
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemo-selective cross reaction of two enals via carbene-catalyzed dual activation†
Xiaolin
Peng
a,
Jun
Xu
bc,
Tingting
Li
a,
Yonggui Robin
Chi
 ac and
Zhichao
Jin
*a
ac and
Zhichao
Jin
*a
aLaboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering Ministry of Education, Guizhou University, Huaxi District, Guiyang 550025, China
bSchool of Pharmacy, Guiyang College of Traditional Chinese Medicine, Huaxi District, Guiyang 550025, China
cDivision of Chemistry & Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: zcjin@gzu.edu.cn
First published on 18th September 2020
Abstract
A dual catalytic chemo-selective cross-coupling reaction of two enals is developed. One enal (without α-substitution) is activated by an NHC catalyst to form an acylazolium enolate intermediate that undergoes Michael-type addition to another enal molecule bearing an alkynyl substituent. Mechanistic studies indicate that non-covalent interactions between the alkynyl enal and the NHC·HX catalyst play important roles in substrate activation and enantioselectivity control. Many of the possible side reactions are not observed. Our reaction provides highly chemo- and diastereo-selective access to chiral lactones containing functionalizable 1,3-enyn units with excellent enantioselectivities (95 to >99% ee).
The development of chemo-selective reactions of two or more substrates bearing similar functional groups remains a classic challenge in organic synthesis.1 Enals (α,β-unsaturated aldehydes) are common building blocks that offer multiple useful modes of reactions. For instance, enals are readily used as Michael acceptors in many reactions including organic catalytic reactions mediated by amines.2 In the area of N-heterocyclic carbene (NHC) organocatalysis,3 enals are used as precursors of several NHC-bound intermediates, including Breslow acyl anion intermediates,4 homoenolate intermediates,5 enolate intermediates,6 and acylazolium intermediates.7 Somewhat surprisingly, on the other hand, there is little success in using enals as Michael acceptors to react with any of these NHC-bound intermediates.8 Elegant studies in this direction are from Scheidt, in which they showed that in the presence of an NHC catalyst, a homo coupling reaction of enals (with one enal molecule as the Michael acceptor) occurred effectively (Fig. 1a, top side).8a,c Berkessel reported an intramolecular reaction of two enal moieties (in one molecule) to form a bicyclic lactone adduct in the presence of an achiral NHC catalyst (Fig. 1a, bottom side).8b To the best of our knowledge, the intermolecular Michael addition reaction of two different enal substrates mediated by NHC catalysts has not been reported.9 Possible reasons for the difficulties of enals to behave as effective Michael acceptors likely include: (a) the relatively low electrophilicity of the α,β-unsaturated bonds of enals under the typical NHC catalytic conditions and (b) the presence of competing reactions involving both the alkene and aldehyde moieties of enals.
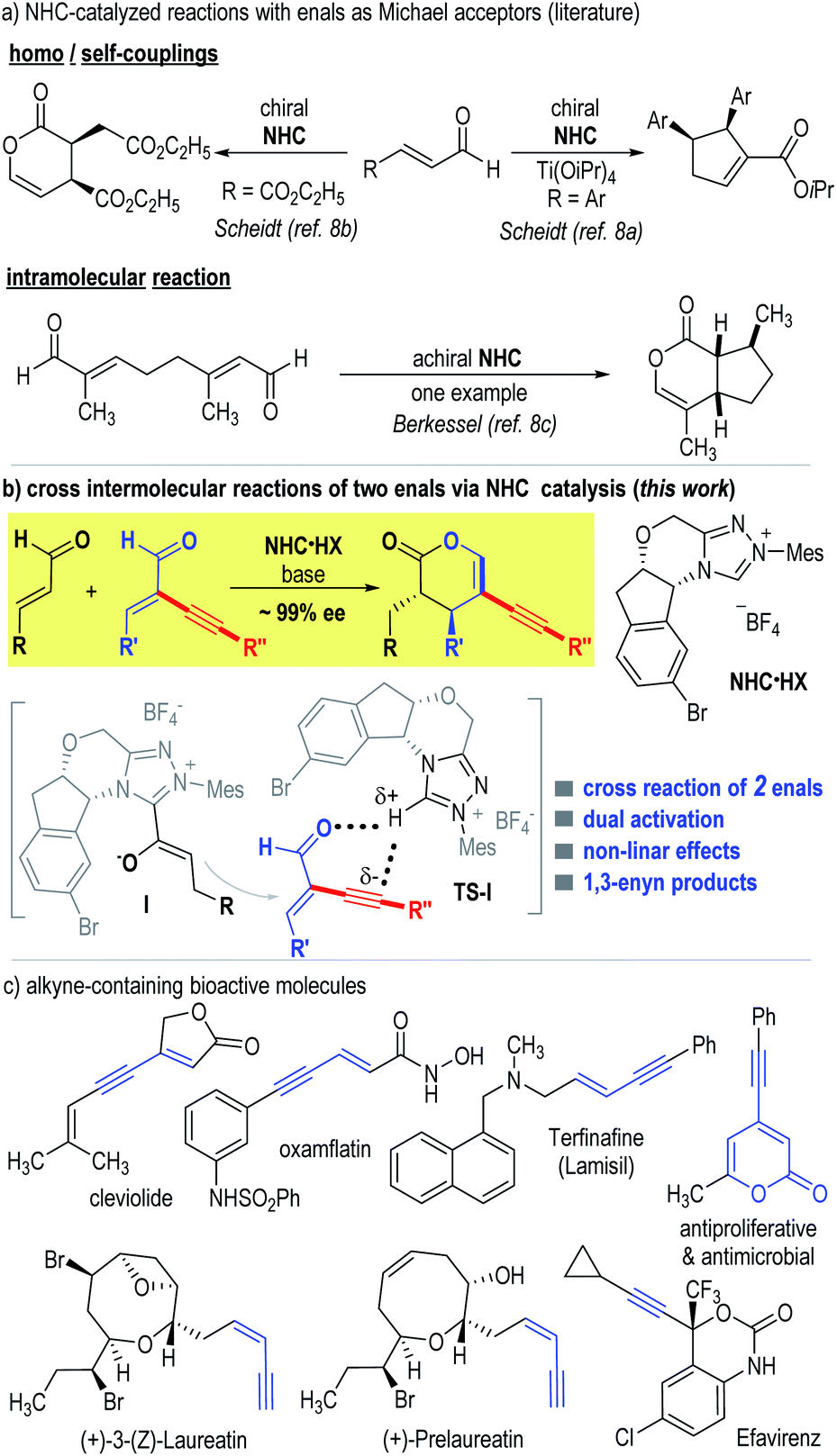 | ||
| Fig. 1 NHC-catalyzed reactions (a) with enals as Michael acceptors, (b) via cross intermolecular reactions of two enals, and (c) bio-active molecules bearing alkyne units. | ||
Here we disclose the first cross intermolecular reaction of two enals catalyzed by NHC catalysts (Fig. 1b). We envisioned that installation of an alkynyl substituent at the α-position of an enal can likely promote its reactivity as a Michael acceptor.10 The presence of an α-substituent can interrupt π-conjugations and thus minimize its reactivity via the corresponding enal-derived enolate/homoenolate intermediate formed with NHC, as shown by Bode, Glorius and others.6b,11 In addition, the alkynyl substituent can promote hydrogen-bonding interactions to increase the electrophilicity of the enal to react as a Michael acceptor, as observed in Jørgensen's amine-catalyzed reactions.12 In our present study, a non-linear effect was observed regarding enantiomeric excesses of the NHC catalyst and the catalytic reaction product. The reaction enantioselectivity was also found to be sensitive to solvents and bases. These results suggested that the NHC and its azolium salt pre-catalyst (NHC·HX) played dual roles in our reaction: one is to activate the α-unsubstituted enal via the formation of the NHC-bound enolate intermediate,6 the other is to activate the α-alkynyl substituted enal via the acidic proton of the chiral NHC·HX (Fig. 1b, intermediate I & transition state TS-I).13 With respect to applications, carbon–carbon triple bonds are found in a good number of bioactive molecules such as cleviolide, (+)-prelaureatin, and oxamflatin (Fig. 1c).14 We demonstrated that our products containing these alkynyl units could be readily transformed into a diverse set of molecules.
Cinnamaldehyde 1a and α-alkynyl enal 2a were chosen as the model substrates to search for suitable cross coupling reaction conditions (Table 1). The reactions were first carried out with Et3N as the base and THF as the solvent. When aminoindanol derived azoium salt A15 was used as the NHC pre-catalyst, the desired formal [4 + 2] product (3a) was obtained in a very encouraging yield (52%) with excellent ee and dr values (entry 1). The reactions appeared to be very sensitive to the structure of the NHC pre-catalysts, as similar azolium salts with N-phenyl or N–C6F5 substituents (B16 and C17) were completely ineffective, leading to no product formation (entries 2 & 3). Additional studies on the NHC pre-catalysts finally revealed that introduction of a Br substituent in the indane phenyl ring of the catalyst (D)18 led to 3a in 85% yield with 99% ee as nearly a single diastereomer (entry 4). Replacing Et3N with DIEA led to similar results (entry 5). Very interestingly, when the bases were replaced with DABCO or K3PO4, a significant drop in the enantioselectivity was observed (entries 6 & 7; see the ESI† for more details). Changing the solvent from THF to CHCl3 or EtOAc has moderate effects on reaction yields (entries 8 & 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | NHC | Base | Solvent | Yieldb (%) | Eec (%) | Drd |
| a Unless otherwise specified, the reactions were carried using 1a (0.15 mmol), 2a (0.1 mmol), NHC (0.02 mmol), base (0.05 mmol) and solvent (1.0 mL) at rt for 24 h. b Isolated yield of 3a. c The ee values were determined via HPLC on a chiral stationary phase. d Dr values were determined via1H NMR of the crude reaction mixture. | ||||||
| 1 | A | Et3N | THF | 52 | 99 | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | B | Et3N | THF | 0 | — | — |
| 3 | C | Et3N | THF | 0 | — | — |
| 4 | D | Et3N | THF | 85 | 99 |
>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 5 | D | DIEA | THF | 83 | 98 | >20:1 |
| 6 | D | DABCO | THF | 72 | 67 | >20:1 |
| 7 | D | K3PO4 | THF | 80 | 79 | >20:1 |
| 8 | D | Et3N | CHCl3 | 64 | 97 | >20:1 |
| 9 | D | Et3N | EtOAc | 68 | 99 | >20:1 |
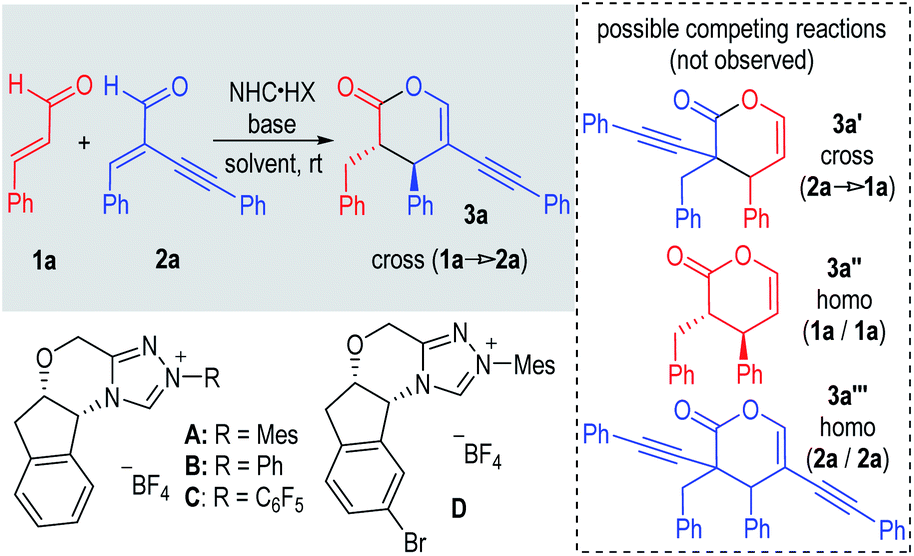
Our reactions are highly chemo-selective. Under all these conditions (Table 1), several possible side products were not formed. For example, possible adducts with enal 2a as the enolate precursor (to form 3a′ or 3a′′′) were not observed. This is not a complete surprise as α-substituted enals are unreactive azolium enolate intermediate precursors under NHC catalysis.11 Our results showed that mixing of enal 2a with highly reactive electrophiles (such as alkylidene diketone; see the ESI† for more details) did not lead to any formal [2 + 4] addition product. Interestingly, the simple enal 1a did not behave as a Michael acceptor under our conditions, as homo-coupling adduct 3a′′ was not observed. In Scheidt's elegant study, the introduction of a Lewis acid additive is necessary to activate one molecule of the enal to react as a Michael acceptor.8a
Our further control experiments showed that when the α-alkynl substituent of 2a was replaced with an alkyl (e.g., Fig. 2, 2a1), vinyl (2a2), phenyl (2a3) or cyano (2a4) unit, the corresponding cross [2 + 4] reactions were not observed, with most of the starting materials remaining unchanged (for more details, see the ESI†). It is clear that the alkynl unit present in enal 2a played more important roles than simply blocking the enal α-carbon to interrupt the π-conjugations. Although attempts to identify key intermediates (and possible non-covalent interactions) between the NHC catalysts and the two enals did not lead to conclusive mechanistic pictures, our experiments did show strong non-linear effects with respect to the optical purities of the NHC pre-catalyst and the reaction product (Fig. 3, see the ESI† for more details).
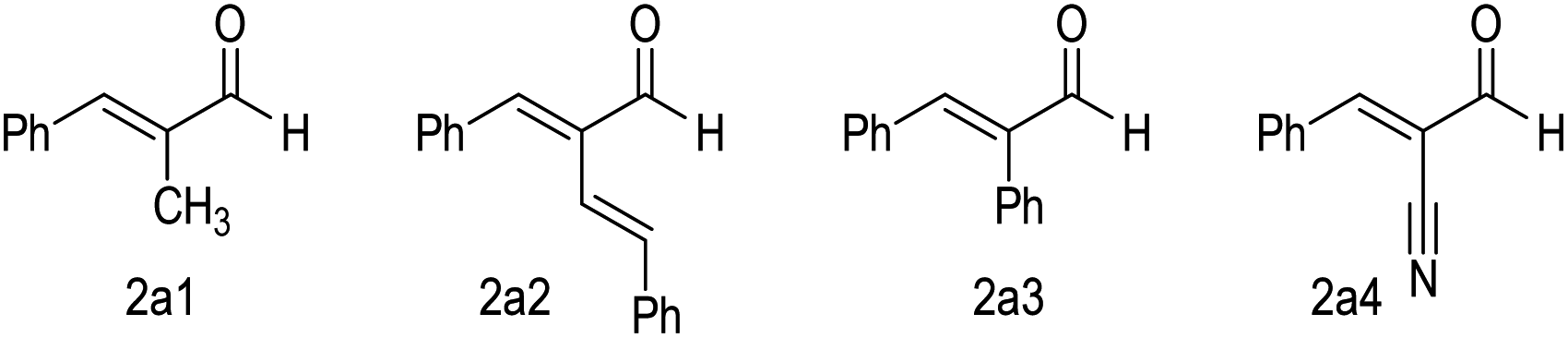 | ||
| Fig. 2 Unsuccessful α-substituted enal substrates for the NHC catalytic chemo-selective cross [2 + 4] reactions. | ||
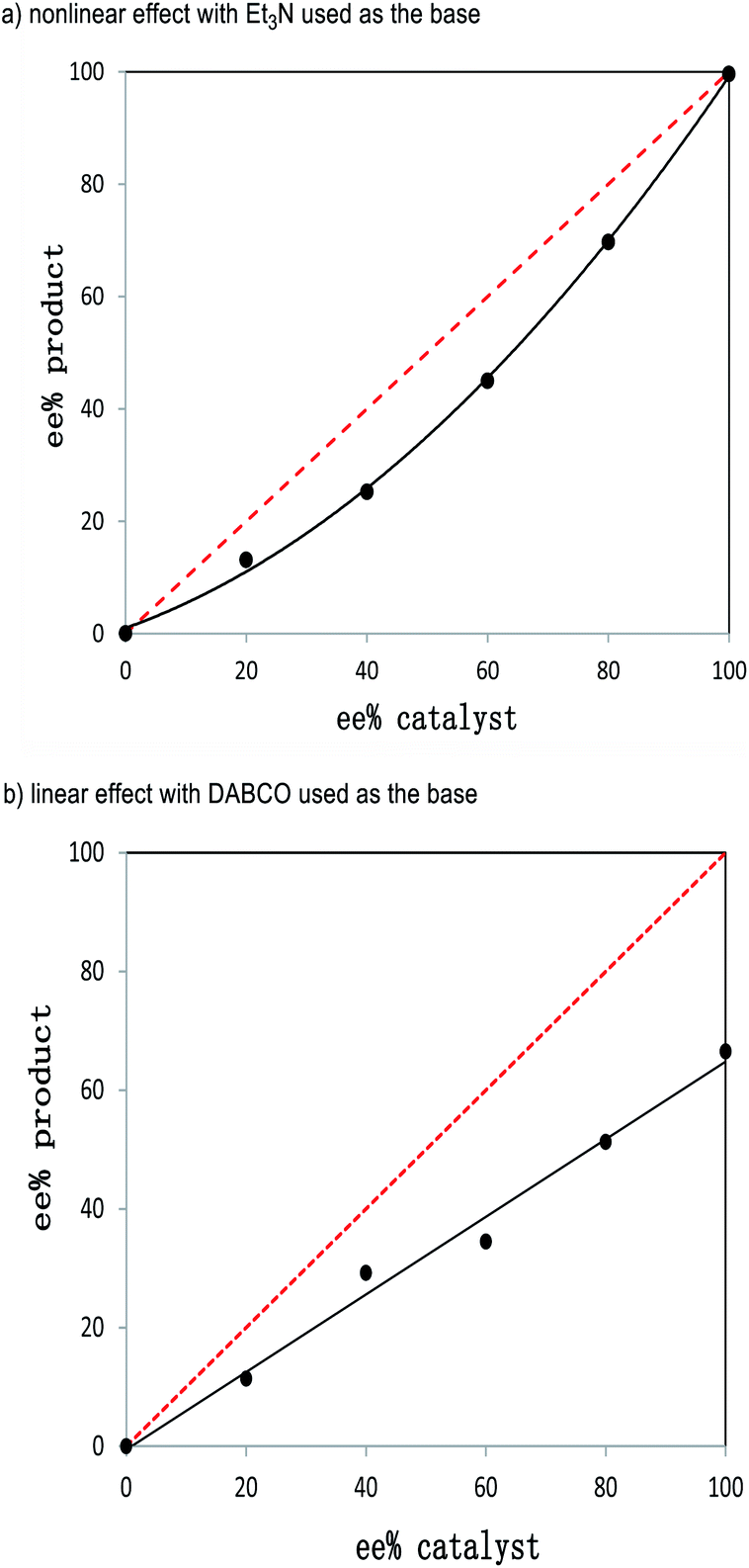 | ||
| Fig. 3 Nonlinear effects with respect to the product ee and the catalyst ee values using different bases: (a) Et3N and (b) DABCO. | ||
Specifically, the reaction of 1a and 2a was studied by varying the enantiomeric purities of the NHC pre-catalyst D under the optimized reaction conditions as indicated in Table 1, entry 4 (Fig. 3). The ee values of the products and the ee values of the catalysts showed an obvious negative nonlinear effect (Fig. 3a). This nonlinear effect suggests that at least two catalysts are involved in the enantio-differentiating step of our reaction.19 It appears both of the enals (1a and 2a) are activated by NHC and/or its salt (NHC·HX) in our formal [2 + 4] reaction. It is well established that cinnamaldehyde (1a) can be activated by NHC to form an acylazolium enolate intermediate.6 We therefore propose that the other enal (2a) bearing an alkynyl unit is activated by the acidic proton from NHC·HX via non-covalent interactions. These non-covalent interactions between 2a and NHC·HX could be further supported by the “linear-effect” shown by the ee values of the products and the catalysts when using DABCO as the base (Fig. 3b). In this case, only one catalyst was involved in the enantio-differentiating step of our reaction, since the non-covalent H-bonding interactions between 2a and NHC·HX could be broken by a stronger base (e.g., DABCO, K3PO4, see the ESI† for details) existing in the catalytic system. Similar activation of the α-alkynyl enal by a proton was proposed in Jørgensen's amine-catalyzed reaction.11 In the field of NHC related catalysis, the use of NHC·HX as a H-bond donating catalyst has been demonstrated by Huang, Scheidt, Guin, and others.13
The non-covalent interactions between the NHC pre-catalyst D and the alkynyl enal 2a can also be supported by 1H NMR analysis (Fig. 4). In the presence of the weak base Et3N, the acidic proton of the NHC pre-catalyst D shows an obvious change in the chemical shift after mixing with the alkynyl enal 2a (Fig. 4, a vs. b). Meanwhile, the chemical shift of the aldehyde proton of the substrate 2a is not changed in the same reaction system (a vs. c). These results support the existence of a non-covalent interaction between the NHC pre-catalyst D and the alkynyl enal 2a in our NHC organocatalytic reaction system (for more details, see the ESI†).
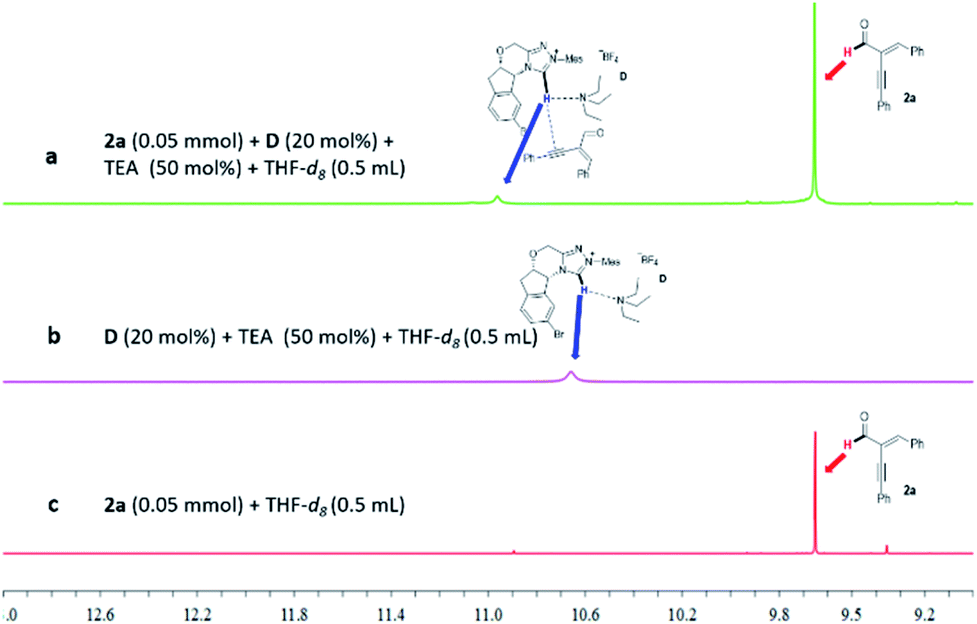 | ||
| Fig. 4 Chemical shift of the acidic proton of the NHC pre-catalyst D under various conditions. | ||
We then examined the substrate scope using different enals (1) to react with 2a under the optimized reaction conditions indicated in Table 1, entry 4 (Scheme 1). Substituents could be installed at each position of the phenyl ring of the cinnamaldehyde 1a, with all the products afforded in moderate to excellent yields with excellent chemo-, enantio- and diastereoselectivities (3b to 3p). The β-phenyl rings of the enal substrates (1) could also be switched to a naphthyl group or heteroaromatic groups. The corresponding products were afforded in excellent enantioselectivities, although the yields or dr values dropped in these cases (3q to 3s). To our delight, aliphatic enals could also be used as the enolate precursors for this NHC catalyzed chemoselective reaction, with the desired products afforded in moderate yields with excellent dr and ee values (3t & 3u).
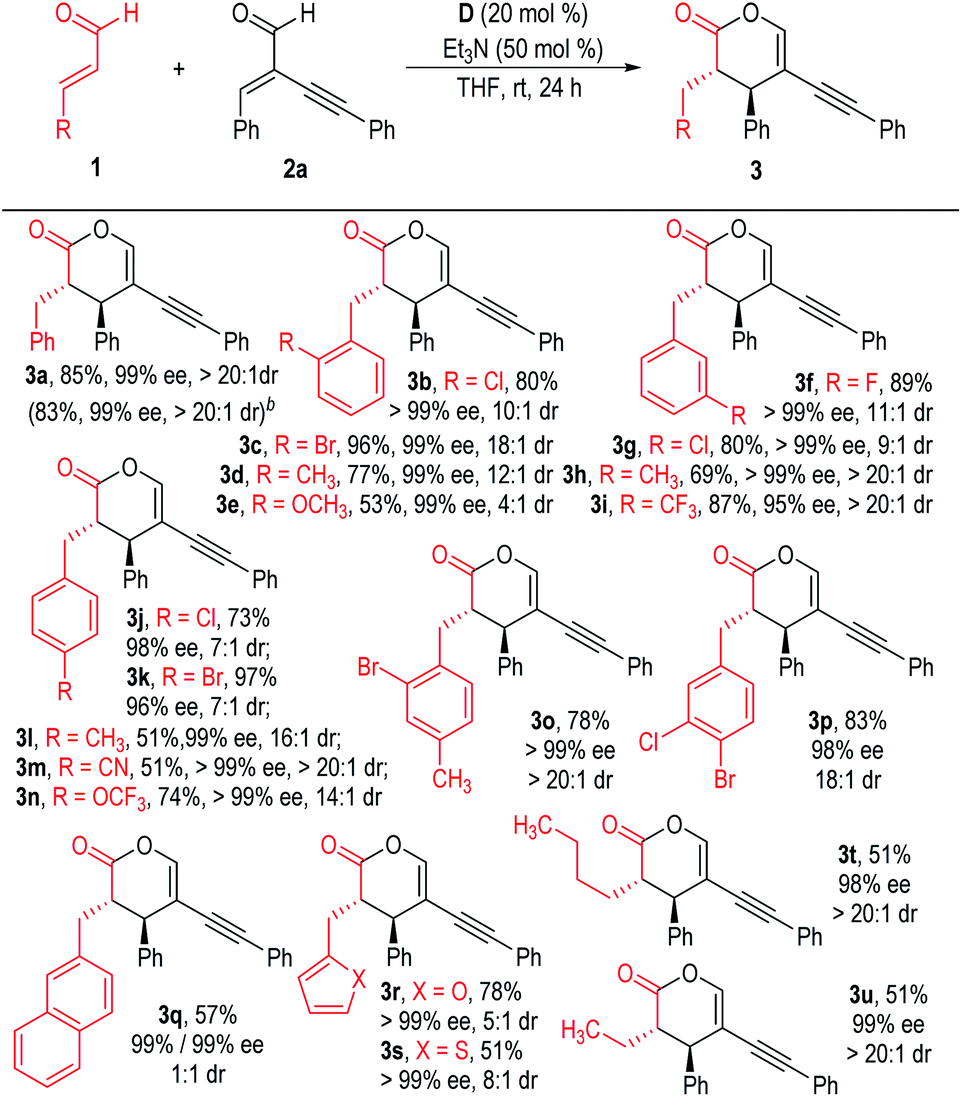 | ||
| Scheme 1 Scope of enals 1. aReaction conditions as stated in Table 1, entry 4. Yields are isolated yields after purification by column chromatography. Er values were determined via HPLC on a chiral stationary phase. bThe reaction was carried out on a 1.0 mmol scale based on 2a. | ||
The scope of the α-alkynyl enal substrates (2) was also examined (Scheme 2). Electron-donating substituents could be well tolerated on the β-phenyl rings of the α-alkynyl enals, with the desired products afforded in good yields with excellent ee values as single diastereomers (4a & 4b). The yields of the [2 + 4] products decreased when installing electron-withdrawing groups at any position of the β-phenyl rings, although the enantioselectivities were not affected (4c to 4f). The β-phenyl rings of the α-alkynyl enal substrates (2) could also be replaced with various heteroaromatic groups without obvious reduction in the product yields or stereoselectivities (4g & 4h). Substituents were also well tolerated on the phenyl rings attached to the alkynyl units of the enal substrates 2, with all the corresponding products afforded in moderate to good yields with excellent optical purities as single diastereomers (4i to 4p). Enal substrates 2 bearing heteroaromatic, aliphatic or terminal α-alkynyl groups also worked well in this reaction and gave the target products in moderate to good yields with excellent enantio- and diastereoselectivities (4q to 4w).
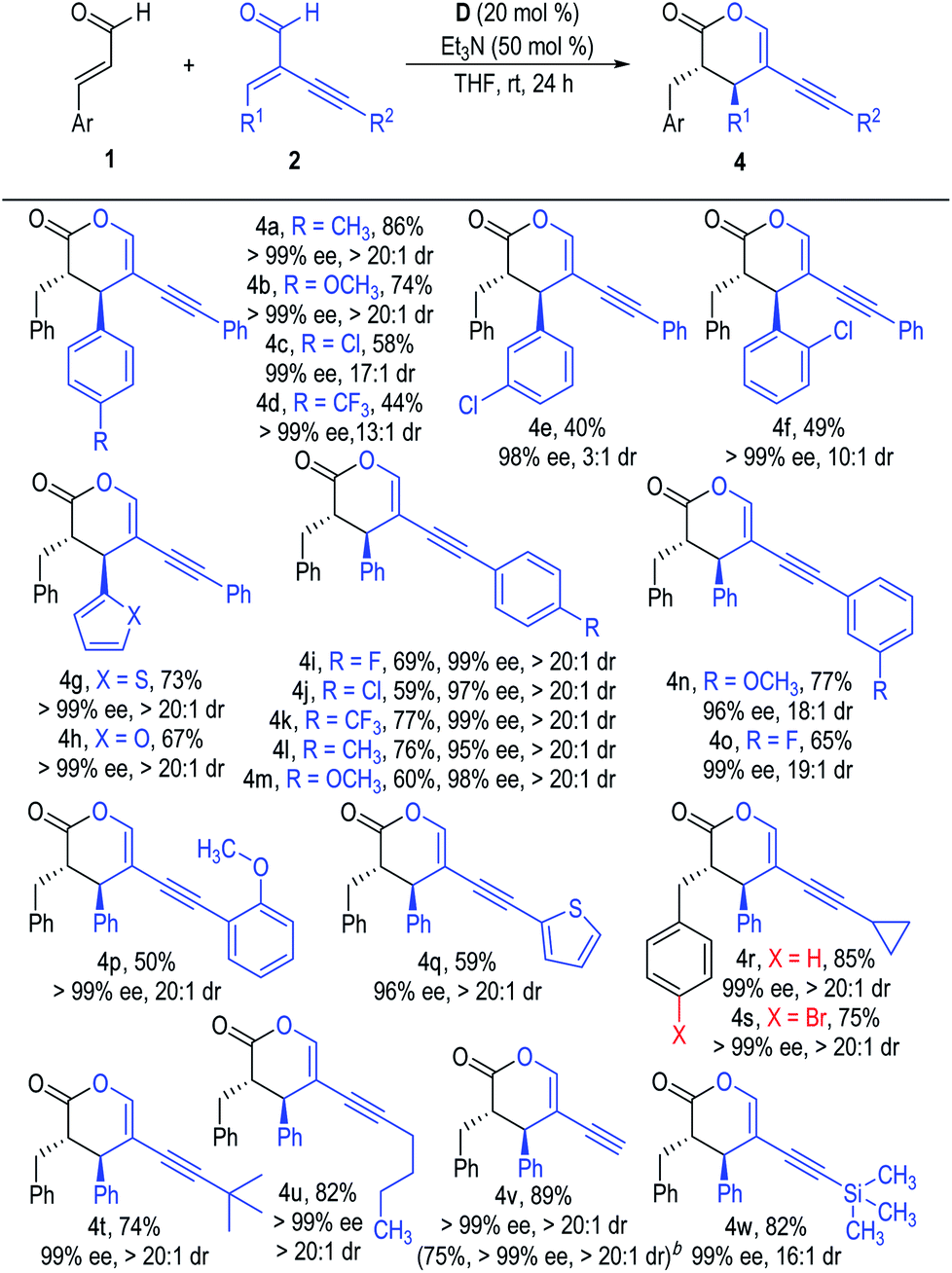 | ||
| Scheme 2 Scope of α-alkynyl enals 2. aReaction conditions as stated in Table 1, entry 4. Yields are isolated yields after purification by column chromatography. Er values were determined via HPLC on a chiral stationary phase. bThe reaction was carried out on a 6.4 mmol scale based on 2v (1.0 g). | ||
As a technical note, this chemo-selective reaction of α,β-unsaturated enals could be carried out on a large scale without reduction of the product ee or dr values, although the yields of the final products slightly dropped (e.g., Scheme 1, 3a & Scheme 2, 4v).
Having examined the reaction scope with both of the enal reactants, we next seek to get additional insights into the reaction mechanism. Hammett studies20 were carried out using alkynyl enal substrates 2 bearing various p-substituents on the phenyl groups of the alkynyl units (Fig. 5). Alkynyl enal substrates 2 bearing 4-F (2i), 4-Cl (2j), 4-CF3 (2k), 4-CH3 (2l), and 4-OCH3 (2m) groups were chosen as the target substrates to evaluate their relative reaction rates compared with the alkynyl enal 2a. Kinetic studies showed that the substrates 2 bearing electron-withdrawing groups reacted faster than those bearing electron-donating groups (Fig. 5a). The Hammett plot of the relative reaction rates of the substrates 2i to 2m gave a positive slope (ρ = 1.0128). Therefore, a negatively charged transition state should be built up in the rate determining step of this [2 + 4] cycloaddition process. This is in accordance with the non-covalent H-bonding interactions that we have proposed to exist between the acidic proton of the NHC-precatalyst D and the alkynyl unit of the enal substrate 2 (Fig. 1b, TS-I, see the ESI† for more details).
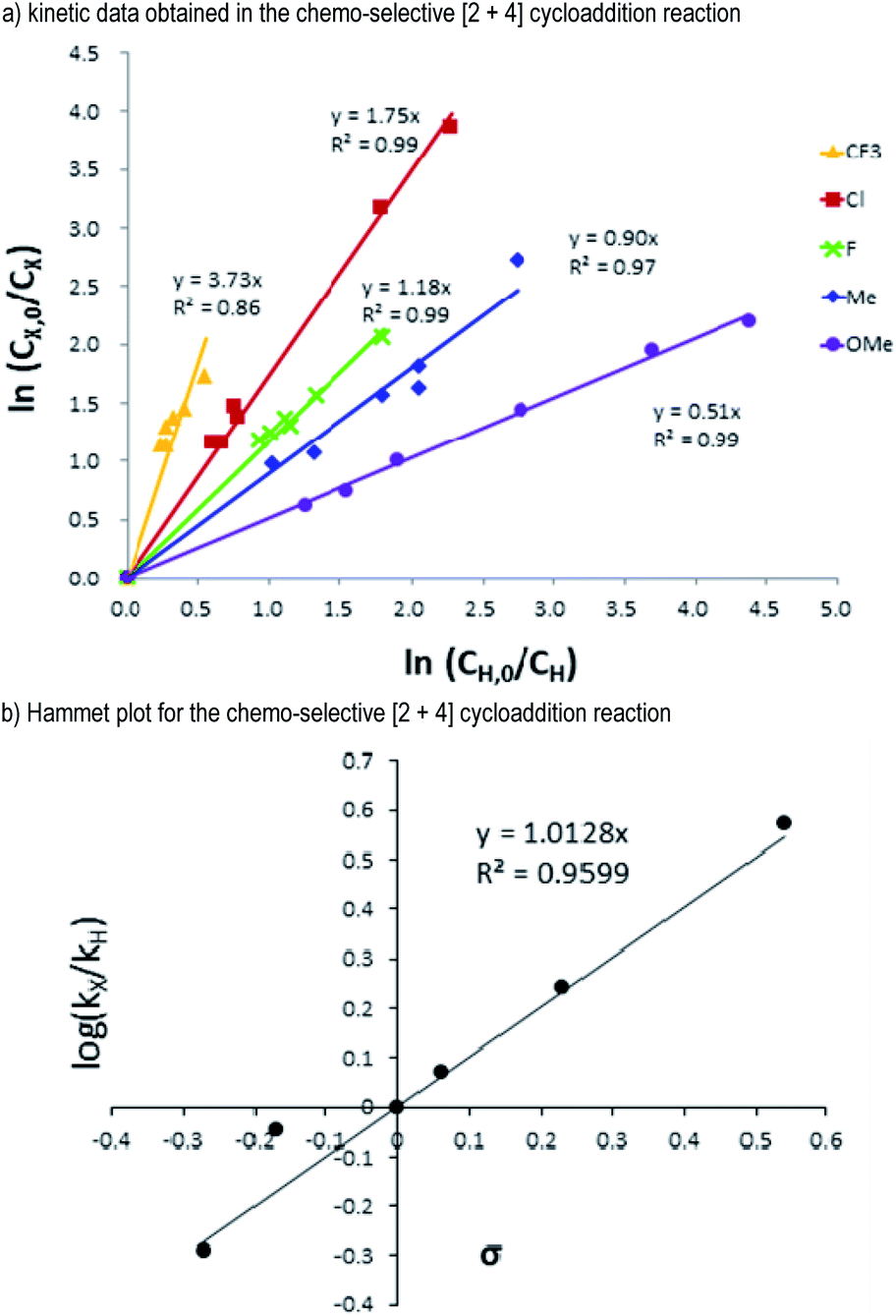 | ||
| Fig. 5 (a) Kinetic data and (b) Hammet plot for the competitive [2 + 4] cycloaddition reactions based on the σ values. | ||
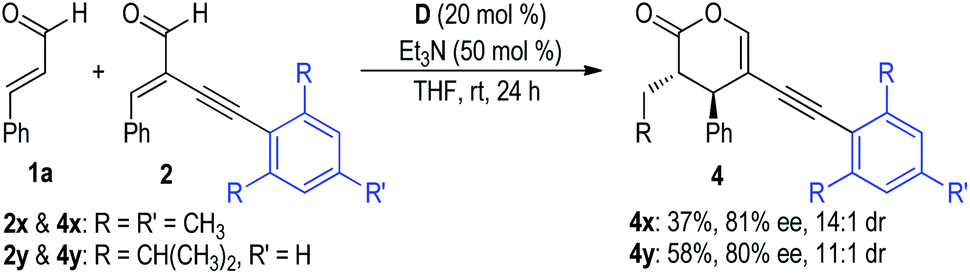
Additionally, substrates 2x and 2y bearing steric bulky substituted phenyl groups on the alkynyl units were further examined for this NHC dual catalytic [2 + 4] cycloaddition reaction (Fig. 6). It is not surprising that the corresponding reaction products 4x and 4y were only afforded in poor yields with moderate ee values. Because the alkynly groups of the substrates 2x and 2y were shielded by the bulky mesityl and 2,6-diisopropylphenyl groups, the H-bonding interactions between the NHC pre-catalyst D and the alkynyl groups cannot be efficiently formed in these cases.
 | ||
| Fig. 6 Reactions with enals 2 bearing bulky alkynyl substituents. | ||
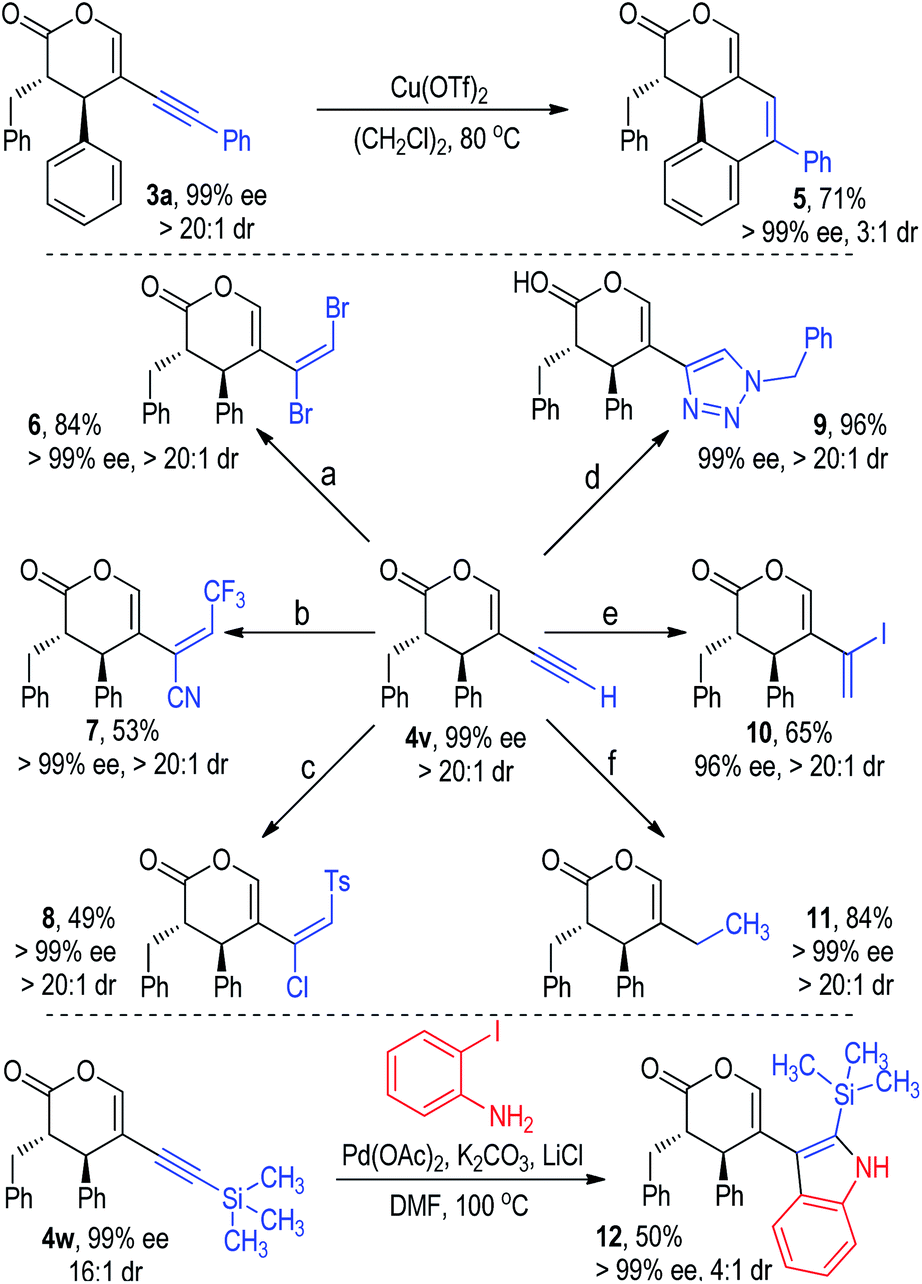
The chiral alkynyl pyranone products obtained from this methodology are rich in functionalities for further synthetic transformations (Fig. 7). For instance, the alkynyl group in 3a could react with the adjacent phenyl group under the catalysis of Cu(OTf)2 to give tricyclic product 5 in a good yield without reduction of the optical purity.21 The terminal alkylnyl group in 4v could participate in various addition reactions and afford a variety of multi-functionalized alkene products in moderate to excellent yields with excellent ee values as single diastereomers (e.g., 6, 7, 8, 10).22 A click reaction between the alkynyl group in 4v and benzyl azide led to the formation of the chiral triazole product 9 in almost quantitative yield with excellent optical purity as a single diastereomer.22d The ethynyl group in 4v could be selectively reduced to an ethyl group with a Pd/C and CaCO3 catalyst in a hydrogen atmosphere (11). Pyranone 4w bearing a 2-trimethylsilylethynyl group could be coupled with 2-iodoaniline to give the indole product 12 in a moderate yield and diastereoselectivity with an excellent ee value.23
 | ||
| Fig. 7 Synthetic transformations of the chiral pyranone products. aCuBr2, CH3CN, r.t.,1 h; bTogni reagent, TMSCN, Cu(OAc)2, terpyridine, CH3CN, 70 °C, 5 h; cTosNHNH2, FeCl3, TBHP, CH3CN, 80 °C, 8 h; dBnN3, sodium L-ascorbate, DCM/H2O (v/v = 1/1), r.t., 12 h; eNaI, TMSCl, H2O, CH3CN, r.t., 4 h; fPd/C, CaCO3, H2 (balloon), EtOH, r.t., 2 h. | ||
Conclusions
In summary, we have developed a chemo-selective intermolecular cross-coupling reaction of two different enals via a dual activation process. One of the enals is activated by NHC to form an acylazolium enolate intermediate. The other enal (α-alkynl substituted enal) is presumably activated by NHC·HX to behave as a Michael acceptor. Non-linear effects with respect to catalyst and product ee values suggest the involvement of more than one catalyst in the enantio-differentiation step of our reaction. Our reaction provides chiral lactones containing functionalizable 1,3-enyn units with excellent diastereo- and enantioselectivities. Synthetic transformations of our catalytic reaction products give a diverse set of functional molecules. Further studies in developing challenging chemo-selective reactions, as well as mechanistic studies, are being pursued in our laboratories.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (21772029, 21801051, and 21961006), The 10 Talent Plan (Shicengci) of Guizhou Province ([2016]5649), the Guizhou Province Returned Oversea Student Science and Technology Activity Program [(2014)-2], the Science and Technology Department of Guizhou Province ([2018]2802, [2019]1020), the Program of Introducing Talents of Discipline to Universities of China (111 Program, D20023) at Guizhou University, the Guizhou Province First-Class Disciplines Project [(Yiliu Xueke Jianshe Xiangmu)-GNYL(2017)008], Guizhou University of Traditional Chinese Medicine, Guizhou University (China), and the Singapore National Research Foundation under its NRF Investigatorship (NRF-NRFI2016-06), the Ministry of Education, Singapore, under its MOE AcRF Tier 1 Award (RG108/16, RG5/19, RG1/18), MOE AcRF Tier 3 Award (MOE2018-T3-1-003), the Agency for Science, Technology and Research (A*STAR) under its A*STAR AME IRG Award (A1783c0008, A1783c0010), GSK-EDB Trust Fund, and Nanyang Research Award Grant, Nanyang Technological University.Notes and references
- (a) A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307–1370 CrossRef CAS; (b) R. A. Shenvi, D. P. O'Malley and P. S. Baran, Acc. Chem. Res., 2009, 42, 530–541 CrossRef CAS; (c) I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 CrossRef CAS; (d) E. Roulland, Angew. Chem., Int. Ed., 2011, 50, 1226–1227 CrossRef CAS; (e) D. Kaiser, A. Bauer, M. Lemmerer and N. Maulide, Chem. Soc. Rev., 2018, 47, 7899–7925 RSC.
- For selected reviews, see: (a) A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef; (b) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570–1581 CrossRef CAS; (c) D. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS; (d) E. Arceo and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 5290–5292 CrossRef CAS; (e) P. Chauhan, S. Mahajan and D. Enders, Acc. Chem. Res., 2017, 50, 2809–2821 CrossRef CAS; (f) L. Klier, F. Tur, P. H. Poulsen and K. A. Jørgensen, Chem. Soc. Rev., 2017, 46, 1080–1102 RSC; (g) L. Zhu, D. Wang, Z. Jia, Q. Lin, M. Huang and S. Luo, ACS Catal., 2018, 8, 5466–5484 CrossRef CAS; (h) G. J. R. Rodríguez, N. M. Rezayee, A. V. Albalat and K. A. Jørgensen, Chem. Rev., 2019, 119, 4221–4260 CrossRef; (i) F. An, B. Maji, E. Min, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2020, 142, 1526–1547 CrossRef CAS.
- For selected recent reviews on NHC catalysis: (a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS; (c) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS; (d) D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 3, 53–57 RSC; (e) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS; (f) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (g) J. Douglas, G. Churchill and A. D. Smith, Synthesis, 2012, 44, 2295–2309 CrossRef CAS; (h) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686–11698 CrossRef CAS; (i) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617–1639 CrossRef CAS; (j) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC; (k) S. J. Connon, Angew. Chem., Int. Ed., 2014, 53, 1203–1205 CrossRef CAS; (l) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS; (m) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696–707 CrossRef CAS; (n) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC; (o) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS; (p) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS; (q) X. Y. Chen, Q. Liu, P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 3862–3873 CrossRef CAS; (r) Z. Wang, D. Pan, T. Li and Z. Jin, Chem.–Asian J., 2018, 13, 2149–2163 CrossRef CAS; (s) R. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2019, 58, 8628–8630 CrossRef CAS; (t) X.-Y. Chen, Z.-H. Gao and S. Ye, Acc. Chem. Res., 2020, 53, 690–702 CrossRef CAS; for a recent book on NHC organocatalysis, see: (u) A. T. Biju, N-Heterocyclic carbenes in organocatalysis, Wiley-VCH, Weinheim, Germany, 2019 Search PubMed.
- For pioneering studies on the generation of Breslow intermediates, see: (a) T. Ukai, R. Tanaka and T. J. Dokawa, Pharm. Soc. Jpn., 1943, 63, 296–300 CrossRef CAS; (b) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
- For pioneering studies on the NHC-catalyzed homoenolate activation of enals, see: (a) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS; (b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS.
- For pioneering studies on the NHC-catalyzed enolate activation of enals, see: (a) M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418–8420 CrossRef CAS; (b) C. Burstein, S. Tschan, X. Xie and F. Glorius, Synthesis, 2006, 14, 2418–2439 Search PubMed.
- For pioneering studies on the NHC-catalyzed oxidative LUMO activation of enals, see: (a) K. Zeitler, Org. Lett., 2006, 8, 637–640 CrossRef CAS; (b) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176–14177 CrossRef CAS; (c) S. D. Sarkar and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 9266–9269 CrossRef. For selected reviews, see: (d) C. Zhang, J. F. Hooper and D. W. Lupton, ACS Catal., 2017, 7, 2583–2596 CrossRef CAS; (e) S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, Acc. Chem. Res., 2019, 52, 425–436 CrossRef CAS.
- (a) D. T. Cohen, B. Cardinal-David, J. M. Roberts, A. A. Sarjeant and K. A. Scheidt, Org. Lett., 2011, 13, 1068–1071 CrossRef CAS; (b) V. R. Yatham, W. Harnying, D. Kootz, J. M. Neudörfl, N. E. Schlörer and A. Berkessel, J. Am. Chem. Soc., 2016, 138, 2670–2677 CrossRef CAS; (c) E. R. Miller, M. T. Hovey and K. A. Scheidt, J. Am. Chem. Soc., 2020, 142, 2187–2192 CrossRef CAS.
- For NHC catalytic reactions between aldehydes and enals, see: (a) S. R. Yetra, T. Kaicharla, S. S. Kunte, R. G. Gonnade and A. T. Biju, Org. Lett., 2013, 15, 5202–5205 CrossRef CAS; (b) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS; (c) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS; (d) S. S. Sohn and J. W. Bode, Org. Lett., 2005, 7, 3873–3876 CrossRef CAS; (e) R. Ide, R. Kyan, T. P. Le, Y. Kitagawa, K. Sato, N. Mase and T. Narumi, Org. Lett., 2019, 21, 9119–9123 CrossRef CAS.
- (a) T. Yao, X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 11164–11169 CrossRef CAS; (b) Y. Xiao and J. Zhang, Angew. Chem., Int. Ed., 2008, 47, 1903–1906 CrossRef CAS; (c) X. Yu, B. Du, K. Wang and J. Zhang, Org. Lett., 2010, 12, 1876–1879 CrossRef CAS.
- (a) S. S. Sohn and J. W. Bode, Org. Lett., 2005, 7, 3873–3876 CrossRef CAS; (b) A. Chan and K. A. Scheidt, Org. Lett., 2005, 7, 905–908 CrossRef CAS; (c) A. G. Kravina, J. Mahatthananchai and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9433–9436 CrossRef CAS.
- P. H. Poulsen, Y. Li, V. H. Lauridsen, D. K. B. Jorgensen, T. A. Palazzo, M. Meazza and K. A. Jørgensen, Angew. Chem., Int. Ed., 2018, 57, 10661–10665 CrossRef CAS.
- For a pioneering study using NHCs as chiral non-covalent organic catalysts, see: (a) J. Chen and Y. Huang, Nat. Commun., 2014, 5, 3437 CrossRef. For selected examples, see: (b) E. M. Phillips, M. Riedrich and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 13179–13181 CrossRef CAS; (c) J. Chen, S. Meng, L. Wang, H. Tang and Y. Huang, Chem. Sci., 2015, 6, 4184–4189 RSC; (d) L. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 15414–15418 CrossRef CAS; (e) S. Santra, A. Porey, B. Jana and J. Guin, Chem. Sci., 2018, 9, 6446–6450 RSC; (f) S. Lu, J.-Y. Ong, H. Yang, S. B. Poh, X. Liew, C. S. D. Seow, M. W. Wong and Y. Zhao, J. Am. Chem. Soc., 2019, 141, 17062–17067 CrossRef CAS; (g) S. Santra, U. Maji and J. Guin, Org. Lett., 2020, 22, 468–473 CrossRef CAS.
- (a) G. J. Hollingworth and J. B. Sweeney, Synlett, 1993, 7, 463–465 CrossRef; (b) I. J. S. Fairlamb, L. R. Marrison, J. M. Dickinson, F.-J. Lu and J. P. Schmidt, Bioorg. Med. Chem., 2004, 12, 4285–4429 CrossRef CAS; (c) K. Hyoungsu, L. Hyunjoo, L. Dongjoo, K. Sanghee and K. Deukjoon, J. Am. Chem. Soc., 2007, 129, 2269–2274 CrossRef; (d) Y. Zhou, Y. Zhang and J. Wang, Org. Biomol. Chem., 2016, 14, 6638–6650 RSC; (e) T. T. Talele, J. Med. Chem., 2020, 63, 5625–5633 CrossRef CAS.
- M. He, G. J. Uc and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 15088–15089 CrossRef CAS.
- M. S. Kerr, J. R. Alaniz and T. Rovis, J. Am. Chem. Soc., 2002, 124, 10298–10299 CrossRef CAS.
- M. S. Kerr and T. Rovis, J. Am. Chem. Soc., 2004, 126, 8876–8877 CrossRef CAS.
- C. Zhao, F. Li and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 1820–1824 CrossRef CAS.
- T. Satyanarayana, S. Abraham and H. B. Kaga, Angew. Chem., Int. Ed., 2009, 48, 456–494 CrossRef CAS.
- (a) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS; (b) E. Casper, J. Thomas, R.-R. Sergio and F. Peter, ACS Catal., 2013, 3, 294–302 CrossRef; (c) Y. Ayana, A. Yoshinori and C. Naoto, J. Org. Chem., 2014, 79, 11922–11932 CrossRef; (d) M. R. Brennan, D. Kim and A. R. Fout, Chem. Sci., 2014, 5, 4831–4839 RSC.
- Y.-L. Wang, W.-M. Zhang, J.-J. Dai, Y.-S. Feng and H.-J. Xu, RSC Adv., 2014, 4, 61706–61710 RSC.
- (a) J. Xiang, R. Yuan, R. Wang, N. Yi, L. Lu, H. Zou and W. He, J. Org. Chem., 2014, 79, 11378–11382 CrossRef CAS; (b) Y.-T. He, Q. Zhao, X. Wang, J.-Y. Liu, P.-F. Xu and Y.-M. Liang, Chem. Commun., 2015, 51, 13209–13212 RSC; (c) X. Li, X. Shi, M. Fang and X. Xu, J. Org. Chem., 2013, 78, 9499–9504 CrossRef CAS; (d) Q. Gu, H. H. A. Mamari, K. Graczyk, E. Diers and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 3868–3871 CrossRef CAS; (e) L. W. C. Lawrence and A. K. Yudin, Org. Lett., 2009, 11, 1281–1284 CrossRef; (f) Y. Kim and C.-J. Li, Green Synthesis & Catalysis, 2020, 1, 1–11 Search PubMed.
- W.-B. Liu, D. P. Schuman, Y.-F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 6867–6879 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1980067. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03297b |
| This journal is © The Royal Society of Chemistry 2020 |