
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriazole-based osmium(II) complexes displaying red/near-IR luminescence: antimicrobial activity and super-resolution imaging†
Kirsty L.
Smitten
 a,
Paul A.
Scattergood
*b,
Charlotte
Kiker
a,
Jim A.
Thomas
*a and
Paul I. P.
Elliott
*b
a,
Paul A.
Scattergood
*b,
Charlotte
Kiker
a,
Jim A.
Thomas
*a and
Paul I. P.
Elliott
*b
aDepartment of Chemistry, University of Sheffield, Brook Hill, Sheffield, S3 7HF, UK. E-mail: james.thomas@sheffield.ac.uk
bDepartment of Chemistry & Centre for Functional Materials, University of Huddersfield, Queensgate, Huddersfield, HD1 3DH, UK. E-mail: p.i.elliott@hud.ac.uk; p.scattergood@hud.ac.uk
First published on 7th August 2020
Abstract
Cellular uptake, luminescence imaging and antimicrobial activity against clinically relevant methicillin-resistant S. aureus (MRSA) bacteria are reported. The osmium(II) complexes [Os(N^N)3]2+ (N^N = 1-benzyl-4-(pyrid-2-yl)-1,2,3-triazole (12+); 1-benzyl-4-(pyrimidin-2-yl)-1,2,3-triazole (22+); 1-benzyl-4-(pyrazin-2-yl)-1,2,3-triazole (32+)) were prepared and isolated as the chloride salts of their meridional and facial isomers. The complexes display prominent spin-forbidden ground state to triplet metal-to-ligand charge transfer (3MLCT) state absorption bands enabling excitation as low as 600 nm for fac/mer-32+ and observation of emission in aqueous solution in the deep-red/near-IR regions of the spectrum. Cellular uptake studies within MRSA cells show antimicrobial activity for 12+ and 22+ with greater toxicity for the meridional isomers in each case and mer-12+ showing the greatest potency (32 μg mL−1 in defined minimal media). Super-resolution imaging experiments demonstrate binding of mer- and fac-12+ to bacterial DNA with high Pearson's colocalisation coefficients (up to 0.95 using DAPI). Phototoxicity studies showed the complexes exhibited a higher antimicrobial activity upon irradiation with light.
Introduction
Over recent decades considerable attention has been paid to the use of complexes of RuII, IrIII and ReI d6 transition metal ions as luminescent probes for biological imaging.1–7 The long-lived triplet metal-to-ligand charge transfer (3MLCT) excited states responsible for the emission from these complexes can also sensitise singlet oxygen production which provides a basis for applications in photodynamic therapy (PDT) and the development of theranostic probes.8–11 A noticeable drawback of these systems is their relatively high photoexcitation energy, which can result in unintended cellular damage during imaging and also limits the depth of tissue penetration. Consequently, cell-permeant luminescent imaging agents which display both low-energy electronic absorption features and deep-red phosphorescence, thus permitting excitation and luminescence imaging within the biologically-transparent region of the visible spectrum, are attractive synthetic targets.12–14Osmium(II) complexes offer several advantages over their ruthenium(II) analogues. The high spin–orbit coupling constant associated with OsII facilitates formally forbidden direct transitions from the ground state into 3MLCT excited states, resulting in associated absorbance bands of appreciable intensity in their optical spectra, often extending out to 700 nm.15 Excitation into these low energy absorptions results in deep-red to near-infrared (NIR) phosphorescence (λem ≈ 650–900 nm) and given that they are also highly kinetically inert, OsII complexes offer significant potential as cellular imaging agents.16 However, despite these favourable attributes, the use of Os(II) complexes as phosphorescent biological probes is still relatively scarce.
The Keyes group has reported the cellular uptake and imaging of a polyarginine-conjugated osmium(II) imidazophenanthroline complex17 as well as live-cell imaging of a mitochondria-targeting OsII bis-(4-carboxyphenyl-terpyridine)-derived complex.18 The Zhang group has investigated a benzimidazole-containing OsII complex as a NIR emissive lysosomal tracker,19 as well as a related complex that enables NIR luminescence imaging of the RNA within the nucleus of live cells.20 Very recently Zhu et al. have also exploited the deep-red luminescence of [Os(phen)2(dppz)]2+ (phen = 1,10-phenanthroline, dppz = dipyridophenazine) for correlative luminescence and transmission electron microscopy (TEM) imaging of nuclear DNA.21 In this context, the Scattergood and Elliott groups have previously investigated triazole-containing OsII complexes that show lysosomal/endosomal and mitochondrial localisation as optical probes,22,23 whilst the Thomas group has looked at DNA targeting polypyridyl OsII complexes as high-resolution contrast probes for TEM.24
Yet, whilst there has been interest in OsII complexes as potential anti-cancer therapeutics,19,25–32 examples of their use in bacteria are far rarer. Despite Dwyer and co-workers reporting the antimicrobial activity of RuII polypyridyl complexes almost 70 years ago,33,34 interest in the antibiotic activity of d6 metal-ion complexes has only returned to some prominence over the last decade.35–41 However, the use of luminescent complexes for bacterial cell imaging remains a rarity. Following on from its earlier report that a luminescent di-nuclear RuII complex was taken up by bacteria,42 the Thomas group recently reported on the antimicrobial activity of di-nuclear analogues against a range of Gram positive and Gram negative bacteria. In these reports, several microscopy techniques were used to investigate cellular internalisation and explore the nature of the interaction of the metal complexes with the bacteria.43,44 Massi and co-workers have reported cationic IrIII-tetrazolato complexes which show good antimicrobial activity against Gram-positive D. radiodurans,45 whilst charge-neutral members of the same family have also been utilised as molecular probes for the live imaging of B. cereus.46 Whilst there have been some scarce reports of OsII complexes showing inhibition of both Gram-positive and Gram-negative bacteria,47 to the best of our knowledge, there has yet to be any report that combines luminescence imaging with antimicrobial studies on OsII systems despite their favourable photophysical properties.
Herein we report on the anti-microbial activity of the individual isomers of a family of water-soluble homoleptic OsII complexes featuring 1,2,3-triazole-containing ligands – Scheme 1. Strikingly, it was found that the meridional isomers of these architectures display much greater growth inhibition of Gram-positive bacteria over their facial geometrical isomers. Using the deep-red phosphorescence of these complexes we explore their cellular internalisation and identify their intracellular interaction with DNA.
 | ||
| Scheme 1 Synthesis of 12+–32+. | ||
Results and discussion
Synthesis and characterisation
The previously reported complex [Os(pytz)3](PF6)2 [12+](PF6)2 was prepared through an established procedure,23 which was also used to prepare the related homoleptic complexes [Os(pymtz)3](PF6)2 [22+](PF6)2 and [Os(pyztz)3][PF6]2 [32+](PF6)2. Briefly, [OsCl6][NH4]2 was heated under reflux in ethylene glycol with 3 equivalents of the appropriate ligand, which after counterion metathesis with NH4PF6 yielded the hexafluorophosphate salts of the complexes as deep-red coloured solids (Scheme 1).Due to the asymmetry of the triazole-containing ligands, analysis by 1H NMR spectroscopy (Fig. S1–S8†) reveals the isolated products to be composed of a mixture of meridional and facial isomers, with formation of the former being favoured in all cases with mer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fac ratios of 1.4:1 (12+), 2:1 (22+) and 3:1 (32+). Whilst separation of these isomers by column chromatography is challenging, we found that efficient separation is possible on a small scale by employing preparative thin layer chromatography. Metathesis of counterions yielded chloride salts of the separated fac- and mer-isomers of 12+–32+ which display excellent aqueous solubility.
:fac ratios of 1.4:1 (12+), 2:1 (22+) and 3:1 (32+). Whilst separation of these isomers by column chromatography is challenging, we found that efficient separation is possible on a small scale by employing preparative thin layer chromatography. Metathesis of counterions yielded chloride salts of the separated fac- and mer-isomers of 12+–32+ which display excellent aqueous solubility.
The hexafluorophosphate salts of the complexes were analysed by cyclic voltammetry (Fig. 1 and Table 1). The cyclic voltammogram traces for each pair of mer- and fac-isomers are similar, showing that geometric isomerism has little impact on the electronic properties of the complexes. All complexes exhibit one electrochemically reversible oxidation process attributed to the OsII/III redox couple. Consistent with the increased electron-withdrawing character of each heterocyclic ligand, the potential of this process anodically shifts upon exchange of the pyridyl moiety for pyrimidine and pyrazine. A similar trend was reported recently for a series of bis-terdentate 1,2,3-triazole-based OsII complexes featuring pyridyl and pyrazinyl donors.48
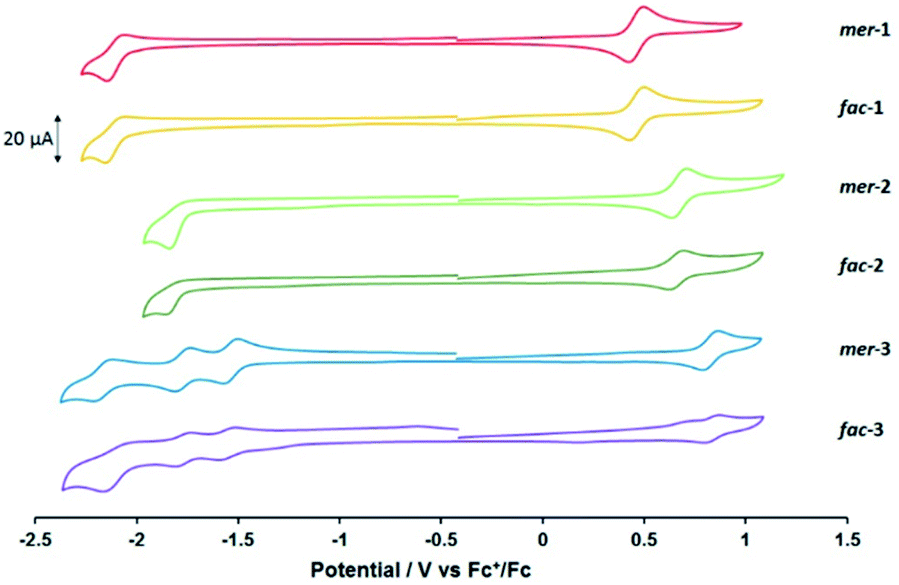 | ||
| Fig. 1 Cyclic voltammograms recorded at room temperature at 100 mV s−1 for 1.0 mmol dm−3 MeCN solutions of mer- and fac-12+–32+ as their hexafluorophosphate salts. Solutions contained 0.2 mol dm−3 NBu4PF6 as supporting electrolyte. All potentials are shown against the Fc+/Fc couple (E1/2 = 0 V). | ||
| Complex | E ox/V | E red/V |
|---|---|---|
| a Measured at room temperature at a scan rate of 100 mV s−1. Potentials are shown versus the Fc+/Fc couple. Anodic–cathodic peak separations (ΔEa,c) for reversible processes are shown in millivolts within brackets. ΔEa,c for Fc+/Fc was typically 70 mV. b Incompletely-reversible. c Irreversible process, cathodic peak potential is quoted. | ||
| mer-12+ | +0.46 (73) | −2.10 (87)b |
| fac-12+ | +0.46 (76) | −2.10 (100)b |
| mer-22+ | +0.67 (80) | −1.84c |
| fac-22+ | +0.66 (70) | −1.85c |
| mer-32+ | +0.82 (78) | −1.54 (69), −1.77 (73), −2.17 (98)b |
| fac-32+ | +0.83 (68) | −1.54 (63), −1.77 (73), −2.10 (118)b |
All the complexes display at least one reduction process within the available electrochemical solvent window. For mer- and fac-12+, a reduction which is not completely chemically reversible is observed at −2.10 V vs. Fc+/Fc. By comparison with previous studies on complexes of OsII and RuII containing this ligand this process is attributed to a ligand-based reduction.23 Substitution of the pytz ligand with pymtz for both mer- and fac-22+ results in the appearance of an irreversible reduction process at more positive potential. This shift is of similar magnitude to the anodic shift in the OsII/III couple relative to 12+, resulting in minimal change in the HOMO–LUMO energy gap. Introduction of a pyrazinyl-donor into the ligand set leads to more complex reductive electrochemistry, with the appearance of two distinct reversible processes at −1.54 and −1.77 V followed by an incompletely chemically reversible couple in the region of −2.1 V vs. Fc+/Fc. These processes are assigned to the successive one-electron reductions of the three coordinated pyztz ligands. Here the anodic shift for the ligand-based reduction is larger than for the Os(II/III) oxidation resulting in a narrowing of the HOMO–LUMO gap.
Density functional theory (DFT) calculations on the fac- and mer-isomers of 12+ to 32+, carried out using the Orca 4.2 software package,49,50 confirm the electrochemical data (see Fig. S9, S10 and ESI for details†); in all cases the HOMO has predominantly Os d-orbital character whilst the LUMO is spread over the three ligands (mostly localised over the 6-membered heterocycles) and is of π* character. In agreement with the anodic shift in the oxidation and reduction potentials, the HOMO and LUMO of fac/mer-22+ are both stabilised compared to their pyridine-containing analogues resulting in near-identical HOMO–LUMO gaps (fac-12+/22+ 3.51 eV; mer-12+/22+ 3.48 eV). For fac/mer-32+ the HOMO and LUMO are again stabilised with respect to the pyrimidine-containing complexes but with the latter stabilised to a greater extent, such that the HOMO–LUMO gap is reduced (fac-32+ 3.36 eV; mer-32+ 3.28 eV).
UV-Visible absorption spectra were recorded for the mer and fac chloride salts of 12+–32+ in aqueous solution (Fig. 2, Table S1†). Sharp and intense bands observed for all the complexes between 220–300 nm are assigned to singlet ligand-centred (1LC) transitions, whilst bands beyond 350 nm are of MLCT character.
 | ||
| Fig. 2 (A) UV-Visible electronic absorption spectra recorded for aqueous solutions of chloride salts mer- and fac-12+–32+. (B) Normalised photoluminescence spectra recorded for chloride salts mer- and fac-12+–32+ in aerated aqueous solution (λex = 500 nm). | ||
Complexes mer/fac-12+ and mer/fac-2+ display a series of bands between 350–420 nm which are attributed to spin-allowed 1MLCT excitations. Weaker bands, still of appreciable intensity, are observed between 430 and 580 nm and are assigned to the expected, formally spin-forbidden, direct excitations to the 3MLCT state.15,51 In agreement with the electrochemical data, indicating a similar HOMO–LUMO gap for mer/fac-12+ and mer/fac-22+ (vide supra), these bands are near coincident with each other. The analogous transitions for mer- and fac-32+ are, however, red-shifted in line with the electrochemical and DFT data.
Time-dependent DFT calculations were carried out for the ground states of all complexes to determine vertical excitation energies and simulate the optical absorption spectra (Fig. S11 and Table S2†). The calculated data are in good agreement with the experimentally measured spectra. Excitations to the S1 states for 12+ and 22+ are near coincident appearing between 468 (fac) and 474 (mer) nm whilst those for the isomers of 32+ are red-shifted to 494 (fac) and 511 nm (mer). In all cases these correspond to transitions predominantly consisting of HOMO → LUMO excitations but have relatively small oscillator strengths and will therefore have little physical significance. The calculated data show that the more intense transitions between 350 and 450 nm correspond to excitations from orbitals of primarily metal d-orbital character (HOMO-2 to HOMO) with population of ligand-localised orbitals (LUMO up to LUMO+5) confirming the 1MLCT character of the absorption bands in this region. The lowest energy spin-forbidden singlet to triplet transitions appear below the lowest singlet states with the T1 excitations for 32+ appearing at 560 (fac) and 590 (mer) nm. In line with expectation, analysis of the compositions of these transitions confirms 3MLCT character.
The chloride salts of all three complexes are luminescent in aerated aqueous solution, exhibiting broad unstructured bands in the red/near-infrared regions of the spectrum attributed to 3MLCT-based emission (Fig. 2B). Again, mer- and fac-isomerism has almost no effect on the band position within each pair of complexes. In agreement with both the electrochemical and electronic absorbance data (vide supra), phosphorescence observed for mer/fac-32+ is the most red-shifted in the series. Moderate luminescence lifetimes are observed in aerated aqueous solutions ranging from 83 ns for mer-32+ to 365 ns for mer-12+ (Table 2) with relatively low emission quantum yields of between 0.6 to 3.9%. Trends in both lifetime and quantum yields are in excellent agreement with the energy gap law52,53 and typical for similar triazole-containing complexes of OsII.23,48 Quantum yields increase upon deoxygenating the aqueous solutions, supporting assignment to an emissive MLCT state of triplet character. Significantly, the low-energy spin-forbidden absorption envelope of the complexes enables direct population of their 3MLCT states, with emission readily observed using excitation wavelengths as low as 560 (mer/fac-12+ and 22+) and 600 nm (mer/fac-32+).
| λ em /nm | Φ em , /% (τ/ns) | Φ em , /% (τ/ns) | |
|---|---|---|---|
| a λ ex = 500 nm. b Aerated aqueous solution. c N2-equilibrated aqueous solution. d Relative to [Ru(bpy)3][PF6]2, Φ = 1.8% in aerated MeCN. | |||
| mer-12+ | 621 | 3.9 (365) | 7.9 (748) |
| fac-12+ | 623 | 3.8 (350) | 8.4 (715) |
| mer-22+ | 637 | 2.3 (191) | 2.9 (242) |
| fac-22+ | 640 | 2.5 (171) | 3.2 (217) |
| mer-32+ | 696 | 0.6 (83) | 0.6 (92) |
| fac-32+ | 698 | 0.7 (98) | 0.8 (110) |
The geometries of the lowest-lying triplet states for the complexes were optimised in U-DFT calculations starting from the optimised ground state geometries. In each case the lower energy singly-occupied molecular orbital (SOMO, Fig. S12†) has predominantly Os d-orbital character whilst the excited electron in SOMO+1 is localised primarily on the 6-membered ring of one of the three ligands. This distribution of the SOMO and SOMO+1 confirms the 3MLCT character of these T1 states further affirmed by Mulliken spin densities of 0.88 to 0.96 for the Os atom.
Cell studies
The cellular uptake properties of the compounds by a range of multi-drug and pan-drug resistant Gram-negative and Gram-positive bacteria were then investigated. In these studies, we chose to investigate a methicillin resistant, clinical isolate strain of the Gram-positive bacterium Staphylococcus aureus, MRSA. We compared this to three Gram-negative bacteria strains: a uropathogenic multidrug resistant EC958 ST131 strain of Escherichia coli, a multidrug resistant clinical isolate strain of Pseudomonas aeruginosa – PA2017 and a multidrug resistant clinical isolate strain of Acinetobacter baumannii – AB184. AB184 was selected as it is the most prevalent clonal group of A. baumannii in the United Kingdom. In addition, AB184 and PA2017 belong to bacteria groups identified by the World Health Organisation as PRIORITY 1: CRITICAL for developing new antibiotics.54The minimum inhibitory concentration, MIC, of the complexes was obtained in both nutrient rich media Mueller–Hinton-II (MH-II) and defined media – Glucose Defined Minimal Media (GDMM) – in the case of Gram-negative strains and Chemical Defined Media (CDM) for Gram-positive strains. This is the minimum concentration of compound required to halt bacterial cell proliferation. While GDMM has been used in previous studies on metal complexes55,56 MH-II is closer to relevant biological conditions and is the bacterial growth medium recommended by the European Committee on Antimicrobial Susceptibility Testing. Data is summarised in Table 3.
| mer-12+ | fac-12+ | mer-22+ | fac-22+ | mer-32+ | fac-32+ | Controla | |
|---|---|---|---|---|---|---|---|
| a Ampicillin used as control. | |||||||
| Defined minimal media | |||||||
| EC958 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| PA2017 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| MRSA | 32 | 256 | 64 | 128 | >512 | >512 | >512 |
| AB184 | >512 | >512 | 256 | >512 | 256 | >512 | >512 |
|
|||||||
| Mueller–Hinton-II | |||||||
| EC958 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| PA2017 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| MRSA | 48 | 256 | 128 | 256 | >512 | >512 | >512 |
| AB184 | >512 | >512 | 256 | >512 | >512 | >512 | >512 |
As with the majority of metal-based antimicrobial compounds39,45,57 a higher activity against Gram-positive bacteria strains was observed, with all compounds showing no detectable activity on the Gram-negative strains. Strikingly, although complexes 12+ and 22+ exhibit activity against the methicillin resistant strain of S. aureus, in both cases their mer isomer is most active, with mer-12+ being comparable to standard antibiotics. More significantly still, this activity is not reduced in a pathogenic strain resistant to methicillin and ampicillin.
In recent studies, the antimicrobial activity of the direct RuII analogue of 12+ has been investigated. The Crowley group reported that both the mer and fac isomers of its hexafluorophosphate salt showed very low activity against wild-type S. aureus.39 This observation is probably due to the low water solubility of such salts as a more recent study on the unseparated isomers of this complex, as a chloride salt, showed good activity against the non-pathogenic Gram-positive bacteria Bacillus subtilis and Staphylococcus epidermidis.58 The Crowley group have also varied the identity of the triazole substituent within these RuII systems, reporting that those bearing n-hexyl and n-octyl groups are the most potent against wild-type S. aureus.39 The chloride salts of these two complexes were then investigated against a wider panel of bacteria, which revealed that this activity was retained in pathogenic MRSA strains. However, as both the hexafluorophosphate and chloride salts display similar MIC figures against wild-type S. aureus, their activity is attributed to an optimisation of lipophilicity, rather than solubility. Promising antimicrobial activity has also been observed in related RuII analogues incorporating isoquinoline appended triazole ligands,58 and in quinoline-based RuII systems featuring tetrazole moieties.59 Interestingly, in the latter case, the fac-isomers are more active than their mer-analogues. Although structurally similar to the OsII systems reported herein, the subtle increase in hydrophobicity achieved through inclusion of the extended aromatic units within the coordinated ligands is likely to be responsible for the improved potency of both of these architectures against both Gram-positive and Gram-negative bacteria, certainly a similar effect is observed in RuII complexes incorporating the alkylated ligands described above.39
To determine whether the complexes cause bacterial cell death or just halt bacterial proliferation, estimates of minimum bactericidal concentration, MBC, were obtained and these data are summarised in Table 4. As with the MIC experiments, the mer isomers are the most active with mer-12+ exhibiting the highest potency. The MBC values of all compounds lie within a 4-fold window of the MIC values, revealing that the compounds are classically bactericidal.
| mer-12+ | fac-12+ | mer-22+ | fac-22+ | mer-32+ | fac-32+ | Controla | |
|---|---|---|---|---|---|---|---|
| a Ampicillin used as control. | |||||||
| Defined minimal media | |||||||
| EC958 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| PA2017 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| MRSA | 48 | 256 | 96 | 256 | 512 | 512 | >512 |
| AB184 | >512 | >512 | 512 | 512 | 512 | 512 | >512 |
|
|||||||
| Mueller–Hinton-II | |||||||
| EC958 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| PA2017 | >512 | >512 | >512 | >512 | >512 | >512 | >512 |
| MRSA | 128 | 256 | 256 | 256 | 512 | 512 | >512 |
| AB184 | >512 | >512 | 256 | >512 | >512 | >512 | >512 |
Given that osmium complexes have been more widely targeted as photodynamic therapy agents,19,28,60 the phototoxic effects of the compounds towards the pathogenic, Gram-positive bacteria S. aureus, MRSA strain were also studied (see Fig. S13†). Bacterial cell survival following exposure to each compound was determined upon exposure to different light fluences (0, 8 and 48 J cm−2) and compared to a control in the dark. These data show that, for the majority of complexes, low light fluences do not result in increased antibacterial activity. However, at higher light fluences a detectable but small increase in activity (phototoxic index: ∼2) was observed. As with the previous studies mer-12+ exhibited the highest phototoxicity. As mer-12+ appeared to be the most promising therapeutic in all the assays, we further explored its activity and mechanism of action and compared to its corresponding fac-isomer.
By exploiting the emission properties of 12+ we investigated its cellular uptake and localisation using Structured Illumination Microscopy (SIM),61–63 a super-resolution technique that allows for improved resolutions up to 100 nm. SIM imaging with S. aureus (MRSA strain) indicated that mer-12+ is cell-permeant (Fig. 3). These images indicate that, 60 minutes after exposure to mer-12+, staining to cell membranes and bacterial DNA occurs (see Fig. S14† for intracellular luminescence intensity plots). However, after 120 minutes, accumulation within bacterial DNA predominates which is particularly apparent in 3D projections. The DNA appears to be condensed which may be an indication of DNA damage.
 | ||
| Fig. 3 Uptake of complex mer-12+ by S. aureus MRSA cells monitored through SIM microscopy. S. aureus (MRSA strain) cells were incubated with MIC concentrations of mer-12+ for 60 (A) and 120 (B) minutes. Left: cross-section through the centre of the cell where peak intensity was observed. Right: 3D surface plots of selected cells in the area of the white box shown in cross-section image, revealing membrane and nuclear staining at 60 min and solely nuclear staining at 120 min. Emission was collected in the A458 channel upon excitation with the 405 nm laser. Cells were fixed using paraformaldehyde (4%) and washed in PBS. Cells were mounted using Slowfade Gold Antifade. | ||
The uptake properties of fac-12+ in the same MRSA strain was also investigated (see Fig. S15†). As with mer-12+, fac-12+ initially stains both the membranes and intracellular DNA of the bacteria, although there is significantly less membrane localisation. Again, after two hours, it largely accumulates within the bacterial DNA. Intriguingly, measured distances between the membrane, DNA and cell septum for compounds stained with the fac-isomer were smaller on average than those for cells exposed to mer-12+, suggesting that uniquely mer-12+ causes cell swelling, perhaps as a consequence of the cell death mechanism it provokes.
To confirm DNA localisation, co-staining experiments were carried out with the DNA stain DAPI (4′,6-diamidino-2-phenylindole, Fig. 4).64 These images show a direct overlay between the emission of DAPI and both complexes within the bacterial DNA, which is clearly seen in the corresponding 3D surface projections and also reflected in their high Pearson's coefficients (fac-12+ = 0.95; mer-12+ = 0.92). As with the previous imaging experiments, it is clear that cells stained with the mer-isomer are noticeably swollen.
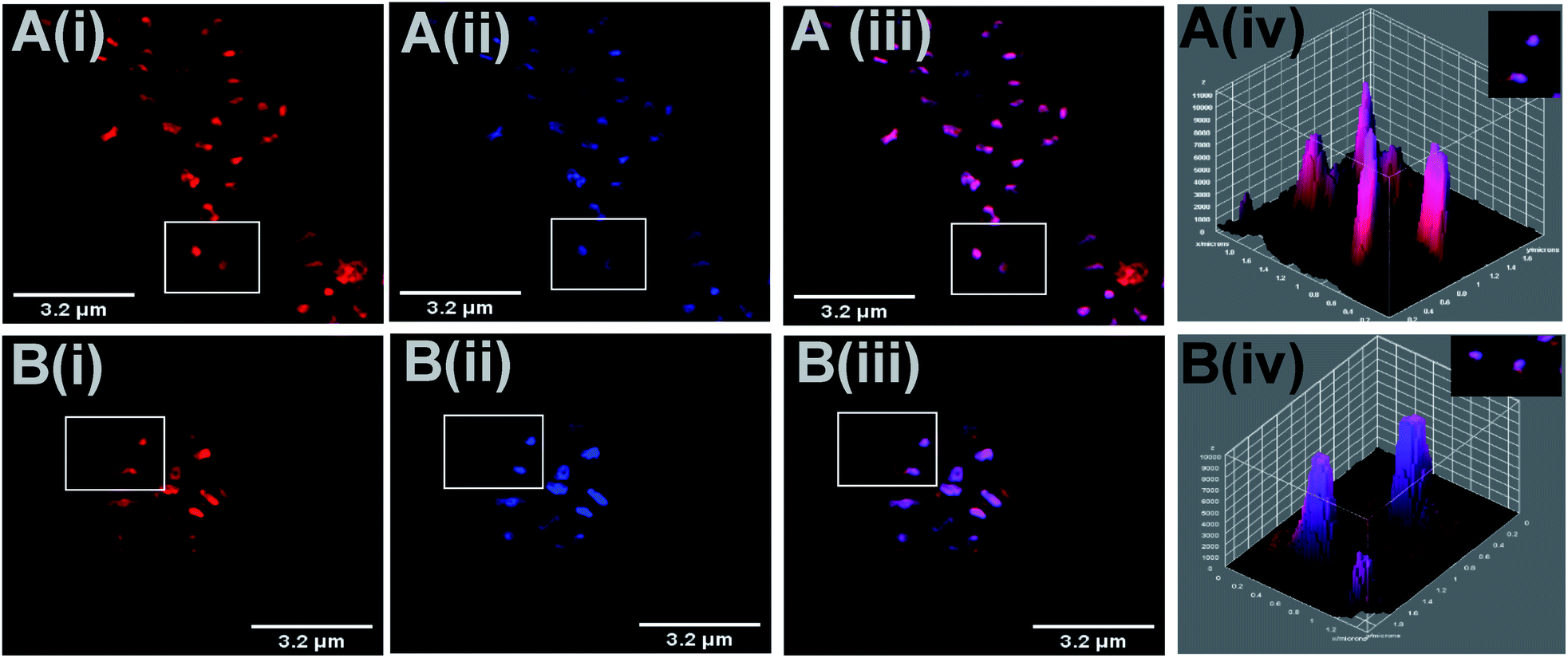 | ||
| Fig. 4 3D Z-stack projection, SIM colocalisation study using S. aureus MRSA after 1 hour incubation of cells with MIC concentrations of fac-12+ (A) and mer-12+ (B). (i) Image obtained on excitation of 12+ collected in the A568 channel. (ii) Images obtained by simultaneous staining with DAPI, collected in the DAPI channel. (iii) Overlay of both channels. (iv) 3D surface plots for the selected cells shown in the corresponding white boxes of the colocalisation images, showing emission intensity for 12+ and DAPI. | ||
Cell-free binding titrations on both isomers of 12+ using their DNA-induced increase in steady-state emission (see Fig. S16†), reveal they both bind to duplex DNA with relatively low affinities of Kb(fac-12+) = 4.1 × 105 M−1 and Kb(mer-12+) = 9.8 × 104 M−1. However, as the DNA binding affinity of fac-12+ is actually slightly higher than the more therapeutically active mer-12+ it seems that this is, at best, only one factor in the cell death mechanism. It has been suggested that the mechanism of action of the previously discussed RuII analogues of 12+, is due to membrane disruption and no DNA localisation was observed. However, these systems incorporate more lipophilic ligands and it is known that such physiochemical changes enhance binding to membranes over nucleic acids.65,66
To investigate any difference in internalisation, the uptake properties of both isomers were investigated using ICP-AES (inductively coupled plasma atomic emission spectroscopy, Fig. 5). Uptake experiments were conducted in the absence of glucose over the course of an hour, as the imaging studies showed both compounds accumulate within bacteria over this period.
 | ||
| Fig. 5 ICP-AES accumulation data for the uptake of osmium by S. aureus (MRSA) (left) and E. coli (EC958) (right), in the absence of glucose after exposure to mer-12+ (blue) and fac-12+ (green). [Fe] levels were calculated as a control (red). Os and Fe levels are expressed as metal (g) per cell. Compounds were added at MIC concentration in GDMM. Cells were washed with 0.5% (v/v) nitric acid to remove unbound complex. Error bars represent three independent biological repeats ± SD. | ||
The ICP-AES experiments reveal that overall the mer-isomer is preferentially taken up by S. aureus. In addition, the uptake profiles of the isomers are different; initially, fac-12+ concentrations increase quite rapidly but uptake plateaus between 10 and 20 minutes. In contrast, the uptake of mer-12+ is rapid for 5 minutes and although the uptake rate then decreases, the concentration of mer-12+ continues to rise for the entire 60 minutes of the experiment. The differential uptake of mer-12+ and fac-12+ is consistent with their observed activities and the brighter emission intensity of mer-12+ within cells.
The experiment was repeated with E. coli, EC958, to investigate whether any change in uptake between Gram-positive and Gram-negative bacteria affected the observed differences in activity. As illustrated in Fig. 5, both complexes show levels of uptake in E. coli which reflects their lowered activity in this Gram-negative species. Indeed, SIM experiments with EC958, showed little or no detectable uptake of either complex, as their emission was too weak to produce image reconstructions.
Conclusions
We have presented the synthesis and photophysical study of new osmium(II)-based complexes and their cellular uptake and antimicrobial activities against clinically relevant bacterial strains. The meridional isomer of 12+ exhibited the highest antimicrobial activity which was competitive with conventional antibiotics and was active against methicillin- and ampicillin-resistant pathogenic S. aureus. Further, these antimicrobial studies were combined with super-resolution imaging studies using the red phosphorescence for 12+ which demonstrated a high degree of bacterial DNA localisation. As 12+ undergoes quenching of emission in the presence of air through generation of 1O2 the light-induced toxicity was investigated, resulting in the determination of a modest phototoxic index of 2. Fascinatingly, the fac and mer isomers of 12+ display distinct differences in their biological properties, with mer-12+ exhibiting both higher antibacterial activity and brighter cell staining properties. These differences are due to the higher cell uptake of this isomer compared to fac-12+.Complexes of osmium(II) have attractive photophysical properties for biological and biomedical applications with prominent low energy absorption bands for direct ground state to 3MLCT state transitions and deep-red to near-IR emission. This enables increased depth of penetration in tissues for excitation with low energy light, imaging and 1O2 sensitisation. The results presented here therefore demonstrate the significant potential for Os(II) complexes in (photo)therapeutic antimicrobial applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. I. P. E. and P. A. S. acknowledge the University of Huddersfield for supporting this research as well as Dr Steve Andrews (3M Business and Innovation Centre, University of Huddersfield) for computational resources used in this work. KS is grateful to the BBSRC for a Ph.D. studentship through the White Rose Structural Biology DTP. Imaging work was performed at the Wolfson Light Microscopy Facility, using a DeltaVision/GE OMX optical microscope, funded by MRC grant MK/K0157531/1.Notes and references
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215–310 RSC.
- K. K.-W. Lo, W.-K. Hui, C.-K. Chung, K. H.-K. Tsang, D. C.-M. Ng, N. Zhu and K.-K. Cheung, Coord. Chem. Rev., 2005, 249, 1434–1450 CrossRef CAS.
- K. K.-W. Lo, M.-W. Louie and K. Y. Zhang, Coord. Chem. Rev., 2010, 254, 2603–2622 CrossRef CAS.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- M. P. Coogan and V. Fernández-Moreira, Chem. Commun., 2013, 50, 384 RSC.
- K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 36, 1–51 Search PubMed.
- J. D. Knoll, B. A. Albani and C. Turro, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS PubMed.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- J. Liu, C. Zhang, T. W. Rees, L. Ke, L. Ji and H. Chao, Coord. Chem. Rev., 2018, 363, 17–28 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2018, 119, 797–828 CrossRef PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- G. Lemercier, M. Four and S. Chevreux, Coord. Chem. Rev., 2018, 368, 1–12 CrossRef CAS.
- F. Felix, J. Ferguson, H. U. Güdel and A. Ludi, Chem. Phys. Lett., 1979, 62, 153–157 CrossRef CAS.
- P. Zhang and H. Huang, Dalton Trans., 2018, 47, 14841–14854 RSC.
- A. Byrne, C. N. Dolan, R. D. Moriarty, A. Martin, U. Neugebauer, R. J. Forster, A. Davies, Y. Volkov and T. E. Keyes, Dalton Trans., 2015, 44, 14323–14332 RSC.
- K. S. Gkika, A. Byrne and T. E. Keyes, Dalton Trans., 2019, 48, 17461–17471 RSC.
- P. Zhang, Y. Wang, K. Qiu, Z. Zhao, R. Hu, C. He, Q. Zhang and H. Chao, Chem. Commun., 2017, 53, 12341–12344 RSC.
- C. Ge, H. Huang, Y. Wang, H. Zhao, P. Zhang and Q. Zhang, ACS Appl. Bio Mater., 2018, 1, 1587–1593 CrossRef CAS.
- R. Huang, F.-P. Feng, C.-H. Huang, L. Mao, M. Tang, Z.-Y. Yan, B. Shao, L. Qin, T. Xu, Y.-H. Xue and B.-Z. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 3465–3473 CrossRef CAS PubMed.
- S. A. E. Omar, P. A. Scattergood, L. K. McKenzie, H. E. Bryant, J. A. Weinstein and P. I. P. Elliott, Molecules, 2016, 21, 1382–1412 CrossRef PubMed.
- S. A. E. Omar, P. A. Scattergood, L. K. McKenzie, C. Jones, N. J. Patmore, A. J. H. M. Meijer, J. A. Weinstein, C. R. Rice, H. E. Bryant and P. I. P. Elliott, Inorg. Chem., 2018, 57, 13201–13212 CrossRef CAS.
- A. Wragg, M. R. Gill, C. J. Hill, X. Su, A. J. H. M. Meijer, C. Smythe and J. A. Thomas, Chem. Commun., 2014, 50, 14494–14497 RSC.
- J. Maksimoska, D. S. Williams, G. E. Atilla-Gokcumen, K. S. M. Smalley, P. J. Carroll, R. D. Webster, P. Filippakopoulos, S. Knapp, M. Herlyn and E. Meggers, Chem. –Eur. J., 2008, 14, 4816–4822 CrossRef PubMed.
- N. P. E. Barry, F. Edafe, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 2816 RSC.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS PubMed.
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 6759 RSC.
- S. H. V. Rijt, H. Kostrhunova, V. Brabec and P. J. Sadler, Bioconjugate Chem., 2011, 22, 218–226 CrossRef PubMed.
- K. Suntharalingam, T. C. Johnstone, P. M. Bruno, W. Lin, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2013, 135, 14060–14063 CrossRef CAS PubMed.
- Y. Fu, M. J. Romero, L. Salassa, X. Cheng, A. Habtemariam, G. J. Clarkson, I. Prokes, A. Rodger, G. Costantini and P. J. Sadler, Angew. Chem., Int. Ed., 2016, 55, 8909–8912 CrossRef CAS PubMed.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Dalton Trans., 2018, 47, 9934–9974 RSC.
- F. P. Dwyer, E. C. Gyarfas, W. P. Rogers and J. H. Koch, Nature, 1952, 170, 190–191 CrossRef CAS PubMed.
- F. P. Dwyer, A. Shulman, G. M. Laycock and S. Dixson, Aust. J. Exp. Biol. Med. Sci., 1969, 47, 203–218 CrossRef CAS PubMed.
- A. Regiel-Futyra, J. M. Dąbrowski, O. Mazuryk, K. Śpiewak, A. Kyzioł, B. Pucelik, M. Brindell and G. Stochel, Coord. Chem. Rev., 2017, 351, 76–117 CrossRef CAS.
- X. Li, A. K. Gorle, M. K. Sundaraneedi, F. R. Keene and J. G. Collins, Coord. Chem. Rev., 2018, 375, 134–147 CrossRef CAS.
- A. Bolhuis, L. Hand, J. E. Marshall, A. D. Richards, A. Rodger and J. Aldrich-Wright, Eur. J. Pharm. Sci., 2011, 42, 313–317 CrossRef CAS PubMed.
- L. Lu, L.-J. Liu, W. C. Chao, H.-J. Zhong, M. Wang, X. P. Chen, J. J. Lu, R. N. Li, D.-L. Ma and C.-H. Leung, Sci. Rep., 2015, 5, 14544–14549 CrossRef CAS PubMed.
- S. V. Kumar, S. Ø. Scottwell, E. Waugh, C. J. McAdam, L. R. Hanton, H. J. L. Brooks and J. D. Crowley, Inorg. Chem., 2016, 55, 9767–9777 CrossRef CAS PubMed.
- P. Srivastava, M. Shukla, G. Kaul, S. Chopra and A. K. Patra, Dalton Trans., 2019, 48, 11822–11828 RSC.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Chem. Sci., 2020, 11, 2627–2639 RSC.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS.
- K. L. Smitten, H. M. Southam, J. Bernardino de la Serna, M. R. Gill, P. J. Jarman, C. G. W. Smythe, R. K. Poole and J. A. Thomas, ACS Nano, 2019, 13, 5133–5146 CrossRef CAS PubMed.
- K. L. Smitten, S. D. Fairbanks, C. C. Robertson, J. Bernardino de la Serna, S. J. Foster and J. A. Thomas, Chem. Sci., 2020, 11, 70–79 RSC.
- V. Fiorini, I. Zanoni, S. Zacchini, A. L. Costa, A. Hochkoeppler, V. Zanotti, A. M. Ranieri, M. Massi, A. Stefan and S. Stagni, Dalton Trans., 2017, 46, 12328–12338 RSC.
- A. M. Ranieri, C. Caporale, V. Fiorini, A. Hubbard, P. Rigby, S. Stagni, E. Watkin, M. I. Ogden, M. J. Hackett and M. Massi, Chem. –Eur. J., 2019, 25, 10566–10570 CrossRef CAS PubMed.
- E. Menéndez-Pedregal, Á. Manteca, J. Sánchez, J. Díez, M. P. Gamasa and E. Lastra, Eur. J. Inorg. Chem., 2015, 2015, 1424–1432 CrossRef.
- P. A. Scattergood, J. Roberts, S. A. E. Omar and P. I. P. Elliott, Inorg. Chem., 2019, 58, 8607–8621 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 73–78 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, 33–36 Search PubMed.
- E. M. Kober and T. J. Meyer, Inorg. Chem., 1982, 21, 3967–3977 CrossRef CAS.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS.
- E. M. Kober, J. V. Caspar, R. S. Lumpkin and T. J. Meyer, J. Phys. Chem., 1986, 90, 3722–3734 CrossRef CAS.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, F. R. Burkert, O. Cars, G. Cornaglia, S. Gandra, C. G. Giske, D. A. Goff, M. Guzman Blanco, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, K. Leder, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, A. Pan, D. L. Paterson, M. Paul, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, G. E. Thwaites, J. W. van der Meer, S. Vega, A. Wechsler-Fördös, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef.
- J. Flatley, J. Barrett, S. T. Pullan, M. N. Hughes, J. Green and R. K. Poole, J. Biol. Chem., 2005, 280, 10065–10072 CrossRef CAS.
- H. M. Southam, J. A. Butler, J. A. Chapman and R. K. Poole, Adv. Microb. Physiol., 2017, 71, 1–96 CrossRef CAS.
- F. Li, Y. Mulyana, M. Feterl, J. M. Warner, J. G. Collins and F. R. Keene, Dalton Trans., 2011, 40, 5032 RSC.
- N. W. Kreofsky, M. D. Dillenburg, E. M. Villa and J. T. Fletcher, Polyhedron, 2020, 177, 114259 CrossRef CAS.
- N. Monti, S. Zacchini, M. Massi, A. Hochkoeppler, L. Giorgini, V. Fiorini, A. Stefan and S. Stagni, Appl. Organomet. Chem., 2020, 34, e5806 CrossRef CAS.
- S. Lazic, P. Kaspler, G. Shi, S. Monro, T. Sainuddin, S. Forward, K. Kasimova, R. Hennigar, A. Mandel, S. McFarland and L. Lilge, Photochem. Photobiol., 2017, 93, 1248–1258 CrossRef CAS PubMed.
- M. Gustafsson, J. Microsc., 2000, 198, 82–87 CrossRef CAS.
- L. Shao, P. Kner, E. H. Rego and M. G. L. Gustafsson, Nat. Methods, 2011, 8, 1044–1046 CrossRef CAS.
- R. Fiolka, L. Shao, E. H. Rego, M. W. Davidson and M. G. L. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5311–5315 CrossRef CAS PubMed.
- J. Kapuscinski, Biotech. Histochem., 1995, 70, 220–233 CrossRef CAS PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03563g |
| This journal is © The Royal Society of Chemistry 2020 |