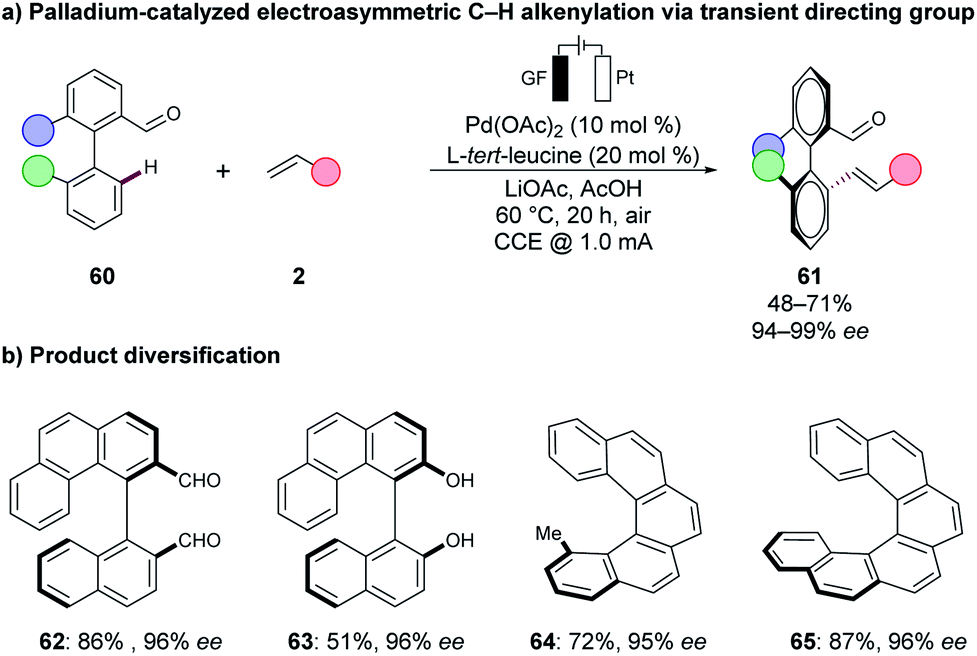 | ||
| Scheme 21 Palladaelectro-catalyzed atroposelective synthesis. | ||
Furthermore, the concept has mainly been applied to C(sp2)–H bonds, while transformations of C(sp3)–H by means of electrocatalysis are less explored.135 On a different note, sensitive reactions may not tolerate polar protic solvent conditions, hence, additional electrolytes are necessary to reduce the electrochemical cell resistance. Here, notable innovative concepts, including the combination of electrochemistry with recently introduced findings in micellar catalysis,136,137 ionic liquids138 or flow chemistry75,139 are expected to gain further prominence. Moreover, the recent merger of photochemistry with electrosynthesis enabled homogenous photoelectrocatalysis with outstanding redox potentials, thus avoiding drastic chemical redox reagents.140 This innovative strategy was successfully implemented in selective C–H alkylations of heteroarenes,141 as well as in otherwise challenging C–H trifluoromethylations of various (het)-arenes.142
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support by the DFG (Gottfried-Wilhelm-Leibniz) and the Alexander-von-Humboldt foundation (fellowship to R. C. S.) is gratefully acknowledged.Notes and references
- T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends Chem., 2019, 1, 63–76 CrossRef.
- B. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- P. T. Anastas and J. B. Zimmerman, Chem, 2016, 1, 10–12 CAS.
- S. A. Matlin, G. Mehta, H. Hopf and A. Krief, Nat. Chem., 2016, 8, 393–398 CrossRef CAS PubMed.
- F. Cavani, G. Centi, S. Perathoner and F. Trifiró, Sustainable Industrial Chemistry, Wiley-VCH, Weinheim, 2009 Search PubMed.
- UN-sustainability goals, https://www.undp.org/content/undp/en/home/sustainable-development-goals.html, accessed May 2020.
- M. Beller and C. Bolm, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, Wiley-VCH, Weinheim, 2017 Search PubMed.
- A. Behr and L. Johnen, in Handbook of Green Chemistry: GreenSynthesis, ed. C.-J. Li and P. T. Anastas, Wiley-VCH, Weinheim, 2012, vol. 7, pp. 69–92 Search PubMed.
- W. Steffen, K. Richardson, J. Rockström, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpenter, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sörlin, Science, 2015, 347, 1259855 CrossRef PubMed.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed.
- R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- O. Baudoin, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202001224.
- C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031–1058 CAS.
- L. Ackermann, A. R. Kapdi, H. K. Potukuchi and S. I. Kozhushkov, in Handbook of Green Chemistry, ed. C.-J. Li, Wiley-VCH, Weinheim, 2012, pp. 259–305 Search PubMed.
- A. d. Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2008 Search PubMed.
- L. G. Habgood, T. B. Gunnoe and L. Ackermann, Catalytic Hydroarylation of Carbon-Carbon Multiple Bonds, Wiley-VCH, Weinheim, 2018 Search PubMed.
- L. Ackermann, Modern Arylation Methods, Wiley-VCH, Weinheim, 2009 Search PubMed.
- S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed.
- F. Ferlin, S. Santoro, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 2510–2514 RSC.
- P. Gandeepan, N. Kaplaneris, S. Santoro, L. Vaccaro and L. Ackermann, ACS Sustainable Chem. Eng., 2019, 7, 8023–8040 CrossRef CAS.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed.
- R. D. Little and K. D. Moeller, Chem. Rev., 2018, 118, 4483–4484 CrossRef CAS PubMed.
- H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed.
- J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed.
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed.
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- R. F. Service, Science, 2019, 365, 1236–1239 CrossRef CAS PubMed.
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS PubMed.
- J. Yoshida and S. Suga, in Handbook of Green Chemistry, ed. C.-J. Li, Wiley-VCH, Weinheim, 2012, pp. 369–385 Search PubMed.
- Y. Qiu, J. Struwe and L. Ackermann, Synlett, 2019, 30, 1164–1173 CrossRef CAS.
- N. Sauermann, T. H. Meyer and L. Ackermann, Chem.–Eur. J., 2018, 24, 16209–16217 CrossRef CAS PubMed.
- S. Tang, L. Zeng and A. Lei, J. Am. Chem. Soc., 2018, 140, 13128–13135 CrossRef CAS PubMed.
- L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed.
- K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed.
- T. H. Meyer, J. C. A. de Oliveira, D. Ghorai and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 10955–10960 CrossRef CAS PubMed.
- S.-K. Zhang, J. Struwe, L. Hu and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 3178–3183 CrossRef CAS PubMed.
- R. C. Samanta, J. Struwe and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 14154–14159 CrossRef CAS PubMed.
- Y. Kawamata, J. C. Vantourout, D. P. Hickey, P. Bai, L. Chen, Q. Hou, W. Qiao, K. Barman, M. A. Edwards, A. F. Garrido-Castro, J. N. deGruyter, H. Nakamura, K. Knouse, C. Qin, K. J. Clay, D. Bao, C. Li, J. T. Starr, C. Garcia-Irizarry, N. Sach, H. S. White, M. Neurock, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6392–6402 CrossRef CAS PubMed.
- C. Li, Y. Kawamata, H. Nakamura, J. C. Vantourout, Z. Liu, Q. Hou, D. Bao, J. T. Starr, J. Chen, M. Yan and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 13088–13093 CrossRef CAS PubMed.
- S. T. Wismann, J. S. Engbæk, S. B. Vendelbo, F. B. Bendixen, W. L. Eriksen, K. Aasberg-Petersen, C. Frandsen, I. Chorkendorff and P. M. Mortensen, Science, 2019, 364, 756–759 CrossRef CAS PubMed.
- T. Wu, B. H. Nguyen, M. C. Daugherty and K. D. Moeller, Angew. Chem., Int. Ed., 2019, 58, 3562–3565 CrossRef CAS PubMed.
- J. Tsuji and M. Minato, Tetrahedron Lett., 1987, 28, 3683–3686 CrossRef CAS.
- C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef CAS.
- Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef CAS PubMed.
- F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310–11311 CrossRef CAS PubMed.
- C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC.
- Y. Qiu, W.-J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828–5832 CrossRef CAS PubMed.
- Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5818–5822 CrossRef CAS PubMed.
- L. Yang, R. Steinbock, A. Scheremetjew, R. Kuniyil, L. H. Finger, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 11130–11135 CrossRef CAS PubMed.
- L. Massignan, X. Tan, T. H. Meyer, R. Kuniyil, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 3184–3189 CrossRef CAS PubMed.
- F. Xu, Y.-J. Li, C. Huang and H.-C. Xu, ACS Catal., 2018, 8, 3820–3824 CrossRef CAS.
- R. Mei, J. Koeller and L. Ackermann, Chem. Commun., 2018, 54, 12879–12882 RSC.
- W.-J. Kong, Z. Shen, L. H. Finger and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 5551–5556 CrossRef CAS PubMed.
- Z.-J. Wu, F. Su, W. Lin, J. Song, T.-B. Wen, H.-J. Zhang and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 16770–16774 CrossRef CAS PubMed.
- Y. Qiu, A. Scheremetjew and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 2731–2738 CrossRef CAS PubMed.
- W.-J. Kong, L. H. Finger, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 6342–6346 CrossRef CAS PubMed.
- W.-J. Kong, L. H. Finger, A. M. Messinis, R. Kuniyil, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 17198–17206 CrossRef CAS PubMed.
- Y. Zhang, J. Struwe and L. Ackermann, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202005257.
- Q.-L. Yang, Y.-K. Xing, X.-Y. Wang, H.-X. Ma, X.-J. Weng, X. Yang, H.-M. Guo and T.-S. Mei, J. Am. Chem. Soc., 2019, 141, 18970–18976 CrossRef CAS PubMed.
- Y. Qiu, M. Stangier, T. H. Meyer, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14179–14183 CrossRef CAS PubMed.
- K. Hirano and M. Miura, Chem. Lett., 2015, 44, 868–873 CrossRef CAS.
- G. Cera and L. Ackermann, Top. Curr. Chem., 2016, 374, 57 CrossRef PubMed.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS PubMed.
- W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS.
- R. Mei, U. Dhawa, R. C. Samanta, W. Ma, J. Wencel-Delord and L. Ackermann, ChemSusChem, 2020, 13, 3306–3356 CrossRef CAS PubMed.
- T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245–1262 CrossRef CAS.
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed.
- S. M. Khake and N. Chatani, Trends Chem., 2019, 1, 524–539 CrossRef.
- J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19–30 CrossRef CAS.
- N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed.
- C. Tian, T. H. Meyer, M. Stangier, U. Dhawa, K. Rauch, L. H. Finger and L. Ackermann, Nat. Protoc., 2020, 15, 1760–1774 CrossRef CAS PubMed.
- N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090–5094 CrossRef CAS PubMed.
- R. Mei, X. Fang, L. He, J. Sun, L. Zou, W. Ma and L. Ackermann, Chem. Commun., 2020, 56, 1393–1396 RSC.
- T. H. Meyer, J. C. A. Oliveira, S. C. Sau, N. W. J. Ang and L. Ackermann, ACS Catal., 2018, 8, 9140–9147 CrossRef CAS.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383–2387 CrossRef CAS PubMed.
- R. Mei, N. Sauermann, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2018, 140, 7913–7921 CrossRef CAS PubMed.
- R. Mei, W. Ma, Y. Zhang, X. Guo and L. Ackermann, Org. Lett., 2019, 21, 6534–6538 CrossRef CAS PubMed.
- S. C. Sau, R. Mei, J. Struwe and L. Ackermann, ChemSusChem, 2019, 12, 3023–3027 CrossRef CAS PubMed.
- S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed.
- L. Zeng, H. Li, S. Tang, X. Gao, Y. Deng, G. Zhang, C.-W. Pao, J.-L. Chen, J.-F. Lee and A. Lei, ACS Catal., 2018, 8, 5448–5453 CrossRef CAS.
- X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195–4199 CrossRef CAS PubMed.
- Y. Cao, Y. Yuan, Y. Lin, X. Jiang, Y. Weng, T. Wang, F. Bu, L. Zeng and A. Lei, Green Chem., 2020, 22, 1548–1552 RSC.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- S.-K. Zhang, R. C. Samanta, A. Del Vecchio and L. Ackermann, Chem.–Eur. J., 2020 DOI:10.1002/chem.202001318.
- S.-K. Zhang, R. C. Samanta, N. Sauermann and L. Ackermann, Chem.–Eur. J., 2018, 24, 19166–19170 CrossRef CAS PubMed.
- Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494 CrossRef CAS PubMed.
- S. Kathiravan, S. Suriyanarayanan and I. A. Nicholls, Org. Lett., 2019, 21, 1968–1972 CrossRef CAS PubMed.
- C. Tian, U. Dhawa, A. Scheremetjew and L. Ackermann, ACS Catal., 2019, 9, 7690–7696 CrossRef CAS.
- C. Zhu, M. Stangier, J. C. A. Oliveira, L. Massignan and L. Ackermann, Chem.–Eur. J., 2019, 25, 16382–16389 CrossRef CAS PubMed.
- K.-J. Jiao, D. Liu, H.-X. Ma, H. Qiu, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 6520–6524 CrossRef CAS PubMed.
- G. S. Kumar, A. Peshkov, A. Brzozowska, P. Nikolaienko, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 6513–6519 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- C. Tian, U. Dhawa, J. Struwe and L. Ackermann, Chin. J. Chem., 2019, 37, 552–556 CrossRef CAS.
- S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601–1612 RSC.
- Y. Gu and F. Jérôme, Green Chem., 2010, 12, 1127–1138 RSC.
- F. Harnisch and U. Schröder, ChemElectroChem, 2019, 6, 4126–4133 CrossRef CAS.
- T. H. Meyer, G. A. Chesnokov and L. Ackermann, ChemSusChem, 2020, 13, 668–671 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- L. M. Reid, T. Li, Y. Cao and C. P. Berlinguette, Sustainable Energy Fuels, 2018, 2, 1905–1927 RSC.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- B. H. Nguyen, R. J. Perkins, J. A. Smith and K. D. Moeller, Beilstein J. Org. Chem., 2015, 11, 280–287 CrossRef CAS PubMed.
- L. A. Anderson, A. Redden and K. D. Moeller, Green Chem., 2011, 13, 1652–1654 RSC.
- Y. Yang and J.-W. Lee, Chem. Sci., 2019, 10, 3905–3926 RSC.
- N. W. J. Ang, J. C. A. de Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 12842–12847 CrossRef CAS PubMed.
- U. Dhawa, C. Tian, T. Wdowik, J. C. A. Oliveira, J. Hao and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 13451–13457 CrossRef CAS PubMed.
- Q. Zhang, X. Chang, L. Peng and C. Guo, Angew. Chem., Int. Ed., 2019, 58, 6999–7003 CrossRef CAS PubMed.
- J. F. Rusling, Acc. Chem. Res., 1991, 24, 75–81 CrossRef CAS.
- S. R. Yetra, T. Rogge, S. Warratz, J. Struwe, W. Peng, P. Vana and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 7490–7494 CrossRef CAS PubMed.
- M. Kathiresan and D. Velayutham, Chem. Commun., 2015, 51, 17499–17516 RSC.
- M. Elsherbini and T. Wirth, Acc. Chem. Res., 2019, 52, 3287–3296 CrossRef CAS PubMed.
- J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020 DOI:10.1021/acscentsci.0c00549.
- H. Yan, Z.-W. Hou and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef CAS PubMed.
- Y. Qiu, A. Scheremetjew, L. H. Finger and L. Ackermann, Chem.–Eur. J., 2020, 26, 3241–3246 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |