
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery and biosynthesis of bosamycins from Streptomyces sp. 120454†
Zi Fei
Xu‡
a,
Sheng Tao
Bo‡
a,
Mei Jing
Wang
a,
Jing
Shi
a,
Rui Hua
Jiao
a,
Yang
Sun
ac,
Qiang
Xu
a,
Ren Xiang
Tan
*ab and
Hui Ming
Ge
 *ac
*ac
aState Key Laboratory of Pharmaceutical Biotechnology, Institute of Functional Biomolecules, School of Life Sciences, Nanjing University, 210023, P. R. China. E-mail: rxtan@nju.edu.cn; hmge@nju.edu.cn
bState Key Laboratory Cultivation Base for TCM Quality and Efficacy, Nanjing University of Chinese Medicine, Nanjing 210023, P. R. China
cChemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing 210023, P. R. China
First published on 11th August 2020
Abstract
Nonribosomal peptides (NRPs) that are synthesized by modular megaenzymes known as nonribosomal peptide synthetases (NRPSs) are a rich source for drug discovery. By targeting an unusual NRPS architecture, we discovered an unusual biosynthetic gene cluster (bsm) from Streptomyces sp. 120454 and identified that it was responsible for the biosynthesis of a series of novel linear peptides, bosamycins. The bsm gene cluster contains a unique monomodular NRPS, BsmF, that contains a cytochrome P450 domain at the N-terminal. BsmF (P450 + A + T) can selectively activate tyrosine with its adenylation (A) domain, load it onto the thiolation (T) domain, and then hydroxylate tyrosine to form 5-OH tyrosine with the P450 domain. We demonstrated a NRPS assembly line for the formation of bosamycins by genetic and biochemical analysis and heterologous expression. Our work reveals a genome mining strategy targeting a unique NRPS domain for the discovery of novel NRPs.
Introduction
Nonribosomal peptides (NRPs), which are biosynthesized by multidomain enzymes known as nonribosomal peptide synthetases (NRPSs), are one of the largest and most diverse classes of natural products with a variety of biological functions including cytotoxic, antibiotic, antiviral and immunosuppressive activities.1,2 Different to ribosomal peptide synthetases, which only utilize 20 proteinogenic amino acids as substrates, NRPSs can recruit hundreds of building blocks including proteinogenic and nonproteinogenic amino acids, hydroxyl acids, or even fatty acids, and assemble them into peptides.3 A minimal NRPS module includes: an adenylation (A) domain, a thiolation (T) domain and a condensation (C) domain. The A domain activates amino acids through adenylation of carboxylic acids, and then installs substrates as covalent thioesters to the phosphopantetheinyl groups of the T domain. The C domain subsequently mediates the peptide bond formation in two adjacent modules. A mature oligopeptide is usually released by the terminal thioesterase (TE) domain to form either linear or cyclic peptides through hydrolysis or cyclization. Besides these most common domains observed in NRPS machinery, amino acid building blocks or elongated peptides tethered on the T domain can be shuttled to other protein domains embedded in NRPS modules, such as methylation (M), formylation (F), epimerization (E), oxidation (Ox), cyclization (Cy), and reduction (R) domains for additional structural modifications.4,5Recent advances of microbial genome sequencing together with bioinformatic tools have significantly accelerated the discovery process of new NRPs. Taking advantage of the NRPS biosynthetic machinery, we have successfully identified and activated a cryptic orphan NRPS gene cluster, in which all four NRPSs are stand-alone monomodule enzymes instead of canonical multimodule NRPS megaenzymes, resulting in the discovery of novel NRPs named ashimides.6 Furthermore, genome mining targeting unusual N-terminal modification of biosynthetic genes led us to characterize a series of rare cinnamoyl-containing NRPs with one showing potent activity towards the stimulator of interferon genes (STING) protein.7 During our continuing efforts to discover novel/bioactive NRPs through genome mining,6,7 we became interested in an unusual NRPS biosynthetic gene cluster (named bsm) from the genome of Streptomyces sp. 120454. The unique feature for the bsm cluster is that a cytochrome P450 monooxygenase domain, which shows moderate sequence identity towards P450 hydroxylase HmtN (31%) and EryK (30%) in himstatin8 and erythromycin9 biosynthesis, respectively, is embedded in a NRPS (BsmF) enzyme. In contrast to the canonical NRPS domains described above, a P450 monooxygenase domain has never been observed in NRPS megaenzymes. Basic Local Alignment Search Tool (BLAST) search in a public genomic database using BsmF as a query sequence resulted in no hits,10 indicating that this gene cluster may be responsible for the production of a novel type of NRP. In the present work, we identified and characterized six novel linear NRPs encoded by the bsm gene cluster, bosamycins A–F (1–6) through a genome mining strategy. The biosynthetic pathway for bosamycins was revealed through in vivo gene deletion, in vitro biochemical reactions, and heterologous expression.
Results and discussion
Isolation and structural characterization of bosamycins
Streptomyces sp. 120454 was fermented on different media commonly used for secondary metabolite production in actinomycetal strains.11–14 Metabolic analysis through LC-MS and DAD-HPLC showed several putative peptide ion peaks with molecular weights ranging from 900–1200 Da and maximum UV absorbance at 280 and 220 nm (Fig. 1). Large scale fermentation for this strain resulted in isolation of compounds 1–6. The planar structures of 1–6 were characterized by extensive analyses of their HRESIMS, MS/MS and NMR data, and their absolute configuration was determined by advanced Marfey's method (Fig. S2, S17–S22 and S37–S86†).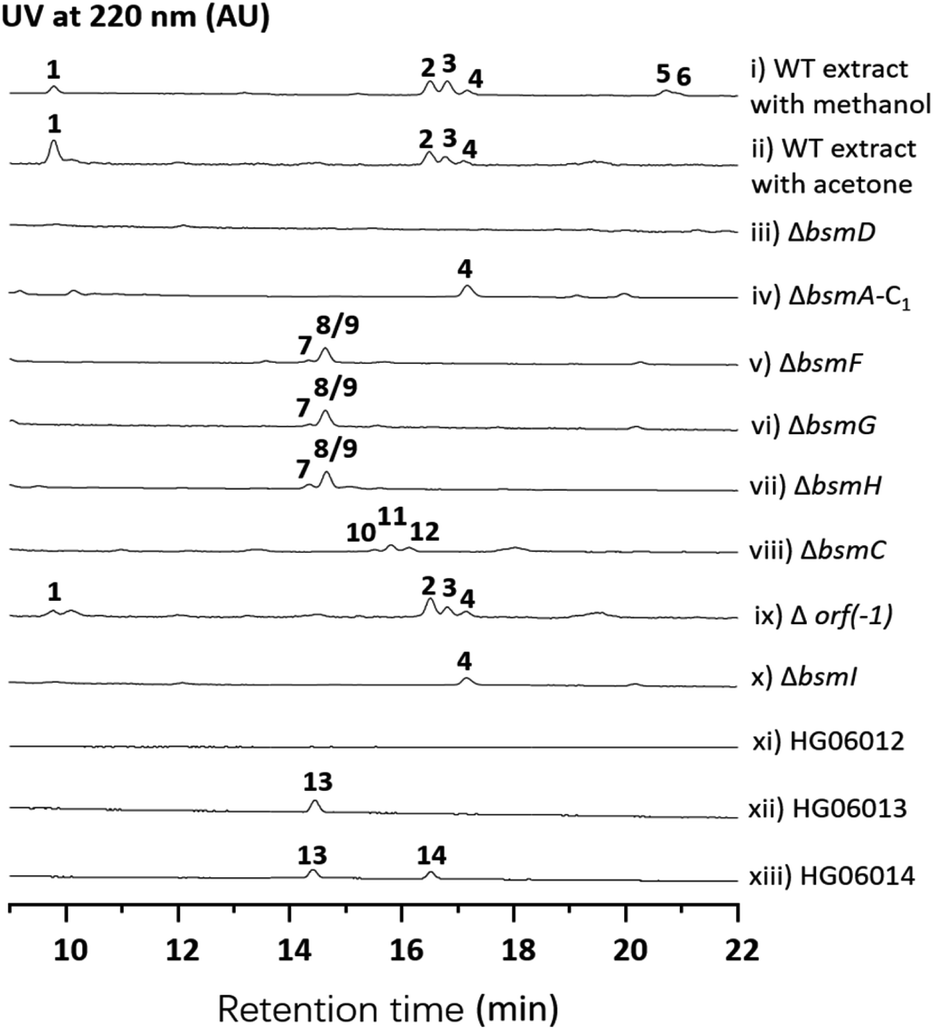 | ||
| Fig. 1 HPLC analysis of metabolite extracts from wild-type and recombinant strains. | ||
Compound 1 was obtained as a white amorphous solid. The molecular formula of 1 was determined to be C40H57N7O15 by its HRESIMS data (Fig. S37†). The typical signals for amide protons and carbons in 1H and 13C NMR spectra suggested the peptide nature of 1. The structure of 1 was determined to be a heptapeptide containing Tyr (1), Leu (2), Ser (1), and Gly (1), together with two nonproteinogenic amino acids including 3-hydroxyl aspartic acid (β-OHAsp) and 5-methoxyl tyrosine (5-OMeTyr) through detailed analysis of its 1H, 13C, DEPT, COSY, HMBC and HSQC NMR data. Based on the HMBC and NOESY NMR correlations, a linear Tyr1-Leu2-β-OH-Asp3-Ser4-5-OMeTyr5-Leu6-Gly7 heptapeptide was determined for compound 1. The gross structure of 1 was further supported by MS/MS analysis (Fig. S17†). According to modified Marfey's analysis, a linear heptapeptide D-Tyr1-L-Leu2-L-erythro-β-OH-Asp3-L-Ser4-D-5-OMeTyr5-L-Leu6-Gly7 was elucidated for 1 (Fig. S2†). Compounds 2–4 have an additional N-carbamoyl, N-carboxyl carbamoyl and N-acetyl L-tyrosine residue at the N-terminus of 1, respectively (Tables S5–S7 and Fig. S45–S70†). Compounds 5 and 6 (Tables S8 and S9, and Fig. S71–S86†), which are methylated derivatives of 3 and 4, respectively, are only observed when using methanol as a solvent during extraction (Fig. 1 and traces i and ii), suggesting that both compounds could be artefacts. Bosamycin E (5) showed inhibitory activity towards SHP2 (Src homology 2-containing protein tyrosine phosphatase 2), a major phosphatase involved in growth factor and cytokine-mediated signalling, with an IC50 value of 24.25 μM (Fig. S3†), whereas other compounds were inactive.
Identification and characterization of the bsm gene cluster
With bosamycins A–F (1–6) in hand, we sought to analyse if their structures can be correlated with the bsm biosynthetic gene cluster. The bsm cluster consists of 12 open reading frames (orfs) spanning a 41 kb contiguous DNA region. Intriguingly, three giant NRPS megaenzymes (BsmA, BsmB, and BsmD) and a single module NRPS (BsmF) in the bsm gene cluster contain nine modules that are expected to incorporate nine amino acids to assemble a nonapeptide,15–18 which is not consistent with the isolated 8-mer peptide backbone of 2–6. Other genes within the bsm gene cluster encode a non-heme iron α-ketoglutarate dependent oxygenase (BsmC),19 a methyltransferase (BsmH) and a hydrolase (BsmG), indicating that the NRPS-generated peptide may undergo several tailoring steps. In addition, the MbtH-like protein encoding gene bsmE is located adjacent to bsmF. The function of MbtH-like proteins has previously been demonstrated to improve the NRPS's production (Table 1).20–22 To verify if the gene cluster is related to the biosynthesis of bosamycins, we knocked out the bsmD gene through in-frame deletion. The resulting ΔbsmD mutant strain was then fermented. In comparison with the wild-type strain, the ΔbsmD mutant completely abolished the production of bosamycins (Fig. 1 and trace iii), confirming its role in bosamycin biosynthesis.| Gene | aab | Protein homologue | Accession number | Coverage/identity (%) | Organism |
|---|---|---|---|---|---|
| a The sequence has been deposited in GenBank with accession number MN509472. b Number of amino acids. | |||||
| orf(−3) | 714 | ATPase | KOU35251 | 98/84 | S. sp. WM6378 |
| orf(−2) | 42 | Hypothetical protein | — | No hit | — |
| orf(−1) | 630 | ATP-binding protein | ARI56342.1 | 98/97 | S. sp. S8 |
| bsmI | 84 | Acyl carrier protein | QCX41947 | 85/37 | S. novoguineensis |
| bsmA | 2605 | Nonribosomal peptide synthetase | QBG38784 | 99/47 | S. atratus |
| bsmB | 3198 | Nonribosomal peptide synthetase | EFE66253 | 99/52 | S. viridosporus |
| bsmC | 313 | Dioxygenase | KFF83983.1 | 91/35 | Pseudomonas syringae |
| bsmD | 3950 | Nonribosomal peptide synthetase | AEH41793 | 94/50 | S. griseoflavus |
| bsmE | 69 | MbtH protein | AJV88378 | 95/55 | S. drozdowiczii |
| bsmF | 1136 | P450–A–T | — | No hit | — |
| bsmG | 272 | Alpha/beta hydrolase | A1JSF8 | 84/27 | Yersinia enterocolitica |
| bsmH | 330 | O-methyltransferase | ABX71118 | 96/36 | S. rishiriensis |
| orf(+1) | 247 | ABC transporter | KIX49568 | 99/98 | S. griseus |
| orf(+2) | 298 | ABC transporter | KIX49569 | 99/95 | S. griseus |
Biosynthesis of 5-OH tyrosine (5-OHTyr)
Cytochrome P450 monooxygenases are widely distributed in NRPS biosynthetic pathways,23,24 where they catalyse an array of impressive oxidations of nonactivated C–H bonds as exemplified in the biosynthesis of ether bond linkage in vancomycin,25 β-hydroxytyrosine in skyllamycin,26,27 and novobiocin,28 β-hydroxytrophan in echinomycin,29 and β-hydroxyhistidine in nikkomycins.30 In these cases, P450s utilized the PCP-bound amino acids as substrates; in contrast, free amino acids or small molecule surrogates are not efficient substrates, indicating that the PCP is necessary for the substrate recognition, and highlighting the importance of protein–protein interactions between P450s and PCPs.31,32 Indeed, crystal structure analysis indicated that an additional X-domain or PCP binding site is indispensable for recruiting stand-alone P450s to the NRPS for ether bond formation or β-hydroxylation in glycopeptide biosynthesis.33,34Different to the more common stand-alone P450s in NRPS biosynthesis, the bsm cluster encodes a monomodular NRPS (P450–A–T) with an extra cytochrome P450 monooxygenase domain fused at the N-terminal. As a domain, a P450 monooxygenase has never been a part of a NRPS megaenzyme. The only somewhat related example is LtxB in lyngbyatoxin biosynthesis, where a P450 is fused to an N-terminal MbtH-like domain with only about 80 amino acid residues.35 Prediction of the amino acid activation specificity of the A domain in BsmF revealed that tyrosine is the preferred substrate (Table S3†). Because only eight modules are required for bosamycin biosynthesis, BsmF is very likely involved in the synthesis of a modified amino acid building block, e.g. 5-OMeTyr, using tyrosine as the starting material.
To test the function of the unique P450 containing NRPS enzyme (BsmF), we generated a ΔbsmF mutant. LC-MS analysis of the metabolic extract indicated that the ΔbsmF strain completely lost the production of 1–4, but clearly accumulated two new peaks (7–9) albeit with lowered titers (Fig. 1 and trace v). Compound 7 has a molecular formula of C49H65N9O17, based on the HRESIMS data (Fig. S23†), and has 30 Da less than 2. Because only minute amounts of compounds 7–9 could be obtained, we performed comparative MS/MS analysis to solve their structures. The MS/MS fragmentation of 7 gave the characteristic b and y ion signals. The b1, b2, b3, b4, and b5 signals of 7 are identical to those found in 2, whereas y3, y4, y5, y6, y7, b6 and b7 signals have 30 Da less than the corresponding signals in 7 (Fig. 2), indicating that the only variation between 2 and 7 is at the 6th amino acid residue. The observed mass difference of 30 Da suggested that the 5-OMeTyr residue in 2 was replaced by a Tyr in 7 (Fig. 2). Similarly, 8 and 9 also had a Tyr instead of a 5-OMeTyr residue at the 6th amino acid residue according to comparative MS/MS fragmentation analysis (Fig. S24 and S25†). To further prove that the P450 domain is essential for the tyrosine hydroxylation, we site mutated three key residues that are crucial for its activity and generated three single-alanine mutants (BsmFT281A, BsmFF380A, and BsmFC387A) (Fig. S12†).36,37 All these variants failed to produce 2–4, but generated 7–9 (Fig. S8,† and traces vi–viii), demonstrating that the P450 domain is responsible for the hydroxylation of PCP-bound Tyr. Different to standalone P450s that act on PCP bound amino acids which contain 23 highly conserved fingerprint residues across 6 different regions,31,32 BsmF-P450 has a low degree of conservation of these residues (Table S14†), indicating that BsmF-P450 may use a different binding mode that is seen for OxyD or Sky32 type P450s.31,32 The lower similarity may also be related to the different site of modification of the amino acid, as BsmF-P450 acts on the aromatic ring rather than the β-position. In addition, alignment of the BsmF sequence with standalone P450s and canonical A-T regions indicated that two long linker regions are among three domains (Fig. S13†), providing flexible sites for domain–domain interactions.
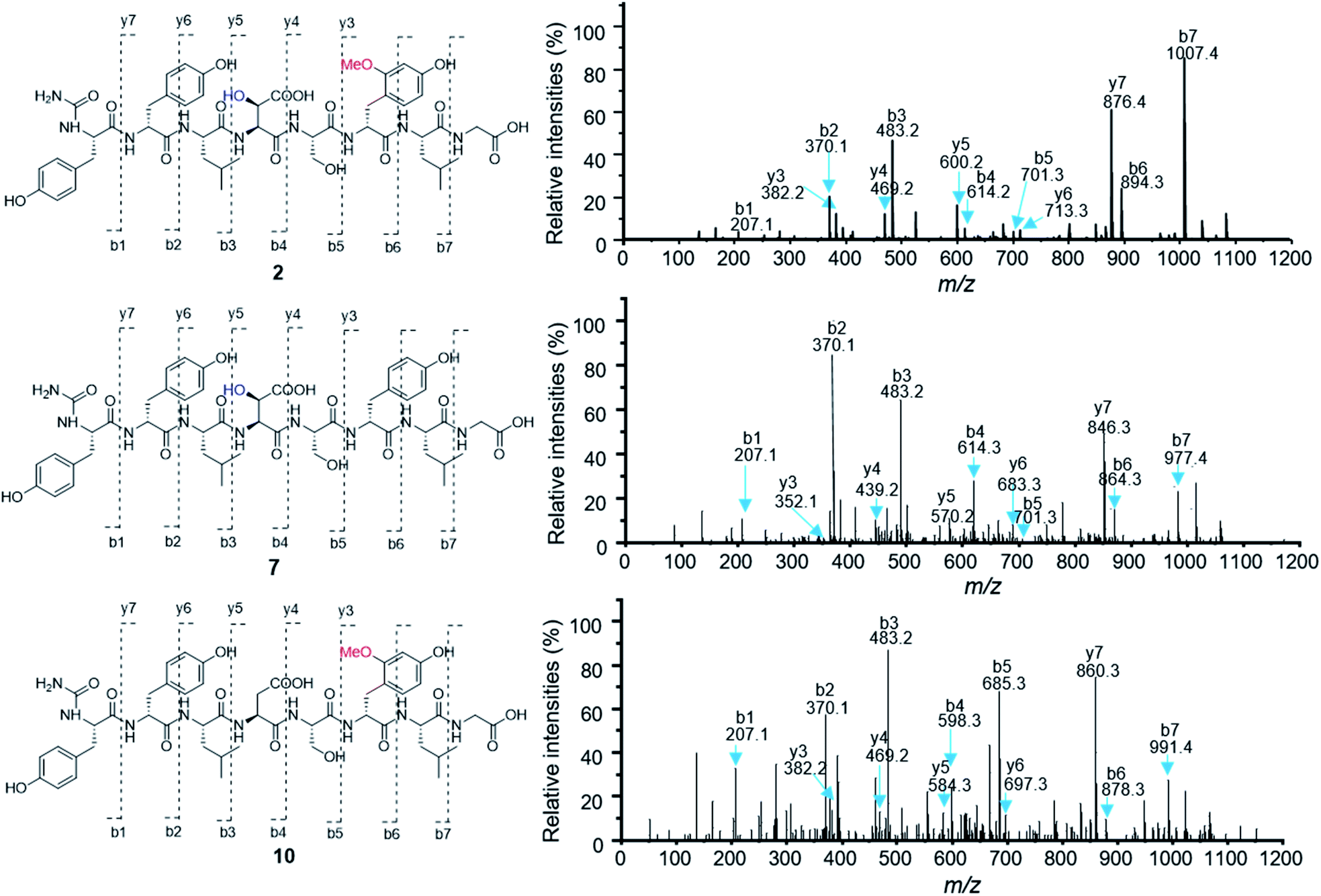 | ||
| Fig. 2 Comparative MS/MS analysis of compounds 2, 7 and 10. | ||
Biosynthesis of 5-OMe tyrosine (5-OMeTyr)
Next, we intended to understand the roles of hydrolase (BsmG) and methyltransferase (BsmH), and constructed the corresponding gene mutants. Similar to ΔbsmF mutants, the resulting mutants (ΔbsmH and ΔbsmG) failed to produce 2–4, instead accumulated 7–9 in low titers (Fig. 1 and traces vi and vii), indicating that both genes are related to 5-OMeTyr biosynthesis. However, whether the O-methylation occurs at the PCP-bound or free 5-OHTyr remains elusive. In NRPS biosynthesis, the on-line methylation is usually catalysed by the M (methyltransferase) domain that is embedded in the NRPS megaenzyme, whereas,38 the off-line methylation is often mediated by a stand-alone methyltransferase.39,40 To examine if methylation is before or after hydrolysis of PCP bound 5-OHTyr, we chemically synthesized 5-OMeTyr and 5-OHTyr and fed them individually to ΔbsmF, ΔbsmG, and ΔbsmH mutants. Most of these chemical complementation experiments can restore the wild-type production, except for feeding 5-OHTyr to the ΔbsmH mutant (Fig. S6 and S7†), indicating that the methylation occurs at free 5-OHTyr. Therefore, BsmG would release the PCP bound 5-OHTyr through hydrolysis, and BsmH then catalyses the methylation on free 5-OHTyr to form 5-OMeTyr, which will then be incorporated into the NRPS assembly line. The upload, offload and upload processes for 5-OMeTyr biosynthesis and reincorporation in bosamycin biosynthesis are reminiscent of the biosynthesis of β-hydroxlated tyrosine in balhimycin biosynthesis.15,16 In this pathway, the activation of a tyrosine by a single module NRPS (BpsD) with an A and a PCP domain allows the hydroxylation of the PCP-bound tyrosine to β-hydroxyltyrosine by a separate cytochrome P450 (OxyD).17 Hydrolysis of β-hydroxyltyrosine from BpsD is accomplished by a hydrolase (Bph),18 and the free β-hydroxyltyrosine is then loaded by the main NRPS.To further verify the role of BsmH, we overproduced and reconstituted its enzymatic activity in vitro. Free 5-OHTyr was incubated with BsmH in the presence of SAM. From HPLC analysis, the production of 5-OMeTyr was observed. In contrast, no product was detected when the reaction was conducted using boiled BsmH as a catalyst (Fig. 3). Taken together, the results support our proposed pathway and timing for 5-OMeTyr biosynthesis.
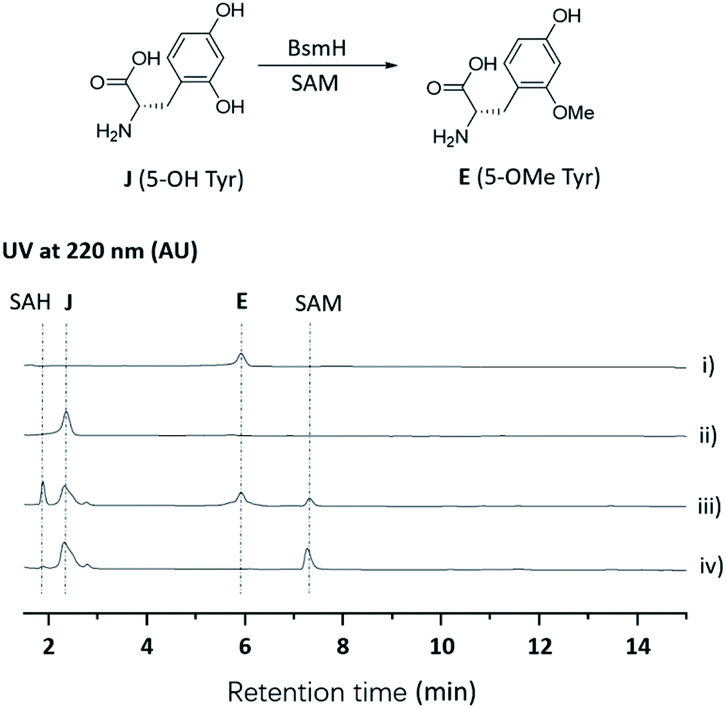 | ||
| Fig. 3 HPLC analysis of the BsmH-catalyzed reaction. (i) Standard of E (5-OMe Tyr); (ii) standard of J (5-OH Tyr); (iii) J (5-OH Tyr) was incubated with BsmH and SAM for 2 h; (iv) J (5-OH Tyr) was incubated with boiled BsmH and SAM for 2 h. | ||
Biosynthesis of β-OH aspartic acid (β-OH Asp) building blocks
Besides 5-OMeTyr, bosamycins contain another unnatural amino acid building block, L-erythro-β-OH-Asp residue. Enzyme catalysed β-hydroxylation has already been observed in Asn, Glu and Trp in the NRP biosynthesis.41–43 Previous research by Butler and co-workers on non-heme Fe(II)/α-KG dependent β-hydroxylases revealed that stand-alone β-hydroxylases acting on Asp loaded by a C-A-T-E or C-A-T module in bacterial siderophore biosynthesis have a strictly conserved GGDSI motif in the thiolation domains,44 whereas β-hydroxylases found in nonsiderophore peptides including SyrP, ThaF and NupP in the biosynthesis of syringomycin,19 thanamycin,45 and nunamycin,46 respectively, have a common GGHSL motif. Bioinformatic analysis revealed that bsmC encodes a non-heme α-KG dependent dioxygenase showing 70% identity towards SyrP, and the corresponding T domain has a conserved GGHSL motif (Fig. S15†). BsmC is most likely responsible for the β-hydroxylation of Asp.To test this hypothesis, we inactivated the bsmC gene in the wild-type strain through in-frame deletion. The resulting ΔbsmC strain was then fermented in the production medium, together with the wild-type strain as a control, to investigate the role of bsmC in bosamycin biosynthesis. LC-MS analysis showed that all metabolites produced in the wild-type strain were abolished in the ΔbsmC strain, but three minor peaks (10–12) were clearly generated (Fig. 1, and trace viii). The fragmentation ions for b1, b2, b3, y3 and y4 signals of 10 are identical to those of 2, whereas b4, b5, b6, b7, y5, y6 and y7 signals have 16 Da less than the corresponding signals for 2 (Fig. 2), indicating that the β-OH Asp residue in 2 is replaced by Asp in 10. Similarly, structures of 11 and 12 were elucidated as deshydroxyl derivatives of 3 and 4, respectively, through MS/MS fragmentation analysis (Fig. S34 and S35†). Moreover, chemical complementation of β-OHAsp did not restore the wild-type production (Fig. S9,† trace ii). Collectively, these data indicated that BsmC is a hydroxylase that acts on NRPS bound Asp.
Verification of A domain substrate specificity
Based on the above analysis, three giant NRPSs (BsmA, BsmB and BsmD) consisting of eight modules are responsible for the assembly of 8-mer peptides 2–4. In contrast, the remaining NRPS (BsmF) is related to recruitment of tyrosine and synthesis of 5-OHTyr. Bioinformatic analysis of the substrate specificity-conferring residues indicated that the predicted amino acid residues activated by A domains in modules 1, 2, 4, 5, 6, 8 and 0 are consistent with or similar to the composition of the bosamycin structure (Table S3†).47 However, A domains in modules 3 and 7 are predicted to accept Phe instead of Leu as their preferred substrate (Table S3†). One of the putative reasons is their similar hydrophobic nature as also observed in noursamycin biosynthesis.48We performed an in vitro adenylation activity assay towards A1, A4, A6 and A0 domains by monitoring pyrophosphate production.49 After several attempts, we successfully overproduced and obtained soluble proteins for truncated versions of A1, A4-T4, C6-A6-T6, and A0-T0 from E. coli BL21(DE3) (Fig. S11†). The activities of these reconstituted A domains against amino acids are listed in Fig. 4. It shows that L-Tyr is the preferred substrate for the A1 domain, indicating that an N-terminal modification is required in bosamycin biosynthesis. In line with our above results that BsmC hydroxylates the PCP-bound L-Asp and feeding β-OH Asp cannot restore the wild-type production, the A4 domain prefers to select L-Asp instead of L-erythro-β-OH-Asp as the substrate. A0 and A6 domains were found to activate L-Tyr and L-5-OMe Tyr, respectively (Fig. 4), supporting our data that tyrosine hydroxylation occurs at BsmF and 5-OMeTyr is the building block incorporated by BsmD. In addition, the A6 domain also shows 37% relative activity towards L-Tyr (Fig. 4), which is in accordance with the observed desmethoxy derivatives 7–9 produced from the ΔbsmF, ΔbsmG or ΔbsmH mutant (Fig. 1 and traces v–vii).
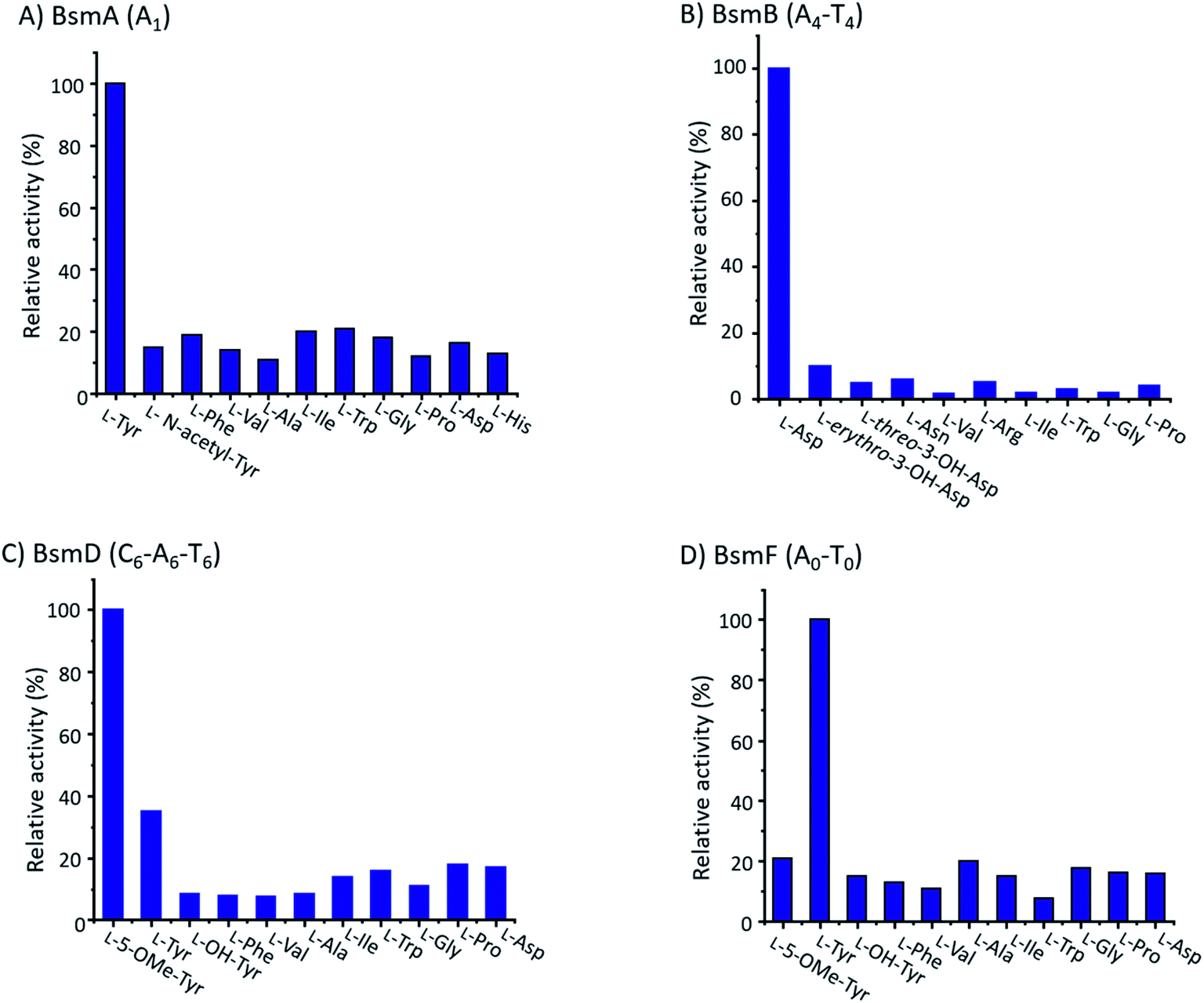 | ||
| Fig. 4 The relative adenylation activities for (A) BsmA (A1), (B) BsmB (A4-T4), (C) BsmD (C6-A6-T6), and (D) BsmF (A0-T0). | ||
BsmI and C1 domains are involved in N-terminal modification
Modifications of the N-terminal which is considered as an end cap are important for protecting the N-terminus from degradation. Compounds 2–4 have three different N-terminal modifications including carbamoyl, carboxyl carbamoyl and acetyl groups. Similar modifications have also been observed in actinoramides A and B, which have carbamoyl and acetyl modifications, respectively. Ryan and coworkers showed that a carbamoyltransferase (PadQ) and an acetyltransferase (EctA) are responsible for transferring a carbamoyl and acetyl groups in actinoramides A and B, respectively.50 However, no PadQ homologous enzymes could be found in S. sp. 120454 genomic sequence data. Close to the bsmA gene reside orf(−1) and bsmI genes encoding an ATP-binding protein and a peptidyl carrier protein, respectively. We envisioned that these two gene products may resemble adenylation and thiolation domains, respectively, in NRPS biosynthesis, which displays selectivity for activation of carboxyl carbamic acid or carbamic acid. We then individually inactivated these two genes. HPLC analysis of culture extracts revealed that Δorf(−1) still can produce 1–4 at a titer comparable to the wild-type strain (Fig. 1 and trace ix), whereas the ΔbsmI mutant strain failed to produce all carbamoyl or carboxyl carbamoyl-type peptides (2 and 3), but generated an acetylated peptide (4) (Fig. 1 and trace x).The starter C domain (C1 domain) in BsmA contains the conserved catalytic motif HHxxxD (Fig. S16†),4 and thus it would condense the BsmI-bounded carbamoyl or carboxyl carbamoyl groups with L-tyrosine that is tethered onto the thiolation domain of module 1. We specifically deleted the C1 domain region corresponding to 14th–294th amino acid residues in bsmA through in-frame deletion. The resulting mutant, ΔbsmA-C1, only produced 4 (Fig. 1 and trace iv), which is similar to the ΔbsmI strains. These results indicated that BsmI and C1 domains are necessary for N-carbamoylation but not required for N-acetylation, and the adenylation enzyme for activation of carbamoyl acid could be encoded outside the gene cluster.
Heterologous expression of the bsm gene cluster
To investigate if the proposed bsm gene cluster is sufficient for the production of bosamycins, we sought to express the complete bsm gene cluster in a heterologous host. A cosmid genomic library of S. sp. 120454 was thus constructed using a Streptomyces integrative vector pJTU2554.51,52 One positive cosmid, pHG06015, that covers part of the bsm cluster spanning from bsmI to bsmE (Scheme 1 and panel B) was obtained. As we already demonstrated that the three remaining genes bsmF, bsmG and bsmH are solely responsible for the biosynthesis of 5-OMe Tyr, and lacking these genes resulted in the production of desmethoxyl derivatives (Fig. 1 and traces v–vii), we then introduced the pHG06015 cosmid into the S. albus J1074 strain. LC-MS analysis of metabolic extracts showed that the recombinant strain produced a new compound 13 with a molecular weight of m/z 1009.4564 [M + H]+ (Fig. 1 and trace xii). Structure elucidation showed that 13 has no N-terminal cap and methoxyl group at the 6th tyrosine residue (Table S10 and Fig. S87–S94†).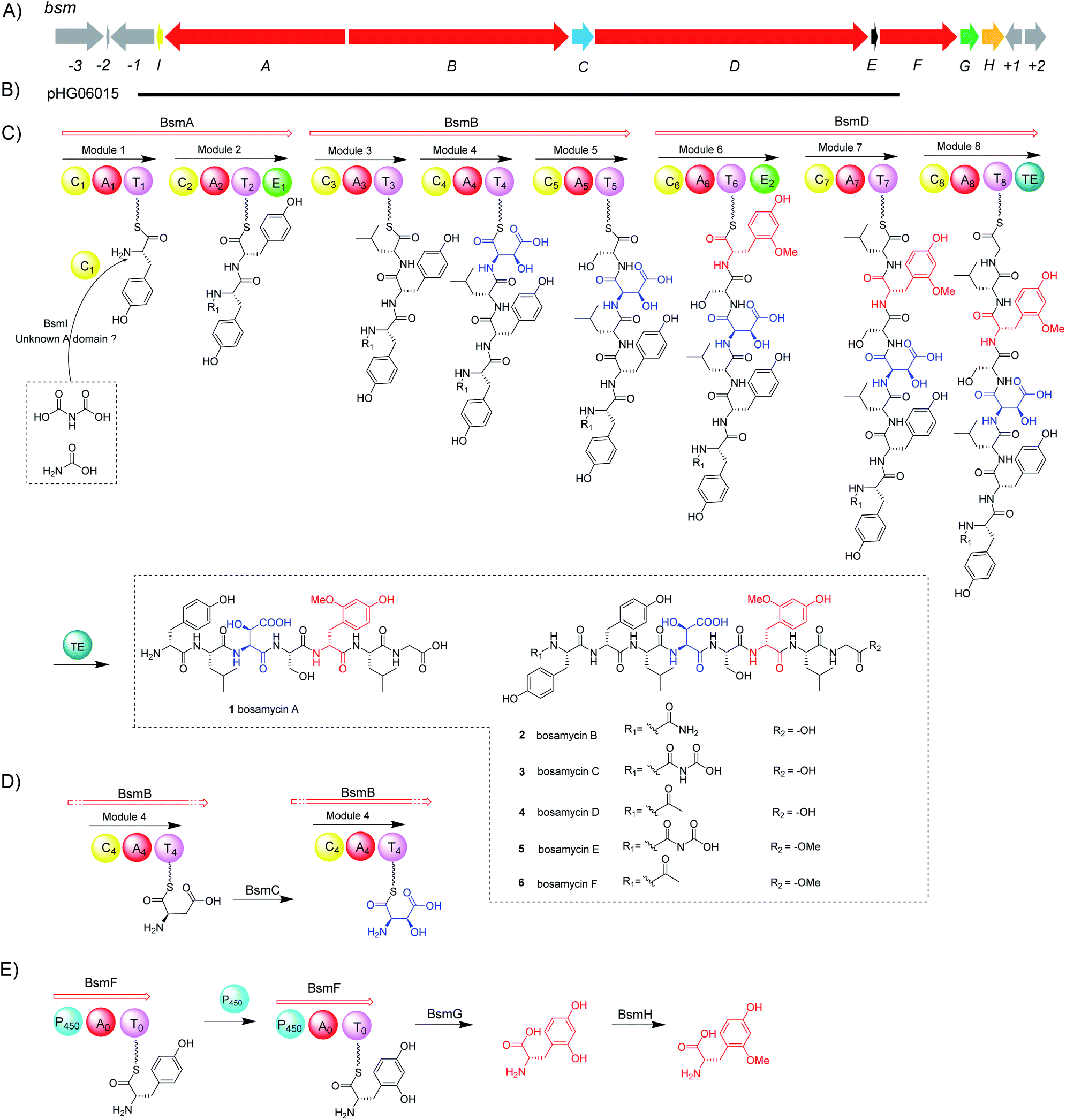 | ||
| Scheme 1 Biosynthesis of bosamycins. (A) Genetic organization of the bsm gene cluster, (B) relative location of cosmid pHG06015, (C) proposed biosynthetic pathway for bosamycins, (D) biosynthesis of 3-OH aspartic acid, and (E) biosynthesis of 5-OMe tyrosine. A, adenylation domain; T, thiolation domain; C, condensation domain; E, epimerization domain; TE, thioesterase domain; and P450, cytochrome P450 monooxygenase domain. | ||
To complete the biosynthetic pathway of bosamycins in a heterologous host, we further cloned bsmF, bsmG and bsmH genes together into an E. coli/Streptomyces shuttle vector, pUWL201-OriT, under the control of an ermE* promoter,53 yielding pHG06016. Upon introducing pHG06016 into the S. albus J1074/pHG06015 strain, we observed the production of a new compound 14 (Fig. 1 and trace xiii). Structure elucidation by NMR and HRESIMS established the structure of 14 to be L-Tyr1-D-Tyr2-L-Leu3-L-erythro-β-OH-Asp4-L-Ser5-D-5-OMeTyr6-L-Leu7-Gly8 (Table S11 and Fig. S95–S102†). Given the fact that 14 lacks the N-modification, we concluded that the N-terminal modification enzymes are encoded outside the bsm gene cluster in the S. sp. 120454 strain.
Conclusions
In conclusion, our genome mining targeting a noncanonical NRPS architecture reveals a unique NRPS enzyme where a cytochrome P450 domain is embedded. The successful characterization of bosamycins as new linear peptides demonstrates the feasibility of our genome mining strategy in natural product discovery. The biosynthesis of bosamycins was revealed through in vivo gene deletion, in vitro biochemical assay, and heterologous expression of the bsm gene cluster. We demonstrated that the rare P450-containing NRPS (BsmF) is involved in the biosynthesis of nonproteinogenic amino acid intermediate 5-OHTyr in a PCP-dependent manner. Our findings therefore expand the functional diversity of NRPSs, and the prospect of using unusual NRPSs as beacons for novel NRP discovery.Experimental section
General experimental procedures
1D and 2D NMR spectra were acquired on a Bruker Avance III HD400 or a Bruker Avance 600 spectrometer. The high resolution electrospray ionization mass spectrometry (HRESIMS) data were obtained on an Agilent 6530 spectrometer with a Poroshell 120 column (Agilent Technologies). All the analytical and semi-preparative HPLC analyses were performed on an Agilent 1260 HPLC system with a DAD detector equipped with a Poroshell 120 and a Zorbax Eclipse XDB column (Agilent Technologies), respectively. PCR amplifications were performed on a Bio-Rad S1000™ Thermal Cycler using Phanta Super-Fidelity DNA Polymerase. Recombinant proteins were purified on a GE AKTA pure system with a 5 mL HisTrap HP column (GE life sciences).Gene inactivation and complementation
To construct a plasmid for inactivating bsmD, upstream and downstream homologous arms were amplified from genomic DNA of S. sp. 120454 with primers listed in Table S3.† After DNA purification, the homologous arms were subcloned into a linearized pKC1139 plasmid, yielding plasmid pHG06003. Then, the pHG06003 plasmid was transformed into E. coli ET12563/pUZ8002 and further conjugated into a wild-type strain of 120454. The mutants showing an apramycin sensitive phenotype were confirmed by diagnostic PCR analysis. Similar procedures were performed to obtain other gene mutants.For bsmC gene complementation, the plasmid pSET152-KasOp* was digested using SpeI and EcoRI and the fragments for gene complementation were PCR-amplified from genomic DNA of S. sp. 120454 with primers listed in Table S3,† yielding pHG06009. pHG06009 was then transferred to the ΔbsmC mutant strain by conjugation to afford the gene complementation strain. Similar procedures were carried out to obtain other complemented strains.
Strain fermentation and analysis of metabolites from S. sp. 120454 wild-type and mutant strains
Fresh spores of the S. sp. 120454 wild-type strain were inoculated into TSB seed medium (17.0 g tryptone, 3.0 g soytone, 2.5 g glucose, 5.0 g sodium chloride and 2.5 g Na2HPO4 in 1 L water, pH 7.0) and cultivated for 24 h. The seed culture was then inoculated into B medium (dextrin (40 g), tomato paste (7.5 g), NZ amine (2.5 g), and primary yeast (5 g) in 1 L distilled water, pH 7.0) at 30 °C. After cultivation for 7 days, the fermentation broth was filtered and absorbed with a XAD-16 resin. The resin was washed with water and eluted with acetone. The HPLC analysis was performed using a 25 min solvent gradient from 20% to 30% acetonitrile in water supplied with 0.1 TFA at a flow rate of 0.5 mL min−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to Prof. Yasuhiro Igarashi (Toyama Prefectural University) for providing authentic erythro-L-OH Asp, and Prof. Ben Shen (The Scripps Research Institute) for providing Streptomyces sp. 120454. This work was financially supported by the NSFC (81925033, 21861142005, 81773591, 81673333, 81803380, and 2171101213), MOST (2019YFC0312500, 2018YFC1706200 and 2018YFA0902000), and Fundamental Research Funds for the Central Universities (14380092, 14380113, and 143801389).Notes and references
- M. A. Marahiel, Nat. Prod. Rep., 2016, 33, 136–140 RSC.
- C. T. Walsh, Nat. Prod. Rep., 2015, 33, 127–135 RSC.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- M. Strieker, A. Tanovic and M. A. Marahiel, Curr. Opin. Struct. Biol., 2010, 20, 234–240 CrossRef CAS PubMed.
- J. Shi, Y. J. Zeng, B. Zhang, F. L. Shao, Y. C. Chen, X. Xu, Y. Sun, Q. Xu, R. X. Tan and H. M. Ge, Chem. Sci., 2019, 10, 3042–3048 RSC.
- J. Shi, C. L. Liu, B. Zhang, W. J. Guo, J. Zhu, C. Y. Chang, E. J. Zhao, R. H. Jiao, R. X. Tan and H. M. Ge, Chem. Sci., 2019, 10, 4839–4846 RSC.
- H. Zhang, J. Chen, H. Wang, Y. Xie, J. Ju, Y. Yan and H. Zhang, FEBS Lett., 2013, 587, 1675–1680 CrossRef CAS PubMed.
- C. Savino, L. C. Montemiglio, G. Sciara, A. E. Miele, S. G. Kendrew, P. Jemth, S. Gianni and B. Vallone, J. Biol. Chem., 2009, 284, 29170–29179 CrossRef CAS PubMed.
- S. F Altschul, W. Gish, W. Miller, E. W. Myers and D. J Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef.
- M. Elibol, Process Biochem., 2004, 39, 1057–1062 CrossRef CAS.
- Y. H. Wang, B. Yang, J. Ren, M. L. Dong, D. Liang and A. L. Xu, Process Biochem., 2005, 40, 1161–1166 CrossRef CAS.
- M. E. Rateb, W. E. Houssen, W. T. Harrison, H. Deng, C. K. Okoro, J. A. Asenjo, B. A. Andrews, A. T. Bull, M. Goodfellow, R. Ebel and M. Jaspars, J. Nat. Prod., 2011, 74, 1965–1971 CrossRef CAS PubMed.
- M. Falzone, E. Martens, H. Tynan, C. Maggio, S. Golden, V. Nayda, E. Crespo, G. Inamine, M. Gelber and R. Lemence, Appl. Microbiol. Biotechnol., 2013, 97, 9535–9539 CrossRef CAS PubMed.
- J. Recktenwald, R. Shawky, O. Puk, F. Pfennig, U. Keller, W. Wohlleben and S. Pelzer, Microbiology, 2002, 148, 1105–1118 CrossRef CAS PubMed.
- O. Puk, D. Bischoff, C. Kittel, S. Pelzer, S. Weist, E. Stegmann, R. D. Süssmuth and W. Wohlleben, J. Bacteriol., 2004, 186, 6093–6100 CrossRef CAS PubMed.
- E. Stegmann, H. J. Frasch and W. Wohlleben, Curr. Opin. Microbiol., 2010, 13, 595–602 CrossRef CAS PubMed.
- S. Mulyani, E. Egel, C. Kittel, S. Turkanovic, W. Wohlleben, R. D. Sussmuth and K. H. van Pee, ChemBioChem, 2010, 11, 266–271 CrossRef CAS PubMed.
- G. M. Singh, P. D. Fortin, A. Koglin and C. T. Walsh, Biochemistry, 2008, 47, 11310–11320 CrossRef CAS PubMed.
- B. Boll, T. Taubitz and L. Heide, J. Biol. Chem., 2011, 286, 36281–36290 CrossRef CAS PubMed.
- M. Wolpert, B. Gust, B. Kammerer and L. Heide, Microbiology, 2007, 153, 1413–1423 CrossRef CAS PubMed.
- S. Lautru, D. Oves-Costales, J. L. Pernodet and G. L. Challis, Microbiology, 2007, 153, 1405–1412 CrossRef CAS PubMed.
- L. M. Alkhalaf, S. M. Barry, D. Rea, A. Gallo, D. Griffiths, J. R. Lewandowski, V. Fulop and G. L. Challis, J. Am. Chem. Soc., 2019, 141, 216–222 CrossRef CAS PubMed.
- A. Greule, J. E. Stok, J. J. De Voss and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 757–791 RSC.
- K. Woithe, N. Geib, K. Zerbe, D. B. Li, M. Heck, S. Fournier-Rousset, O. Meyer, F. Vitali, N. Matoba, K. Abou-Hadeed and J. A. Robinson, J. Am. Chem. Soc., 2007, 129, 6887–6895 CrossRef CAS PubMed.
- S. Pohle, C. Appelt, M. Roux, H. P. Fiedler and R. D. Süssmuth, J. Am. Chem. Soc., 2011, 133, 6194–6205 CrossRef CAS PubMed.
- S. Uhlmann, R. D. Süssmuth and M. J. Cryle, ACS Chem. Biol., 2013, 8, 2586–2596 CrossRef CAS PubMed.
- H. Chen, B. K. Hubbard, S. E. O'Connor and C. T. Walsh, Chem. Biol., 2002, 9, 103–112 CrossRef CAS PubMed.
- K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS PubMed.
- M. Steffensky, A. Mühlenweg, Z. X. Wang, S. M. Li and L. Heide, Antimicrob. Agents Chemother., 2000, 44, 1214–1222 CrossRef CAS PubMed.
- M. J. Cryle, A. Meinhart and I. Schlichting, J. Biol. Chem., 2010, 285, 24562–24574 CrossRef CAS PubMed.
- K. Haslinger, C. Brieke, S. Uhlmann, L. Sieverling, R. D. Süssmuth and M. J. Cryle, Angew. Chem., Int. Ed., 2014, 53, 8518–8522 CrossRef CAS PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- M. Peschke, M. Gonsior, R. D. Süssmuth and M. J. Cryle, Curr. Opin. Struct. Biol., 2016, 41, 46–53 CrossRef CAS PubMed.
- D. J. Edwards and W. H. Gerwick, J. Am. Chem. Soc., 2004, 126, 11432–11433 CrossRef CAS PubMed.
- H. Yeom, S. G. Sligar, H. Li, T. L. Poulos and A. J. Fulco, Biochemistry, 1995, 34, 14733–14740 CrossRef CAS PubMed.
- T. W. Ost, C. S. Miles, A. W. Munro, J. Murdoch, G. A. Reid and S. K. Chapman, Biochemistry, 2001, 40, 13421–13429 CrossRef CAS PubMed.
- K. J. Labby, S. G. Watsula and S. Garneau-Tsodikova, Nat. Prod. Rep., 2015, 32, 641–653 RSC.
- J. Liu, B. Wang, H. Z. Li, Y. C. Xie, Q. L. Li, X. J. Qin, X. Zhang and J. H. Ju, Org. Lett., 2015, 17, 1509–1512 CrossRef CAS PubMed.
- E. Kim, Y. H. Shin, T. H. Kim, W. S. Byun, J. Cui, Y. E. Du, H. J. Lim, M. C. Song, A. S. Kwon, S. H. Kang, J. Lee, S. K. Shin, J. Jang, D. C. Oh and Y. J. Yoon, Biomolecules, 2019, 9, 672–686 CrossRef CAS PubMed.
- J. M. Neary, A. Powell, L. Gordon, C. Milne, F. Flett, B. Wilkinson, C. P. Smith and J. Micklefield, Microbiology, 2007, 153, 768–776 CrossRef CAS PubMed.
- M. Strieker, E. M. Nolan, C. T. Walsh and M. A. Marahiel, J. Am. Chem. Soc., 2009, 131, 13523–13530 CrossRef CAS PubMed.
- K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS PubMed.
- Z. L. Reitz, C. D. Hardy, J. Suk, J. Bouvet and A. Butler, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19805–19814 CrossRef CAS PubMed.
- C. W. Johnston, M. A. Skinnider, M. A. Wyatt, X. Li, M. R. Ranieri, L. Yang, D. L. Zechel, B. Ma and N. A. Magarvey, Nat. Commun., 2015, 6, 8421–8431 CrossRef CAS PubMed.
- C. F. Michelsen, J. Watrous, M. A. Glaring, R. Kersten, N. Koyama, P. C. Dorrestein and P. Stougaard, mBio, 2015, 6, e00079 CrossRef PubMed.
- M. Rottig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed.
- C. M. Mudalungu, W. J. von Torne, K. Voigt, C. Ruckert, S. Schmitz, O. N. Sekurova, S. B. Zotchev and R. D. Sussmuth, J. Nat. Prod., 2019, 82, 1478–1486 CrossRef CAS PubMed.
- J. Kottur and D. T. Nair, Nucleic Acids Res., 2018, 46, 5875–5885 CrossRef CAS PubMed.
- Y. L. Du, D. S. Dalisay, R. J. Andersen and K. S. Ryan, Chem. Biol., 2013, 20, 1002–1011 CrossRef CAS PubMed.
- L. Li, Z. Xu, J. Wu, Y. Zhang, X. He, T. M. Zabriskie and Z. Deng, ChemBioChem, 2008, 9, 1286–1294 CrossRef CAS PubMed.
- J. Shi, X. Xu, E. J. Zhao, B. Zhang, W. Li, Y. Zhao, R. H. Jiao, R. X. Tan and H. M. Ge, Org. Lett., 2019, 21, 6825–6829 CrossRef CAS.
- J. D. Rudolf, L. B. Dong, T. Huang and B. Shen, Mol. BioSyst., 2015, 11, 2717–2726 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03469j |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |