
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraordinary electrochemical stability and extended polaron delocalization of ladder-type polyaniline-analogous polymers†
Xiaozhou
Ji
 a,
Mingwan
Leng
a,
Haomiao
Xie
a,
Chenxu
Wang
b,
Kim R.
Dunbar
a,
Yang
Zou
*c and
Lei
Fang
*ab
a,
Mingwan
Leng
a,
Haomiao
Xie
a,
Chenxu
Wang
b,
Kim R.
Dunbar
a,
Yang
Zou
*c and
Lei
Fang
*ab
aDepartment of Chemistry, Texas A&M University, College Station, TX 77843-3255, USA. E-mail: fang@chem.tamu.edu
bDepartment of Materials Science and Engineering, Texas A&M University, College Station, TX 77843-3255, USA
cShenzhen Key Laboratory of Polymer Science and Technology, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, China. E-mail: yangzou@szu.edu.cn
First published on 6th August 2020
Abstract
Electrochemical stability and delocalization of states critically impact the functions and practical applications of electronically active polymers. Incorporation of a ladder-type constitution into these polymers represents a promising strategy to enhance the aforementioned properties from a fundamental structural perspective. A series of ladder-type polyaniline-analogous polymers are designed as models to test this hypothesis and are synthesized through a facile and scalable route. Chemical and electrochemical interconversions between the fully oxidized pernigraniline state and the fully reduced leucoemeraldine state are both achieved in a highly reversible and robust manner. The protonated pernigraniline form of the ladder polymer exhibits unprecedented electrochemical stability under highly acidic and oxidative conditions, enabling the access of a near-infrared light-absorbing material with extended polaron delocalization in the solid-state. An electrochromic device composed of this ladder polymer shows distinct switching between UV- and near-infrared-absorbing states with a remarkable cyclability, meanwhile tolerating a wide operating window of 4 volts. Taken together, these results demonstrate the principle of employing a ladder-type backbone constitution to impart superior electrochemical properties into electronically active polymers.
Introduction
Conjugated ladder polymers, in which the macromolecular backbones are constituted of uninterrupted fused cyclic π-systems, represent a highly promising class of organic electronic materials. The covalently locked coplanar conformation of a conjugated ladder-type backbone,1 in contrast with conventional non-ladder polymers with torsional rotation, allows for extensive intrachain delocalization of molecular orbitals and transport of quasi-particles such as charges, excitons, polarons, and spins.2–4 The rigid coplanar conformation can also enhance interchain electronic coupling of conjugated ladder polymers due to the small reorganization energy of a rigid system upon electron transfer or photoexcitation,5–8 which is important for solid-state materials properties. In addition, a significantly higher activation energy is required to break the double-stranded backbones of a ladder polymer, translating to high stability that is important for applications under harsh conditions.9–11 These advantageous features render conjugated ladder polymers promising candidates for next generation organic electronic materials.Polyaniline (PANI) derivatives, which represent one of the most extensively studied classes of conjugated polymers, are particularly intriguing for the incorporation of the ladder-type constitution. PANI derivatives feature reversible redox transformation, high conductivity, feasible synthesis, and good processability,12–18 hence are widely employed in conductive protective coating,19 electrochromism,13 and energy storage.20 However, PANI shares some of the common drawbacks with other organic electronic materials, such as (1) low stability of molecular constitutions and degraded performance in devices over time and (2) limited intrachain delocalization ranges of states or quasi-particles, such as polarons and excitons. One of the primary structural origins of these issues can be identified as the single-stranded and torsionally rotating bonds in the PANI polymer backbone, which gives rise to low barriers of bond scission and disordered chain conformation in the solid-state. For example, the fundamental investigation and practical application of pernigraniline salt – the fully oxidized and protonated form of PANI – are severely limited due to its unstable nature. Iminium bond hydrolysis of pernigraniline salt takes place readily under acidic and oxidative conditions,21,22 leading to irreversible scission of the conjugated polymer chain and consequently loss in electrical conductivity, charge storage ability, and redox properties.15,23 Several engineering approaches have been developed to improve the apparent stability of devices fabricated from conventional PANI, such as introducing external protection,24 applying limited voltage sweep windows and pretreatment at low voltages,15 and compositing PANI with carbon or inorganic nanomaterials.15,23,25 A few examples on the modification of the chemical constitution of PANI have also shown to be effective in enhancing the stability.26–30 In these cases, however, the intrinsically labile iminium bonds remain as a potential weakness that impacts the wider application of these PANI derivatives, especially when harsh electrochemical conditions are involved.
Recently, we reported the significantly improved acid stability of the pernigraniline salt-like structure in a series of ladder oligomers, in which the entropy-driven decomposition is inhibited by the ladder-type double stranded covalent bonds. In addition, unprecedented polaron delocalization in the solid-state was observed. In this context, we envisioned that the implementation of a ladder-type constitution in a polymeric PANI derivative could fundamentally address the unsolved issues of electrochemical stability and state delocalization of these polymers for their practical future applications. Herein, we report the feasible synthesis of a series of PANI-derived ladder polymers, their extraordinary electrochemical stability under harsh conditions, and extensive polaron delocalization properties. The intrinsic electrochemical stability and conductivity were further demonstrated by the excellent performance and cyclability of the polymer as the active material in a near-infrared (NIR) electrochromic device.
Results and discussion
Ladder polymers can often be synthesized by a step-wise approach: the first strand of bonds is formed through a conventional linear polymerization, followed by a ladderization step in which the second strand of covalent bonds is formed along the backbone through annulation.2 The ladder-type PANI derivative (LPANI) in this study was synthesized (Fig. 1a) from a non-ladder polymer precursor P1 (Mn = 25.9 kg mol−1, Đ = 2.23), which could be synthesized using a scalable approach involving imine-condensation polymerization followed by in situ oxidation.9,31 As a PANI-analogue, P1 is composed of alternating fluorene and diester-functionalized aniline repeating units. Two n-dodecyl groups were installed onto the 9-position of each fluorene unit to ensure solubility of the ladder polymer product for subsequent synthesis and investigations. To facilitate the ring-fusing ladderization reaction, the ester groups in P1 were converted into tertiary alcohol groups in P2 by treating the polymer with p-tolyl Grignard reagent (see the ESI†). Subsequently, Friedel–Crafts cyclization between the fluorene units and the tertiary alcohol group was promoted by boron trifluoride to afford the ladder polymer product LLB, which represents the fully reduced form of LPANI. This reaction was designed and optimized in a way that only the 3-position of the fluorene unit was activated for the cyclization, facilitated by (i) the combined electronic effect of the fluorene moiety itself and the amino group on the 2-position and (ii) the steric effect of the two dodecyl groups that prevents the 1-position from reacting. In addition, the centrosymmetric positioning of the tertiary alcohol functional groups ensured a linear extension of the resulting rigid ladder polymer, instead of a helical extension if the repeating units are non-centrosymmetric.32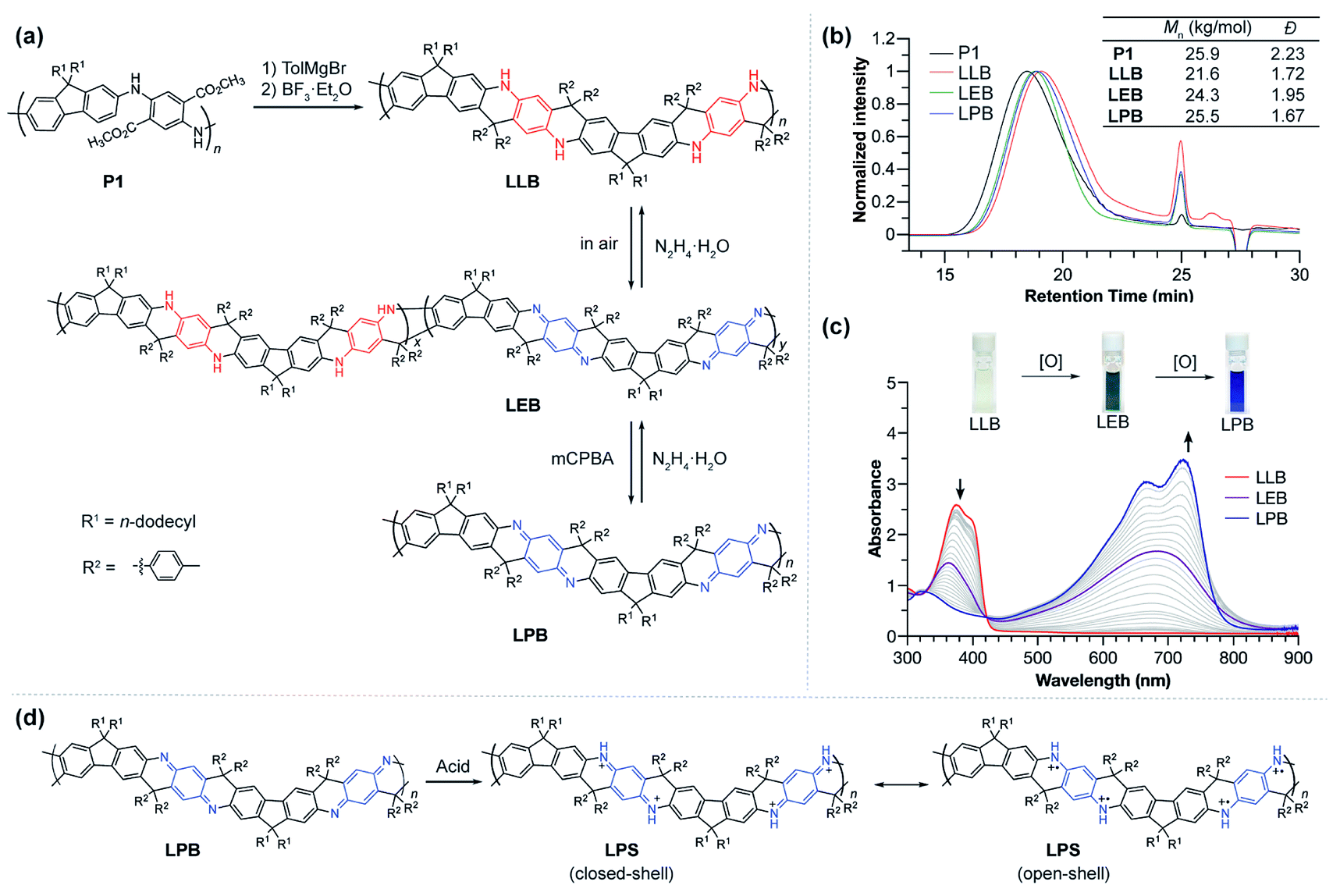 | ||
| Fig. 1 (a) Synthesis of LPANI from polymer precursor P1, and the redox interconversion of its LLB, LEB, and LPB forms. (b) Size exclusion chromatogram, calculated Mn, and polydispersity (Đ) values. (c) UV-vis spectra of the oxidation process from LLB to LEB to LPB in THF; (inset) photographic images of LLB, LEB and LPB solutions. (d) Protonation of LPB with MSA and structural formulae of the closed-shell and open-shell resonance forms of the protonated LPS form. | ||
LLB can be considered a ladder-type analogue of the leucoemeraldine base – the fully reduced form of conventional PANI. Upon exposure to air, ∼50% of the benzenoid phenylene-1,4-diamine units in LLB can be oxidized to the quinonoid cyclohexadiene-1,4-diimine unit, to afford a green compound LEB, which is analogous to the emeraldine base form of conventional PANI. Complete oxidation of either LLB or LEB can be achieved quantitatively by treating the solution with m-chloroperoxybenzoic acid (mCPBA) to afford a blue compound LPB, which is the ladder-type analogue of fully oxidized PANI, i.e. the pernigraniline base. The reductive conversion from LPB through LEB to LLB was also clean and quantitative upon the treatment of reducing reagents, such as hydrazine or ascorbic acid. These well-defined, reversible oxidation and reduction processes indicated the highly robust nature of LPANI. In addition, LPB can be protonated on the cyclohexadiene-1,4-diimine units under strong acidic conditions, such as 4.0 mol L−1 methanesulfonic acid (MSA), to afford LPS, which is a ladder-type counterpart of the unstable, PANI-derived pernigraniline salt (Fig. 1d). As expected, LPS exhibits much higher stability under acidic conditions than conventional pernigraniline salt.
Thanks to the presence of n-dodecyl groups R1 on the fluorene unit and the p-tolyl groups R2 on the backbone, LPANI is well-soluble in common organic solvents such as THF and chloroform regardless of the oxidation state, enabling comprehensive solution-phase characterization and processing. The molar mass values of all three forms of LPANI were measured by size exclusion chromatography (Fig. 1b), showing similar polydispersity (Đ = 1.67–1.95) and a slight increase in measured Mn from LLB (21.6 kg mol−1) to LPB (25.5 kg mol−1). Considering the higher rigidity of the oxidized cyclohexadiene-1,4-diimine unit than that of the reduced phenylene-diamine unit, the higher apparent Mn of LPB and LEB is in a good agreement with the hypothesis that LPB has a more rigid backbone and hence a larger radius of gyration, according to the Mark–Houwink equation.33,34
The chemical oxidation of LLB to LPB was monitored with UV-vis spectroscopy (Fig. 1c). With an increasing amount of oxidant (mCPBA) added into LLB, the absorption peak of LLB at 375 nm decreased and a broad peak centered at 680 nm emerged as a result of the generation of conjugated cyclohexadiene-1,4-diimine unit moieties. The clear isosbestic point during the titration demonstrated the well-defined, clean transformation between these different redox states. In comparison, conventional PANI does not exhibit such a clear isosbestic point when transforming from the leucoemeraldine base to pernigraniline base, likely due to the presence of configurational isomers and poor solubility of the pernigraniline base.35 In addition, by comparing the spectra of LEB obtained by air oxidation with the titration plot, the oxidation ratio of LEB was determined to be ∼50% (Fig. S1†). The photo-absorption peak of LPB is narrow and exhibits a clear vibrational progression, indicating a well-defined configuration and a rigid conformation at room temperature. This result is in sharp contrast with the spectrum of a conventional non-ladder pernigraniline base, which shows a broad absorption peak with no vibrational features due to the fast trans-/cis- isomerization and torsional rotation.35–37 These pronounced differences between LPB and the conventional pernigraniline base suggest the important impacts of a ladder-type constitution on extending the orbital/state delocalization as well as on inhibiting isomerization and conformational disorder.
In order to fully elucidate the constitutional structures of LPANI, two small molecular model compounds, SLB and SPB, were synthesized and characterized with 1H, 13C, HSQC, and HMBC NMR spectroscopy (Fig. S13–S20†). The 13C NMR peaks for SLB and SPB were fully assigned to their structures, so that they can be used as references to analyze the spectra of LPANI. 1H and 13C NMR spectra of LLB, LEB, and LPB were obtained in d8-THF at room temperature. Compared to the broad and heavily overlapping 1H NMR signals (Fig. S23 and S25†), the 13C NMR peaks of LLB and LPB are sharp and well-defined. They matched well with those of SLB and SPB, respectively, so that full assignments of these polymer peaks were accomplished (Fig. 2). The clearly identified NMR signals of LLB indicated high efficiency and regioselectivity of the Friedel–Crafts cyclization reaction, and the absence of observable defects during the ladderization step. Comparing the 13C NMR spectra of LLB and LPB, it is notable that the signals of the central ring (C5, C6, C9, and C12) downfield shifted dramatically in the spectrum of LPB due to the more electron-deficient nature of the oxidized cyclohexadiene-1,4-diimine units compared to the electron-rich form of phenylene-1,4-diamine in LLB. Less distinct shifts were observed for carbons on the fluorene units (C10, C11, and C13) and the sp3–C bridge (C14). Apart from the backbone, the p-tolyl side groups (C7 and C8) also showed distinguishable chemical shift changes, serving as a good probe for identifying different oxidation states. LEB can be considered a statistic copolymer of the phenylene-1,4-diamine- and the cyclohexadiene-1,4-diimine-containing repeating units. Not surprisingly, its 13C NMR spectrum exhibited clearly both sets of peaks found in those of LLB and LPB. The functional group transformation among these compounds is further confirmed by FT-IR spectra. The characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (1681 cm−1) and CC (1604 cm−1) stretching bands of cyclohexadiene-1,4-diimine units were observed in LEB and LPB while absent in LLB (Fig. S2†).
N (1681 cm−1) and CC (1604 cm−1) stretching bands of cyclohexadiene-1,4-diimine units were observed in LEB and LPB while absent in LLB (Fig. S2†).
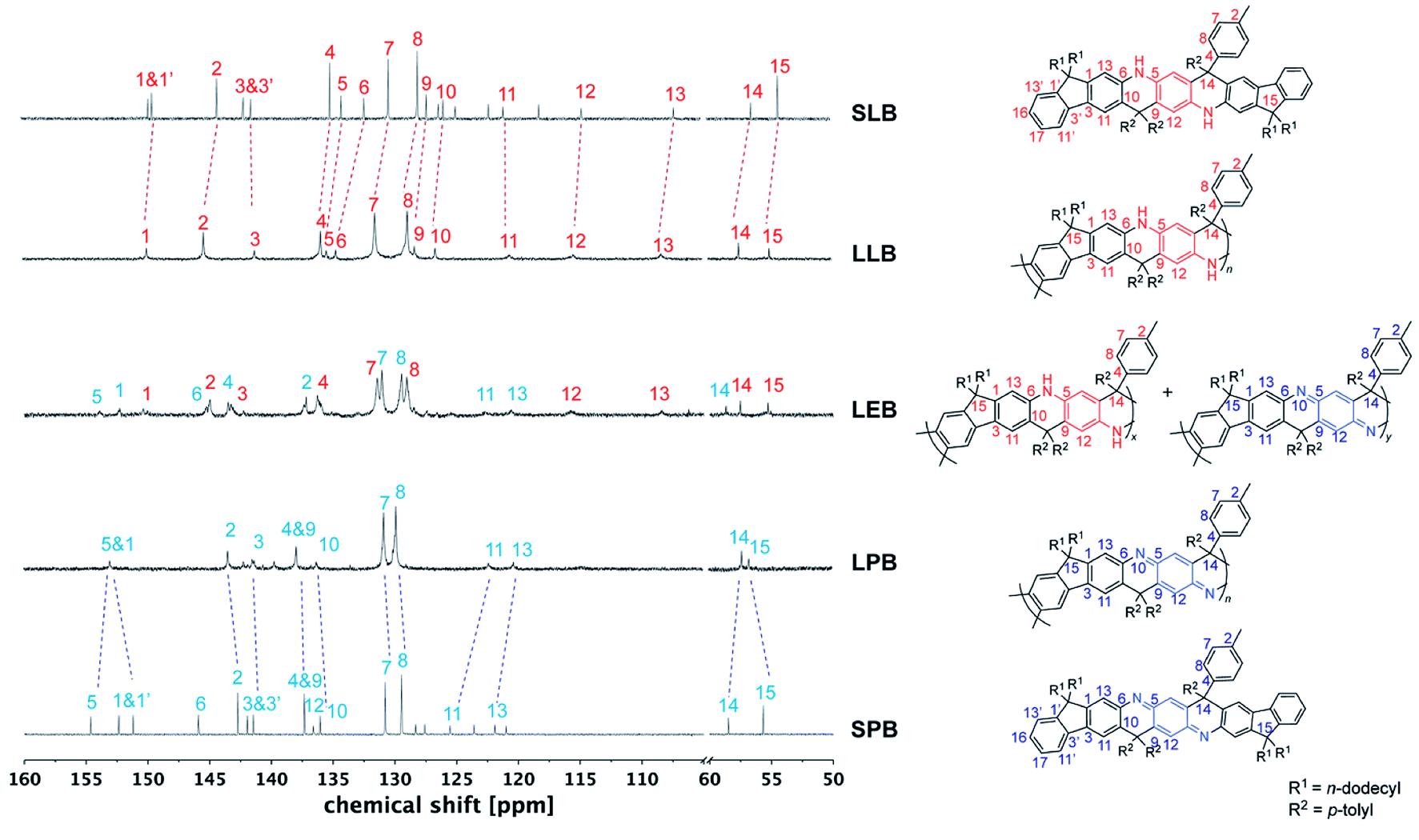 | ||
| Fig. 2 13C NMR spectra of small molecule model SLB and SPB, and varied oxidation states of LPANI (d8-THF at 298 K). | ||
The electrochemical interconversion of different oxidation states of LPANI was investigated in both the solution phase and solid state by cyclic voltammetry (CV) experiments. LEB was selected as the focus of the analysis as the polymer backbone contains both the phenylene-1,4-diamine unit (unit A in Fig. 3), which can be oxidized, and the cyclohexadiene-1,4-diimine unit (unit D in Fig. 3), which can be reduced (Fig. 3a). Referring to the cyclic voltammograms of SLB and SPB (Fig. S3†), the step-wise oxidation and reduction processes of a thin film of LEB were identified. In an inert electrolyte (TBAPF6) solution, these processes are electrochemically irreversible although the chemical redox processes were confirmed to be reversible. The electrochemical irreversibility was attributed to the deprotonation of the fully oxidized form C into C′ and protonation of the fully reduced form F into A shown in Fig. 3a. Differently, with high concentration electrolytes of Brønsted acid or LiClO4, reversible oxidation processes were observed among different oxidation states, because the oxidized unit C was kept protonated or lithiated upon oxidation (Fig. 3b and c). These results demonstrated the intrinsically reversible nature of the interconversion between A and C, which was important for the subsequent electrochromic device with Li+ electrolyte. CV of LLB and LPB was also conducted, showing only the anticipated oxidation process of A and reduction process of D, respectively (Fig. S4†). The presence of multiple redox peaks in these cyclic voltammograms indicated good electronic coupling between neighboring units along the ladder-type polymer backbones, i.e., the oxidation/reduction of one unit made it much more difficult to oxidize/reduce the neighboring unit due to strong electronic coupling and molecular orbital delocalization. The fully reversible oxidation and reduction processes of LPANI under protonated or lithiated conditions demonstrated its excellent robustness within a wide potential range, and set the foundation for its electrochemical applications.
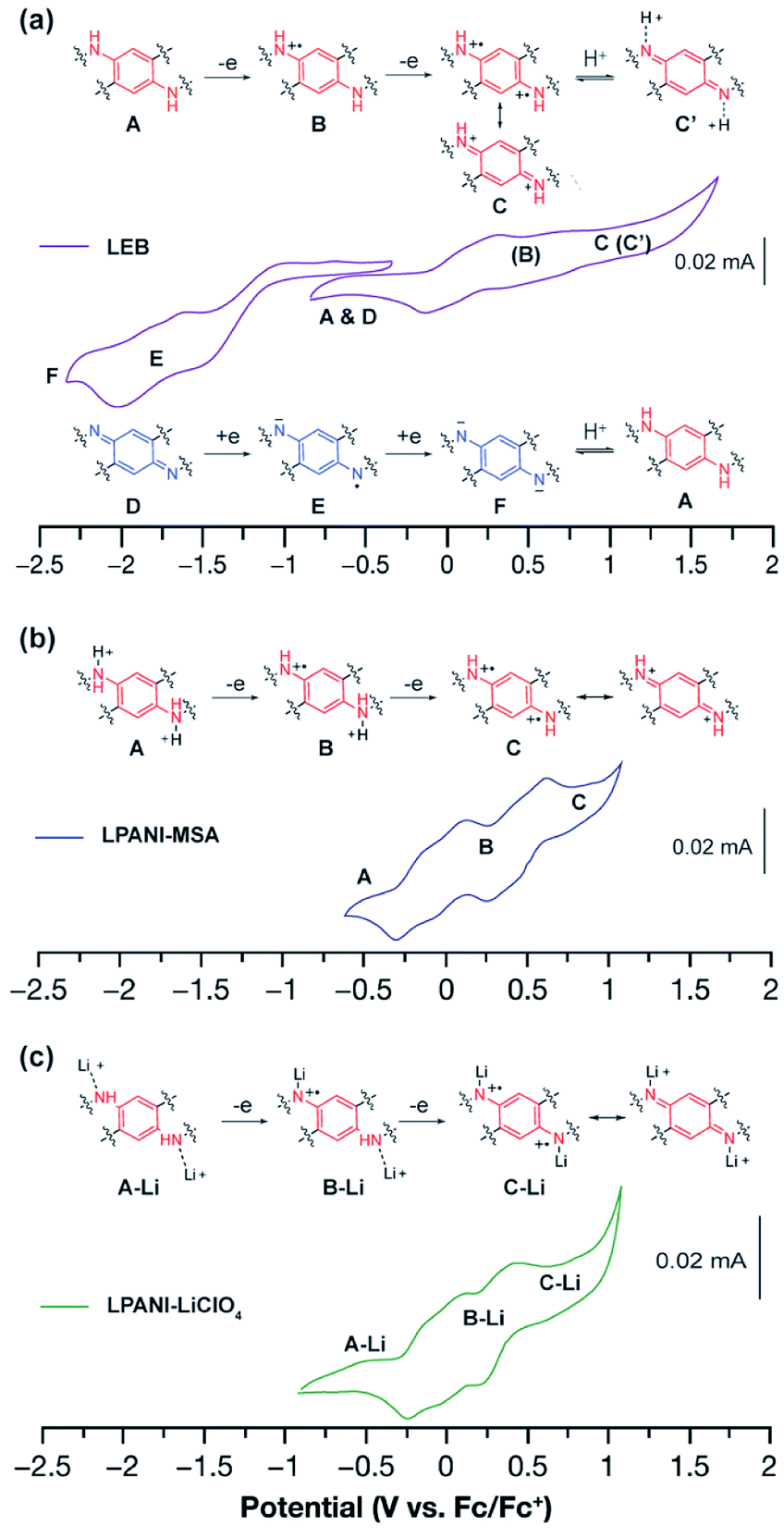 | ||
| Fig. 3 Cyclic voltammograms of (a) the LEB film in acetonitrile with 0.1 mol L−1 TBAPF6. (b) LPANI solution in THF with 0.15 mol L−1 MSA. (c) LPANI solution in THF with 1.2 mol L−1 LiClO4. In all cases, a Pt wire was used as the counter electrode and Ag/AgCl was used as the reference electrode. | ||
The electrochemical stability of LPANI was studied under highly acidic and oxidative conditions, which is of great importance for applications including energy storage and electrochromic devices.38 It is well known that, under acidic conditions, electrochemically oxidized conventional PANI (in its pernigraniline salt form) readily degrades and leads to deterioration of the electrochemical cycles.39,40 In this work, conventional PANI and LPANI were both coated on carbon fabric cloth as the working electrodes for the test. In acetonitrile solution with 0.5 mol L−1 of sulfuric acid, a controlled potential at +1.0 V (vs. Ag/AgCl) was maintained for variable time periods (0–60 min), during which polymers on the working electrode were fully oxidized and maintained in the form of pernigraniline salt at the oxidation potential. After each controlled-potential period, a CV experiment was performed and the peak current intensity was used to evaluate the relative amount of remaining electroactive species.41,42 A rapid decreasing of CV current intensity of conventional PANI was observed (Fig. 4a), showing degradation by ca. 40% in the first 10 min. The degradation process was also found to be accelerated with increased oxidative potentials (Fig. S5†). In contrast, the cyclic voltammograms of LPANI remained almost unchanged after applying controlled potential at +1.0 V under these harsh conditions for up to 60 min (Fig. 4b). Although slight decomposition started to take place when the potential was increased to +2.0 V (Fig. S6†), the much enhanced electrochemical and acidic stability of LPANI suggested that the long-standing issues of hydrolysis and redox side-reactions of PANI derivatives could be intrinsically inhibited by the fully ladder-type constitution.
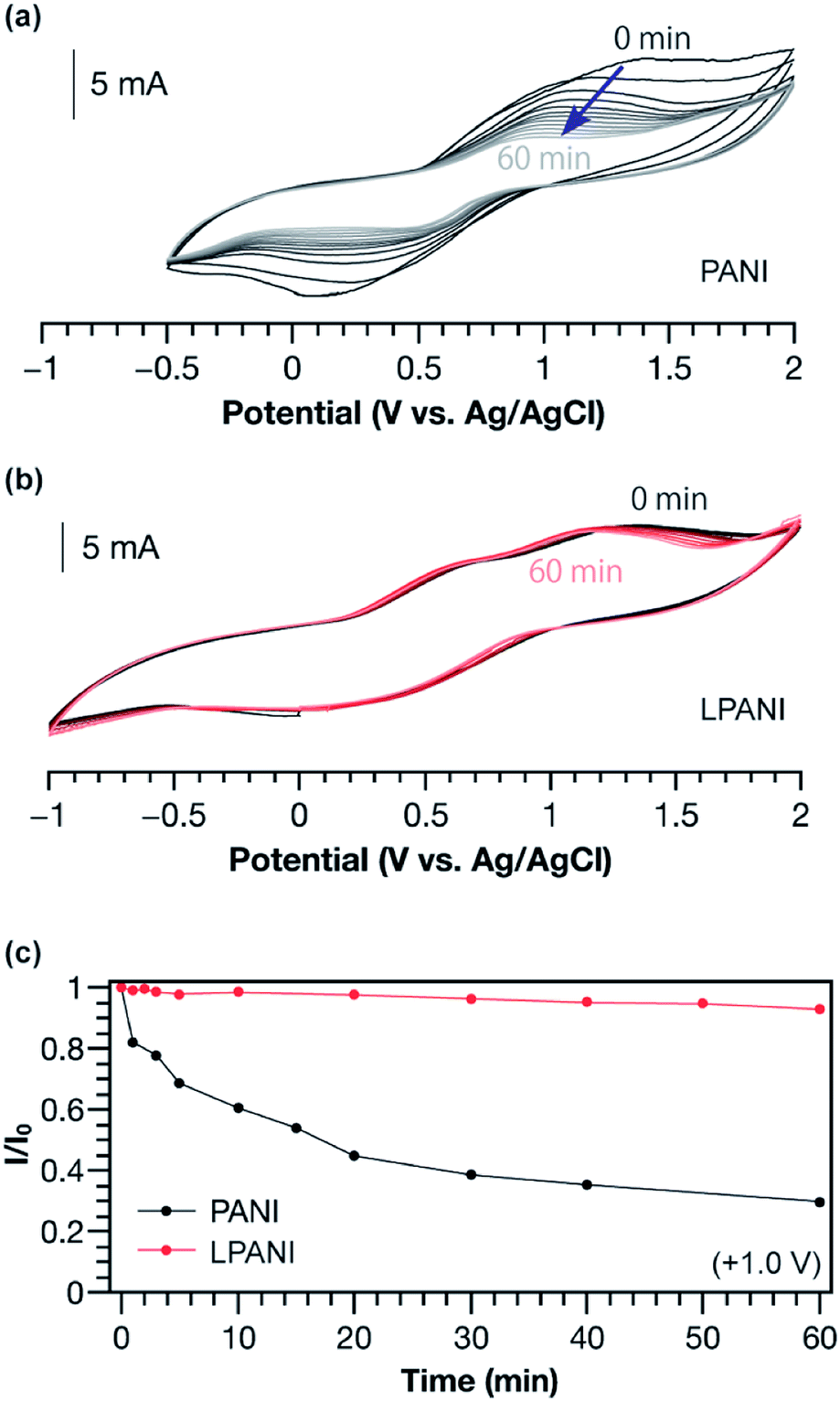 | ||
| Fig. 4 Cyclic voltammograms of (a) conventional PANI and (b) LPANI after applying +1.0 V potential for a variable amount of time (0–60 min). (c) Plot of the current intensities in (a) and (b) vs. the time of holding at +1.0 V. The experiment was performed on working electrodes (conventional PANI or LPANI solid deposited on carbon fabric cloth) in acetonitrile with 0.5 mol L−1 sulfuric acid. A Pt wire was used as the counter electrode and Ag/AgCl was used as the reference electrode. | ||
The superior stability of LPANI in the LPS state set the stage for comprehensive investigations of this acid-doped species with ensured structural integrity. The absorption spectrum of LPS exhibited a significant red-shift of the band compared to that of LPB, into the NIR region (λmax ∼ 1400 nm) (Fig. 5a). Solid-state LPS was prepared by mixing LPB with an excess amount of non-volatile acid such as p-toluenesulfonic acid (PTSA). A spin-cast thin film of the LPS/PTSA mixture exhibited a further red-shift of the absorption peak to λmax ∼ 1650 nm, likely as a result of intermolecular electronic coupling between the ladder polymer chains. Despite in highly acidic media, LPS was remarkably stable in either solution-phase or solid-state, with little change in the absorption spectra for over 48 hours under ambient conditions (Fig. S7†). Such an excellent stability under highly acidic conditions is unprecedented among conventional non-ladder pernigraniline derivatives.
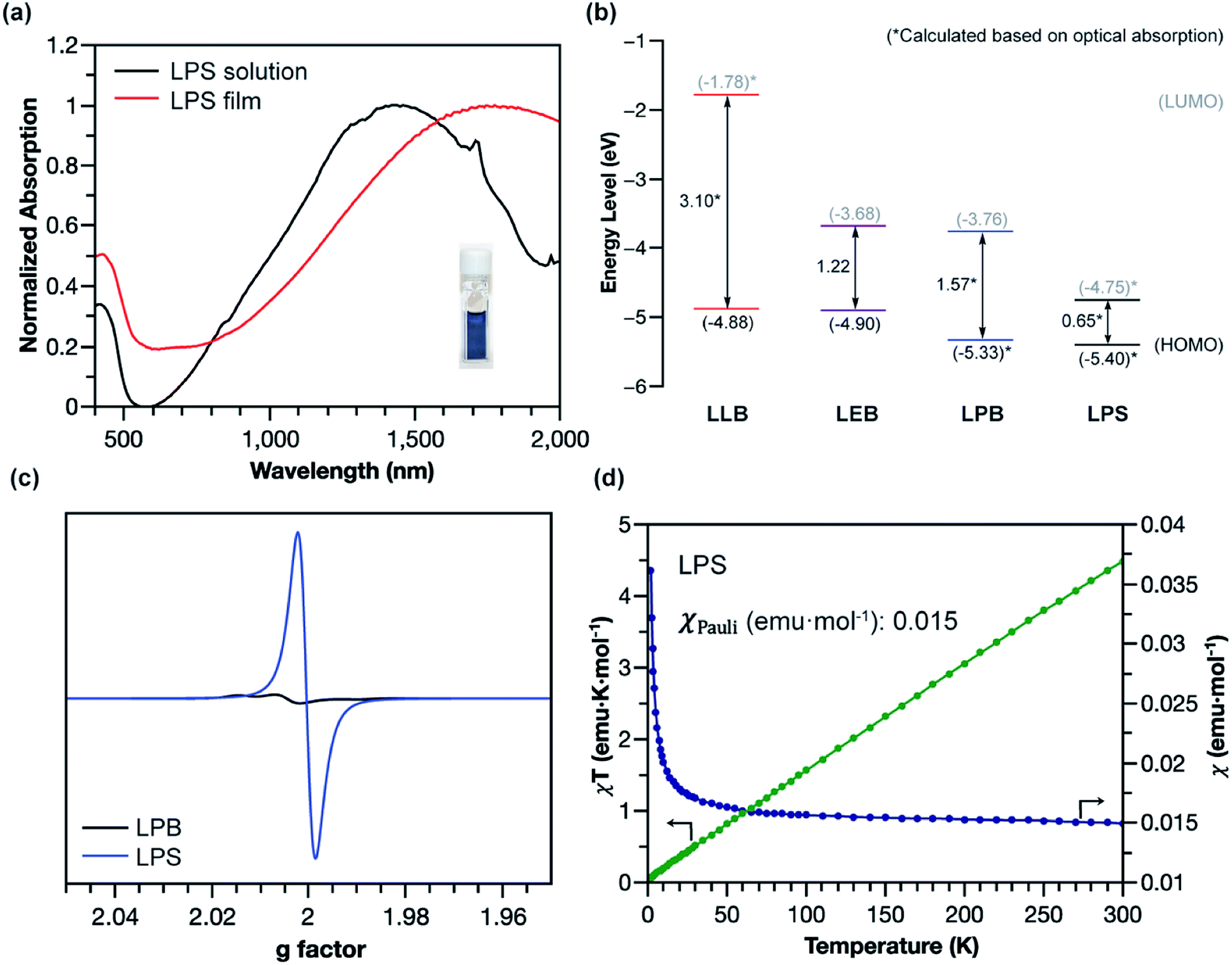 | ||
| Fig. 5 (a) Visible-NIR absorption spectra of LPS in solution and in thin film. (b) HOMO/LUMO energy level diagrams of LLB, LEB, LPB, and LPS. (c) EPR spectra of LPB and the LPS solid. (d) Temperature-varied magnetic susceptibility of the LPS solid. | ||
The electronic structures of the varied states of LPANI were elucidated experimentally. Based on the CV data and UV-vis-NIR absorption spectra, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of LLB, LEB, LPB, and LPS were determined (Fig. 5b). The estimated bandgaps of LLB, LEB, and LPB are 3.10, 1.22, and 1.57 eV, respectively, which were all lower by 0.6–0.9 eV than those of the corresponding conventional PANI derivatives.43 The uniformly lower bandgaps indicated that the coplanar conformation of the ladder polymer backbone greatly extended the MO delocalization and π-conjugation. A drastically deeper LUMO level of LPS than that of LPB was a result of acid-doping of the cyclohexadiene-1,4-diimine units.44,45 The small energy gap resulted in a better admixture of the HOMO and LUMO in the ground state, leading to significant natural orbital occupancies in the lowest unoccupied natural orbitals and, therefore, open-shell diradical character.46–50 As shown in the electron paramagnetic resonance (EPR) spectra (Fig. 5c and S8a†), LPS possessed a much higher spin concentration compared to LPB, as a result of the acid-induced transition from the closed-shell to open-shell resonance. The acid-doping was also expected to lead to polaron delocalization, which was evidenced by DC conductivity of the solid pellet of acid-mixed LPS (Fig. S10†). Magnetic susceptibility measurement of the LPS solid by using a superconducting quantum interference device (SQUID) revealed a dominant temperature-independent Pauli paramagnetism (χPauli) at 0.015 emu per mole of repeating unit (Fig. 5d and S9a†), indicative of a delocalized nature of the radical cations (polarons) in the solid-state. Compared to the emeraldine salt of conventional PANI, which demonstrated both Pauli paramagnetism and Curie paramagnetism, LPS featured one order of magnitude higher χPauli value and absence of Curie paramagnetism, corresponding to a significantly enhanced polaron delocalization range and higher density of states of polarons at the Fermi level.51 The χPauli of LPS was also significantly higher than those measured on shorter oligomeric analogues,11 suggesting that the longer ladder polymer chains allowed for the extension of polaron delocalization. The acid-doped salt of LEB (LES) was also characterized with EPR and SQUID (Fig. S8b and S9b†). Dominant Pauli paramagnetism was observed with χPauli at 0.0023 emu per mole of repeating unit, although much lower than that of LPS as a result of lower density of the charge carrier. This value, however, is much higher than that of the conventional emeraldine salt (∼10−4 emu per mole of repeating unit), indicating an enhanced polaron delocalization in LES similar to that in LPS.
The outstanding electrochemical stability, fully reversible redox transformation, and conductive nature enabled the application of LPANI in highly robust electrochromic devices. Organic electrochromic materials undergoing reversible optical changes upon electrochemical oxidation or reduction have attracted a great deal of interest in diverse applications.52 Compared with inorganic materials, organic molecules, especially polymers, exhibit advantages in high coloration efficiency, excellent processability, and fast switching ability.53 PANI represents an important class of organic electrochromic materials that can cover visible and NIR regions and has been applied in various fields, such as smart windows, displays, and camouflage.54 The switching between the bleached and colored states of PANI corresponds to the interconversion between leucoemeraldine and half-oxidized emeraldine salt states. The rapid degradation of the fully oxidized pernigraniline salt, however, limits the range of operational voltage, hence imposing an intractable challenge on the practical deployment of robust and recyclable devices based on PANI.38 In this study, LPANI was employed as the active material in a sandwiched electrochromic device, which was fabricated with two ITO–glass substrates, a spin-cast LPANI thin film, and lithium gel electrolyte (PMMA/PC/LiClO4) (Fig. 6a). When −2.0 V voltage was applied on the LPANI layer, the device showed a pale yellow color with an absorption peak at 378 nm, corresponding to the LLB state. As the voltage switched to +2.0 V, the device turned a deep blue color and a broad absorption peak evolved in the NIR region (λmax = 905 nm), corresponding to the lithiated LPS state (Fig. 6b). The color-switching cyclability was studied by applying alternating working voltage at −2.0 V and +2.0 V for 30 s at each stage, leading to fully reversible transitions of the absorption between the LLB and LPS states, in agreement with the solid-state CV results (Fig. 3c). No observable degradation of absorption intensity at 378 nm was observed over 200 cycles, while only less than 2% decay was observed for the absorption at 845 nm (Fig. 6c). Such an excellent cyclability allowed for a reliable kinetic analysis of the electrochromic processes based on data from multiple cycles (Fig. S11†). Both the oxidation and reduction processes fit in a pseudo-first-order model to give the apparent rate constants of 0.10 and 0.39 s−1, respectively (Fig. 6d). The slower oxidation rate is likely a result of the rate-determining lithiation process which requires a slow process of lithium diffusing to the oxidized cyclohexadiene-1,4-diimine unit.55 In contrast, during reduction, the phenylene-1,4-diamine unit state was recovered through the reductive charge transfer followed by a rapid protonation step so that a higher rate constant was observed. Overall, LPANI showed unprecedented electrochromic cyclability among PANI-like polymers over a wide operation voltage window (spanning for 4 V), thanks to the highly reversible interconversion between LLB and LPS states and the extraordinary stability of LPS under highly oxidative conditions. This result further demonstrated the superior electrochromic properties endowed by the ladder-type backbone constitution. It is anticipated that, with the understanding of the electrochromic mechanism and kinetics, further chemical and device optimization can lead to devices with desired electrochromic performance in terms of response rate and color contrast. The design principle of such a robust electrochemical transition can also benefit the development of electrochemical energy storage applications using conjugated polymers.
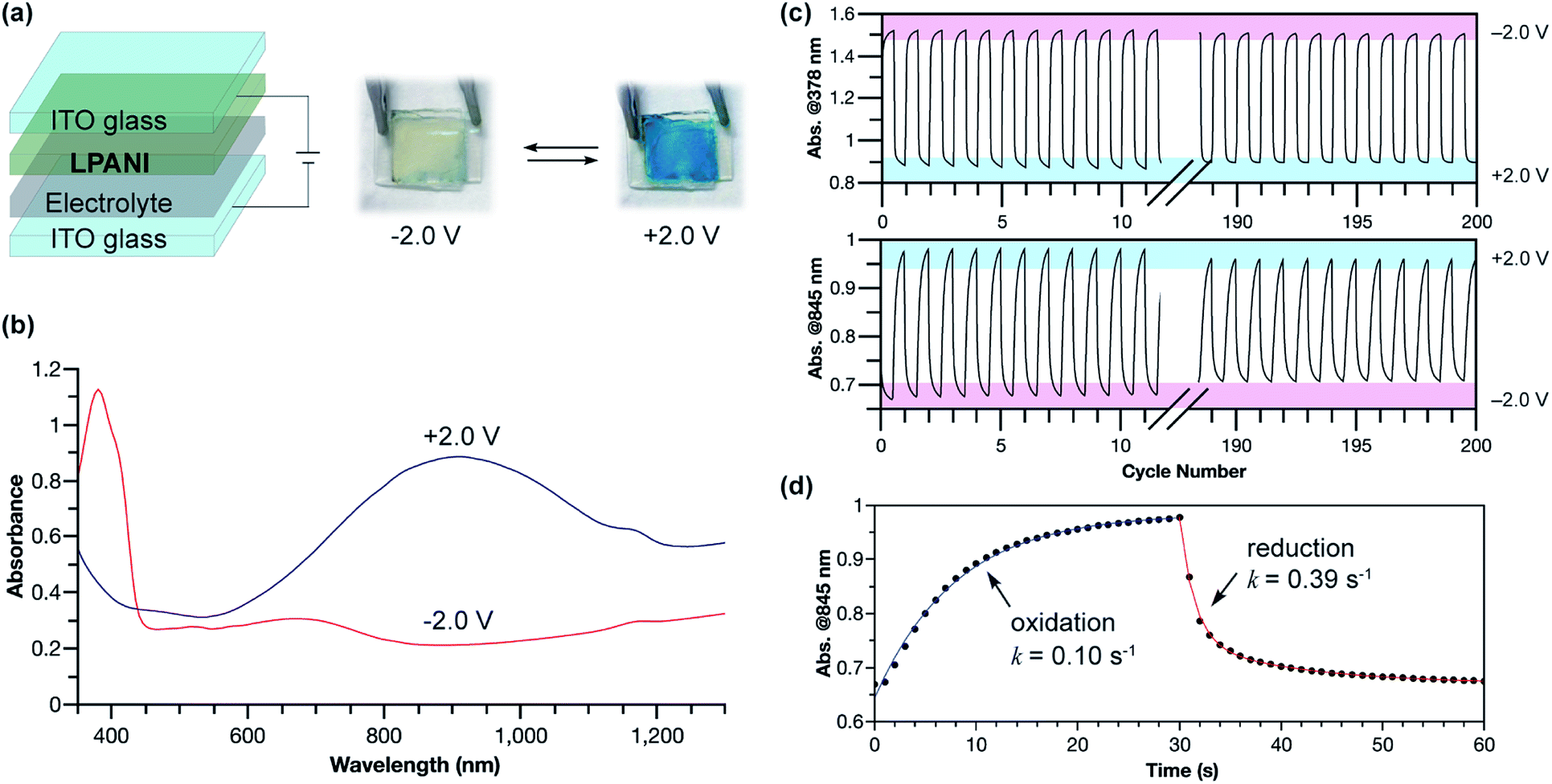 | ||
| Fig. 6 (a) Architecture and photographic images of the electrochromic device at −2.0 V and +2.0 V, respectively. (b) Absorption spectra of the electrochromic device when applying −2.0 V or +2.0 V for 60 s. (c) Absorbance at 378 nm and 845 nm with voltage swept between −2.0 V and +2.0 V for 200 cycles. (d) Time-dependent absorbance changes at 845 nm; dots: experimental data spots; lines: pseudo-first-order fitting. | ||
Conclusion
In summary, this work demonstrates that a ladder-type backbone constitution can be incorporated into polyaniline analogues through a feasible and scalable synthesis. A ladder-type polyaniline-analogous polymer with a low defect level was obtained through highly efficient and regioselective cyclization. The ladder-type constitution imparts unprecedented stability and extensive state delocalization to the resulting LPANI derivatives. Specifically, LPANI derivatives exhibit significantly superior electrochemical stability compared to non-ladder PANI derivatives, evidenced by the highly reversible transformation among different oxidation states and extraordinary robustness under harsh electrochemical conditions. The excellent stability also allows for the investigation and application of the strong NIR absorption and Pauli paramagnetism of the acidified and fully oxidized LPS form, revealing its low bandgap and the presence of extended polaron delocalization in the solid-state. The unique combination of these functions enables the fabrication of a robust and highly recyclable NIR electrochromic device using LPANI as the active material. Taken together, this work establishes the principle of using a ladder-type constitution to address the long-standing challenges of stability and state delocalization seen in the field of organic electronic materials. This strategy should be formulated in the future design of electronically active polymers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support by the Robert A. Welch Foundation (A-1898), the Qatar National Priority Research Program (NPRP11S-1204-170062), and the National Natural Science Foundation of China (Grant No. 51703131). KRD gratefully acknowledges funding from the National Science Foundation under CHE-1808779 and the Robert A. Welch Foundation under Grant A-1449 for summer support for HX. Use of the TAMU Materials Characterization Facility is acknowledged.Notes and references
- J. Lee, A. J. Kalin, T. Yuan, M. Al-Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC.
- C. Zhu, A. J. Kalin and L. Fang, Acc. Chem. Res., 2019, 52, 1089–1100 CrossRef CAS PubMed.
- Z. Cai, M. A. Awais, N. Zhang and L. Yu, Chem, 2018, 4, 2538–2570 CAS.
- Z. Cai, W.-Y. Lo, T. Zheng, L. Li, N. Zhang, Y. Hu and L. Yu, J. Am. Chem. Soc., 2016, 138, 10630–10635 CrossRef CAS PubMed.
- T. P. Silverstein, J. Chem. Educ., 2012, 89, 1159–1167 CrossRef CAS.
- B. Carlotti, Z. Cai, H. Kim, V. Sharapov, I. K. Madu, D. Zhao, W. Chen, P. M. Zimmerman, L. Yu and T. Goodson III, Chem. Mater., 2018, 30, 4263–4276 CrossRef CAS.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 CrossRef CAS PubMed.
- D. Yuan, Y. Guo, Y. Zeng, Q. Fan, J. Wang, Y. Yi and X. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4958–4962 CrossRef CAS PubMed.
- Y. Zou, X. Ji, J. Cai, T. Yuan, D. J. Stanton, Y.-H. Lin, M. Naraghi and L. Fang, Chem, 2017, 2, 139–152 CAS.
- S. Che, J. Pang, A. J. Kalin, C. Wang, X. Ji, J. Lee, D. Cole, J.-L. Li, X. Tu and Q. Zhang, ACS Mater. Lett., 2019, 2, 49–54 CrossRef.
- X. Ji, H. Xie, C. Zhu, Y. Zou, A. U. Mu, M. Al-Hashimi, K. R. Dunbar and L. Fang, J. Am. Chem. Soc., 2020, 142, 641–648 CrossRef CAS PubMed.
- E. Genies, A. Boyle, M. Lapkowski and C. Tsintavis, Synth. Met., 1990, 36, 139–182 CrossRef CAS.
- R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147–156 RSC.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581–2590 CrossRef CAS PubMed.
- K. D. Fong, T. Wang and S. K. Smoukov, Sustainable Energy Fuels, 2017, 1, 1857–1874 RSC.
- A. G. MacDiarmid, Synth. Met., 1997, 84, 27–34 CrossRef CAS.
- A. E. Pullen and T. M. Swager, Macromolecules, 2001, 34, 812–816 CrossRef CAS.
- S. Virji, J. Huang, R. B. Kaner and B. H. Weiller, Nano Lett., 2004, 4, 491–496 CrossRef CAS.
- A. Mirmohseni and A. Oladegaragoze, Synth. Met., 2000, 114, 105–108 CrossRef CAS.
- A. MacDiarmid, L. Yang, W. Huang and B. Humphrey, Synth. Met., 1987, 18, 393–398 CrossRef CAS.
- A. Pud, Synth. Met., 1994, 66, 1–18 CrossRef CAS.
- A. Malinauskas and R. Holze, J. Appl. Polym. Sci., 1999, 73, 287–294 CrossRef CAS.
- L. M. Santino, Y. Lu, S. Acharya, L. Bloom, D. Cotton, A. Wayne and J. M. D'Arcy, ACS Appl. Mater. Interfaces, 2016, 8, 29452–29460 CrossRef CAS PubMed.
- F. F. Bazito, L. T. Silveira, R. M. Torresi and S. I. C. de Torresi, Phys. Chem. Chem. Phys., 2008, 10, 1457–1462 RSC.
- G. Ćirić-Marjanović, Synth. Met., 2013, 177, 1–47 CrossRef.
- H.-J. Yen, H.-Y. Lin and G.-S. Liou, Chem. Mater., 2011, 23, 1874–1882 CrossRef CAS.
- N. Sun, S. Meng, D. Chao, Z. Zhou, Y. Du, D. Wang, X. Zhao, H. Zhou and C. Chen, Polym. Chem., 2016, 7, 6055–6063 RSC.
- H.-J. Yen and G.-S. Liou, Chem. Mater., 2009, 21, 4062–4070 CrossRef CAS.
- M. M. Nobrega, C. H. Silva, V. R. Constantino and M. L. Temperini, J. Phys. Chem. B, 2012, 116, 14191–14200 CrossRef CAS PubMed.
- S. Xiong, S. Li, X. Zhang, R. Wang, R. Zhang, X. Wang, B. Wu, M. Gong and J. Chu, J. Electron. Mater., 2018, 47, 1167–1175 CrossRef CAS.
- F. Li, Y. Zou, S. Wang, L. Fang and J. L. Lutkenhaus, Macromol. Rapid Commun., 2017, 38, 1700067 CrossRef PubMed.
- M. Daigle and J.-F. o. Morin, Macromolecules, 2017, 50, 9257–9264 CrossRef CAS.
- W.-F. Su, Polymer Size and Polymer Solutions, in Principles of Polymer Design and Synthesis, Springer, Berlin, Heidelberg, 2013 Search PubMed.
- H. W. Lai, S. Liu and Y. Xia, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3075–3081 CrossRef CAS.
- J. De Albuquerque, L. Mattoso, R. Faria, J. Masters and A. MacDiarmid, Synth. Met., 2004, 146, 1–10 CrossRef CAS.
- J. Albuquerque, L. Mattoso, D. T. Balogh, R. M. Faria, J. Masters and A. MacDiarmid, Synth. Met., 2000, 113, 19–22 CrossRef CAS.
- A. G. MacDiarmid, S. K. Manohar, J. G. Masters, Y. Sun, H. Weiss and A. J. Epstein, Synth. Met., 1991, 41, 621–626 CrossRef CAS.
- R. Mažeikien and A. Malinauskas, Eur. Polym. J., 2002, 38, 1947–1952 CrossRef.
- A. Watanabe, K. Mori, Y. Iwasaki, Y. Nakamura and S. Niizuma, Macromolecules, 1987, 20, 1793–1796 CrossRef CAS.
- H. Tang, Y. Ding, C. Zang, J. Gu, Q. Shen and J. Kan, Int. J. Electrochem. Sci., 2014, 9, 7239–7252 Search PubMed.
- A. Zhang, C. Cui and J. Y. Lee, Synth. Met., 1995, 72, 217–223 CrossRef CAS.
- C. Cui, X. Su and J. Y. Lee, Polym. Degrad. Stab., 1993, 41, 69–76 CrossRef CAS.
- W. Huang and A. MacDiarmid, Polymer, 1993, 34, 1833–1845 CrossRef CAS.
- A. Varela-Álvarez, J. A. Sordo and G. E. Scuseria, J. Am. Chem. Soc., 2005, 127, 11318–11327 CrossRef PubMed.
- R.-X. Wang, L.-F. Huang and X.-Y. Tian, J. Phys. Chem. C, 2012, 116, 13120–13126 CrossRef CAS.
- T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186–2199 RSC.
- J. E. Baker, J. J. Dressler, A. Cárdenas Valdivia, R. Kishi, E. T. Strand, L. N. Zakharov, S. N. MacMillan, C. J. Gómez-García, M. Nakano, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2020, 142, 1548–1555 CrossRef PubMed.
- G. Tan and X. Wang, Acc. Chem. Res., 2017, 50, 1997–2006 CrossRef CAS PubMed.
- X. Hu, H. Chen, L. Zhao, M. Miao, J. Han, J. Wang, J. Guo, Y. Hu and Y. Zheng, Chem. Commun., 2019, 55, 7812–7815 RSC.
- K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen and H. Sun, J. Am. Chem. Soc., 2020, 142, 4329–4340 CrossRef CAS PubMed.
- V. Krinichnyi, A. Konkin and A. Monkman, Synth. Met., 2012, 162, 1147–1155 CrossRef CAS.
- S. Wang, E. K. Todd, M. Birau, J. Zhang, X. Wan and Z. Y. Wang, Chem. Mater., 2005, 17, 6388–6394 CrossRef CAS.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- J. Tarver, J. E. Yoo and Y.-L. Loo, Chem. Mater., 2010, 22, 2333–2340 CrossRef CAS.
- D. Ge, L. Yang, Z. Tong, Y. Ding, W. Xin, J. Zhao and Y. Li, Electrochim. Acta, 2013, 104, 191–197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03348k |
| This journal is © The Royal Society of Chemistry 2020 |