
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of a putative sensory [FeFe]-hydrogenase provides new insight into the role of the active site architecture†
Henrik
Land
 a,
Alina
Sekretareva
a,
Ping
Huang
a,
Holly J.
Redman
a,
Brigitta
Németh‡
a,
Nakia
Polidori§
a,
Lívia S.
Mészáros
a,
Moritz
Senger
bc,
Sven T.
Stripp
*c and
Gustav
Berggren
*a
a,
Alina
Sekretareva
a,
Ping
Huang
a,
Holly J.
Redman
a,
Brigitta
Németh‡
a,
Nakia
Polidori§
a,
Lívia S.
Mészáros
a,
Moritz
Senger
bc,
Sven T.
Stripp
*c and
Gustav
Berggren
*a
aMolecular Biomimetics, Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, SE-75120, Uppsala, Sweden. E-mail: gustav.berggren@kemi.uu.se
bPhysical Chemistry, Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, SE-75120, Uppsala, Sweden
cBioinorganic Spectroscopy, Department of Physics, Freie Universität Berlin, Arnimallee 14, DE-14195, Berlin, Germany. E-mail: sven.stripp@fu-berlin.de
First published on 21st September 2020
Abstract
[FeFe]-hydrogenases are known for their high rates of hydrogen turnover, and are intensively studied in the context of biotechnological applications. Evolution has generated a plethora of different subclasses with widely different characteristics. The M2e subclass is phylogenetically distinct from previously characterized members of this enzyme family and its biological role is unknown. It features significant differences in domain- and active site architecture, and is most closely related to the putative sensory [FeFe]-hydrogenases. Here we report the first comprehensive biochemical and spectroscopical characterization of an M2e enzyme, derived from Thermoanaerobacter mathranii. As compared to other [FeFe]-hydrogenases characterized to-date, this enzyme displays an increased H2 affinity, higher activation enthalpies for H+/H2 interconversion, and unusual reactivity towards known hydrogenase inhibitors. These properties are related to differences in active site architecture between the M2e [FeFe]-hydrogenase and “prototypical” [FeFe]-hydrogenases. Thus, this study provides new insight into the role of this subclass in hydrogen metabolism and the influence of the active site pocket on the chemistry of the H-cluster.
Introduction
Hydrogenase enzymes play a central role in hydrogen metabolism, where they catalyze the interconversion between protons and molecular hydrogen (H2). The [FeFe]-hydrogenases are generally considered the most active, operating close to the thermodynamic limit with reported H2 production rates exceeding 9000 s−1.1,2 Consequently, they have been intensively studied, both for their biotechnological potential and as a model system for the design of synthetic catalysts.3,4 Phylogenetically, [FeFe]-hydrogenases can be broadly divided into four main groups, denoted group A, B, C, and D, which in turn contain numerous subclasses.5–9 Considering the well-conserved nature of the auxiliary proteins involved in cofactor assembly (HydEFG),6 they all arguably share a dependence on the same hexanuclear iron cofactor, the “H-cluster”. This biologically unique cofactor consists of a canonical [4Fe–4S] cluster ([4Fe–4S]H) connected to a low valent dinuclear iron complex ([2Fe]H).10–13 The [2Fe]H subsite is coordinated by CO and CN− ligands, and bridged by an azadithiolate ligand (adt = −SCH2NHCH2S−). The overwhelming majority of biochemically characterized [FeFe]-hydrogenases belong to group A, with a primary focus on the “prototypical” [FeFe]-hydrogenases, e.g., Cr HydA1 from Chlamydomonas reinhardtii,14,15Dd HydAB from Desulfovibrio desulfuricans,11,16,17CpI from Clostridium pasteurianum,10,18 as well as the multimeric electron bifurcating [FeFe]-hydrogenase from Thermotoga maritima.18–20 Studies of these enzymes form the foundation for our understanding of [FeFe]-hydrogenase biochemistry. Spectroscopy has identified numerous redox and protonation states of the H-cluster, around which various mechanistic proposals have been put forth.21–24 In short, the active-ready resting state (Hox) features a mixed valence Fe(II)Fe(I) form of the [2Fe]H subsite and an oxidized [4Fe–4S]H cluster (2+). One-electron reduction results in either the Hred′ or Hred state, where Hred′ features a reduced [4Fe–4S]H cluster while Hred features a reduced and protonated [2Fe]H subsite.25 Further reduction results in the formation of the so-called Hhyd state featuring a terminal hydride on the [2Fe]H subsite.26–28 Protonation of Hhyd results in H2 release, potentially proceeding via a discrete intermediate (HhydH+),27,29 and returns the H-cluster to the Hox state. Additionally, CO can reversibly bind to the H-cluster, giving rise to the inhibited Hox-CO and Hred′-CO states.30Considering the diverse nature of [FeFe]-hydrogenase, both with regards to structure as well as function, it is clear that characterization of representative examples from other subclasses is necessary to complete our understanding of this enzyme family and H-cluster chemistry. It has repeatedly been shown that [FeFe]-hydrogenases can operate at minimal over-potentials, albeit specific enzymes generally display a bias for either H+ reduction or H2 oxidation.31–34 Indeed, even in the relatively narrow selection of enzymes studied to-date significant differences in catalytic rates, stability of different H-cluster states and reactivity towards inhibitors (e.g., CO and O2) have been observed.22,31,35–39 On a fundamental level, further insight into subclass-specific reactivities is critical for our understanding of hydrogen metabolism, and elucidating the interplay between the H-cluster and the protein. It will also serve to strengthen efforts related to biotechnological energy applications and potentially facilitate the development of selective antibiotics.8,9
We recently reported the whole-cell characterization of an [FeFe]-hydrogenase from the thermophilic firmicute Thermoanaerobacter mathranii in E. coli.40 The enzyme belongs to the hitherto uncharacterised M2e subclass, which displays a number of well-conserved differences in amino acid sequence as compared to the prototypical group A hydrogenases; namely in the active site cavity and the proton transfer pathway (Fig. 1).41 The M2e subclass has been proposed to form a distinct group of [FeFe]-hydrogenases, group D, and their physiological function is unknown.5 Still, it is phylogenetically most closely related to the M2f subclass of group C, which has been identified as “putative sensory” and includes the recently characterized Tm HydS enzyme from Thermotoga maritima.5,7,8,38 In addition to the H-domain, which harbors the H-cluster, both subclasses feature an N-terminal domain with two [4Fe–4S] cluster-binding motifs as well as a C-terminal domain with a four-cysteine motif (Cx2Cx4Cx16C) indicative of iron–sulfur (FeS) cluster binding. Additionally, the highly conserved cysteine residue initiating the proton transfer pathway from the adt amine in group A [FeFe]-hydrogenase is not conserved in the M2e and M2f subclasses (C299, CpI numbering is used throughout the text unless otherwise noted). Indeed, all residues that have previously been shown to be crucial for proton transfer in group A are missing in these subclasses (Fig. 1).42–44 Moreover, the genes encoding M2e and M2f enzymes are often located on the same operon as group A hydrogenases.5 In light of these similarities, we have now denoted the enzyme as Tam HydS (previously Tam HydA40). However, there are key differences between the two subclasses. In contrast to the M2f enzymes, the M2e subclass does not feature a Per-Arnt-Sim (PAS) domain, which was used to propose the sensing function of group C, as it is usually involved in the regulation of histidine kinases associated with signal transduction.5 In addition, the active site architecture displays distinct differences between the two subclasses (Fig. 1). The two methionine residues framing the diiron site, M497 and M353, are exchanged against serine and glycine in Tm HydS; and leucine and serine, respectively, in Tam HydS. These methionines are considered critical for modulating the reactivity of prototypical [FeFe]-hydrogenases,31,41 and it is noteworthy that the exchange of M353 to a hydroxyl donor (serine or threonine) appears to be a well-conserved property of M2e enzymes. On the other hand, M497 shows a low level of conservation, both for group C and D enzymes.
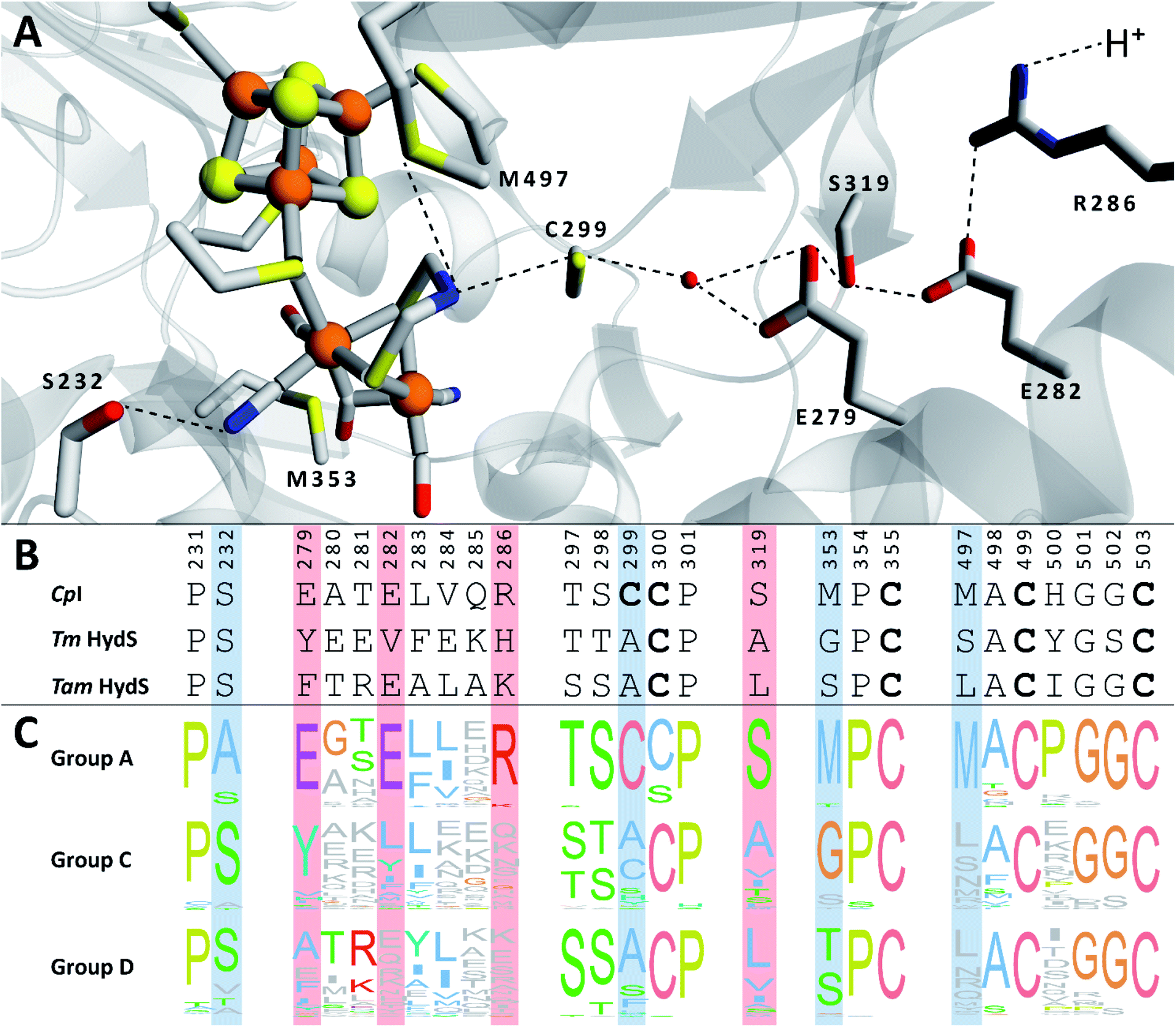 | ||
| Fig. 1 (A) Structural view of the active site and proton transfer pathway of a prototypical [FeFe]-hydrogenase. Structure and numbering based on CpI (PDB ID: 4XDC). Shown amino acid residues are either involved in interactions with the H-cluster or in the proton transfer pathway and show large variations between groups A, C and D. Potential interactions are shown with dashed lines. (B) Amino acid sequence comparison of CpI, Tm HydS and Tam HydS (CpI numbering) based on a ClustalΩ sequence alignment80 of sequences retrieved from Greening et al. 2016 (ref. 7) and homology modeling. H-cluster interacting cysteine residues are highlighted in bold. (C) Normalized consensus logos of [FeFe]-hydrogenase groups A, C and D generated in Jalview using the sequence alignment in (B). Coloring is based on the Clustal X color scheme. Amino acid residues involved in H-cluster interaction and proton transfer that show variation between the groups are highlighted in blue and red, respectively. | ||
Herein, we report the first detailed characterization of an M2e enzyme, and by extension a group D [FeFe]-hydrogenase. The aforementioned Tam HydS enzyme has been isolated following heterologous expression, and characterized through electron paramagnetic resonance (EPR) spectroscopy, attenuated total reflection Fourier-transform infrared (ATR FTIR) spectroscopy, and protein film electrochemistry (PFE). Despite lacking several amino acid residues considered critical for the activity of prototypical [FeFe]-hydrogenase, Tam HydS shows reversible H+/H2 interconversion close to the thermodynamic potential, with a slight bias for H+ reduction. These findings show that the active site pocket can be significantly altered while still retaining the catalytic function of the H-cluster. However, in contrast to previously characterized [FeFe]-hydrogenases, an over-potential is observed at low temperatures; and the overall catalytic rates are low. Moreover, the M2e enzyme displays significant differences in its reactivity towards CO and O2, as compared to previously characterized [FeFe]-hydrogenases. We propose that the catalytic rates are influenced by intramolecular proton transfer. The study, furthermore, highlights the importance of the active site pocket in modulating the reactivity towards known inhibitors, and the stability of specific H-cluster states. Based on the aforementioned properties of Tam HydS which resemble those of known regulatory [NiFe]-hydrogenases, in combination with analysis on a genome level, we hypothesize that group D [FeFe]-hydrogenases serve a sensory rather than catalytic function.
Results and discussion
Isolation and characterization of apo-Tam HydS
Aerobic heterologous expression of Tam HydS in E. coli, in the absence of the HydEFG maturation enzymes, has been shown to result in soluble apo-protein that could be activated in vivo using [2Fe]H subsite mimics.40 In this study, anaerobic isolation of apo-Tam HydS resulted in a purified protein (Fig. S1†) containing 13.9 ± 0.85 Fe/protein. Additional reconstitution of the FeS clusters resulted in a final Fe content of 16.1 ± 0.29 Fe/protein, as expected from the incorporation of four [4Fe–4S] clusters. Isolation of apo-Tam HydS also under aerobic conditions provided a protein with an Fe content of 10.6 ± 0.06 Fe/protein.All three different purified forms of apo-Tam HydS displayed hydrogenase activity in in vitro sodium dithionite/methyl viologen (NaDT/MV2+) enzymatic assays, following anaerobic incubation with the synthetic [2Fe]H analogue [Fe2(adt)(CO)4(CN)2]2− ([2Fe]adt). Activation of aerobically and anaerobically purified apo-Tam HydS samples resulted in H2 production activities of 0.068 ± 0.001 and 0.077 ± 0.009 μmol H2 per min per mg, respectively. A further increase in the H2 production activity, to 0.090 ± 0.005 μmol H2 per min per mg was observed after activation of the reconstituted samples (Fig. 2).
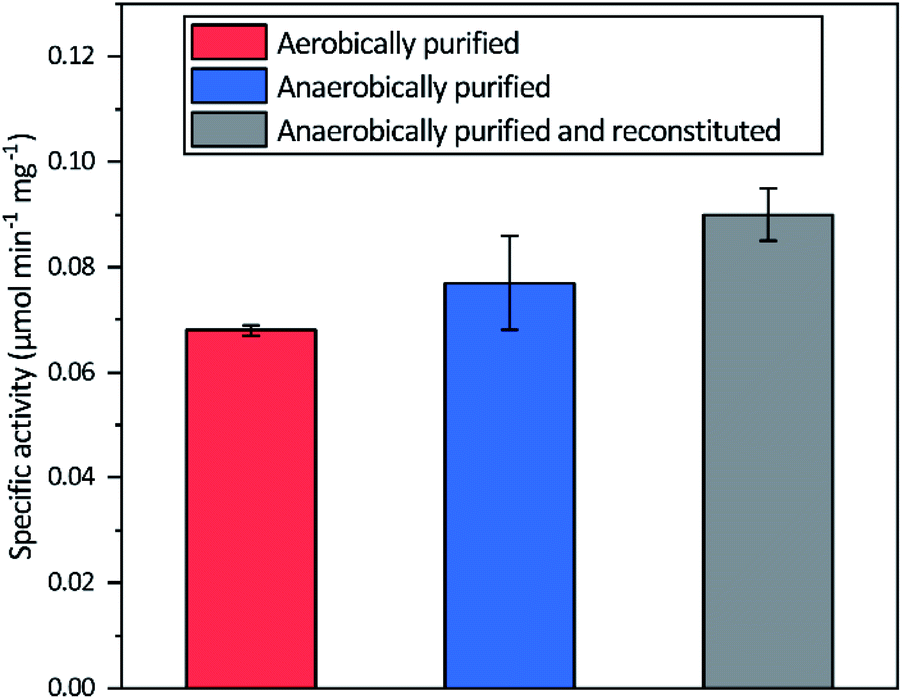 | ||
| Fig. 2 Specific H2 production activities of Tam HydS isolated under different conditions and activated in vitro with [2Fe]adt. The Fe content of the different preparations prior to insertion of [2Fe]adt were 10.9 ± 0.06 (aerobic purification), 13.9 ± 0.85 (anaerobic purification) and 16.1 ± 0.29 (reconstituted) Fe/protein. Reactions were performed in sodium phosphate buffer (100 mM, pH 6.8) with Triton X-100 (1% v/v), methyl viologen (10 mM) and sodium dithionite (100 mM). | ||
X-Band EPR spectra recorded on samples of as-isolated and reconstituted apo-Tam HydS are essentially silent, indicating that all FeS-clusters are present at a [4Fe–4S]2+ oxidation state with spin s = 0, thus diamagnetic. Reduction of the reconstituted enzyme by NaDT at pH 8 brought the sample to an EPR active state, displaying a broad rhombic EPR spectrum near the g ∼ 2 region, typical of s = 1/2 [4Fe–4S]+ species (Fig. 3). Spin quantification resulted in two spin centers per protein, suggesting that two of the [4Fe–4S] clusters are susceptible to NaDT reduction.
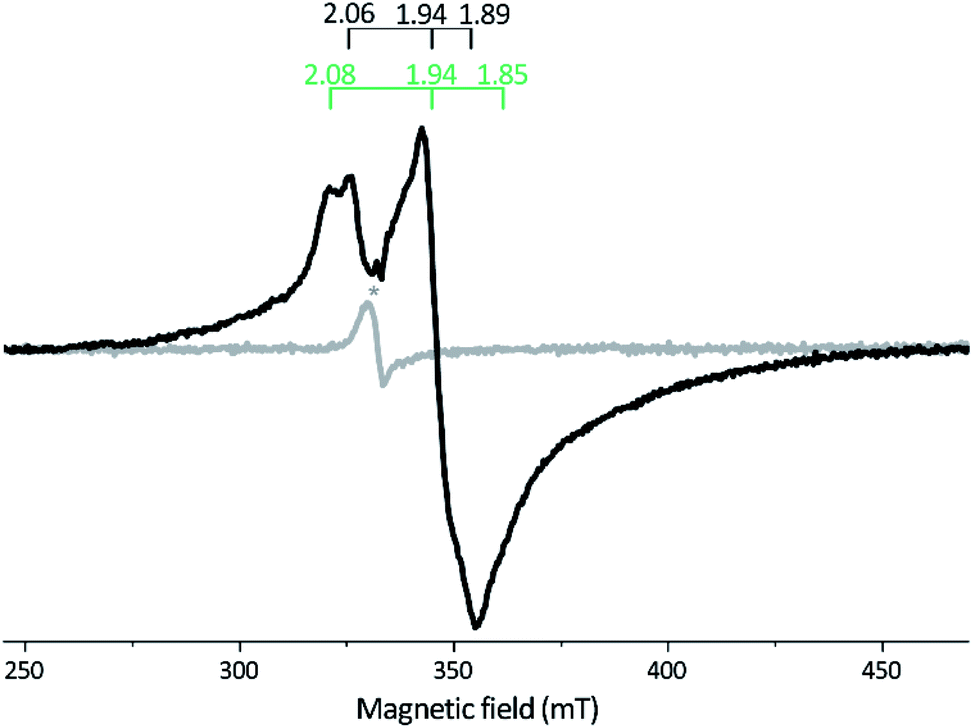 | ||
| Fig. 3 X-band EPR spectra of reconstituted apo-Tam HydS (50 μM). Gray spectrum: as prepared; black spectrum: NaDT reduced. The observed rhombic signal in the reduced sample is typical for [4Fe–4S]+ clusters. The g-tensors are indicated for the two contributing components, identified through comparison to NaDT reduced holo-Tam HydS (see Fig. S3†). The weak signal marked with * appearing at g = 2.02 is attributed to a trace amount of [3Fe–4S]2+. EPR settings: T = 17 K; modulation frequency 100 kHz, amplitude 15 G; microwave frequency 9.4 GHz, power 8 mW. | ||
The iron quantification supports the presence of four [4Fe–4S] clusters in the reconstituted protein, including the active-site [4Fe–4S]H cluster. This assignment is further supported by EPR spectroscopy. Albeit only two of the clusters were clearly discernable, no signal attributable to [2Fe–2S] clusters was observed upon reduction, and only traces of a [3Fe–4S] cluster signal were present in the non-reduced samples.45,46 Similar results were recently reported for the putative sensory M2f enzyme Tm HydS, and it appears to be a shared trait between the two subclasses.38 This FeS cluster composition is also supported by the linear increase in activity as iron content is increased up to 16 Fe/protein. Although the H2 production activity of Tam HydS, observed in in vitro assays, is low compared to most known prototypical [FeFe]-hydrogenases, it is similar to Tm HydS.38 As the two N-terminal [4Fe–4S] clusters are well conserved across numerous [FeFe]-hydrogenases, including the aforementioned Dd HydAB as well as the M2 enzyme from Megasphaera elsdenii this arguably represents the catalytic electron transfer pathway also in the M2e enzymes.21,32,39 In contrast, the biochemical and physiological role of the [4Fe–4S] cluster coordinated by the C-terminal Cx2Cx4Cx16C motif is unknown. As it is located between the H-domain and the PAS domain in M2f [FeFe]-hydrogenases, it is likely to be involved in a signaling process.
EPR characterization of holo-Tam HydS
The in vitro enzymatic assays revealed that treating apo-Tam HydS with [2Fe]adt resulted in spontaneous formation of holo-Tam HydS on a minute time-scale, similar to what was previously observed under whole-cell conditions.40 In the absence of reductant (NaDT), this treatment is expected to yield an oxidized form of the H-cluster, either the Hox state or the Hox-CO state.13,47–49 Both states are paramagnetic and best described as [4Fe–4S]H2+–[2Fe(II,I)] (s = 1/2) species, thus EPR spectroscopy can be employed to monitor H-cluster assembly. It is well established that the EPR spectra of Hox and Hox-CO generally display rhombic and axial signals, respectively, with small anisotropy and spin transitions at g ≈ 2.22Expecting a mixture of Hox and Hox-CO, the X-band EPR spectra collected on solution samples of as-prepared holo-Tam HydS, generated under an inert argon atmosphere, displayed a surprisingly complex pattern. As seen in Fig. 4A (spectrum a), at least seven features were resolved. The spectrum displayed characteristics suggesting the presence of Hox and Hox-CO but the unusual complexity of the spectrum shows that more than two species contribute to the overall spectrum. Spin quantification of a representative spectrum resulted in 0.64 spin per protein, indicating that a fraction of the enzyme also resided in an EPR silent state, assigned by FTIR spectroscopy to the Hred state (see below). The relaxation behavior of the signal(s) for holo-Tam HydS was estimated by monitoring the dominant g ≈ 2.04–2.02 feature (Fig. S4†). All components indicated in the spectrum followed similar saturation trends, and displayed low P1/2 values (73 μW and 1.15 mW at 15 and 21 K, respectively). This suggests that spin relaxation is a slow process, most likely due to isolation of the H-cluster from the lattice.
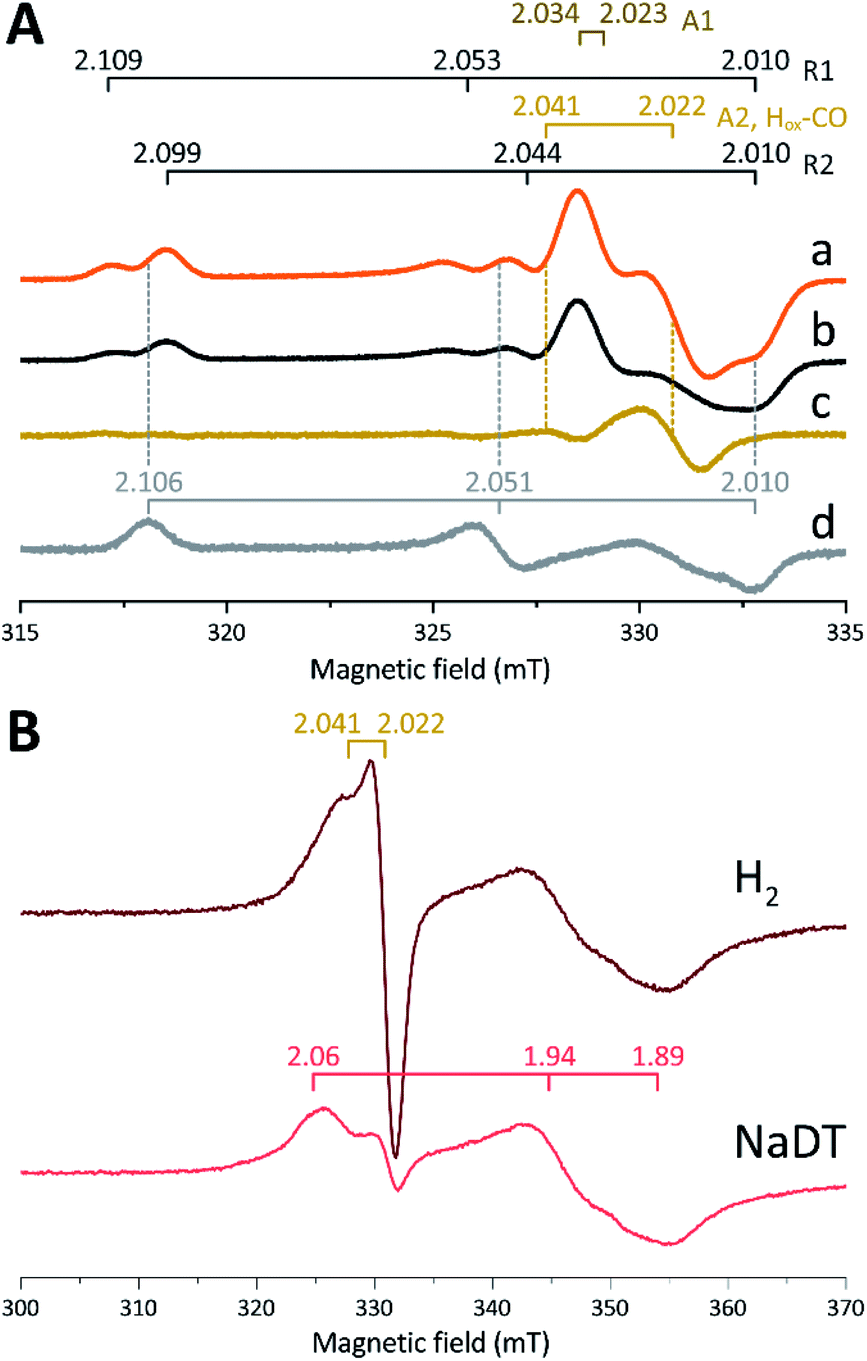 | ||
| Fig. 4 (A) Signals observed for holo-Tam HydS and attributed to the H-cluster, (a) holo-Tam HydS activated with [2Fe]adt as isolated; (b) holo-Tam HydS flushed by N2 to remove Hox-CO contributions (contains components R1, R2, A1 and A2); (c) difference spectra obtained from subtraction of spectrum (b) from (a) revealing an axial signal attributed to Hox-CO (“component A2, Hox-CO”); (d) Tam HydS activated with [2Fe]pdt (pdt-Tam HydS, as isolated). EPR settings: T 21 K; modulation frequency 100 kHz, amplitude 10 G; microwave frequency 9.4 GHz, power 16 μW. (B) Reduction of holo-Tam HydS activated with [2Fe]adt by H2 and NaDT resulting in disappearance of the H-cluster signals and appearance of a rhombic signal attributed to a [4Fe–4S]+ cluster. EPR settings: T 10 K; modulation frequency 100 kHz, amplitude 15 G; microwave frequency 9.4 GHz, power 80 μW. g-values indicated in the figure on horizontal bars, for details see main text. | ||
Flushing of holo-Tam HydS solutions with N2 prior to freezing resulted in approx. 50% decrease of total spin density, reflected in a minor decrease in amplitude of the low-field features (g ≈ 2.1) while a pseudo-axial component represented by a feature at g = 2.022 was almost completely lost (Fig. 4A, spectrum b). Subtraction of the spectrum obtained following N2 flushing (spectrum b) from the spectrum of the as-prepared sample (spectrum a) provided a “pure” pseudo-axial spectrum (Fig. 4A, spectrum c) with g‖ = 2.041 and g⊥ = 2.022. This signal is assigned to the Hox-CO state (Fig. 4A, component “A2, Hox-CO”). Similarly, the Hox-CO component was diminished in samples of holo-Tam HydS isolated following H-cluster assembly in the presence of NaDT (Fig. S5†). The residual spectrum (spectrum b) appears to feature two sets of rhombic signals (components R1 and R2), in combination with an additional narrow axial signal (component A1). The g‖ = 2.034 tensor of the latter signal is readily apparent while the g⊥ tensor is tentatively assigned to 2.023 from simulations (indicated in dark brown in Fig. 4A, see Fig. S5† for simulation details). With regards to the rhombic signals, the g-values are 2.109 and 2.099 for the two features at the lower field wing, 2.053 and 2.044 in the centre region and 2.010 in the high field wing. The overall spectral shape was highly similar between biological repeats prepared at pH 8. The relative signal amplitudes observed in spectrum (b) were also retained at pH 5 (Fig. S5†) but this acidification resulted in a 2–3 gauss downshift of one set of the rhombic EPR signals, which facilitated separation into two sets of separate g-tensors (gzyx = 2.109, 2.053, 2.010, “component R1”; and gzyx = 2.099, 2.044, 2.010, “component R2”; Fig. 4A). It should be noted that the assignment of gx = 2.010 is speculative due to its overlap with the adjacent axial signal.
To further clarify the EPR spectrum observed for Tam HydS, the holo-enzyme was generated using the modified cofactor [Fe2(pdt)(CO)4(CN)2]2− ([2Fe]pdt, pdt = −SCH2CH2CH2S−; pdt-Tam HydS). It has been shown for both group A and C [FeFe]-hydrogenases that replacing the amine-bridgehead of the adt ligand with a methylene group destabilizes the Hox-CO state.13,30,38,50,51 Thus, analogous samples where [2Fe]pdt replaced [2Fe]adt were examined by EPR under the same recording conditions. The obtained spectrum showed a rhombic anisotropy with g-values of 2.106, 2.051 and 2.010 (Fig. 4A, spectrum d). This signal is attributed to the formation of a single pure Hox state, in good agreement with FTIR spectroscopy (Fig. S6†), as well as earlier studies of Tam HydS under whole-cell conditions.40 A comparison between holo-Tam HydS generated with [2Fe]adt (Fig. 4A, spectra a and b) and [2Fe]pdt (Fig. 4A, spectrum d) reveals that the rhombic features observed in spectra (a) and (b) display a significant overlap with the signal observed for pdt-Tam HydS (indicated with dashed lines in Fig. 4A). Consequently, the rhombic components R1 and R2 are assigned to two distinct Hox-like species. The non-overlapping features of the spectrum correspond to the pseudo-axial components (A1 and A2), in agreement with the assignment of A2 to the Hox-CO state.
Reduction of holo-Tam HydS with NaDT resulted in disappearance of the aforementioned H-cluster signals with concomitant appearance of a broader rhombic EPR spectrum (Fig. 4B, spectrum NaDT). The loss of the Hox-CO and Hox-like signals is attributed to a one-electron reduction of the H-cluster to the diamagnetic Hred state (see below). The new signal partially resembles that observed for reduced apo-Tam HydS (Fig. 3), and spin quantification showed one spin per protein. Thus, one [4Fe–4S] cluster, with an EPR signature of gzyx = 2.06, 1.94 and 1.89, is susceptible to NaDT reduction in holo-Tam HydS. Subtraction of the holo-Tam HydS signal from that of apo-Tam HydS revealed another broad rhombic EPR signal (gzyx = 2.08, 1.94 and 1.85, see Fig. S3,† green spectrum). As this signal was present in the apo-protein but lost upon H-cluster formation, it is tentatively attributed to the [4Fe–4S]H cluster of apo-Tam HydS. Reduction of holo-Tam HydS with H2 provided a similar result compared to reduction with NaDT, although a larger fraction of the Hox-CO state remained (Fig. 4B, spectrum H2).
In summary, the combined EPR data from as-prepared and gas-flushed solution samples reveal an unusually complex mixture of oxidized states. Still, three of the contributing species can be assigned with relatively high certainty. Comparison of as-prepared and N2 flushed samples show that a standard Hox-CO species can form also in holo-Tam HydS. Conversely, the “split” Hox-like signal observed in holo-Tam HydS is suggestive of the formation of two distinguishable Hox-like states. Based on FTIR spectroscopy (see below), R2 is attributed to a state highly similar to the well-known Hox state of prototypical [FeFe]-hydrogenases (gzyx = 2.099, 2.044, 2.010), and the second rhombic EPR signal, R1, to a state similar to HoxH (gzyx = 2.109, 2.053, 2.010). A similar downshift of the Hox-signal upon formation of HoxH has recently been reported for Cr HydA1.9 As comparing spectra of samples prepared at mildly basic and acidic conditions did not reveal significant changes in the relative amplitudes of the rhombic signals their interconversion appears to be more complicated than an acid–base equilibrium. The structural details of this HoxH-like state in Tam HydS remains to be fully elucidated. Still, both appear catalytically competent, as they were both lost upon exposure to H2.
FTIR characterization of holo-Tam HydS
The H-cluster of holo-Tam HydS was further investigated using ATR FTIR spectroscopy at different pH values and in the presence of H2, N2, CO, or O2. The enzyme adopted Hred as a semi-stable resting state under our experimental conditions (20 °C, N2 atmosphere with approx. 1% H2, hydrated protein films at pH 7). A quantitative enrichment of Hox was achieved only after 20–30 h of continuous purging with pure N2. In contrast to what is generally reported for prototypical [FeFe]-hydrogenases,52 the Hox-state (≈65%) accumulated together with a small fraction (≈25%) of an alternative state displaying an Hox-like spectrum at higher frequencies (Fig. S7†). This hypsochromically-shifted signature is attributed to an HoxH-like state, albeit this state is generally not observed at neutral pH in prototypical [FeFe]-hydrogenases. This mixture of Hox-like states and their relative ratio is in agreement with the observation of two rhombic EPR signals under similar conditions (Fig. 4). The Hred to Hox transition was further analyzed by ATR FTIR spectro-electrochemistry, revealing two unusual properties of the Tam HydS enzyme (Fig. S8†). The reaction displayed a significant over-potential requirement, and while the quasi-reversible nature of the process prevented an exact assignment of the Hox/Hred reduction potential it was clearly shifted in an anodic direction as compared to previously studied prototypical [FeFe] hydrogenase, with Ems of ≈−350 to −450 mVs reported for Cr HydA1 and Dd HydAB.39,53,54 During the reductive scan, Hox/Hred interconversion in Tam HydS was observed at Em ≈ −300 mV vs. SHE, while the re-oxidation did not occur until a potential of approx. +200 mV was applied (at pH 8). No intermediates were observed in the process. Thus, Hred appears to be both kinetically and thermodynamically stabilized in Tam HydS. A relatively anodic Hox/Hred midpoint potential has been reported also for the putative sensory Group C hydrogenase Tm HydS.38Fig. 5A shows the IR signatures of Hox, Hox-CO, and Hred observed for Tam HydS. The assignment of the spectra to specific H-cluster states was facilitated by their overall similarities to spectra previously reported for prototypical [FeFe]-hydrogenases. Still, the frequencies of the terminal CO/CN− ligands are upshifted in comparison to Cr HydA1 and Dd HydAB and closer to CpI and CaI from C. acetobutylicum.31,37,52,55 The high-frequency CO band of Hox-CO (2026 cm−1) indicates a constrained geometry.51 In contrast to these upshifts, the μCO band of Hox (1788 cm−1) and Hox-CO (1786 cm−1) was found at lower frequencies than typically observed. These latter differences, as compared to group A and C [FeFe]-hydrogenases, are likely attributable to the M393S variation in Tam HydS as this residue is in close contact with the bridging μCO ligand. A distinct feature in the μCO region for the Hred state could not be discerned. At low pH and high concentrations of NaDT, accumulation of HoxH over Hox was achieved (Fig. 5B), although the protein film never fully converted into HoxH (Fig. S7†). Note that the signature of HoxH at low pH is in excellent agreement with the hypsochromically shifted Hox-like spectrum observed at pH 8. In contrast to what has been reported for prototypical [FeFe]-hydrogenases, no accumulation of Hhyd was observed, e.g., when low pH samples were exposed to H2.56 Moreover, Hsred and Hred′ were never detected. Table 1 summarizes the IR signature of all observed H-cluster states.
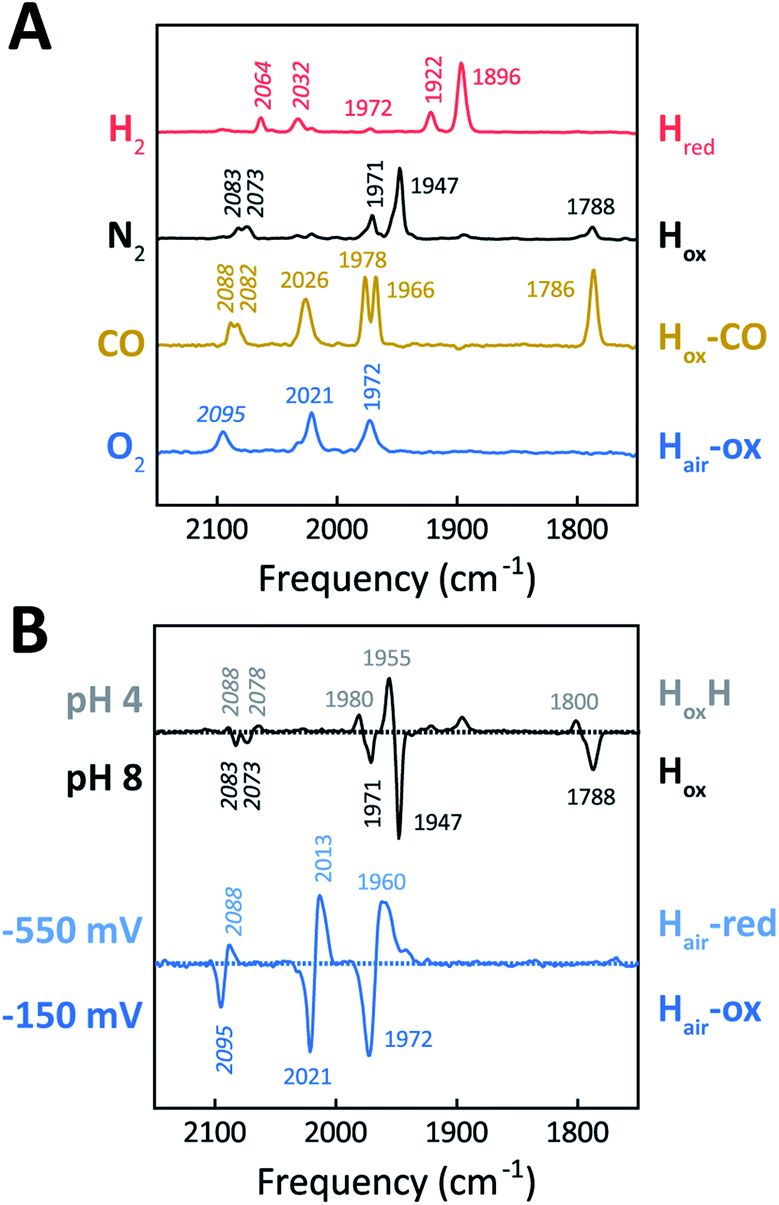 | ||
| Fig. 5 ATR FTIR characterization of holo-Tam HydS. All data recorded at RT. (A) Infrared signature of the H-cluster in Tam HydS in the presence of H2 (red, Hred), N2 (black, Hox and HoxH), CO (brown, Hox-CO), and after reaction with O2 (blue, Hair). (B) The upper difference spectrum shows accumulation of HoxH (positive bands, grey, pH 4) over Hox (negative bands, black, pH 8) under N2. Note: accumulation of HoxH also required NaDT addition. In the lower difference spectrum, FTIR spectro-electrochemistry was used to accumulate Hair-red (positive bands, light blue, −550 mV vs. SHE) over Hair-ox (negative bands, blue, −150 mV). | ||
| CN− (cm−1) | CO (cm−1) | |||||
|---|---|---|---|---|---|---|
| Hred | 2064 | 2032 | 1972 | 1922 | 1896 | |
| Hox | 2083 | 2073 | 1971 | 1947 | 1788 | |
| HoxH | 2088 | 2078 | 1980 | 1955 | 1800 | |
| Hox-CO | 2088 | 2082 | 2026 | 1978 | 1966 | 1786 |
| Hair-ox | 2095 | 2021 | 1972 | |||
| Hair-red | 2088 | 2013 | 1960 | |||
The absorbance spectra of the H-cluster states identified in Fig. 5 were fitted and used to describe the interconversion reactions as a function of gas composition and time. For comparative purposes, analogous experiments were performed with the prototypical [FeFe]-hydrogenases Dd HydAB or Cr HydA1. Fig. 6A depicts the rapid conversion of Hox into Hred for Tam HydS and Dd HydAB at 1%, 10%, and 100% H2 over N2. Dd HydAB was chosen for comparison because it shows a similar, albeit not identical, composition of reduced H-cluster states under H2 (Fig. S10†). The identity of Hred as resting state in Tam HydS is illustrated by the pronounced persistence of Hred when H2 was removed from the gas phase (t > 16.5 min) whereas Dd HydAB immediately converted into Hox. This accumulation of Hox is a consequence of auto-oxidation, i.e. due to H2 release. In the next step, the influence of temperature on the Hred to Hox transition of Tam HydS and Dd HydAB was investigated. We addressed the kinetics of auto-oxidation for five temperature points in the range between 20–40 °C. The enzymes were reduced in the presence of 1% H2 and subjected to pure N2 for 10 min, before they were re-reduced with 1% H2. Fig. 6B depicts the changing population of Hox in Tam HydS as a function of gas, time, and temperature. Higher temperature increased the rate of Hox formation, upon removal of H2 from the atmosphere, and induced a higher percentage of Hox accumulation (i.e., after 10 min). The same set of experiments was performed for Dd HydAB. Albeit apparent instability of the H-cluster in Dd HydAB at T > 30 °C prevented a complete study, the net-oxidation rate in Dd HydAB is significantly higher than in Tam HydS (Fig. S11†). Moreover, it should be noted that an increase in temperature also resulted in an increase in steady-state concentration of Hox already when Tam HydS equilibrated under 1% H2, highlighting a positive entropy contribution for the Hred to Hox transition (Fig. 6B).
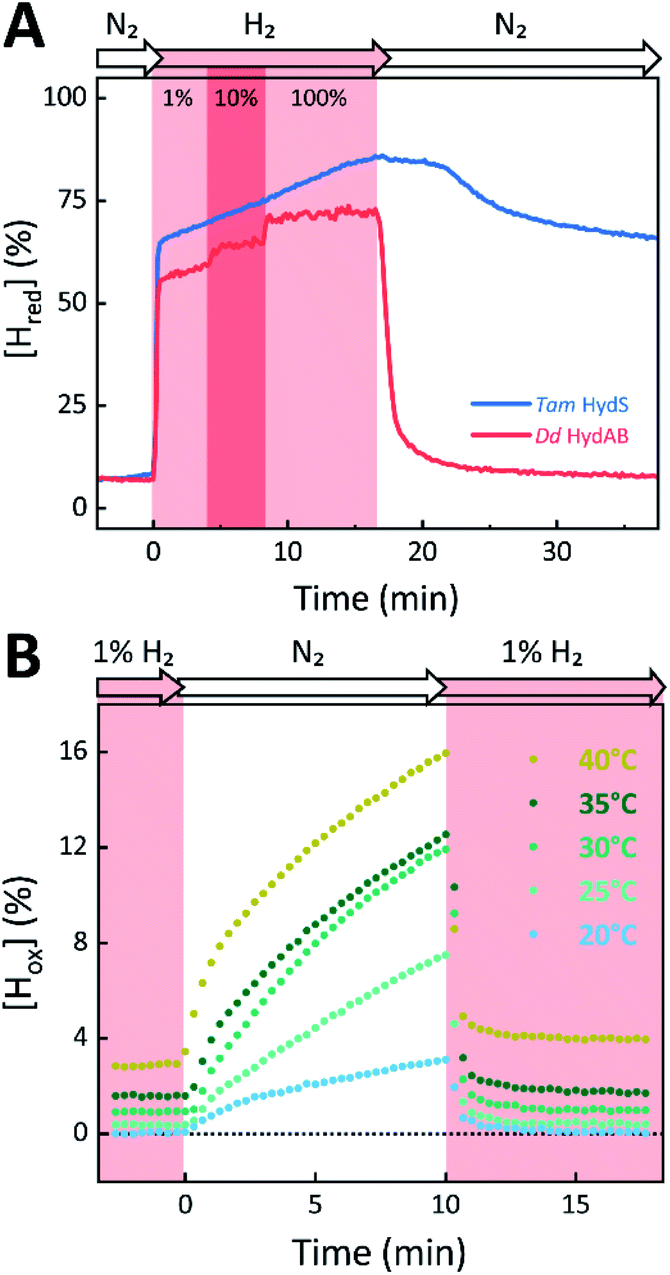 | ||
| Fig. 6 The reactivity of Tam HydS and prototypical [FeFe]-hydrogenase (Dd HydAB) towards H2 (A) and temperature dependence of the auto-oxidation activity (B) monitored by time-resolved ATR FTIR spectroscopy. (A) Kinetic traces of Hred for Tam HydS (blue) and Dd HydAB (red) that show the reaction with different concentrations of H2 (RT). Note the persistence of Hred in the absence of H2 for Tam HydS. (B) Change of [Hox] as a function of gas, time, and temperature. Representative data set for Tam HydS, recorded at pH 7. The equilibrium under 1% H2 is slightly shifted in favour of Hox over Hred as temperature is increased, and so does rate of [Hox] formation as the atmosphere is changed to pure N2. | ||
The reactivity towards known [FeFe]-hydrogenase inhibitors was probed by exposing protein films to CO or O2. Fig. 7A depicts the conversion of Hox into Hox-CO for Tam HydS and Dd HydAB at 1%, 10%, and 100% CO over N2. Here, Tam HydS displays a notable lack of CO inhibition. Even under 100% CO, only 60% of the H-cluster population converted into Hox-CO. Similar trends were observed with 10% H2 in the N2 carrier gas (Fig. S10†). In the absence of CO gas, Hred recovered quickly. Adjusted for the CO-insensitive contamination of ∼30% Hinact (Fig. S10†), Dd HydAB showed immediate, complete, and enduring CO inhibition. Fig. 7B depicts the reaction of oxidized Tam HydS and Cr HydA1 with 1 atm air. We chose Cr HydA1 for comparison because Dd HydAB partly converted into unready states like Hinact in the presence of O2 whereas O2 exposure rapidly destroyed the H-cluster in Cr HydA1 (Fig. S12†). In contrast, Tam HydS converted into an unprecedented species, denoted Hair, and the formation of this state was observed regardless of whether the H-cluster resided in the Hred or Hox state upon O2 exposure (Fig. S9†). As seen in Fig. 5A (blue spectrum) this new state featured two bands in the CO region and one band in the CN− region of the spectrum, suggestive of partial degradation of the [2Fe]H subsite. Moreover, it was found to be unreactive towards N2, H2, and CO. EPR samples collected of Tam HydS exposed to air did not reveal any discernable EPR signal, apart from minor features at g ≈ 4.3 and 2.02, attributable to small amounts of Fe3+ ions (“junk iron”) and [3Fe–4S] cluster species, respectively. Similarly, a NaDT reduced anaerobic sample of Hair was also essentially EPR silent, albeit trace amounts of a [4Fe–4S]+ species became discernable (Fig. S13†). A mononuclear version of the [2Fe]H subsite has previously been observed by X-ray crystallography in the prototypical CpI [FeFe]-hydrogenase, following extended O2 exposure of the enzyme in cristallo.57 The overall FTIR spectral features in combination with 13CO isotope editing clearly supports the assignment of a mononuclear Fe(CO)2CN species (Fig. S14†). Spectro-electrochemistry also suggests that this mononuclear complex is bound to the [4Fe–4S]H cluster and that the modified H-cluster displays at least one redox transition, enabling accumulation of “Hair-red” and “Hair-ox” (Fig. 5B, S14 and S15†).
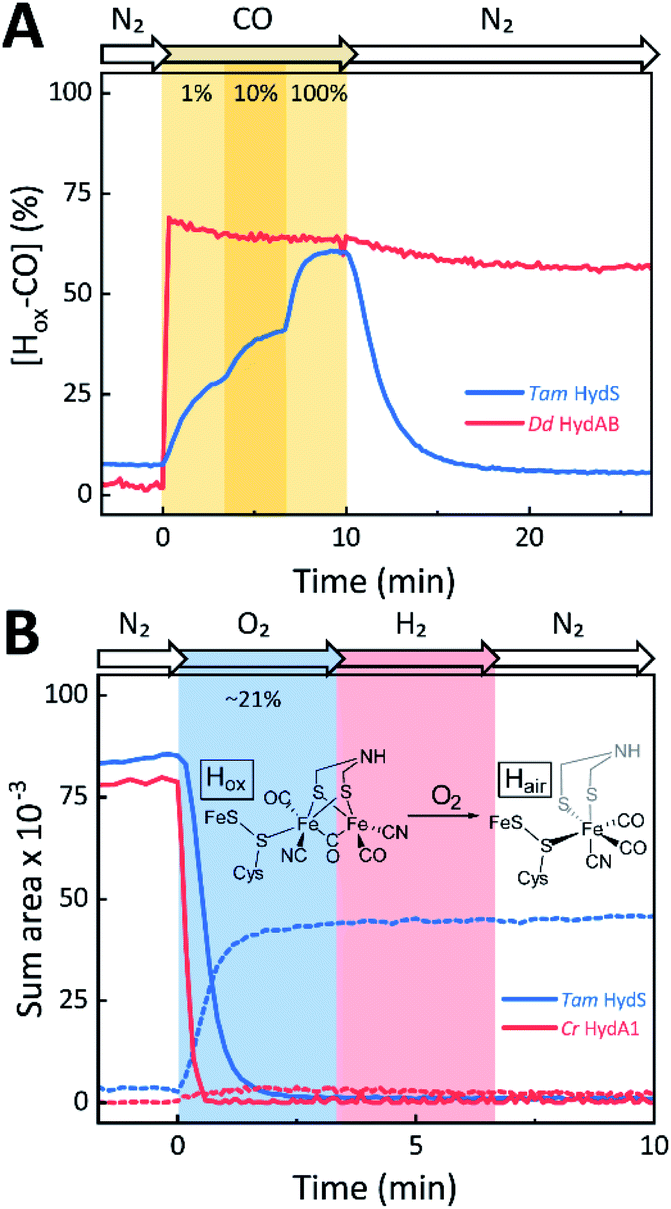 | ||
| Fig. 7 The reactivity of Tam HydS and prototypical [FeFe]-hydrogenase (Dd HydAB or Cr HydA1) towards CO (A) and O2 (B) monitored by time-resolved ATR FTIR spectroscopy. (A) Kinetic traces of Hox-CO for Tam HydS (blue) and Dd HydAB (red) that show the reaction with different concentrations of CO. About 30% of the Dd HydAB sample were arrested in the CO-insensitive Hinact state. Thus, the observed accumulation of ∼70% Hox-CO can be considered complete. (B) Kinetic traces for Tam HydS (blue) and Cr HydA1 (red) that show the reaction with ∼21% O2 (air). Solid traces depict Hox. While nearly 100% of the H-cluster is lost in Cr HydA1, a stable Fe(CO)2CN species prevails in Tam HydS (dashed traces). The proposed reaction between the oxidized H-cluster (Hox) and O2 is depicted. FeS = [4Fe–4S]H. | ||
The catalytic properties of Tam HydS
The catalytic properties of holo-Tam HydS were investigated using protein film electrochemistry (PFE), revealing pH and temperature dependent catalytic currents for both H+ reduction and H2 oxidation (Fig. 8A). Four different procedures were tested for electrode immobilization of the enzyme with the best method being absorption on a pyrolytic graphite electrode in the presence of the polycationic polymyxin B sulfate (Fig. S16†). The observation that the enzyme favors interaction with a positively charged surface suggests that it has a negative net surface charge at the pH of immobilization (pH 7, theoretical pI = 5.87, ExPASy ProtParam tool).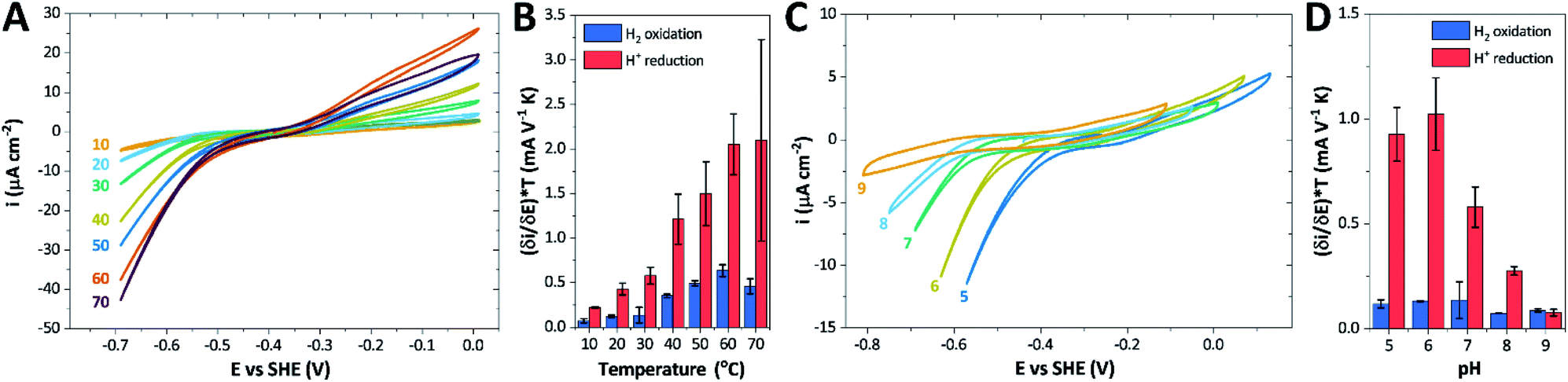 | ||
| Fig. 8 CVs obtained at a rotating disc PGE modified with Tam HydS under 1 atm H2 at (A) various temperatures in the 10–70 °C range at pH 7 and (C) various pH values from 5 to 9 at 30 °C. The scan rate is 2 mV s−1, the rotation rate is 3000 rpm. The data shown in (A) and (C) are obtained from single films cycled up and down in temperature and pH, respectively. Film stability was verified at the end of each experiment by returning the solution to its starting state (pH 7 solution at 30 °C). Dependence of the high driving force slopes of the voltammograms (eqn (1)) times the temperature for H2 oxidation and H+ reduction on (B) temperature and (D) pH. Error bars show standard deviation between three films. | ||
It is important to note that in all experiments even at large over-potentials for both H+ reduction and H2 oxidation the catalytic current does not reach a steady-state value, but increases almost linearly with over-potential. Such behavior has been rationalized by disorder among the adsorbed enzyme molecules, resulting in a dispersion of interfacial electron transfer rate constants.58–60 In this case, the steady-state limiting current (ilim) can be estimated from a linear fit of the high driving force part of the cyclic voltammograms (CVs), where the slope (∂i/∂E) is:60,61
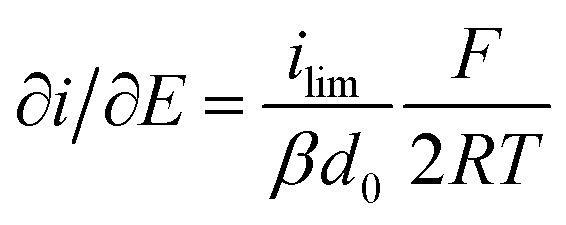 | (1) |
Eqn (1) predicts that the product of the slope and temperature is proportional to the limiting current and therefore to the activity.
We first evaluated the enzyme affinity towards H2. It has been noted earlier that 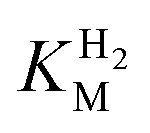 values determined from PFE experiments can be potential-dependent.62,63 Therefore, we recorded CVs at various concentrations of H2 in a broad potential window (Fig. S17†). For
values determined from PFE experiments can be potential-dependent.62,63 Therefore, we recorded CVs at various concentrations of H2 in a broad potential window (Fig. S17†). For 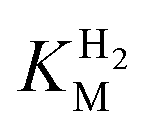 estimation it is important to measure the current response under conditions where it is limited by the catalytic rate of the enzyme, i.e. proportional to the catalytic rate rather than mass transport or interfacial electron transfer.64 Thus,
estimation it is important to measure the current response under conditions where it is limited by the catalytic rate of the enzyme, i.e. proportional to the catalytic rate rather than mass transport or interfacial electron transfer.64 Thus, 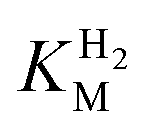 values at various over-potentials were calculated, and the measurements were performed at 30 and 60 °C. Moreover, to ensure that the catalytic rate is not limited by mass transport, CVs were recorded at rotation rates of 2000 and 3000 rpm at each concentration of H2. At over-potentials starting from 200 mV (30 °C) and 100 mV (60 °C), calculated
values at various over-potentials were calculated, and the measurements were performed at 30 and 60 °C. Moreover, to ensure that the catalytic rate is not limited by mass transport, CVs were recorded at rotation rates of 2000 and 3000 rpm at each concentration of H2. At over-potentials starting from 200 mV (30 °C) and 100 mV (60 °C), calculated 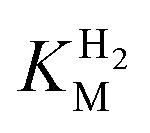 values were identical within error (Table 2), indicating that the observed current is dominated by the catalytic reaction. Similar
values were identical within error (Table 2), indicating that the observed current is dominated by the catalytic reaction. Similar 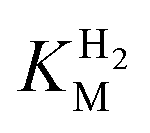 values were obtained from linear fits of the high driving force part of the cyclic voltammograms at various H2 concentrations, further confirming prevalence of the catalytic reaction over interfacial electron transfer at high driving forces (Table S3†).
values were obtained from linear fits of the high driving force part of the cyclic voltammograms at various H2 concentrations, further confirming prevalence of the catalytic reaction over interfacial electron transfer at high driving forces (Table S3†).
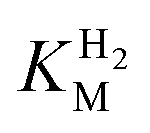 -values for Tam HydS determined at various overpotentials and temperatures at pH 7
-values for Tam HydS determined at various overpotentials and temperatures at pH 7
| Over-potential (mV) |
|
|
|---|---|---|
| 30 °C | 60 °C | |
| a The errors for over-potentials 100–400 mV show standard deviation between three films. | ||
| 100 | 0.05 ± 0.01 | 0.15 ± 0.05 |
| 200 | 0.08 ± 0.02 | 0.12 ± 0.02 |
| 300 | 0.10 ± 0.02 | 0.13 ± 0.03 |
| 400 | 0.10 ± 0.03 | 0.15 ± 0.05 |
| Average | 0.09 ± 0.03 | 0.14 ± 0.05 |
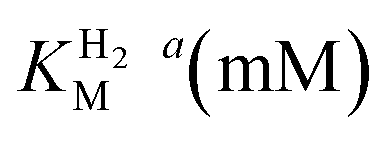
We further scrutinized the effect of temperature and pH on the catalytic activity of holo-Tam HydS under conditions when the catalytic current for H2 oxidation is not limited by mass transport (3000 rpm rotation speed and 1 atm H2). Fig. 8A and C displays CVs recorded at various temperatures (10–70 °C) at pH 7, and different pH values at 30 °C, respectively. Fig. 8B shows the temperature dependence of the CVs at high driving force (eqn (1)) for H+ reduction and H2 oxidation. Fig. 8D shows the corresponding data as a function of pH. When pH is decreased, the catalytic activity towards H+ reduction increases and the catalytic wave is shifted to more anodic potentials, consistent with higher H+ concentration (Fig. 8C). Conversely, the magnitude of the catalytic current for H2 oxidation does not vary smoothly with pH. The oxidation process is pH independent between pHs 8–9, increases at pH 6–7 and remains stable at this higher current down to pH 5. Moreover, over-potential for both H2 oxidation and H+ reduction is lowest at pH 5 (Fig. S18†). The origin of this pH switching behavior for H2 oxidation is currently not identified.
Temperature was found to have a strong influence on H2 oxidation and H+ reduction between 10–60 °C at pH 7 (Fig. 8A). Increasing the temperature not only resulted in higher overall currents, but also a significant decrease in over-potential. At room temperature, an over-potential of 50–60 mVs was observed in both catalytic directions, in contrast to previously characterized group A [FeFe]-hydrogenases. With the exception of specific mutants the latter enzymes generally display rapid increase in currents around the thermodynamic midpoint potential.44,65,66 The over-potential requirement of Tam HydS is in line with the quasi-reversible nature of the Hox/Hred transition observed by FTIR spectro-electrochemistry (Fig. S8†). At temperatures of 50–60 °C the over-potential decreased to approx. 10 mV. The decrease of catalytic currents at 70 °C is attributed to the deactivation of the protein, since we did not observe any significant protein loss during the experiment at temperatures up to 60 °C (Fig. S19†). It should also be noted that no oxidative inactivation67,68 was observed when cycling up to ±0 mV vs. SHE. Stability and increased catalytic activity of the protein at elevated temperatures is not surprising considering the thermophilic nature of T. mathranii.69 The activation enthalpies (ΔH‡) of the H2 oxidation and H+ reduction reactions were estimated through Eyring plots based on the change of the high potential slope as a function of temperature (Fig. 9), and found to be similar in both catalytic directions (Table 3). In the case of the prototypical [FeFe]-hydrogenases Cr HydA1 and CaI (Table 3),70 distinctly lower activation enthalpies (ΔH‡) for either H+ reduction or H2 oxidation, respectively, have been reported, suggesting that Tam HydS is exceptionally well balanced for bidirectional catalysis. Moreover, albeit the activation enthalpies are higher for Tam HydS than Cr HydA1 and CaI, they are still significantly lower than what has been reported from the E. coli [NiFe]-hydrogenases Ec Hyd1 and Ec Hyd2 (Table 3). Thus, the low specific activity observed for Tam HydS (Fig. 2) cannot be explained by differences in activation enthalpies alone. Rather, the low catalytic rate of Tam HydS is governed by mass transfer, e.g. proton- or H2 transfer within the protein. Impaired proton transfer could also, at least partially, explain the over-potential observed at low temperature.44 Finally, it is noteworthy that films prepared of the enzyme exposed to air, to induce the formation of the Hair state, displayed limited capacity for H+ reduction but a complete loss of H2 oxidation function (Fig. S20†). The catalytic properties of Hair was further supported by in vitro assays, showing a H+ reduction activity approximately hundred-fold lower than the native enzyme.
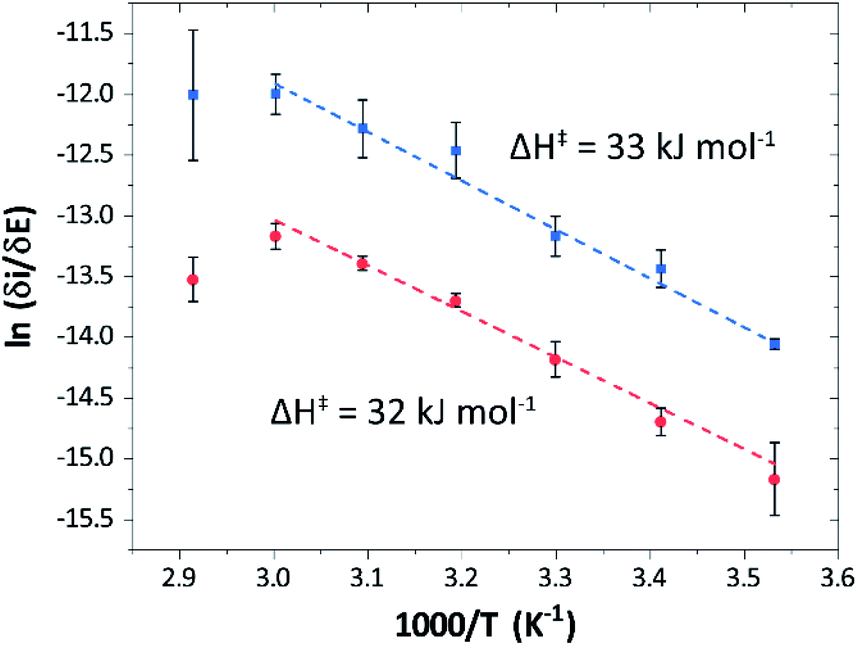 | ||
| Fig. 9 Eyring plots for H+ reduction (red) and H2 oxidation (blue) with linear fits (dashed lines). Plots prepared using estimated ilim based on data from Fig. 8B (data observed at 70 °C excluded from the linear fit). | ||
| Enzyme | ΔH‡ (kJ mol−1) | |
|---|---|---|
| H+ reduction | H2 oxidation | |
| a Experimental data obtained from ref. 62, determined at pH 6, 30 °C. b Only reached at high over-potential. c [NiFe]-hydrogenase E. coli Hyd1. d [NiFe]-hydrogenase E. coli Hyd2. | ||
| Tam HydS (this work) | 32 ± 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
33 ± 2 |
| CaIa | 29 | 19 |
| Cr HydA1a | 20b | 26b |
| Ec Hyd1a,c | — | 48 |
| Ec Hyd2a,d | 65 | 37 |
Conclusions
This report represents the first biochemical and biophysical characterization of a group D [FeFe]-hydrogenase. As described herein, Tam HydS features a number of properties similar to regulatory [NiFe]-hydrogenases,71 including a relatively low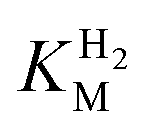 and an increased tolerance against CO inhibition. This is in line with a potential sensory function, as expected from its close relationship to the putatively sensory group C [FeFe]-hydrogenases. Still, a
and an increased tolerance against CO inhibition. This is in line with a potential sensory function, as expected from its close relationship to the putatively sensory group C [FeFe]-hydrogenases. Still, a 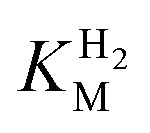 of around 0.1 mM is 5–10 times higher than what has been reported for regulatory [NiFe]-hydrogenases,71,72 but is arguably in agreement with the overall higher
of around 0.1 mM is 5–10 times higher than what has been reported for regulatory [NiFe]-hydrogenases,71,72 but is arguably in agreement with the overall higher 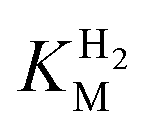 and
and 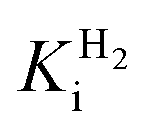 reported for prototypical [FeFe]-hydrogenase.73 Moreover, analysis on a genomic level shows that Tam HydS is encoded upstream of a heterotrimeric bifurcating group A [FeFe]-hydrogenase. Thus, we hypothesize that M2e represents an alternative type of sensory [FeFe]-hydrogenase. This raises the question of how signal transduction is achieved. In the case of group C putative sensory [FeFe]-hydrogenase, a PAS-domain is fused to the H-domain. Similarly, to the best of our knowledge, all regulatory [NiFe]-hydrogenases identified to-date feature the so-called HoxJ subunit, encoding a PAS domain-like sequence. A PAS-domain cannot be identified in the operon encoding for Tam HydS but the gene is flanked by a histidine kinase-like ATPase and an AraC family transcriptional regulator. These genes are commonly involved in signal transduction and regulation of transcription, which further suggests a regulatory role for the M2e hydrogenases.74,75
reported for prototypical [FeFe]-hydrogenase.73 Moreover, analysis on a genomic level shows that Tam HydS is encoded upstream of a heterotrimeric bifurcating group A [FeFe]-hydrogenase. Thus, we hypothesize that M2e represents an alternative type of sensory [FeFe]-hydrogenase. This raises the question of how signal transduction is achieved. In the case of group C putative sensory [FeFe]-hydrogenase, a PAS-domain is fused to the H-domain. Similarly, to the best of our knowledge, all regulatory [NiFe]-hydrogenases identified to-date feature the so-called HoxJ subunit, encoding a PAS domain-like sequence. A PAS-domain cannot be identified in the operon encoding for Tam HydS but the gene is flanked by a histidine kinase-like ATPase and an AraC family transcriptional regulator. These genes are commonly involved in signal transduction and regulation of transcription, which further suggests a regulatory role for the M2e hydrogenases.74,75
In the context of H-cluster function, Tam HydS displays a number of diverging properties. As reflected in the tolerance towards CO, the unusual mixture of Hox-like states, reaction enthalpies (ΔH‡) that were practically identical for both H+ reduction and H2 oxidation, and a stabilization of the Hred-state. Moreover, it is noteworthy that despite its high tolerance towards CO Tam HydS reacted rapidly with O2 to form a stable mononuclear version of the [2Fe]H subsite (Hair). This argues against gas diffusion (mass transfer) to the active site as the main defense mechanism towards CO inhibition, which we instead attribute to the unusual hydrogen-bonding network around the H-cluster afforded by variations of e.g. Cys299, Met353 and Met497 (Fig. 1). Indeed, an increased resistance towards CO has also been reported for the putative-sensory group C [FeFe]-hydrogenase Tm HydS, featuring different alterations of the same residues.38 The influence of the hydrogen-bonding network on CO affinity has also been suggested by studies of cofactor variants and mutants of Cr HydA1, in which the nearby cysteine was replaced by an alanine residue.51
The effect of O2 on prototypical group A [FeFe]-hydrogenases has been intensively studied, nevertheless formation of Hair has not been reported for any other [FeFe]-hydrogenase. Instead, the reaction generally causes complete H-cluster degradation or formation of reversibly inhibited states like Hinact by coordination of thiol ligands to the [2Fe] subsite.37,76 Whether Hair has a physiological role remains uncertain. However, as this state appears unreactive towards H2 while retaining a limited H2 production capacity, it is unlikely to be relevant in H2 sensing. Still, its formation provides a striking example of how the active site environment modulates the reactivity of the H-cluster. It should be noted that formation of Hair is dependent on O2 as the equivalent state does not seem to be formed under oxidizing electrochemical conditions. As O2 acts as a chemical oxidant it can lead to an alternative pathway that remains to be fully elucidated. Arguably, access to electrons and protons has a large influence on the reaction between the H-cluster and O2 and several factors need to be considered to explain the formation of Hair in Tam HydS.77,78 It has been shown that in the case of [NiFe]-hydrogenase, O2 can be efficiently reduced to H2O through rapid electron injection to the [NiFe] cofactor involving an unusual [4Fe–4S] cluster.79 The presence of a C-terminal [4Fe–4S] cluster and a disrupted proton transfer pathway differentiates M2e enzymes from prototypical [FeFe]-hydrogenases. Still, formation of Hair was not reported in the case of Tm HydS which features the same differences.38 This suggests that the reaction between the H-cluster and O2 is significantly influenced by other variations in the active site architecture.
We presume that the lack of a cysteine in position 299 also influences the catalytic activity of Tam HydS, as hindered proton release could stabilize Hred kinetically, following reduction of the H-cluster with H2. A similar stabilization of Hred has been noted for Tm HydS.38 However, while the latter enzyme is suggested to form an unusual, unprotonated, CO-bridged form of Hred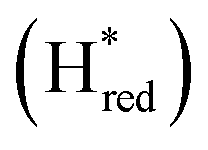 , Tam HydS appears to form an Hred state highly similar to that of prototypical [FeFe]-hydrogenase. Also, the complete lack of the well-conserved proton transfer pathway found in group A [FeFe]-hydrogenases raises the question of where an alternative pathway could reside. In the homology model of Tam HydS, a conserved glutamate residue close to the H-cluster (Glu252) is located in a preferable position for initiating proton transfer (Fig. S21†). However, there is no clear continuation of this potential pathway. In addition to changes in the proton transfer pathway, variation of Met353 and Met497 certainly also modulates the electronic properties of the H-cluster. It has been previously proposed that Met353 is important for tuning
, Tam HydS appears to form an Hred state highly similar to that of prototypical [FeFe]-hydrogenase. Also, the complete lack of the well-conserved proton transfer pathway found in group A [FeFe]-hydrogenases raises the question of where an alternative pathway could reside. In the homology model of Tam HydS, a conserved glutamate residue close to the H-cluster (Glu252) is located in a preferable position for initiating proton transfer (Fig. S21†). However, there is no clear continuation of this potential pathway. In addition to changes in the proton transfer pathway, variation of Met353 and Met497 certainly also modulates the electronic properties of the H-cluster. It has been previously proposed that Met353 is important for tuning 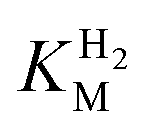 as well as the catalytic bias of prototypical [FeFe]-hydrogenase, in favor of either H+ reduction or H2 oxidation.31,41 Still, the exact contributions of the Met353Ser variation on the activity of Tam HydS remain to be fully elucidated.
as well as the catalytic bias of prototypical [FeFe]-hydrogenase, in favor of either H+ reduction or H2 oxidation.31,41 Still, the exact contributions of the Met353Ser variation on the activity of Tam HydS remain to be fully elucidated.
In closing, this study shows that the Tam HydS M2e enzyme displays significant differences in reactivity as compared to previously studied group A as well as group C [FeFe]-hydrogenases. Thus, in addition to proposing a biological function for the group D [FeFe]-hydrogenases, it underscores how mutations of known [FeFe]-hydrogenases need to be complemented with further studies of the biodiversity to fully realize the chemical space of this fascinating family of enzymes. Mapping out the reactivity of these diverse enzymes is certainly critical for our understanding of hydrogen metabolism and envisioned biotechnological applications. It is also of high relevance in the context of bioinspired catalyst design, as it will provide new model systems for elucidating the influence of the protein environment and the outer coordination sphere on the reactivity of the H-cluster.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Novo Nordisk Foundation (HL contract no. NNF19OC0055613), the European Research Council under the European Union's Seventh Framework Programme (grant agreement No. 714102 to GB) and Horizon 2020 research and innovation programme (Marie Sklodowska-Curie grant agreement No. 897555 to MS), and the Deutsche Forschungsgemeinschaft through the priority program 1927 (grant agreement No. 1554/5-1 to STS). A. S. acknowledges support from the Knut and Alice Wallenberg Foundation (KAW 2015.0418).Notes and references
- C. Madden, M. D. Vaughn, I. Díez-Pérez, K. A. Brown, P. W. King, D. Gust, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2012, 134, 1577–1582 CrossRef CAS.
- E. C. Hatchikian, N. Forget, V. M. Fernandez, R. Williams and R. Cammack, Eur. J. Biochem., 1992, 209, 357–365 CrossRef CAS.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef CAS.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- M. Calusinska, T. Happe, B. Joris and A. Wilmotte, Microbiology, 2010, 156, 1575–1588 CrossRef CAS.
- J. Meyer, Cell. Mol. Life Sci., 2007, 64, 1063–1084 CrossRef CAS.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, ISME J., 2016, 10, 761–777 CrossRef CAS.
- S. L. Benoit, R. J. Maier, R. G. Sawers and C. Greening, Microbiol. Mol. Biol. Rev., 2020, 84, e00092 CrossRef CAS.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS.
- C. Kamp, A. Silakov, M. Winkler, E. J. Reijerse, W. Lubitz and T. Happe, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 410–416 CrossRef CAS.
- T. Happe and J. D. Naber, Eur. J. Biochem., 1993, 214, 475–481 CrossRef CAS.
- S. P. J. Albracht, W. Roseboom and E. C. Hatchikian, J. Biol. Inorg Chem., 2006, 11, 88–101 CrossRef CAS.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, J. Biol. Inorg Chem., 2006, 11, 102–118 CrossRef CAS.
- B. Bennett, B. J. Lemon and J. W. Peters, Biochemistry, 2000, 39, 7455–7460 CrossRef CAS.
- M. F. Verhagen, T. O'Rourke and M. W. Adams, Biochim. Biophys. Acta, Bioenerg., 1999, 1412, 212–229 CrossRef CAS.
- N. Chongdar, K. Pawlak, O. Rüdiger, E. J. Reijerse, P. Rodríguez-Maciá, W. Lubitz, J. A. Birrell and H. Ogata, J. Biol. Inorg Chem., 2020, 25, 135–149 CrossRef CAS.
- D. W. Mulder, E. M. Shepard, J. E. Meuser, N. Joshi, P. W. King, M. C. Posewitz, J. B. Broderick and J. W. Peters, Structure, 2011, 19, 1038–1052 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, J. Am. Chem. Soc., 2020, 142, 222–232 CrossRef CAS.
- M. Haumann and S. T. Stripp, Acc. Chem. Res., 2018, 51, 1755–1763 CrossRef CAS.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS.
- L. S. Mészáros, P. Ceccaldi, M. Lorenzi, H. J. Redman, E. Pfitzner, J. Heberle, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 4608–4617 RSC.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS.
- J. H. Artz, O. A. Zadvornyy, D. W. Mulder, S. M. Keable, A. E. Cohen, M. W. Ratzloff, S. G. Williams, B. Ginovska, N. Kumar, J. Song, S. E. McPhillips, C. M. Davidson, A. Y. Lyubimov, N. Pence, G. J. Schut, A. K. Jones, S. M. Soltis, M. W. W. Adams, S. Raugei, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2020, 142, 1227–1235 CrossRef CAS.
- G. Caserta, C. Papini, A. Adamska-Venkatesh, L. Pecqueur, C. Sommer, E. Reijerse, W. Lubitz, C. Gauquelin, I. Meynial-Salles, D. Pramanik, V. Artero, M. Atta, M. del Barrio, B. Faivre, V. Fourmond, C. Léger and M. Fontecave, J. Am. Chem. Soc., 2018, 140, 5516–5526 CrossRef CAS.
- C. Gauquelin, C. Baffert, P. Richaud, E. Kamionka, E. Etienne, D. Guieysse, L. Girbal, V. Fourmond, I. Andre, B. Guigliarelli, C. Leger, P. Soucaille and I. Meynial-Salles, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 69–77 CrossRef CAS.
- J. N. Butt, M. Filipiak and W. R. Hagen, Eur. J. Biochem., 1997, 245, 116–122 CrossRef CAS.
- S. Morra, M. Arizzi, F. Valetti and G. Gilardi, Biochemistry, 2016, 55, 5897–5900 CrossRef CAS.
- C. Baffert, M. Demuez, L. Cournac, B. Burlat, B. Guigliarelli, P. Bertrand, L. Girbal and C. Léger, Angew. Chem., Int. Ed., 2008, 47, 2052–2054 CrossRef CAS.
- P. Rodríguez-Maciá, E. J. Reijerse, M. van Gastel, S. DeBeer, W. Lubitz, O. Rüdiger and J. A. Birrell, J. Am. Chem. Soc., 2018, 140, 9346–9350 CrossRef.
- N. Chongdar, J. A. Birrell, K. Pawlak, C. Sommer, E. J. Reijerse, O. Rüdiger, W. Lubitz and H. Ogata, J. Am. Chem. Soc., 2018, 140, 1057–1068 CrossRef CAS.
- P. Rodríguez-Maciá, K. Pawlak, O. Rüdiger, E. J. Reijerse, W. Lubitz and J. A. Birrell, J. Am. Chem. Soc., 2017, 139, 15122–15134 CrossRef.
- H. Land, P. Ceccaldi, L. S. Mészáros, M. Lorenzi, H. J. Redman, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2019, 10, 9941–9948 RSC.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Nat. Commun., 2018, 9, 4726 CrossRef.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, J. Am. Chem. Soc., 2019, 141, 17394–17403 CrossRef CAS.
- O. Lampret, J. Duan, E. Hofmann, M. Winkler, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20520–20529 CrossRef CAS.
- M.-E. Pandelia, W. Nitschke, P. Infossi, M.-T. Giudici-Orticoni, E. Bill and W. Lubitz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6097–6102 CrossRef CAS.
- M. Rousset, Y. Montet, B. Guigliarelli, N. Forget, M. Asso, P. Bertrand, J. C. Fontecilla-Camps and E. C. Hatchikian, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11625–11630 CrossRef CAS.
- G. Caserta, A. Adamska-Venkatesh, L. Pecqueur, M. Atta, V. Artero, S. Roy, E. Reijerse, W. Lubitz and M. Fontecave, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 1734–1740 CrossRef CAS.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS.
- B. Németh, C. Esmieu, H. J. Redman and G. Berggren, Dalton Trans., 2019, 48, 5978–5986 RSC.
- A. Adamska-Venkatesh, T. R. Simmons, J. F. Siebel, V. Artero, M. Fontecave, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2015, 17, 5421–5430 RSC.
- J. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, T. Happe, E. Hofmann, U.-P. Apfel, M. Winkler, M. Senger, M. Haumann and S. T. Stripp, ACS Catal., 2019, 9, 9140–9149 CrossRef CAS.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, J. Biol. Inorg Chem., 2006, 11, 102–118 CrossRef CAS.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS.
- M. W. Ratzloff, J. H. Artz, D. W. Mulder, R. T. Collins, T. E. Furtak and P. W. King, J. Am. Chem. Soc., 2018, 140, 7623–7628 CrossRef CAS.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8, 16115 CrossRef CAS.
- J. Esselborn, L. Kertess, U.-P. Apfel, E. Hofmann and T. Happe, J. Am. Chem. Soc., 2019, 141, 17721–17728 CrossRef CAS.
- A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458–11462 CrossRef CAS.
- A. K. Jones, E. Sillery, S. P. J. Albracht and F. A. Armstrong, Chem. Commun., 2002, 866–867 RSC.
- C. Léger, A. K. Jones, S. P. J. Albracht and F. A. Armstrong, J. Phys. Chem. B, 2002, 106, 13058–13063 CrossRef.
- Where ilim is the limiting current, β is a decay constant, d0 is a range of the tunneling distances between the electrode and the entry point for electrons in the enzyme, F is the Faraday constant, R is the gas constant, and T is temperature.
- C. Léger, S. Dementin, P. Bertrand, M. Rousset and B. Guigliarelli, J. Am. Chem. Soc., 2004, 126, 12162–12172 CrossRef.
- G. Goldet, A. F. Wait, J. A. Cracknell, K. A. Vincent, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 11106–11113 CrossRef CAS.
- C. Léger, S. J. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones and F. A. Armstrong, Biochemistry, 2003, 42, 8653–8662 CrossRef.
- O. Lampret, A. Adamska-Venkatesh, H. Konegger, F. Wittkamp, U.-P. Apfel, E. J. Reijerse, W. Lubitz, O. Rüdiger, T. Happe and M. Winkler, J. Am. Chem. Soc., 2017, 139, 18222–18230 CrossRef CAS.
- K. Pandey, S. T. A. Islam, T. Happe and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3843–3848 CrossRef CAS.
- K. A. Vincent, A. Parkin, O. Lenz, S. P. J. Albracht, J. C. Fontecilla-Camps, R. Cammack, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 18179–18189 CrossRef CAS.
- V. Fourmond, C. Greco, K. Sybirna, C. Baffert, P.-H. Wang, P. Ezanno, M. Montefiori, M. Bruschi, I. Meynial-Salles, P. Soucaille, J. Blumberger, H. Bottin, L. De Gioia and C. Léger, Nat. Chem., 2014, 6, 336–342 CrossRef CAS.
- H. S. Jayasinghearachchi, P. M. Sarma and B. Lal, Int. J. Hydrogen Energy, 2012, 37, 5569–5578 CrossRef CAS.
- S. V. Hexter, F. Grey, T. Happe, V. Climent and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11516–11521 CrossRef CAS.
- M. Bernhard, T. Buhrke, B. Bleijlevens, A. L. De Lacey, V. M. Fernandez, S. P. J. Albracht and B. Friedrich, J. Biol. Chem., 2001, 276, 15592–15597 CrossRef CAS.
- P. A. Ash, J. Liu, N. Coutard, N. Heidary, M. Horch, I. Gudim, T. Simler, I. Zebger, O. Lenz and K. A. Vincent, J. Phys. Chem. B, 2015, 119, 13807–13815 CrossRef CAS.
- V. Fourmond, C. Baffert, K. Sybirna, S. Dementin, A. Abou-Hamdan, I. Meynial-Salles, P. Soucaille, H. Bottin and C. Léger, Chem. Commun., 2013, 49, 6840–6842 RSC.
- P. M. Wolanin, P. A. Thomason and J. B. Stock, Genome Biol., 2002, 3, reviews3013.1–3013.8 CrossRef.
- M. T. Gallegos, R. Schleif, A. Bairoch, K. Hofmann and J. L. Ramos, Microbiol. Mol. Biol. Rev., 1997, 61, 393–410 CAS.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS.
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Léger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS.
- S. Mebs, R. Kositzki, J. Duan, L. Kertess, M. Senger, F. Wittkamp, U.-P. Apfel, T. Happe, S. T. Stripp, M. Winkler and M. Haumann, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 28–41 CrossRef CAS.
- H. S. Shafaat, O. Rüdiger, H. Ogata and W. Lubitz, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 986–1002 CrossRef CAS.
- F. Sievers, A. Wilm, D. Dineen, T. J. Gibson, K. Karplus, W. Li, R. Lopez, H. McWilliam, M. Remmert, J. Söding, J. D. Thompson and D. G. Higgins, Mol. Syst. Biol., 2011, 7, 539 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03319g |
| ‡ Current address: Department of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, USA. |
| § Current address: Institute of Molecular Biosciences, University of Graz, Humboldtstrasse 50, 8010 Graz, Austria. |
| This journal is © The Royal Society of Chemistry 2020 |