
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic interaction of fluoroquinolones with magnesium ions monitored using bacterial outer membrane nanopores†
Jiajun
Wang‡
 *a,
Jigneshkumar Dahyabhai
Prajapati‡
c,
Ulrich
Kleinekathöfer
c and
Mathias
Winterhalter
b
*a,
Jigneshkumar Dahyabhai
Prajapati‡
c,
Ulrich
Kleinekathöfer
c and
Mathias
Winterhalter
b
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: jiajunwang@nju.edu.cn
bDepartment of Life Sciences and Chemistry, Jacobs University Bremen, 28759 Bremen, Germany
cDepartment of Physics and Earth Sciences, Jacobs University Bremen, 28759, Germany
First published on 31st August 2020
Abstract
Divalent ions are known to have a severe effect on the translocation of several antibiotic molecules into (pathogenic) bacteria. In the present study we have investigated the effect of divalent ions on the permeability of norfloxacin across the major outer membrane channels from E. coli (OmpF and OmpC) and E. aerogenes (Omp35 and Omp36) at the single channel level. To understand the rate limiting steps in permeation, we reconstituted single porins into planar lipid bilayers and analyzed the ion current fluctuations caused in the presence of norfloxacin. Moreover, to obtain an atomistic view, we complemented the experiments with millisecond-long free energy calculations based on temperature-accelerated Brownian dynamics simulations to identify the most probable permeation pathways of the antibiotics through the respective pores. Both, the experimental analysis and the computational modelling, suggest that norfloxacin is able to permeate through the larger porins, i.e., OmpF, OmpC, and Omp35, whereas it only binds to the slightly narrower porin Omp36. Moreover, divalent ions can bind to negatively charged residues inside the porin, reversing the ion selectivity of the pore. In addition, the divalent ions can chelate with the fluoroquinolone molecules and alter their physicochemical properties. The results suggest that the conjugation with either pores or molecules must break when the antibiotic molecules pass the lumen of the porin, with the conjugation to the antibiotic being more stable than that to the respective pore. In general, the permeation or binding process of fluoroquinolones in porins occurs irrespective of the presence of divalent ions, but the presence of divalent ions can vary the kinetics significantly. Thus, a detailed investigation of the interplay of divalent ions with antibiotics and pores is of key importance in developing new antimicrobial drugs.
Introduction
The widespread presence of multidrug resistance (MDR) combined with the lack of new antimicrobial agents entering the market leads to an urgent need for novel antibiotics.1–3 One of the current bottlenecks in the development of molecules active against Gram-negative bacteria is their low permeability across the bacterial cell wall, composed of an outer membrane (OM) and inner membrane (IM).4,5 Methods to investigate the accumulation of antimicrobial agents inside a cell can be in vivo viability assay,6 liposome swelling assay,7 mass spectrometry and fluorometry related approaches,8–10 and electrophysiology.5,11Nanopore based single molecule tools could already investigate a single nucleotide segment,12,13 and nucleotide base lesion analogues,14,15 peptides16–19etc. Here we introduce outer membrane nanopores, which are pore forming proteins in the OM and especially general diffusion channels also called “porins”, e.g., OmpF and OmpC from E. coli, facilitate the diffusion of hydrophilic nutrients and are slightly cation selective as found in both in situ as well as in silico characterization.6,20,21 These porins are known to have a significant role in the influx of several classes of antibiotics, as shown by recent studies based on viability assays,22 electrophysiological characterization23–25 and in silico studies.26,27 Therefore, down-regulating the expression of these porins is often a prime mechanism chosen by bacteria to achieve resistance to many drugs.6 It is impossible to study the porin interaction with the permeating antibiotics using the conventional electrophysiological approach, patch clamp, due to the presence of lipopolysaccharides (LPSs) causing patch leakage. The emerging field of nanopores focuses on using single artificial or biological channels to study molecular interaction, recognition or translocation using ion current fluctuation.18,28,29 Here, we reconstituted a single porin into a lipid membrane, and monitored the dynamics of antibiotic passage. It is interesting to note that LPSs as a part of the barrier can be studied via fusion of outer membrane vesicles to create a native microenvironment.30
In this study, we use nanopore electrochemistry to characterize the transport across single porins. A nanopore provides a confinement effect to amplify the amperometric response of single molecules.31,32 Outer membrane nanopores achieved already defined inter-molecular competition of different antimicrobial agents inside the pore lumen.33 To this end, one can titrate antibiotic molecules on one side and the subsequent permeation of individual antibiotic molecules into the channel constriction transiently interrupts the ionic current.25 To obtain a molecular view, molecular dynamics (MD) simulations using enhanced sampling methods, e.g., umbrella sampling or metadynamics simulations, have been used in the past.34,35 Such simulations have been tremendously useful to understand the transport mechanisms of solutes through nanopores, but remained computationally very exhaustive.26,36 In this context, we employ our recently developed temperature accelerated Brownian dynamics (TABD) approach27,37 to investigate the passage of fluoroquinolones and especially norfloxacin through bacterial outer membrane porins from E. coli (OmpF and OmpC) and E. aerogenes (Omp35 and Omp36).38 Quinolones belong to the commonly prescribed classes of antimicrobials in clinics. These synthetic broad-spectrum antibiotics are effective by inhibiting the action of DNA gyrase/topoisomerase on bacterial DNA and have to effectively cross the outer as well as the inner membrane to reach their site of action at a lethal dose. To complement the experiments more rigorously, we also applied external electric fields during the TABD simulations, which have only been combined rarely with free energy calculations in the past26,39,40
To obtain an enhanced understanding of antibiotic uptake, we compare the pathways of norfloxacin (Fig. 1C) through four major porins, i.e., two from E. coli and two from E. aerogenes. Inspection of the crystal structures38 of these pores results in very similar architectures including the distribution of charged amino acids in the constriction region (CR) (Fig. 1A and B). However, both viability assays41,42 and biophysical characterization43,44 suggest that the interactions of antibiotic molecules with OmpF and the slightly smaller OmpC differ significantly. This illustrates that slight structural differences inside the CR can cause significant changes in the biophysical behaviour. Moreover, divalent ions have a significant effect on the antibiotic activity; they even bind to the CR altering the ionic preference of the porin. The potential binding site of a magnesium ion to a norfloxacin molecule is depicted in Fig. 1C. It has been found that tri-valent ions could influence the surface charge distribution at the nanopore opening thus altering the capture rate for single molecule analysis.45 Herein, we particularly focus on the effect of divalent ions on the translocation of small molecules inside the nanopore.46,47
 | ||
| Fig. 1 Structural features of the OM porins and the norfloxacin molecule. (A) The structural alignment of the four trimeric OM porins OmpF (PDB ID: 2ZFG),48 OmpC (PDB ID: 2J1N),49 Omp35 (PDB ID: 5O78),38 and Omp36 (PDB ID: 5O9C)38 is illustrated in cartoon representation. (B) Top view of the monomers in cartoon representation with loop L3 which is folded into the constriction region highlighted in magenta. The negatively charged residues (C atoms in green and O atoms in red) located on loop L3 and the positively charged residues (C atoms in yellow and N atoms in blue) on the opposite barrel wall are shown as sticks. (C) 2D structure of norfloxacin in its zwitterionic configuration. The binding site of divalent ions near the carboxyl group is indicated as well.47 | ||
As will be shown below, our experimental and computational results suggest that norfloxacin passes through OmpF, Omp35 and OmpC but only binds and bounces back from the slightly narrower Omp36 channel. Divalent ions such as magnesium can bind at negatively charged residues in the CR of the porins reversing their ion selectivity. At the same time, magnesium ions can also chelate with the fluoroquinolone molecules inducing difference in the dipole moment of the respective complex causing a significant alteration in the interaction with the pores. These two options will be analyzed and discussed in detail in the present study. Such an investigation will also help to better understand the differences in translocation in electrophysiology experiments with/without divalent ions and whole cell assays in which divalent ions are usually present.
Results and discussion
Ion conductance of OmpF, OmpC, Omp35 and Omp36
As the first step, the ion permeation through all four porins in the absence and presence of divalent ions has been studied (see the ESI† for Materials and methods). The conductance of the porins was measured by reconstituting single channels into lipid bilayers in the presence of a symmetric salt concentration on both sides of the membrane. Subsequently, transmembrane potentials of ±50 mV were applied and the ion flux through the porins was measured (see Fig. S1† in the ESI). In agreement with previous studies,25 we obtained conductance values in 1 M KCl buffered at pH 7 of about 4.1 ± 0.4 nS (OmpF), 2.7 ± 0.3 nS (OmpC), 4.3 ± 0.5 nS (Omp35), and 3.1 ± 0.2 nS (Omp36). Note that the higher conductance values of OmpF and Omp35 compared to OmpC and Omp36 suggest a larger pore size which is consistent with the crystal structures. It is interesting to note that Omp36 having a slightly smaller diameter than OmpC shows a higher conductance.38Ion selectivity of OmpF, OmpC, Omp35 and Omp36
Previous ion selectivity characterization experiments39 for OmpF and OmpC were repeated for consistency while for the Omp35 and Omp36 porins zero current membrane potential measurements were performed to estimate the ion selectivity (see Section 1.2 of the ESI† for Materials and methods). To this end, we started with symmetrical 10 mM salt concentrations on both sides of the membrane, titrated the concentrated salt solution to the ground side (cis, side of protein addition) and measured the reversal potential. In Fig. 2 the reversal potentials measured for the Omp35 and Omp36 porins versus the concentration gradient of the different salts are displayed. Monovalent cations (KCl and NaCl) resulted in positive whereas MgCl2 resulted in negative potentials across the membrane, suggesting cation and anion selectivity in the respective cases. The quite similar behaviour of the different pores is not surprising since sequence alignment of E. aerogenes Omp35 and Omp36 to OmpF and OmpC from E. coli shows a high similarity especially at the L3 loop which forms the CR in the porins.50 Moreover, the observed inversion of the pore selectively in the presence of the salt MgCl2 is in agreement with previous studies in the case of OmpF.48,51,52 | ||
| Fig. 2 (A) Zero-current membrane potential of the bilayer in the presence of Omp35 and Omp36 porins measured as a function of the salt (KCl, NaCl and MgCl2) gradient across the membrane. The respective salt concentration at the beginning was 10 mM on both sides and then gradually titrated at the cis (potential extracellular) side to reach the desired concentration. Average results of three experiments with different membranes at 20 °C and pH 7 are shown. (B) For 1 M MgCl2 the permeability ratio of anions to cations (PCl/PMg) is estimated for all four proteins from MD simulations performed without an external field. (C) The top three binding sites for Mg2+ ions which have been identified by MD simulations for all four proteins are denoted as sites 1, 2 and 3 (magenta spheres). The position of the binding sites inside the constriction region is depicted by the respective sphere, whereas the size of the sphere represents the probability of occurrence of these sites, i.e., the larger the sphere, the higher the probability. In the case of OmpF, one binding site of Mg2+ ions was observed in the crystal structure (yellow sphere) and is identical to site 3 observed in the simulations. The acidic residues inside the constriction region are highlighted by van der Waals spheres. | ||
To gain further molecular-level understanding, unbiased molecular dynamics (MD) simulations have been carried for all four porins embedded in a POPE bilayer in the presence of 1 M MgCl2 (see Section 1.4 of the ESI† for Materials and methods). Note that these simulations were carried out mainly to find the binding sites for Mg2+ ions inside the constriction region of these pores rather than to describe the ionic transport quantitatively. Thus, the choice of the POPE lipids in the simulations which differ from the lipopolysaccharides naturally found in the outer membrane and from the experimentally used DPhPC, is expected to only have a minimal effect on the properties in the constriction region of these pores. The estimated permeability ratios of anions to cations were found to have values higher than unity clearly showing that all four pores become anion selective in the presence of 1 M MgCl2 (see Fig. 2B). Moreover, we have also identified the top three binding spots of Mg2+ ions inside the CR and named them site 1, 2 and 3 (see Fig. 2C). In the case of OmpF, OmpC and Omp36, the Mg2+ ions tend to bind at very similar positions near the three acidic residues of loop L3. These residues are denoted as top, middle and bottom residues based on their position from the EC to the PP side. Thus, site 1 is near the top acidic residue (e.g. D121 in OmpF), site 2 is between the top and the middle residue (e.g. D121 and E117 in OmpF), and site 3 is between the middle and the bottom residue (e.g. E117 and D113 in OmpF). Remarkably, site 3 is identical to the one observed in the crystal structure of OmpF (PDB ID: 2ZFG) and found to be the most probable in the simulations.48 Omp35 also has sites 2 and 3 like the other porins but site 1 was found at a different position due to the presence of an additional acidic residue (E20) on the barrel wall (see Fig. 1B). This Mg2+ binding site is also found to be the most abundant one in Omp35. Moreover, the insight obtained in terms of the binding sites was further used in the BD simulations (described below) to understand the influence of Mg2+ ions on the norfloxacin translocation through porins.
Permeation of norfloxacin through OmpF, OmpC, Omp35 and Omp36
The protocol to measure norfloxacin translocation is similar to that described above for ion permeation (see Section 1.2 of the ESI† for Materials and methods). After reconstitution of a single trimer into a planar bilayer, application of a transmembrane voltage of 50 mV to 1 M KCl created silent current traces further used as a control (Fig. S2 in the ESI†). Addition of 0.25 mM norfloxacin on the cis (corresponding to the extracellular) side induced short ion current blockages.To gain insight into the mode of translocation, the bias voltages were varied in the range from −150 to +150 mV. The association rates (kon) and residence times (τ) were estimated as described previously53 and are shown in Fig. 3. In agreement with previous measurements for norfloxacin and OmpF, kon increases exponentially upon raising the magnitude of the transmembrane potential at negative polarities.26 Moreover, the voltage-dependent increase in the kon rate was very similar for OmpF (or OmpC) and its ortholog Omp35 (or Omp36), suggesting that the molecules tend to approach the CR in a similar manner. The second parameter important for the interpretation of the data is the voltage dependence of the residence time (τ). In agreement with previous measurements,26 when increasing the negative applied voltage in magnitude, OmpF shows a very steep decrease of the residence time from ∼750 to ∼50 μs, whereas for OmpC and Omp35 we have observed only a slight reduction in residence time from ∼100 to ∼50 μs. In contrast, for Omp36 the residence time increased rapidly from ∼100 to ∼1000 μs with increasingly negative voltages indicating that the molecule is not able to permeate through the constriction region likely due to the steric limitations imposed by the smaller diameter of the Omp36 pore.38 At positive voltages, one can observe a voltage-dependent increase in the kon rates for the studied porins; however, the values are slightly lower than the ones observed at negative voltages. Interestingly, a decrease in the residence time (τ) can only be observed for the OmpF and OmpC porins, whereas the values remain almost constant for Omp35 and Omp36. This finding suggests that with the application of positive voltages the permeation of norfloxacin is enhanced for OmpF and OmpC. In contrast, the permeation through Omp35 and Omp36 is slower (or maybe even vanishing) at positive voltages.
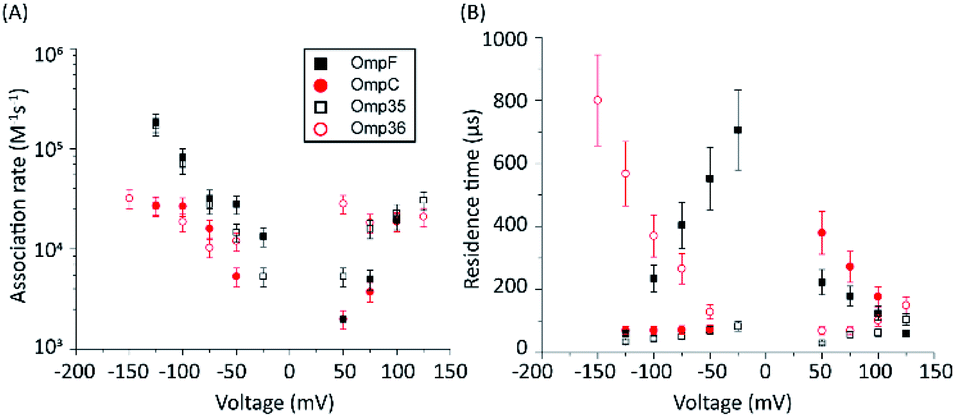 | ||
| Fig. 3 Voltage-dependent interaction of 0.25 mM norfloxacin with the respective porins. Addition of norfloxacin at the cis side, i.e., the putative extracellular side. (A) Association rate kon. (B) Residence time. Experimental conditions were 1 M KCl, 10 mM HEPES, and pH 7.0. | ||
Next, we have carried out temperature accelerated Brownian dynamics (TABD) simulations in the absence of external voltages to provide atomic-level understanding of the transport mechanisms of norfloxacin through all four porins (see the ESI† for Materials and methods). These BD simulations were carried out by treating only the antibiotic molecule in an explicit manner, whereas the contributions of the remaining components, i.e., protein, lipids and water, were taken into account using potential maps rather simulating them on an atomic level explicitly. Such a reduction of the simulated system enables the computation of free energy surfaces in a much faster manner compared to all-atom MD simulations by a factor of 1000 or more.37 The reconstructed free energy surfaces as a function of the collective variables (CVs), i.e., the position z along the pore axis and the orientation-related variable zij of norfloxacin,27,37 are illustrated in Fig. 4. The CV z represents the position of the molecule along the channel axis, where values of −25, 0 and 20 Å correspond to molecule locations at the EC (extracellular) side, the CR and the PP (periplasmic) side, respectively. The values of CV zij represent the orientation with limiting values of +8 and −8 Å which respectively correspond to the carboxyl group and the amino group facing the PP space, denoted as orientation I and II, respectively. The permeation pathways with these orientations are denoted as path I and II, respectively. For all four porins, we have observed that norfloxacin molecules are most likely to enter the EC side with orientation II (amino group ahead) and then orient in two different ways as they move further towards the CR, i.e., they either keep the same orientation or completely reorient to obtain orientation I (carboxyl group ahead). In general, we observed that the molecules tend to approach the CR with orientation II much more easily than with orientation I. However, further crossing through the CR is less likely to occur with orientation II in the case of OmpF, OmpC, and Omp35 as the energy barrier is much higher than that in the case of orientation I. In conclusion, path I is the route of choice in all three porins (see Fig. 4). Interestingly, both paths are found to be disconnected in the case of the Omp36 porin suggesting that the energy barriers are very high inside the CR and that permeation of norfloxacin is least likely to occur through this pore. Notably, the broken paths in the case of OmpC and Omp36 suggest high energy barriers, i.e., so high that they are not sampled with the present simulation parameters. For these positions and orientations one should be able to sample the respective paths by using higher artificial temperature for the biased CVs as shown in our previous study on the ciprofloxacin translocation through OmpC.27
 | ||
| Fig. 4 2D FESs as a function of the CVs z and zij from TABD simulations. The FESs for norfloxacin permeation through the four OM porins, i.e., OmpF (A), OmpC (B), Omp35 (C) and Omp36 (D), in the absence of any ionic solution. The arrows indicate the most probable path which the molecules most likely take while permeating through the respective pore. The molecules can adopt two different orientations inside the constriction region depicted as path I (solid arrows) and path II (dotted arrows). In the case of OmpC (B) and Omp36 (D) these paths are broken which is indicated by red crosses. (E) Representative conformations of norfloxacin translocation through OmpF at the EC vestibule, the CR (for path I and II) and the PP vestibule. The norfloxacin molecule is shown in stick representation (C atoms in green, N atoms in blue, O atoms in red and the F atom in pink) and the protein in cartoon representation with loop L3 highlighted in magenta. | ||
Furthermore, we have carried out TABD simulations for all four proteins in the presence of various external voltages, i.e., at −200, −100, +100 and +200 mV, to illustrate voltage-dependent effects on the permeation paths and to complement the experimental results (see Fig. S3 in the ESI†). The likelihood of a permeation event using path II, i.e., the amino group ahead, in the absence of any applied voltage is largest for OmpF, followed by Omp35 and OmpC and smallest for Omp36 (see Fig. 4A). In analogy to the experiments, the application of negative voltages should enhance the probability for such an orientation along the pore due to the zwitterionic nature of the molecule with a strong dipole of ∼44 D. As described earlier, the rapid decrease of the residence time observed with increasingly negative voltages in OmpF during experiments suggests that the permeation becomes much easier with orientation II (see Fig. 3). Apparently, such a behaviour can be observed for simulations at −100 and −200 mV where we clearly see a progressive decrease in the energy barrier for path II and an increase in the case of path I (see Fig. S3A in the ESI†). In contrast, very small decreases in the residence time with increasingly negative voltages in the case of Omp35 and OmpC during experiments indicate that the translocation is not efficient with orientation II and remains basically unaltered with increasing external voltages. Interestingly, we have observed an increase in the sampling along path II for both porins with the application of negative voltages in the simulations; however, the change in the energy barrier height is insignificant (see Fig. S3B & C in the ESI†). This finding suggests that the intrinsic lack of favourable interactions of the molecule with pore-lining residues along path II in OmpC and Omp35 is the primary reason why no change in the residence time (τ) was observed during the experiments. In the case of Omp36, a very rapid increase in the residence time was observed in the bilayer experiments, and a very large region of path II was not sampled in the absence of an external potential in the simulation. Moreover, we did not observe any significant changes along path II with application of external potentials (see Fig. S3D in the ESI†), leading to the conclusion that Omp36 most likely does not allow the transport of norfloxacin, at least in orientation II.
On the other hand, the likelihood of permeation in orientation I, i.e., the carboxyl group facing the PP space, in the absence of any external potentials is largest for OmpF, followed by OmpC and Omp35 and is smallest for Omp36. This orientation is expected to be enhanced at positive voltages. Path I can be seen to be favourable for OmpF and OmpC in the absence of external fields. A rapid decrease observed in the residence time during experiments clearly supports the fact that a translocation event is more likely for orientation I with increasing positive voltages. With application of positive voltages during the simulations, one can clearly see that path I remains intact with a slight reduction in the energy barriers and path II eventually gets disconnected for both porins when the voltage reaches +200 mV (see Fig. S3A & B in the ESI†). In the case of Omp35, we have observed that in certain regions of the EC side (at z = −10 to −5 Å) along path I, the molecules face a quite high energy barrier in the absence of an external potential. With the application of positive voltages, the path becomes slightly more accessible, but the energy barrier remains much higher than that in the case of OmpF/C (see Fig. S3C in the ESI†). Again, this intrinsic high energy barrier likely is the reason why no decrease in the residence time was observed in the experiments with the increasing positive voltages. Nonetheless, the molecule can still permeate at rates lower than that of OmpF/C. In Omp36, again a large region of path I is not sampled suitably which means that the respective energies are too high for the present simulation parameters. This finding stays the same in the presence of external fields (see Fig. S3D in the ESI†). Surprisingly, no change in the residence time was observed in the experiments at positive voltages compared to the rapid increase at negative voltages. Based on the high energy barriers observed in the simulations and the unchanged residence times in the experiment, we believe that norfloxacin cannot or only very rarely permeates through Omp36.
Our observation that norfloxacin can permeate faster through OmpF than through OmpC is consistent with earlier studies.54 It is also in agreement with the fact that in minimum inhibitory concentration (MIC) assays for E. coli the deletion of OmpF leads to higher MIC values than for the case of OmpC.55 On the other hand, there have been so far no studies which clearly distinguish the role of Omp35 and Omp36 in quinolone uptake. However, it was shown that E. aerogenes is much less susceptible to ciprofloxacin and some other quinolones compared to E. coli.56 Therefore, it is quite likely that the low permeability through the Omp35 and Omp36 porins is the reason for the poor activity of norfloxacin and other quinolones in E. aerogenes bacteria.
Interaction of norfloxacin with divalent ions
To characterize the affinity of Mg2+ ions with fluoroquinolone molecules, fluorescence assays have been performed (see the ESI† for Materials and methods). In the presence of divalent ions, norfloxacin and other fluoroquinolones chelate to form complexes. In agreement with the literature,57 at pH 7.0 we observed a shift in the fluorescence while changing the MgCl2 concentration from 0.4 to 1.6 μM (see Fig. S4 in the ESI†) suggesting that fluoroquinolones (norfloxacin, ciprofloxacin and enrofloxacin) form complexes with the Mg ions. In contrast, at pH 5, only a slight increase in the fluorescence intensity was observed (see Fig. S4 in the ESI†). This result confirms that at pH 5 (pKa = 6.1) no complexes are formed pointing to a protonation of the carboxyl group of the antibiotic molecules.58Permeation of norfloxacin in the presence of divalent ions
In a series of experiments described in the following, the role of divalent ions in the permeation of norfloxacin has been investigated. OmpF and Omp36 have been taken as examples since their pathways for the translocation without divalent ions differed the most. At voltages in the range from −25 mV to −125 mV, the increase in the kon rates was observed for both porins in the presence of divalent cations which is similar to the findings observed in the absence of these ions. As shown in Fig. 5A, at −125 mV the kon rates for OmpF increased from 0.15 in the absence of divalent ions to 6 and 7 × 105 s−1 M−1 in the presence of 5 mM Ca2+ and Mg2+ ions, respectively. Similarly, the kon rates for Omp36 increased from 0.12 to 2 and 4 × 105 s−1 M−1 at −125 mV as shown in Fig. 5C. The increased kon rates for both porins are assumed to be due to the conjugation of norfloxacin with the divalent ions leading to a complex with +2e net charge. Due to this net charge, the complex gets pulled from the cis to trans side of the channel when negative potentials are applied on the trans side leading to an increased permeation rate. | ||
| Fig. 5 Voltage-dependent association rates kon (A) and residence time (τ) (B) on the cis side for the interaction of norfloxacin with the OmpF porin in the presence of 1 M KCl (black squares), 1 M KCl with 5 mM MgCl2 (red dots) and 1 M KCl with 5 mM CaCl2 (blue triangles). Panels (C) and (D) are for the case of the Omp36 porin with the same buffer conditions as in (A) and (B). | ||
Such pulling effects for OmpF can be observed in the presence of Ca2+ and Mg2+ ions as the residence time τ for norfloxacin decreased from 400 μs to 60 μs and from 200 μs to 60 μs, respectively, when the magnitude of the negative voltage is increased from −25 mV to −125 mV (see Fig. 5B). In the case of Omp36, however, increasing the magnitude of the negative voltage causes an increase of the residence time from 60 μs to 2 ms and 7 ms in the presence of Ca2+ and Mg2+ ions, respectively (see Fig. 5D). Both cases demonstrated that the addition of divalent ions causes a stronger pulling effect irrespective of the pathway. Moreover, there are no drastic alternations for both porins at positive voltages. The lower kon rates and residence times τ indicate that the positively charged chelate complexes interact less with the porins.
To get further atomistic insight into the influence of divalent ions, we have carried out TABD simulations in the three most probable scenarios based on the ability of divalent ions to complex with the porin and/or the norfloxacin molecule, i.e., (i) only with norfloxacin, (ii) only with the porin, and (iii) with both, norfloxacin and the porin (see Section 1.5 of the ESI† for Materials and methods). All these simulations together should be able to provide significant insight because in experiments one probably finds a mixture of all three cases. The estimated FESs for OmpF and Omp36 are shown in Fig. 6. In the case of OmpF, both paths can be seen, i.e., path I and II with the carboxyl and the amino group ahead, respectively, when the norfloxacin molecule is complexed with an Mg2+ ion (see Fig. 6A). The barriers for both paths actually changed into wells with the well for path I being deeper. In the case of a Mg2+ ion only attached to the pore but not to the norfloxacin molecule (see Fig. 6B), the respective path is slightly less accessible but path I remains intact. Finally, when Mg2+ ions were placed on both, i.e., the norfloxacin molecule and the pore, leading to positive charges on both, path II is broken using the present simulation parameters and the sampling around path I is significantly reduced (see Fig. 6C). It is highly likely that path I would also disappear if more divalent ions are present inside the CR. Since the dipole moment of norfloxacin is reduced by about 70% when complexed with a Mg2+ ion, the polarity of external potentials will not have a strong influence on the orientation preference (path I or II). Interestingly, one can observe a very strong interaction near the constriction region when the molecule follows path I (between z = −10 to 0 Å) in all three scenarios, especially for the conjugated norfloxacin molecule, indicating that divalent ions intrinsically enhance the affinity of norfloxacin to the CR independent of where it binds. However, the permeation through the CR would become difficult when divalent ions bind to both, the molecule and the pore, which is likely to occur. Therefore, one of the complexes has to break in order to make permeation more feasible. Based on the experimental data obtained at negative and positive voltage polarities, we argue that the complex with norfloxacin remains quite intact compared to the one with the pore. Following this argument, we have observed an increase in the kon rate and a decrease in the residence time τ with negative applied voltages since the molecule in its conjugated form with net charge +2e gets pulled along the pore.
 | ||
| Fig. 6 2D FESs as a function of the CVs z and zij determined using TABD simulations for OmpF and Omp36 and obtained to understand the influence of Mg2+ ions on the norfloxacin translocation. The FESs for OmpF have been obtained for three different scenarios, i.e., Mg2+ ion(s) bound to (A) only norfloxacin, (B) only the porin, and (C) both, norfloxacin and the porin. Panels D, E and F show the same but for Omp36. As in Fig. 4, the two possible pathways with opposite orientation inside the constriction region are depicted as path I and path II with solid and dashed arrows, respectively, and broken paths are marked by red crosses. | ||
In the case of Omp36, the results in the CR along both possible paths for all three scenarios in terms of Mg2+ ion binding (see Fig. 6D–F) indicate only a rare permeation likelihood of norfloxacin. On the other hand, a strong interaction of norfloxacin can be observed near the CR (at z = −10 to −5 Å) similar to the case of OmpF. This result supports the very high residence times τ observed in the experiments displayed in Fig. 5. Overall, it can be concluded that the molecules can strongly interact within the CR but permeation is unlikely to happen across Omp36 also in the presence of divalent cations.
Moreover, in the experiments using OmpC and Omp35 we have seen an increase in the kon rates (see Fig. S5 in the ESI†). The change in the residence times, however, was not drastic, i.e., a slight increase for OmpC from 90 to 200 μs and a moderate decrease for Omp35 from 100 to 60 μs. These findings differ from the drastic changes observed for OmpF and Omp36. Furthermore, the simulations also suggest that Mg2+ ions clearly influence the permeation paths for OmpC and Omp35 (see Fig. S6 in the ESI†) enforcing strong interactions with the pore when complexed with the molecule. In general, the behavior was quite similar to that of OmpF, i.e., either path I or II was accessible depending on where Mg2+ ions bind. A major difference was observed in Omp35 when the Mg2+ ions bind to the pore only. In this case, path II becomes more accessible. Due to the tendency of Mg2+ ions in Omp35 to bind at different positions compared to that in other porins, i.e., site 1 which was found near residue E20, the molecule orients with the amino group facing the PP space (path II). Overall, based on the experiments and the simulations, we suggest that permeation of norfloxacin should be possible through OmpC and Omp35 in the presence of divalent cations without drastic change in kinetics.
Dependence of the interaction kinetics on the Mg2+ concentration
In the following set of experiments, we analyzed the effect of the Mg2+ ion concentration on the kon rate and the residence time τ for norfloxacin interactions with OmpF and Omp36. As shown in Fig. 7, the increase of the Mg2+ ion concentration from 10 μM to 5 mM induces an increase of the kon rates for both, OmpF (2 to 10 × 104 s−1 M−1) and Omp36 (5 to 10 × 104 s−1 M−1). The τ values for OmpF (400 to 60 μs) keep decreasing while they increase for Omp36 (from 1 to 2 ms). Interestingly, this trend is similar to that observed in voltage-dependent experiments shown in Fig. 5. | ||
| Fig. 7 OmpF and Omp36 interaction with 250 μM norfloxacin (cis addition) as a function of the MgCl2 concentration at pH 7 with a −100 mV potential. The association rate kon calculated from the single channel interactions is shown in the upper panel and the residence time (|) is plotted in the lower panel. | ||
Analysing the magnesium concentration dependence in more detail shows that the kon rates for both porins increase in a steady manner. The rise is small in the concentration range from 10 to 250 μM and then becomes drastically larger until 5 mM. Notably, this sudden change in the kinetic constants appears when the concentration of Mg2+ ions becomes equivalent to that of the norfloxacin molecules, i.e., 250 μM. This coincidence indicates that if the concentration of Mg2+ is lower than that of norfloxacin, fewer chelates are formed and therefore less change in the kon values can be observed at negative applied voltages. But as the concentration of the Mg2+ ions surpasses that of the norfloxacin molecules, the probability to form chelates is rapidly increased resulting in a fast enhancement of the kon values. On the other hand, the concentration-dependent decrease in the residence time τ for OmpF supports the fact that the chelate formation increases the permeability of norfloxacin through OmpF. It must be noted that the increase in the number of Mg2+ ions also increases their tendency to bind to pores, which is expected to slow down the permeation of norfloxacin as shown in simulations. However, the steady decrease in the residence time suggests otherwise. This must be due to our earlier claim that the conjugated form of the molecule remains intact and the Mg2+ ions get cleaved from the CR of the pore more easily during the permeation process through OmpF, especially because of the presence of external voltages. Interestingly, the τ values for Omp36 increase for increasing Mg2+ ion concentrations below 250 μM and remain unchanged above this concentration. It is quite clear that with the formation of more chelates outside the pore, more molecules approach the pore and bind strongly to the CR, but as the molecules do not permeate through the pore, further addition of Mg2+ ions does not improve the binding ability with the pore. Overall, considering the results for both porins, it is evident that magnesium ions clearly change the kinetics of norfloxacin molecules, irrespective of the fact if they permeate or just bind to the pore.
Conclusion
The permeation of norfloxacin across four homologous channels from E. coli (OmpF and OmpC) and E. aerogenes (Omp35 and Omp36) has been studied with a special focus on the effect of additional divalent ions. Experiments have been conducted in the absence and presence of divalent ions in order to illustrate their influence on the permeation process. The effect of divalent ions is investigated due to the physiological relevance to the bacterial OM, e.g., divalent ions are present in LPS membranes and thus also within the porins surrounded by LPS molecules. As shown in the crystallographic structure of OmpF, these divalent ions can bind to the pore lining residues. Moreover, atomistic insights into the underlying permeation pathways have been obtained by virtue of free energy calculations using the TABD simulation technique.As one important point we addressed which of the four studied porins facilitates the permeation of norfloxacin molecules in the absence of divalent ions. Based on the experimental and simulation results (see Table S1 in the ESI†), we conclude that OmpF, which has the largest pore diameter, allows the most efficient permeation of norfloxacin irrespective of the magnitude and polarity of external voltages. Its orthologue Omp35 and homologue OmpC are less efficient and might not facilitate the permeation of norfloxacin at all external voltages. The porin Omp36 does not seem to be suitable for norfloxacin transport due to its pore diameter which is slightly smaller than that of the other three porins. Nonetheless, the molecule approaches and binds to the Omp36 pore in a similar fashion as to the three proteins. At the same time, norfloxacin cannot translocate through Omp36 but bounces back to the same membrane side from which it originated.
Furthermore, we have demonstrated that divalent ions have a strong affinity with the norfloxacin molecule and CR residues of the pore using fluorescence assays and reversal potential measurements, respectively. With the introduction of divalent ions in single channel experiments, we have observed a strong increase in the association rates for norfloxacin with all four porins at one polarity of external voltages, indicating the improved approachability of the molecule to the CR. Such a behaviour was also observed during the simulations in which the molecule binds with a strong affinity to the conjugated form, especially when the carboxyl group of the molecule with bound Mg2+ faces the CR. The subsequent crossing from the CR to the periplasmic vestibule remains possible in the case of three porins, i.e., OmpF, OmpC and Omp35, as already observed in the absence of divalent ions. Based on our findings, we suggest that the complexes of divalent ions with norfloxacin molecules have a stronger internal binding than the ions with the CR of the pores during the permeation process. For Omp36, a crossing of the CR is still not possible even though divalent ions improve the binding affinity. Overall, we conclude that divalent ions improve the affinity of norfloxacin with all the pores. Moreover, they do not alter the permeation process through OmpF, OmpC and Omp35 as well as the binding and bouncing back process in the case of Omp36, though changes in the kinetic rates can clearly be seen. Based on the outcomes for norfloxacin, we suggest that a similar alteration in the kinetics for other fluoroquinolone antibiotics can be expected as they retain the same chemical scaffold. The fluorescence test for ciprofloxacin and enrofloxacin has already confirmed that these compounds bind to divalent ions (Fig. S4 in the ESI†). Moreover, our study suggests that all OmpF and OmpC orthologues must have binding pockets for divalent ions inside their constriction region as they share a similar architecture; therefore, the transport of all other classes of antibiotics is expected to be affected. Hence, more studies in this direction will be needed in the future to generalize the role of divalent ions in porin-mediated antibiotic uptake.
In terms of technical advancement, we have presented the application of external voltages in free energy calculations to support experimental outcomes thoroughly. Moreover, we also demonstrate the influence of divalent ions on antibiotic permeation which has often been neglected in the past. On the one hand, MD simulations allow more accurate and realistic simulations of OM porins, e.g. by inclusion of lipopolysaccharides and ions. However, on the other hand, influence of voltages is not easy to study in MD simulations due to a tremendous need for computational resources. Therefore, TABD simulations can be used alone or in parallel with MD simulations to gain better atomistic insight in the future. Additionally, such simulations could have a great impact in the future to complement single channel electrophysiology experiments which are based on applying external bias potentials. With the combination of electrophysiology experiments and molecular simulations, one can improve the understanding of permeation mechanisms of existing antibiotics through OM porins at the atomistic scale which will help to improve the design of next generation antimicrobials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Prof. Jean-Marie Pagès and Prof. Muriel Masi (Faculté de Médecine, Aix-Marseille Université, Marseille, France) for providing the purified proteins Omp35 and Omp36. The authors would like to thank Prof. Yi-Tao Long (Nanjing University, Nanjing, China) for deep discussion on the nanopore confined electrochemistry. This work has received support from the Innovative Medicines Initiative Joint Undertaking under Grant Agreement No. 115525, and resources which are composed of financial contribution from European Union seventh framework programme (FP7/2007–2013) and EFPIA companies. We further acknowledge support from EU FP7-PEOPLE-2013-ITN; the Marie-Skłodowska Curie Translocation network Nr. 607694; the National Natural Science Foundation of China (61901171); Deutsche Forschungsgemeinschaft (DFG) through project KL1299/9-2.Notes and references
- P. Fernandes, Nat. Biotechnol., 2006, 24, 1497–1503 CrossRef CAS.
- P. S. Hoffman, Antibiotics (Basel, Switz.), 2020, 9, 213 Search PubMed.
- L. Martínez-Martínez, A. Pascual and G. A. Jacoby, Lancet, 1998, 351, 797–799 CrossRef.
- R. A. Stavenger and M. Winterhalter, Sci. Transl. Med., 2014, 6, 228ed227 Search PubMed.
- J. Vergalli, I. V. Bodrenko, M. Masi, L. Moynié, S. Acosta-Gutiérrez, J. H. Naismith, A. Davin-Regli, M. Ceccarelli, B. van den Berg, M. Winterhalter and J.-M. Pagès, Nat. Rev. Microbiol., 2020, 18, 164–176 CrossRef CAS.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS.
- H. Nikaido, W. Liu and E. Y. Rosenberg, Antimicrob. Agents Chemother., 1990, 34, 337–342 CrossRef CAS.
- R. Iyer, Z. Ye, A. Ferrari, L. Duncan, M. A. Tanudra, H. Tsao, T. Wang, H. Gao, C. L. Brummel and A. L. Erwin, ACS Infect. Dis., 2018, 4, 1336–1345 CrossRef CAS.
- E. Dumont, J. Vergalli, L. Conraux, C. Taillier, A. Vassort, J. Pajović, M. Réfrégiers, M. Mourez and J.-M. Pagès, J. Antimicrob. Chemother., 2019, 74, 58–65 CAS.
- H. Prochnow, V. Fetz, S. K. Hotop, M. A. García-Rivera, A. Heumann and M. Brönstrup, Anal. Chem., 2019, 91, 1863–1872 CrossRef CAS.
- J. A. Bafna, E. Sans-Serramitjana, S. Acosta-Gutiérrez, I. V. Bodrenko, D. Hörömpöli, A. Berscheid, H. Brötz-Oesterhelt, M. Winterhalter and M. Ceccarelli, ACS Infect. Dis., 2020, 6, 1855–1865 CrossRef CAS.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS.
- C. Cao, Y. L. Ying, Z. L. Hu, D. F. Liao, H. Tian and Y. T. Long, Nat. Nanotechnol., 2016, 11, 713–718 CrossRef CAS.
- R. P. Johnson, A. M. Fleming, R. T. Perera, C. J. Burrows and H. S. White, J. Am. Chem. Soc., 2017, 139, 2750–2756 CrossRef CAS.
- J. Wang, M.-Y. Li, J. Yang, X.-Y. Wu, J. Huang, Y.-L. Ying and Y.-T. Long, ACS Cent. Sci., 2020, 6, 76–82 CrossRef CAS.
- Y.-L. Ying, J. Yang, F.-N. Meng, S. Li, M.-Y. Li and Y.-T. Long, Research, 2019, 2019, 1050735 CrossRef.
- U. Lamichhane, T. Islam, S. Prasad, H. Weingart, K. R. Mahendran and M. Winterhalter, Eur. Biophys. J., 2013, 42, 363–369 CrossRef CAS.
- H. Ouldali, K. Sarthak, T. Ensslen, F. Piguet, P. Manivet, J. Pelta, J. C. Behrends, A. Aksimentiev and A. Oukhaled, Nat. Biotechnol., 2020, 38, 176–181 CrossRef CAS.
- F. Piguet, H. Ouldali, M. Pastoriza-Gallego, P. Manivet, J. Pelta and A. Oukhaled, Nat. Commun., 2018, 9, 966 CrossRef.
- A. Alcaraz, E. M. Nestorovich, M. Aguilella-Arzo, V. M. Aguilella and S. M. Bezrukov, Biophys. J., 2004, 87, 943–957 CrossRef CAS.
- W. Im and B. Roux, J. Mol. Biol., 2002, 322, 851–869 CrossRef CAS.
- J. Chevalier, M. Mallea and J.-M. Pages, Biochem. J., 2000, 348, 223–227 CrossRef CAS.
- D. Vikraman, R. Satheesan, K. S. Kumar and K. R. Mahendran, ACS Nano, 2020, 14, 2285–2295 CrossRef CAS.
- C. Danelon, E. M. Nestorovich, M. Winterhalter, M. Ceccarelli and S. M. Bezrukov, Biophys. J., 2006, 90, 1617–1627 CrossRef CAS.
- J. Wang, J. Bafna, S.-P. Bhamidimarri and M. Winterhalter, Angew. Chem., Int. Ed., 2019, 58, 4737–4741 CrossRef.
- H. Bajaj, S. Acosta Gutierrez, I. Bodrenko, G. Malloci, M. A. Scorciapino, M. Winterhalter and M. Ceccarelli, ACS Nano, 2017, 11, 5465–5473 CrossRef CAS.
- J. D. Prajapati, C. J. Fernández Solano, M. Winterhalter and U. Kleinekathöfer, J. Chem. Theory Comput., 2017, 13, 4553–4566 CrossRef CAS.
- D. Deamer, M. Akeson and D. Branton, Nat. Biotechnol., 2016, 34, 518–524 CrossRef CAS.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, S. Garaj, A. Hibbs, X. Huang, S. B. Jovanovich, P. S. Krstic, S. Lindsay, X. S. Ling, C. H. Mastrangelo, A. Meller, J. S. Oliver, Y. V. Pershin, J. M. Ramsey, R. Riehn, G. V. Soni, V. Tabard-Cossa, M. Wanunu, M. Wiggin and J. A. Schloss, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS.
- J. Wang, R. Terrasse, J. A. Bafna, L. Benier and M. Winterhalter, Angew. Chem., Int. Ed., 2020, 59, 8517–8521 CrossRef CAS.
- R.-J. Yu, S.-M. Lu, S.-W. Xu, Y.-J. Li, Q. Xu, Y.-L. Ying and Y.-T. Long, Chem. Sci., 2019, 10, 10728–10732 RSC.
- Y.-L. Ying, J. Wang, A. R. Leach, Y. Jiang, R. Gao, C. Xu, M. A. Edwards, A. D. Pendergast, H. Ren, C. K. T. Weatherly, W. Wang, P. Actis, L. Mao, H. S. White and Y.-T. Long, Sci. China: Chem., 2020, 63, 589–618 CrossRef CAS.
- J. Wang, N. Fertig and Y.-L. Ying, Anal. Bioanal. Chem., 2019, 1–7 Search PubMed.
- K. R. Pothula, C. J. Solano and U. Kleinekathofer, Biochim. Biophys. Acta, 2016, 1858, 1760–1771 CrossRef CAS.
- F. Samsudin and S. Khalid, J. Phys. Chem. B, 2019, 123, 2824–2832 CrossRef CAS.
- V. K. Golla, E. Sans-Serramitjana, K. R. Pothula, L. Benier, J. A. Bafna, M. Winterhalter and U. Kleinekathofer, Biophys. J., 2019, 116, 258–269 CrossRef CAS.
- C. J. Solano, J. D. Prajapati, K. R. Pothula and U. Kleinekathöfer, J. Chem. Theory Comput., 2018, 14, 6701–6713 CrossRef CAS.
- S. Acosta-Gutierrez, L. Ferrara, M. Pathania, M. Masi, J. Wang, I. Bodrenko, M. Zahn, M. Winterhalter, R. A. Stavenger and J.-M. Pagès, ACS Infect. Dis., 2018, 4, 1487–1498 CrossRef CAS.
- R. Reale, N. J. English, J. A. Garate, P. Marracino, M. Liberti and F. Apollonio, J. Chem. Phys., 2013, 139, 205101 CrossRef.
- L. M. Napolitano, I. Bisha, M. De March, A. Marchesi, M. Arcangeletti, N. Demitri, M. Mazzolini, A. Rodriguez, A. Magistrato, S. Onesti, A. Laio and V. Torre, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3619–E3628 CrossRef CAS.
- M. Masi and J.-M. Pagès, Open Microbiol. J., 2013, 7, 22–33 CrossRef CAS.
- L. S. Redgrave, S. B. Sutton, M. A. Webber and L. J. Piddock, Trends Microbiol., 2014, 22, 438–445 CrossRef CAS.
- J. D. Prajapati, C. J. F. Solano, M. Winterhalter and U. Kleinekathofer, J. Phys. Chem. B, 2018, 122, 1417–1426 CrossRef CAS.
- J. Cama, H. Bajaj, S. Pagliara, T. Maier, Y. Braun, M. Winterhalter and U. F. Keyser, J. Am. Chem. Soc., 2015, 137, 13836–13843 CrossRef CAS.
- Z.-L. Hu, M.-Y. Li, S.-C. Liu, Y.-L. Ying and Y.-T. Long, Chem. Sci., 2019, 10, 354–358 RSC.
- H. Alkaysi, M. Abdel-Hay, M. S. Salem, A. Gharaibeh and T. Na'was, Int. J. Pharm., 1992, 87, 73–77 CrossRef CAS.
- B. D. Bax, P. F. Chan, D. S. Eggleston, A. Fosberry, D. R. Gentry, F. Gorrec, I. Giordano, M. M. Hann, A. Hennessy and M. Hibbs, Nature, 2010, 466, 935–940 CrossRef.
- E. Yamashita, M. V. Zhalnina, S. D. Zakharov, O. Sharma and W. A. Cramer, EMBO J., 2008, 27, 2171–2180 CrossRef CAS.
- A. Baslé, G. Rummel, P. Storici, J. P. Rosenbusch and T. Schirmer, J. Mol. Biol., 2006, 362, 933–942 CrossRef.
- C. Bornet, N. Saint, L. Fetnaci, M. Dupont, A. Davin-Regli, C. Bollet and J. M. Pages, Antimicrob. Agents Chemother., 2004, 48, 2153–2158 CrossRef CAS.
- B. van den Berg, S. Prathyusha Bhamidimarri, J. Dahyabhai Prajapati, U. Kleinekathöfer and M. Winterhalter, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E2991–E2999 CrossRef CAS.
- E. García-Giménez, A. Alcaraz and V. M. Aguilella, Biochem. Res. Int., 2012, 2012, 245786 Search PubMed.
- G. Schwarz, C. Danelon and M. Winterhalter, Biophys. J., 2003, 84, 2990–2998 CrossRef CAS.
- K. R. Mahendran, M. Kreir, H. Weingart, N. Fertig and M. Winterhalter, J. Biomol. Screening, 2010, 15, 302–307 CrossRef CAS.
- P. G. Mortimer and L. J. Piddok, J. Antimicrob. Chemother., 1993, 32, 195–213 CrossRef CAS.
- J. Sirot, M. H. Nicolas-Chanoine, H. Chardon, J. L. Avril, C. Cattoen, J. C. Croix, H. Dabernat, T. Fosse, J. C. Ghnassia, E. Lecaillon, A. Marmonier, M. Roussel-Delvallez, C. J. Soussy, A. Trevoux, F. Vandenesch, C. Dib, N. Moniot-Ville and Y. Rezvani, Clin. Microbiol. Infect., 2002, 8, 207–213 CrossRef CAS.
- J. Vergalli, E. Dumont, B. Cinquin, L. Maigre, J. Pajovic, E. Bacqué, M. Mourez, M. Réfrégiers and J.-M. Pagès, Sci. Rep., 2017, 7, 9821 CrossRef.
- C. Gu and K. Karthikeyan, Environ. Sci. Technol., 2005, 39, 9166–9173 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03486j |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2020 |