DOI:
10.1039/D0SC03528A
(Perspective)
Chem. Sci., 2020,
11, 10571-10593
From CO2 activation to catalytic reduction: a metal-free approach
Received
25th June 2020
, Accepted 19th August 2020
First published on 20th August 2020
Abstract
Over exploitation of natural resources and human activities are relentlessly fueling the emission of CO2 in the atmosphere. Accordingly, continuous efforts are required to find solutions to address the issue of excessive CO2 emission and its potential effects on climate change. It is imperative that the world looks towards a portfolio of carbon mitigation solutions, rather than a single strategy. In this regard, the use of CO2 as a C1 source is an attractive strategy as CO2 has the potential to be a great asset for the industrial sector and consumers across the globe. In particular, the reduction of CO2 offers an alternative to fossil fuels for various organic industrial feedstocks and fuels. Consequently, efficient and scalable approaches for the reduction of CO2 to products such as methane and methanol can generate value from its emissions. Accordingly, in recent years, metal-free catalysis has emerged as a sustainable approach because of the mild reaction conditions by which CO2 can be reduced to various value-added products. The metal-free catalytic reduction of CO2 offers the development of chemical processes with low cost, earth-abundant, non-toxic reagents, and low carbon-footprint. Thus, this perspective aims to present the developments in both the reduction and reductive functionalization chemistry of CO2 during the last decade using various metal-free catalysts.
 Sreejyothi P | Sreejyothi P. obtained her dual BS-MS degree majoring in Chemical Sciences from the Indian Institute of Science Education and Research (IISER) Kolkata in 2016. In the same year, she started her PhD under the supervision of Prof. Swadhin K. Mandal at IISER Kolkata. She has extensive expertise in handling low-valent main group compounds and her current research interest deals with CO2 activation using low-valent phosphorus compounds. |
 Swadhin K. Mandal | Dr Swadhin K. Mandal is currently a Professor in the Department of Chemical Sciences at the Indian Institute of Science Education and Research Kolkata. He obtained his Doctoral Degree under supervision of Prof. S. S. Krishnamurthy from the Indian Institute of Science, Bangalore. He was a Postdoctoral Fellow in the Department of Chemistry at the University of California, Riverside, USA with Prof. Robert C. Haddon and an Alexander von Humboldt Fellow at the University of Göttingen, Germany with Prof. Herbert W. Roesky. His current research interests include organometallic catalysis using base metals and main-group metals with emphasis on developing transition metal-mimicking catalysts. He was awarded the prestigious Shanti Swarup Bhatnagar Prize in Chemical Sciences for 2018. |
1. Introduction
The dream of a decarbonized economy is realistically far from reality since the global energy-related CO2 emissions over the last 120 years have continuously increased with a few sporadic dips.1a For example, the global CO2 emissions are expected to drop by 8% in 2020, which will be the lowest since 2010.1b This sharp decline in CO2 emission comes at the expense of the COVID-19 pandemic, which has resulted in a severe health crisis and tremendous economic hardship globally, restricting human activities and hence is not sustainable.
In 2019, the carbon dioxide emissions from anthropogenic activity reached 34 Gt,1c and Fig. 1 presents the steady increase in CO2 emission over the last two decades. The CO2 emissions from power plants and other industrial facilities are considered primarily as a waste product and liability. Accordingly, to address the issue of CO2 emissions and their potential effects on climate change, society must continue to accelerate the exploitation of new energy technologies. It has been predicted that the CO2 emissions need to decline by at least 50% to restrict an increase in the global average temperature by 2 °C by the year 2050.2 Consequently, single technology solution is not expected to resolve this issue, rather the world will require a portfolio of carbon mitigation solutions relying on low-carbon or zero-carbon energy sources. Thus, one of the options that may be beneficial in achieving this target is the carbon capture and storage (CCS), which is being considered worldwide.3a–c CCS technologies have the potential to mitigate a significant amount of CO2; however, the storage of captured carbon faces multiple challenges in terms of economic viability. Hence, recently, a relatively new alternative of carbon capture and recycling (CCR) has been gaining momentum. CCR involves the transformation of CO2 into fuels and chemicals, while creating an added value, and thus can compensate the cost of its capture. In this regard, CO2 is considered an attractive carbon feedstock in chemical synthesis since it is non-toxic, increasingly abundant, and environmentally well-distributed. Among the chemical processes, the reduction of CO2 into various chemicals such as formic acid and methanol or light hydrocarbons such as methane is an extremely useful conversion owing to the wide applications of these products such as hydrogen carriers and their direct use as fuels. Thus, the reduction of CO2 into various energy intensive chemicals and fuels is considered to have the most potential for chemical transformation of CO2.4,5 However, it should be noted that the current use of CO2 in chemical synthesis is very limited,4,6 which may be overcome by addressing two major issues. Firstly, the high kinetic and thermodynamic stability of CO2 requires suitable catalysts for its reduction. Traditionally, over the years, various homogenous and heterogeneous methods have been developed for the catalytic reduction of CO2 mainly using transition metal-based catalysts under harsh reaction conditions.7–11 The transition metals used in the catalytic reduction of CO2 are usually rare, and hence expensive, and also toxic to the environment. Secondly, the economics of the catalytic process must be competitive with the products obtained from the petrochemical industry. This may be achieved by replacing the expensive transition metals with metal-free catalysts operating under mild conditions. An ideal catalyst will ensure that the activation energy remains low, and thus the overall carbon balance is not compromised by the thermal loading since the energy input should be as carbon-free as possible. In recent years, metal-free catalysis along with transition metal-based catalysis have emerged as a sustainable approach because of the milder reaction conditions under which CO2 can be reduced into various value-added products using boranes and silanes as reductants. The metal-free catalytic reduction of CO2 can allow the development of chemical processes with low cost, earth-abundant, non-toxic reagents, and a significantly reduced carbon-footprint.
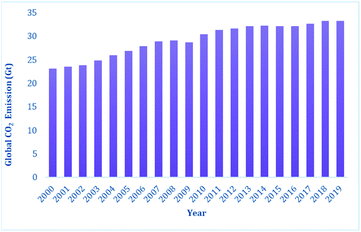 |
| | Fig. 1 Energy-related global CO2 emission in the last two decades.1c | |
In this regard, it is worth mentioning that excellent reviews are available in literature, which either highlighted reduction or reductive functionalization of CO2.6,11–20 However, a detailed article covering the activation, reduction and reductive functionalization of CO2 in an integrated way using metal-free catalysts and reducing agents such as boranes, silanes and hydrogen is lacking. Thus, this perspective aims at documenting these developments in both the reduction and reductive functionalization chemistry of CO2 during the last decade using metal-free catalysts. We begin by highlighting the various activation modes of CO2 by major classes of metal-free systems. The various conceptual aspects of CO2 activation are described and correlated with how metal-free systems can mimic transition metals. This understanding is considered to be fundamental in the design of appropriate catalysts. Subsequently, two aspects of the reduction chemistry of CO2 are widely covered. The first part deals with recent developments in the reduction of CO2 into formic acid, methanol and methane. The second part documents the catalytic reduction of CO2 together with its functionalization, forming new C–N bonds to yield N-formylamines, N-formylamides, N-methylamines and aminals.
2. Activation of CO2 by various metal-free systems
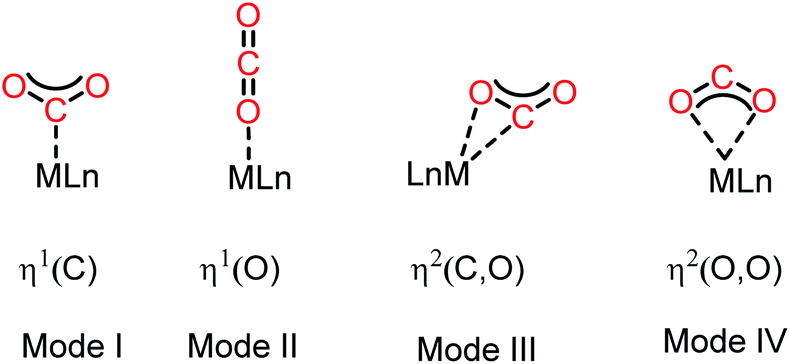
The activation of CO2 molecules is an uphill task owing to its high thermodynamic and kinetic stability.21 Nevertheless, this process is considered to be the key step in the design of any catalytic process for the conversion of CO2 into value-added products. Although CO2 has a zero dipole moment, it is a charge-separated molecule with an inherent difference in polarity between its oxygen and carbon atoms. Thus, in contrast to other inert molecules, such as N2, the presence of this internal dipole along the C–O bonds makes the CO2 molecule ambiphilic in nature. The HOMO of the CO2 molecule is centred on the oxygen atoms, while its LUMO is located on the carbon atom. Consequently, the carbon centre is susceptible to nucleophilic attack, while the oxygen centres can interact with electrophiles. Traditionally, transition metal-based catalysts have been considered as the most efficient for activation of CO2 owing to their ability to interact with CO2 molecules in various ways. In this regard, the most common strategy for CO2 activation involves coordination with metal ions. The commonly observed binding modes of CO2 with metals are demonstrated in Fig. 2.22 One of the most common binding modes involves η1–(C) bonding, in which the metal activates CO2 upon interaction with its C centre (Mode I, Fig. 2). This binding mode is generally observed in electron-rich metals, where electron transfer is facilitated by metal coordination with the electrophilic carbon centre. In this type of coordination with electron-rich metal ions, the LUMO of CO2 gets populated, resulting in the re-hybridization of the C atom and a transformation in its geometry from linear to bent. The other mode of interaction involves one of the oxygen atoms with a metal ion generating the η1–(O) binding mode (Mode II, Fig. 2). In this interaction, an electron-deficient metal ion binds with the nucleophilic ‘O’ centre of CO2. The third type of interaction (Mode III, Fig. 2) corresponds to the bidentate binding mode, in which the metal ion functions as an ambiphilic centre coordinating in an η2(C, O) fashion, where the metal ion binds with both the carbon and oxygen centres. Here, the metal ion acts in a synergistic fashion, in which a suitable metal orbital filled with electrons interacts with the C-centred LUMO, while the O-centred HOMO interacts with the empty d-orbital of the metal ion. Mode IV bonding represents the η2 (O,O) bonding mode (Fig. 2), which is usually known as the “metal carboxylate” type of binding.
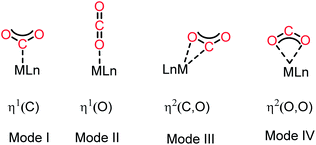 |
| | Fig. 2 Common binding modes of the CO2 molecule with metal (considering a single metal). | |
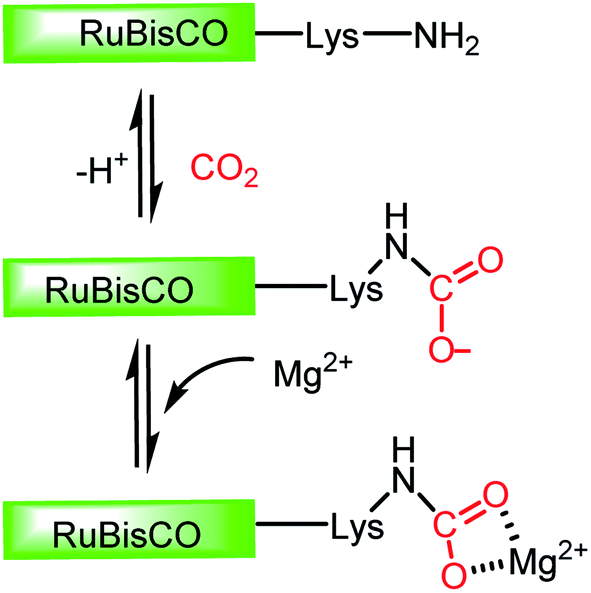
In this regard, it should be noted that catalytic processes involving CO2 without a metal is challenging. Consequently, researchers assumed that CO2 cannot be activated under metal-free conditions. However, recent developments have made it possible to overcome this challenge, where the transition metal-free activation of CO2 has been demonstrated. The most prominent example of metal-free CO2 activation can be cited from the nature itself, where an enzyme known as ribulose-1,5-bisphosphate carboxylase oxygenase (RuBisCO) can fix atmospheric CO2 during photosynthesis.23 The active site of the RuBisCO enzyme consists of the amino acid lysine, which is where the activation of CO2 occurs. During this enzymatic process, the carbamate intermediate is formed through capture of CO2 molecule by the nucleophilic amine group present in the active site of RuBisCO (Fig. 3). This understanding clearly highlights the affinity of the nitrogen lone pair of electrons to form an adduct with CO2. During this interaction with the CO2 molecule, the electron density is transferred from the nitrogen to the low lying empty molecular orbital (LUMO) of CO2, resulting in the bending of its linear structure in order to minimize the energy of the transition state. This nucleophilic interaction by the nitrogen of the amine group is similar to the metal binding Mode I, as mentioned in Fig. 2. Infact, the activation modes of CO2 by metal ions can be considered as strategic guidelines for conceiving metal-free activation. To date, most of the metal-free activations follow the Mode 1- or Mode III-type interaction, as described in Fig. 2. However, this knowledge from nature clearly inspires the logical extension using various nitrogen bases for the activation of CO2 as a viable route to replace the expensive and rare transition metals. To date, the reported metal-free activation of CO2 can be divided into three distinct subclasses: (a) organic nucleophile-mediated activation (including nitrogen and phosphorus bases and N-heterocyclic carbenes (NHCs)); (b) ionic liquid (IL)-mediated activation and (c) ambiphilic activation by frustrated Lewis acid base pairs (FLPs). Organic nucleophiles are probably the most studied metal-free systems with regard to CO2 activation owing to their availability and structural diversity, including alkyl/aryl mono- and polyamines, N-heterocyclic carbenes, amidine-like and guanidine-like derivatives. Among the organic bases, amidines and guanidines show better activity towards CO2 activation. For example, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) are the most recognised organic bases (Fig. 4), which are widely used in various transformations for the activation of the CO2 molecule. The interaction between DBU and CO2 molecule can be traced back to 1978 when Iwatani et al. reported the formation of a white solid upon the exposure of liquid DBU to CO2 gas, which was characterized by IR spectroscopy and elemental analysis.24 The high basicity of DBU is beneficial in the adduct formation with CO2 by nucleophilic attack. In 2002, Franco and co-workers used this adduct for the synthesis of N-alkyl carbamates.25 In 2004, Pérez et al. attempted to crystallize the DBU–CO2 adduct by exposing a DBU solution in acetonitrile (ACN) to CO2, but failed to isolate the desired crystals.26 However, they were able to characterize the DBU–CO2 adduct by NMR spectroscopy and the 13C NMR spectrum in D2O, which displayed a resonance at δ 160.7 ppm. Up to 2010, the solid-state characterization of this nitrogen base-CO2 adduct remained unsuccessful, until Villiers et al. first isolated the TBD–CO2 adduct and characterised it via single crystal X-ray crystallography.27 Dissolving TBD in anhydrous THF, and subsequent purging with CO2 resulted in the formation of an off-white powder, which was crystallized at 70 °C under a CO2 atmosphere. The N–C(CO2) bond length was measured to be 1.48 Å, which is slightly longer than the typical N–C bond length (∼1.35 Å) observed in various carbamates. The 13C NMR spectrum recorded in CD3CN revealed that the 13C resonance arising from the CO2 molecule displayed a downfield shift at δ 154.4 ppm. The reaction was carried out under anhydrous conditions in order to avoid the possible formation of bicarbonate. Subsequently, various nitrogen bases were used for the activation of CO2. Some of the representative examples of these isolated adducts (1–4) are presented in Fig. 4.
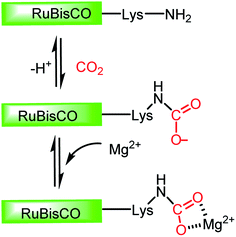 |
| | Fig. 3 Schematic representation of CO2 activation in the active site of RuBisCo. | |
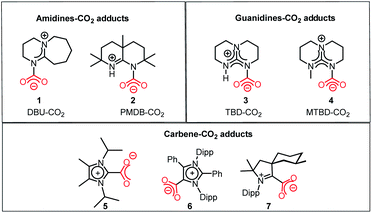 |
| | Fig. 4 Representative examples of isolated and well-characterized nitrogen base-CO2 (1–4) adducts and NHC–CO2 adducts (5–7). | |
In addition to nitrogen bases, N-heterocyclic carbenes (NHCs) emerged as another class of nucleophiles for the activation of CO2. Soon after the isolation of the first NHC by Arduengo in 1991, NHC became the most versatile ligand for organometallic catalysis.28 By virtue of their singlet ground state, the lone pair of electrons resides in a sp2-hybridized orbital on the carbene carbon atom along with an unoccupied p orbital. The lone pair of electrons in the carbene centre makes NHCs nucleophilic, and hence they can act as very efficient σ donors. In 1999, Kuhn and co-workers first introduced NHCs to capture CO2 molecules, where they demonstrated the formation of a zwitterionic adduct (5, Fig. 4) and characterised this adduct crystallographycally.29a Later in 2004, Louie and co-workers demonstrated the reversible binding of CO2 with NHC, in which the addition of 1 atm CO2 to 1,3-dimesitylimidazol-2-ylidene (IMes), or 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) resulted in the formation of the corresponding zwitterionic imidazolium carboxylates.29b The IPr–CO2 adduct was characterised via X-ray crystallography, and almost similar bond lengths of both C–O bonds in the adduct indicate that the negative charge is distributed equally. The 13C NMR spectrum in CD2Cl2 showed a peak at δ 152.3 ppm, which was assigned to the 13C resonance arising from the carboxylate group. Following this study, CO2 adducts (6 and 7, Fig. 4) with various newer versions of NHCs such as abnormal NHC (aNHC)30 and cyclic(alkyl)(amino) carbene (CAAC)31 were isolated and characterized.
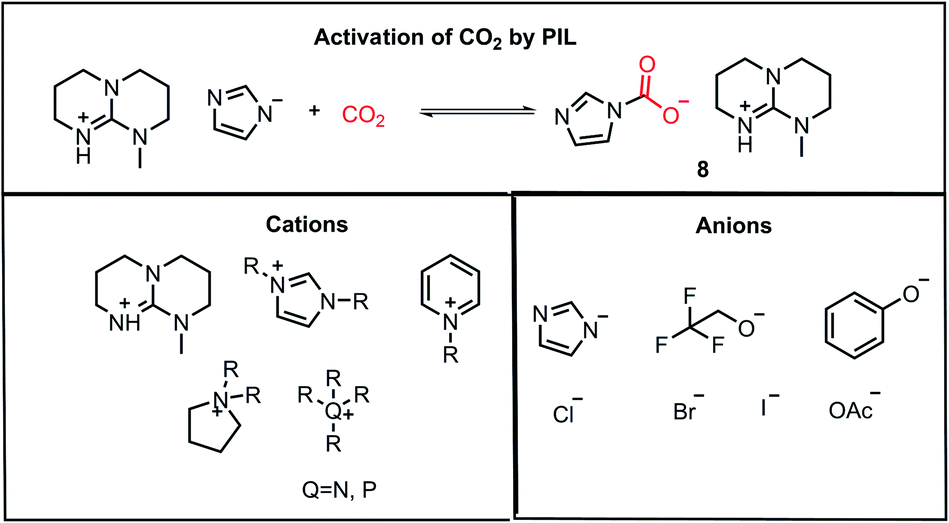
Another class of compounds commonly used for the activation of CO2 molecules is ionic liquids (ILs). ILs are comprised of a cation and anion with one of the ions as an organic species possessing a delocalized charge. Ionic liquids exhibit high thermal stability and volatility, which can sustain both high temperature and pressure in the reaction medium, unlike organic solvents. Furthermore, the properties of ionic liquids can be tuned by varying the substituents on the organic part of the counterion, and hence they are often termed as “designer solvents”. In 2002, Davis and co-workers reported the chemisorption of CO2 on an amino group-functionalized IL.32 In 2010, Lei and Dai developed anion-functionalized protic ionic liquids (PIL).33 In their work, the PILs were formed via the combination of different super bases such as 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD) with partially fluorinated alcohols/imidazoles/phenols, etc. These strong bases have very high proton affinity, deprotonating even weak proton donors to form thermodynamically stable PILs. These PILs could capture CO2 efficiently (8, Fig. 5). The nucleophilic character of ILs is exploited during the activation of CO2, where the accumulated negative charge on the CO2 molecule after its activation is stabilized by ion-pairing interactions with the cationic part of the IL, as shown in 8. The major ILs used for the activation of CO2 are depicted in Fig. 5.33
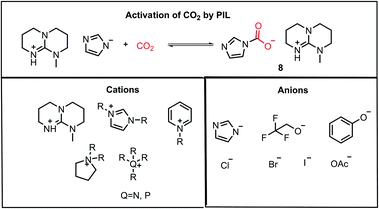 |
| | Fig. 5 Activation of CO2 by PIL and combination of cations and anions for the construction of various ILs used for CO2 activation. | |
Another common mode of metal-free CO2 activation is observed through frustrated Lewis pairs (FLPs). The concept of FLPs was developed from the initial observation by H. C. Brown in 1942 while studying the interaction between lutidine and BMe3,34 and later from the seminal work by Wittig and Benz,35 Tochtermann,36 Piers,37 Oestreich38 and many others. Based on the information gained through the years, FLPs are defined as a combination of a Lewis acid (LA) and Lewis base (LB) that is unable to form a classical acid–base adduct because of steric or geometric restrictions. The area of FLP research propelled in 2006 from the breakthrough made by Stephan and co-workers when Mes2P(C6F4)B(C6F5)2 was observed to reversibly bind with molecular hydrogen under mild reaction conditions.39 The activation of H2 was carried out using an FLP following the concept of synergistic activation, which is similar to the ambiphilic activation mode of a transition metal for a small molecule such as H2. In the case of FLP-mediated activation, the electrons from the bonding orbitals of H2 are transferred to the LUMO of the LA, while in the case of metal-mediated activation, the bonding electrons are transferred to a suitable empty d orbital (Fig. 6, A and C). Simultaneously, the electrons from the HOMO of the LB are transferred to the anti-bonding orbital of H2, while in the case of metal-mediated activation, the d electrons are transferred to the anti-bonding orbital of the H2 molecule which results in the cleavage of the H–H bond (Fig. 6, B and D). This concept was logically extended to the activation of CO2 by FLPs. FLPs are capable of activating carbon dioxide since they are ambiphilic, allowing nucleophilic activation by the Lewis base (LB) at the carbon, and subsequent electrophilic activation by the Lewis acid (LA) at the oxygen atoms (Fig. 6, F). This bifunctional interaction was shown to be the key feature for the transition metal-mediated CO2 activation process (Fig. 6, E and mode III, Fig. 2). The first report on FLP CO2 activation appeared in 2009 by Stephen, Erker and co-workers, in which they reported a phosphino–borane LA–LB system for the formation of a zwitterionic product after activating the CO2 molecule.40 Specifically, B(C6F5)3 and PtBu3 were added to bromobenzene under CO2 atmosphere, which resulted in the formation of a white precipitate. It was found that the electrophilic C centre binds with the Lewis basic phosphorus unit, while the nucleophilic O of CO2 binds with the Lewis acid boron, forming stable carboxylate adduct 9 (Scheme 1).
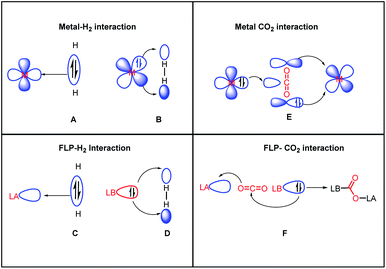 |
| | Fig. 6 Activation of H2 and CO2, highlighting the resemblance between transition metal-mediated activation and FLP-mediated activation. | |
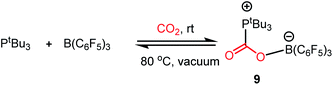 |
| | Scheme 1 Activation of CO2 by P/B FLP, forming a carboxylate adduct. | |
3. Metal-free catalytic reduction of CO2
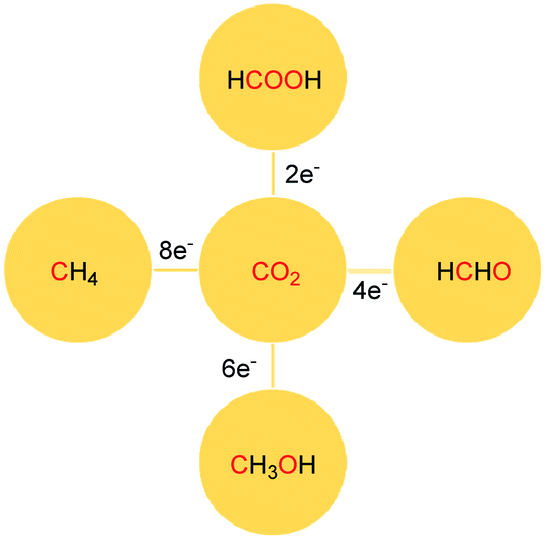
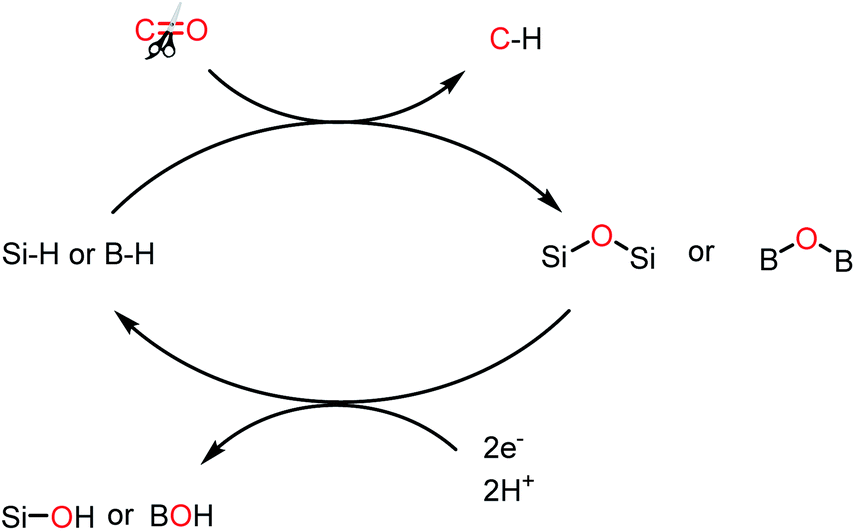
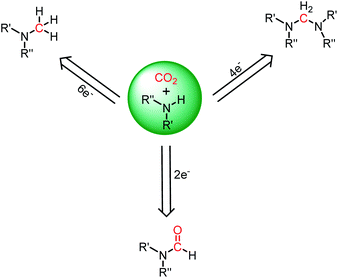
The reduction of CO2 to various value-added products leads to a decrease in the formal oxidation state of carbon centre of CO2 is of great interest. CO2 can be reduced sequentially to various energy intensive chemicals and fuels. For example, the 2e-, 4e-, 6e- and 8e-reduction of CO2 can lead to the formation of formic acid, formaldehyde, methanol and methane, respectively (Scheme 2). Traditionally, these reductions have been carried out using transition metal-based catalysts.41–43 In recent years, catalytic metal-free CO2 reduction has emerged as an alternative to the expensive transition metal-mediated process. The previous section described various metal-free systems that can activate CO2 under ambient conditions, opening up the possibility of the catalytic reduction of CO2 under metal-free conditions.
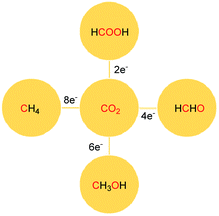 |
| | Scheme 2 Sequential reduction of CO2 by 2e, 4e, 6e and 8e leads to the formation of various chemicals and fuels. | |
3.1. Frustrated Lewis pairs for CO2 reduction
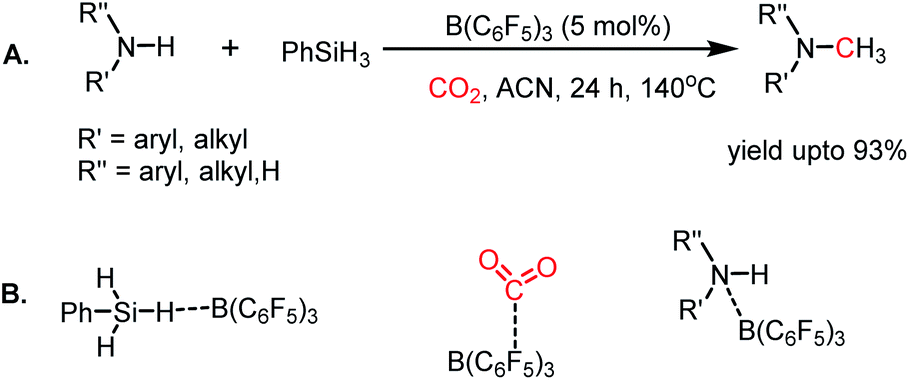
Since the pioneering discovery in 2009 by Stephen, Erker and co-workers40 on CO2 activation by a phosphino–borane FLP system, CO2 reduction has taken a quantum leap in the last decade. The first FLP-mediated CO2 activation followed by its reduction into methanol was reported in 2009 by Ashley and O'Hare.44 The reduction of CO2 into methanol was accomplished using hydrogen gas in a stoichiometric amount. Their work took advantage of the fact that FLP can split the hydrogen molecule,39 and subsequently the hydrogen activation is integrated with the CO2 activation step to realize the thermodynamically challenging conversion. In this work, the combination of B(C6F5)3 and TMP (TMP = 2,2,6,6-tetramethylpiperidine) in presence of H2 and CO2 resulted in the formation of a formatoborate complex, [TMPH]-[HCO2B(C6F5)3] (11), under heating (Scheme 3A). This observation can be explained considering stepwise activation of hydrogen and CO2 molecules. The reaction between an equimolar amount of TMP and B(C6F5)3 in the presence of H2 yields [TMPH][HB(C6F5)3] (10), where the H2 molecule is heterolytically cleaved. This splitting of the hydrogen molecule by an FLP composed of TMP/B(C6F5)3 was previously proposed by Sumerin et al.45 Subsequently, heating 10 in toluene above 110 °C under 1 atm CO2 led to the formation of the formatoborate complex [TMPH][HCO2B(C6F5)3] (11, Scheme 3B). A carbonyl stretch at 1662 cm−1 was observed in the IR spectrum of complex 11 and the 13C NMR spectrum revealed a resonance at δ 169.9 ppm, confirming the incorporation of CO2. Furthermore, the structure of 11 was established by single crystal X-ray study. Finally, purging CO2 in a mixture of TMP/B(C6F5)3 in C7D8 under an H2 atmosphere resulted in their quantitative conversion into [CH3OB(C6F5)3][TMPH] (15) after 6 days of heating at 160 °C. The formation of methanol in ∼17–25% yield was observed after vacuum distillation. This may be explained considering following steps presented in Scheme 3B. Specifically, upon heating, 11 reversibly decomposes to 10, which stays in equilibrium with TMP, B(C6F5)3 and H2. The free B(C6F5)3 present in the medium can be arrested by the acyl oxygen of 11 to produce intermediate 12. This activated formate intermediate 12 can be reduced by 1 equivalent of 10 to form acetal 13, releasing B(C6F5)3. Due to the instability of acetal in solution, the [TMPH]+ counterion serves as the H+ donor and 13 is converted into 14 and [TMPH]+[B(C6F5)3OH]−. Next, 14 undergoes reduction in the presence of 10 in the medium to form 15. The formation of methanol can be explained by considering the reaction of 15 with TMP or its conjugate acid. Even though this work represents the first reduction of CO2 to methanol using an FLP-based system under a low pressure of CO2 (1–2 atm), it suffers from a serious drawback since it is not a catalytic process. Thus, to address this shortcoming, Piers and co-workers reported that the addition of a hydrosilane to the reaction mixture in the presence of excess B(C6F5)3 transforms the process into a catalytic one.46 In the presence of excess B(C6F5)3 and triethylsilane, the B(C6F5)3/Et3SiH adduct 16 is generated, which can rapidly hydrosilate the formatoborate complex 11 to form formatosilane compound 17, regenerating [TMPH]+[HB(C6F5)3]−10 (Scheme 4), thus making this process catalytic. Subsequently, the formatosilane 17 is further hydrosilated by 16 sequentially to (Et3SiO)2CH2 (18), Et3SiOCH3 (20) and CH4 (21), leaving (Et3Si)2O (19) as a by-product.
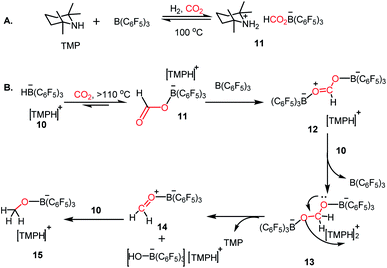 |
| | Scheme 3 (A) CO2 activation in the presence of TMP/B(C6F5)3/H2. (B) Reduction of CO2 with H2 by FLP composed of TMP/B(C6F5)3. | |
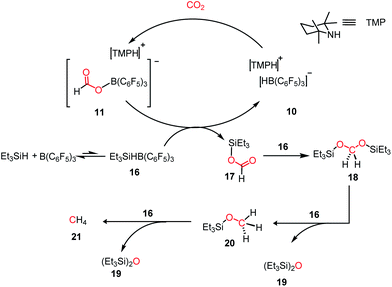 |
| | Scheme 4 Converting stoichiometric CO2 reduction into catalytic process by adding excess B(C6F5)3 and Et3SiH to the TMP/B(C6F5)3 system. | |
Although, this reaction was catalytic, it still lacked good efficiency. The low efficiency was attributed to multiple factors such as the nature of the ion pair formed in the reaction medium47 and the high entropy of the FLP system, as pointed by Fontaine and coworkers,12,48 which originates from the use of discrete molecules of LA, LB and CO2. Thus, to favour the reaction entropically, the LA and LB centres were assembled within the same molecule. In this regard, in 2013, an aryl bridged phosphine–borane system was introduced for the catalytic hydroboration of CO2 into methoxyborane, which could further be hydrolysed to methanol.48 In this work, the phosphine–borane 1-Bcat-2-PPh2–C6H4 (cat = catechol) (22) functions as an ambiphilic system for the reduction of carbon dioxide in presence of various hydroboranes, resulting in the formation of CH3OBR2 or (CH3OBO)3 as the product. The TON (turnover numbers) and TOF (turnover frequencies) reported were as high as >2950 and 853 h−1, respectively (Scheme 5A). In a typical catalytic run, exposing 1-Bcat-2-PPh2–C6H4 to 1 atm of CO2 at room temperature in the presence of 100 equiv. of HBcat resulted in the formation of a white precipitate. The white precipitate was characterized as catBOBcat. Analyzing the solution by 1H NMR spectroscopy showed the presence of a new singlet at δ 3.37 ppm, which was attributed to CH3OBcat. Moreover, this FLP reduced CO2 in the presence of BH3·SMe2 at 70 °C with a TOF of 973 h−1 and TON of up to 2950, which is comparable to that of transition metal catalysts. This was the first report where BH3 was introduced as a hydrogen source for the catalytic reduction of CO2. A detailed DFT study was conducted to understand the mechanism of the reaction.49 The reaction proceeds via a three-step process, in which CO2 is first reduced to formatoborate, followed by formaldehyde, and then the final product methoxyborane (Scheme 5B). Four different modes of activation of borane were considered during this calculation and the most feasible one involved the simultaneous activation and binding of both borane and CO2 to the catalyst (Scheme 5B). As the first step, the Lewis basic phosphorus atom present in the FLP activates the boron centre of the hydroborane by forming an adduct, and the enhanced nucleophilicity of the borane causes hydride to transfer to the electrophilic carbon of CO2, forming a formate anion. Further, the formate anion can bind with the boron centre of the catalyst in a concerted way, thus resulting in intermediate 23 (see Scheme 5B). However, this type of activation contradicts the classical FLP-mediated CO2 activation. This simultaneous activation results in a four-coordinated boron centre observed in 23, which can stay in equilibrium with compound 24. The next step involves the formation of formaldehyde intermediate 26 (Scheme 5B) upon reduction by hydroborane through intermediate 25 (see Scheme 5B). In the next step, in the presence of hydroborane, 26 is further reduced to the corresponding methoxyborane (27), while regenerating the catalyst 22. In subsequent study with 13C labeled CO2,50 it was identified that 26 plays the role of an active catalyst instead of a reaction intermediate, as portrayed in Scheme 5B. The ambiphilic nature of the catalyst where LA–LB is present in the same molecule plays a key role in the efficacy of this FLP-based catalyst.
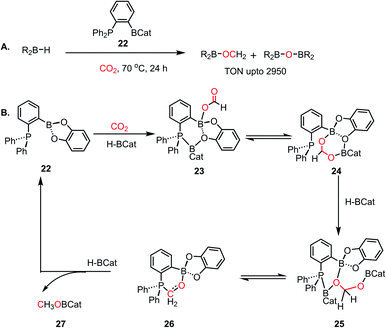 |
| | Scheme 5 (A) Catalytic reduction of CO2 using ambiphilic B/P FLP catalyst. (B) Plausible catalytic cycle using ambiphilic B/P FLP catalyst based on DFT calculation. | |
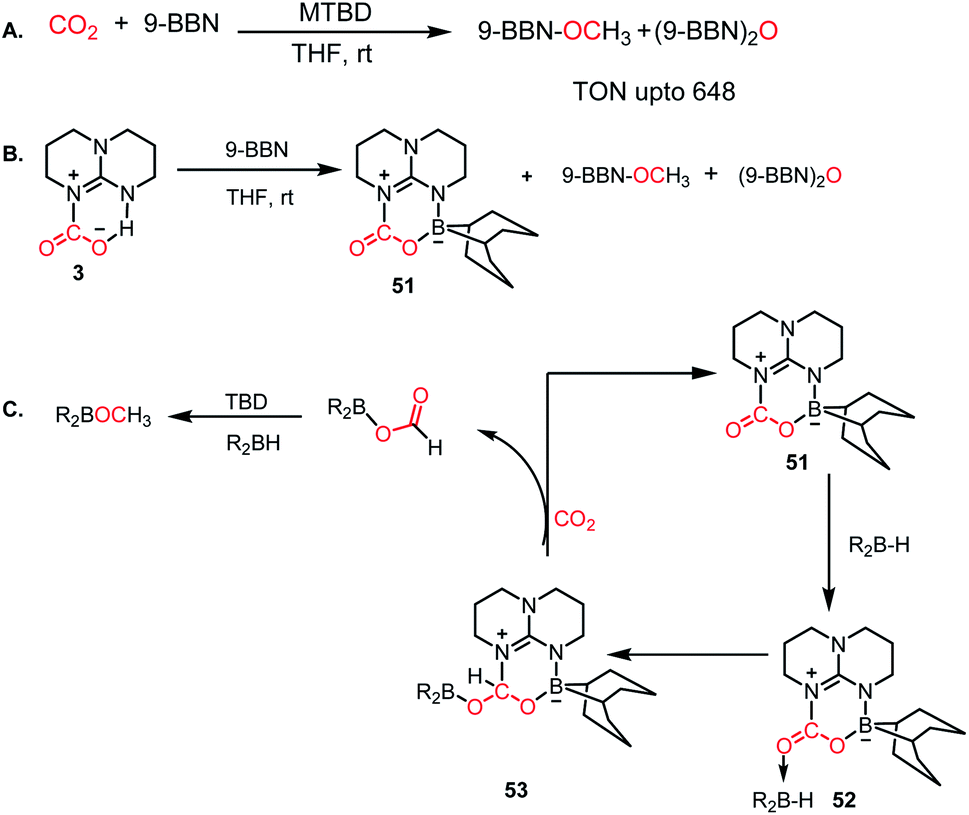
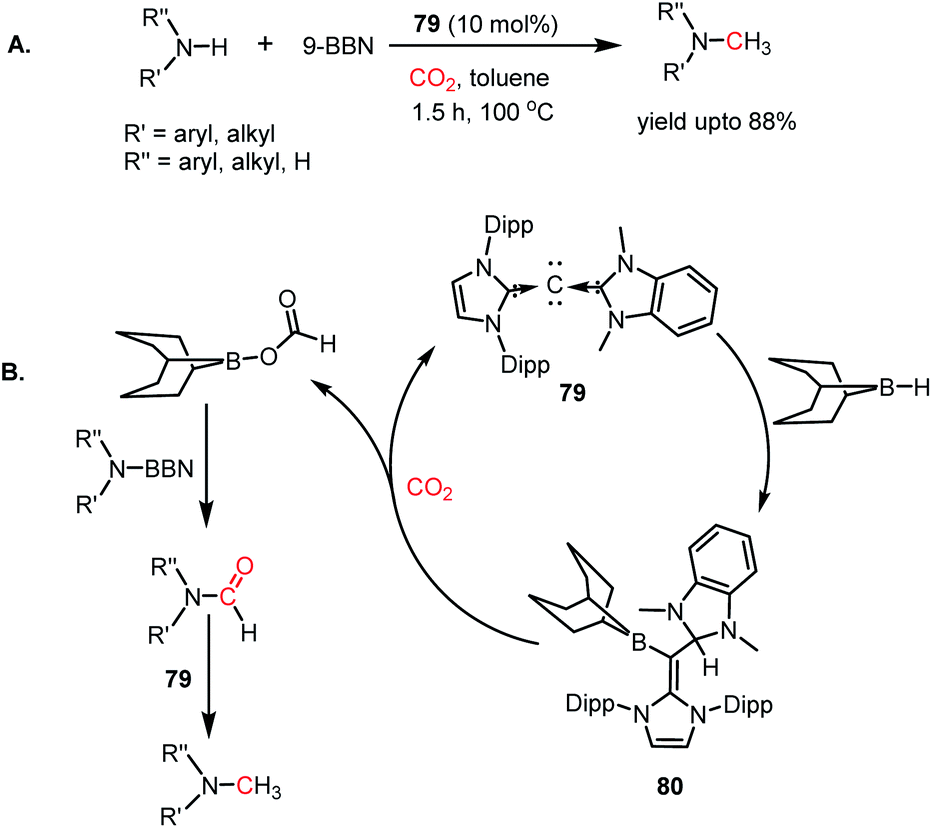
In 2014, Stephan's group reported an intermolecular P/B-based FLP for the catalytic reduction of CO2 (Scheme 6A).51 In their report, a stoichiometric reaction between 9-BBN, phosphine (R3P; R = tBu, 4-MeC6H4) and CO2 yielded (R3PCH2O)(HC(O)O)B(C8H14) (28) (see Scheme 6B), in which CO2 is incorporated both in the form of formate and a phosphonium methoxy unit, and its structure was characterized by X-ray crystallography. This control experiment established the CO2 activation step using the P/B FLP. This observation prompted them to extend their study in a catalytic direction, where in the presence of a catalytic amount of tBu3P, CO2 was successfully reduced using 9-BBN at 60 °C to methoxyborane with the highest TON of 5500 (Scheme 6A). When the catalytic reaction was performed in the presence of 9-BBN, 4 mol% tBu3P, using CO2, it yielded various reduction intermediates. The first step involves the formation of a boron formate intermediate 30via intermediate 29, which is then converted into diolate derivative 31 (Scheme 6C). Subsequently, 31 can further react with R3P via the elimination of a B–O–B dimer to generate compound 32. Compound 32 can react with boron formate 30 to generate compound 28, which was characterized via X-ray crystallography, or it can be further reduced to methoxyborane by reaction with 9-BBN (Scheme 6C).
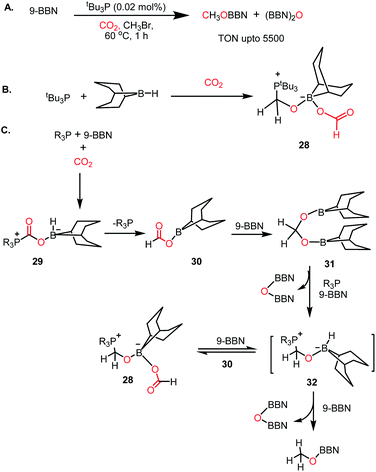 |
| | Scheme 6 (A) Intermolecular B/P FLP for the catalytic reduction of CO2. (B) Activation of CO2 by B/P FLP. (C) Plausible mechanistic cycle for B/P FLP-mediated CO2 reduction. | |
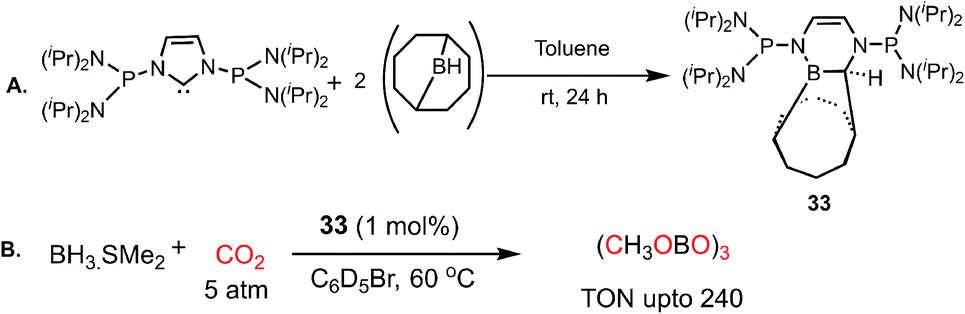
In 2012, Stephan's group reported an adduct of NHC with 9-borabicyclo[3.3.1]nonane (9-BBN) in the presence of PtBu3, which could be used for the activation of H2.52 Inspired by this work, the same group introduced an intramolecular FLP by reacting a phosphine-bearing NHC derivative with 9-BBN. The reaction proceeds through C–N bond cleavage of NHC and insertion into the B–C bond of 9-BBN resulting in ring expansion (33, Scheme 7A).53 This adduct consists of a strong basic site (P center) and weak Lewis acidic site (B center), making it a suitable intramolecular FLP, 33. This FLP was used for the catalytic hydroboration of CO2 in the presence of a boron hydride (Scheme 7B), exhibiting a TON of 240 and affording methoxyborane.
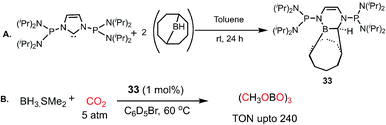 |
| | Scheme 7 (A) Preparation of intramolecular P/B FLP system. (B) Catalytic CO2 reduction using the P/B FLP 33. | |
In 2016, Cantat and co-workers introduced another class of FLPs, in which a nitrogen-stabilized silylium cation was used for the activation of CO2.54 It should be noted that being isoelectronic with boranes, silyl cations are considered as a good replacement for boranes as Lewis acids. A series of base-stabilized silylium species was prepared, which formed an N/Si FLP–CO2 adduct (34) upon exposure to CO2. This adduct with CO2 was characterized by X-ray study. These silicon species were found to be active catalysts in the hydroboration of CO2 to methoxyborane with various boranes, giving the highest TOF of 3.7 h−1 (Scheme 8). In these FLPs, the silicon centre exhibits affinity towards oxygen, while the N center binds with the C of CO2 during the activation process.
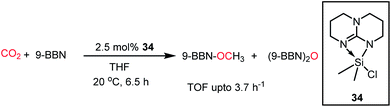 |
| | Scheme 8 Reduction of CO2 using N/Si FLP system 34. | |
In 2018, the viability of commercially available alkylic carbodiimides (CDIs) was demonstrated when they were treated with boranes such as 9-BBN or BH3 in the catalytic hydroboration of CO2.55 The catalytically-active FLP species were formed in situ via the reaction of CDIs with borane and CO2. The CDIs react with boron hydrides to form boron amidinates,56 which have potential to act as an FLP owing to the presence of N/B combinations. Commercially available diisopropylcarbodiimide (CDI, 35) was used as a catalyst for reduction of CO2 using various hydride sources such as 9-BBN and BH3·SMe2 under mild conditions (1 atm CO2, 25–60 °C) to give a mixture of methoxyborane and bis(boryl)acetal (Scheme 9A). Various stoichiometric reactions were carried out to elucidate the reaction mechanism, and it was proposed that the first step involves the hydroboration of 35 by borane to yield boron amidinate (36), which further reacts with CO2 to form a 1,2 addition intermediate (37), as shown in Scheme 9B. Further reaction with another unit of 9-BBN led to the formation of a formaldehyde adduct (38). All these intermediates (36–38) were identified and characterized via X-ray crystallography. Complex 38 was identified as the active catalyst, which reduces CO2 to methoxyborane and bis(boryl)acetal upon the sequential addition of 9-BBN. It may be noted that in the case of BH3·SMe2, the products of CO2 reduction are (OBOMe)3 and B(OMe)3 (Scheme 9A).
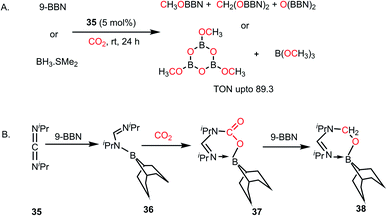 |
| | Scheme 9 (A) Catalytic reduction of CO2 using CDI, forming in situ FLP, which acts as a catalyst. (B) Formation of the active catalyst 38 for CO2 reduction using CDI. | |
Thus far, we have discussed CO2 reduction using various FLP-based catalysts in presence of hydrosilane or hydroborane as a reducing agent. Although these processes exhibit good catalytic efficacy, there is always room for further improvement since these reducing agents are expensive. In this regard, H2 gas, owing to its low cost, can be a good replacement for the expensive silane/borane reagent. Consequently, the development of metal-free systems that can activate both CO2 and H2 simultaneously and selectively is highly rewarding, although very challenging. In 2015, Stephan and Fontaine et al. introduced an intramolecular B/N-based FLP, where CO2 was reduced to formates, acetals and methoxides in the presence of molecular H2 (Scheme 10A).57 An ambiphilic B/N FLP 39 was developed, which upon exposure to H2 gas at 80 °C resulted in the formation of 40 (Scheme 10B) with the sequential elimination of mesitylene, which was evident from the 1H NMR spectroscopy of the reaction mixture. This observation suggests the possibility of a protodeborylation process as a result of H2 activation. Further, compound 40 generated upon H2 activation can act in dual fashion for the activation of CO2 together with its reduction. For example, 40 can act as an ambiphilic FLP for CO2 capture, and subsequently the BH2 group present in 40 may function as a reducing agent. Although the selectivity of the process is somewhat compromised, resulting in a mixture of products, this result suggests that an appropriate strategy in designing FLPs can provide an avenue for the design of metal-free catalysts for H2/CO2 chemistry. Accordingly, this work represents a unique way where H2 was used as a reductant for the reduction of CO2, avoiding borane/silane as reductant.
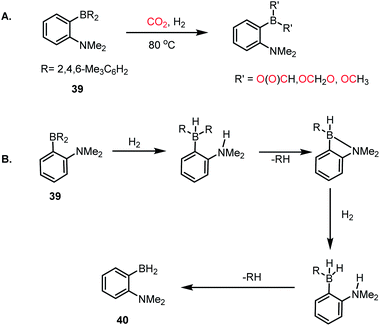 |
| | Scheme 10 (A) CO2 reduction in the presence of a B/N FLP using molecular H2. (B) Heterolytic cleavage of H2 in the presence of a B/N FLP, resulting in 40. | |
3.2 NHC mediated CO2 reduction
As mentioned earlier, N-heterocyclic carbenes (NHCs), owing to their strong nucleophilicity, are known to form imidazolium carboxylates upon reaction with CO2,29 which was explored towards the catalytic reduction of CO2 to form various value-added products. In 2009, Ying and co-workers first reported the hydrosilylation of CO2 in the presence of an NHC to form methanol under ambient conditions.58 This was the first time a metal-free system was involved in the catalytic reduction of CO2 to methanol. It was observed that the 1,3-bis(2,4,6-trimethylphenyl)imidazolium carboxylate (41) under a CO2 atmosphere resulted in the hydrosilylation of CO2 by diphenylsilane at room temperature within 6 h (Scheme 11A). 1H NMR spectroscopy and GC-MS studies identified the presence of diphenylformoxysilane, diphenyldiformoxysilane, bis(silyl)acetal and silylmethoxide in the reaction mixture, which gave insight into the mechanistic pathway of this fascinating conversion. A peak at δ 3.5 ppm in the 1H NMR spectrum of the reaction mixture in DMF-d7 confirmed the formation of silyl methoxide, which can be easily hydrolysed to methanol. To understand the reaction mechanism, various control reactions were carried out using isotopically labelled 13CO2 gas. The 13C {1H} NMR spectroscopic studies revealed the appearance of new peaks at around δ 160 ppm, indicating the formation of formoxy and di-formoxy silane. The peak at δ 85 ppm was ascribed to the silyl acetal group and the resonance at δ 50 ppm was assigned to silyl methoxide. During the course of the reaction, the resonance at δ 85 ppm diminished together with an increase in the intensity of the peak at δ 50 ppm, indicating the conversion of acetals into methoxide. This catalytic conversion led to the highest TON of 1850 and TOF of 25.5 h−1 under ambient conditions (Scheme 11A). Finally, treatment of the reduced products with NaOH/H2O produced methanol. The detailed mechanistic understanding remained unclear; however, a plausible cycle was proposed, in which imidazolium carboxylate 41 first attacks the silicone centre of diphenylsilane, which facilitates the hydride transfer in a concerted manner producing formoxysilane 42 (Scheme 11B). This releases a free NHC, which captures CO2 to regenerate 41. Formoxysilane 42 can be reduced by hydrosilanes to form the silyl methoxide and the reduction continues until all the hydrosilanes are consumed (Scheme 11B). This mechanistic proposal later attracted considerable attention when Wang and co-workers came up with a detailed mechanistic proposal based on DFT calculations.59 In their study, it was shown that the first step involves the activation of the Si–H of silane by NHC, which results in the transfer of hydride to the electrophilic carbon centre of CO2 to form a formate ion. Based on their calculations, the prior activation of the carbon centre of CO2 by NHC coordination was found to be energetically less favourable than silane activation. Further reduction of formate by additional equivalents of silane resulted in the formation of methoxyborane. They proposed a mechanistic path, where NHC directly coordinates to the silane centre instead of CO2. However, Zhang and co-workers further came up with a detailed study to reveal the underlying mechanism.60 The stoichiometric reaction performed between NHC and silane did not show any change in chemical shift values, even after prolonging the reaction for 2 weeks. Hence the possibility of NHC coordination to silane was discarded. In their study, it was uncovered that the reaction takes place through a three-step cascade pathway. The first step of the cascade mechanism involves the interaction of diphenylsilane with imidazolium carboxylate 41 under a CO2 atmosphere to form a formoxysilane intermediate (42, Scheme 11C) via a five-membered transition state 43. This step represents the rate-limiting step since it possesses the highest energy barrier. The next step proceeds with the hydride transfer from diphenylsilane to the formoxysilane intermediate and the formation of bis(silyl)acetal (44, Scheme 11C), whereas the third step involves the formation of the methoxy silane product by reacting with another molecule of silane, while eliminating silyl ether (Scheme 11C).
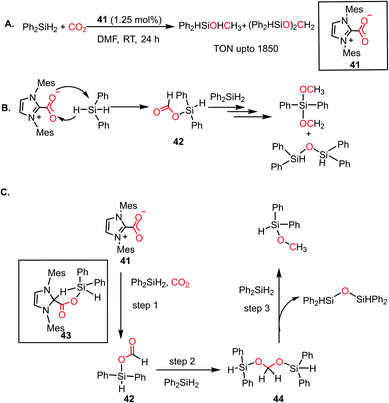 |
| | Scheme 11 (A) Hydrosilylation of CO2 using NHC as a metal-free catalyst. (B) Reduction of CO2 to methoxide in the presence of NHC carboxylate 41. (C) Three-step cascade pathway for the reduction of CO2 using NHC-carboxylate 41. | |
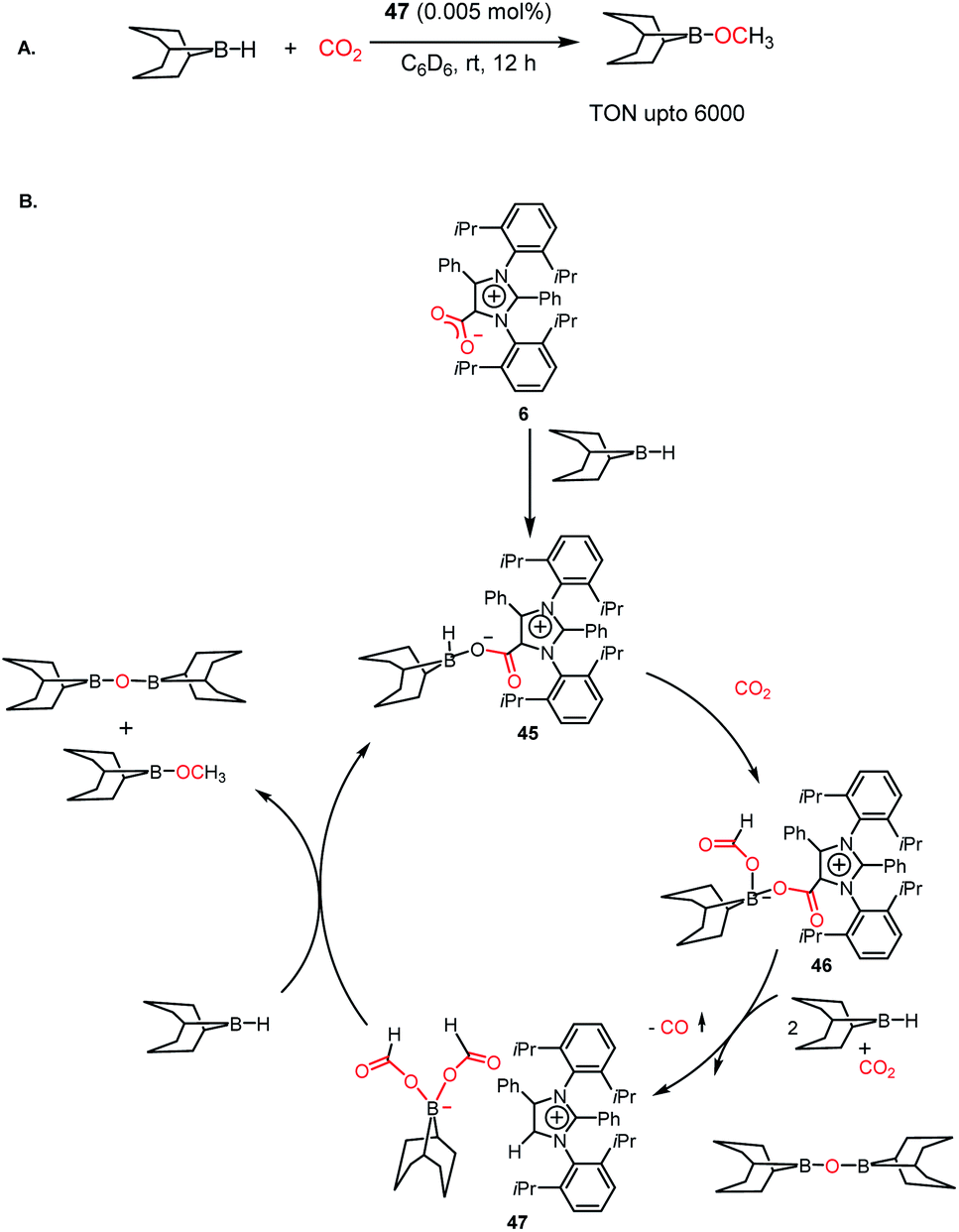
The efficiency of NHC in the reduction of CO2 prompted the use of an abnormal NHC (aNHC), exhibiting superior nucleophilicity over normal NHC.30 As mentioned in the previous section, the synthesis of the aNHC–CO2 adduct (6) was reported by Bertrand and co-workers in 2009.30 Later, the introduction of this adduct in synthesis of methanol was carried out by Mandal and co-workers in 2016 via the hydroborylative reduction of CO2.61 The use of an abnormal N-heterocyclic carbene and corresponding CO2 adduct was reported for the reduction of CO2 to methoxyborane under ambient conditions (1 atm CO2, rt) using various hydride donors such as 9-BBN, HBPin, CatBH, BH3·SMe2 and BH3·THF. The stoichiometric reaction between the aNHC–CO2 adduct and 9-BBN under a CO2 atmosphere resulted in the formation of a colourless solution, which was identified as a boron diformate (47), as established by various spectroscopic studies, and finally its solid-state structure was determined by X-ray crystallography. The 1H NMR spectrum showed two singlets at δ 8.51 and 8.33 ppm with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 intensity, which corresponds to formate and C5–H of aNHC, respectively. The reduction of CO2 was performed in the presence of 9-BBN using 47 as a catalyst, leading to a TON of 6000, which is among the highest TON reported using a metal-free catalyst under ambient conditions (Scheme 12A). Based on a series of control experiments together with DFT studies, the mechanistic cycle was proposed, in which aNHC–CO2 (6) upon reaction with 9-BBN forms adduct 45, activating the B–H bond of borane. Subsequently, 45 further undergoes an insertion reaction with CO2, which results in the formation of a four-coordinated boron species 46. The addition of two molecules of 9-BBN to 46 results in the formation of a zwitterionic borondiformate 47 together with the release of a B–O–B dimer. Subsequently, another molecule of 9-BBN reacts with 47, which results in the regeneration of 45 together with the formation of a methoxide derivative (Scheme 12B).
:1 intensity, which corresponds to formate and C5–H of aNHC, respectively. The reduction of CO2 was performed in the presence of 9-BBN using 47 as a catalyst, leading to a TON of 6000, which is among the highest TON reported using a metal-free catalyst under ambient conditions (Scheme 12A). Based on a series of control experiments together with DFT studies, the mechanistic cycle was proposed, in which aNHC–CO2 (6) upon reaction with 9-BBN forms adduct 45, activating the B–H bond of borane. Subsequently, 45 further undergoes an insertion reaction with CO2, which results in the formation of a four-coordinated boron species 46. The addition of two molecules of 9-BBN to 46 results in the formation of a zwitterionic borondiformate 47 together with the release of a B–O–B dimer. Subsequently, another molecule of 9-BBN reacts with 47, which results in the regeneration of 45 together with the formation of a methoxide derivative (Scheme 12B).
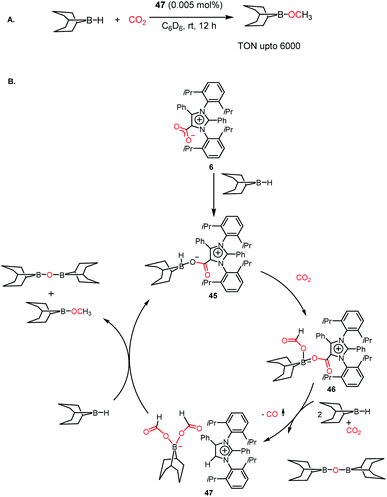 |
| | Scheme 12 (A) Catalytic reduction of CO2 in the presence of aNHC borondiformate 47. (B) Plausible mechanistic cycle for the reduction of CO2 using aNHC. | |
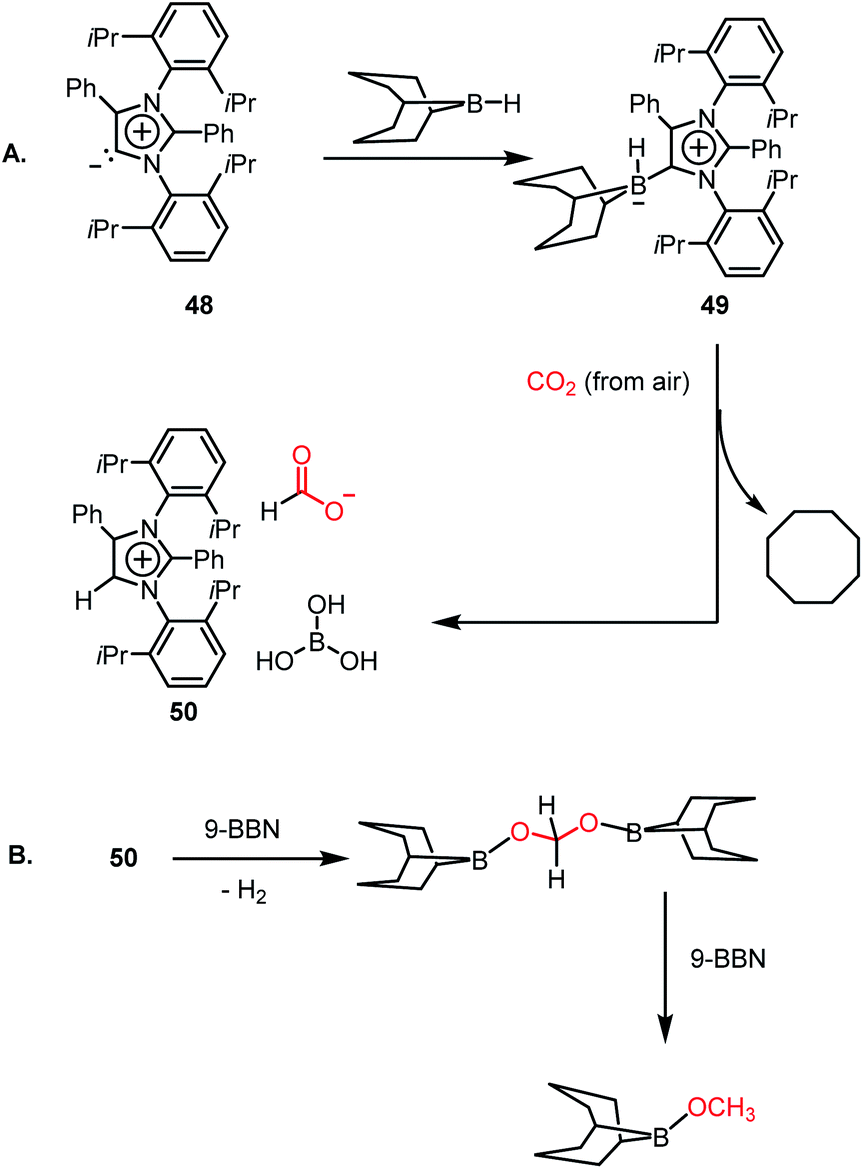
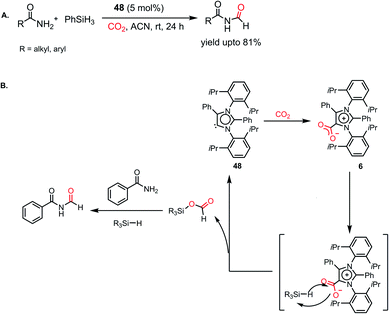
However, all these catalytic systems reported thus far demand commercially available, often greater than 99.99% pure CO2, which is available from a cylinder, to carry out the reduction. Hence, the development of catalytic systems that can capture CO2 from the air and perform the reduction is of great interest. In 2019, Mandal and co-workers reported the use of an abnormal N-heterocyclic carbene borane adduct (49) obtained from aNHC 48 by reacting it with 9-BBN to capture atmospheric CO2, and its reduction into formic acid and methanol under ambient conditions (Scheme 13A).62 Upon the exposure of adduct 49 in benzene-d6 to air overnight, it showed a sharp colour change from light yellow to green. 1H NMR spectroscopic analysis of the new compound showed two singlets at δ 8.53 and 8.55 ppm with the equal intensity, which were attributed to the formate anion and C5–H of the aNHC moiety, respectively indicating formation of 50. 13C NMR spectroscopic studies also supported the presence of a formate group, which is evident from the appearance of a resonance at δ 169.2 ppm. This observation clearly indicates the activation of atmospheric CO2. The 11B NMR spectrum of 50 showed a singlet at δ 20.8 ppm, supporting the presence of free boric acid, which was observed in the crystal lattice of 50 (Scheme 13A). The formation of boric acid may be attributed to hydrolysis of 9-BBN by the moisture present in air with the elimination of a cyclooctane molecule. X-ray studies illustrated that the exposure of CO2 to adduct 49 led to the incorporation of CO2 as a formate anion, resulting in the formation of 50 (Scheme 13A). Upon further treatment with NaOH solution, 50 formed sodium formate. On the other hand, in the presence of air, this aNHC–BBN adduct 50 underwent reduction with 10 equivalents of 9-BBN to form boronmethoxide together with the release of H2 gas (Scheme 13B). This work represents the first metal-free system where CO2 is captured from the air under a low concentration of CO2 (∼400 ppm) and further reduced to formate or methoxide.
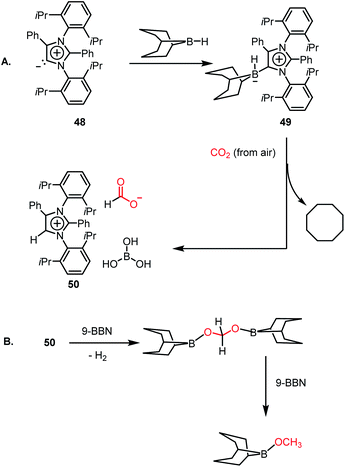 |
| | Scheme 13 Capture of CO2 from the air under ambient conditions by aNHC–BBN adduct 49 and its reduction to formate (A) and methoxide (B). | |
3.3. CO2 reduction by various other metal-free catalysts
Besides FLP- and NHC-mediated conversion, several other methods have been reported for the metal-free reduction of CO2. Among them, nitrogen bases are known for the catalytic reduction of CO2 into methoxyborane in the presence of a hydroborane. In 2014, Cantat and co-workers introduced TBD, Me-TBD (MTBD) and DBU and other nitrogen bases for the metal-free reduction of CO2 in the presence of 9-BBN or CatBH at room temperature to achieve a TON of up to 648 (Scheme 14A).63 NMR spectrometric monitoring of the reaction mixture revealed that over time, CO2 is reduced to borylformate, which undergoes further reduction to acetal, and subsequently to methoxyborane. For long time, TBD has been known to activate CO2, which forms a TBD–CO2 (3) adduct.27 Hence, the formation of the TBD–CO2 adduct is expected to be the first step in the reduction cycle. The stoichiometric reaction between 3 and 9-BBN in THF resulted in the formation of an adduct 51 together with other reduced products (Scheme 14B). X-ray study of 51 showed that the acidic NH proton of 3 is replaced with a dehydrido 9-BBN unit. 51 can be considered an intramolecular N/B FLP system captured with a CO2 molecule. Based on further control reactions, a plausible scheme was proposed, in which the CO2 molecule in 51 acts a Lewis base and coordinates with hydroborane to form adduct 52, which in turn facilitates the hydride transfer from borane to carbon, forming 53. In the presence of CO2, 53 is regenerates to 51, leaving boron formate, which is further reduced to methoxyborane. In the case of MTBD, it was found that the reaction proceeds with the activation of borane, followed by the incorporation of CO2.
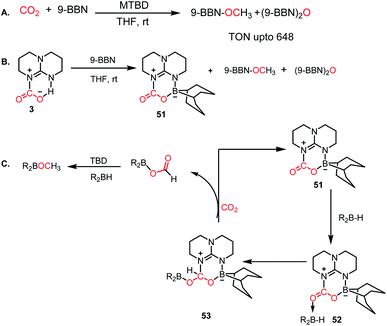 |
| | Scheme 14 (A) Catalytic CO2 reduction by MTBD. (B) Formation of TBD–CO2–BBN adduct 51. (C) Plausible mechanistic cycle. | |
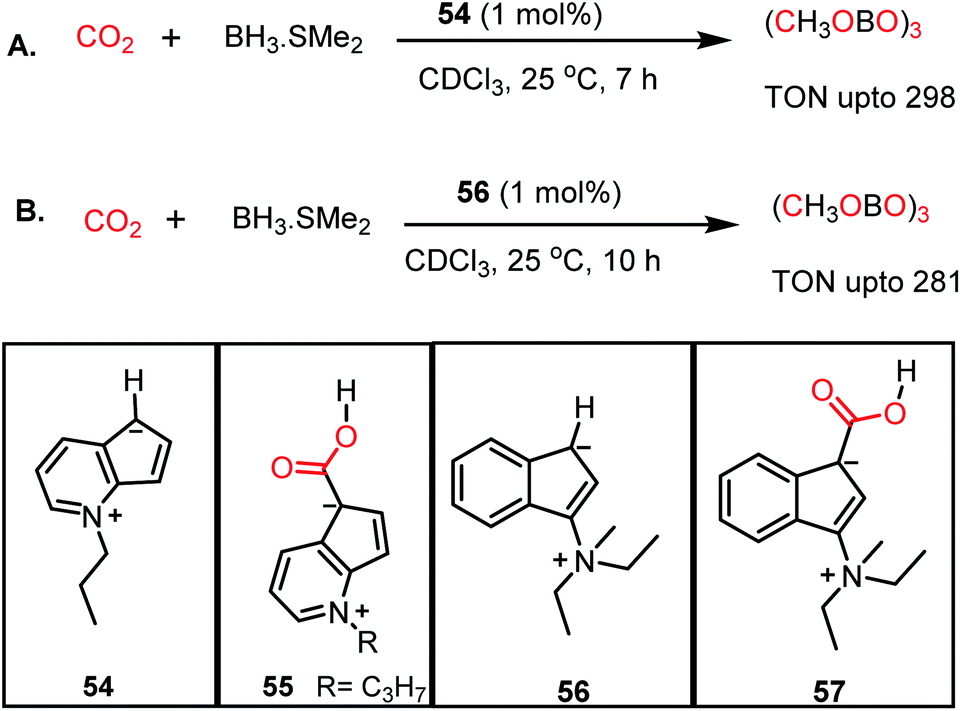
Similar to nitrogen bases, a carbanion such as N-methyl-4,5-diazafluorenide (54) was used for the catalytic reduction of CO2 by Song and co-workers.64 The reaction between 54 and CO2 resulted in the formation of a carboxylic acid adduct (55) (Scheme 15). The carboxylic acid proton of 55 showed a resonance at δ 11.02 ppm in DMSO-d6, whereas the 13C NMR spectrum showed a resonance at δ 166.4 ppm attributable to the carbon of CO2. The reduction of CO2 with various hydroboranes such as HBPin, 9-BBN, BH3·SMe2, and HBCat in the presence of 1 mol% 54 resulted in the formation of the corresponding methoxyboranes (Scheme 15A). In this case, the catalytic activity originates from the charged carbanionic form, which is actually in resonance with the uncharged form. The charged species can interact with the CO2 molecule by forming an adduct. To avoid the contribution from the uncharged form of resonance, the same group introduced zwitterionic complex 56 (Scheme 15B).65 Upon treatment of 56 with 1 atm CO2 under ambient conditions, an immediate colour change was observed, and the product was confirmed to be the CO2 adduct (57) by X-ray crystallography. The catalyst efficiently performed hydroboration of CO2 using 9-BBN or BH3·SMe2 under ambient conditions, resulting in a TON of 281 (Scheme 15B).
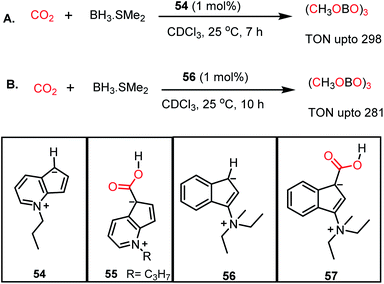 |
| | Scheme 15 Catalytic reduction of CO2 using a carbanions; (A) 54 and (B) 56. | |
In addition to nucleophiles, electrophilic reagents such as Lewis acids also can act as catalysts for CO2 reduction. For example, in 2016, Okuda and co-workers reported the use of a weak Lewis acid BPh3 for the selective hydrosilylation of CO2 to silyl formate.66 It should be noted that the selective reduction to silyl formate is a challenging task owing to the affinity of carbonyls to undergo over reduction. In the presence of PhMeSiH2 and 10 mol% BPh3 n acetonitrile, CO2 was selectively hydrosilylated to PhMeSiH(O2CH) at 40 °C (Scheme 16). It was proposed that BPh3 plays a dual role in this catalytic process, where it activates the hydride of silane by hydrogen bonding with the electrophilic boron centre. BPh3 also can activate CO2 by interaction between the B of BPh3 and O of CO2 (Scheme 16). This report highlighted that weaker non-fluorinated borane was efficient for the catalytic reduction of CO2 into formate, opening the possibility of using Lewis acid-based catalysts.
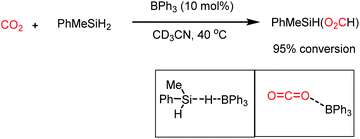 |
| | Scheme 16 Selective hydrosilylation of CO2 into silylformate using a Lewis acid (BPh3) as the catalyst. | |
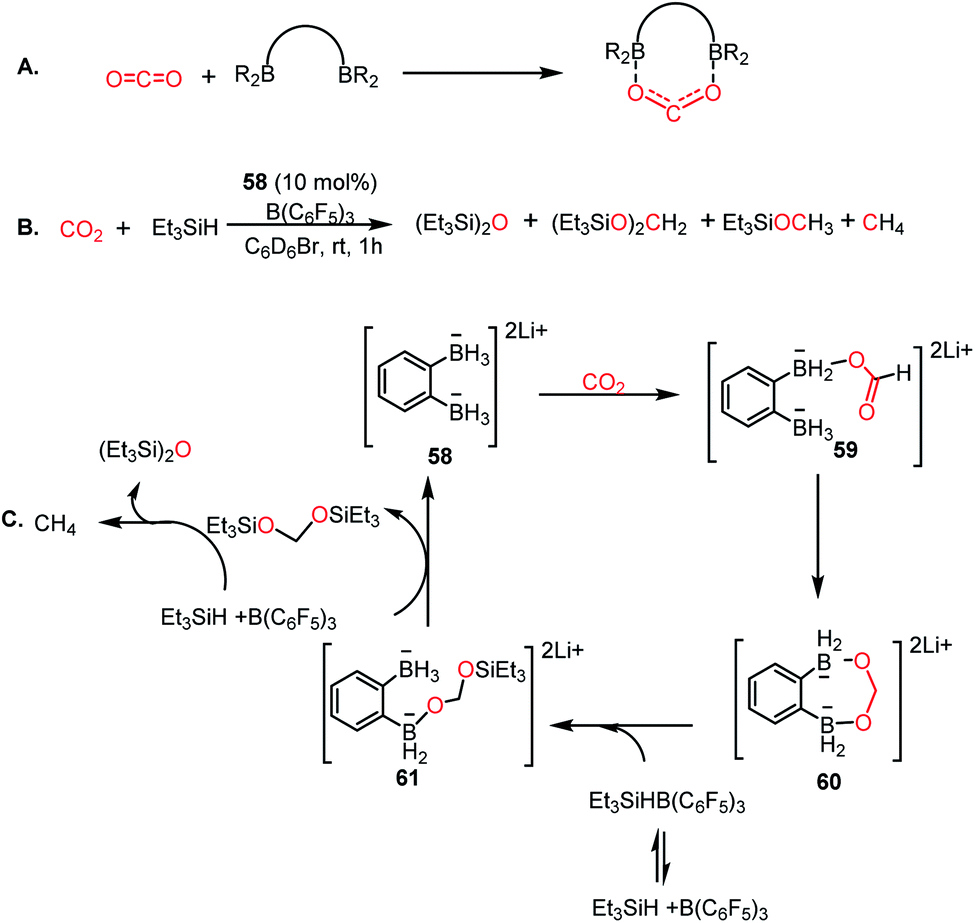
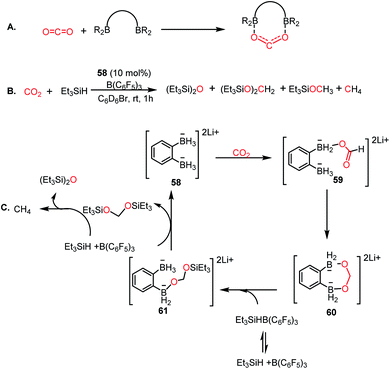
In 2018, Wegner and co-workers introduced the concept of using a bidentate bis(borane) for the catalytic reduction of CO2 into methane and methanol.67 The presence of two oxygen atoms in CO2 favours a bidentate mode of binding to the pair of intramolecularly integrated boron centres (Scheme 17A). Lithium o-phenylbisborate (58) was used as a catalyst in the presence of triethylsilane and B(C6F5)3 to reduce CO2 into a mixture of CH4, (Et3SiO)2CH2 and Et3SiOMe (Scheme 17B). The presence of CH4 was confirmed by the singlet at δ 0.14 ppm via1H NMR spectroscopy. The necessity of the bisborane catalyst was judged by a control reaction, which was performed using the monoborohyride LiPhBH3, leading to a dramatic reduction in the efficiency. The reaction was selective towards methanol when the reducing agent was changed to HBpin instead of silane. Based on various control experiments, a plausible mechanism was proposed (Scheme 17C). Catalyst 58 reacts with CO2 to form a precipitate, the NMR spectroscopic characterization of which was not possible because of its poor solubility. However, the IR spectroscopic analysis showed a strong absorption at 1592 cm−1, which accounts for the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group, suggesting the formation of a CO2 adduct with the catalyst. The next step involves intramolecular hydride transfer to generate formate 59 (Scheme 17C). This can further be reduced by the second BH3 group of 59 to form acetal 60 (Scheme 17C). Next, the silane cleaves the acetal to form a disilylacetal (Scheme 17C) via intermediate 61, which in turn can be reduced to methane.
O group, suggesting the formation of a CO2 adduct with the catalyst. The next step involves intramolecular hydride transfer to generate formate 59 (Scheme 17C). This can further be reduced by the second BH3 group of 59 to form acetal 60 (Scheme 17C). Next, the silane cleaves the acetal to form a disilylacetal (Scheme 17C) via intermediate 61, which in turn can be reduced to methane.
 |
| | Scheme 17 (A) Activation of CO2 using bidentate binding mode. (B) Catalytic reduction of CO2 using a bis-boronhydride ligand in the presence of a silane. (C) Plausible mechanistic cycle for the reduction of CO2. | |
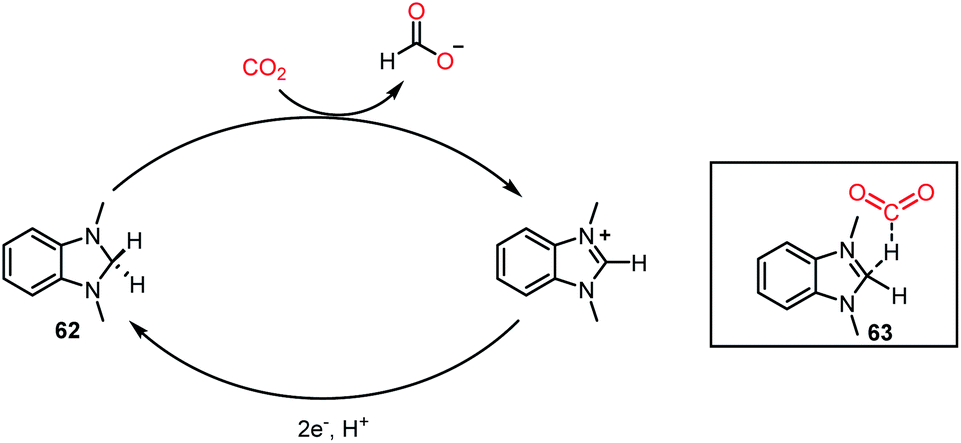
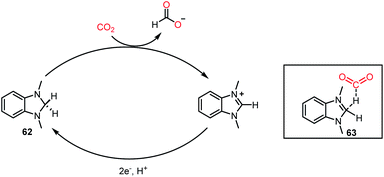
Recently, a different class of catalysts was introduced by Lim, Musgrave and co-workers, where a benzimidazole hydride was used for the activation and reduction of CO2.68 Thus far, a sacrificial reductant together with a catalyst was mandatory to carry out the reduction of CO2. In sharp contrast, an organic hydride donor was used to reduce CO2 to HCOO−, and the organic hydride donor could be regenerated via an electrochemical method, opening the possibility to act as a reductant and catalyst simultaneously. Organic hydrides can chemically reduce CO2 to HCOO−, but these reactions are limited to a stoichiometric version because of the limitation in regenerating the active catalyst. Interestingly, here, this challenge was addressed via an electrochemical method using metal electrodes. The reaction was performed using catalyst 62 in the presence of 20 psig of CO2, and KBF4 in DMSO-d6 at 50 °C for 18 h, resulting in 66% yield of formate (Scheme 18). It was anticipated from DFT calculations that the interaction between CO2 and hydride occurs through transition state 63. The presence of salts provides a suitable polar environment to stabilize the ionic products and shifts the equilibrium towards product formation. This report represented the first approach where an organic hydride donor was used in the reduction of CO2 to formate and integrated in the catalytic cycle by regenerating the active catalyst with the help of electrochemistry. Besides the catalytic systems described above, the reduction of CO2 has been performed using various other catalysts such as phosphazene,69 NaBH4,70 carbenoid compound,71 and formamidinates.72
 |
| | Scheme 18 Activation of CO2 by an organic hydride donor. | |
4. Metal-free reductive functionalization of CO2
In addition to the reduction of CO2 into formic acid, methanol and methane, the utilization of CO2 may find application by undergoing reduction together with functionalization by forming new C–N bonds. As it is known, the major products of CO2 functionalization without formal reduction include urea, polycarbonates, and salicylic acid. However, since the functionalization of CO2 does not require any formal reduction in the carbon centre, energy storage cannot be achieved efficiently. Thus, to maintain energy economy, Cantat and co-workers introduced a diagonal approach, in which CO2 undergoes simultaneous reduction and functionalization in the presence of a reducing agent and functionalizing reagent to form a new C–N bond.73 The major type of reductive functionalization involves the reduction of CO2 in the presence of an amine to synthesize various energy storage materials such as formamides, aminals and methylamines, which are the 2-, 4-, and 6-electron reduced products of CO2, respectively (Scheme 19). It is worth to mentioning that the synthesis of formamides via the reduction of CO2 in the presence of amines was achieved by Vaska and co-workers in 1988.74 Subsequently, various metal-free catalysts such as nitrogen bases, NHC, FLP, IL and others were introduced for this reductive functionalization of CO2, and the following sections describe these developments.
 |
| | Scheme 19 Reductive functionalization of CO2 with amines. | |
4.1 Formylation of amines using CO2
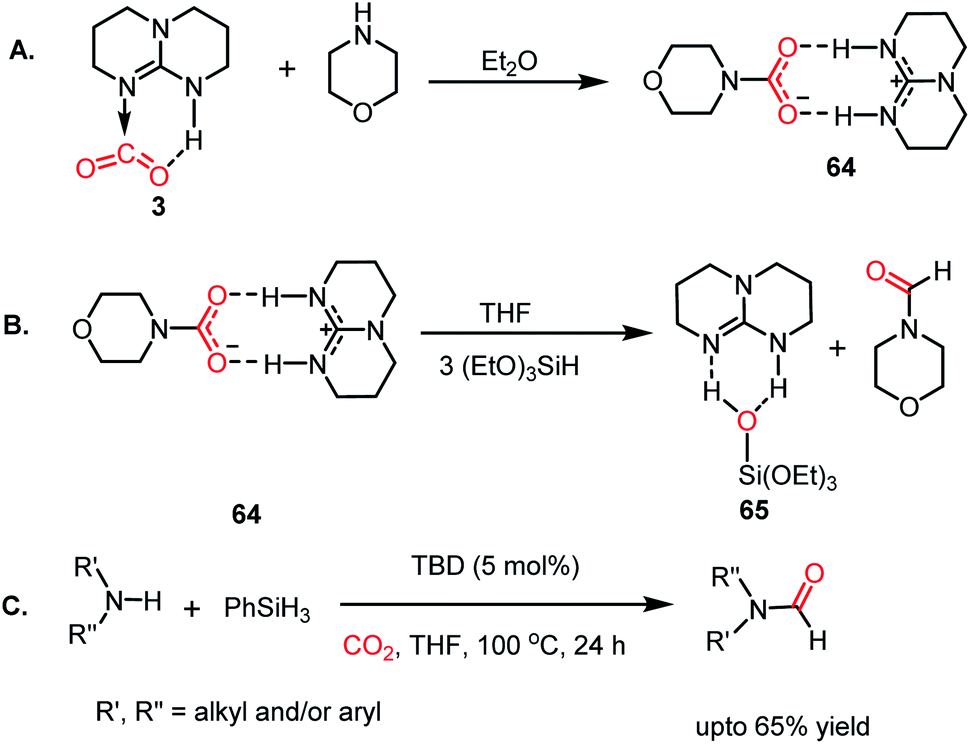
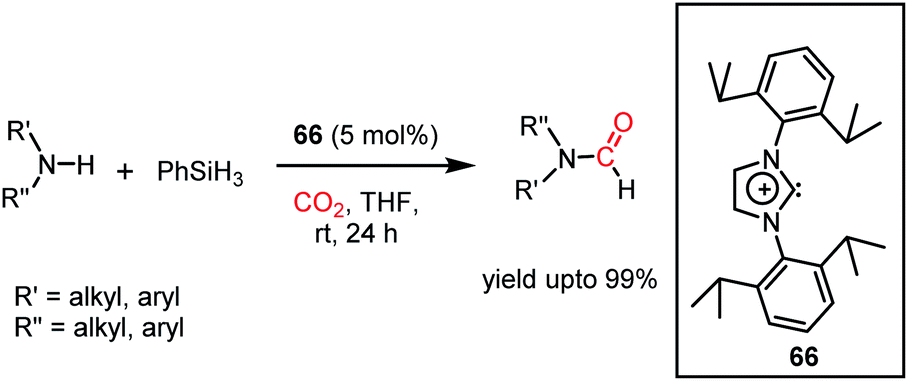
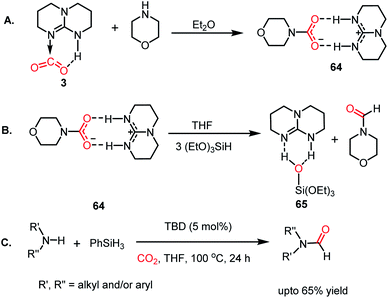
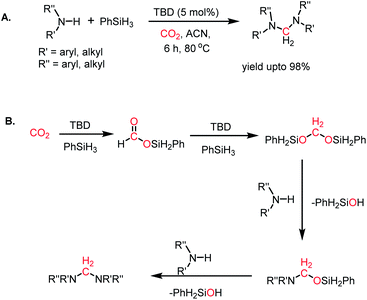
The formylation of amines using CO2 produces formamides. Formamides represent the 2e-reduction products of CO2 together with the formation of a new C–N bond, which are considered as one of the primary building blocks in organic synthesis. Traditionally, formamides are often synthesized from carbon monoxide under harsh reaction conditions.75 The use of CO can be effectively replaced by CO2 together with an appropriate reducing agent. In this regard, various transition metal catalysts have emerged using H2 as a reductant. However, the harsh reaction conditions and poor selectivity with the use of H2 as a reductant prompted the use of hydrosilanes as a milder alternative owing to their similar reduction potential.76 In addition, the use of efficient metal-free catalysts with mild reaction conditions is preferable over expensive transition metals. The last decade has witnessed the development of efficient metal-free catalysts for the formylation of amines using CO2, which are described herein. In 2012, Cantat and co-workers introduced TBD for the first metal-free catalytic reductive formylation of amines using a silane and CO2.73 Their preliminary computational study indicated that to carry out the functionalization of CO2 using silanes, a functionalizing group (such as morpholine) with greater affinity towards carbon and less affinity towards silicon is required. The reaction between a secondary amine such as morpholine and TBD–CO2 (3) in the presence of diethyl ether results in the formation of a hydrogen-bonded carbamate (64, Scheme 20A), in which CO2 is transferred from TBD to morpholine, as confirmed by X-ray crystallography. Further, the reaction was performed using carbamate 64 and three equivalents of (EtO)3SiH in THF, which resulted in the reduction of the carbon centre to form the corresponding formamide together with the formation of 65 (Scheme 20B). This result represents the first report on the reduction of carbamates using silane as a reducing agent. Here, silicon plays a dual role, in which it promotes the reduction of the carbon centre and facilitates the cleavage of the C–O bond of CO2. Subsequently, to develop a catalytic strategy for the reductive functionalization of amines, a series of control reactions was performed. Initially, different nitrogen bases such as TBD, MTBD, DBU, DABCO and DMAP were screened, where TBD exhibited the highest catalyitc efficiency. It was found that solvent-free conditions showed better activity. In the presence of 5 mol% TBD, CO2 and PhSiH3, morpholine was completely consumed to produce N-formylmorpholine at 100 °C in 24 h. Using the optimized reaction conditions, various amines underwent reductive formylation, which include aromatic and aliphatic amines to give up to 65% yield (Scheme 20C). This work represents the first report on the reductive functionalization of amines using CO2 under metal-free conditions. However, this system is specific to amines and high temperature (100 °C) is needed since TBD is not nucleophilic enough to activate organic silanes. This drawback was addressed by introducing a stronger nucleophilic NHC for the formylation of amines.77 Among the various NHC catalysts screened (IPr, IMes, s-IPr, s-IMes, Cl2-IPr and ItBu), IPr (66) showed the highest reactivity, which was over 2000 times greater than that for the formylation of morpholine using TBD, CO2, and PhSiH3 at rt (Scheme 21). The scope of the formylation of various amines was tested using the optimized reaction conditions, and it was observed that the reaction proceeded smoothly for primary and secondary aliphatic amines, aromatic amines, imines, hydrazines, and N-heterocycles. The NHC outperformed the yield of the desired product (87%) compared to that by TBD (10%) in the case of iPr2NH, which indicates that the steric crowding around the nitrogen centre becomes less significant due to the strong nucleophilicity of the NHC. To develop a cost effective and sustainable system, less expensive and abundant silanes were also screened, and consequently, polymethylhydrosiloxane (PMHS), which is a by-product of the silicon industry, emerged as an excellent choice, giving 90% N-formylmorpholine. Since PMHS is nontoxic and moisture stable, it is an ideal replacement for the expensive PhSIH3. PMHS displayed good reactivity towards aliphatic and aromatic amines, hydrazines and heterocycles to form N-formylated products. This work represented the first metal-free system using PMHS as a reducing agent for the formylation reaction with CO2.
 |
| | Scheme 20 (A) Formation of carbamate from TBD–CO2 adduct and morpholine. (B) Reduction of carbamate to formamide in the presence of a silane. (C) Optimized catalytic condition for the formylation of amines using TBD as a catalyst. | |
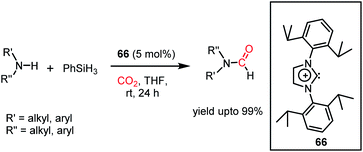 |
| | Scheme 21 Reductive functionalization of CO2 with amines using NHC 66 as a catalyst under ambient conditions. | |
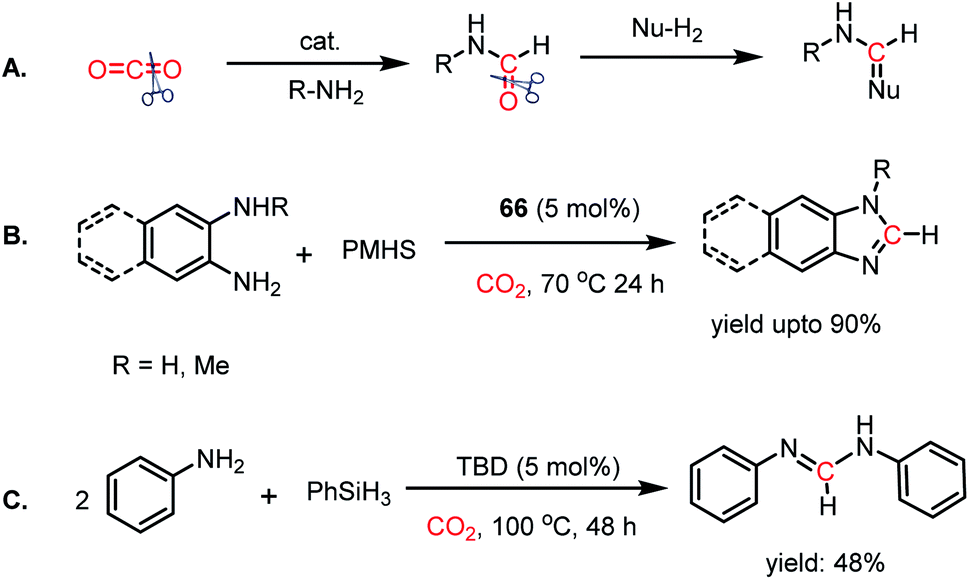
Thus far, the 2e-reductive functionalization of CO2 to form formylated products has been reported. However, the utilization of CO2 as a building block of C1 chemicals is possible when the absolute deoxygenation of CO2 occurs to prepare a wide array of chemicals. In 2013, Cantat and co-workers introduced a cascade reductive functionalization, which led to the complete deoxygenation of CO2.78 This resulted in the synthesis of various heterocycles, which have wide applications in the pharmaceutical industry. In the presence of IPr or TBD as a catalyst, CO2 underwent reductive functionalization with o-diamines, and subsequent intramolecular condensation resulted in the formation of benzimidazole, quinazolinone, formamidines or their derivatives. This one pot reaction may be viewed as “cut and paste” approach, where four C–O bonds of CO2 are chopped off to build four new bonds (CN, C–N and C–H, Scheme 22A). However, at room temperature, this reaction was not selective towards heterocycles and a mixture of mono and diformylated products was also formed together with heterocycles.
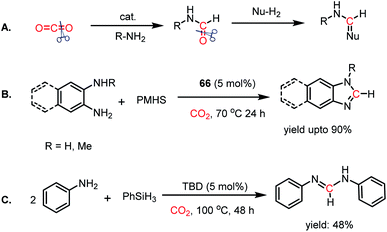 |
| | Scheme 22 (A) Deoxygenation of CO2via cascade reductive functionalization. (B) Reductive functionalization of CO2 using diamines and IPr (66) as a catalyst. (C) Deoxygenation of CO2via the four-component approach. | |
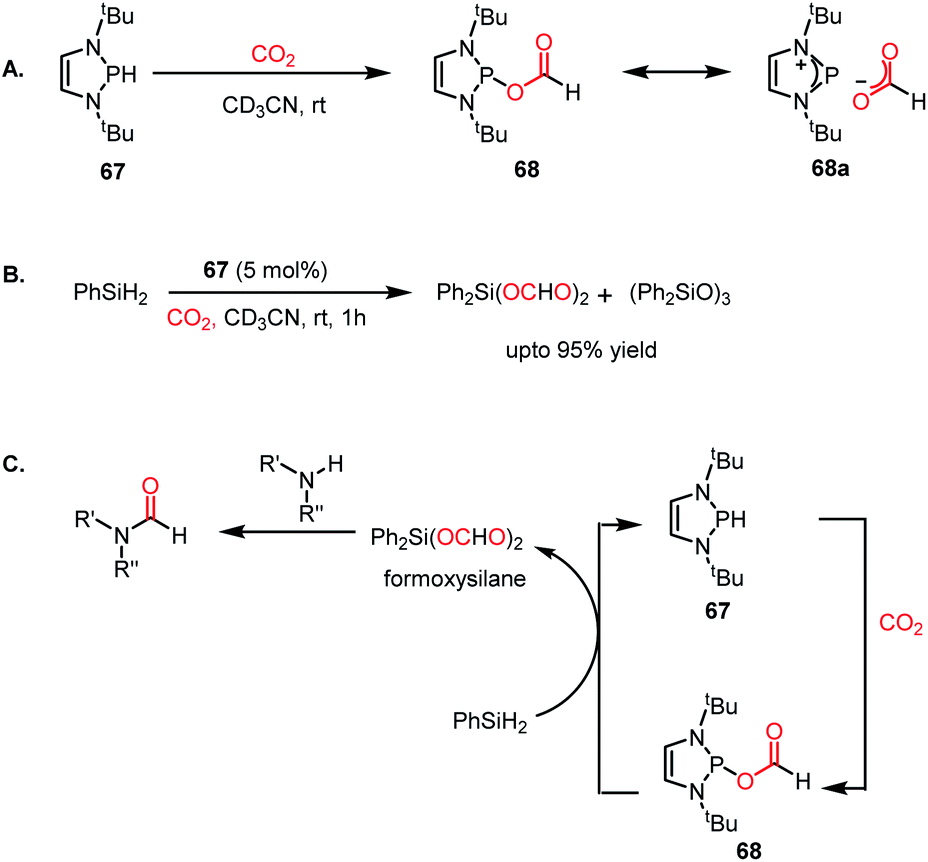
To address this problem, an elevated temperature (70 °C) was applied to achieve selectivity. In the presence of 5 mol% IPr (66) and 1 atm CO2, 1,2 diamines were efficiently transformed into benzimidazole derivatives with a yield of up to 90% using PMHS as a reducing agent at 70 °C for 24 h (Scheme 22B). Further, a 4-component reaction was accomplished, where two functionalizing agents were linked by a linker as a result of intermolecular reactions (Scheme 22C). In 2015, Kinjo et al. introduced a phosphorus-based catalyst for the reductive functionalization of CO2.79 In their work, 1,3,2-diazaphospholene (67) was introduced for the activation of CO2. In the presence of CO2 in acetonitrile-d3 under ambient conditions, 67 underwent a rapid colour change from yellow to deep brown. The peak in the 31P NMR spectrum showed a downfield shift from δ 57.8 ppm to 111.5 ppm, whereas the 1H and 13C NMR spectra showed new peaks at δ 8.10 ppm and 164.2 ppm, indicating the presence of a formate (68, Scheme 23A), which was further established by X-ray crystallography. The formation of 68 represents the first hydrophosphination of CO2. The down-fielded 31P NMR chemical shift and X-ray structure revealed that the P–O bond distance (1.808(1) Å) is 11% longer than that of the typical P–O single bond (1.63 Å), suggesting a significant contribution from the zwitterionic form (68a) in resonance with 68 (Scheme 23A). This ionic property of the bond that induces zwitterionic character perhaps facilitates the formate group transfer to the appropriate receptor. The reaction between 67 and a silane in a CO2 atmosphere led to the formation of a formoxysilane (Scheme 23B). Encouraged by this result, the reductive functionalization of CO2 in the presence of an amine was attempted. It was found that the combination of 5 mol% of 67 with diphenylsilane in the presence of acetonitrile at rt for 4 h resulted in the successful formylation of a variety of amines, which include both primary and secondary amines, with 61–99% yield. A plausible cycle was proposed, in which the first step involves the incorporation of CO2 in the P–H bond, which in turn transfers the formate group to silane to form formoxysilane and regenerates 67. Formoxysilane upon reaction with amines forms formamides and other by-products (Scheme 23C). This work represents the first hydrophosphination of CO2.
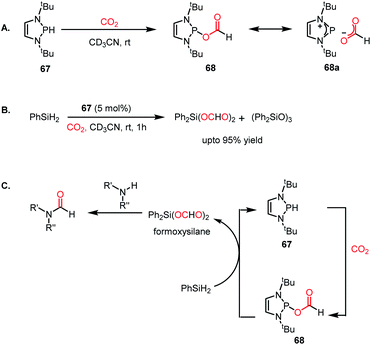 |
| | Scheme 23 (A) Activation of CO2 by 1,3,2-diazaphospholene 67. (B) Hydrosilylation of CO2 in the presence of 67. (C) Plausible mechanistic cycle for the formylation of amines using 67 as a catalyst. | |
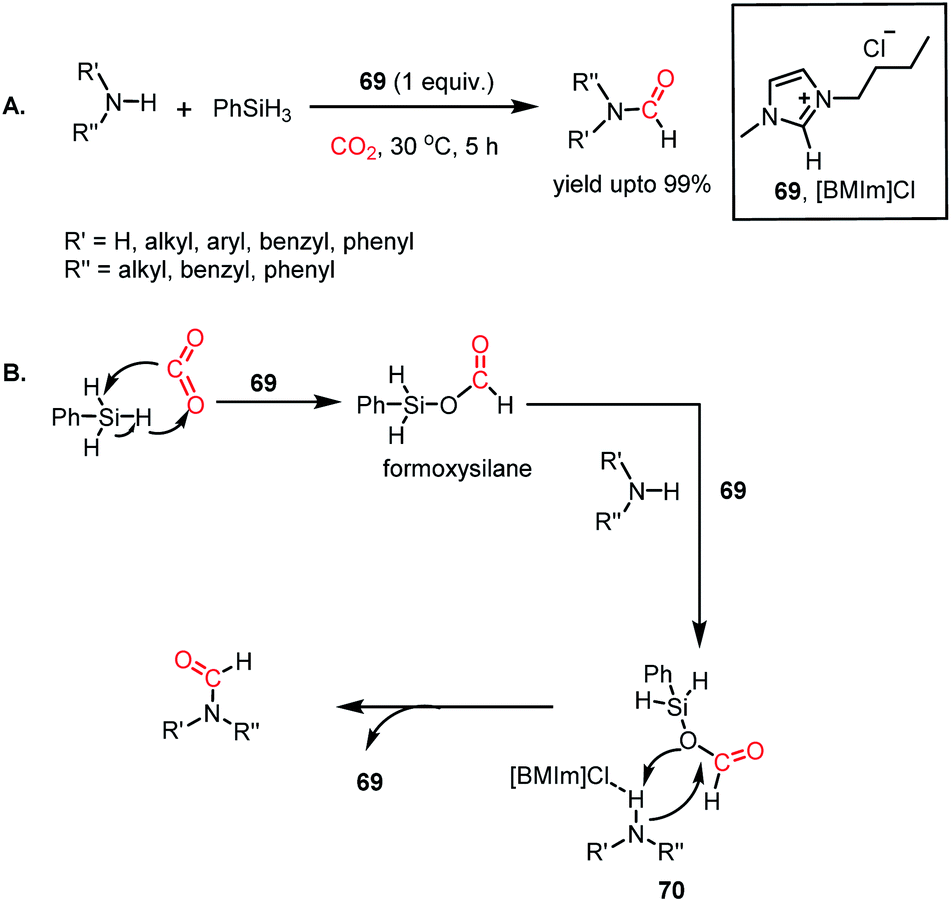
In 2015, Liu and co-workers introduced ionic liquids for the formylation of amines for the first time.80 It was found that imidazolium-based ILs could perform the N-formylation of various amines with CO2 under ambient conditions using PhSiH3 as a reductant. Consequently, 1-butyl-3-methylimidazolium chloride ([BMIm]Cl, 69) emerged as an efficient catalyst for the formylation of various aromatic and aliphatic amines, resulting in up to 99% yield (Scheme 24A). The reaction between 69 and phenylsilane displayed a noticeable change in the NMR spectra, which indicates that the first step involves the activation of the Si–H bond of phenylsilane by 69. Further, the reaction between phenylsilane and 69 in a CO2 atmosphere led to a signal in the 13C NMR spectrum at δ 163.0 ppm and δ 8.27 ppm in the 1H NMR spectrum. These signals were attributed to the formation of a formoxysilane, which is the key intermediate of the reaction. This result suggests that the activation of phenylsilane by 69 favours facile insertion of CO2. Earlier, Wang and co-workers reported this kind of silane activation for the NHC-mediated hydrosilylation of CO2.59 On the other hand, the reaction between 69 and N-methylaniline experienced a change in signal for the N–H proton of the amine. This observation may be attributed to the hydrogen bonding between the N–H proton of the amine and the anionic fragment of IL 69.
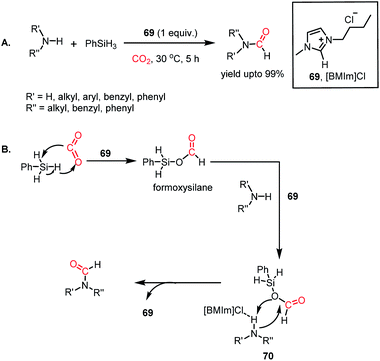 |
| | Scheme 24 (A) Ionic liquid (69)-mediated amine formylation with CO2. (B) Plausible mechanistic steps for amine formylation using 69. | |
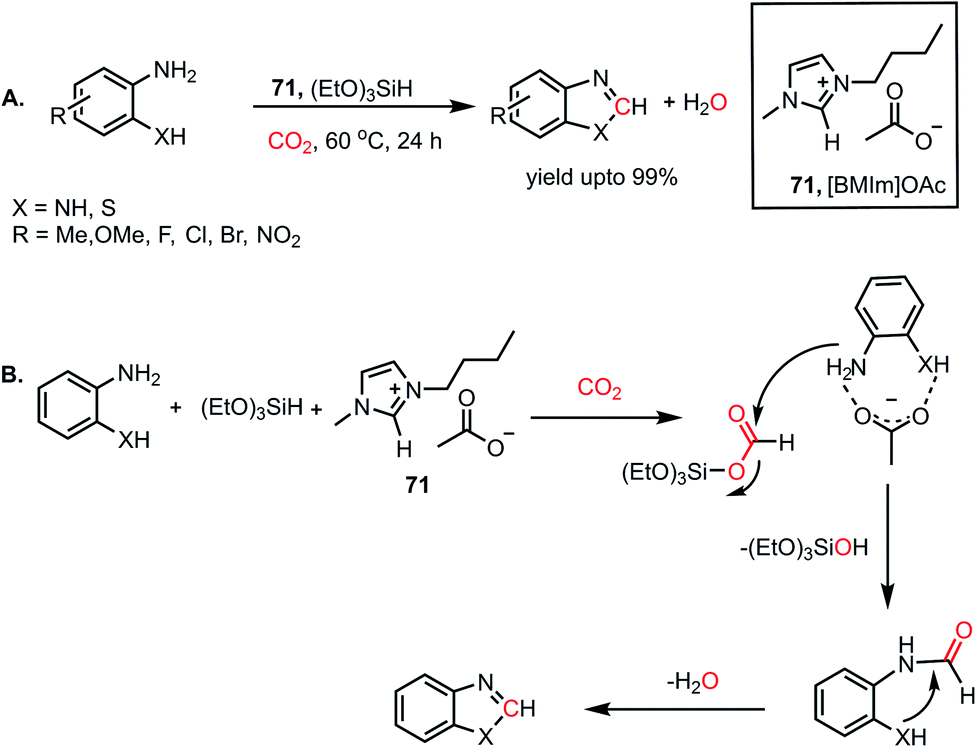
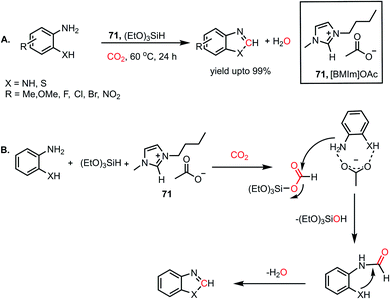
Combining these observations, it can be concluded that 69 plays a dual role for the activation of phenylsilane, resulting in the insertion of CO2 and activation of amines via hydrogen bonding. Based on these experimental evidence, a possible cycle was proposed, where the first step involves activation of the Si–H bond of phenylsilane by 69, which leads to the insertion of CO2 to form formoxysilane. Further, the N–H bond of the amine is involved in hydrogen bonding with 69, and thus the amine nitrogen participates in a nucleophilic attack on the carbon atom of formoxysilane to yield the desired product via70 (Scheme 24B). This report represents the first ionic liquid-mediated reductive functionalization of CO2. In 2015, the same group introduced ionic liquids in cascade reductive functionalization using aminophenols for the construction of new C–S bonds to form various benzothiazole derivatives.81 Sulphur-containing organic compounds exhibit numerous applications in the agriculture, pharmaceutical and chemical industries.82 Benzothiazoles in particular are useful as enzyme inhibitors, anti-inflammatory agents, antioxidants and fluorescent materials. Hence there is a high demand for developing cost-effective methods for the synthesis of various benzothiazoles. Accordingly, an efficient protocol was developed using an ionic liquid, 1-butyl-3-methylimidazolium acetate ([Bmim][OAc], 71), for the cyclization of 2-aminophenols in the presence of CO2 and hydrosilane under ambient conditions. Using (EtO)3SiH and 0.5 MPa CO2, different substituted 2-aminophenols and diamines were transformed into various benzothiazoles (up to yield 99%) and benzimidazoles (up to 96%) at 60 °C (Scheme 25A). The NMR spectroscopic analysis of the control reactions revealed strong hydrogen bonding interaction between 71, 2-aminophenol and silane. A plausible mechanistic scheme was proposed, in which the first step involves activation of CO2 by the ionic liquid to the carboxylate adduct, which further interacts with the activated silane to form formoxysilane. Subsequently, formoxysilane undergoes nucleophilic attack by the hydrogen-bonded amine to form the formylated product, which in turn takes part in intramolecular condensation to obtain the desired product (Scheme 25B). Here, IL 71 acts as a tri-functional catalyst, which activates CO2, amine and silane to develop an efficient strategy for the synthesis of various heterocycles.
 |
| | Scheme 25 (A) Ionic liquid 71-mediated reductive functionalization of amines with CO2 for the synthesis of heterocycles. (B) Plausible mechanistic steps for the reductive functionalization of diamines by IL 71. | |
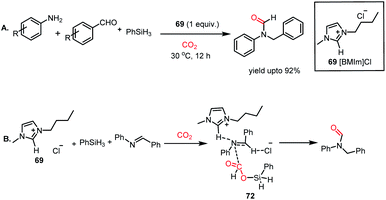
Next, the ionic liquid-mediated reductive functionalization of CO2 resulted in the synthesis of N,N-disubstituted formamides (NNFAs). NNFA represents a class of formamides with wide applications in the pharmaceutical field, but their synthesis remains a challenging task. In 2017, Liu and co-workers further extended their ionic liquid-mediated reductive functionalization of CO2 strategy towards the synthesis of NNFAs.83 In their work, 1-butyl-3-methylimidazolium chloride ([BMIm]Cl, 69)-mediated three-component coupling between CO2, primary amines and aldehydes was developed to synthesize NNFA in the presence of phenylsilane as a reducing agent under ambient conditions. 69 was used to perform the coupling between aryl amines and aryl aldehydes followed by reductive formylation in the presence of 10 atm CO2 and PhSiH3 at 30 °C to produce unsymmetrical N,N formamides in 92% yield (Scheme 26A). 69 was reused consecutively five times without loss in its activity and the process was easily scaled to gram scale. The scope of this reaction was further extended to aliphatic amines and aldehydes to synthesize various N,N-formamides with alkyl substitution. In the presence of 69, CO2 interacts with phenylsilane to form formoxysilane, which in turn reacts with benzylideneaniline, an in situ generated product by reaction between amine and aldehyde, resulting in the formation of the final product via72 (Scheme 26B).
 |
| | Scheme 26 (A) Ionic liquid 69-mediated synthesis of N,N-formamides. (B) Plausible mechanistic cycle for the synthesis of NNFAs. | |
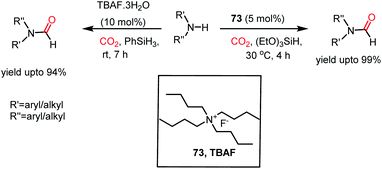
Thus far, we have seen how both the cation and anion of ILs are required simultaneously for the reductive functionalization of CO2. In contrast, tetrabutylammonium salt as a catalyst necessitates only the involvement of its anion, whereas its cation only serves as a counterion. The steric crowding of the tetrabutylammonium cation minimizes the cation–anion interaction and allows the anion to be available in the medium to take part in reaction. In 2016, Dyson and co-workers introduced tetrabutylammonium fluoride as an efficient catalyst for the N-formylation of amines with CO2 in the presence of phenylsilane under ambient conditions.84 It was found that in the presence of 10 mol% TBAF·3H2O, various aromatic and aliphatic amines underwent formylation using phenylsilane and 0.1 MPa CO2 at room temperature to achieve a yield of up to 94% (Scheme 27). Moreover, this was demonstrated for the synthesis of 11C-labelled drug molecules using 11CO2. In the same year, He and co-workers also reported the use of TBAF for the N-formylation of aromatic and aliphatic amines using triethoxysilane at 30 °C to attain a maximum yield of 99% (Scheme 27).85
 |
| | Scheme 27 Fluoride anion-mediated N-formylation of amines. | |
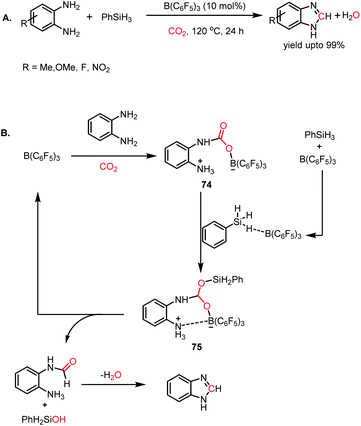
To date, we have seen that the nucleophilicity of catalysts plays a pivotal role in activating CO2 and its catalytic reductive functionalization. Besides nucleophilic reagents, the report by Sun and co-workers first described the electrophile-mediated catalytic reductive functionalization of CO2.86 B(C6F5)3 was introduced as a catalyst for the synthesis of benzimidazoles from o-phenylenediamines. In the first step, the interaction of o-phenylenediamine with B(C6F5)3 generates an FLP based on the amine/borane pair. In the presence of 10 mol% B(C6F5)3 and 1 MPa of CO2, various aryl diamines underwent formylation followed by condensation to form the corresponding benzimidazoles (Scheme 28A). A plausible mechanism was proposed, in which first B(C6F5)3 interacts with diamine to form an N/B FLP and CO2 interacts with the in situ formed FLP to form a CO2 adduct (74). Secondly, B(C6F5)3 activated PhSiH3 which in turn coordinated with 74 to form intermediate 75 which lead to the release of formamide and regeneration of B(C6F5)3. Formamide underwent subsequent condensation to release benzimidazole as the final product (Scheme 28B). In addition, several other methods were also employed for the N-formylation of amines using CO2 such as thiazolium carbene,87 N-heterocyclic olefins,88 tetrabutylammonium-carboxylates,89 acetates,90 phosphorus ylides,91 γ-valerolactone,92 and ionic liquids.93,94 Moreover, it was found that the catalytic formylation of amines is also possible in the presence of polar solvents such as DMF95 and DMSO.96 This can be attributed to the high solubility of CO2 and the enhanced basicity of amines in polar aprotic solvents.
 |
| | Scheme 28 (A) B(C6F5)3-mediated catalytic N-formylation of o-diamines. (B) Plausible mechanistic cycle for the catalytic N-formylation of diamines. | |
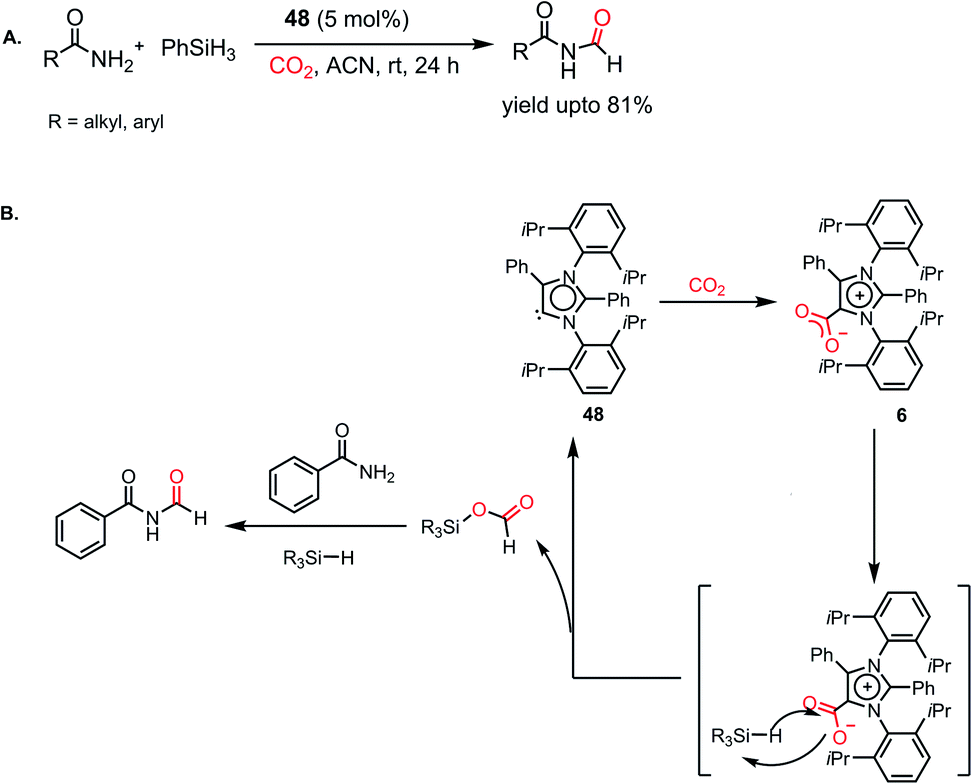
All the above-mentioned reports required an amine functional group to carry out the reduction. The high basicity of amines makes them suitable candidates for the reductive functionalization of CO2. On the contrary, amides represent very weak bases and less reactive in comparison to amines because of the conjugation of their nitrogen lone pair of electrons with the C=O group. In 2018, Mandal and co-workers accomplished the N-formylation of primary amides by CO2 using an abnormal N-heterocyclic carbene for the first time.97 Previously, DFT calculations showed that the HOMO of aNHC is higher in energy compared to isomeric NHC, which makes it more nucleophilic,26 and hence can be involved in hydrogen bonding with the amide proton, facilitating its interaction with silane. It was found that 5 mol% aNHC (48) resulted in the reductive functionalization of primary amides with 1 atm CO2 using phenylsilane. Various aromatic and aliphatic amides underwent facile N-formylation with a yield of up to 81% (Scheme 29A). Moreover, this protocol was successful in synthesizing the precursors for two natural products, alatamide and lansiumamide, which exhibit strong larvicidal activity. The reaction initiates with the formation of the aNHC–CO2 adduct (6). The stoichiometric reaction between 6 and triphenylsilane in acetonitrile in the absence of CO2 displayed a singlet at δ 8.71 ppm in 1H NMR spectroscopy and a signal at δ 168.1 ppm in 13C NMR spectroscopy, which confirmed the formation of a formoxysilane intermediate. Upon interaction with amide, formoxysilane transfers the formyl group to the amide molecule in the presence of another unit of silane to yield the desired product (Scheme 29B).
 |
| | Scheme 29 (A) aNHC-mediated N-formylation of primary amides. (B) Plausible mechanistic cycle for the formylation of amides. | |
4.2 Aminals from CO2
Aminals represent the 4e-reduced species of CO2, in which carbon is reduced to C(0) from C(+4) via the C(+2) species. Arresting the C(0) species is a challenging task since it tends to undergo over reduction into C(−2) species. Therefore, only a few reports are currently available that address the metal-free reductive functionalization of CO2 to aminals.98,99 In 2015, Cantat and co-workers reported the first metal-free catalyst for the 4e-reductive functionalization of CO2.98 In the presence of 10 mol% TBD and CO2, secondary aromatic amines underwent reduction by PhSiH3 at 80 °C in acetonitrile, resulting in aminals with 98% yield (Scheme 30A).
 |
| | Scheme 30 (A) Catalytic reductive functionalization of CO2 with amines to form aminals using TBD. (B) Mechanistic steps for aminal formation. | |
Moreover, this strategy was extended to obtain unsymmetrical aminals using two different amines. The reaction proceeds via the hydrosilylation of CO2 in the presence of TBD to form silyl acetals. The silyl acetal further undergoes sequential nucleophilic attack by amines to form the desired product (Scheme 30B).
4.3 Catalytic N-methylation by CO2
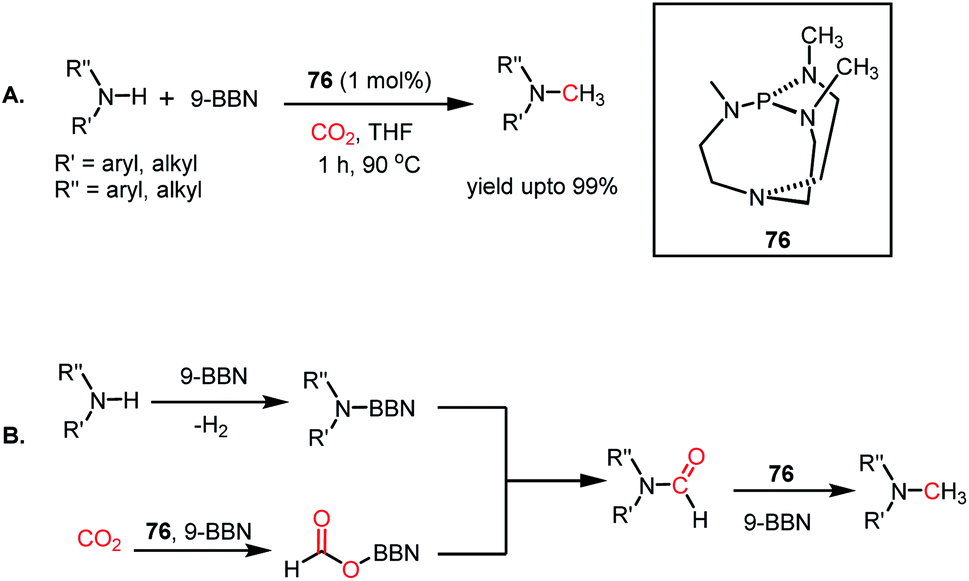
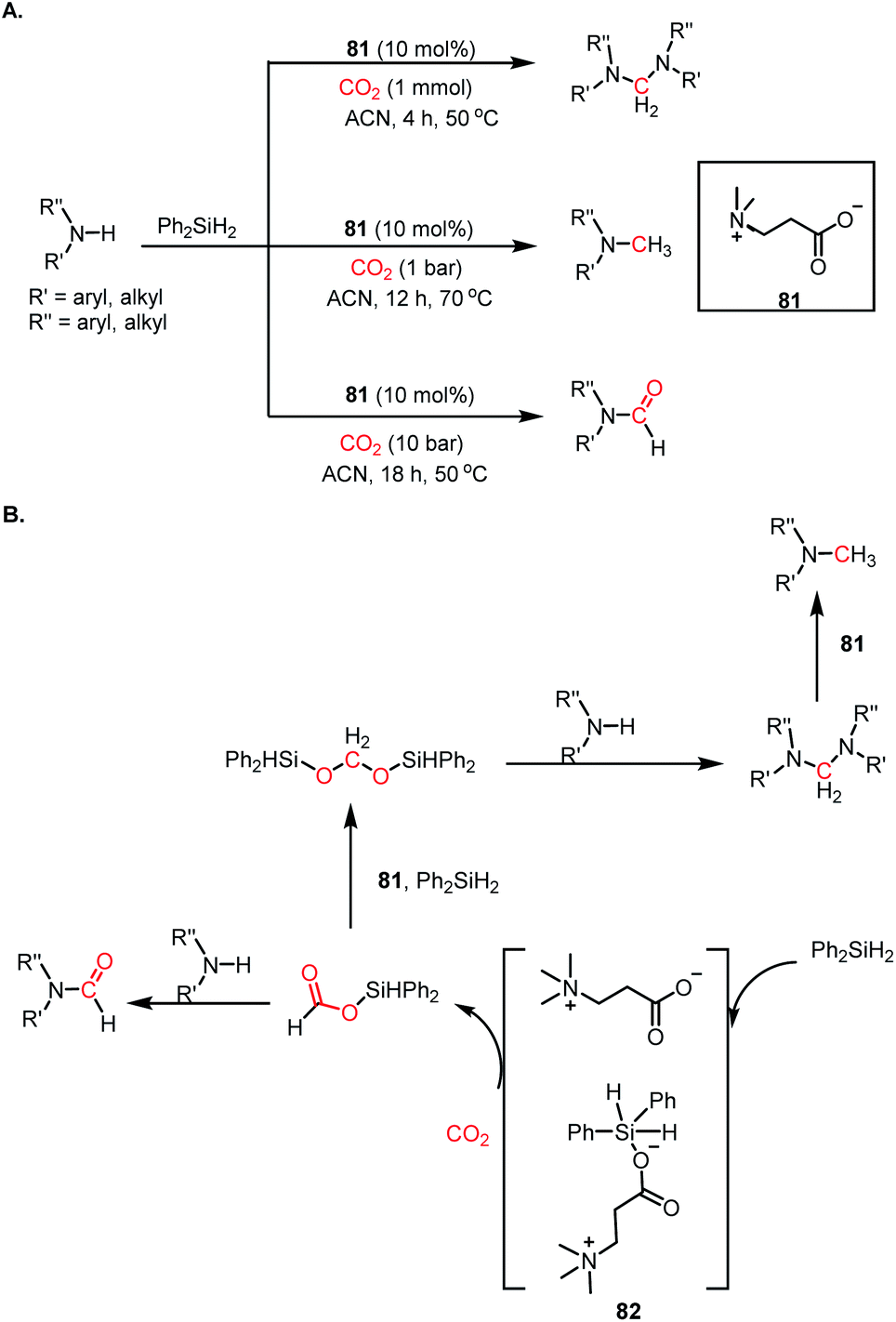
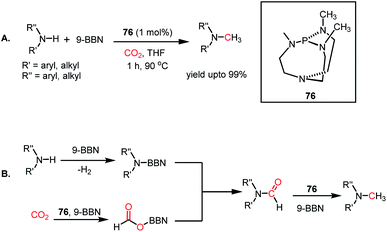
The use of CO2 as a reagent for methylation represents a 6e-reduction process. CO2 can undergo reductive functionalization with amines to formylamines, which upon further reduction, leads to methylamines. Earlier, the N-methylation of amines by CO2 used various transition metal catalysts such as Ru, Cu, and Zn.100–102 In addition to the use of expensive metals, these systems required harsh reaction conditions, such as high pressure and temperature. In 2014, Cantat and co-workers introduced the first metal-free catalytic methylation of amines using CO2, in which a phosphine was used as the catalyst (Scheme 31A).103 The affinity of phosphine toward borane to form a phosphine–borane adduct facilitates the hydride transfer process, and hence hydroboranes were chosen as the reductants for this transformation. Proazaphosphatrane (76) is known to have superior basicity over classical phosphines owing to the pπ–dπ back bonding from the lone pairs of electrons from nitrogen centres. 76 catalysed the N-methylation of aromatic and aliphatic secondary amines in the presence of CO2 and 9-BBN at 90 °C with yield up to 99%. Furthermore, in the presence of excess borane, primary amines also underwent N-methylation. This study was further extended to the one-pot reduction and reductive functionalization, in which the nitro group undergoes reduction to form an amine in situ, which is further reduced to methylamine in the presence of CO2. It was found that amines undergo reduction by 9-BBN to afford N-borylated amine via the elimination of H2 (Scheme 31B). Simultaneously, the formoxyborane formed by the reduction of CO2 in the presence of 9-BBN and the catalyst interacts with N-borylated amine to form N-formyl-amine, forming a new C–N bond. N-Formyl-amine undergoes further reduction to yield the methylamine. The elevated temperature favours the reaction to undergo reductive functionalization over reduction.
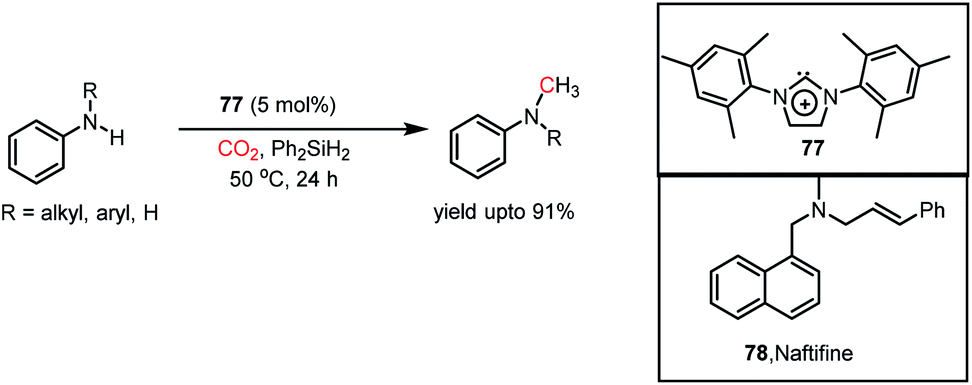
 |
| | Scheme 31 (A) Methylation of amines by proazaphosphatrane 76 in the presence of CO2. (B) Plausible mechanistic steps for the catalytic methylation of amines by 76 using CO2. | |
In the same year, Dyson and co-workers introduced N-heterocyclic carbenes as metal-free catalysts for the N-methylation of amines using CO2.104 It was found that IMes (77) could perform the N-methylation of various aromatic, heteroaromatic, and aliphatic amines with CO2 in the presence of Ph2SiH2 as a reducing agent at 50 °C with a yield of up to 91% (Scheme 32). This catalytic protocol was efficient in preparing an antifungal agent naftifine 78 with an overall yield of 58% (Scheme 32). Moreover, this method exhibited good functional group tolerance, in which NO2, CN, ester, double and triple bonds were well-tolerated.
Instead of bivalent carbene possessing a pair of electrons, it was thought carbodicarbene (CDC), having an additional pair of electrons on the C(0) centre, may have enhanced nucleophilicity for the activation of CO2. In 2008, the first CDC molecules were synthesized,105,106 and subsequently, these CDC molecules were successfully introduced with suitable metal ions in catalysis for various organic transformations.107 In 2015, Ong and co-workers developed a series of unsymmetrical carbodicarbenes.108 These CDCs were utilized as metal-free catalysts in the N-methylation of amines using CO2 in the presence of 9-BBN (Scheme 33A). In the presence of 10 mol% CDC 79 and 9-BBN, primary and secondary amines underwent N-methylation using CO2 in toluene at 100 °C to give a yield of up to 90%. The stoichiometric reaction between 79 and 9-BBN resulted in the formation of the CDC–BBN adduct 80, which was characterised via X-ray crystallography. It was found that the hydrogen of 9-BBN migrated to the carbodicarbene, which was also confirmed by the signal at δ 5.26 ppm in the 1H NMR spectrum. The catalytic reaction performed using 80 also yielded the desired product in 90% yield, which indicates that 80 is a catalytic intermediate. In the presence of CO2, the 9-BBN unit present in 80 migrates to CO2 to form boronformate, which reacts with N-borylated amine formed in situ to produce N-formylated amines. In the presence of 79, the N-formylated amine undergoes further reduction to yield N-methylated amines (Scheme 33B).
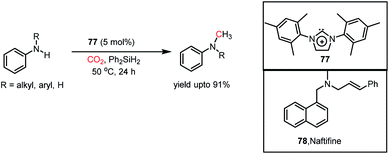 |
| | Scheme 32 Catalytic methylation of amines by CO2 using 77. | |
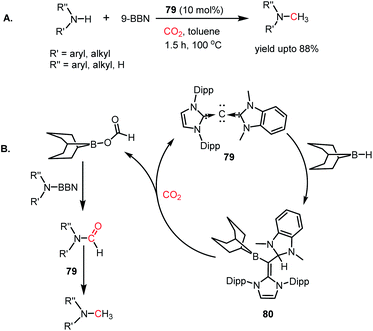 |
| | Scheme 33 (A) Methylation of amines using CO2 in the presence of carbodicarbene 79. (B) Plausible mechanistic cycle for the methylation of amines. | |
In addition to nucleophiles, electrophiles were also used for the methylation of amines. In 2015, Liu and co-workers introduced B(C6F5)3 for the reductive functionalization of CO2 to form methylamines.109 5 mol% B(C6F5)3 in the presence of phenylsilane resulted in the N-methylation of primary and secondary amines in 93% yield using a CO2 pressure of 0.5 MPa (Scheme 34A). The reaction proceeds via a synergistic pathway, in which B(C6F5)3 plays a triple role in activating all the reagents. B(C6F5)3 activates phenylsilane via a non-covalent interaction between the hydride and B centre (Scheme 34B). On the other hand, the presence of polar C–F bonds in B(C6F5)3 makes it suitable for electrostatic interaction with the carbon centre of CO2, leading to its activation. Simultaneously, amine can interact with B(C6F5)3 owning to the strong electrostatic interaction between nitrogen and boron, which was evident from NMR spectroscopy. These synergistic activations led to the formylation of amine, which in turn underwent reduction to generate methylamines. In addition to these reports, the catalytic methylation of amines was also carried out using DBU.110 Lei and co-workers reported that an aprotic polar solvent such as DMF can also promote the N-methylation reaction.111
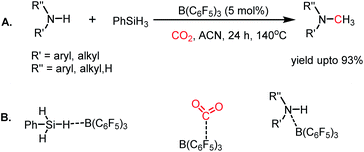 |
| | Scheme 34 (A) Methylation of amines by CO2 in the presence of B(C6F5)3. (B) Triple role of B(C6F5)3 for the activation of all the reagents. | |
The previous section described how different metal-free catalysts were used to enable the reductive functionalized products. However, none of the them could perform the hierarchical reduction of CO2, in which a single catalyst can control the kinetics to obtain a range of reduced products sequentially. In this regard, in 2017, He and co-workers introduced glycine betaine (GB) 81 as a metal-free catalyst, which represents a zwitterionic salt consisting of tetraammonium cation and carboxylate anion.112 By regulating the CO2 pressure, 81 successfully produced formamides or aminals or methylamine. It was found that in the presence of 10 bar of CO2 at 50 °C, amines underwent N-formylation, whereas 1 bar CO2 at 70 °C gave methylamines. Aminals were formed when CO2 was restricted to one equiv. with respect to amine in a closed system at 50 °C (Scheme 35A). It was proposed that the first step involves the nucleophilic attack by the oxygen centre of the catalyst to the silicon centre of hydrosilane to form a hypervalent silicon species (82) (Scheme 35B). Next, the active hydride inserts a molecule of CO2 to form a silylformate, regenerating the catalyst. At a high pressure of CO2 (10 bar), amines interact with silylformates to form the formylated product. The excess pressure of CO2 enhances the rate of formation of silylformate, consuming all the silanes, and hence no further reduction is possible. In the presence of excess silane, silylformate undergoes further reduction to form silylacetal, which in the presence of amine is transformed to aminals via condensation. Aminals may be transformed into methylamines in the presence of additional silane and catalyst (Scheme 35B). Subsequently, Nguyen et al. also reported the hierarchical reductive functionalization of CO2 with amines using N,N,N′,N'-tetramethylguanidine (TMG) as the catalyst with less control over the selectivity.113
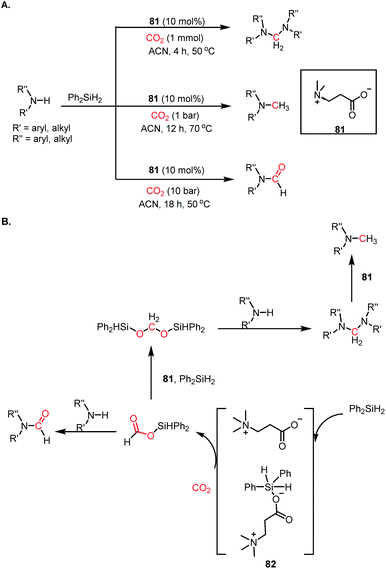 |
| | Scheme 35 (A) Hierarchical reductive functionalization of CO2 with amines using 81. (B) Plausible mechanistic cycle of the hierarchical reductive functionalization of CO2. | |
5. Future outlook and conclusions
During the last decade, we have witnessed the emergence of metal-free catalysts as efficient alternatives to transition metal-based catalysts for CO2 reduction. The stoichiometric activation of CO2 to catalytic reduction has come a long way, and thus, herein, we attempted to showcase this journey. Specifically, there are a few methods available for the catalytic reduction of CO2 into various chemicals and fuels on the laboratory scale under completely metal-free conditions. Moreover, these reductive transformations take place under very mild conditions. This is attributed to the combination of metal-free catalysts with silane or borane having optimal reduction potential to reduce the C–O bond. However, to translate these successes from the bench to market, one must pay serious attention to a number of factors. The marketability of these reduction processes will ultimately depend on their competitiveness between the market values of these products set by the petrochemical industry, which can be only addressed by making the process sustainable. Obviously, the catalytic reduction of thermodynamically inert C1 feedstock such as CO2 for the production of fuels and chemicals is a significant challenge in catalysis for several reasons.
Firstly, the choice of the hydride donor or the reductant is very important, which is largely driven by its commercial availability, cost, and reactivity together with its selectivity and ease of handling. In this regard, Cantat in his excellent review discussed various hydride donors and their advantages/disadvantages in reduction chemistry.114 Accordingly, dihydrogen, which is cheap and obtained through fossil technology, has been extensively utilized on the industrial scale, but its use for CO2 reduction generally requires harsh reaction conditions and transition metal-based catalysts, making this process economically less attractive. Thermodynamically, the reduction of CO2 producing C–H bonds requires only low energy since the redox potential of CO2 couples falls within the range of 0.17 to −0.47 V vs. NHE (25 °C; pH = 0).114 Thus, the reduction of CO2 does not require a strong reductant such as the commercially available LiAlH4 having quite a negative redox potential of E0 (Al3+, H2/AlH4−) = −1.78 V (vs. NHE).115 This negative redox potential leads to a large overpotential, and thus a high energy cost. Consequently, the use of common reducing agents such as LiAlH4 and NaBH4 in the large-scale reduction of CO2 is not very attractive. In addition, their use is not atom-economic, which leads to the formation of a stoichiometric amount of waste.
On the other hand, hydrosilane or hydroborane as a reductant is very attractive from both thermodynamic and kinetic viewpoints. Their redox potential falls just in the optimum range (−0.6 V to 0 V (E0(B(OMe)3/HB(OMe)2) = −0.54 V and E0((Me3Si)2O)/Me3SiH) = −0.1 V vs. NHE),114 and matches very well with the required energy input for C–O bond reduction. Moreover, they are commercially available. Especially, polymethylhydrosiloxane (PMHS), which is a byproduct of the silicon industry, and tetramethyldisiloxane (TMDS) are cheap and non-toxic with great ease in handling since they are stable in moisture and air. The optimal reduction potential of these reductants brings significant advantages in achieving enhanced chemoselectivity. However, the major pitfall of the hydrosilylation/hydroboration method arises from the formation of strong Si–O and B–O bonds, driving the reactions towards the forward direction. The formation of oxidized products such as siloxanes or boroxanes is the major drawback, making the process non-atom economic. The currently available recycling process of these siloxanes or boroxanes to hydrosilanes or hydroboranes is chemically highly energy intensive. Consequently, hydrosilanes and hydroboranes are considered as disposable hydrides. In this regard, an electrochemical approach over the chemical route may be attractive, leading to the electro-organo-recycling of these hydrides. The utilization of silanes or boranes in CO2 reduction chemistry will remain in its infancy unless cost-efficient methods are developed to regenerate them (Scheme 36).
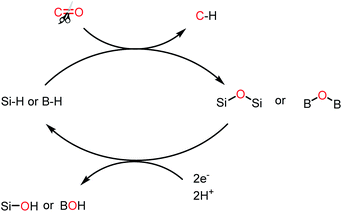 |
| | Scheme 36 Use of silane and borane for the reduction of CO2 will be sustainable if they can be recycled in an energy efficient way. | |
Additionally, for translating the metal-free catalytic CO2 reduction into an industrial process, one must heterogenize the process by immobilizing the catalyst on a solid support. The heterogenization of the metal-free catalyst will make the process sustainable in terms of recyclability. In this regard, a few heterogenous catalysts have been developed thus far. In 2016, Zhang and co-workers reported a solid poly-N-heterocyclic carbene for the reductive functionalization of CO2 in the presence of amine.116 Later, two more reports emerged using porous organic polymers117 and nitrogen-doped graphene nanosheets.118 These initial reports on heterogeneous processes using metal-free catalysts are certainly encouraging towards translating this laboratory process to a scaled-up process.
Finally, as a note of caution, one must pay attention to the environmental sustainability by considering the life cycle assessment (LCA)119,120 of the CCR process. The practical potential of this process relies on its overall energy efficiency, including both the capture of CO2 and its utilization, which require energy. The supply of energy may lead to indirect CO2 emission. Thus, the anticipated environmental advantages of CCR may not be guaranteed always, for example, a tediously accomplished CCR process may finally be environmentally less sustainable than a classical fossil-based route. Thus, whether or not a specific CCR process is favorable from the viewpoint of environmental sustainability, one must consider its life cycle assessment (LCA), which takes into account the entire life cycle of products and processes, from the extraction of raw materials to the final disposal of waste, with respect to environmental impacts such as fossil resource depletion, global warming, and toxicities.
Moreover, as mentioned in the beginning, only a limited extent of total CO2 emissions are currently utilized in chemical conversion to value-added products, although this can be potentially expanded. To achieve this goal, one must improve the scope of chemicals available from the reduction of CO2, which presently remains very narrow in comparison to that currently available from fossil technology. For example, the basic functional groups encountered frequently in organic chemistry, such as ketones, amides and esters, featuring a carbon atom that is reduced, are unavailable from carbon dioxide currently. Thus, this chemical reduction is certainly welcome in the future to broaden the horizon of CO2 reduction chemistry. To translate these initial successes into practical applications, multidisciplinary approaches combining metal-free catalysts immobilized on a suitable support together with the recycling of hydrosilanes or hydroboranes through electrochemical methods can offer an appealing strategy in the catalytic reduction chemistry of CO2.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We thank the STARS Program (Grant No. STARS/APR2019/CS/473/FS) of India. SP thanks DST-Inspire for research fellowship.
References
-
(a)
IEA, Annual change in global energy-related CO2 emissions, 1900-2020, IEA, Paris, https://www.iea.org/data-and-statistics/charts/annual-change-in-global-energy-related-CO2-emissions-1900-2020 Search PubMed;
(b)
https://www.iea.org/articles/the-impact-of-the-covid-19-crisis-on-clean-energy-progress
;
(c)
Global CO2 emissions in 2019, IEA, Paris, https://www.iea.org/articles/global-CO2-emissions-in-2019 Search PubMed.
- C. Figueres, C. Le Quéré, A. Mahindra, O. Bäte, G. Whiteman, G. Peters and D. Guan, Nature, 2018, 564, 27–30 CrossRef CAS , Intergovernmental Panel on Climate Change. Global Warming of 1.5 °C (IPCC, 2018).
-
(a) D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS;
(b) H. De Coninck and S. M. Benson, Annual Review of Environment and Resources, 2014, 243–270 CrossRef;
(c) T. Bruhn, H. Naims and B. Olfe-Kräutlein, Environ. Sci. Policy, 2016, 60, 38–43 CrossRef CAS.
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS.
- C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS.
- S. Bontemps, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2014, 136, 4419–4425 CrossRef CAS.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- F.-G. Fontaine, M.-A. Courtemanche and M.-A. Légaŕe, Chem.–Eur. J., 2014, 20, 2990–2996 CrossRef CAS.
- F.-G. Fontaine and D. W. Stephan, Current Opinion in Green and Sustainable Chemistry, 2017, 3, 28–32 CrossRef.
- X.-F. Liu, X. Y. Li, C. Qiao and L.-N. He, Synlett, 2018, 29, 548–555 CrossRef CAS.
- H. Zhou and X. Lu, Sci. China: Chem., 2017, 60, 904–911 CrossRef CAS.
- M. Hulla and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 1002–1017 CrossRef CAS.
- Y. Zhang, T. Zhang and S. Das, Green Chem., 2020, 22, 1800–1820 RSC.
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS.
- F. J. Fernández-Alvarez, A. M. Aitani and L. A. Oro, Catal. Sci. Technol., 2014, 4, 611–624 RSC.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- D. H. Gibson, Coord. Chem. Rev., 1999, 185–186, 335–355 CrossRef CAS.
- J. R. Ellis, Nature, 2010, 463, 164–165 CrossRef.
- M. Iwatani, K. Kudo, N. Sugita and Y. Takezaki, Sekiyu Gakkaishi, 1978, 21, 290–296 CrossRef CAS.
- E. R. Pérez, M. O. da Silva, V. C. Costa, U. P. Rodrigues-Filho and D. W. Franco, Tetrahedron Lett., 2002, 43, 4091–4093 CrossRef.
- E. R. Pérez, R. H. A. Santos, M. T. P. Gambardella, L. G. M. d. Macedo, U. P. Rodrigues-Filho, J.-C. Launay and D. W. Franco, J. Org. Chem., 2004, 69, 8005–8011 CrossRef.
- C. Villiers, J.-P. Dognon, R. Pollet, P. Thuéry and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–3468 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. J. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
-
(a) N. Kuhn, E. Niquet, M. Steimann and I. Walker, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1999, 54, 427–433 CAS;
(b) H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112–113 RSC.
- E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 326, 556–559 CrossRef CAS.
- G. Kuchenbeiser, M. Soleilhavoup, B. Donnadieu and G. Bertrand, Chem.–Asian J., 2009, 4, 1745–1750 CrossRef CAS.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS.
- C. M. Wang, H. M. Luo, D. E. Jiang, H. R. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978–5981 CrossRef CAS.
- H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325–329 CrossRef CAS.
- G. Wittig and E. Benz, Chem. Ber., 1959, 92, 1999–2013 CrossRef CAS.
- W. Tochtermann, Angew. Chem., Int. Ed. Engl., 1966, 5, 351–371 CrossRef CAS.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef.
- P. G. Jessop, F. Jo and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS.
- D. H. Gibson, Chem. Rev., 1996, 96, 2063–2096 CrossRef CAS.
- A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskela, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS.
- A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS.
- A. Berkefeld, W. E. Piers, M. Parvez, L. Castro, L. Maron and O. Eisenstein, Chem. Sci., 2013, 4, 2152–2162 RSC.
- M.-A. Courtemanche, M.-A. Légaŕe, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2013, 135, 9326–9329 CrossRef CAS.
- M.-A. Courtemanche, M.-A. Légaŕe, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708–10717 CrossRef CAS.
- R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS.
- T. Wang and D. W. Stephan, Chem. Commun., 2014, 50, 7007–7010 RSC.
- J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS.
- T. Wang and D. W. Stephan, Chem.–Eur. J., 2014, 20, 3036–3039 CrossRef CAS.
- N. von Wolff, G. Lefevre, J.-C. Berthet, P. Thuéry and T. Cantat, ACS Catal., 2016, 6, 4526–4535 CrossRef CAS.
- A. Ramos, A. Antiñolo, F. Carrillo-Hermosilla, R. Fernández-Galán, A. Rodríguez-Diéguez and D. García-Vivó, Chem. Commun., 2018, 54, 4700–4703 RSC.
- R. Boese, R. Köster and M. Yalpani, Z. Naturforsch., B: J. Chem. Sci., 1994, 49, 1453–1458 CAS.
- M. A. Courtemanche, A. P. Pulis, É. Rochette, M. A. Légaré, D. W. Stephan and F. G. Fontaine, Chem. Commun., 2015, 51, 9797–9800 RSC.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS.
- F. Huang, G. Lu, L. Zhao, H. Li and Z.-X. Wang, J. Am. Chem. Soc., 2010, 132, 12388–12396 CrossRef CAS.
- S. N. Riduan, J. Y. Ying and Y. Zhang, ChemCatChem, 2013, 5, 1490–1496 CrossRef CAS.
- S. C. Sau, R. Bhattacharjee, P. K. Vardhanapu, G. Vijaykumar, A. Datta and S. K. Mandal, Angew. Chem., Int. Ed., 2016, 55, 15147–15151 CrossRef CAS.
- S. C. Sau, R. Bhattacharjee, P. K. Hota, P. K. Vardhanapu, G. Vijaykumar, R. Govindarajan, A. Datta and S. K. Mandal, Chem. Sci., 2019, 10, 1879–1884 RSC.
- C. Das Neves Gomes, E. Blondiaux, P. Thuéry and T. Cantat, Chem.–Eur. J., 2014, 23, 7098–7106 CrossRef.
- Y. Yang, M. Xu and D. Song, Chem. Commun., 2015, 51, 11293–11296 RSC.
- Y. Yang, L. Yan, Q. Xie, Q. Liang and D. Song, Org. Biomol. Chem., 2017, 15, 2240–2245 RSC.
- D. Mukherjee, D. F. Sauer, A. Zanardi and J. Okuda, Chem.–Eur. J., 2016, 22, 7730–7733 CrossRef CAS.
- Z. Lu, H. Hausmann, S. Becker and H. A. Wegner, J. Am. Chem. Soc., 2015, 137, 5332–5335 CrossRef CAS.
- C.-H. Lim, S. Ilic, A. Alherz, B. T. Worrell, S. S. Bacon, J. T. Hynes, K. D. Glusac and C. B. Musgrave, J. Am. Chem. Soc., 2019, 141, 272–280 CrossRef CAS.
- M.-A. Courtemanche, M.-A. Légaré, E. Rochette and F.-G. Fontaine, Chem. Commun., 2015, 51, 6858–6861 RSC.
- K. Fujiwara, S. Yasuda and T. Mizuta, Organometallics, 2014, 33, 6692–6695 CrossRef CAS.
- S. Y.-F. Ho, C.-W. So, N. Saffon-Merceron and N. Mézailles, Chem. Commun., 2015, 51, 2107–2110 RSC.
- W. Huang, T. Roisnel, V. Dorcet, C. Orione and E. Kirillov, Organometallics, 2020, 39, 698–710 CrossRef CAS.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS.
- S. Schreiner, J. Y. Yu and L. Vaska, J. Chem. Soc., Chem. Commun., 1988, 602–603 RSC.
- C. J. Gerack and L. McElwee-White, Molecules, 2014, 19, 7689–7713 CrossRef.
- R. Walsh, Acc. Chem. Res., 1981, 14, 246–252 CrossRef CAS.
- O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934–2937 CrossRef CAS.
- O. Jacquet, C. D. N. Gomes, M. Ephritikhine and T. Cantat, ChemCatChem, 2013, 5, 117–120 CrossRef CAS.
- C. C. Chong and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 12116–12120 CrossRef CAS.
- L.-D. Hao, Y.-F. Zhao, B. Yu, Z.-Z. Yang, H.-Y. Zhang, B.-X. Han, X. Gao and Z.-M. Liu, ACS Catal., 2015, 5, 4989–4993 CrossRef CAS.
- X. Gao, B. Yu, Z.-Z. Yang, Y.-F. Zhao, H.-Y. Zhang, L.-D. Hao, B.-X. Han and Z.-M. Liu, ACS Catal., 2015, 5, 6648–6652 CrossRef CAS.
- Y. Xi, B. Dong, E. J. McClain, Q. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Peterson and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 4657–4661 CrossRef CAS.
- Z.-G. Ke, L.-D. Hao, X. Gao, H.-Y. Zhang, Y.-F. Zhao, B. Yu, Z.-Z. Yang, Y. Chen and Z.-M. Liu, Chem.–Eur. J., 2017, 23, 9721–9725 CrossRef CAS.
- M. Hulla, F. D. Bobbink, S. Das and P. J. Dyson, ChemCatChem, 2016, 8, 3338–3342 CrossRef CAS.
- X.-F. Liu, R. Ma, C. Qiao, H. Cao and L.-N. He, Chem.–Eur. J., 2016, 22, 16489–16493 CrossRef CAS.
- Z. Zhang, Q. Sun, C. Xia and W. Sun, Org. Lett., 2016, 18, 6316–6319 CrossRef CAS.
- S. Das, F. D. Bobbink, S. Bulut, M. Soudan and P. J. Dyson, Chem. Commun., 2016, 52, 2497–2500 RSC.
- V. B. Saptal and B. M. Bhanage, ChemSusChem, 2016, 9, 1980–1985 CrossRef CAS.
- X. F. Liu, C. Qiao, X. Y. Li and L. N. He, Green Chem., 2017, 19, 1726–1731 RSC.
- T. Murata, M. Hiyoshi, M. Ratanasak, J.-ya. Hasegawa and T. Ema, Chem. Commun., 2020, 56, 5783–5786 RSC.
- H. Zhou, G. X. Wang, W. Z. Zhang and X. B. Lu, ACS Catal., 2015, 5, 6773–6779 CrossRef CAS.
- J. Song, B. Zhou, H. Liu, C. Xie, Q. Meng, Z. Zhang and B. X. Han, Green Chem., 2016, 18, 3956–3961 RSC.
- X.-Y. Li, H.-C. Fu, X.-F. Liu, S.-H. Yang, K.-H. Chen and L.-N. He, Catal. Today DOI:10.1016/j.cattod.2020.01.030.
- W. F. Zhao, X. P. Chi, H. Li, J. He and S. Yang, Green Chem., 2019, 21, 567–577 RSC.
- T.-X. Zhao, G.-W. Zhai, J. Liang, P. Li, X.-B. Hu and Y.-T. Wu, Chem. Commun., 2017, 53, 8046–8049 RSC.
- H. Lv, Q. Xing, C. Yue, Z. Lei and F. Li, Chem. Commun., 2016, 52, 6545–6548 RSC.
- P. K. Hota, S. C. Sau and S. K. Mandal, ACS Catal., 2018, 8, 11999–12003 CrossRef CAS.
- X. Frogneux, E. Blondiaux, P. Thuery and T. Cantat, ACS Catal., 2015, 5, 3983–3987 CrossRef CAS.
- X.-Y. Li, S.-S. Zheng, X.-F. Liu, Z.-W. Yang, T.-Y. Tan, A. Yu and L.-N. He, ACS Sustainable Chem. Eng., 2018, 6, 8130–8135 CrossRef CAS.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS.
- O. Santoro, F. Lazreg, Y. Minenkov, L. Cavallo and C. S. J. Cazin, Dalton Trans., 2015, 44, 18138–18144 RSC.
- E. Blondiaux, J. Pouessel and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 12186–12190 CrossRef CAS.
- S. Das, F. D. Bobbink, G. Lanurenczy and P. J. Dyson, Angew. Chem., Int. Ed., 2014, 53, 12876–12879 CrossRef CAS.
- C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206–3209 CrossRef CAS.
- A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210–3214 CrossRef.
- C. Pranckevicius, L. Fan and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 5582–5589 CrossRef CAS.
- W. Chen, J. Shen, T. Jurca, C. Peng, Y. Lin, Y. Wang, W. Shih, G. P. A. Yap and T. Ong, Angew. Chem., Int. Ed., 2015, 54, 15207–15212 CrossRef CAS.
- Z. Yang, B. Yu, H. Zhang, Y. Zhao, G. Ji, Z. Ma, X. Gao and Z. Liu, Green Chem., 2015, 17, 4189–4193 RSC.
- G. Li, J. Chen, D. Y. Zhu, Y. Chenand and J.-B. Xia, Adv. Synth. Catal., 2018, 360, 2364–2369 CrossRef CAS.
- H.-Y. Niu, L.-J. Lu, R.-Y. Shi, C.-W. Chiang and A.-W. Lei, Chem. Commun., 2017, 53, 1148–1151 RSC.
- X. Liu, X. Li, C. Qiao, H. Fu and L. He, Angew. Chem., Int. Ed., 2017, 56, 7425–7429 CrossRef CAS.
- R. L. Nicholls, J. A. McManus, C. M. Rayner, J. A. Morales-Serna, A. J. P. White and B. N. Nguyen, ACS Catal., 2018, 8, 3678–3687 CrossRef CAS.
- C. Chauvier and T. Cantat, ACS Catal., 2017, 7, 2107–2115 CrossRef CAS.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- S. N. Riduan, S. Nurhanna, J.-Y. Ying and Y.-G. Zhang, J. Catal., 2016, 343, 46–51 CrossRef CAS.
- H. Lv, W.-L. Long and F.-W. Li, Chem.–Eur. J., 2018, 24, 16588–16594 CrossRef CAS.
- Q.-J. Shen, X.-H. Chen, Y.-Y. Tan, J.-Z. Chen, L.-M. Chen and S.-Z. Tan, ACS Appl. Mater. Interfaces, 2019, 11, 38838–38848 CrossRef CAS.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- L. J. Müller, A. Kätelhön, S. Bringezu, S. McCoy, S. Suh, R. Edwards, V. Sick, S. Kaiser, R. Cuéllar-Franca, A. El Khamlichi, J. H. Lee, N. von der Assen and A. Bardow, Energy Environ. Sci., 2020 10.1039/d0ee01530j.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*