
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic studies toward longeracemine: a SmI2-mediated spirocyclization and rearrangement cascade to construct the 2-azabicyclo[2.2.1]heptane framework†
Keita
Komine
,
Kyle M.
Lambert
 ,
Quentin R.
Savage
,
Joshua B.
Cox
and
John L.
Wood
*
,
Quentin R.
Savage
,
Joshua B.
Cox
and
John L.
Wood
*
Department of Chemistry and Biochemistry, Baylor University, One Bear Place 97348, Waco, TX 76798, USA. E-mail: john_l_wood@baylor.edu
First published on 30th July 2020
Abstract
Longeracemine, a member of the Daphniphyllum family of alkaloids contains a novel carbon framework featuring a highly functionalized 2-azabicyclo[2.2.1]heptane core as part of an overall 5/6/5/5/6/5 skeleton. A synthetic intermediate containing the core of longeracemine has been efficiently prepared by employing a stereoselective SmI2-mediated cascade reaction to advance a 7-azabicyclo[2.2.1]heptadiene to a 2-azabicyclo[2.2.1]heptene that is functionally poised for conversion to the natural product.
Introduction
Since the isolation of daphnimacrine in 1909 by Yagi,1a over 320 members of the Daphniphyllum family have been isolated from evergreen shrubs and trees from southeast Asia.1Daphniphyllum alkaloids have been shown to exhibit a wide variety of biological activities including cytotoxicity against a number of cancer cell lines, antioxidant properties, inhibition of platelet aggregation, insecticidal, and anti-HIV activity.1c,2g In addition to their promising pharmacological profiles, these alkaloids are structurally diverse and possess a plethora of polycyclic fused structures, and as such, have gained a wide-interest in the synthetic community as highly sought-after and attractive targets for total synthesis.2 The Daphniphyllum alkaloids isolated to date, have been found to contain 35 unique carbon frameworks,2g and longeracemine, which was isolated in 2013 by Di and co-workers from the fruits of Daphniphyllum longeracemosum,3 possesses a novel structure consisting of a 2-azabicyclo[2.2.1]heptane core. Longeracemine's bicyclic system features a tertiary amine, eight alkyl substituents, and three contiguous quaternary carbons within an overall 5/6/5/5/6/5 skeleton (Fig. 1).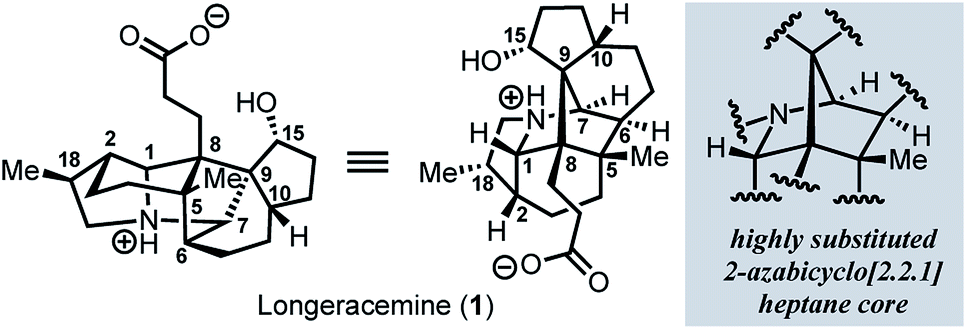 | ||
| Fig. 1 Longeracemine (1) and the highly substituted 2-azabicyclo-[2.2.1]heptane core. | ||
Recently, we reported an efficient assembly of the 2-azabicyclo[2.2.1]heptane core of 1 through an intramolecular [4+2] cycloaddition of 3H-pyrroles (Scheme 1).4 Exposure of diols 2a-b to Swern oxidation, amine condensation, and an acid-mediated [4+2] cycloaddition afforded the desired bicycles 4a and 4b in 33% and 42% yields, respectively, in single pot transformations; 2c failed to afford 4c (Scheme 1a). Employing optimized conditions, the reaction of a more functionalized diol (5), to access 7, which is better poised for elaboration to longeracemine (1) compared to 4a-c was explored (Scheme 1b). However, the [4+2] cycloaddition was found to be highly substrate dependent and the reaction of 5 failed to provide desired compound 7.4 Based on these results, an alternative approach was required to construct the 2-azabicyclo[2.2.1]heptane framework. Herein, we report that a novel SmI2-mediated cascade reaction, involving a spirocyclization and rearrangement, affords a 2-azabicyclo[2.2.1]heptene framework that is suitably functionalized for advancement to 1 (Scheme 1c). The reaction proceeds in good yield, with excellent regio- and stereoselectivity.
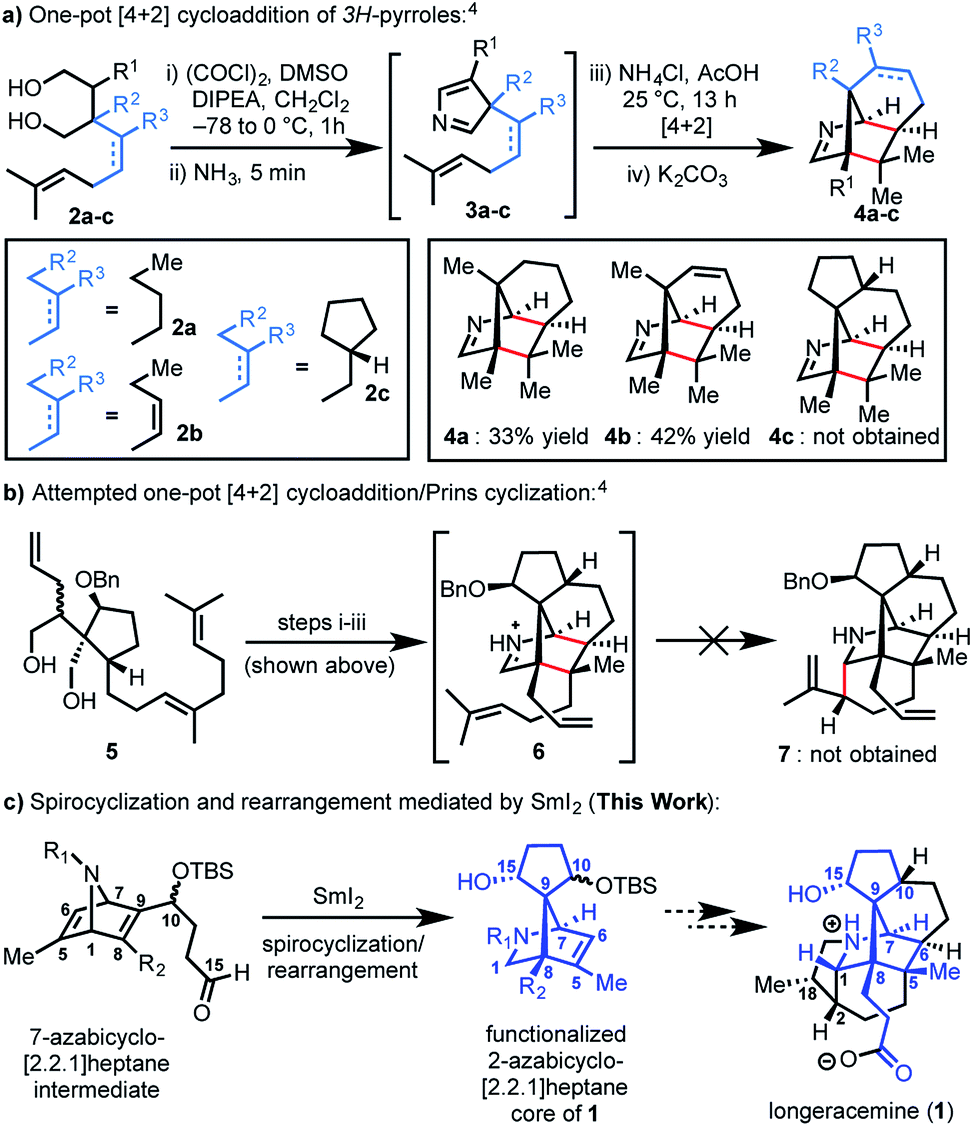 | ||
| Scheme 1 (a) Our previous work involving an intramolecular [4+2] cycloaddition; (b) our attempted tandem [4+2] cycloaddition/Prins cyclization; (c) SmI2 mediated spirocyclization/rearrangement to afford the 2-azabicyclo[2.2.1]heptane core of 1; DMSO = dimethylsulfoxide, DIPEA = diisopropylethylamine, Bn = benzyl, TBS = tert-butyldimethylsilyl. | ||
Results and discussion
In 1958, Cristol and co-workers reported that the addition of aryl mercaptans to norbornadiene under radical conditions generates homoallyl radical 8, which undergoes a reversible ring closure to form nortricyclene radical 9, resulting in a (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture of nortricyclane and norbornene products; products resulting from rearranged norbornene radical 10 were never observed (Scheme 2a).5 More recently, Hodgson and co-workers have reported a selective radical rearrangement of 7-azabicyclo[2.2.1]heptadienes that is guided by the increased stability of a resultant α-nitrogen radical versus a secondary alkyl radical (cf.13a to 10; Scheme 2b).6,7 In one instance, Hodgson demonstrated that treatment of vinyl sulfone 11 with 2-iodoethanol and an excess of tributyltin hydride in presence of a catalytic amount of triethylborane and oxygen afforded the rearranged adduct 13 in 72% yield.7g Given the range of substrates evaluated and seemingly robust nature of this transformation,7 we envisioned that this radical rearrangement could be performed in more complex systems and perhaps prove useful for construction of the bicyclic framework of longeracemine (1).
:1) mixture of nortricyclane and norbornene products; products resulting from rearranged norbornene radical 10 were never observed (Scheme 2a).5 More recently, Hodgson and co-workers have reported a selective radical rearrangement of 7-azabicyclo[2.2.1]heptadienes that is guided by the increased stability of a resultant α-nitrogen radical versus a secondary alkyl radical (cf.13a to 10; Scheme 2b).6,7 In one instance, Hodgson demonstrated that treatment of vinyl sulfone 11 with 2-iodoethanol and an excess of tributyltin hydride in presence of a catalytic amount of triethylborane and oxygen afforded the rearranged adduct 13 in 72% yield.7g Given the range of substrates evaluated and seemingly robust nature of this transformation,7 we envisioned that this radical rearrangement could be performed in more complex systems and perhaps prove useful for construction of the bicyclic framework of longeracemine (1).
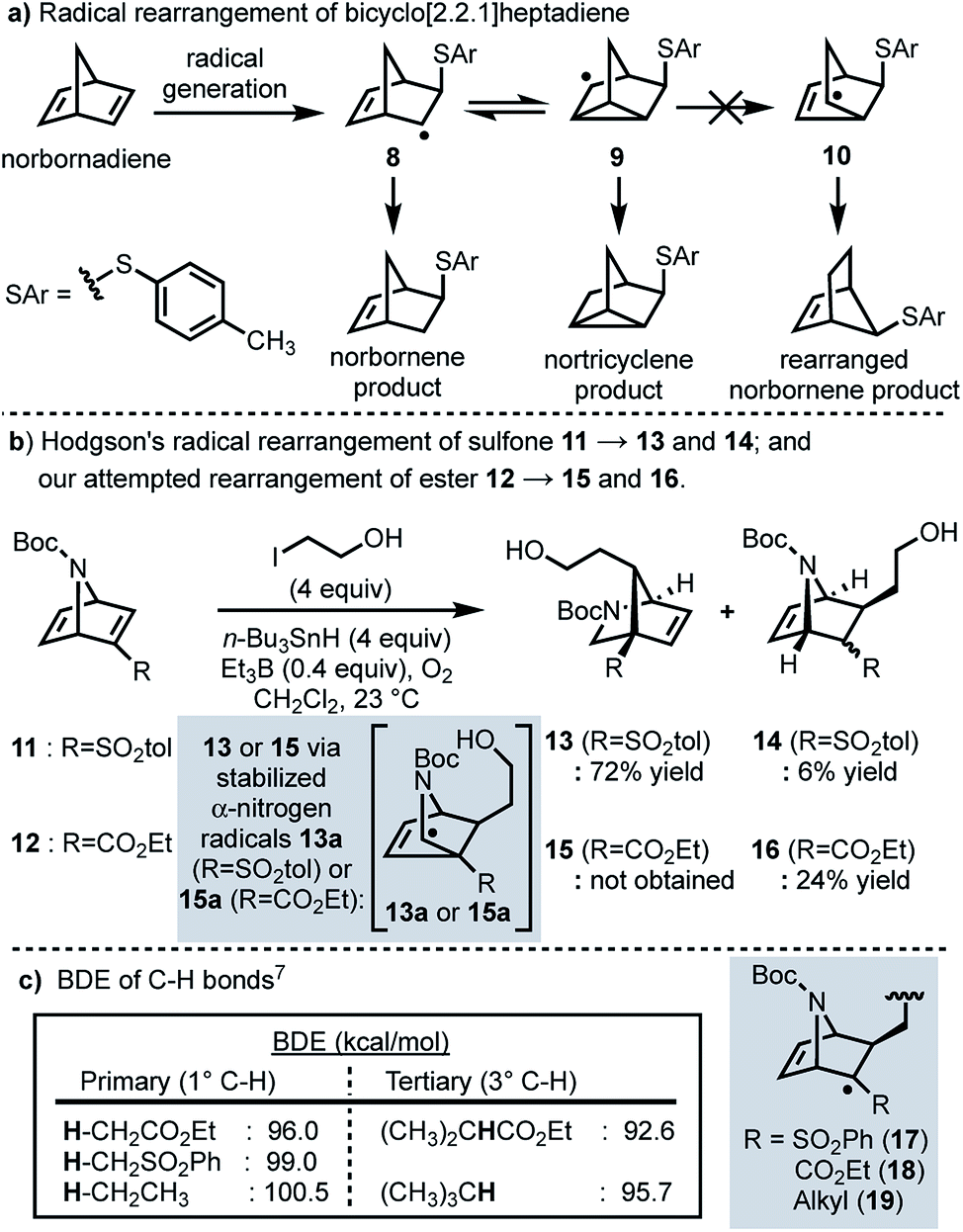 | ||
| Scheme 2 (a) Radical rearrangement of bicyclo[2.2.1]heptadiene; (b) radical rearrangements of 7-azabicyclo[2.2.1]heptadienes; (c) BDEs of relevant C–H bonds. | ||
In considering the application of Hodgson's rearrangement to 1, we first focused our attention on the sulfone moiety. Although advancing the derived neopentyl sulfones might be possible in a synthesis, substrates wherein sulfur is replaced by carbon were envisioned as much more viable. Thus, in an initial study we evaluated whether this rearrangement could be performed on the respective ester. To this end we prepared ethyl ester 12 which, upon exposure to Hodgson's conditions, was found to exclusively afford the non-rearranged product 16 in 24% yield (Scheme 2b). The failure of the intermediate radical derived from 12 to undergo rearrangement and furnish 15, compared to the efficient conversion of 11 to 13, clearly indicated that the electron-withdrawing nature of the substituent alpha to the intermediate radical (sulfone vs. ester) was important as well, not just the increased stability of the terminal α-nitrogen radical (i.e.13aversus10). Thus, we considered the differences in the known bond dissociation energies (BDE) of C–H bonds adjacent to ester, sulfone, and alkyl moieties (Scheme 2c).8 The BDE of a primary Cα–H bond for an ester is lower than that of a sulfone or an alkyl group; 96.0 vs. 99.0 and 100.5 kcal mol−1, respectively (Scheme 2c). The BDE of the Cα–H bond of a tertiary sulfone has, to our knowledge, not been reported, however, by analogy one might anticipate it as being similar to the C–H bond of a tertiary alkyl group (95.7 kcal mol−1; Scheme 2c). Thus, we anticipated that replacing the sulfone with an alkyl group (cf., 17 and 19) would result in a slightly more reactive intermediate, thereby facilitating the desired rearrangement.
Based on these preliminary results, as illustrated retrosynthetically in Scheme 3, we envisioned that longeracemine (1) would be derived from ketone 20, which would, in turn, arise from iodide 21via tandem radical cyclization (Scheme 3).9 To access iodide 21, we considered an approach from 22 that called for homologation at the C10 ketone with acetonitrile10 and iodination at C4. For the stereoselective synthesis of 22, we seek to employ vinyl iodide 23 in a SnBu3H or SmI2-mediated 1,5-hydrogen translocation to afford an α-aminoalkyl radical which would be followed by a 5-exo-trig cyclization.11 Vinyl iodide 23 would arise from deprotection and N-alkylation of 25 with 24, available from 26.12 Key intermediate 25 would, in turn, arise from 29via a tandem radical spirocyclization/rearrangement sequence triggered by initial formation of a Sm-derived ketyl radical.13,14 Spirocyclization of the ketyl in a 5-exo-trig-fashion would afford tertiary radical 28 which, in contrast to the analogous radical derived from 12, was anticipated to undergo rearrangement to intermediate cyclopropane 27. Subsequent homolysis of the C1–C5 bond in 27 and hydrogen atom abstraction,7b,15 or further reduction of the stabilized α-nitrogen radical by excess SmI2 to the secondary Sm-anion16 followed by protonation, would then deliver 25. We anticipated that 29 could be prepared from N-Boc pyrrole 30 and dienophile 31 by a Diels–Alder reaction.
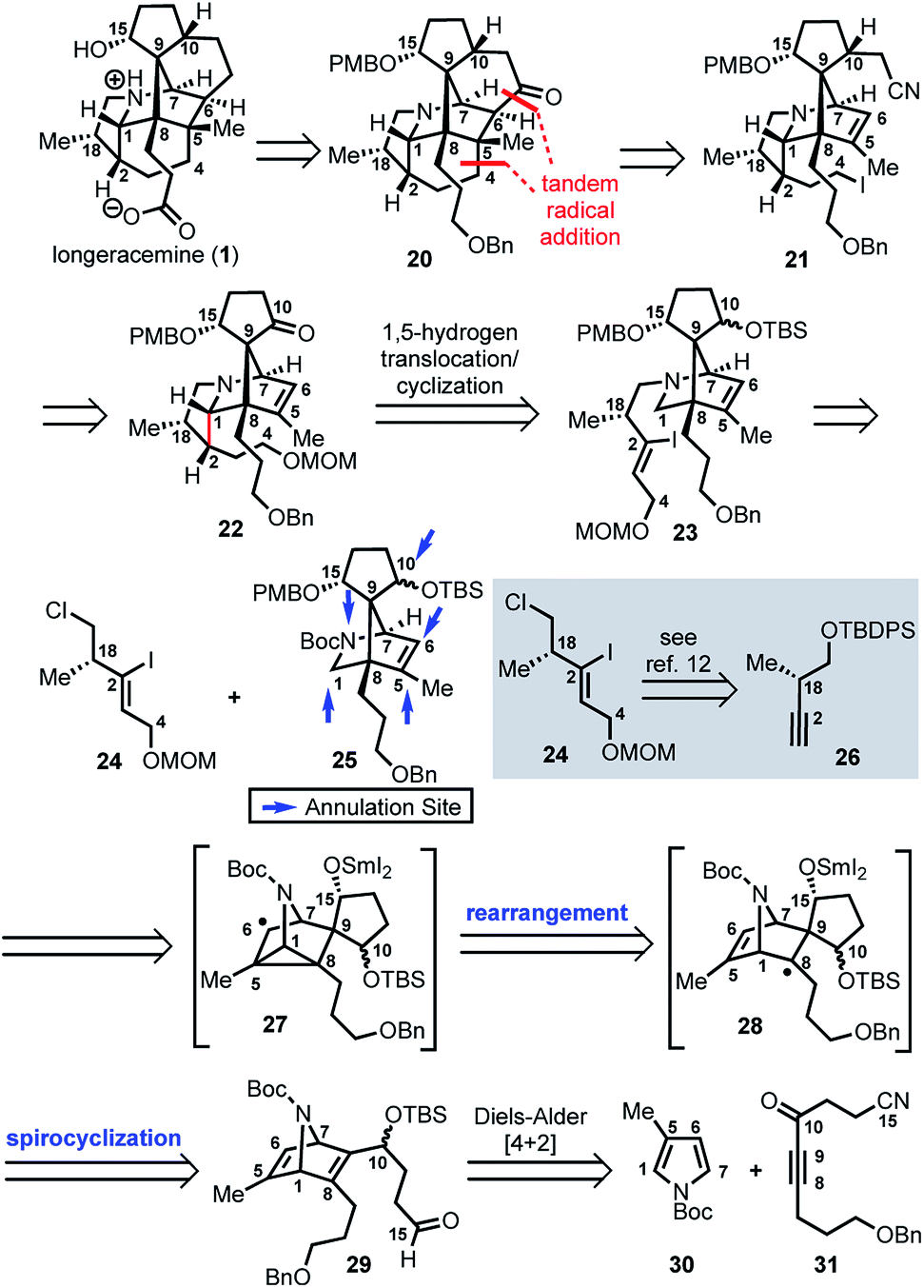 | ||
| Scheme 3 Retrosynthesis of longeracemine (1); PMB = p-methoxybenzyl, Boc = t-butoxycarbonyl. | ||
Our synthetic efforts began with the preparation of aldehyde 29via a Diels–Alder reaction (Scheme 4). Ozonolysis of commercially available nitrile 32, followed by nucleophilic alkynylation with 33,17 and oxidation of the resulting alcohol furnished 31. Next, we examined various thermal and Lewis acid conditions to promote the [4+2] cycloaddition of pyrroles 30, 34, and 35; however, desired bicycle 36 was not obtained.18,19 Since pyrroles are generally poor dienes for the Diels–Alder reaction and can react with dienophiles to afford Michael addition products, we sought to alter the electron-withdrawing nature of the dienophile (Table 1).20,21
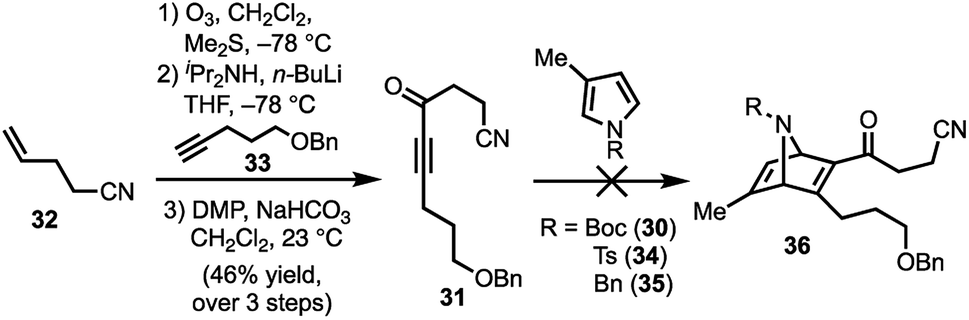 | ||
| Scheme 4 Attempt to perform the Diels–Alder reaction of 31 with pyrroles 30, 34, and 35; DMP = Dess–Martin periodinane. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Temperature | Pyrrole (30, 34, 35) | Alkyne (37) | Yielda (%) |
|
a Yield (%) refers to the isolated yields of the mixture; ratios of the products (38:39) were determined by 1H NMR analysis of the mixture; Ts = p-toluenesulfonyl.
|
|||||
| 1 | Boc (30) | 90 °C | 2 equiv. | 1 equiv. | 39% (4:1) |
| 2 | Ts (34) | 90 °C | 2 equiv. | 1 equiv. | 14% (4:1) |
| 3 | Boc (30) | 90 °C | 1 equiv. | 2 equiv. | 55% (5:1) |
| 4 | Ts (34) | 90 °C | 1 equiv. | 2 equiv. | 21% (3:1) |
| 5 | Ts (34) | 110 °C | 1 equiv. | 2 equiv. | 0% |
| 6 | Bn (35) | 90 °C | 1 equiv. | 2 equiv. | 0% |
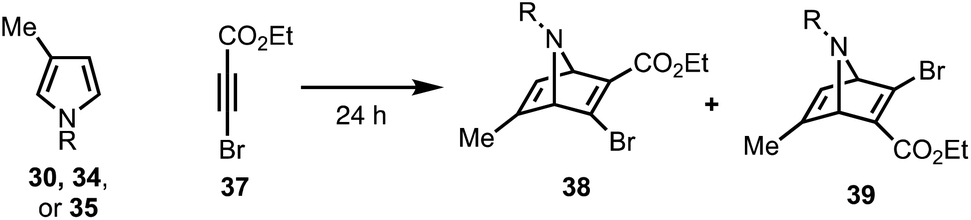
To this end, we examined the use of ethyl 3-bromopropiolate 37, under thermal conditions (Table 1). Treatment of 3022 or 3423 with 3724 at 90 °C under neat conditions, afforded mixtures (4:1) of N-Boc and N-Ts protected regiomers 38 and 39 in 39% and 14% yield, respectively (entries 1–2), as well as Michael adducts. Reversing the ratio of pyrrole to alkyne from 2:1 to 1:2, respectively, improved the overall yields (entries 3–4) while increasing the temperature proved ineffective (entry 5). Notably, the use of a relatively electron-rich N-benzyl pyrrole (35) failed to furnish any cycloaddition products (entry 6).
With N-Boc-38 in hand, we moved to prepare key intermediate 29 (Scheme 5). The regiomeric mixture containing N-Boc-38 was subjected to an organocopper coupling25 with 40 to afford 41 in 88% yield. Ester 41 was then converted to aldehyde 42 by DIBAL-H reduction and subsequent Dess–Martin oxidation of the resulting alcohol. At this point, chromatography allowed removal of the unwanted regiomer and isolation of 42. Next, aldehyde 42 was subjected to Grignard reagent 43 in the presence of CeCl3, which suppresses 1,4-addition and facilitates production of the desired alcohol 44 (71% yield).26 Protection of the secondary alcohol in 44 with TBSOTf, followed by deprotection of the PMB-ether (DDQ) and Dess–Martin oxidation of the resulting alcohol delivers aldehyde 29.
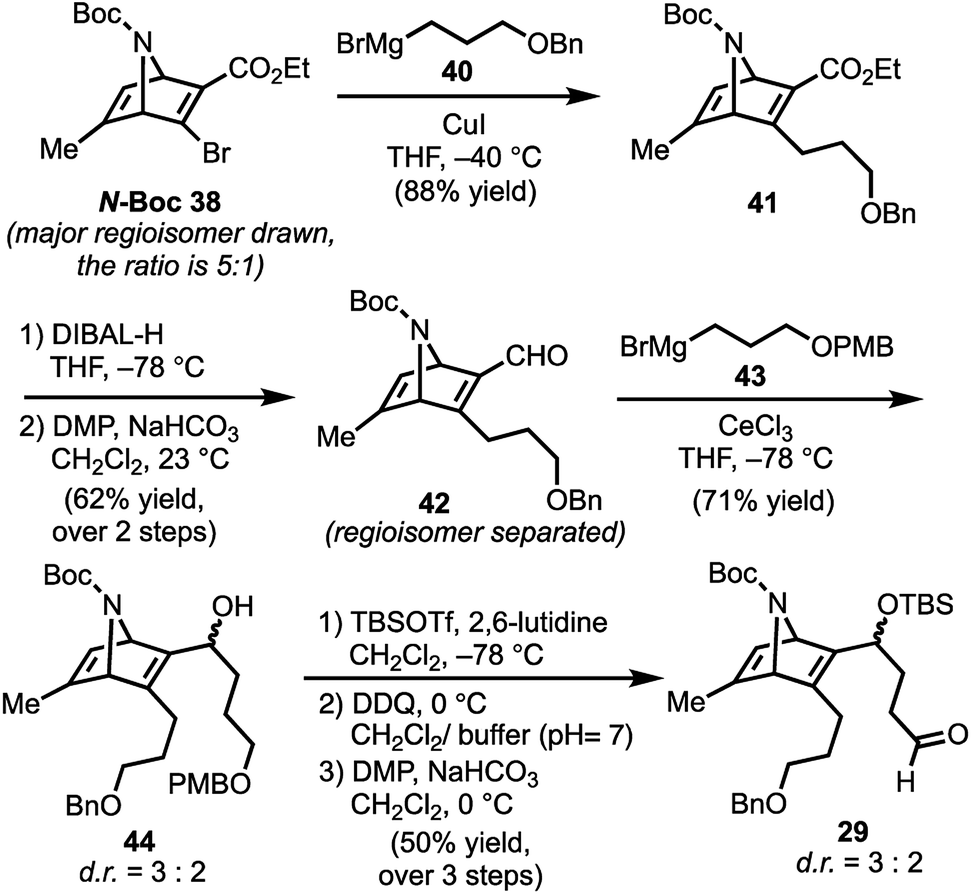 | ||
| Scheme 5 Preparation of key intermediate 29; DIBAL-H = diisobutylaluminum hydride, TBSOTf = tert-butyldimethylsilyl trifluoromethanesulfonate; DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. | ||
With 29 in hand, we were able to investigate the key SmI2-mediated spirocyclization/rearrangement cascade (Scheme 6). In the event, treatment of 29 with three molar equivalents of SmI2 in THF at 23 °C for 1 h furnished desired 2-azabicyclo[2.2.1]heptenes 48a and 48b in good yield with excellent site selectivity, and in a stereoselective fashion. We were delighted to find that reaction proceeds under these very mild conditions, and exclusively affords the C-15 hydroxyl stereochemistry necessary to access 1 as evident by 1H NOESY analysis; a strong NOE correlation is observed between the C15 hydrogen and the equatorial C1 hydrogen in both 48a and 48b.27 Mechanistically, we believe that ketyl radical 45 is generated via reaction of SmI2 with aldehyde 29 (Scheme 6). Subsequent 5-exo-trig spirocyclization of 45 furnishes tertiary radical 28 wherein the newly formed stereogenic center derives from the ketyl radical (45) engaging the olefin from the least hindered face with the C(15) hydrogen over the ring system thereby avoiding steric repulsion between the bicycle and the OSmI2 moiety (cf., 45-disfavored and 45-favored). Finally, the intermediate tertiary radical (28) undergoes a 3-exo-trig cyclization to radical 27, which then rapidly fragments7b to afford the stabilized α-nitrogen radical 46. Termination of the sequence can occur through two possible pathways, which efficiently delivers 48a and 48b as a pair of inconsequential epimers (3:2) at C10 in a combined isolated yield of 70%. One pathway involves the further reduction of 46 in the presence of excess SmI2,16 to dipole-stabilized Sm-anion 4728 followed by protonation (“path A”; Scheme 6). A second, more direct pathway involves hydrogen atom abstraction by 46 (“path B”; Scheme 6). Experimentally, quenching of the terminal reactive species was attempted with D2O and CH3OD at −78 °C to effect deuterium exchange, however, no deuterium incorporation was ever observed and 48a and 48b were cleanly afforded; thus, H-atom abstraction from the THF solvent (path B; Scheme 6) cannot be ruled out at this time. Additionally, when the sequence is conducted with 2 molar equivalents of SmI2versus 3 molar equivalents, 48a and 48b are produced a significantly reduced yield of 42%, lending support for “path A” over “path B”, which would not require an excess of SmI2. Regardless, of the details of the termination sequence, the SmI2-mediated cascade stereoselectively forges two adjacent all-carbon quaternary centers and sets the correct stereochemistry at the C-15 hydroxyl necessary to access 1. Protection of the C15 secondary alcohol as a triethylsilyl (TES) ether can be accomplished using triethylsilyl trifluoromethanesulfonate and 2,6-lutidine; concomitant removal of the Boc group, which conveniently reveals the secondary amine, as demonstrated with 48a → 49, and sets the stage for further advancement of 49 to 1 (Scheme 7).27
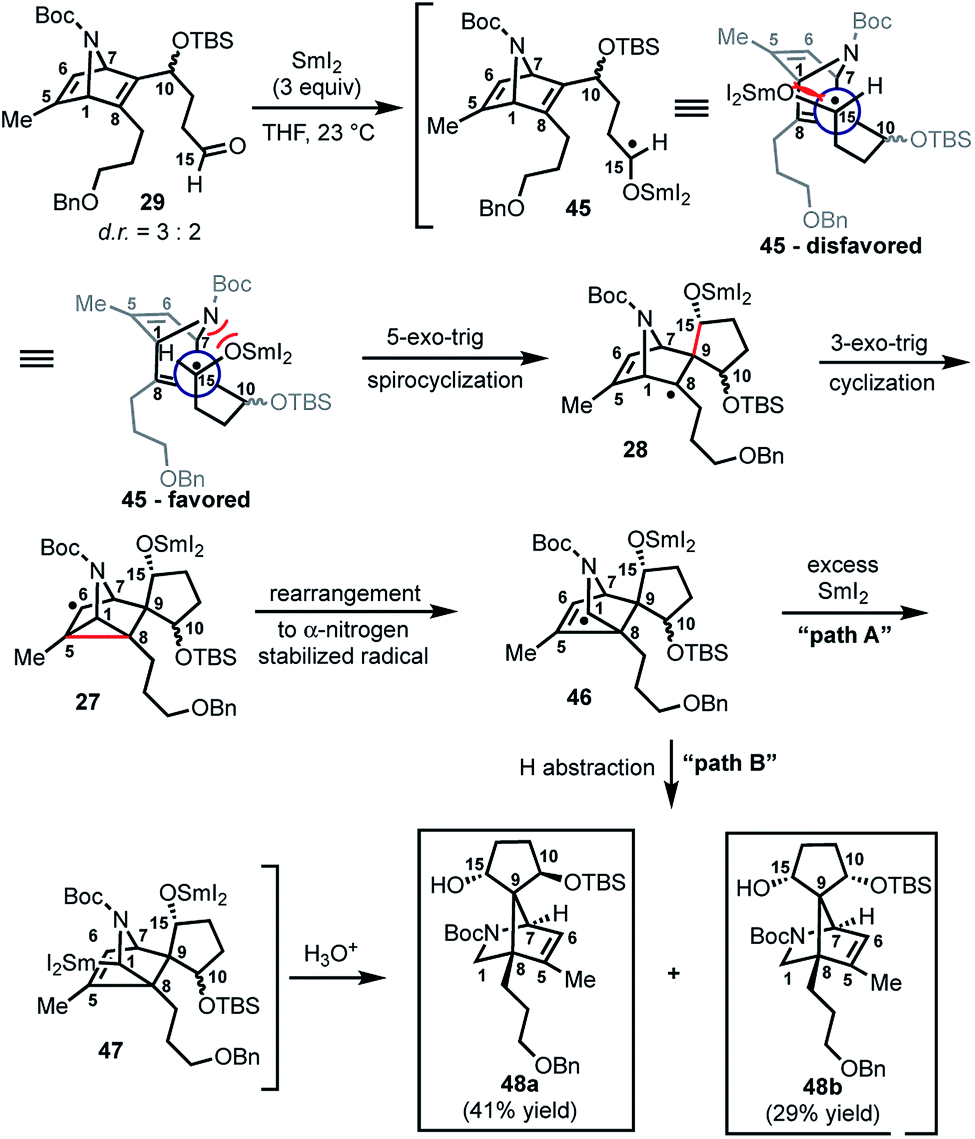 | ||
| Scheme 6 SmI2-mediated spirocyclization/rearrangement cascade to construct key bicyclic framework 25. | ||
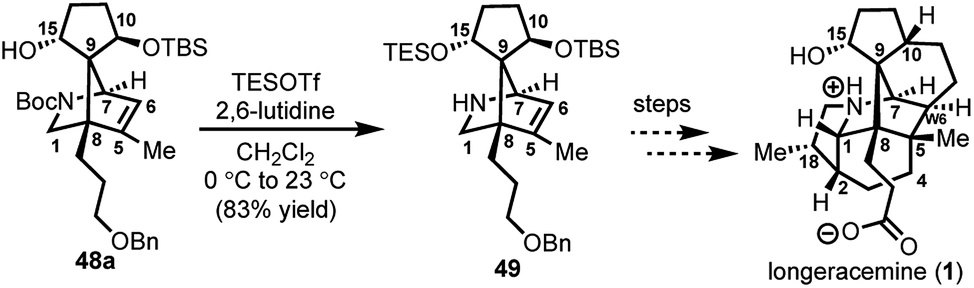 | ||
| Scheme 7 C15 hydroxyl protection of 48a and concomitant Boc removal to afford 49; TESOTf = triethylsilyl trifluoromethanesulfonate. | ||
Conclusions
In conclusion, we have accessed the 2-azabicyclo[2.2.1]heptane framework of longeracemine (1) via a novel SmI2 spirocyclization/rearrangement cascade. The reaction proceeds under mild conditions, in good yield, with excellent site selectivity, and in a stereoselective fashion. Our efforts to implement this rearrangement in a complex setting revealed that success is dependent on the stability of the radical intermediate (e.g., 17–19, and 28), which can be estimated by considering the BDE of corresponding C–H bond. Investigation of the biological activity of the intermediates produced in this synthetic sequence along with efforts to complete their advancement to longeracemine (1) are currently underway and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from Baylor University, the Welch Foundation (Chair, AA-006), the Cancer Prevention and Research Institute of Texas (CPRIT, R1309), the National Science Foundation (NSF, CHE-1764240), and the National Institute of General Medical Sciences of the National Institutes of Health (NIGMS-NIH, R01GM136759). K. M. L. is grateful for an NIH NRSA postdoctoral fellowship (NIGMS-NIH, F32GM129969).Notes and references
- (a) S. Yagi, Kyoto Pharmaceutical Science Journal, 1909, 6, 208–222 Search PubMed; (b) J. Kobayashi and T. Kubota, Nat. Prod. Rep., 2009, 26, 936–962 RSC; (c) H. Wu, X. Zhang, L. Ding, S. Chen, J. Yang and X. Xu, Planta Med., 2013, 79, 1589–1598 CrossRef CAS PubMed.
- For examples of recent total syntheses of Daphniphyllum alkaloids, see: (a) W. Zhang, M. Ding, J. Li, Z. Guo, M. Lu, Y. Chen, L. Liu, Y.-H. Shen and A. Li, J. Am. Chem. Soc., 2018, 140, 4227–4231 CrossRef CAS PubMed; (b) H. Shi, I. N. Michaelides, B. Darses, P. Jakubec, Q. N. N. Nguyen, R. S. Paton and D. J. Dixon, J. Am. Chem. Soc., 2017, 139, 17755–17758 CrossRef CAS PubMed; (c) A. Shvartsbart and A. B. Smith, J. Am. Chem. Soc., 2015, 137, 3510–3519 CrossRef CAS PubMed; (d) Z. Lu, Y. Li, J. Deng and A. Li, Nat. Chem., 2013, 5, 679–684 CrossRef CAS PubMed; (e) X. Chen, H.-J. Zhang, X. Yang, H. Lv, X. Shao, C. Tao, H. Wang, B. Cheng, Y. Li, J. Guo, J. Zhang and H. Zhai, Angew. Chem., Int. Ed., 2018, 57, 947–951 CrossRef CAS PubMed; for reviews, see: (f) B. Kang, P. Jakubec and D. J. Dixon, Nat. Prod. Rep., 2014, 31, 550–562 RSC; (g) A. K. Chattopadhyay and S. Hanessian, Chem. Rev., 2017, 117, 4104–4146 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhang, T.-Q. Yang, Y.-T. Di and X.-J. Hao, RSC Adv., 2013, 3, 9658–9661 RSC.
- J. B. Cox and J. L. Wood, Tetrahedron, 2018, 74, 4539–4549 CrossRef CAS.
- (a) S. J. Cristol, G. D. Brindell and J. A. Reeder, J. Am. Chem. Soc., 1958, 80, 635–640 CrossRef CAS; (b) S. J. Cristol and D. I. Davies, J. Org. Chem., 1964, 29, 1282–1284 CrossRef CAS; (c) C. R. Warner, R. J. Strunk and H. G. Kuivila, J. Org. Chem., 1966, 31, 3381–3384 CrossRef CAS; (d) D. C. Nonhebel, Chem. Soc. Rev., 1993, 22, 347–359 RSC; (e) A. Studer and M. Bossart, Tetrahedron, 2001, 57, 9649–9667 CrossRef CAS.
- D. J. Pasto, R. Krasnansky and C. Zercher, J. Org. Chem., 1987, 52, 3062–3072 CrossRef CAS.
- (a) D. M. Hodgson, C. R. Maxwell and I. R. Matthews, Synlett, 1998, 1349–1350 CrossRef CAS; (b) D. M. Hodgson, M. W. P. Bebbington and P. Willis, Chem. Commun., 2001, 889–890 RSC; (c) D. M. Hodgson, C. R. Maxwell, R. Wisedale, I. R. Matthews, K. J. Carpenter, A. H. Dickenson and S. J. Wonnacott, J. Chem. Soc., Perkin Trans. 1, 2001, 3150–3158 CAS; (d) D. M. Hodgson, M. W. P. Bebbington and P. Willis, Org. Lett., 2002, 4, 4353–4356 CrossRef CAS PubMed; (e) D. M. Hodgson, M. W. P. Bebbington and P. Willis, Org. Biomol. Chem., 2003, 1, 3787–3798 RSC; (f) D. M. Hodgson, M. L. Jones, C. R. Maxwell, O. Ichihara and I. R. Matthews, Org. Biomol. Chem., 2003, 1, 3787–3798 RSC; (g) D. M. Hodgson, S. Hachisu and M. D. Andrews, Org. Lett., 2005, 7, 815–817 CrossRef CAS PubMed; (h) D. M. Hodgson, S. Hachisu and M. D. Andrews, Synlett, 2005, 1267–1270 CrossRef CAS; (i) D. M. Hodgson and J.-M. Galano, Org. Lett., 2005, 7, 2221–2224 CrossRef CAS PubMed; (j) D. M. Hodgson, S. Hachisu and M. D. Andrews, J. Org. Chem., 2005, 70, 8866–8876 CrossRef CAS PubMed; (k) D. M. Hodgson and L. H. Winning, Synlett, 2006, 2476–2479 CrossRef CAS; (l) D. M. Hodgson, M. L. Jones, C. R. Maxwell, A. R. Cowley, A. L. Thompson, O. Ichihara and I. R. Matthews, Tetrahedron, 2009, 65, 7825–7836 CrossRef CAS.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- R. Tsang and B. Fraser-Reid, J. Am. Chem. Soc., 1986, 108, 2116–2117 CrossRef CAS.
- D. Kim, B.-M. Park and J. Yun, Chem. Commun., 2005, 1755–1757 RSC and references cited therein.
- (a) J. Robertson, M. A. Peplow and J. Pillai, Tetrahedron Lett., 1996, 5825–5828 CrossRef CAS; (b) M. Murakami, M. Hayashi and Y. Ito, Appl. Organomet. Chem., 1995, 385–397 CrossRef and references cited therein; (c) K. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC and references cited therein.
- (a) E. O. Onyango, J. Tsurumoto, N. Imai, K. Takahashi, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2007, 46, 6703–6705 CrossRef CAS PubMed; (b) B. M. Trost and J. P. N. Papillon, J. Am. Chem. Soc., 2004, 126, 13618–13619 CrossRef CAS PubMed.
- For examples of total syntheses using SmI2, see: (a) C. Beemelmanns and H.-U. Reissig, Angew. Chem., Int. Ed., 2010, 49, 8021–8025 CrossRef CAS PubMed; (b) T. Xu, C.-C. Li and Z. Yang, Org. Lett., 2011, 13, 2630–2633 CrossRef CAS PubMed; (c) J. Y. Cha, J. T. S. Yeoman and S. E. Reisman, J. Am. Chem. Soc., 2011, 133, 14964–14967 CrossRef CAS PubMed; (d) S. Breitler and E. M. Carreir, Angew. Chem., Int. Ed., 2013, 52, 11168–11171 CrossRef CAS PubMed; (e) N. J. Fazakerley, M. D. Helm and D. J. Procter, Chem.–Eur. J., 2013, 19, 6718–6723 CrossRef CAS PubMed; (f) I. Paterson, M. Xuan and S. M. Dalby, Angew. Chem., Int. Ed., 2014, 53, 7286–7289 CrossRef CAS PubMed; (g) S. Pan, B. Gao, J. Hu, J. Xuan, H. Xie and H. Ding, Chem.–Eur. J., 2016, 22, 959–970 CrossRef CAS PubMed; (h) J. Gong, H. Chen, X.-Y. Liu, Z.-X. Wang, W. Nie and Y. Qin, Nat. Commun., 2016, 7, 12183–12188 CrossRef CAS PubMed; (i) E. P. Farney, S. S. Feng, F. Schäfers and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 1267–1270 CrossRef CAS PubMed; (j) J. C. Leung, A. A. Bedermann, J. T. Njardarson, D. A. Spiegel, G. K. Murphy, N. Hama, B. M. Twenter, P. Dong, T. Shirahata, I. M. McDonald, M. Inoue, N. Taniguchi, T. C. McMahon, C. M. Schneider, N. Tao, B. M. Stoltz and J. L. Wood, Angew. Chem., Int. Ed., 2018, 57, 1991–1994 CrossRef CAS PubMed.
- For reviews on the use of SmI2 in synthesis, see: (a) D. J. Edmonds, D. Johnston and D. J. Procter, Chem. Rev., 2004, 104, 3371–3403 CrossRef CAS PubMed; (b) K. C. Nicolaou, S. P. Ellery and J. S. Chen, Angew. Chem., Int. Ed., 2009, 48, 7140–7165 CrossRef CAS PubMed; (c) M. Szostak, M. Spain and D. J. Procter, Chem. Soc. Rev., 2013, 42, 9155–9183 RSC; (d) M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS PubMed; (e) D. J. Procter, R. A. Flowers II and T. Skrydstrup, Organic Synthesis using Samarium Diiodide: A Practical Guide, RSC Publishing, Cambridge, 2009 Search PubMed.
- J. H. Rigby and C. Pigge, Tetrahedron Lett., 1996, 37, 2201–2204 CAS.
- (a) C. Beemelmanns and H.-U. Reissig, Pure Appl. Chem., 2011, 83, 507–518 CAS; (b) D. P. Curran and M. J. Totleben, J. Am. Chem. Soc., 1992, 114, 6050–6058 CAS.
- E. D. Slack, C. M. Gabriel and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 14051–14054 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, P. S. Baran, Y.-L. Zhong, H.-S. Choi, W. H. Yoon, Y. He and K. C. Fong, Angew. Chem., Int. Ed., 1999, 38, 1669–1675 CrossRef CAS; (b) N. Waizumi, T. Itoh and T. Fukuyama, J. Am. Chem. Soc., 2000, 122, 7825–7826 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- Z. Chen and M. L. Trudell, Chem. Rev., 1996, 96, 1179–1193 CrossRef CAS PubMed.
- Ethynyl p-tolyl sulfones have high reactivity toward pyrroles, however, such dienophiles are not suitable to access to 1.
- T. Ikawa, A. Takagi, Y. Kurita, K. Saito, K. Azechi, M. Egi, K. Kakiguchi, Y. Kita and S. Akai, Angew. Chem., Int. Ed., 2010, 49, 5563–5566 CrossRef CAS PubMed.
- P. Folly, PhD dissertation, University of Fribourg, Fribourg, Switzerland, 2000.
- F. Beltran, I. Fabre, I. Ciofini and L. Miesch, Org. Lett., 2017, 19, 5042–5045 CrossRef CAS PubMed.
- C. Zhang and M. L. Trudell, Tetrahedron, 1998, 54, 8349–8354 CrossRef CAS.
- T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima and Y. Kamiya, J. Am. Chem. Soc., 1989, 111, 4392–4398 CrossRef CAS.
- See the ESI† for further details.
- (a) P. Beak and W. J. Zajdel, J. Am. Chem. Soc., 1984, 106, 1010–1018 CrossRef CAS; (b) W. F. Bailey, P. Beak, S. T. Kerrick, S. Ma and K. B. Wiberg, J. Am. Chem. Soc., 2002, 124, 1889–1896 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectral data, FTIR data, and HRMS data. See DOI: 10.1039/d0sc03422c |
| This journal is © The Royal Society of Chemistry 2020 |