
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantifying internal charge transfer and mixed ion-electron transfer in conjugated radical polymers†
Shaoyang
Wang
a,
Alexandra D.
Easley
b,
Ratul M.
Thakur
a,
Ting
Ma
a,
Junyeong
Yun
a,
Yiren
Zhang
c,
Christopher K.
Ober
c and
Jodie L.
Lutkenhaus
 *ab
*ab
aArtie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, TX, USA. E-mail: jodie.lutkenhaus@tamu.edu
bDepartment of Materials Science and Engineering, Texas A&M University, College Station, TX, USA
cMaterials Science and Engineering, Cornell University, Ithaca, New York, USA
First published on 31st August 2020
Abstract
Macromolecular radicals are receiving growing interest as functional materials in energy storage devices and in electronics. With the need for enhanced conductivity, researchers have turned to macromolecular radicals bearing conjugated backbones, but results thus far have yielded conjugated radical polymers that are inferior in comparison to their non-conjugated partners. The emerging explanation is that the radical unit and the conjugated backbone (both being redox active) transfer electrons between each other, essentially “quenching” conductivity or capacity. Here, the internal charge transfer process is quantified using a polythiophene loaded with 0, 25, or 100% nitroxide radicals (2,2,6,6-tetramethyl-1-piperidinyloxy [TEMPO]). Importantly, deconvolution of the cyclic voltammograms shows mixed faradaic and non-faradaic contributions that contribute to the internal charge transfer process. Further, mixed ion-electron transfer is determined for the 100% TEMPO-loaded conjugated radical polymer, from which it is estimated that one triflate anion and one propylene carbone molecule are exchanged for every electron. Although these findings indicate the reason behind their poor conductivity and capacity, they point to how these materials might be used as voltage regulators in the future.
Introduction
As a potential next-generation cathode material for energy storage, organic radical polymers offer fast charging, high cycling stability without the need for transition metal elements. In these polymers, redox-active nitroxide radicals are attached to nonconjugated polymer backbones as pendant groups. The radical is capable of rapid electron transfer, because it does not involve bond breaking or formation. The most-studied organic radical polymer is poly(2,2,6,6-tetramethylpiperidinyloxy-4-yl methacrylate) (PTMA),1–3 in which the nitroxide radical is a 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) pendant group that is sterically protected by bulky methyl groups. Although PTMA exhibits a fast redox reaction, the long-range electronic conductivity in PTMA is rather low, on the order of 10−6 -10−11 S cm−1.4–6 Therefore, most cathodes contain 30 to 90 wt% conductive carbon additive to promote charge transfer at the expense of diluting active materials and energy density.1,7–9 More recently, conjugated radical polymers (CRPs) have been proposed as an alternative means to boost conductivity and increase capacity,10 but no success has yet been achieved due to internal charge transfer between the radical moiety and the conjugated backbone.11,12To further understand the issue, we first describe the basic properties of non-conjugated PTMA, which has a theoretical capacity of 111 mA h g−1 and a relatively high redox potential of 3.6 V vs. Li/Li+.1 During oxidation, the free radical loses an electron, forms an oxoammonium cation, and is doped by an anion; during reduction, the oxoammonium cation gains an electron, the free radical is regenerated, and the group is de-doped. Electron transfer in PTMA occurs via a hopping mechanism between free radicals and adjacent oxoammonium cations.13 Specifically, the heterogeneous electron transfer (polymer electrode to current collector) rate constant (k0) is on the order of 10−1 to 10−2 cm2 s−1, and the electron self-exchange reaction rate constant (kex) is on the order of 108 M−1 s−1.13–16
In contrast, conjugated polymers facilitate charge transfer through delocalized π electrons in the conjugated backbone and achieve long-range conductivity, exhibiting redox activity through doping and de-doping of the backbone.17–19 On the other hand, CRPs build upon typical conjugated polymer backbones such as polythiophene,20–22 polypyrrole,23,24 PEDOT,25 polydithienopyrrole,11,12 and polyacetylene26,27 by installing redox active groups (usually TEMPO). Most CRPs exhibit lower capacity and conductivity than expected (24–115 mA h g−1 and 10−13 to 10−2 S cm−1). Reasons cited include: electropolymerization leading to partly soluble (oligomeric) materials that cause capacity decay;28 the redox potential of the conjugated backbone depends on the degree of doping, which often overlaps with the redox potentials of nitroxide radicals;29 and the rapid redox kinetics of nitroxide radicals compete with the sluggish redox kinetics of the conjugated backbone.10
To summarize, the reason for the poor performance of CRPs thus far is related to the internal transfer of electrons between the redox-active side group and the redox-active conjugated backbone. The direction of electron transfer depends upon the relative redox potentials of the two species, as we have demonstrated previously for polythiophene–TEMPO and polydithienopyrrole–TEMPO CRPs.11,12 Specifically, we attached TEMPO pendant radicals to polythiophene (P3HT) backbones, whose redox potential was above the TEMPO radical (3.88 V and 3.6 V vs. Li/Li+, respectively) to obtain P3HT–TEMPO.11 Internal charge transfer was observed by monitoring the open circuit potential (OCP) after charging, in which the OCP dropped quickly from 4.2 V to 3.9 V within the first 1000 s.11 This coincided with some unoxidized TEMPO transferring an electron to de-dope the oxidized polythiophene, as confirmed using spectroelectrochemistry.11 As a result, the OCP relaxed to that of the species with the lower redox potential – TEMPO, 3.6 V vs. Li/Li+.11 We have also observed similar internal charge transfer in TEMPO-bearing poly(dithieno[3,2-b:2′,3′-d]pyrrole) (poly(DTP–TEMPO)).12 The OCP of poly(DTP–TEMPO) had a sharp decrease to from 4.1 V to 3.6 V within the first hour, and eventually relaxed to 3.0–3.1 V – the redox potential of the DTP unit.12 Again, it seems that the internal charge transfer occurs so that the lowest redox potential is achieved.
In the above-mentioned studies, the polymers were synthesized using electropolymerization, leading to insoluble CRPs with poor control. This approach prevented traditional characterization such that the molar mass or regioregularity could not be examined. This prevented quantification of the internal charge transfer mechanism and reduced the phenomena to a qualitative understanding. This knowledge is important so that one might design these polymers as charge or voltage regulators in one case or as materials for energy storage in another case.
Fortunately, access to controlled regioregular P3HT–TEMPO polymers with various TEMPO loadings (Fig. 1) has been recently reported by Zhang et al.30 TEMPO units were clicked onto a polythiophene backbone made from controlled amounts of 2,5-dibromo-3-hexylthiophene (HT) and 2,5-dibromo-3-(6-bromohexyl)thiophene (BrHT), modified with an azide.30 The final TEMPO-bearing P3HT was denoted as P3HT–TEMPO-X, in which X refers to the intended percent radical loading. The authors found that the solid-state conductivity decreased with increasing TEMPO loading for two reasons.30 First, bulky TEMPO groups twisted the polymer backbone, making it less planar and reducing the conjugation length.30 As a result, intrachain electron transfer through the backbone was impeded.30 Second, the steric hinderance of TEMPO side groups disrupted polymer packing and crystallization.30 At low TEMPO loading (25%), P3HT–TEMPO was semi-crystalline, which allowed for interchain electron transfer; at high TEMPO loading (50–75%), P3HT–TEMPO formed amorphous aggregates, causing a higher energy barrier for interchain electron transfer.30 Although the solid-state conductivity was examined for P3HT–TEMPO-X, the electrochemical behavior – and likewise the internal charge transfer – remained unknown.
 | ||
| Fig. 1 Chemical structure of P3HT–TEMPO-X. | ||
Here, we quantify the internal electron transfer behavior of regioregular P3HT, P3HT–TEMPO-25 and P3HT–TEMPO-100 in the presence of nonaqueous electrolyte, along with the coupled ion transport. The general electrochemical behavior was probed using cyclic voltammetry and galvanostatic charge–discharge of the solution-cast polymers. To characterize the relative capacity contributions from the different redox-active moieties (i.e. TEMPO radicals and the conjugated backbone), galvanostatic charge–discharge at various cutoff voltages was performed. The internal charge transfer was characterized through both OCP monitoring and decoupling of the faradaic and non-faradaic contributions for the first time. This is further quantified using electrochemical quartz crystal microbalance with dissipation monitoring (EQCM-D), which examines in situ ion transport associated with doping of the two redox active species, for the first time. These results provide a fundamental understanding of electron transfer within the CRPs, and in return, it will guide future design of redox polymers exhibiting both high conductivity and fast charging.
Materials and methods
Materials
P3HT, P3HT–TEMPO-25 and P3HT–TEMPO-100 were provided by Zhang et al., as synthesized in their recent publication.30 The reported regioregularities of P3HT–TEMPO-25 and P3HT–TEMPO-100 were 89% and 93%, respectively, and the actual radical concentration in P3HT–TEMPO-25 was only 20% versus 80% in P3HT–TEMPO-100.30 The molecular weights of P3HT–TEMPO-25 and P3HT–TEMPO-100 were 5.9 and 9.2 kDa, respectively. The solid-state conductivities of P3HT, P3HT–TEMPO-25 and P3HT–TEMPO-100 were 7 × 10−5, 2 × 10−7, and 3.8 × 10−11 S cm−1, respectively.30 All other chemicals were used as received from Sigma-Aldrich unless otherwise noted.Three-electrode cell
Indium tin oxide-coated glass (ITO, Delta Technologies) was used as the substrate for the working electrode. 1 mg of P3HT and P3HT–TEMPO-25 were each dissolved in 1 ml of chloroform. 1 mg of P3HT–TEMPO-100 was dissolved in 1 ml of 1,4-dioxane with heat. 60–100 μL of the solution was drop-cast onto ITO-coated glass and the mass loading varied from 0.3 to 0.5 mg cm−2. Typical active areas were 2.5–3.5 cm2. The counter and reference electrode were both lithium foils. The electrolyte was 0.5 M lithium triflate (LiCF3SO3) in ethylene carbonate/diethyl carbonate (EC/DEC, 3/7 by volume). The electrodes were assembled in a three-neck flask under inert conditions in an argon-filled glove box.Electrochemical tests
All electrochemical tests were conducted using a Solartron 1260/1280. The polymer film was conditioned at 10 mV s−1 for 10 cycles first, followed by cyclic voltammetry at 0.5, 1, 2, 5, 10, 25, and 50 mV s−1, 3 cycles at each scan rate. Subsequently, galvanostatic charge–discharge was conducted at 5, 10, and 20 μA cm−2 from 3 V to 4.2 V vs. Li/Li+, three cycles at each current density. To isolate the contribution of the TEMPO radical and the conjugated backbone, the cell was charged from 3 V to various potential cutoffs at 3.5, 3.6, 3.7, 3.8, 4.0 and 4.2 V, three cycles under each voltage. Considering the difference in capacity and charge–discharge profiles, P3HT was charged at a higher current of 5 μA cm−2, whereas P3HT–TEMPO-25 and P3HT–TEMPO-100 were charged at 2 μA cm−2. Electron impedance spectroscopy (EIS) was performed over a frequency range of 10 mHz to 1 MHz with a perturbation voltage of 10 mV and the cell was tested at 3.6 V, 3.8 V and 4.0 V. At last, the cell was charged to 4.2 V and held at this potential for 30 min. The open circuit potential (OCP) was monitored for 6 h after removing the bias. The electrochemical cell was stored under inert conditions for the course of the experiments.EQCM-D
Gold-coated, planar AT-cut quartz crystals (QSX 338) with a fundamental frequency of 4.95 MHz were used as substrates for EQCM-D (Biolin Scientific). Detailed cleaning and substrate preparation procedures are reported in our previous publications.31,32 The stock solution for spin-coating was prepared by dissolving 10 mg of P3HT in 1 ml chloroform, and 10 mg of P3HT–TEMPO-100 in 1 ml 1,4-dioxane with medium heat. (P3HT–TEMPO-25 was not presented because it yielded inconsistent results). A thin layer of the polymer was deposited on the sensor by spin-coating 40 μL of the stock solution at 500 rpm for 1 min. Electrochemical measurements were done in a three-electrode cell in which the working electrode was the polymer-coated quartz crystal, the counter electrode was Pt plate, and the quasi-reference electrode (QRE) was silver wire. The electrolyte was 0.5 M LiCF3SO3 in propylene carbonate. The frequency and dissipation response in EQCM-D was monitored during cyclic voltammetry at various scan rates. Electrode mass was modelled using the Voigt model and the parameters were the same as our previous publication.31 The charge was normalized by the active area of the sensor (1.13 cm2).Results and discussion
The fundamental electrochemical responses of the three polymers were first compared to examine the effect of radical loading on the redox behavior. Cyclic voltammograms of P3HT, P3HT–TEMPO-25, and P3HT–TEMPO-100 at 0.5 mV s−1 are plotted in Fig. 2a–c. P3HT exhibited one oxidation peak at 3.84 V and two reduction peaks at 3.80 V and 3.36 V vs. Li/Li+(Fig. 2a); this response is consistent with prior reports for P3HT.33–35 P3HT–TEMPO-100 showed a similar current response as that of PTMA,1 in which a reversible redox couple at E1/2 = 3.68 V vs. Li/Li+ was observed (Fig. 2c); thus, this peak is assigned to the nitroxide radical. On the other hand, P3HT–TEMPO-25 displayed mixed behavior, in which a rectangular capacitive response was superimposed on a redox peak (E1/2 = 3.69 V vs. Li/Li+) (Fig. 2b). The capacitive response is attributed to the P3HT backbone, and the peak is attributed to the TEMPO functional group.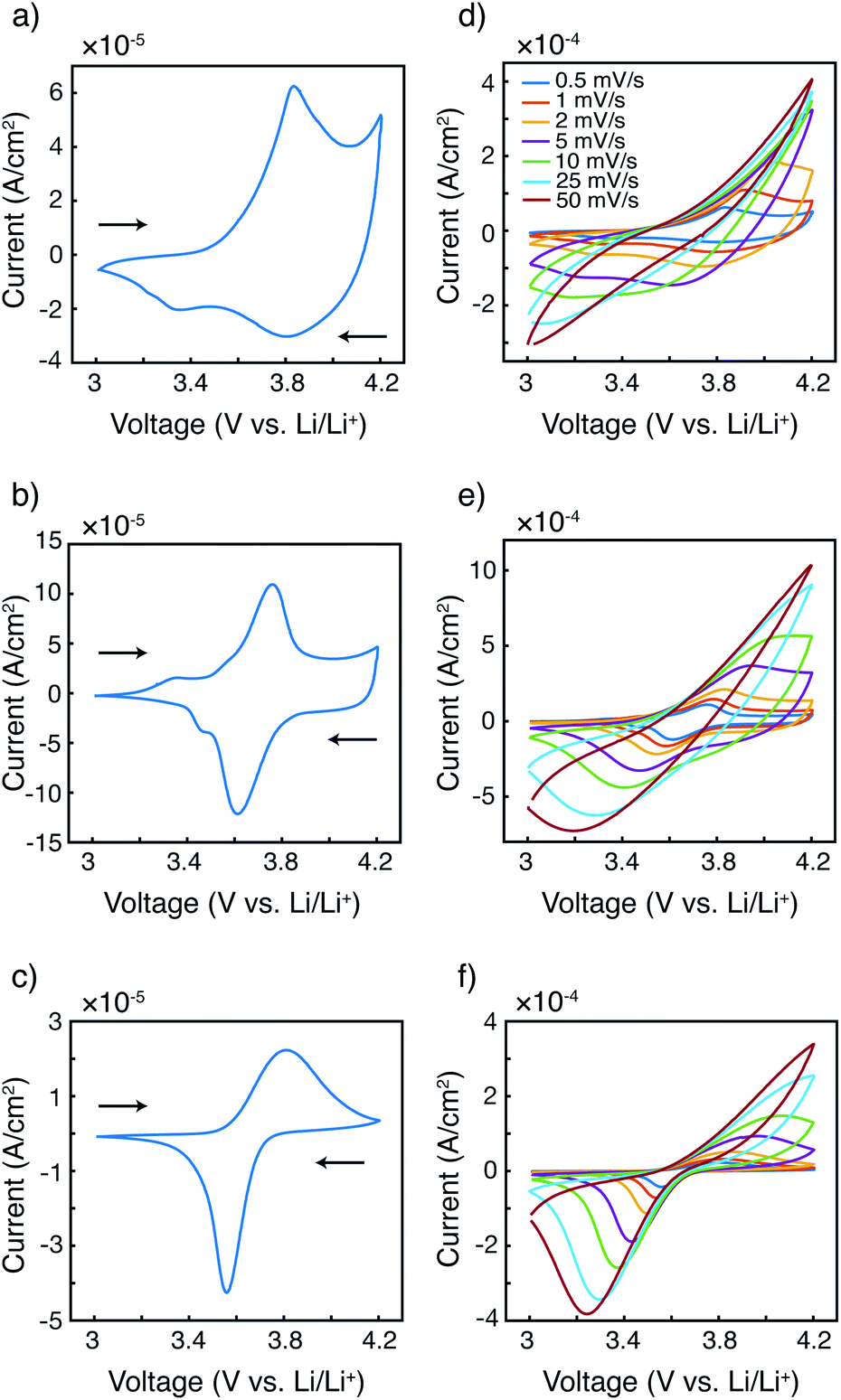 | ||
| Fig. 2 Cyclic voltammetry at 0.5 mV s−1 for (a) P3HT, (b) P3HT–TEMPO-25, and (c) P3HT–TEMPO-100. Overlapped cyclic voltammograms of (d) P3HT, (e) P3HT–TEMPO-25, and (f) P3HT–TEMPO-100 at scan rates of 0.5 to 50 mV s−1. The working electrode was made by drop-casting the polymer solution on indium tin oxide (ITO) coated glass. The counter and reference electrodes were both lithium ribbons. The electrolyte was 0.5 M LiCF3SO3 in EC/DEC (3/7 by volume). | ||
As scan rate increases, the redox peaks become less obvious and the cyclic voltammograms become distorted for all three polymers (Fig. 2d–f). Specifically, the anodic peaks disappear when the scan rate is above 2 mV s−1, 10 mV s−1, and 10 mV s−1 for P3HT, P3HT–TEMPO-25, and P3HT–TEMPO-100, respectively. On the contrary, most cathodic peaks remain present with increasing scan rate. The cathodic peaks persist up to a scan rate of 5 mV s−1 for P3HT and persist for all scan rates for P3HT–TEMPO-25 and P3HT–TEMPO-100. Such asymmetry between anodic and cathodic scans is commonly seen in conjugated polymers.34,36,37
Notably, the shape of the cathodic current response is largely affected by the radical loading. As the TEMPO loading increased from P3HT–TEMPO-25 to P3HT–TEMPO-100, the cathodic peak became sharper and closer to the current response of homopolymer PTMA.1 This reflects that the process shifted from delocalized electron transport to localized electron hopping under higher radical loading.
In P3HT–TEMPO-100, electron transfer likely occurs via hopping between adjacent TEMPO sites, because the bulky TEMPO radicals impede polymer packing which then prevents charge transfer along the conjugated polythiophene backbone. In P3HT–TEMPO-25, charge transfer occurs both through the TEMPO radicals and along the conjugated backbone. When the radical loading is relatively low, the bulky TEMPO groups are spatially distant, which permits partial charge transfer through the conjugated backbone; this mixed behavior results in both the capacitive and the redox characters in Fig. 2b.
The relationship between peak current and scan rate was next inspected to further understand the redox mechanism. Only the cathodic scan was examined because the anodic scans were distorted at higher scan rates. The CV distortion might be due to limited ion diffusion in the relatively thick polymer films prepared from drop-casting. Fig. 3 shows the cathodic peak current vs. scan rate1/2 (Fig. 3a–c) and vs. scan rate (Fig. 3d–f). Because P3HT does not show a clear reduction peak at higher scan rates, the cathodic current at 3.80 V was plotted instead. The current response of P3HT shows a linear relationship with neither scan rate1/2 nor scan rate, probably because of the distorted response even in the cathodic scans. The cathodic peak current values of P3HT–TEMPO-25 and P3HT–TEMPO-100 were linearly proportional to scan rate1/2, except at the highest two scan rates (25 and 50 mV s−1). This indicates that charge transfer in both CRPs is diffusion-controlled at lower scan rates, consistent with our previous observation in non-regioregular TEMPO bearing polythiophenes.11
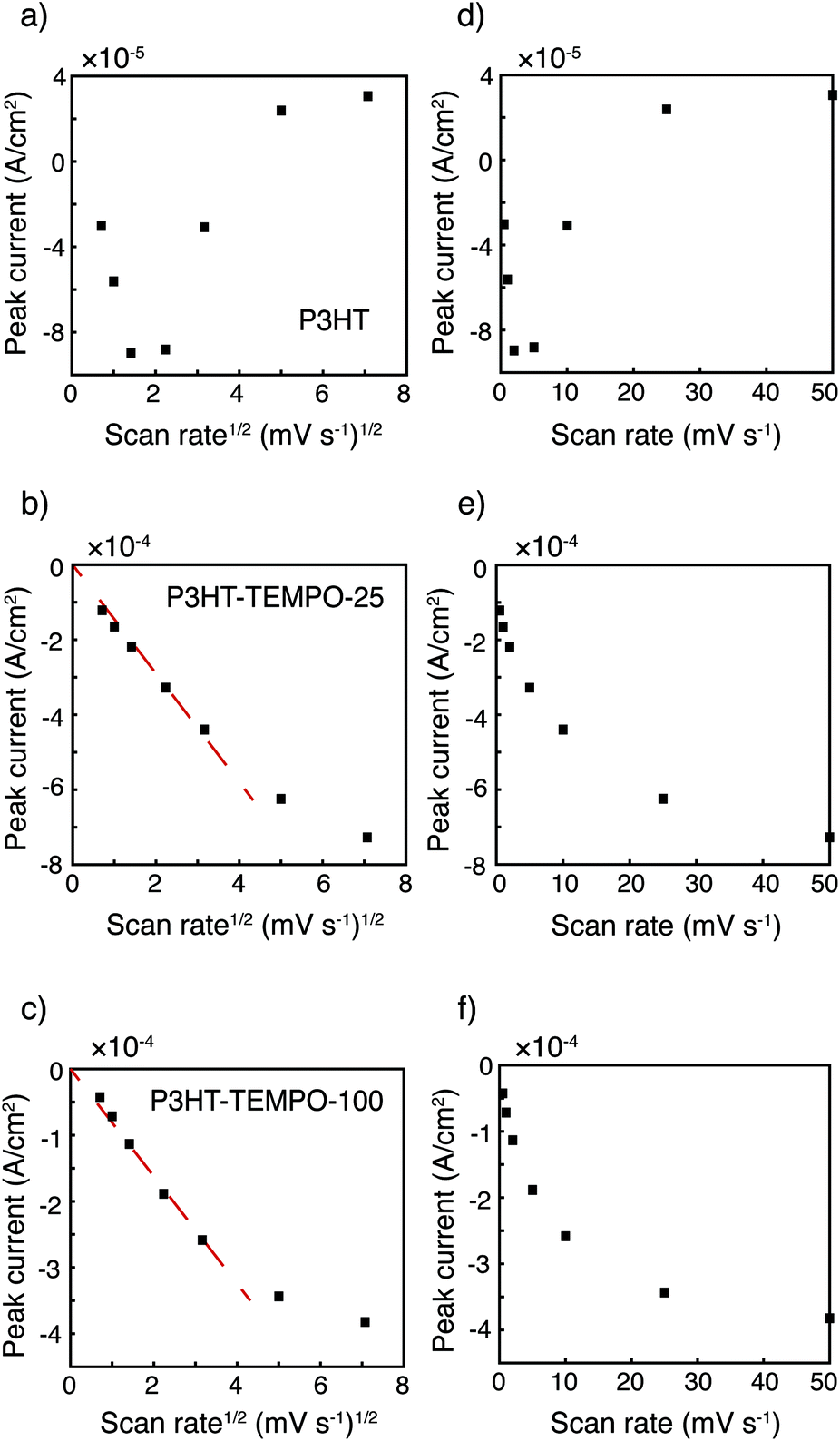 | ||
| Fig. 3 Peak current vs. scan rate1/2 for (a) P3HT, (b) P3HT–TEMPO-25, and (c) P3HT–TEMPO-100 and vs. scan rate for (d) P3HT, (e) P3HT–TEMPO-25, and (f) P3HT–TEMPO-100. Data are analyzed from cyclic voltammograms shown in Fig. 2. The dashed lines indicate linear regions. | ||
Cyclic voltammograms at 10 mV s−1 were further deconvoluted to estimate the faradaic vs. non-faradaic contributions to the total charge transfer for the three polymers (Fig. 4, and ESI†) following the method of Sathiya et al.38 For this analysis, we utilized CVs from thin films obtained with EQCM-D (see below) to reduce diffusion limitations. It is clearly seen that the faradaic process was the major charge transfer mechanism for P3HT (Fig. 4a). For P3HT-TEMPO-25, we were not able to reliably deconvolute the CVs, as any attempts at linear fits resulted in R2 values lower than 0.8; this could be a result of internal charge transfer, but other methods are needed for such a claim. As for P3HT-TEMPO-100, the deconvolution was much better-behaved (Fig. 4b), in which a sharp non-faradaic peak associated with TEMPO competes with faradaic processes assigned to P3HT. In comparing P3HT and P3HT-TEMPO-100, the presence of the TEMPO radical causes the charge transfer to be dominated by the TEMPO group in its active potential window, leaving faradaic processes to occur at higher potentials. This difference could be ascribed to internal charge transfer as well as the rapid TEMPO redox kinetics. Fig. S2† displays the respective charge contributions, obtained from the integrated, deconvoluted cyclic voltammograms.
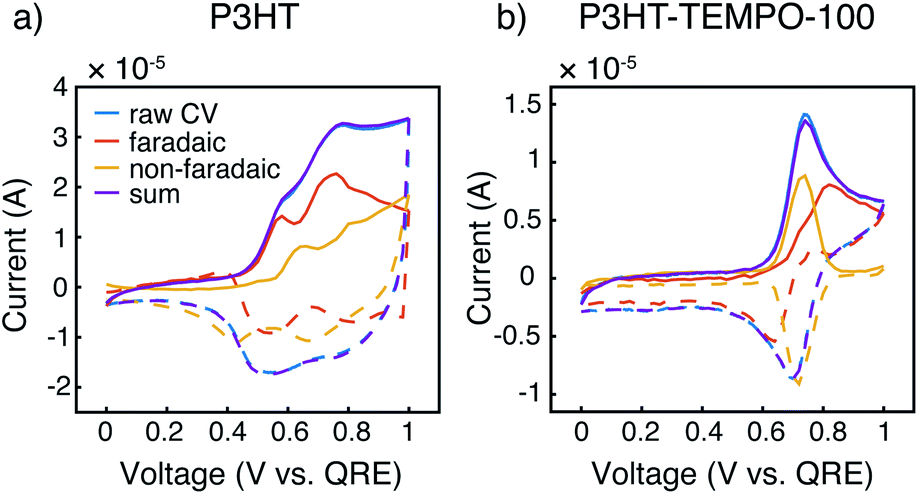 | ||
| Fig. 4 Cyclic voltammetry deconvolutions for (a) P3HT and (b) P3HT-TEMPO-100 at 10 mV s−1. The anodic currents are solid lines, and the cathodic currents are dashed lines. The faradaic and non-faradaic contributions to the total current (blue) are red and yellow, respectively. The sum of calculated faradaic and non-faradaic contributions is purple. The measurements were conducted in an EQCMD chamber, in which the working electrode was P3HT/P3HT–TEMPO-100-coated quartz crystal, the counter electrode was Pt plate, and the quasi-reference electrode was silver wire. The electrolyte was 0.5 M LiCF3SO3 in propylene carbonate. | ||
Galvanostatic charge–discharge was next applied to examine each polymer's response to a constant current, Fig. 5. P3HT exhibited a sloping voltage profile (Fig. 5a), which is typical for conjugated polymers.19 The minor plateau between 4.1 to 4.2 V is likely from slow charging under a low current density of 5 μA cm−2. At current densities of 10 and 20 μA cm−2, the plateau was not observed. P3HT–TEMPO-100 shows a similar charge–discharge profile consistent with homopolymer PTMA,39 having a flat plateau from 3.6 to 3.7 V and little capacity contribution from voltages above and below the plateau (Fig. 5c). Again, P3HT–TEMPO-25 exhibited mixed behavior, in which both a sloping discharge profile and plateau were observed (Fig. 5b); this short plateau appeared between 3.6 and 3.8 V, and then the voltage increased gradually above 3.8 V. For P3HT–TEMPO-25, this mixed behavior shows that the low TEMPO loading permits electron transfer through both the conjugated backbone and the radical sites. Also, it is notable that this voltage plateau is more obvious in the discharge profile than in the charge profile; this observation is consistent with the more distinct cathodic peaks in the cyclic voltammograms (Fig. 2e).
 | ||
| Fig. 5 Charge–discharge profiles for (a) P3HT, (b) P3HT–TEMPO-25, and (c) P3HT–TEMPO-100. | ||
The capacity values also vary with radical loading. P3HT shows the highest capacity under all current densities. Specifically, the areal discharge capacities of P3HT were 9.6, 8.8 and 7.8 μAh cm−2 at 5, 10 and 20 μA cm−2, respectively (Fig. 5a). Although bearing a second redox-active moiety, P3HT–TEMPO-100 yielded lower areal discharge capacities of 2.9, 2.6 and 2.4 μAh cm−2 at 5, 10 and 20 μA cm−2, respectively (Fig. 5c). On the other hand, P3HT–TEMPO-25 exhibited a moderate discharge capacity of 4.2 μAh cm−2 at 5 μA cm−2, but its capacity drastically dropped to 1.6 and 0.4 μAh cm−2 at 10 and 20 μA cm−2, respectively, having the lowest areal capacities among the three (Fig. 5b). We note that the samples are thin and have low mass loadings (0.3–0.5 mg cm−2), so capacity is best displayed using an areal basis. However, we also display specific capacity in Fig. 5 for comparison to the reader.
Interestingly, the trend in radical loading with capacity is counterintuitive. Specifically, the theoretical capacities for P3HT, P3HT–TEMPO-25, and P3HT–TEMPO-100 are 161.4, 160.9, and 159.5 mA h g−1 (assuming one electron transferred per P3HT unit and another one transferred per TEMPO unit). However, the obtained capacities, in order from lowest to highest, were P3HT–TEMPO-100 < P3HT–TEMPO-25 < P3HT, respectively. Electrochemical impedance spectroscopy of the three polymers also revealed a supporting trend, in which the charge transfer resistance at 4.0 V vs. Li/Li+ was highest for P3HT–TEMPO-100 and lowest for P3HT, Fig. S3–S5 and Table S1.† This is explained by the phenomena of internal charge transfer between the loaded TEMPO and the conjugated backbone.11,12 The effect of radical loading on internal transfer has not been examined before, and these results show that even with 25% TEMPO loading, internal transfer still occurs.
The capacity contributions from the nitroxide group and the polythiophene backbone were estimated by charging the polymers to various potential cutoffs from 3.5 to 4.2 V. Fig. 6a–c shows the calculated areal capacities at each cutoff voltage for P3HT, P3HT–TEMPO-25, and P3HT–TEMPO-100, respectively; Fig. 6d shows the normalized charge capacity, which is the ratio of charging capacity at a particular cutoff voltage over the highest capacity among all tested voltages. In other words, the normalized charge capacity reflects the capacity contributions from each cutoff voltage to the “total” charging capacity.
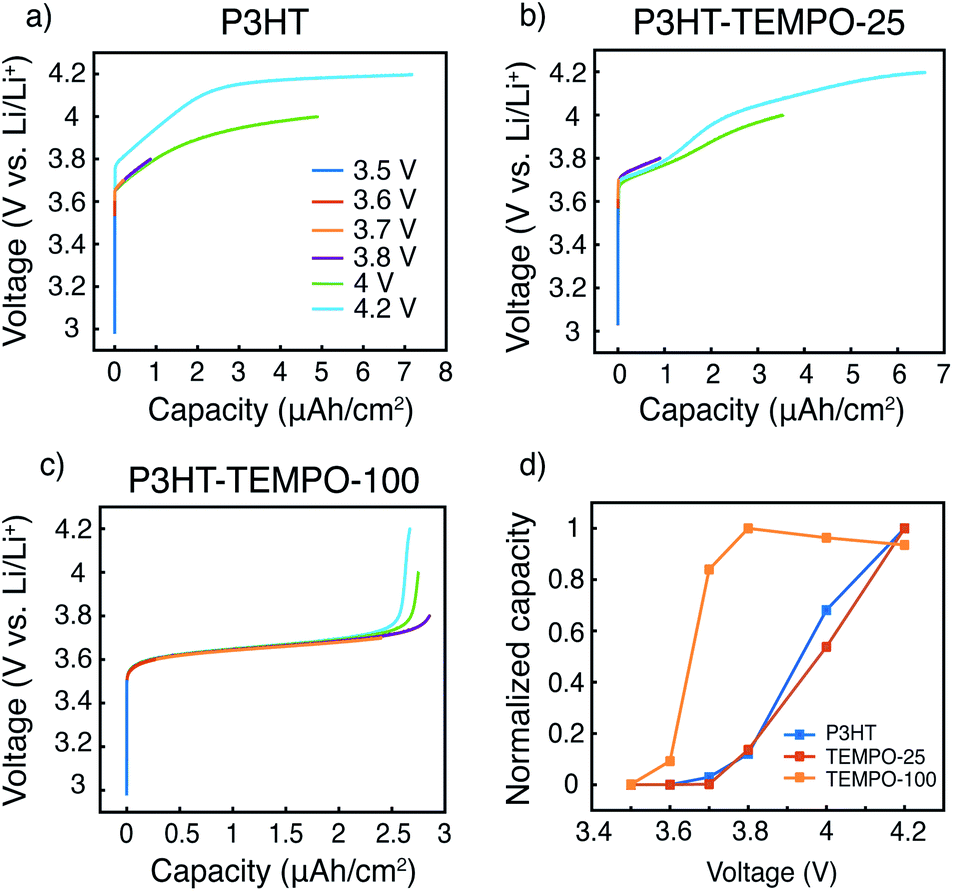 | ||
| Fig. 6 Galvanic charging to various potential cutoffs for (a) P3HT–TEMPO-25, and (b) P3HT–TEMPO-100. Normalized specific capacities of (c) P3HT–TEMPO-25, and (d) P3HT–TEMPO-100. | ||
The charging profile of P3HT increases with potential and the highest charging capacity is obtained at 4.2 V (Fig. 6a). The capacity at 3.8 V is 12% of the total capacity. Further charging to 4 V leads to 68% of the “total” capacity. The voltage plateau from 4.1 to 4.2 V is again a result of long charging time from a small current density (5 μA cm−2), and it contributes to 32% of the “total” capacity. The charging profile in P3HT is consistent with the charge discharge profile presented in Fig. 5a. Thus, at a higher discharge current density, the capacity contribution above 4 V is reduced.
In P3HT–TEMPO-100, the highest capacity is obtained at 3.8 V, and this value is used as “total” capacity (Fig. 6c). The plateau from 3.6–3.7 V dominates the charge transfer process and contributes to 84% of the total capacity. Similar capacity contributions are also reported in our previous study for non-regioregular TEMPO-bearing polythiophene.11 Further charging from 3.7 to 4.2 V only accounts for 16% of the overall capacity. Note that the capacity at 4.2 V is less than that at 3.8 V. This is a result from internal charge transfer, which appears to be more severe in the P3HT–TEMPO-100 case. In P3HT–TEMPO-25, the charging curve shows a short plateau from 3.7 to 3.8 V, and above that the capacity increases with voltage (Fig. 6b). This is consistent with the charging profile shown earlier in Fig. 5b. Charging to 3.8 V and 4.0 V accounts for 14% and 54% of the total capacity, respectively.
At last, the open circuit potential (OCP) was monitored for 6 hours to investigate internal charge transfer (Fig. 7). The three polymers were charged to 4.2 V, and the potential was held constant for 30 min. After removing the bias, the OCP for P3HT gradually decayed to 3.93 V. On the contrary, the OCP for P3HT-TEMPO-100 sharply decreased to 3.8 V in the first 130 s and then the OCP stabilized to 3.67 V after 6 hours, consistent with the redox potential of TEMPO radicals. P3HT–TEMPO-25 exhibited a gradual OCP decay similar to P3HT, but the OCP eventually stabilized to 3.8 V, which is the redox potential of polythiophene.
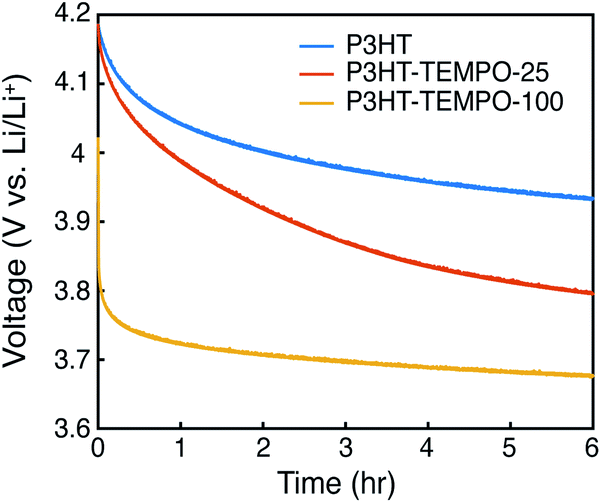 | ||
| Fig. 7 Open circuit potential monitoring for 6 hours. | ||
The rapid OCP drop in P3HT–TEMPO-100 reflects internal charge transfer from the nitroxide radical to the conjugated polythiophene backbone. At 4.2 V, the majority of nitroxide radicals and polythiophene backbone are oxidized and doped. Since the oxidation reaction does not proceed to full completion, there remains a small amount of unoxidized TEMPO that is capable of transferring one electron to reduce the doped polythiophene backbone. Thus, following 6 h of relaxation, the OCP reflects the redox potential of TEMPO. A similar OCP response was also reported in our earlier studies.11
These results clearly show that regioregularity has a minor (if any) effect on electron transfer and internal charge transfer. The electrochemical responses of the regioregular P3HT–TEMPO-100 (93% regioregularity) were very similar to the non-regioregular TEMPO bearing P3HT11 synthesized using electropolymerization, indicating that electron transfer in P3HT–TEMPO-100 still occurs via a hopping mechanism. Under a high radical loading, the regioregular backbone was twisted and distorted by the bulky TEMPO side chains, which impedes the formation of crystalline regions.30 As a result, the polymers with high TEMPO loading remain amorphous regardless of the backbone regioregularity, and delocalized electron transfer through the conjugated backbone is prohibited. Both regioregular and non-regioregular P3HT–TEMPOs exhibited internal charge transfer, which will occur as long as the two redox-active moieties have different redox potentials because electron transfer from a species with lower redox potential to another species with higher redox potential is thermodynamically favorable. Thus, electrons transfer from TEMPO (E1/2 = 3.6 V vs. Li/Li+), having a lower redox potential, to the polythiophene backbone (E1/2 = 3.8 V vs. Li/Li+) after charging the CRP.
We conclude that the extent of internal charge transfer is dominated by the TEMPO loading, rather than the regioregularity. In support of this, we discuss radical coupling with respect to TEMPO loading and the OCP decay results displayed in Fig. 7. Zhang et al. measured the radical concentration in the P3HT–TEMPO-25 and P3HT–TEMPO-100 using electron paramagnetic resonance (EPR).30 P3HT–TEMPO-25 showed a hyperfine-induced triplet similar to TEMPO small molecules, indicating that the loaded TEMPO radicals are scattered through the polymer chain and local radical concentration is rather low.30 On the contrary, P3HT–TEMPO-100 showed a broad EPR peak resulting from spin–spin interactions between closely installed TEMPO radicals.30 The scattered vs. dense loading for P3HT–TEMPO-25 vs. P3HT–TEMPO-100 manifests as differences in the decay of the OCP after charging and equilibration to 4.2 V. The sharp potential decay is not observed for P3HT–TEMPO-25, because only a small amount of the polythiophene is reduced by unoxidized TEMPO radicals in P3HT–TEMPO-25, and the majority of the polythiophene remains in the doped state. As a result, the OCP decayed faster than that for P3HT, but the OCP still stabilized near the redox potential of polythiophene (3.8 V), indicating that there was less internal charge transfer. Thus, internal charge transfer in P3HT–TEMPO-25 was limited by the low concentration of un-oxidized TEMPO units. In contrast, P3HT–TEMPO-100 exhibited sharp OCP decay and stabilization to the redox potential of the TEMPO units. Altogether, the extent of internal charge transfer is balanced by which redox units are majority-minority components.
At last, we conducted EQCM-D to examine the doping mechanism in the CRP to compare with prior findings for homopolymer PTMA.31Fig. 8 shows the raw EQCM-D data collected during cyclic voltammetry for P3HT-TEMPO-100. During oxidation, the TEMPO radical was oxidized to an oxoammonium cation. To maintain charge neutrality, anions in the electrolyte (in this case, CF3SO3−) dope the created oxoammonium cation. In consideration of the polythiophene backbone, doping may also occur, also accompanying the uptake of a CF3SO3− anion. The doping process is reflected by a decrease in frequency and an increase in dissipation, which corresponds to an increase in mass increase and electrode softening, respectively. During reduction, the polymer electrode was reduced and de-doped, accordingly, the mass decrease and electrode stiffening were reflected by the reverse trend in frequency and dissipation. The step change observed for PTMA–TEMPO-100 is consistent with the EQCM-D response of homopolymer PTMA.31 This result further confirms that charge transfer in P3HT–TEMPO-100 is dominated by the hopping mechanism associated with the TEMPO unit.
 | ||
| Fig. 8 Frequency (f3) and dissipation (D3) responses of the third overtone from EQCM-D during cyclic voltammetry for P3HT–TEMPO-100. Two cycles were performed at each indicated scan rate, followed by equilibration in the liquid electrolyte for 100 s. The working electrode was P3HT–TEMPO-100-coated quartz crystal. The counter and reference electrode were Pt plate and silver wire, respectively. The electrolyte was 0.5 M LiCF3SO3 in propylene carbonate. | ||
To gain more insight into the doping mechanism, we examined the shape of the cyclic voltammogram and calculated the mass change with the cumulative charge transfer (Fig. 9 and S6†). In our previous report on homopolymer PTMA, we showed that two doping processes exist – doping by Li-ion expulsion and anion uptake.31 The two processes corresponded to two cyclic voltammetry peaks, in which doping by Li-ion expulsion occurred at a lower potential. However, Fig. 9a only shows a single peak, indicating that one doping mechanism is dominating over the other, namely anion uptake. Using viscoelastic modeling of the raw data, the change in electrode mass was overlaid with the corresponding cyclic voltammogram at 25 mV s−1. The mass change coincided well with the redox peak, confirming significant mass changes affiliated with the TEMPO redox process.
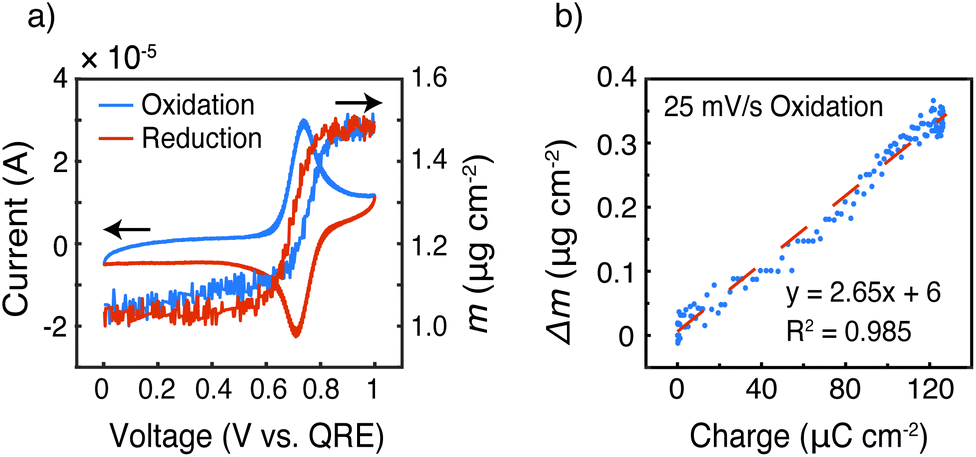 | ||
| Fig. 9 (a) Cyclic voltammetry of P3HT–TEMPO-100 at 25 mV s−1 overlapped with electrode mass, and (b) change in electrode mass versus charge passed during oxidation at 25 mV s−1via EQCM-D from 0.595 to 1.0 V vs. QRE. | ||
Finally, the change in mass was plotted against the integrated current, taken from the oxidation scan in the voltage range of 0.595 to 1.0 V vs. QRE, to examine ion transport coinciding with those regions in which internal charge transfer had been observed, Fig. 9b. The mass increased linearly with the charge transferred, and the slope of this linear relationship (Δm/Q) gave a measure of the doping process. Assuming that each TEMPO radical is doped by an anion from the bulk electrolyte (because it appears to be the dominating species), the theoretical Δm/Q value is simply the ratio of the molecular weight of the balancing anion (in this case, CF3SO3−) over Faraday's constant, yielding 1.55 mg C−1. In Fig. 9b, the slope of Δm vs. Q was 2.65 mg C−1, which means that either (1) doping occurs mainly via anion uptake with solvent participation or (2) the electrolyte may transfer in ion pairs with or without solvent. We speculate the former process is dominating because individual anions are likely easier to transport than ion pairs or clusters. In that case, we calculate by a mass balance that 1 propylene carbonate molecule accompanies each triflate anion during the oxidation process at this specific scan rate.
Conclusions
This work quantified internal charge transfer in CRPs consisting of polythiophene backbones and TEMPO side groups. This was accomplished by using CRPs of varying TEMPO loadings (0, 25, 100%). P3HT–TEMPO-100's behaviour was dominated by the TEMPO radical due to internal transfer. Specifically, this was distinguished by the mixed faradaic and non-faradaic charge contributions from the conjugated backbone and radical sidegroups. P3HT–TEMPO-25 exhibited features of both the conjugated backbone and the TEMPO radical, suggesting that radical loading strongly influences the electron transfer behaviour. From comparison, we conclude that the extent of internal charge transfer depends on the radical loading, similar to the consideration of limiting and excess “reactants”, as exhibited by the final OCPs during relaxation monitoring. Finally, coupled electron-ion transport was observed in situ for the first time for P3HT–TEMPO-100; it was suggested that ∼1 propylene carbonate molecule accompanies a triflate ion upon each doping event of the CRP during cyclic voltammetry at 25 mV s−1.All together, these findings confirm the underlying reasons for these CRP's generally low conductivity and capacity. On the other hand, these findings suggest that CRPs such as these might be useful as voltage regulating materials, in which the CRP might mitigate voltage excursions through internal charge transfer. For future design, the trade-off between radical loading and electronic conductivity need to be balanced. Novel molecular design that decouples the radical unit and the charge conducting pathway might be desirable to reach a high specific capacity. In addition, the direction of internal charge transfer between the two redox moieties need to be carefully considered.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grant DE-SC0014006 funded by the US Department of Energy, Office of Science (Lutkenhaus). Synthesis work (Ober) was supported by grant DE-SC0014336 funded by the US Department of Energy, Office of Science.References
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Rechargeable batteries with organic radical cathodes, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- P. T. Anastas and J. B. Zimmerman, Innovations in Green Chemistry and Green Engineering Selected Entries from the Encyclopedia of Sustainability Science and Technology, Springer, New York, NY, 2013, ISBN 978-1-4614-5817-3 Search PubMed.
- K. Nakahara, K. Oyaizu and H. Nishide, Organic Radical Battery Approaching Practical Use, Chem. Lett., 2011, 40, 222–227 CrossRef CAS.
- Y. Zhang, A. Park, A. Cintora, S. R. McMillan, N. J. Harmon, A. Moehle, M. E. Flatté, G. D. Fuchs and C. K. Ober, Impact of the synthesis method on the solid-state charge transport of radical polymers, J. Mater. Chem. C, 2018, 6, 111–118 RSC.
- L. Rostro, A. G. Baradwaj and B. W. Boudouris, Controlled radical polymerization and quantification of solid state electrical conductivities of macromolecules bearing pendant stable radical groups, ACS Appl. Mater. Interfaces, 2013, 5, 9896–9901 CrossRef CAS.
- A. G. Baradwaj, S. Wong, J. S. Laster, A. J. Wingate, M. E. Hay and B. W. Boudouris, Impact of the addition of redox-active salts on the charge transport ability of radical polymer thin films, Macromolecules, 2016, 49, 4784–4791 CrossRef CAS.
- L. Bugnon, C. J. Morton, P. Novak, J. Vetter and P. Nesvadba, Synthesis of poly(4-methacryloyloxy-TEMPO) via group-transfer polymerization and its evaluation in organic radical battery, Chem. Mater., 2007, 19, 2910–2914 CrossRef CAS.
- J.-K. Kim, Y. Kim, S. Park, H. Ko and Y. Kim, Encapsulation of organic active materials in carbon nanotubes for application to high-electrochemical-performance sodium batteries, Energy Environ. Sci., 2016, 9, 1264–1269 RSC.
- S. Iwasa, T. Nishi, H. Sato and S. Nakamura, Flexibility and high-rate discharge properties of organic radical batteries with gel-state electrodes, J. Electrochem. Soc., 2017, 164, A884–A888 CrossRef CAS.
- Y. Xie, K. Zhang, M. J. Monteiro and Z. Jia, Conjugated nitroxide radical polymers: synthesis and application in flexible energy storage devices, ACS Appl. Mater. Interfaces, 2019, 11, 7096–7103 CrossRef CAS.
- F. Li, D. N. Gore, S. Wang and J. L. Lutkenhaus, Unusual internal electron transfer in conjugated radical polymers, Angew. Chem., Int. Ed., 2017, 56, 9856–9859 CrossRef CAS.
- F. Li, S. Wang, Y. Zhang and J. L. Lutkenhaus, Electrochemical Energy Storage in Poly(dithieno[3,2-b:2′,3′-d]pyrrole) Bearing Pendant Nitroxide Radicals, Chem. Mater., 2018, 30, 5169–5174 CrossRef CAS.
- K. Sato, R. Ichinoi, R. Mizukami, T. Serikawa, Y. Sasaki, J. Lutkenhaus, H. Nishide and K. Oyaizu, Diffusion-Cooperative Model for Charge Transport by Redox-Active Nonconjugated Polymers, J. Am. Chem. Soc., 2018, 140, 1049–1056 CrossRef CAS.
- K. Oyaizu, Y. Ando, H. Konishi and H. Nishide, Nernstian Adsorbate-like Bulk Layer of Organic Radical Polymers for High-Density Charge Storage Purposes, J. Am. Chem. Soc., 2008, 130, 14459–14461 CrossRef CAS.
- K. Oyaizu and H. Nishide, Radical polymers for organic electronic devices: a radical departure from conjugated polymers?, Adv. Mater., 2009, 21, 2339–2344 CrossRef CAS.
- K. Nakahara, K. Oyaizu and H. Nishide, Electrolyte anion-assisted charge transportation in poly(oxoammonium cation/nitroxyl radical) redox gels, J. Mater. Chem., 2012, 22, 13669–13673 RSC.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Charge transport in high-mobility conjugated polymers and molecular semiconductors, Nat. Mater., 2020, 19, 491–502 CrossRef CAS.
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Organic mixed ionic–electronic conductors, Nat. Mater., 2020, 19, 13–26 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Polymer-Based Organic Batteries, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS.
- T. K. Kunz and M. O. Wolf, Electrodeposition and properties of TEMPO functionalized polythiophene thin films, Polym. Chem., 2011, 2, 640–644 RSC.
- F. Li, Y. Zhang, S. R. Kwon and J. L. Lutkenhaus, Electropolymerized Polythiophenes Bearing Pendant Nitroxide Radicals, ACS Macro Lett., 2016, 5, 337–341 CrossRef CAS.
- S. Almubayedh and M. h. Chahma, Electrosynthesis and Characterization of Stable Radical-Functionalized Oligo/Polythiophenes, New J. Chem., 2015, 39, 738–7741 RSC.
- L. Xu, F. Yang, C. Su, L. Ji and C. Zhang, Synthesis and Properties of Novel TEMPO-Contained Polypyrrole Derivatives as the Cathode Material of Organic Radical Battery, Electrochim. Acta, 2014, 130, 148–155 CrossRef CAS.
- J. Lu, J. Ma, J. Yi, Z. Shen, Y. Zhong, C. Ma and M. Li, Electrochemical Polymerization of Pyrrole Containing TEMPO Side Chain on Pt Electrode and Its Electrochemical Activity, Electrochim. Acta, 2014, 130, 412–417 CrossRef CAS.
- N. Casado, G. Hernández, A. Veloso, S. Devaraj, D. Mecerreyes and M. Armand, PEDOT Radical Polymer with Synergetic Redox and Electrical Properties, ACS Macro Lett., 2016, 5, 59–64 CrossRef CAS.
- J. Qu, T. Katsumata, M. Satoh, J. Wada and T. Masuda, Synthesis and Properties of Polyacetylene and Polynorbornene Derivatives Carrying 2,2,5,5-Tetramethyl-1-pyrrolidinyloxy Moieties, Macromolecules, 2007, 40, 3136–3144 CrossRef CAS.
- T. Katsumata, M. Satoh, J. Wada, M. Shiotsuki, F. Sanda and T. Masuda, Polyacetylene and Polynorbornene Derivatives Carrying TEMPO. Synthesis and Properties as Organic Radical Battery Materials, Macromol. Rapid Commun., 2006, 27, 1206–1211 CrossRef CAS.
- L. Assumma, Y. Kervella, J.-M. Mouesca, M. Mendez, V. Maurel, L. Dubois, T. Gutel and S. Sadki, A New Conducting Copolymer Bearing Electro-Active Nitroxide Groups as Organic Electrode Materials for Batteries, ChemSusChem, 2020, 13, 2419–2427 CrossRef CAS.
- C. K. Chiang, E. A. Blubaugh and W. T. Yap, Electrochemical Studies on Doping of Polyacetylene, Polymer, 1984, 25, 1112–1116 CrossRef CAS.
- Y. Zhang, A. M. Park, S. R. McMillan, N. J. Harmon, M. E. Flatté, G. D. Fuchs and C. K. Ober, Charge Transport in Conjugated Polymers with Pendent Stable Radical Groups, Chem. Mater., 2018, 30, 4799–4807 CrossRef CAS.
- S. Wang, F. Li, A. D. Easley and J. L. Lutkenhaus, Real-time insight into the doping mechanism of redox-active organic radical polymers, Nat. Mater., 2019, 18, 69–75 CrossRef CAS.
- S. Wang, A. M. G. Park, P. Flouda, A. D. Easley, F. Li, T. Ma, G. D. Fuchs and J. L. Lutkenhaus, Solution-processable thermally crosslinked organic radical polymer battery cathodes, ChemSusChem, 2020, 13, 2371–2378 CrossRef CAS.
- R. J. Waltman, J. Bargon and A. Diaz, Electrochemical studies of some conducting polythiophene films, J. Phys. Chem., 1983, 87, 1459–1463 CrossRef CAS.
- X. Chen and O. Inganäs, Three-step redox in polythiophenes: evidence from electrochemistry at an ultramicroelectrode, J. Phys. Chem., 1996, 100, 15202–15206 CrossRef CAS.
- K. S. Ryu, Y. Lee, K.-S. Han and M. G. Kim, The electrochemical performance of polythiophene synthesized by chemical method as the polymer battery electrode, Mater. Chem. Phys., 2004, 84, 380–384 CrossRef CAS.
- J.-W. Jeon, Y. Ma, J. F. Mike, L. Shao, P. B. Balbuena and J. L. Lutkenhaus, Oxidatively stable polyaniline: polyacid electrodes for electrochemical energy storage, Phys. Chem. Chem. Phys., 2013, 15, 9654–9662 RSC.
- J. F. Mike, L. Shao, J.-W. Jeon and J. L. Lutkenhaus, Charge Storage in Decyl-and 3,6,9-Trioxadecyl-Substituted Poly(dithieno[3,2-b:2,3-d]pyrrole) Electrodes, Macromolecules, 2013, 47, 79–88 CrossRef.
- M. Sathiya, A. S. Prakash, K. Ramesha, J. M. Tarascon and A. K. Shukla, V2O5-Anchored Carbon Nanotubes for Enhanced Electrochemical Energy Storage, J. Am. Chem. Soc., 2011, 133, 16291–16299 CrossRef CAS.
- H. Nishide, S. Iwasa, Y.-J. Pua, T. Suga, K. Nakahara and M. Satoh, Organic radical battery: nitroxide polymers as a cathode-active material, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03567j |
| This journal is © The Royal Society of Chemistry 2020 |