
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExamining histone modification crosstalk using immobilized libraries established from ligation-ready nucleosomes†
Diego
Aparicio Pelaz‡
a,
Zhadyra
Yerkesh‡
b,
Sören
Kirchgäßner‡
 a,
Henriette
Mahler‡
c,
Vladlena
Kharchenko
b,
Dulat
Azhibek
b,
Mariusz
Jaremko
b,
Henning D.
Mootz
d,
Łukasz
Jaremko
b,
Dirk
Schwarzer
*a and
Wolfgang
Fischle
*bc
a,
Henriette
Mahler‡
c,
Vladlena
Kharchenko
b,
Dulat
Azhibek
b,
Mariusz
Jaremko
b,
Henning D.
Mootz
d,
Łukasz
Jaremko
b,
Dirk
Schwarzer
*a and
Wolfgang
Fischle
*bc
aInterfaculty Institute of Biochemistry, University of Tübingen, Auf der Morgenstelle 34, D-72076 Tübingen, Germany. E-mail: dirk.schwarzer@uni-tuebingen.de
bBiological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia. E-mail: wolfgang.fischle@kaust.edu.sa
cLaboratory of Chromatin Biochemistry, Max Planck Institute for Biophysical Chemistry, 37077 Göttingen, Germany
dInstitute of Biochemistry, University of Muenster, Corrensstr. 36, 48149 Münster, Germany
First published on 20th August 2020
Abstract
Chromatin signaling relies on a plethora of posttranslational modifications (PTM) of the histone proteins which package the long DNA molecules of our cells in reoccurring units of nucleosomes. Determining the biological function and molecular working mechanisms of different patterns of histone PTMs requires access to various chromatin substrates of defined modification status. Traditionally, these are achieved by individual reconstitution of single nucleosomes or arrays of nucleosomes in conjunction with modified histones produced by means of chemical biology. Here, we report an alternative strategy for establishing a library of differentially modified nucleosomes that bypasses the need for many individual syntheses, purification and assembly reactions by installing modified histone tails on ligation-ready, immobilized nucleosomes reconstituted in a single batch. Using the ligation-ready nucleosome strategy with sortase-mediated ligation for histone H3 and intein splicing for histone H2A, we generated libraries of up to 280 individually modified nucleosomes in 96-well plate format. Screening these libraries for the effects of patterns of PTMs onto the recruitment of a well-known chromatin factor, HP1 revealed a previously unknown long-range cross-talk between two modifications. H3S28 phosphorylation enhances recruitment of the HP1 protein to the H3K9 methylated H3-tail only in nucleosomal context. Detailed structural analysis by NMR measurements implies negative charges at position 28 to increase nucleosomal H3-tail dynamics and flexibility. Our work shows that ligation-ready nucleosomes enable unprecedented access to the ample space and complexity of histone modification patterns for the discovery and dissection of chromatin regulatory principles.
Introduction
Genetic information is stored in form of chromatin in the nuclei of all eukaryotic cells. Chromatin organization is not only important for scaffolding of DNA, but also for orchestrating dynamic changes of genome activity. The basic structural unit of chromatin, referred to as nucleosome, is composed of an octamer of two each of the core histone proteins H2A, H2B, H3, and H4 with a stretch of 147 base pairs of DNA wrapped around. Chromatin-mediated gene regulation relies to a large extent on the unstructured N-terminal extensions of the histones, the so-called histone tails that are protruding outward from the nucleosome complex.1–3 These histone tails can be decorated with a multitude of posttranslational modifications (PTMs) that have been implicated in controlling DNA-dependent biological processes. The most common histone tail PTMs include acetylation and methylation of lysine residues as well as phosphorylation of serine and threonine residues.1–3 The biology of a given PTM depends on not only the modification type but also the defined position within the histone tails. For example, methylated lysine 4 of histone H3 (H3K4me) is mostly associated with chromatin regions of active transcription in euchromatin, whereas H3K9me and H3K27me are linked to transcriptionally silent heterochromatin. The distinct methylation level, mono (me1), di- (me2) or tri- (me3) methylation exerts further signaling function.4A major mechanism of histone tail PTM function is the recruitment of specific binding proteins that mediate downstream biological processes.5 These contain common ‘reader’ domains that have evolved to recognize histone PTMs in sequence-specific contexts.6 Heterochromatin protein 1 (HP1) was the first factor described to exhibit histone methyl-lysine binding activity.7 Genetic, cellular and biochemical evidence link HP1 proteins to the establishment and maintenance of heterochromatin domains in different model systems,7–9 possibly involving intrinsic phase separating properties.10–12 In agreement with a link to heterochromatin, HP1 proteins are conserved from yeast to man. These contain an N-terminal chromodomain that in vitro recognizes H3K9 methylation with a preference for the higher methylation states (i.e. H3K9me1 < H3K9me2 < H3K9me3; we refer to H3K9me in cases where such specificity is not entirely determined or functionally relevant).13 To what degree this binding property is contributing to the in vivo function of the factor is actively investigated, as other histone PTMs, RNA and protein–protein interaction might contribute to HP1 chromatin localization.
The fact that histone tails can be modified simultaneously by multiple PTMs has given rise to the idea that extensive crosstalk between modifications modulates their biological function.14 For example, we have described earlier that HP1 binding to H3K9me is attenuated by phosphorylation of the adjacent H3S10 residue.15,16 Assigning defined histone PTM patterns to the recruitment of chromatin factors is an ongoing challenge. On a cellular level, crosslinking based strategies for physically linking factors to chromatin with consecutive MS-based methods for determining modification patterns has been attempted.17 The approach, however, suffers from low throughput and limited resolution. Alternatively, affinity purification schemes using cellular extracts have been applied to map the interactome of synthetic histone tail peptides carrying defined modification patterns. The molecular mechanisms of the interaction of specific reader factors with defined histone PTM patterns have been analyzed using qualitative, quantitative and structural approaches.18–20 While informative, the histone tail-based experimental schemes cannot take the context of nucleosomes, the natural substrates of chromatin binding factors into account. In consequence, we and others have extended the affinity purification approaches for mapping the interactomes of histone PTMs to reconstituted chromatin templates carrying defined modification patterns.21,22 These and direct biochemical studies have reported that the context of chromatin can modulate the interaction of histone PTM reader proteins with their targets.1–3
The major obstacle for mapping the interaction properties of modification patterns with reading factors in chromatin context is the limited access to libraries of differentially modified nucleosomes. Designer nucleosomes with defined PTM patterns are commonly generated by protein semisynthesis of synthetic histones tail peptides that are fused to distinct recombinant globular domains of the core histones.23–26 The individually reconstituted semisynthetic histones are then assembled with the remaining core histones into nucleosomes and used for biochemical studies. In tour de force work, Muir and coworkers have extended this approach to assembling libraries of 64 and 115 modified nucleosomes where the DNA used for individual reconstitution was barcoded. This allowed readout of the interaction of chromatin factors with their targets using next-generation sequencing (NGS).27,28 While this approach has proven useful, the synthesis of designer nucleosomes in large quantities and multiple variations remains highly challenging. The central bottleneck is the nucleosomal reconstitution process. It is both time and material consuming and needs to be conducted for each nucleosome separately.
Results and discussion
Sortase-mediated ligation of ligation-ready nucleosomes for generating a library of H3-modified nucleosomes
To bypass the need for reconstituting nucleosomes on the basis of individually synthesized recombinant designer histones, we looked for a chemoselective ligation strategy that could work in the context of preassembled nucleosomes containing N-terminally truncated histones. We will refer to as ligation-ready nucleosomes to this approach in the following. The pivotal requirements in this context were high-yielding ligation reactions and simple removal procedures of excess peptides since laborious purification steps appeared incompatible with massive parallel semisynthesis of nucleosomes. The bacterial transpeptidase Sortase A recognizes a conserved LPxTG (x = any amino acid) sorting motif, which is cleaved during catalysis at the threonine residue and ligated to a peptide or protein containing an N-terminal glycine residue.29,30 Since a related motif (APATG) is present in H3 at the interface between the tail and the globular fold, we and others have used sortase-mediated ligation (SML) for semisynthesis of this histone using a ligation site downstream of T32.31–34 The reaction conditions for SML are non-denaturing and mild, just Sortase A, peptide and protein substrates buffered at physiological pH. We, therefore, tested whether this approach could be applied to install the tail in H3 that is incorporated into nucleosomes.To this end, we generated H3-ligation-ready nucleosomes by reconstituting recombinant full-length H2A, H2B and H4, as well as N-terminal truncated H3 (residues 33–135, referred to here as ΔH3) into nucleosomes using the Widom 601![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 147 bp DNA sequence.35 The H3-ligation-ready nucleosomes were further equipped with a biotin handle, HindIII, EcoRI and EcoRV restriction sites as well as Cy5 fluorophore at the 5′ and 3′ end of the DNA, respectively (Fig. 1 and see ESI Fig. S1† for a detailed scheme of our experimental strategies). To investigate whether SML of nucleosomal H3 is indeed possible and could be optimized for high ligation yields, we synthesized H3-tail peptides (residues 1–33) containing either the native sorting motif LPATG or the native H3 sequence APATG at the ligation site (ESI Fig. S2†). The latter sequence can be ligated with an evolved F40 Sortase A mutant that is, however, less active than wildtype (wt) Sortase A.31 We further synthesized the peptides equipped with an ester bond linking the threonine residue with glycolic acid, which was shown to result in more effective SML.36 SML of nucleosomal ΔH3 was performed in solution with all four peptides and subsequently analyzed by western blot. Time courses showed up to 90% conversion of ΔH3 to semi-synthetic full-length H3 when the depsipeptide containing the LPATG motif was used in combination with wt sortase (ESI Fig. S2†). In contrast, reactions with mutant F40 Sortase A and non-depsipeptide substrates did not exceed 50% conversion. We then adopted SML using wt Sortase A and LPATG-depsipeptides to immobilized H3-ligation-ready nucleosomes in streptavidin-coated 96-well plates. This allowed simple removal of excess depsipeptides, Sortase A, and reaction by-products with the supernatant. As we obtained yields of 85 ± 4% for these reactions (ESI Fig. S3†), we adopted this approach for further work, accepting the A29L mutation in the ligation products.
147 bp DNA sequence.35 The H3-ligation-ready nucleosomes were further equipped with a biotin handle, HindIII, EcoRI and EcoRV restriction sites as well as Cy5 fluorophore at the 5′ and 3′ end of the DNA, respectively (Fig. 1 and see ESI Fig. S1† for a detailed scheme of our experimental strategies). To investigate whether SML of nucleosomal H3 is indeed possible and could be optimized for high ligation yields, we synthesized H3-tail peptides (residues 1–33) containing either the native sorting motif LPATG or the native H3 sequence APATG at the ligation site (ESI Fig. S2†). The latter sequence can be ligated with an evolved F40 Sortase A mutant that is, however, less active than wildtype (wt) Sortase A.31 We further synthesized the peptides equipped with an ester bond linking the threonine residue with glycolic acid, which was shown to result in more effective SML.36 SML of nucleosomal ΔH3 was performed in solution with all four peptides and subsequently analyzed by western blot. Time courses showed up to 90% conversion of ΔH3 to semi-synthetic full-length H3 when the depsipeptide containing the LPATG motif was used in combination with wt sortase (ESI Fig. S2†). In contrast, reactions with mutant F40 Sortase A and non-depsipeptide substrates did not exceed 50% conversion. We then adopted SML using wt Sortase A and LPATG-depsipeptides to immobilized H3-ligation-ready nucleosomes in streptavidin-coated 96-well plates. This allowed simple removal of excess depsipeptides, Sortase A, and reaction by-products with the supernatant. As we obtained yields of 85 ± 4% for these reactions (ESI Fig. S3†), we adopted this approach for further work, accepting the A29L mutation in the ligation products.
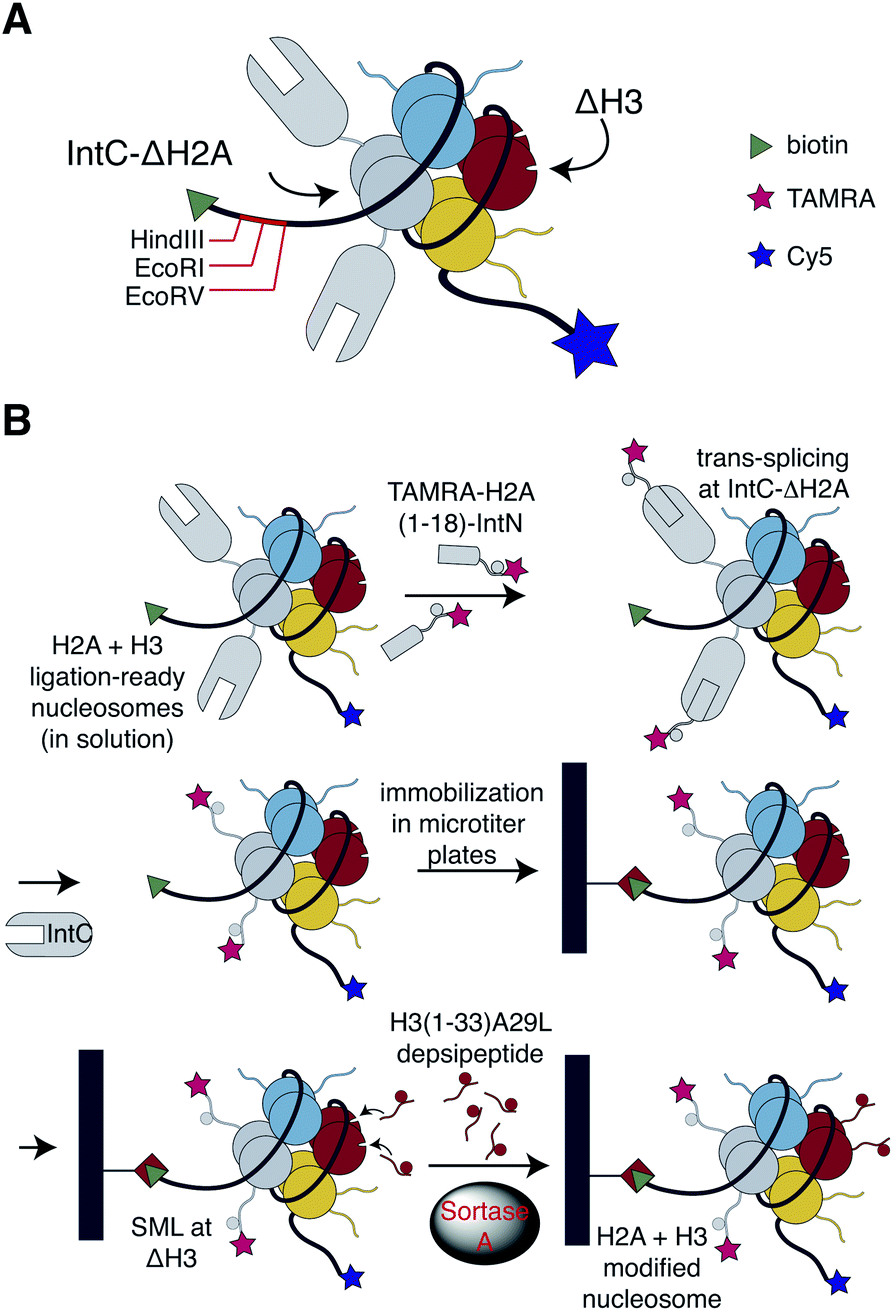 | ||
| Fig. 1 Ligation-ready nucleosomes and assembly strategy of nucleosomal libraries. (A) H2A + H3-ligation-ready nucleosomes contain truncated histone ΔH3 (residues 33–135) and truncated histone ΔH2A (residues 19–129) fused at the N-terminus to the IntC fragment of the M86 mutant of the Ssp DnaB mini-intein. The amino acid sequence GSIE was further inserted to facilitate efficient protein trans-splicing. Histones H2B and H4 were incorporated as full-length wt proteins. Nucleosomes were reconstituted on 147 bp 601 Widom DNA that in addition contained recognition sites for restriction endonucleases HindIII, EcoRI, or EcoRV as well as a biotin handle at the 5′ end for immobilization. A Cy5 fluorophore was added at the 3′ end for visualization and quantification. H3-ligation-ready nucleosomes contained full-length wild type H2A instead of IntC-ΔH2A. (B) The H2A + H3 library was assembled by trans-splicing of IntC-ΔH2A with synthetic peptides derived from the H2A tail containing an N-terminal TAMRA fluorophore and C-terminal IntN sequence (TAMRA-H2A-IntN) in solution. Spliced nucleosomes were immobilized in streptavidin coated 96-well plates, followed by installation of the H3 (residues 1–32, A29L) depsipeptides on-surface by sortase-mediated ligation. The H3-library was generated by on-surface SML of H3(1–33, A29L) depsipeptides with H3-ligation-ready nucleosomes. | ||
To generate a library of immobilized, H3-modified nucleosomes, we synthesized various H3 depsipeptides permutating methylation marks at K4, K9 and K27 in conjunction with phosphorylation of S10 and S28. We focused on dimethylation at the lysine residues as a compromise of covering a spectrum of biological responses. While investigating nucleosomal SML with the library depsipeptides we observed slower reaction kinetics with substrates containing the S28ph modification. We attributed this observation to the negative charge in close proximity to the ligation site and established two separate protocols for SML with S28ph and S28 depsipeptides. Upon optimization of the protocols an average yield of nucleosomal SML of 83 ± 3% was observed in three replicates of establishing a 32 membered library (ESI Fig. S4†). Based on the stoichiometry of two copies of H3 per nucleosome, the library consisted of 69% fully modified nucleosomes, 28% of nucleosomes containing a single H3 tail and 3% of nucleosomes lacking both H3 tails. To further expand the library, we also added H3-depsipeptides with single acetylation marks at K4, K9, and K27, so that the entire library finally covered 35 modification patterns in total.
Intein splicing of ligation-ready nucleosomes for generating a library of H2A- and H3-modified nucleosomes
To enable analysis of possible crosstalk between PTMs on different histones, we extended the library design in a second step towards histone H2A. This core histone is also intensively decorated by PTMs,37 but the knowledge about the biological function of the H2A modifications is limited when compared to H3. To establish H2A-ligation-ready nucleosomes, we needed an orthogonal ligation method to install H2A tails without cross-reactions at ligation-ready H3. To this end, we resorted to protein trans-splicing (PTS)38 and reconstituted nucleosomes with N-terminal truncated H2A fused to the IntC sequence of the M86 mutant of the Ssp DnaB intein (Fig. 1 and ESI Fig. S1†).39 The ligation site was introduced downstream of S18, allowing installation of PTMs at the first 18 residues of H2A. To enable efficient PTS, the residues flanking the junction site of the split intein needed to be optimized resulting in the insertion of four residues (GSIE) into the split site (ESI Fig. S1†). Three modifications of the H2A tail were permutated: H2AS1ph, H2AR3me2asym and H2AK5ac, resulting in eight peptides containing the H2A tail sequences fused to the IntN sequence of M86 Ssp DnaN intein. To facilitate efficient quantification of nucleosomal PTS, the N-termini of the peptides were modified with a fluorophore (TAMRA). Corresponding PTS reactions with the eight H2A-tail peptides were performed in solution and quantified by the ratio of the TAMRA signal of correctly spliced H2A and the Cy5 signal of the nucleosomes, showing an average PTS yield of 78 ± 8% (ESI Fig. S5†). LC-MS of PTS with H2A-ligation-ready nucleosomes further verified efficient installation of the H2A tail with only little C-terminal cleavage of H2A-IntC fusion protein, which is a known side-reaction of PTS (ESI Fig. S6†).40 Despite intense efforts, the PTS strategy did not work well when immobilizing the ligation ready nucleosomes with yields of ligation product below the detection limit (not shown).We then combined the SML and PTS ligation strategies of H3 and H2A using the respective H2A + H3-ligation-ready nucleosomes containing ΔH3 and IntC-ΔH2A. First, we carried out H2A PTS in solution, thereby installing the eight H2A-IntN peptides. This was followed by seeding of the PTS reaction mixture into streptavidin-coated 96-well plates. After removing excess peptide and released IntC from the immobilized nucleosomes by washing, the SML reaction was performed with H3 depsipeptides as described above. For quality control, we confirmed the integrity of selected nucleosomes of the library after the PTS and SML reactions (ESI Fig. S7†). The resulting combined H2A + H3 library contained 280 individually modified nucleosomes. When combining ligation yields and considering the histone stoichiometry, 56% of nucleosomes contained both H3 and at least one H2A tail, while 92% of nucleosomes had at least one H3 and one H2A tail.
Library screening reveals long-range crosstalk of H3 modifications in recruitment of HP1 protein to nucleosomes
With H3 and H2A + H3 libraries in hand we investigated their applicability for profiling the impact of PTM patterns onto the binding of chromatin factors to nucleosomes. We focused on HP1β, one of the three homologous mammalian HP1 proteins that in contrast to HP1α and HP1γ is essential for mouse development.41 Human HP1β (from hereon, referred to HP1) fused at the N-terminus to a 6xHIS-tag and enhanced cyan fluorescent protein (eCFP-HP1) was expressed in bacteria and purified via metal affinity chromatography. In 96-well format, we added 75 μM eCFP-HP1 to each well of the H3 library (approximately 10 pmol of immobilized nucleosomes). After incubation and washing of the resin-bound material, we quantified fluorescence at 433 nm excitation, 476 nm emission (eCFP signal of HP1) using a plate reader. Simultaneously, we recorded fluorescence at 649 nm excitation, 670 nm emission (Cy5 signal of DNA). The latter allowed normalization of the HP1 recovery to the amounts of nucleosomes immobilized in each well of the library. After performing the screen in three fully independent replicates (i.e. independently prepared libraries and eCFP-HP1 protein), four groups of nucleosomes emerged based on their eCFP-HP1 recruitment patterns (Fig. 2). Nucleosomes not containing the H3K9me2 modification showed only background normalized eCFP signals, indicating no recruitment of eCFP-HP1. In contrast, nucleosome species containing H3K9me2 showed elevated normalized eCFP signals in full agreement with the known binding preferences of HP1 proteins.15 Furthermore, H3K9me2S10ph nucleosomes did not recruit eCFP-HP1 as indicated by low normalized eCFP signal intensity, reflecting the attenuation of HP1 H3K9me3-binding by H3S10ph.15 To our surprise, we observed a further group of nucleosomes containing H3K9me2, no modification of H3S10 but phosphorylation of H3S28. These nucleosomes recruited eCFP-HP1 but the normalized eCFP signal intensity was stronger than the eCFP fluorescence of H3K9me2 nucleosomes not containing the additional modification on H3S28. Encouraged by the above finding we also screened the H2A + H3 library for eCFP-HP1 recruitment. This screen was performed in three replicates from two independently prepared libraries exploiting an established library regeneration protocol (ESI Fig. S8†). While these screens did not show a significant impact of H2A-tail modifications onto the recovery of HP1, these verified the enhanced binding of the protein to H3K9me2S28ph nucleosomes. The normalized eCFP signal intensity recorded for H3K9me2S28ph containing nucleosomes was on average 2.3-fold stronger compared to H3K9me2 containing samples (ESI Fig. S9†).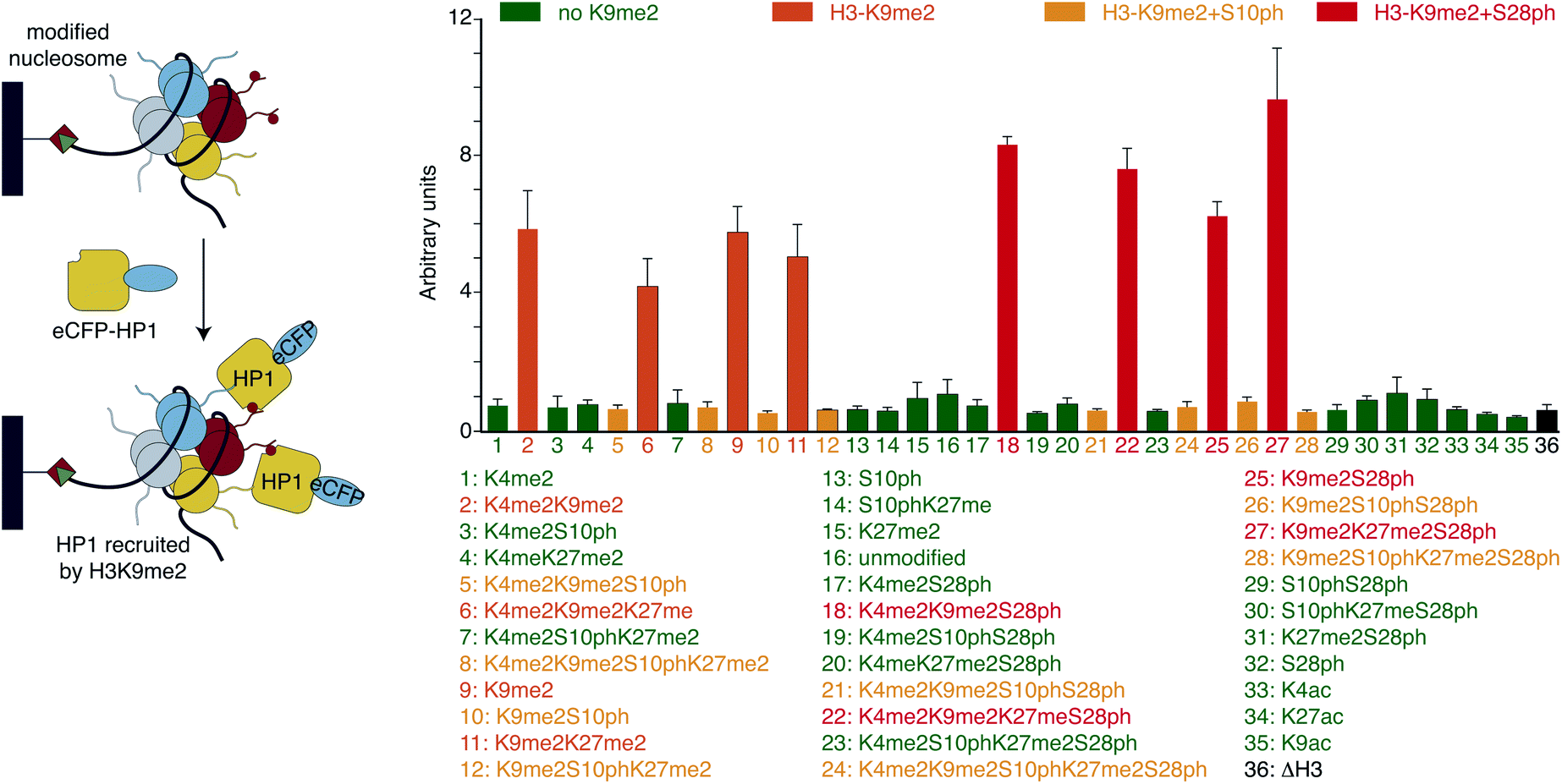 | ||
| Fig. 2 Screening of H3-library for binding of HP1. The H3 library was incubated with eCFP-HP1 followed by washing and detection of the eCFP fluorescence signal. Nucleosomes were grouped into four categories showing distinct HP1 recruitment patterns: green (no H3K9me2) and yellow (H3K9me2S10ph) showed no significant HP1 recruitment with respect to nucleosomes containing unmodified H3. Orange (H3K9me2) showed on average a 4.8-fold and red (H3K9me2S28ph) 7.6-fold enrichment of HP1. Black represents HP1-enrichment on non-ligated H3-ligation-ready nucleosomes. | ||
The enhanced binding of eCFP-HP1 to H3K9me2S28ph nucleosomes could be explained by the dual modification directly impacting on protein binding or the additional H3S28ph indirectly facilitating protein access to the H3K9me2-tail in the context of the nucleosome. In the first case, the observed effect should not be restricted to nucleosomes but be also detectable with isolated modified histone tail peptides. To start discriminating between these possibilities, we investigated eCFP-HP1 recruitment to immobilized peptides carrying H3K9me and H3K9me2S28ph modifications and as control the unmodified H3-tail. Pull-down reactions showed equal binding of eCFP-HP1 to the H3K9me2 and H3K9me2S28ph peptides (ESI Fig. S10†). To further quantify the binding reactions, we prepared HP1 without eCFP tag and carried out isothermal titration calorimetry (ITC) reactions. This verified the results from pull-downs, with the H3K9me2S28ph peptide even showing 1.5-fold weaker binding to HP1 compared to the H3K9me2 peptide (Fig. 3A). The results argued against a direct contribution of H3S28ph to the binding of HP1 to H3K9me2.
To independently verify the preferred recruitment of HP1 to H3K9me3S28ph only in nucleosomal context, we prepared nucleosomes for quantitative measurements. We first ligated the synthetic H3K9me2 or H3K9me2S28ph peptides (1–33) to ΔH3 via SML, followed by purification of the ligation product and assembly of nucleosomes on the same DNA template as used for library preparation (ESI Fig. S11†). Using Microscale Thermophoresis (MST) we determined the Kd of HP1 to H3K9me2 nucleosomes at 2.4 ± 0.2 μM and to H3K9me2S28ph nucleosomes at 1.8 ± 0.1 μM (Fig. 3B). While the actual difference was lower than estimated from the library screening, normalizing to the behavior with free peptide (1.4- fold × 1.5-fold = 2-fold) nevertheless pointed to a putatively significant effect.
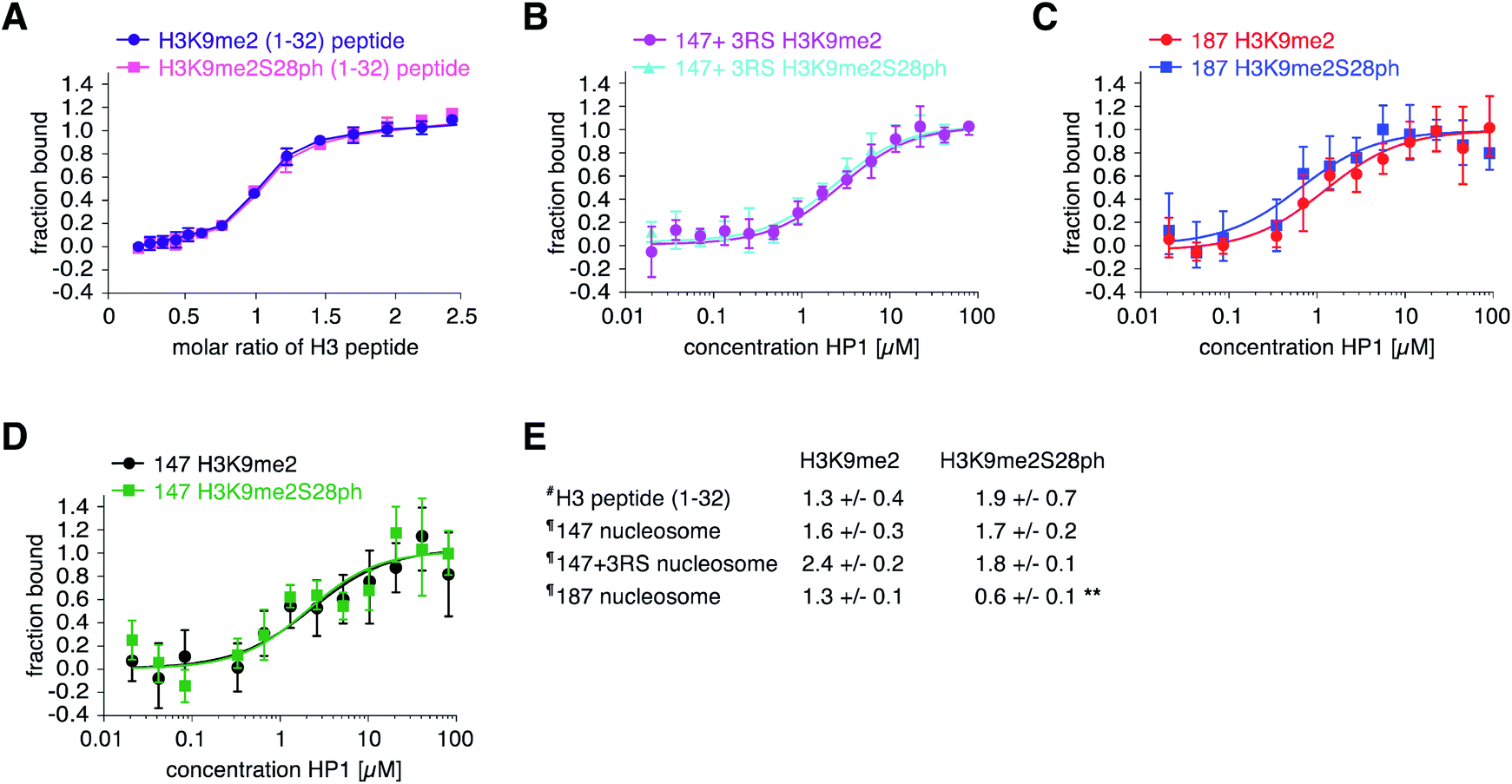 | ||
| Fig. 3 Quantitative binding analysis of HP1 to modified H3 in different biochemical context. (A) The indicated peptides were titrated into a solution of HP1. Reaction heat was measured by isothermal titration calorimetry (ITC) and is plotted (n = 2) in relation to progress of the reaction. Comparable results were obtained with the reverse set-up of the ITC titration. Error bars correspond to standard deviation. (B–D) Titration series of recombinant HP1 with nucleosomes reconstituted with modified H3 on 147 bp plus three restrictions sites at the 5′ end (147 + RS), 187 bp (187) or 147 bp (147) 601 Widom DNA were analysed by microscale thermophoresis (MST). Data are plotted as averages of minimally six independent measurements using multiple independent protein preparations. Error bars correspond to standard deviation. (E) Summary of dissociation constants (Kd in μM) determined as average from multiple, independent ITC (#) or MST (¶) measurements. Error corresponds to the propagated error from the individual measurements. Student's t-test (unpaired, two-tailed) was performed to compare samples; ** corresponds to p < 0.001. | ||
The DNA of the library nucleosomes carries a one-sided linker of 19 bp (Fig. 1). Based on our previous work implying an electrostatic effect of DNA on histone H3-tail behavior,42 we next tested whether having free DNA on both ends of the nucleosome would strengthen the observed effect of H3S28ph onto binding of HP1 to H3K9me2. Indeed, nucleosomes reconstituted with H3K9me2S28ph on 187 bp Widom 601 DNA (i.e. containing 20 bp free linker DNA on both ends) showed a statistically significant 2-fold stronger binding compared to the H3K9me2 counterpart (Fig. 3C). In contrast, no such effect was observed with nucleosomes reconstituted on 147 bp Widom 601 core DNA (i.e. not containing any free linker DNA) (Fig. 3D). Comparing ‘187’ and ‘147’ H3K9me2S28ph nucleosomes showed a significant change in the Kd values (0.6 ± 0.1 μM vs. 1.7 ± 0.2 μM, respectively; p-value = 0.0002), further supporting our notion that the linker DNA governs the preferential recruitment of HP1 to H3K9me2S28ph.
H3S28ph enhances dynamics and flexibility of the nucleosomal H3-tail
To obtain molecular insights into the effect of DNA onto H3S28ph tail behaviour, we conducted NMR measurements on reconstituted 187 bp Widom 601 DNA nucleosomal complexes containing uniformly 15N isotope-labelled H3 together with unlabelled other core histones. As the quantity of materials required for NMR measurements are not accessible by ligation strategies, we resorted to H3S28E instead of H3S28ph, considering that the main effect might be electrostatic in nature and given that S to E substitution has been widely studied as phosphorylation mimicry.43–45 As seen before, 2D [1H/15N] TROSY NMR experiments showed well-resolved resonance cross-peaks corresponding to the main-chain amides of H3-tail residues 3–35 that are pointing outside of the nucleosome.42 In contrast, the folded nucleosome core particle region of isotope-labelled H3 remained invisible in NMR due to the large molecular weight and highly restricted motions of the complex (>220 kDa) that cause signal broadening beyond detection (Fig. 4A). Chemical shift perturbation plot analysis (Δδ) for the tail of H3 and H3S28E nucleosomes showed significant changes in the direct proximity of the mutation site (K27, S28, A29), as expected for an intrinsically disordered protein (IDP) region. Comparatively small changes were observed at the positively charged residues K18 and K9, the latter being barely above noise level (Fig. 4B).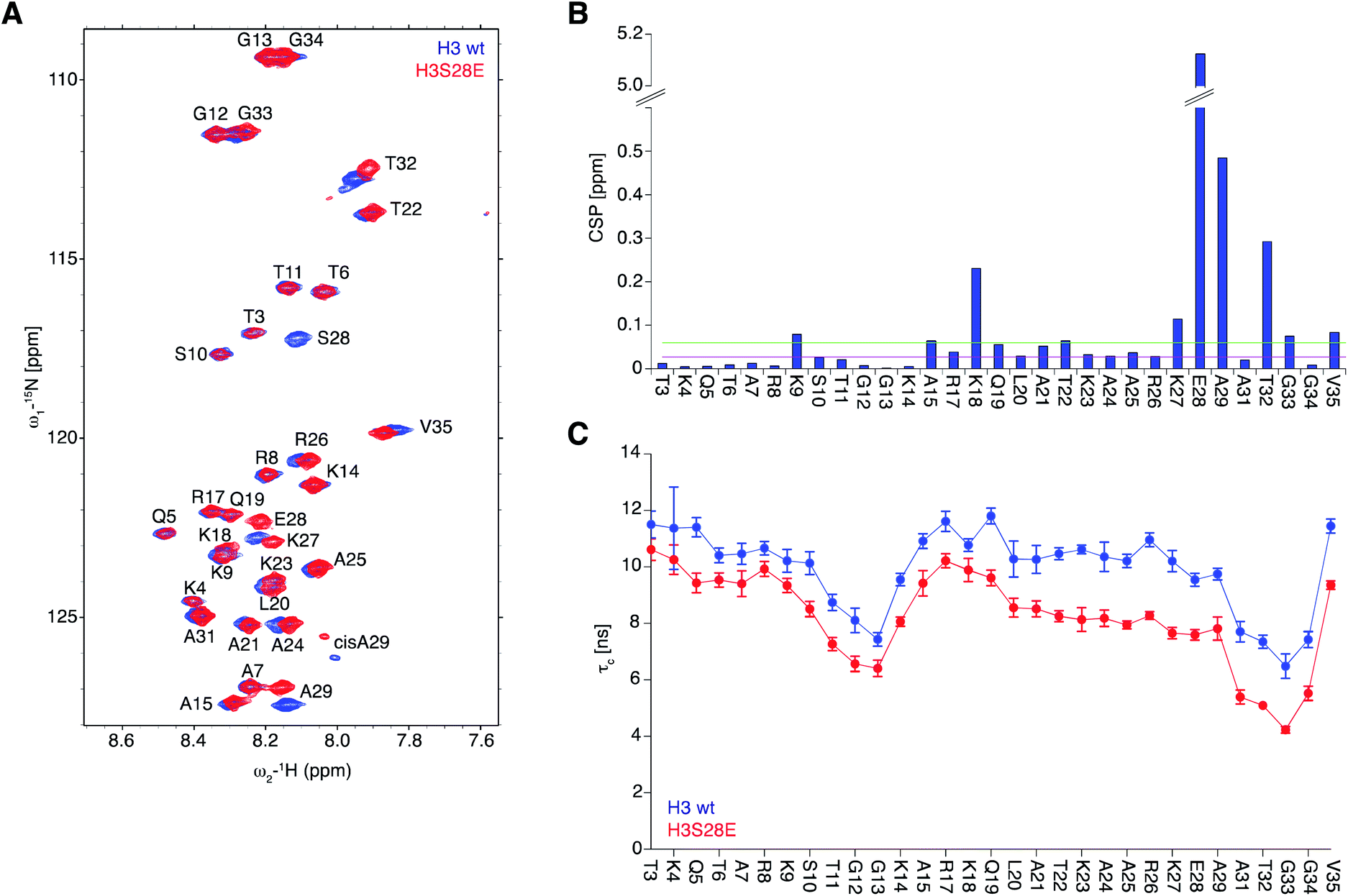 | ||
| Fig. 4 NMR analysis of wt and H3S28E nucleosomes. (A) 2D [1H/15N] TROSY NMR spectra of the H3-tail of nucleosomes reconstituted on 187 bp 601 Widom DNA. (B) Chemical shift perturbation plot (CSP) of main-chain 1H/15N amides. Differences observed for H3 residues 3–35 in the spectrum shown in (A) are plotted. Amide CSPs were calculated as CSPi = [(ΔδH,i)2+(0.25·ΔδN,i)2]0.5, where ΔδH,i and ΔδN,i are detected chemical shift changes of proton and nitrogen, respectively. The 0.05 ppm cut-off (green line) corresponds to the noise (median+1.5*IQR) and the 0.02 ppm cut-off corresponds to the median value (red line). (C) Rotational correlation time (τc) of H3-tail residues determined by NMR relaxation measurements at 22.3 T. Error bars were calculated as described in Experimental Procedures. | ||
To probe the dynamics of H3-tail residues, we collected NMR spin relaxation measurements of 15N–R1, 15N–R2 relaxation rates and steady-state heteronuclear {1H}–15N nuclear Overhauser effects (NOE). Based on the NOE measurements that depict the local picosecond vibrations of the N–H bond vectors, we observed small opposing differences for the first and second half of the H3-tail residues. While these observed differences are at the range of the reported accuracies for such experiments, they nevertheless verify that the H3 and H3S28E solvent and DNA exposed tails remain structurally disordered in nucleosomes. The rotational correlation times (τc) calculated from the longitudinal 15N–R1 and transverse 15N–R2 relaxation rates of individual residues samples the segmental polypeptide chain motions at the nanosecond time scale. We found the average value of τc observed for H3S28E decreased from 10 ns to 8 ns when compared to H3 indicating that S28E substitution enhances the mobility of the entire detectable H3-tail by ca. 20%. While the region closer to the nucleosome (residues 19–35) showed an average of 30 ± 9% increase in dynamics, the effect of the H3S28E substitution on the dynamics of the N-terminal region (3–18 residues) was about 14 ± 5% (Fig. 4C). Overall, the results imply that the introduction of negative charge at S28 makes the H3-tail more flexible, likely via detachment from DNA. This in turn enables stronger HP1 binding to H3K9me.
Conclusions
In this work, we report a novel strategy for generating nucleosomes with defined modification patterns on different core histones in high throughput. The concept of ligation-ready nucleosomes allows chemoenzymatic ligations of histone tails to pre-assembled nucleosomes without going through the cumbersome process of individual reconstitution reactions. We acknowledge that our approach is not completely traceless, yet and in consequence we cannot fully exclude that the introduced ‘ligation scars’ might impact experimental outcomes. Future development will focus on establishing traceless methods for late-stage modification of ligation-ready nucleosomes.We have shown that libraries of immobilized nucleosomes can be used for parallel screening of the binding of chromatin factors to their targets. Our analysis uncovered a previously unknown positive impact of H3S28ph onto the binding of HP1 to H3K9me nucleosomes that we postulate, based on NMR measurements, depends on the modulation of H3-tail-DNA interactions.
Setting up strategies compatible with and orthogonal to the here reported SML and PTS for the tails of the other core histones will allow combining the power of massive parallel peptide synthesis with the novel ligation-ready strategy for generating highly complex libraries of designer nucleosomes. As it is becoming clear that histone tail peptides are only limited surrogates for the complex environment of chromatin, we envision reading out immobilized libraries of modified nucleosomes for the binding of other chromatin factors or with other assays (e.g. enzymatic reactions). The fact that our approach of normalizing nucleosome immobilization via fluorophores enabled the discovery of a 2-fold effect in HP1 recruitment verifies the general usability of the libraries and underscores the sensitivity of their screening readout. We conjure that the H2A + H3 nucleosome libraries that we have created and the ligation-ready strategy for the generation of modified nucleosomes we have introduced here will enable the discovery of unknown chromatin regulatory phenomena depending on patterns of histone PTMs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the priority program SPP 1623 of the Deutsche Forschungsgemeinschaft (DFG FI1513/3-1 to W. F. and SCHW1163/4-1 to D. S.) the Max Planck Society (W. F.), and intramural funds of King Abdullah University of Science and Technology (M. J., L. J. and W. F.). We thank members of the HDM group for help with establishing protein trans-splicing assays.Notes and references
- B. Fierz and M. G. Poirier, Annu. Rev. Biophys., 2019, 48, 321–345 CrossRef CAS.
- W. Fischle, H. D. Mootz and D. Schwarzer, Curr. Opin. Chem. Biol., 2015, 28, 131–140 CrossRef CAS.
- M. M. Muller and T. W. Muir, Chem. Rev., 2015, 115, 2296–2349 CrossRef CAS PubMed.
- N. Justin, V. De Marco, R. Aasland and S. J. Gamblin, Curr. Opin. Struct. Biol., 2010, 20, 730–738 CrossRef CAS PubMed.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, Nat. Struct. Mol. Biol., 2007, 14, 1025–1040 CrossRef CAS.
- S. Rea, F. Eisenhaber, D. O'Carroll, B. D. Strahl, Z. W. Sun, M. Schmid, S. Opravil, K. Mechtler, C. P. Ponting, C. D. Allis and T. Jenuwein, Nature, 2000, 406, 593–599 CrossRef CAS PubMed.
- D. Canzio, A. Larson and G. J. Narlikar, Trends Cell Biol., 2014, 24, 377–386 CrossRef CAS.
- F. Hediger and S. M. Gasser, Curr. Opin. Genet. Dev., 2006, 16, 143–150 CrossRef CAS.
- S. H. Kwon and J. L. Workman, Bioessays, 2011, 33, 280–289 CrossRef CAS.
- A. G. Larson, D. Elnatan, M. M. Keenen, M. J. Trnka, J. B. Johnston, A. L. Burlingame, D. A. Agard, S. Redding and G. J. Narlikar, Nature, 2017, 547, 236–240 CrossRef CAS.
- A. R. Strom, A. V. Emelyanov, M. Mir, D. V. Fyodorov, X. Darzacq and G. H. Karpen, Nature, 2017, 547, 241–245 CrossRef CAS PubMed.
- L. Wang, Y. Gao, X. Zheng, C. Liu, S. Dong, R. Li, G. Zhang, Y. Wei, H. Qu, Y. Li, C. D. Allis, G. Li, H. Li and P. Li, Mol. Cell, 2019, 76, 646–659 e646 CrossRef CAS.
- W. Fischle, Y. Wang, S. A. Jacobs, Y. Kim, C. D. Allis and S. Khorasanizadeh, Genes Dev., 2003, 17, 1870–1881 CrossRef CAS.
- S. Winter and W. Fischle, Essays Biochem., 2010, 48, 45–61 CrossRef CAS PubMed.
- W. Fischle, B. S. Tseng, H. L. Dormann, B. M. Ueberheide, B. A. Garcia, J. Shabanowitz, D. F. Hunt, H. Funabiki and C. D. Allis, Nature, 2005, 438, 1116–1122 CrossRef CAS.
- W. Fischle, Y. Wang and C. D. Allis, Nature, 2003, 425, 475–479 CrossRef CAS PubMed.
- B. M. Zee, A. A. Alekseyenko, K. A. McElroy and M. I. Kuroda, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1784–1789 CrossRef CAS.
- M. Vermeulen, H. C. Eberl, F. Matarese, H. Marks, S. Denissov, F. Butter, K. K. Lee, J. V. Olsen, A. A. Hyman, H. G. Stunnenberg and M. Mann, Cell, 2010, 142, 967–980 CrossRef CAS.
- R. Klingberg, J. O. Jost, M. Schumann, K. A. Gelato, W. Fischle, E. Krause and D. Schwarzer, ACS Chem. Biol., 2015, 10, 138–145 CrossRef CAS PubMed.
- H. C. Eberl, M. Mann and M. Vermeulen, Chembiochem, 2011, 12, 224–234 CrossRef CAS.
- T. Bartke, M. Vermeulen, B. Xhemalce, S. C. Robson, M. Mann and T. Kouzarides, Cell, 2010, 143, 470–484 CrossRef CAS PubMed.
- M. Nikolov, A. Stutzer, K. Mosch, A. Krasauskas, S. Soeroes, H. Stark, H. Urlaub and W. Fischle, Mol. Cell. Proteomics, 2011, 10(11), M110.005371, DOI:10.1074/mcp.M110.005371.
- C. J. A. Leonen, E. Upadhyay and C. Chatterjee, Curr. Opin. Chem. Biol., 2018, 45, 27–34 CrossRef CAS.
- B. Fierz and T. W. Muir, Nat. Chem. Biol., 2012, 8, 417–427 CrossRef CAS.
- B. Fierz, ChemMedChem, 2014, 9, 495–504 CrossRef CAS PubMed.
- C. D. Allis and T. W. Muir, Chembiochem, 2011, 12, 264–279 CrossRef CAS.
- G. P. Dann, G. P. Liszczak, J. D. Bagert, M. M. Muller, U. T. T. Nguyen, F. Wojcik, Z. Z. Brown, J. Bos, T. Panchenko, R. Pihl, S. B. Pollock, K. L. Diehl, C. D. Allis and T. W. Muir, Nature, 2017, 548, 607–611 CrossRef CAS.
- U. T. Nguyen, L. Bittova, M. M. Muller, B. Fierz, Y. David, B. Houck-Loomis, V. Feng, G. P. Dann and T. W. Muir, Nat. Methods, 2014, 11, 834–840 CrossRef CAS.
- N. Pishesha, J. R. Ingram and H. L. Ploegh, Annu. Rev. Cell Dev. Biol., 2018, 34, 163–188 CrossRef CAS.
- L. Schmohl and D. Schwarzer, Curr. Opin. Chem. Biol., 2014, 22, 122–128 CrossRef CAS PubMed.
- K. Piotukh, B. Geltinger, N. Heinrich, F. Gerth, M. Beyermann, C. Freund and D. Schwarzer, J. Am. Chem. Soc., 2011, 133, 17536–17539 CrossRef CAS PubMed.
- M. Wu, D. Hayward, J. H. Kalin, Y. Song, J. W. Schwabe and P. A. Cole, Elife, 2018, 7 DOI:10.7554/eLife.37231.
- A. E. Ringel, A. M. Cieniewicz, S. D. Taverna and C. Wolberger, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5461–E5470 CrossRef CAS.
- Z. A. Wang, C. J. Millard, C. L. Lin, J. E. Gurnett, M. Wu, K. Lee, L. Fairall, J. W. R. Schwabe and P. A. Cole, Elife, 2020, 9 DOI:10.7554/eLife.57663.
- P. T. Lowary and J. Widom, J. Mol. Biol., 1998, 276, 19–42 CrossRef CAS PubMed.
- D. J. Williamson, M. A. Fascione, M. E. Webb and W. B. Turnbull, Angew. Chem., Int. Ed. Engl., 2012, 51, 9377–9380 CrossRef CAS.
- S. C. Galasinski, D. F. Louie, K. K. Gloor, K. A. Resing and N. G. Ahn, J. Biol. Chem., 2002, 277, 2579–2588 CrossRef CAS.
- H. D. Mootz, Chembiochem, 2009, 10, 2579–2589 CrossRef CAS.
- J. H. Appleby-Tagoe, I. V. Thiel, Y. Wang, Y. Wang, H. D. Mootz and X. Q. Liu, J. Biol. Chem., 2011, 286, 34440–34447 CrossRef CAS.
- K. Friedel, M. A. Popp, J. C. J. Matern, E. M. Gazdag, I. V. Thiel, G. Volkmann, W. Blankenfeldt and H. D. Mootz, Chem. Sci., 2019, 10, 239–251 RSC.
- R. Aucott, J. Bullwinkel, Y. Yu, W. Shi, M. Billur, J. P. Brown, U. Menzel, D. Kioussis, G. Wang, I. Reisert, J. Weimer, R. K. Pandita, G. G. Sharma, T. K. Pandita, R. Fundele and P. B. Singh, J. Cell Biol., 2008, 183, 597–606 CrossRef CAS PubMed.
- A. Stutzer, S. Liokatis, A. Kiesel, D. Schwarzer, R. Sprangers, J. Soding, P. Selenko and W. Fischle, Mol. Cell, 2016, 61, 247–259 CrossRef.
- J. Hiscott, P. Pitha, P. Genin, H. Nguyen, C. Heylbroeck, Y. Mamane, M. Algarte and R. Lin, J. Interferon Cytokine Res., 1999, 19, 1–13 CrossRef CAS.
- A. Guerra-Castellano, I. Diaz-Moreno, A. Velazquez-Campoy, M. A. De la Rosa and A. Diaz-Quintana, Biochim. Biophys. Acta, 2016, 1857, 387–395 CrossRef CAS PubMed.
- Z. Chen and P. A. Cole, Curr. Opin. Chem. Biol., 2015, 28, 115–122 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03407j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |