
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFree-standing metal–organic framework (MOF) monolayers by self-assembly of polymer-grafted nanoparticles†
Kyle
Barcus
and
Seth M.
Cohen
 *
*
Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, USA. E-mail: scohen@ucsd.edu
First published on 3rd August 2020
Abstract
We report a general method for the synthesis of free-standing, self-assembled MOF monolayers (SAMMs) at an air–water interface using polymer-brush coated MOF nanoparticles. UiO-66, UiO-66-NH2, and MIL-88B-NH2 were functionalized with a catechol-bound chain-transfer agent (CTA) to graft poly(methyl methacrylate) (PMMA) from the surface of the MOF using reversible addition-fragmentation chain transfer polymerization (RAFT). The polymer-coated MOFs were self-assembled at the air–water interface into monolayer films ∼250 nm thick and capable of self-supporting at a total area of 40 mm2. Mixed-particle films were prepared through the assembly of MOF mixtures, while multilayer films were achieved through sequential transfer of the monolayers to a glass slide substrate. This method offers a modular and generalizable route to fabricate thin-films with inherent porosity and sub-micron thickness composed of a variety of MOF particles and functionalities.
Introduction
Metal–organic frameworks (MOFs) are a specialized class of porous materials known for their high-surface area, defined pore size, and range of chemical functionality.1 Composed of inorganic secondary building units (SBUs) and multitopic organic ligands, MOFs can be fine-tuned for a wide variety of applications including gas storage, catalysis, sensing, and drug delivery.2–7 However, the powdery, crystalline nature of these materials makes their incorporation into many applications difficult to achieve when compared to materials made from polymers. This has created a growing demand for techniques to improve MOF processing and to devise new MOF form factors. Reports show that physically blending MOFs with polymers to make foams8,9 or films10,11 gives composites with improved strength and flexibility; however, incompatibility between the two components can result in defects and low particle loading that diminish the contribution of the MOF filler to the material properties.12–14 As the MOF loading increases, the MOF-like properties of the materials are enhanced, but the weak interactions between the MOF and polymer generally require thicker films to avoid the formation of defects and to provide structural integrity.Postsynthetic polymerization (PSP) is an attractive method to graft polymers on the surface of a MOF that enhances interactions between the MOF surface and the surrounding polymer matrix.15 Several MOF–polymer composites have been prepared by in situ polymerization between monomers and co-functionalized MOFs, and this method affords materials with impressive separation and physical properties when compared to mixed blends of MOFs and polymers.15–19 This copolymerization strategy between monomer and MOF provides a simple route to highly integrated composites, but the inevitable crosslinking of the polymer matrix results in a thermoset-like material which cannot be processed further or easily purified of excess monomer and solvent. To circumvent this, controlled polymerizations from active sites on the MOF surface via a “grafting from” approach have significantly less crosslinking and can show similar matrix compatibility.20–22 However, both of these methods, either polymerizations in the presence of MOF or from the MOF surface, have relied almost exclusively on UiO-66(Zr)-NH2 as a model MOF, taking advantage of the amino group as the anchor for polymer attachment.15,18,19,21,22 This limits the scope of the PSP method because MOFs such as MIL-100, MOF-74, HKUST-1, and NU-1000 contain ligands that are either incompatible or difficult to derivatize with free amines. Therefore, other approaches to the functionalization of these materials (e.g., through coordination of exposed surface metal sites) would avoid the complications and limitations of derivatized ligand synthesis, such that PSP can be generalized to most MOFs.23–25 Surface metal coordination has been used to attach a variety of molecules to the exterior of MOFs, but rarely has it been applied to incorporate initiating sites for polymer growth.20
ZIF-8, one of the most widely studied MOFs, is difficult to synthesize with primary amine groups present on the ligand due to the competing complexation of the amine and imidazole with the metal salt.26 We previously reported a method for PSP from ZIF-8 using postsynthetic exchange (PSE) of an imidazole-bound initiator for atom-transfer radical polymerization (ATRP) to functionalize ZIF-8 nanoparticles.27 This procedure used a modified imidazole possessing an initiator, allowing for surface-modification of the MOF utilizing the strong metal–ligand bonds that hold the structure together. Surface-initiated atom-transfer radical polymerization (SI-ATRP) of methyl methacrylate gave MOF/polymer core–shell particles which could be self-assembled into monolayers at the air–water interface. This was the first example of self-assembled MOF monolayers (SAMMs) constructed from MOFs decorated with polymer brushes. However, the PSE approach for surface functionalization was inherently limited to ZIFs, which contain imidazole linkers necessary for surface PSE to occur, and the exchange conditions required extensive optimization to avoid etching of the MOF particles. Furthermore, it was suspected that poor control over the polymerization, due to the amide-based initiator, gave low molecular weight polymer grafts resulting in monolayer films too fragile to hold their own weight without fracturing (this could only be achieved with multilayer films comprised of >5 layers). The ZIF-8 SAMMs represent an efficient method for forming MOF thin films with high particle loadings without necessitating a readily modifiable MOF ligand. However, these SAMMs were fragile, and an ability to further improve the stability and mechanical properties of the monolayers may expand the potential to use these unusual MOF mono- and multi-layers in applications such as separation and sensing.
Herein, we report a general methodology for the preparation of free-standing SAMMs using a variety of MOF nanoparticles coated with a polymer brush synthesized via surface-initiated reversible addition-fragmentation chain transfer (SI-RAFT) polymerization of methyl methacrylate (Scheme 1). Three MOFs, UiO-66(Zr), UiO-66(Zr)-NH2, and MIL-88B(Fe)-NH2, were surface functionalized using a catechol-modified chain-transfer agent (cat-CTA). The MOFs used in this study were chosen to demonstrate the versatility of cat-CTA to coordinate the surface of MOFs bearing different particle morphologies, sizes, SBUs, and ligand functionalities. Following successful polymerization to high molecular weight, monolayers of the polymer-coated MOFs were prepared through self-assembly at an air–water interface, and suspension of the films on a small loop of copper wire gave self-supporting films spanning an area as large as 7 mm in diameter.
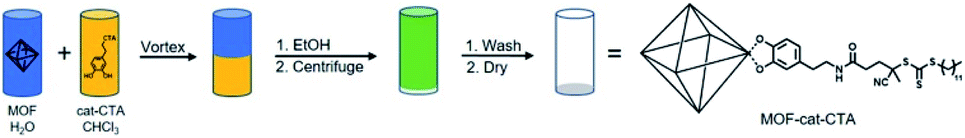 | ||
| Scheme 1 Surface functionalization procedure for coordinating cat-CTA to MOF. MOF particles (UiO-66) are depicted by polyhedron with aqueous and organic solvents represented by the blue and yellow layers, respectively. Mixed (vortexed) solutions represented by green color. | ||
Results and discussion
Synthesis of the catechol-modified RAFT CTA (cat-CTA) was performed using a two-step acylation procedure between a commercially available CTA and dopamine hydrochloride as previously reported (Scheme S1†).28 UiO-66(Zr) truncated octahedra, UiO-66(Zr)-NH2 octahedra, and MIL-88B(Fe)-NH2 hexagonal rods were selected for modification. Due to the low tolerance for particle size dispersity in self-assembled particle-brush systems,29 each MOF was prepared via solvothermal methods that gave relatively narrow size dispersities of the desired morphology.19,30,31 Powder X-ray diffraction (PXRD) and gas sorption analysis with N2 gas were in good agreement with previous reports, and scanning electron microscopy (SEM) images of the MOFs showed discrete particles with controlled morphology and size (Fig. S1–S4†).The surface functionalization of UiO-66(Zr) with cat-CTA was accomplished using a phase-transfer methodology adapted from a previously reported procedure (Scheme 1).32 The MOF particles were dispersed in water and the cat-CTA was dissolved in chloroform. The two solutions were combined and mixed with a vortex mixer for several minutes to functionalize the MOF surface. Ethanol was added to break up the resulting emulsion and the particles were collected by centrifugation, washed several times with THF, and dried under vacuum. A sample of UiO-66(Zr)-CTA was digested in dilute HF and analysed by 1H NMR, which confirmed the presence of the cat-CTA (Fig. S5†). PXRD and N2 gas sorption analysis of the sample indicated the addition of the cat-CTA did not affect the crystallinity or accessible porosity of UiO-66(Zr), and SEM images showed that no particle etching or a change in morphology occurred (Fig. S1 and S2†). The increase in surface area is suspected to be a result of the functionalization procedure involving redispersion in water and washing the particles again. This may act as a second round of activation to remove any residual impurities that may have remained after particle synthesis.
The polymer brush was grown from the surface of UiO-66(Zr) using RAFT polymerization of methyl methacrylate (MMA). RAFT is a simple, controlled-radical polymerization method well-known for its high functional group tolerance and predictable molecular weights.33 To determine the amount cat-CTA coordinated to the surface of UiO-66(Zr), first a ratio of the MOF ligand to cat-CTA was measured using 1H NMR analysis of the digested UiO-66(Zr). This value was then used to calculate the amount of CTA present on the surface of the MOF, and a mass ratio of ∼250 μg of CTA per 10 mg of UiO-66(Zr) was obtained (see ESI† for details). A molar ratio of 5000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MMA to cat-CTA was chosen for a target degree of polymerization (DP) of 2000 and carried out at 70 °C using DMF as a solvent and AIBN as a free radical initiator. The high molar ratio of monomer to target DP is necessary as surface-initiated polymerizations are known to give crosslinked particles at high conversions, resulting in macroscopic gels.34 Unmodified CTA was also added to the polymerization reaction to increase the initial concentration of the transfer agent and improve control over the molecular weight of the polymers growing from the MOF surfaces. Following the polymerization, the particles were recovered by centrifugation and the supernatant was poured into methanol to precipitate the free polymer grown in solution (which was retained for molecular weight analysis by gel permeation chromatography (GPC)). The remaining polymer-coated MOF was washed several times with THF and toluene to remove any unbound polymer and finally suspended in toluene for self-assembly. While PXRD analysis of particles indicated the crystallinity of the MOF was unaffected by the polymerization, the surface area decreased by ∼39% to 846 m2 g−1 (Fig. S1, S6, Table S2†). To correct for the added mass from the nonporous polymer, thermal gravimetric analysis (TGA) of UiO-66(Zr)-PMMA compared to unfunctionalized UiO-66 calculated that the sample is composed of 20% polymer by mass (Fig. S6†). The adjusted surface area would then be ∼1015 m2 g−1, indicating that UiO-66-PMMA still retains 73% of the total surface area of the parent MOF.
:1 MMA to cat-CTA was chosen for a target degree of polymerization (DP) of 2000 and carried out at 70 °C using DMF as a solvent and AIBN as a free radical initiator. The high molar ratio of monomer to target DP is necessary as surface-initiated polymerizations are known to give crosslinked particles at high conversions, resulting in macroscopic gels.34 Unmodified CTA was also added to the polymerization reaction to increase the initial concentration of the transfer agent and improve control over the molecular weight of the polymers growing from the MOF surfaces. Following the polymerization, the particles were recovered by centrifugation and the supernatant was poured into methanol to precipitate the free polymer grown in solution (which was retained for molecular weight analysis by gel permeation chromatography (GPC)). The remaining polymer-coated MOF was washed several times with THF and toluene to remove any unbound polymer and finally suspended in toluene for self-assembly. While PXRD analysis of particles indicated the crystallinity of the MOF was unaffected by the polymerization, the surface area decreased by ∼39% to 846 m2 g−1 (Fig. S1, S6, Table S2†). To correct for the added mass from the nonporous polymer, thermal gravimetric analysis (TGA) of UiO-66(Zr)-PMMA compared to unfunctionalized UiO-66 calculated that the sample is composed of 20% polymer by mass (Fig. S6†). The adjusted surface area would then be ∼1015 m2 g−1, indicating that UiO-66-PMMA still retains 73% of the total surface area of the parent MOF.
Monolayers of UiO-66(Zr)-PMMA were prepared through self-assembly at an air–water interface by dispersing the MOF in toluene at 50–100 mg mL−1 and placing a 10 μL drop on the surface of a layer of water in a 55 mm diameter Petri dish. The drop spread immediately to the edges forming a thin film with an iridescent color (Fig. S6†). A free-standing film was obtained by lifting a copper wire loop up from under the water surface (Video S1†). The loop was fashioned small enough (diameter = 7 mm) that a water drop was trapped in the ring by surface tension with the film floating on the surface. This allowed the monolayer to be lifted without fracturing, and slow evaporation of the water in ambient conditions resulted in a self-supporting membrane of the MOF monolayer (Fig. 1a). Surprisingly, even though the films are almost entirely composed of MOF particles, the free-standing films were translucent and maintained their iridescence. SEM images of the free-standing film showed the particles maintained a tightly packed structure with only small defects present (Fig. 1b and c).
 | ||
| Fig. 1 Free-standing monolayer of self-assembled UiO-66(Zr)-PMMA. (a) Images of the monolayer taken at different angles to show both clarity and iridescence. (b) SEM image of the monolayer surface showing tightly packed UiO-66(Zr)-PMMA particles. Scale bar is 2 μm. (c) SEM image of film from the side, illustrating the continuous monolayer. Scale bar is 5 μm. | ||
Functionalized MOF samples were digested with HF to recover the polymer brush from the MOF surface for molecular weight analysis by GPC and compared to the polymer recovered from the supernatant. The molecular weight of the surface polymer (Mn = 252 kg mol−1, Đ = 1.17) and the free polymer (isolated from the RAFT reaction, vide supra, Mn = 263 kg mol−1, Đ = 1.15) are similar, and the low dispersity of both indicate the polymerization is controlled by the RAFT mechanism. The effect of polymer molecular weight on monolayer formation and particle ordering was investigated by halting the polymerization at several time points (Fig. S12 and S13†). In the first hour, the molecular weight reached only 8.3 kg mol−1, which was too small prevent particle aggregation as shown by SEM (Fig. 2a and b). At two hours, the molecular weight increased to 59 kg mol−1, and monolayers were observed (Fig. 2c). However, these monolayers could not form free-standing films, presumably due to the relatively weak particle interactions leaving large gaps between the MOFs (Fig. 2c). Once the molecular weight reached 138 kg mol−1 at four hours polymerization time, free-standing films of densely packed particles were obtained. These results highlight the importance of polymer length on stabilizing the interparticle interactions and increasing film strength (Fig. 2d).
 | ||
| Fig. 2 Effect of polymer brush molecular weight on order and packing of UiO-66(Zr) particles. (a) 30 minutes, Mn = 6.3 kg mol−1Đ = 2.15. (b) 1 hour, Mn = 8.3 k Đ = 1.8. (c) 2 hours, Mn = 59 kg mol−1Đ = 1.49. (d) 4 hours, Mn = 138 kg mol−1Đ = 1.16. All scale bars are 3 μm. | ||
We next looked to expand the polymerization and self-assembly strategy to other MOF nanoparticles. UiO-66(Zr)-NH2 octahedra and MIL-88B(Fe)-NH2 hexagonal nanorods were chosen to demonstrate the generality of the methodology towards different SBUs, chemical functionalities, and particle morphologies. Functionalization of the surface with cat-CTA using the same methodology was successful, and 1H NMR analysis digested UiO-66(Zr)-CTA showed the amine of the organic linker remained unfunctionalized (Fig. S14†). Following polymerization and digestion of the polymer coated MOFs, GPC analysis of the polymer brush gave molecular weights and dispersities for UiO-66(Zr)-NH2-PMMA (Mn = 215 kg mol−1, Đ = 1.17) and MIL-88B(Fe)-NH2-PMMA (Mn = 190 kg mol−1, Đ = 1.22) that were comparable to UiO-66(Zr). PXRD and gas sorption analysis indicate the effect on crystallinity and surface area of UiO-66(Zr)-NH2-PMMA was comparable to UiO-66(Zr)-PMMA, with the high crystallinity preserved and a total surface area of 692 m2 g−1 (Table S1, Fig. S3, S7†). MIL-88B(Fe)-NH2 is a flexible MOF with pores that remain closed unless swollen with solvent such as DMF; therefore, these materials were non-porous and no change in surface area could be measured under the conditions used for measuring gas sorption.35 Surprisingly, the PXRD of MIL-88B(Fe)-NH2-PMMA changed, giving a diffraction pattern consistent with the open form of the MOF (Fig. S4†).36 While the exact reason for this is unknown, it is possible that the polymer brush makes the complete removal of DMF more difficult and DMF is still present in the interior of these MOF particles. Self-assembly of these MOFs resulted in freestanding monolayers of MIL-88B(Fe)-NH2-PMMA (Fig. 3a); however, the UiO-66(Zr)-NH2-PMMA could only make free-standing films when deposited at a thickness of 2–3 particles (Fig. S15†). This may be due to the substantially smaller particle size of UiO-66(Zr)-NH2 (<100 nm) versus UiO-66(Zr) (∼250 nm) and MIL-88B(Fe)-NH2 (anisotropic shape, >700 × 100 nm). Assuming the films are strengthened by the intercalation of polymer chains in the lateral direction, the smaller particle size of UiO-66(Zr)-NH2 would result in fewer interchain polymer interactions, leading to a less robust film structure.
 | ||
| Fig. 3 Monolayer, multilayers, and mixed monolayers of MOFs. (a) Monolayer of MIL-88B(Fe)-NH2-PMMA. (b) Bilayer of UiO-66(Zr)-PMMA (bottom) and MIL-88B(Fe)-NH2-PMMA (top). (c) Multilayer of UiO-66(Zr)-PMMA (bottom) and MIL-88B(Fe)-NH2-PMMA (middle) and UiO-66(Zr)-NH2-PMMA (top). (d) Mixed monolayer comprised of UiO-66(Zr)-PMMA (larger particles) and UiO-66(Zr)-NH2-PMMA (smaller particles). All scale bars are 1 μm. | ||
One advantage of creating membranes through the self-assembly of individually functionalized particles is that the modularity allows for the components to be assembled in different combinations to create multifunctional composites. To demonstrate this, a monolayer of UiO-66(Zr)-PMMA was coated on a glass slide with SEM tape. After drying, a layer of MIL-88B(Fe)-NH2-PMMA was applied to form a bilayer of both MOFs (Fig. 3b), followed by a layer of UiO-66(Zr)-NH2 to form a multilayer film (Fig. 3c). In addition, mixed monolayers of the MOFs were also fabricated by mixing a toluene suspension of both UiO-66(Zr)-PMMA and UiO-66(Zr)-NH2-PMMA which were then self-assembled together (Fig. 3c). The SEM images show that the large size discrepancy between the two MOFs allows for the smaller UiO-66(Zr)-NH2-PMMA to fill gaps between the larger UiO-66(Zr)-PMMA, resulting in a monolayer that combines the functionalities of both MOFs without adding any extra thickness (Fig. 3d).
Conclusion
In conclusion, we report a general methodology to functionalize MOF nanoparticles with RAFT CTAs for controlled radical polymerization from the MOF surface. These multifunctional, porous nanoparticles were further self-assembled into ultra-thin films of MOF particles and demonstrated to be fully free-standing with inherent iridescence and good optical clarity. Furthermore, multilayered composites were realized through the sequential layering of films, and monolayers of mixed MOFs were synthesized through the self-assembly of MOF mixtures. This system establishes a modular route towards novel multifunctional membranes, and further studies on the effect of coordination, initiator, and polymer type on membrane properties are ongoing.Experimental
Surface functionalization of MOFs with CTA
A vial filled with 10 mL of DI H2O and 200 mg of MOF was sonicated for 30 min to suspend the particles. A 50 mL centrifuge tube was prepared with 4 mg of cat-CTA (3) dissolved in 5 mL of CHCl3 and the solution of suspended particles was added. The biphasic mixture was vortexed for 3 min and 20 mL of EtOH was added to form a homogenous suspension. The particles were collected by centrifugation (6000 rpm, 10 min.), washed through repeated dispersion/centrifugation cycles with EtOH (2 × 25 mL, 30 min each) and THF (25 mL, 12 h), and dried overnight in a vacuum oven at 50 °C. Particle digestion for 1H NMR analysis was performed by sonicating 10–15 mg of dried MOF in a 1 mL plastic centrifuge tube with 400 μL d6-DMSO and minimal (∼6 μL) HF (48% H2O) until a clear solution was obtained.Polymerization of MMA from CTA-Functionalized MOFs
A solution of AIBN in DMF (10 mg mL−1) and free CTA in DMF (100 mg mL−1) were prepared. A 10 mL round bottom flask was charged with 10 mg of CTA@MOF (0.46 μmol cat-CTA, 1 equiv.) and 2 mL of DMF. The flask was sonicated for 30 min to fully suspend the MOFs before methyl methacrylate (2.0 mL, 18.8 mmol, 40000 equiv.) was added and the reaction was sonicated an additional 30 min. From the prepared stock solutions, 7.6 μL AIBN (76 μg, 0.46 μmol, 1 equiv.) and 5.8 μL free CTA (186 μg, 0.46 μmol, 1 equiv.) were both added and the reaction was degassed with argon for 30 min. The flask was briefly sonicated for 5 min before being placed in an oil bath heated to 70 °C. The polymerization was quenched by cooling in liquid nitrogen before opening the flask and adding 6 mL of DMF. The particles were collected by centrifugation (10000 rpm, 20 min) and the supernatant was poured into 10× excess rapidly stirring MeOH to precipitate the polymer. The remaining solids were washed through repeated dispersion/centrifugation cycles with DMF (30 mL, 1 h), THF (2 × 30 mL, 6 h), and toluene (30 mL, 1 h) and suspended in 150 μL of toluene for self-assembly. Particle digestion to isolate the polymer for GPC analysis was performed by sonicating 15–20 mg of dried MOF-PMMA in a 15 mL plastic centrifuge tube with 100 μL d6-DMSO and 20 μL HF (48% H2O) until a clear solution was obtained. 2 mL of 1 M NaOH was added followed by 5 mL of toluene and vigorously mixed before separating the layers by centrifugation. The toluene layer was transferred to a 20 mL vial and dried in a vial under vacuum. The remaining residue was dissolved in 150 μL THF for GPC analysis.
Self-assembly at the air–water interface
The polymer-coated MOFs were suspended in toluene at 50–100 mg mL−1 through sonication. A 10 μL drop of the solution was place on the surface of water in a 55 mm diameter Petri dish. After the toluene evaporated, the SAMM was lifted onto the surface of SEM carbon tape on a glass slide for imaging. For free-standing monolayers, a copper loop was prepared by wrapping copper wire (diameter = 0.5 mm) around a 1 mL plastic syringe barrel. The loop was removed and placed under the water surface, then quickly lifted from underneath the monolayer, suspending a drop of water with the film floating on the surface. The loop was hung to air dry, leaving a thin film of the MOF which was then imaged by SEM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering under Award No. DE-FG02-08ER46519. Imaging work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of U.C. San Diego, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542148).Notes and references
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- H.-C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- Y. Chen, F. Chen, S. Zhang, Y. Cai, S. Cao, S. Li, W. Zhao, S. Yuan, X. Feng, A. Cao, X. Ma and B. Wang, J. Am. Chem. Soc., 2017, 139, 16482–16485 CrossRef CAS PubMed.
- M. L. Pinto, S. Dias and J. Pires, ACS Appl. Mater. Interfaces, 2013, 5, 2360–2363 CrossRef CAS PubMed.
- G. Liu, V. Chernikova, Y. Liu, K. Zhang, Y. Belmabkhout, O. Shekhah, C. Zhang, S. Yi, M. Eddaoudi and W. J. Koros, Nat. Mater., 2018, 17, 283–289 CrossRef CAS PubMed.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS PubMed.
- J. Dechnik, J. Gascon, C. J. Doonan, C. Janiak and C. J. Sumby, Angew. Chem., Int. Ed., 2017, 56, 9292–9310 CrossRef CAS PubMed.
- M. Kalaj, K. C. Bentz, S. Ayala, J. M. Palomba, K. S. Barcus, Y. Katayama and S. M. Cohen, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.9b00575.
- R. Lin, B. Villacorta Hernandez, L. Ge and Z. Zhu, J. Mater. Chem. A, 2018, 6, 293–312 RSC.
- Y. Zhang, X. Feng, H. Li, Y. Chen, J. Zhao, S. Wang, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2015, 54, 4259–4263 CrossRef CAS PubMed.
- K. Xie, Q. Fu, P. A. Webley and G. G. Qiao, Angew. Chem., Int. Ed., 2018, 130, 8733–8738 CrossRef.
- C. Satheeshkumar, H. J. Yu, H. Park, M. Kim, J. S. Lee and M. Seo, J. Mater. Chem. A, 2018, 6, 21961–21968 RSC.
- X. Gao, J. Zhang, K. Huang and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 34640–34645 CrossRef CAS PubMed.
- M. Kalaj, M. S. Denny Jr, K. C. Bentz, J. M. Palomba and S. M. Cohen, Angew. Chem., Int. Ed., 2019, 58, 2336–2340 CrossRef CAS PubMed.
- S. He, H. Wang, C. Zhang, S. Zhang, Y. Yu, Y. Lee and T. Li, Chem. Sci., 2019, 10, 1816–1822 RSC.
- K. A. McDonald, J. I. Feldblyum, K. Koh, A. G. Wong-Foy and A. J. Matzger, Chem. Commun., 2015, 51, 11994–11996 RSC.
- H. Wang, S. He, X. Qin, C. Li and T. Li, J. Am. Chem. Soc., 2018, 140, 17203–17210 CrossRef CAS PubMed.
- S. Wang, C. M. McGuirk, A. d'Aquino, J. A. Mason and C. A. Mirkin, Adv. Mater., 2018, 30, 1800202 CrossRef PubMed.
- R. J. Marshall and R. S. Forgan, Eur. J. Inorg. Chem., 2016, 2016, 4310–4331 CrossRef CAS.
- C. Chen, Z. Wu, Y. Que, B. Li, Q. Guo, Z. Li, L. Wang, H. Wan and G. Guan, RSC Adv., 2016, 6, 54119–54128 RSC.
- K. Y. Cho, H. An, X. H. Do, K. Choi, H. G. Yoon, H.-K. Jeong, J. S. Lee and K.-Y. Baek, J. Mater. Chem. A, 2018, 6, 18912–18919 RSC.
- Y. Katayama, M. Kalaj, K. S. Barcus and S. M. Cohen, J. Am. Chem. Soc., 2019, 141, 20000–20003 CrossRef CAS PubMed.
- O. O. Oyeneye, W. Z. Xu and P. A. Charpentier, RSC Adv., 2015, 5, 76919–76926 RSC.
- N. Bachhar, G. Kumaraswamy and S. K. Kumar, Macromolecules, 2019, 52, 4888–4894 CrossRef CAS.
- M. Kalaj, J. M. Palomba, K. C. Bentz and S. M. Cohen, Chem. Commun., 2019, 55, 5367–5370 RSC.
- S. L. Zhang, B. Y. Guan, H. B. Wu and X. W. D. Lou, Nano-Micro Lett., 2018, 10, 44 CrossRef PubMed.
- W. Zhu, G. Xiang, J. Shang, J. Guo, B. Motevalli, P. Durfee, J. O. Agola, E. N. Coker and C. J. Brinker, Adv. Funct. Mater., 2018, 28, 1705274 CrossRef.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- J. Pyun, T. Kowalewski and K. Matyjaszewski, Macromol. Rapid Commun., 2003, 24, 1043–1059 CrossRef CAS.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef CAS PubMed.
- M. Ma, A. Bétard, I. Weber, N. S. Al-Hokbany, R. A. Fischer and N. Metzler-Nolte, Cryst. Growth Des., 2013, 13, 2286–2291 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03318a |
| This journal is © The Royal Society of Chemistry 2020 |