
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorus-ylides: powerful substituents for the stabilization of reactive main group compounds
Abir
Sarbajna
 ,
V. S. V. S. N.
Swamy
and
Viktoria H.
Gessner
*
,
V. S. V. S. N.
Swamy
and
Viktoria H.
Gessner
*
Faculty of Chemistry and Biochemistry, Chair of Inorganic Chemistry II, Ruhr University Bochum, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: viktoria.gessner@rub.de
First published on 17th July 2020
Abstract
Phosphorus ylides are 1,2-dipolar compounds with a negative charge on the carbon atom. This charge is stabilized by the neighbouring onium moiety, but can also be shifted towards other substituents thus making ylides strong π donor ligands and hence ideal substituents to stabilize reactive compounds such as cations and low-valent main group species. Furthermore, the donor strength and the steric properties can easily be tuned to meet different requirements for stabilizing reactive compounds and for tailoring the properties and reactivities of the main group element. Although the use of ylide substituents in main group chemistry is still in its infancy, the first examples of isolated compounds impressively demonstrate the potential of these ligands. This review summarizes the most important discoveries also in comparison to other substituents, thus outlining avenues for future research directions.
Introduction
The isolation of reactive main group compounds has been a part of extensive research activities in recent years. These research endeavors are driven by efforts to gain a better understanding of chemical bonding and structure–reactivity relations as well as by the ability of such compounds to undergo bond activation reactions. This transition metal-like behaviour has been demonstrated by a variety of main group element species that offer the prospect of applications in homogeneous catalysis.1 While several key compounds in this chemistry were already isolated in the last decades of the 20th century,2 it was only with the beginning of the new millennium that their synthetic potential was recognized. Since then, reactive species, such as heavier alkenes and alkynes and low-valent compounds have been developed from curiosities to isolable and tunable species.3A remarkable breakthrough in this context was reported in 2005 by Power with the activation of dihydrogen by using digermyne 1 (Fig. 1a).4 The facile reaction was possible due to the special bonding situation in the Ge![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ge linkage. A high-lying HOMO and low-lying LUMO allowed a transition metal-like synergistic donation and acceptance of electron density to and from the H–H bond and thus its activation and splitting (Fig. 1b). Two years later, a similar strategy was employed by Bertrand and coworkers using cyclic alkyl(amino)carbenes (CAACs) such as 2.5 They demonstrated the importance of the substituents at the carbene carbon atom for tuning orbital energies. Since then, controlling the energy gap between a filled and vacant orbital has become a general strategy to tailor main group compounds for bond activations,6 which lately has led to the activation of rather inert bonds. As such, Aldridge and Goicoechea reported the reversible C–C bond activation of benzene by using aluminyl complex 3,7 while Braunschweig and coworkers accomplished N2 reduction by using the transient borylene 4.8
Ge linkage. A high-lying HOMO and low-lying LUMO allowed a transition metal-like synergistic donation and acceptance of electron density to and from the H–H bond and thus its activation and splitting (Fig. 1b). Two years later, a similar strategy was employed by Bertrand and coworkers using cyclic alkyl(amino)carbenes (CAACs) such as 2.5 They demonstrated the importance of the substituents at the carbene carbon atom for tuning orbital energies. Since then, controlling the energy gap between a filled and vacant orbital has become a general strategy to tailor main group compounds for bond activations,6 which lately has led to the activation of rather inert bonds. As such, Aldridge and Goicoechea reported the reversible C–C bond activation of benzene by using aluminyl complex 3,7 while Braunschweig and coworkers accomplished N2 reduction by using the transient borylene 4.8
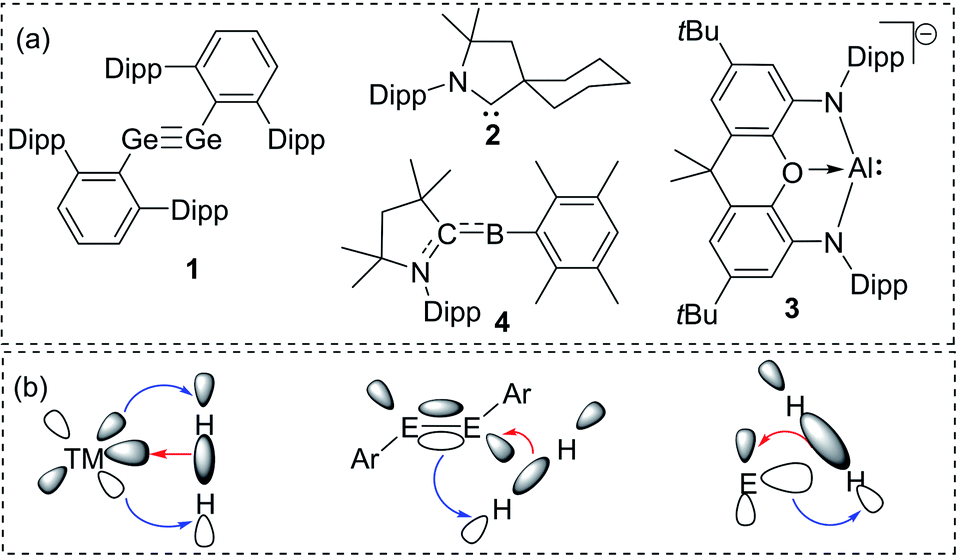 | ||
| Fig. 1 (a) Landmark examples of reactive main group compounds active in bond activations (dipp = 2,6-diisopropylphenyl); (b) H2 activation by using transition and main group element compounds. | ||
These examples impressively demonstrate the potential of reactive main group compounds, and also the importance of the molecular design, i.e. the choice of the substituents at the main group element for their isolation and applications. In this minireview, we focus on the use of phosphorus ylides as substituents in main group chemistry. Unlike traditional aryl or amido substituents, ylides only recently received renewed interest in main group chemistry, which already led to the discovery of unique properties and reactivities. Herein, we highlight important findings and bring them in context with other substituents used in main group chemistry. It must be noted that besides anionic substituents neutral donor ligands (e.g. NHCs and bisylides) have also been used to access reactive main group species and transition metal complexes.9 The use of such ligands is beyond the scope of this review but has been covered in excellent review articles.10
Substituents in main group chemistry
The isolation of reactive main group compounds would not have been feasible without the appropriate choice of substituents. While many remarkable developments have recently been made, further advances are certainly necessary to reach an advanced control of the stability and activities. In order to isolate reactive main group compounds, a balance of thermodynamic and kinetic stabilization is required. In general, large substituents are needed to shield the reactive center from decomposition and other unwanted transformations. Specifically bulky alkyl, aryl and amido groups are often employed,11 particularly with large silyl substituents as used by Lappert in the tetrylenes 5a and 5b12 or more recently by Yamashita in the aluminum anion 6 (Fig. 2).13 Likewise, ortho-substituted aryl groups – either attached to a substituent or as plain aryl groups – provide steric protection and like all bulky substituents further stabilization through London dispersion forces.14 The most popular aryl groups are mesityl (Mes), 2,6-diisopropylphenyl (Dipp) and terphenyl groups as pioneered by Power and coworkers (e.g. in 1).4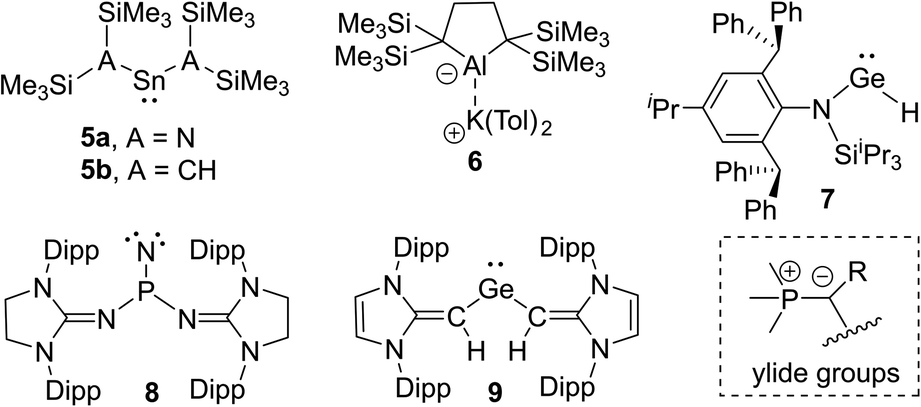 | ||
| Fig. 2 Examples of low-valent main group compounds with different substituents. | ||
The importance of increasing the steric bulk for reactivity control was impressively demonstrated by Jones and coworkers using the extremely bulky amido substituents of type N(Ar*)SiiPr3 (Ar* = 2,6-[C(H)Ph2]2-4-iPrC6H2) such as in 7. These substituents allowed the isolation of low coordinate species, including digermyne iPr3Si(Ar*)NGeGeN(Ar*)SiiPr3, which activates H2 at temperatures as low as −10 °C.15 Because of the extreme bulk of the amide the hydrido-digermene exists in equilibrium with its monomeric metal(II) hydride 7, which catalyses hydroboration reactions.16 A very recent example of the power of steric protection for stabilizing reactive species was reported by Bertrand and coworkers. The careful design of bulky benzo[c]pyrrolidino heterocycles enabled the stabilization and isolation of the first monosubstituted carbene.17
Besides kinetic stabilization, thermodynamic stabilization using ligands with the “right” electronic properties is crucial to access reactive species. A large variety of different substituents have been applied, including special substituents with particular properties, e.g. boryl6d,18 and even metallo substituents.19 For stabilizing low-valent and electron-deficient compounds strong σ- and π-donors are particularly well suited such as aryl or amido groups, but also other σ- and π-donor substituents have emerged in recent years. Prominent examples are N-heterocyclic imines20 and their carbon21,22 and phosphorus23 derivatives. Due to the contribution of an ylidic form to their electronic structure they are strong donors and thus allowed the isolation of a series of reactive main group compounds, e.g. phosphino nitrene 824 and tetrylenes,25 such as germylene 9.26
Phosphorus ylides (Fig. 2), such as the parent methylenetriphenylphosphorane Ph3PCH2, are dipolar compounds with a carbanionic center directly bound to a phosphonium moiety. Due the lone pair at the carbon atom they can act as π-donor substituents, suitable for stabilizing electron-deficient compounds. The R group in the ylide backbone allows the tuning of the steric and electronic properties and hence an advanced control of the reactivity of the main group species. The electronic structure of ylides has been vividly discussed over the years. At first, it was mostly described by two canonical structures – ylene A′ and ylide A (Fig. 3). However, computational studies have shown that the contribution of the ylenic structure A′ is minimal, as it requires (d–p)π interaction with d-orbitals at phosphorus, which are however too high in energy.27 Recently, the canonical structure A′′28 with a donor–acceptor interaction between phosphorus and carbon has found renewed interest. This structure describes ylides as phosphine-stabilized carbenes. An analogous description has also been used for bisylides, particularly carbodiphosphoranes (CDPs).29 However, recent studies showed that in metallated ylides the ionic ylidic structure is more important,30 so this description will be used throughout this review. Despite this unique electronic structure of P-ylides, their use as substituents in main group chemistry is still in its infancy. However, with the recent gram-scale isolation of alkali metal precursors broader applications have opened up.
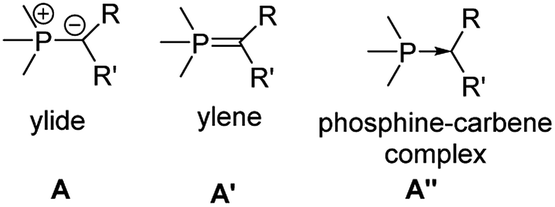 | ||
| Fig. 3 Bonding situation in ylides. | ||
Alkali metal yldiides
Since the first synthesis of ylides over a century ago and their use in Wittig type reactions, this class of compounds has been widely utilized in organic synthesis. In contrast, their metallated congeners, so-called yldiides, have for a long time remained unexplored, despite being ideal reagents to introduce ylide substituents via simple salt metathesis. Schlosser and Corey were the first who synthesized lithium yldiides.31 However, neither any isolation nor structure elucidation was reported probably due to the high sensitivity of these compounds. Until today, only a few alkali metal yldiides have been isolated. In general, strongly electron-withdrawing groups facilitate their preparation via direct metallation of an ylide with strong alkyl or amide metal bases and the isolation of the yldiide. It is noteworthy that in general the number of metal complexes with an yldiide ligand is rather limited, probably also a consequence of the few readily available yldiide precursors. Nonetheless, complexes with mid- and late transition metals and actinides have been reported.32The first isolation of an alkali metal yldiide was accomplished by Bestmann with the synthesis of the cyanido-functionalized sodium yldiide 10.33 Spectroscopic and reactivity studies showed that due to the stabilization of the negative charge by the nitrile functionality both canonical structures 10a and 10b contribute to the electronic structure of the yldiide (Scheme 1). XRD analyses of different alkali metal salts of 11 were later reported and revealed an astonishing structural diversity varying from coordination polymers to monomers depending on the alkali metal and the use of crown ethers as additional ligands.34 The first structure elucidations were reported in the 1990s. Bertrand and coworkers reported the silyl-substituted lithium yldiide 12 which was uniquely synthesized by a 1,2-carbometalation of phosphino(silyl)carbene 11 with n-BuLi.35 In the solid state (Fig. 5), 12 formed a monomeric structure with the lithium atom coordinated by two THF molecules and the ylidic carbon atom, which featured a planar geometry. The short P–C and Si–C bonds indicated the strong stabilization of the negative charge by the silyl and phosphonium groups. Similar observations were made by Niecke with phosphoranylidene ylides 13a and 13b.36 This bond shortening is characteristic of yldiides and the charge accumulation on the ylidic carbon atom. Therefore, the P–C bond length can serve as a measure for the charge concentration and also transfer to other groups.
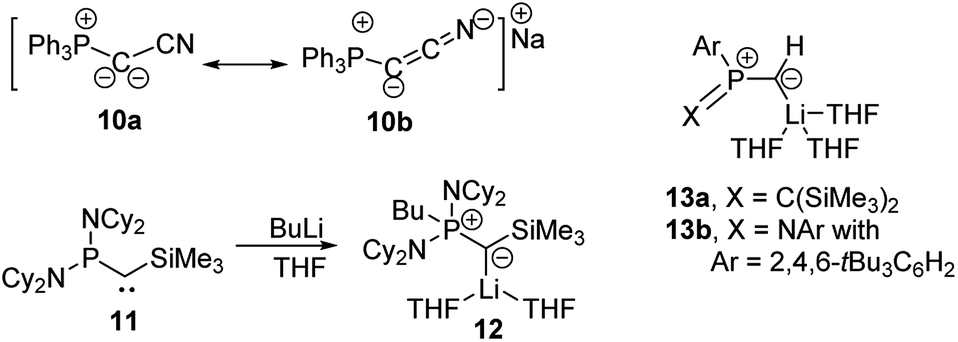 | ||
| Scheme 1 Formation of yldiides 11 and 12 by carbolithiation and metalation and the structure of lithium yldiides 13. | ||
In 2015, our group reported a sulfonyl substituted yldiide 14 which could be prepared on a gram scale by deprotonation of the phosphonium salt 14-H2 (Fig. 4).3714 exhibited a remarkable stability in solution and could be stored for weeks under an inert atmosphere, thus representing an ideal reagent for ylide transfer (see below). The high stability is due to the strong stabilization of the negative charge by the sulfonyl and phosphonium group. While the sodium salt of 14 featured a dimeric structure with two (NaO)4 cubes connected via one common face (Fig. 5), the potassium analogue formed a monomer with 18-crown-6 as an additive. Again, a decrease in the bond lengths was seen in the P–C–S linkage of 14 relative to 14-H2 and 14-H, owing to the increased coulombic interactions with the increased charge on the ylidic carbon atom. Computational studies confirmed the presence of two lone pairs at the carbon atom, one of σ (HOMO−1) and one of π (HOMO) symmetry, thus confirming the strong basicity of 14 and its ability to act as a σ and π donor.
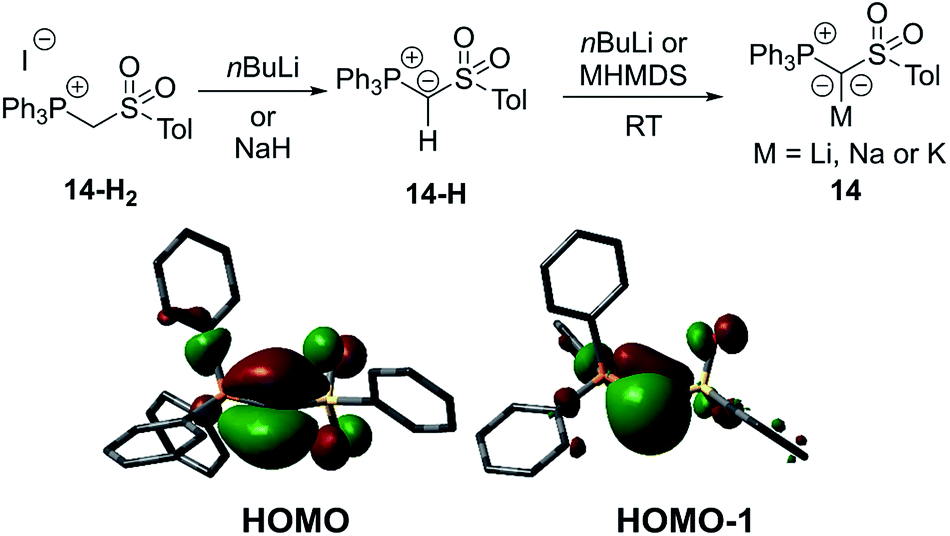 | ||
| Fig. 4 (top) Synthesis and (bottom) the HOMO and HOMO−1 of 14. | ||
 | ||
| Fig. 5 Structures of the lithium yldiide 12 and the sodium salt of 14. | ||
Besides 14 also its PCy3 analogue was recently reported. Introduction of the PCy3 group led to a remarkably reduced acidity of the ylide precursor, thus requiring stronger metal bases (benzyl potassium) for metallation.38 Nonetheless, yldiide 15 was isolable on a gram-scale (Fig. 6). A sulfinyl-stabilized yldiide was reported by Maerten, Baceiredo and coworkers.39 Lithium yldiide 16 was synthesized by deprotonation with n-BuLi and showed a dimeric structure with a Li2O2 core and decreased P–C and C–S bond lengths owing to the increased negative character at the ylidic carbon center.
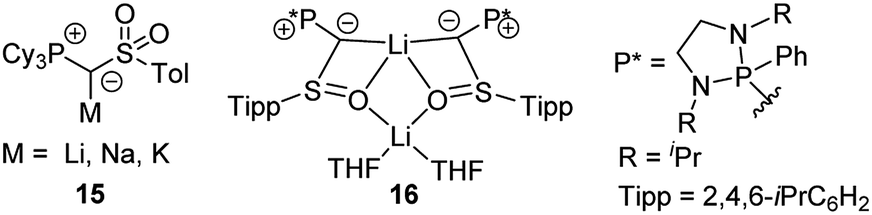 | ||
| Fig. 6 Metalated ylides 15 and 16. | ||
Besides structure elucidation by XRD analysis, phosphorus yldiides can easily be identified by 31P NMR spectroscopy. Upon metalation, a distinct high-field shift of the 31P NMR signal relative to that of the ylide precursor is usually observed. For example, an upfield shift from 14.2 ppm for 14-H to δP = −11.1 ppm for the sodium salt of 14 has been observed. Likewise, the 1JPC coupling constant significantly decreases due to the increased p-character in the P–C linkage, e.g. by ΔJ = 40 Hz in case of the cyano-yldiide 1034 and even by ΔJ = 121 Hz for sulfoxide 16.39
Ylide-stabilized group 13 compounds
As strong donor substituents ylides are supposedly well suited for stabilizing electron-deficient compounds. With their inherent electron-deficiency group 13 compounds should thus be ideal target molecules. Indeed, Bestmann reported the synthesis of ylide-functionalized boranes 17 in the late 1980s by reaction of alkylidenephosphoranes with chloroboranes (Scheme 2).40 Also, the synthesis of the ylide-substituted lithium borate 18 was described, but no further reactivity studies were reported. Breher and coworkers however, recently disclosed an FLP-type reactivity of sterically uncumbered α-borylated ylides, which allowed their application in the activation of small molecules such as CO2 or NH3 and thus suggests a more versatile reactivity of these compounds.40c,d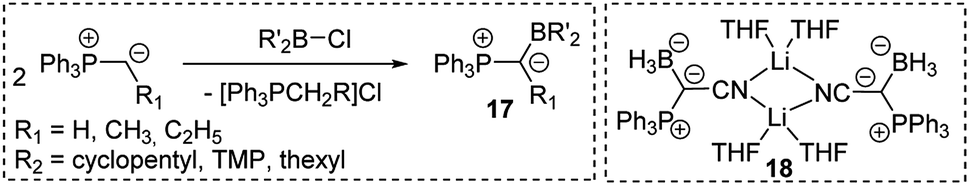 | ||
| Scheme 2 Synthesis of ylide-substituted boranes (thexyl = –C(CH3)2–C(CH3)2H; TMP = 2,2,6,6-tetramethylpiperidino) and the structure of 18. | ||
The ability of ylide substituents to stabilize electron-deficient species was finally proven by means of yldiide 14. The reaction of 14 with BH3·THF delivers the di(ylide)borane 19 which yields the first ylide-stabilized boron cation 20 by hydride abstraction using trityl salts (Fig. 7). The stabilizing effect of the ylide groups was reflected in the high stability of 20 even in boiling toluene and up to 200 °C in the solid state. The molecular structure of the PF6− salt revealed that one of the sulfonyl groups of the ylide ligands coordinates to the boron atom, which is in plane with the two ylide ligands (S(P)C)2B. The short B–Cylide distances (viz. 1.481(7) and 1.510(9) Å) indicated significant π donation from the ylide center. This observation was further reiterated by longer P–C and S–C bonds in the ylide due to the decrease in the negative charge on the ylidic carbon atom. DFT studies confirmed the π interaction, but also a high polarity of the C–B–C linkage as reflected by the Wiberg bond indices, the calculated charges and the frontier orbitals. Consistent with the highly electrophilic boron center 20 reacts with a range of Lewis bases to form the corresponding adducts and with KF to form the corresponding fluoroborane. The high C–B bond polarity is expressed in the reactivity towards primary and secondary amines, which leads to the formation of tris(amino)boranes by N–H activation and subsequent cleavage of the B–C bond.
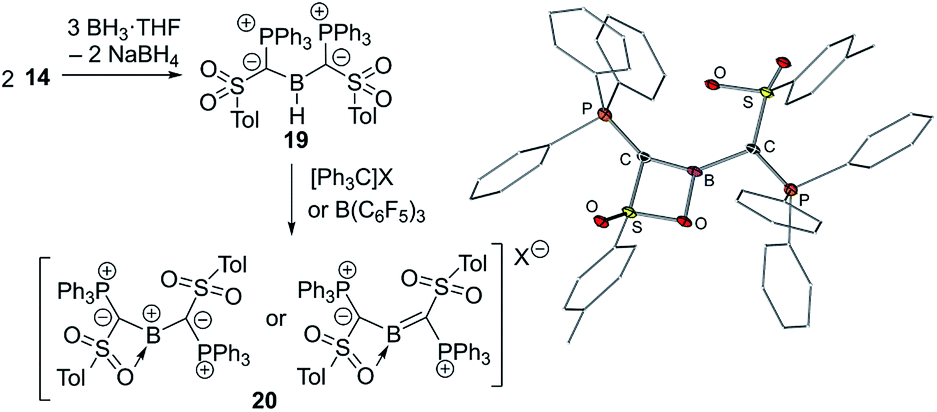 | ||
| Fig. 7 Synthesis of the di(ylidyl)boron cation 20 and its molecular structure (X = [B(3,5-Cl2C6H3)4], [B(3,5-(CF3)2C6H3)4], [B(C6F5)4] or PF6). | ||
Besides 20 no further low-valent group 13 compounds with ylide substituents have been reported to date. However, the potential of ylide substituents in this chemistry was picked up in recent computational studies by Phukan and coworkers.41 They suggested that the use of ylide substituents leads to an improved stability of borylenes and thus could be a promising strategy to access these reactive species.
Ylide-stabilized group 14 compounds
Cyclic amino(ylide)carbenes (CAYCs) (for example 22) have been known for some time, but only stabilized in the coordination sphere of transition metals.42 These complexes were synthesized by cyclization of transition-metal isocyanide complexes and thus limited to few examples. As the donor ability of an ylide exceeds that of an amino substituent, CAYCs were expected to exhibit increased electron-releasing capacities compared to NHCs. In 2008, Kawashima reported the first attempts to isolate a CAYC derived from phosphonium salt 21 (Scheme 3).43 Although the free carbene 22 could not be isolated even at low temperatures, trapping reactions and transfer to transition metals confirmed its formation. IR spectroscopy of the complex [22·Rh(CO)2Cl] proved the superior donor strength of 22 compared with diamino or alkyl(amino)carbenes. This was also confirmed by Fürstner by means of a series of sulphur- and phosphorus-ylide functionalized CAYCs44 and by computational studies.45 Despite several further attempts no stable ylide-substituted carbene has been reported so far.46 This lack is probably due to the high nucleophilicity, which results in decomposition pathways, such as by transfer of aryl substituents at the phosphonium group (e.g. to 23) or intramolecular deprotonations in the α- or β-position of the onium group.47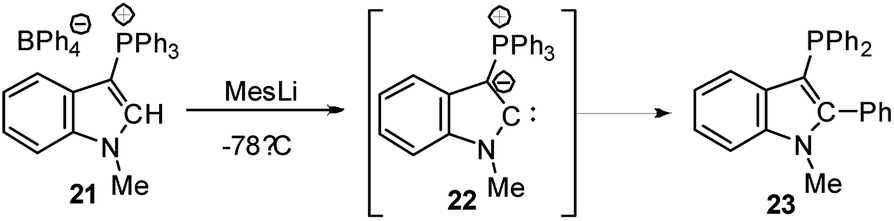 | ||
| Scheme 3 Attempted isolations of CAYC 22. | ||
The unique properties of ylide-substituted tetrylenes have impressively been demonstrated by means of heavier carbene analogues. Driess and coworkers reported the synthesis of the cyclic diylidesilylenes 25 by treatment of α,α′-dibromo-ortho-xylene in the presence of KHMDS (HMDS = hexamethyldisilazide) with SiBr4 (Scheme 4) and subsequent reduction of the dibromosilane 24 with KC8 or Jones' Mg(I) reagent.48,49 Although 25b could not be structurally characterized, solutions of the silylene were stable up to three months. The 29Si NMR signal of 25a (δSi = 213.3 ppm) and the signal of the ylidic carbon atom (approx. 90 ppm) appeared to be significantly downfield shifted compared to those of NHSis and carbanionic compounds, respectively. This was explained by DFT calculations, which revealed an aromatic character and an electron-rich silicon center in 25 as reflected by the resonance structure 25B. The cyclic di(ylide)stannylene analogous to 25a was already reported by Schmidpeter in 1998 by the reaction of the diylide precursor with Sn(HMDS)2 (4). It also featured a monomeric structure in solution with a 119Sn NMR signal appearing as a triplet at δ = 880.0 ppm.50
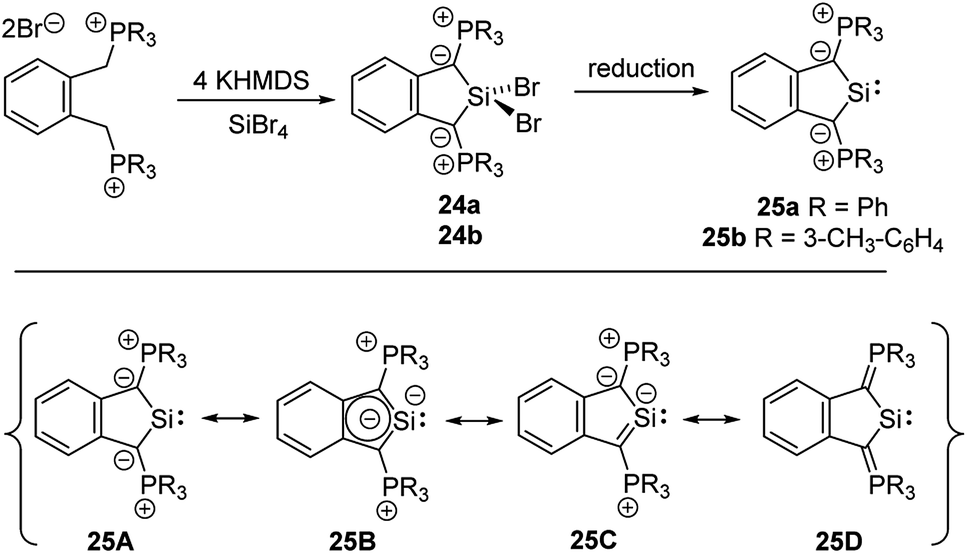 | ||
| Scheme 4 Preparation of cyclic di(ylide)silylenes 25 and their canonical structures. | ||
A further stability increase compared to that of diylidesilylenes 25 was realized by Kato and co-workers by synthesizing a cyclic amino(ylide)silylene. 27 was accessible by reduction of dichlorosilane 26 with elemental potassium in toluene at 80 °C (Scheme 5).5129Si NMR spectroscopy of 27 showed a resonance at δSi = 202.2 ppm, whereas the ylide carbon signal appeared as a doublet at 137.7 ppm in the 13C NMR spectrum, thus suggesting a strong π donation from the ylide to the divalent silicon centre. This was confirmed by the short Si–C bond length [1.798 (2) Å] in the solid-state structure, which featured a planar six-membered ring and an increased Si–N bond length compared to that of diaminosilylenes. Thus, it was concluded that 27 is best described by the canonical structure 27B with a π interaction between the ylide and the silicon center. Accordingly, the silylene exhibited a stronger donor ability compared to NHSis and even NHCs and for example readily reacted with P4 at room temperature through silylene insertion into a P–P bond to give the SiP4 cage 28. Moreover, the high electron density at the silicon atom was used to access the first base-free silanone 29 by simple reaction of 27 with N2O.5229 was isolable at −50 °C, but dimerised upon warming. Structure elucidation again revealed a short C–Si bond due to the strong π donation from the ylide, which favours a canonical structure with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si double bond and thus leads to the possible isolation of the silanone.
Si double bond and thus leads to the possible isolation of the silanone.
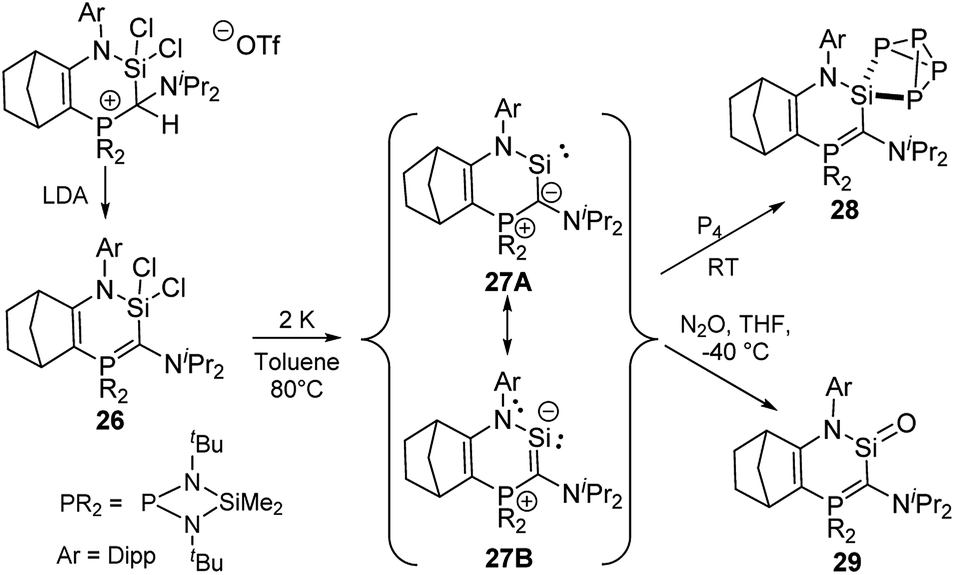 | ||
| Scheme 5 Synthesis of amino(ylide)silylene 27 and its reactivity towards P4 and N2O. | ||
Germylenes 31 analogous to 27 could also be isolated (Scheme 6), but via an unusual isomerization of the phosphine-stabilized germyne 30 which was generated by photolysis of a diazo precursor.53 Both germylenes 31 were stable at room temperature, but no further reactivity studies were reported. Kato, Baceiredo and co-workers further exploited the potential of ylidic donor substituents by studying variants of 27 and 31 with phosphorus and boron instead of carbon as donor atoms.54 This led to a further increase in the electron density at the group 14 elements. Consequently, silylene 32a is a stronger donor than 27 and allowed the generation of the corresponding silanone that exhibited a higher stability than 29.55 Germylene 33a showed an interesting multi-site reactivity. While the neutral Lewis acid B(C6F5)3 preferred the coordination to the germanium centre, the silylium cation SiEt3+ favoured the binding to the phosphorus atom. This bifunctional activity could also be applied in the hydrosilylation of carbon dioxide with triethylsilane using the B(C6F5)3 adduct as the catalyst.56
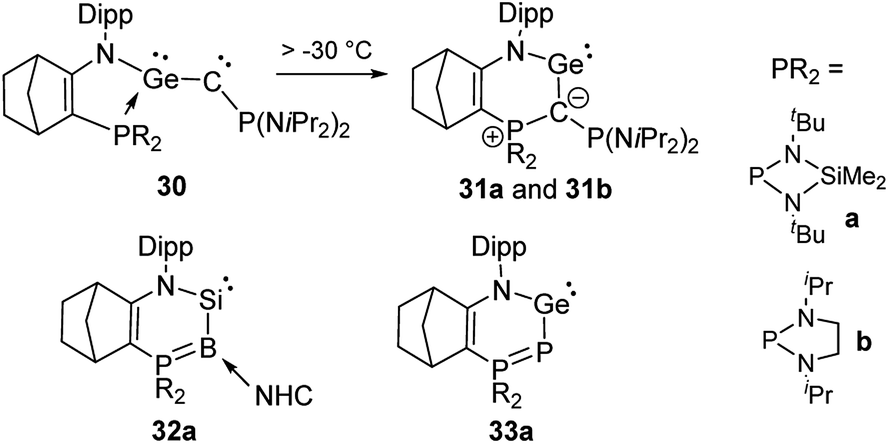 | ||
| Scheme 6 Preparation of cyclic tetrylenes with varying donor atoms. | ||
In 2019, our group reported the first acylic diylide-substituted stannylene and germylene formed by simple reaction of yldiide 14 with half an equivalent of GeCl2·dioxane and SnCl2, respectively (Fig. 8).57 The ease of this synthesis demonstrated the utility of group 1 yldiides in accessing ylide-functionalized main group species. XRD analyses revealed unusual structures of both compounds. In contrast to all other ylide-functionalized tetrylenes, the ylide groups (P–C–S plane) in 34 and 35 arranged perpendicularly to the C–E–C linkage, thus preventing any π-donation from the ylide to the empty p-orbital at Sn/Ge. This results in an unusual bonding situation, in which three lone pairs at the two carbon atoms and the central element are in plane and located next to each other (Fig. 8). Accordingly, the Sn–C and Ge–C bond lengths (2.23 and 2.04 Å, respectively) were found to be in the range of single bonds. This unusual structure was explained by the ability of the sulfonyl group to stabilize the negative charge on the ylidic carbon atom and its coordination to the metal, which demonstrates the impact of the substituents in the ylide backbone on the electronic properties and thus on the reactivity of the tetrylene. A comparison of the HOMO–LUMO energies and calculated Tolman electronic parameter (2032.3 cm−1) of germylene 34 with those of other acyclic germylenes suggested that 34 is the germylene with the highest donor capacity reported so far. The same holds true for stannylene 35. While the stannylene is stable in solution at room temperature, 34 showed limited stability and undergoes C–H activation of one of the PPh3 phenyl groups to form cyclotetragermane 36. DFT studies suggested that the C–H activation occurs across the Ge–C linkage, implying a bifunctional reactivity of ylide-substituted tetrylenes.
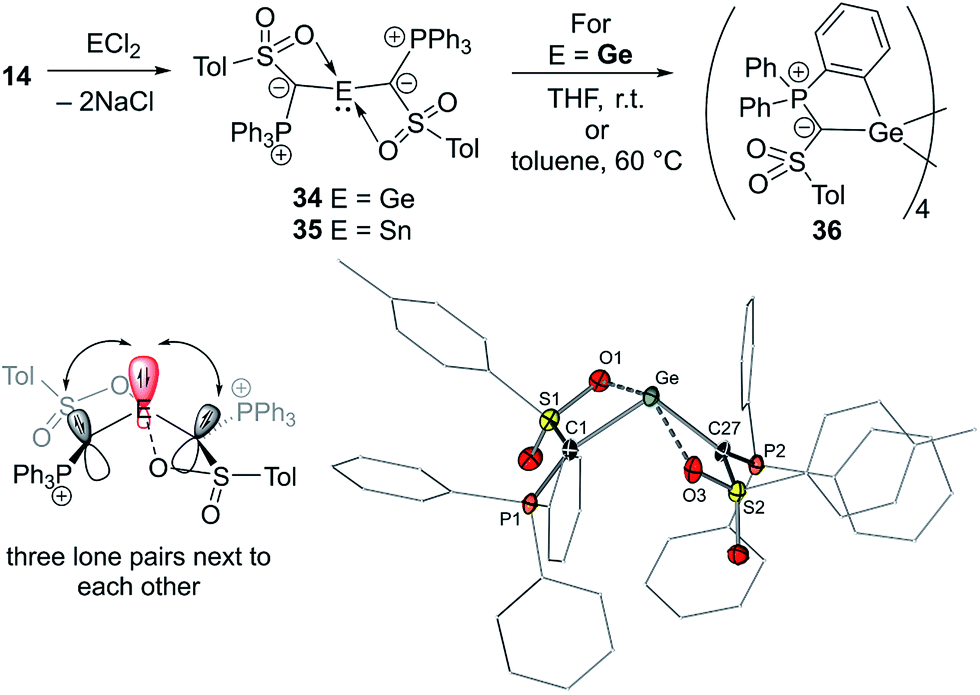 | ||
| Fig. 8 (Top) Synthesis of the diylide germylene and stannylene 34 and 35. (Bottom left) Bonding situation in 34 and 35 and (bottom right) the molecular structure of 34. | ||
Ylide-stabilized group 15 compounds
Ylide-functionalized phosphorus compounds have been intensively studied in the 1990s particularly by Schmidpeter and coworkers who for example focused on the impact of ylide substitution in halophosphines.58 Due to the π-donation (negative hyperconjugation),59 ylide substituents were found to cause a marked polarization of the P–Hal bond. Depending on the other substituents this polarization even results in the spontaneous dissociation of the halide to form phosphenium cations of type 38 (Fig. 9).60 Spontaneous dissociation was for example observed in diylide and amino(ylide) functionalized systems. For all other halophosphines, elongated P–Cl bonds were observed, and the halide could easily be abstracted e.g. by addition of AlCl3. Crystallographic studies of YPHal2 revealed that one of the P–Cl bonds is roughly perpendicularly arranged to the P–C–(R)–P plane which leads to a parallel orientation of the pz orbital at the ylidic carbon atom and the σ* orbital of the P–Cl bond and hence results in an effective charge transfer (Fig. 9B). The same π-donation effects were observed in analogous arsenic compounds and allowed the isolation of di(ylide)-substituted cations such as 39, 40 and 41 which feature a delocalization of the charge within the C–E–C linkage.58b,61,62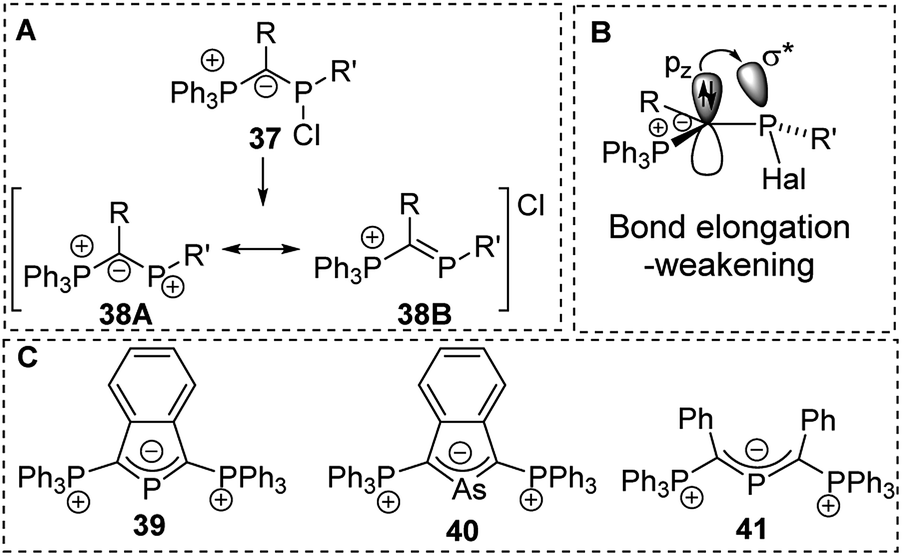 | ||
| Fig. 9 (A) Dissociation of ylide-substituted chlorophosphines to phosphenium cations, (B) orbital interactions in ylide-substituted halophosphines and (C) diylide-substituted group 15 cations. | ||
The preference of certain conformers due to the repulsion between the lone pair at the ylidic carbon atom and the neighbouring phosphorus atom was also found in ylide-substituted phosphines (YPhos, Fig. 7).63 Although these phosphines have been known for quite some time,64,65 their strong donor properties owing to the electron donation from the ylide to the phosphorus centre were only recently recognized. Our group used YPhos ligands as electron-rich phosphines in homogeneous catalysis, which led to highly active gold and palladium catalysts e.g. for hydroamination and C–N as well as C–C coupling reactions at room temperature (Fig. 10).66 The donor strength and the steric properties of the YPhos ligands were found to be highly tunable depending on the nature of the Z substituent in the ylide-backbone. Thus, while offering steric protection through the large phosphonium moiety, donor strengths similar to those of NHCs, which are usually considered to be stronger donors, could be reached.63a Electron-rich phosphines were also reported by Beller, Rivard and Ruiz when using N-heterocyclic olefins (NHOPs)67 as substituents as well as by Dielmann and coworkers using imidazolidin-2-imino substituents (IAPs)68 and by Sundermeyer and coworkers using phosphazenyl groups (PAPs).69 The latter exhibited even higher donor strengths than the YPhos ligands.
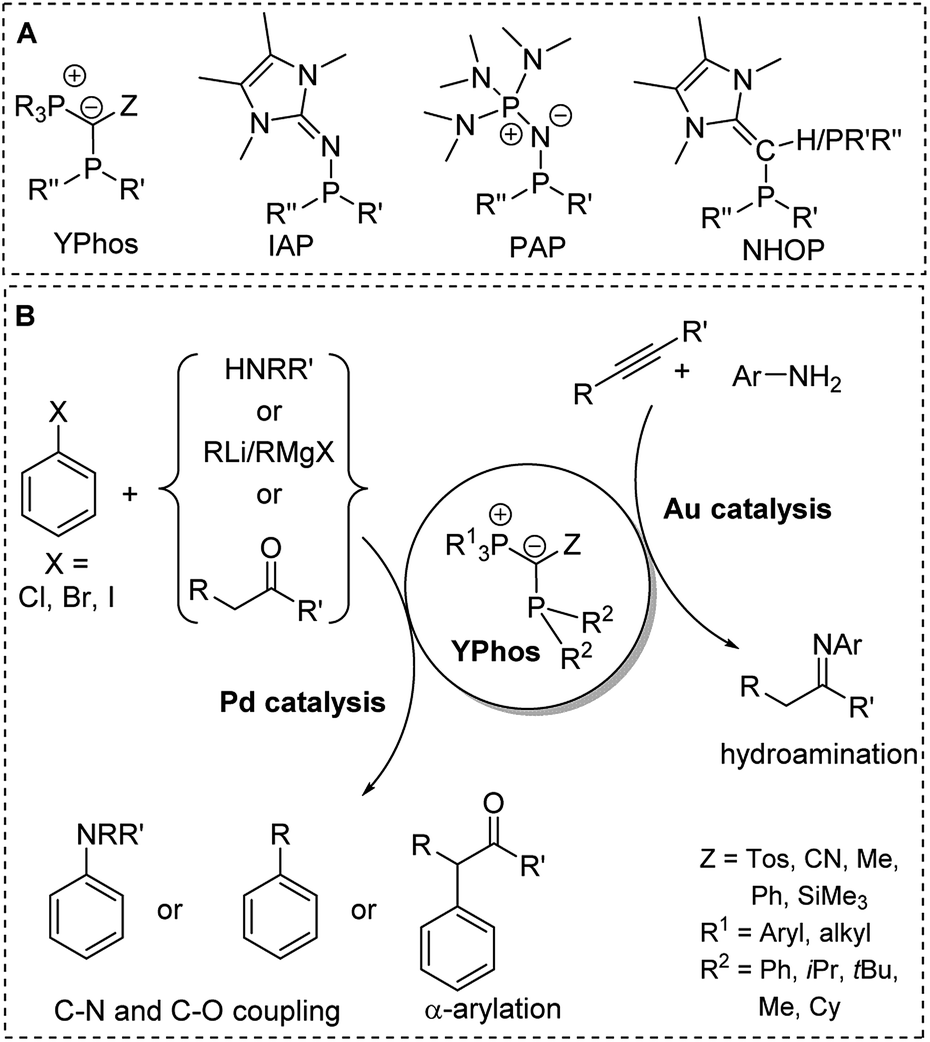 | ||
| Fig. 10 (A) Structures of electron-rich phosphines. (B) Applications of YPhos ligands in homogeneous catalysis (R,R′,R′′ = alkyl). | ||
Conclusions and outlook
Overall, the use of phosphorus ylides as substituents in main group chemistry is still in its infancy. However, the survey of compounds presented in this review article and the breadth of the remarkable results achieved with the structurally related N-heterocyclic vinyl substituents demonstrate that these substituents are well suited for stabilizing reactive compounds and offer a further possibility for tailoring the properties and reactivities of main group metal compounds. Owing to their strongly electron-donating character ylides have particularly been used to stabilize electron-deficient compounds above all cations (borenium and phosphenium) and low-valent species. Specifically the reported group 14 chemistry demonstrates the true potential of these substituents not only for the stabilization of otherwise elusive species (e.g. SiO), but also for imparting new reactivities including ligand-centered reactivity. Therefore, the variation of the substituent in the ylide backbone offers a further opportunity for fine-tuning the donor properties. This concept has been lately used to tailor electron-rich ylide-substituted phosphines for catalysis, which express the electronic and steric flexibility of ylide-substituents. These findings clearly demonstrate the potential of phosphorus ylides as substituents in main group chemistry. The tailoring of the ylide properties for stabilizing reactive compounds is far from being well explored but offers many possibilities for future studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) (Starting Grant: YlideLigands 677749).Notes and references
- (a) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS; (b) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213 CrossRef CAS; (c) R. L. Melen, Chem. Soc. Rev., 2016, 45, 775 RSC; (d) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef CAS PubMed; (e) T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176 RSC; (f) S. Yadav, S. Saha and S. Sen Sakya, ChemCatChem, 2015, 8, 486 CrossRef; (g) D. J. Scott, M. J. Fuchter and A. E. Ashley, Chem. Soc. Rev., 2017, 46, 5689 RSC.
- (a) R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343 CrossRef CAS PubMed; (b) P. J. Davidson and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1973, 317a RSC; (c) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS; (d) A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463 CrossRef CAS; (e) M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587 CrossRef CAS; (f) A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst and R. K. Kallury, J. Chem. Soc., Chem. Commun., 1981, 191 RSC.
- (a) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444 CrossRef CAS PubMed; (b) S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727 RSC.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232 CrossRef CAS PubMed.
- (a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS PubMed; (b) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed.
- (a) A. Jana, C. Schulzke and H. W. Roesky, J. Am. Chem. Soc., 2009, 131, 4600 CrossRef CAS PubMed; (b) Y. Xiong, S. Yao, R. Mìller, M. Kaupp and M. Driess, J. Am. Chem. Soc., 2010, 132, 6912 CrossRef CAS PubMed; (c) W. Wang, S. Inoue, S. Yao and M. Driess, Organometallics, 2011, 30, 6490 CrossRef CAS; (d) A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2012, 52, 568 CrossRef PubMed; (e) J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622 CrossRef CAS PubMed; (f) J. Li, M. Hermann, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 8611 CrossRef CAS PubMed; (g) Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard and P. P. Power, Chem. Commun., 2008, 6042 RSC; (h) Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268 CrossRef CAS PubMed; (i) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134 CrossRef CAS PubMed; (j) M. Arrowsmith, J. Bohnke, H. Braunschweig, M. A. Celik, T. Dellermann and K. Hammond, Chem.–Eur. J., 2016, 22, 17169 CrossRef CAS PubMed.
- (a) J. Hicks, P. Vaski, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000 CrossRef CAS PubMed; (b) J. Hicks, P. Vaski, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92 CrossRef CAS PubMed.
- M.-A. Légaré, G. B. Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896 CrossRef PubMed.
- (a) R. Taakili, C. Lepeti, C. Duhayon, D. A. Valyaev, N. Lugan and Y. Canac, Dalton Trans., 2019, 48, 1709 RSC; (b) R. Taakili, C. Barthes, A. Goëffon, C. Lepetit, C. Duhayon, D. A. Valyaev and Y. Canac, Inorg. Chem., 2020, 59, 7082 CrossRef CAS PubMed.
- (a) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678 CrossRef CAS PubMed; (b) M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Nat. Chem., 2009, 1, 295 CrossRef CAS PubMed; (c) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109(8), 3479 CrossRef CAS PubMed; (d) C. Adam Dyker and G. Bertrand, Nat. Chem., 2009, 1, 265 CrossRef PubMed; (e) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815 CrossRef CAS PubMed; (f) C. J. Carmalt and A. H. Cowley, Adv. Inorg. Chem., 2000, 50, 1 CrossRef CAS; (g) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020 RSC; (h) N. Kuhn and A. Al-Shaikh, Coord. Chem. Rev., 2005, 249, 829 CrossRef CAS; (i) S. Würtemberger-Pietsch, U. Radius and T. B. Marder, Dalton Trans., 2016, 45, 5880 RSC.
- D. B. Kays, Chem. Soc. Rev., 2016, 45, 1004 RSC.
- P. J. Davidson, D. H. Harris and M. F. J. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2268–2274 RSC.
- S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36 CrossRef CAS PubMed.
- D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
- (a) J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622 CrossRef CAS PubMed; (b) T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199 CrossRef CAS PubMed.
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028 CrossRef CAS PubMed.
- R. Nakano, R. Jazzar and G. Bertrand, Nat. Chem., 2018, 10, 1196 CrossRef CAS PubMed.
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, El. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555 CrossRef CAS PubMed.
- (a) S. Takahasi, E. Bellan, A. Baceiredo, N. Saffon-Merceron, S. Massou, N. Nakata, D. Hashizume, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2019, 58, 10310 CrossRef PubMed; (b) H. J. Barnett and A. F. Hill, Angew. Chem., Int. Ed., 2020, 59, 4274 CrossRef CAS PubMed.
- T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327 RSC.
- (a) M. M. D. Roy, M. J. Ferguson, R. McDonald, Y. Zhou and E. Rivard, Chem. Sci., 2019, 10, 6476 RSC; (b) M. K. Sharma, S. Blomeyer, B. Neumann, H.-G. Stammler, A. Hinz, M. van Gastel and R. S. Ghadwal, Chem. Commun., 2020, 56, 3575 RSC; (c) M. K. Sharma, D. Rottschäfer, S. Blomeyer, B. Neumann, H.-G. Stammler, M. van Gastel, A. Hinz and R. S. Ghadwal, Chem. Commun., 2019, 55, 10408 RSC; (d) D. Rottschäfer, B. Neumann, H. -G. Stammler and R. S. Ghadwal, Chem.–Eur. J., 2017, 23, 9044 CrossRef PubMed; (e) L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 273 CrossRef CAS PubMed; (f) D. Rottschäfer, M. K. Sharma, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem.–Eur. J., 2019, 25, 8127 CrossRef PubMed; (g) M. K. Sharma, S. Blomeyer, B. Neumann, H. -G. Stammler and R. S. Ghadwal, Chem.–Eur. J., 2019, 25, 8249 CrossRef CAS PubMed; (h) M. K. Sharma, S. Blomeyer, B. Neumann, H.-G. Stammler, M. van Gastel, A. Hinz and R. S. Ghadwal, Angew. Chem., Int. Ed., 2019, 58, 17599 CrossRef CAS PubMed; (i) D. Rottschäfer, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, J. Org. Chem., 2020 DOI:10.1021/acs.joc.0c00176.
- (a) M. M. D. Roy and E. Rivard, Acc. Chem. Res., 2017, 50, 2017 CrossRef CAS PubMed; (b) R. S. Ghadwal, Dalton Trans., 2016, 45, 16081 RSC.
- (a) M. Balmer, H. Gottschling and C. von Hänisch, Chem. Commun., 2018, 54, 2659 RSC; (b) S. Kundu, S. Sinhababu, A. V. Luebben, T. Mondal, D. Koley, B. Dittrich and H. W. Roesky, J. Am. Chem. Soc., 2018, 140, 151 CrossRef CAS PubMed; (c) M. Balmer, Y. J. Franzke, F. Weigand and C. von Hänisch, Chem.–Eur. J., 2020, 26, 192 CrossRef CAS PubMed.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526 CrossRef CAS PubMed.
- (a) M. W. Lui, C. Merten, M. J. Ferguson, R. McDonald, Y. Xu and E. Rivard, Inorg. Chem., 2015, 54, 2040 CrossRef CAS PubMed; (b) T. Ochiai, T. Szilvási, D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2016, 55, 11619 CrossRef CAS PubMed.
- C. Hering-Junghans, P. Andreiuk, M. J. Ferguson, R. McDonald and E. Rivard, Angew. Chem., Int. Ed., 2017, 56, 6272 CrossRef CAS PubMed.
- (a) H. Lischka, J. Am. Chem. Soc., 1977, 99, 353 CrossRef CAS; (b) D. G. Gilheany, Chem. Rev., 1994, 94, 1339 CrossRef CAS PubMed.
- H. Schmidbaur, Angew. Chem., Int. Ed., 1983, 22, 907 CrossRef.
- (a) R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., 2006, 118, 8206 CrossRef; (b) R. Tonner and G. Frenking, Chem.–Eur. J., 2008, 14, 3260 CrossRef CAS PubMed; (c) R. Tonner and G. Frenking, Chem.–Eur. J., 2008, 14, 3273 CrossRef CAS PubMed.
- L. T. Scharf, D. M. Andrada, G. Frenking and V. H. Gessner, Chem.–Eur. J., 2018, 23, 4432 Search PubMed.
- (a) E. J. Corey and J. Kang, J. Am. Chem. Soc., 1982, 104, 4724 CrossRef CAS; (b) M. Schlosser, T. Kadibelban and G. Steinhoff, Angew. Chem., Int. Ed., 1966, 5, 968 CrossRef CAS.
- For examples: (a) R. Zurawinski, C. Lepetit, Y. Canac, M. Mikolajczyk and R. Chauvin, Inorg. Chem., 2009, 48, 2147 CrossRef CAS PubMed; (b) P. E. Romero, W. E. Piers and R. McDonald, Angew. Chem., Int. Ed., 2004, 43, 6161 CrossRef CAS PubMed; (c) X. Li, A. Wang, L. Wang, H. Sun, K. Harms and J. Sundermeyer, Organometallics, 2007, 26, 1411 CrossRef CAS; (d) P. Rungthanaphatsophon, A. Bathelier, L. Castro, A. C. Behrle, C. L. Barnes, L. Maron and J. R. Walensky, Angew. Chem., Int. Ed., 2017, 56, 12925 CrossRef CAS PubMed.
- H. J. Bestmann and M. Schmidt, Angew. Chem., Int. Ed., 1987, 26, 79 CrossRef.
- C. Schwarz, L. T. Scharf, T. Scherpf, J. Weismann and V. H. Gessner, Chem.–Eur. J., 2019, 25, 2793 CrossRef CAS PubMed.
- S. Goumri-Magnet, H. Gornitzka, A. Baceiredo and G. Bertrand, Angew. Chem., Int. Ed., 1999, 38, 678 CrossRef CAS.
- T. Baumgartner, B. Schinkels, D. Gudat, M. Nieger and E. Niecke, J. Am. Chem. Soc., 1997, 119, 12410 CrossRef CAS.
- (a) T. Scherpf, R. Wirth, S. Molitor, K.-S. Feichtner and V. H. Gessner, Angew. Chem., Int. Ed., 2015, 54, 8542 CrossRef CAS PubMed; ylide 14-H has been reported before: (b) J. Vicente, A. R. Singhal and P. G. Jones, Organometallics, 2002, 21, 5887 CrossRef CAS.
- H. Darmandeh, T. Scherpf, K.-S. Feichtner, C. Schwarz and V. H. Gessner, Z. Anorg. Allg. Chem., 2020, 646, 835 CrossRef CAS.
- A. Garduno-Alva, R. Lenk, Y. Escudié, M. L. González, L. Bousquet, N. Saffon-Merceron, C. A. Toledano, X. Bagan, V. Branchadell, E. Maerten and A. Baceiredo, Eur. J. Inorg. Chem., 2017, 3494 CrossRef CAS.
- (a) H. J. Bestmann and T. Arenz, Angew. Chem., Int. Ed., 1986, 25, 559 CrossRef; (b) H. J. Berstmann, T. Röder, M. Bremer and D. Löw, Chem. Ber., 1991, 124, 199 CrossRef; (c) M. Radius and F. Breher, Chem.–Eur. J., 2018, 24, 15744 CrossRef CAS PubMed; (d) M. Radius, E. Sattler, H. Berberich and F. Breher, Chem.–Eur. J., 2019, 25, 12206 CrossRef CAS PubMed.
- P. Bharadwaz and A. K. Phukan, Inorg. Chem., 2019, 58, 5428 CrossRef CAS.
- For example: G. Facchin, R. Campostrini and R. A. Michelin, J. Organomet. Chem., 1985, 294, C21 CrossRef CAS.
- S. Nakafuji, J. Kobayashi and T. Kawashima, Angew. Chem., Int. Ed., 2008, 47, 1141 CrossRef CAS.
- A. Fürstner, M. Alcarazo, K. Radkowski and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 8302 CrossRef.
- (a) B. Borthakur and a. K. Phukan, Chem.–Eur. J., 2015, 21, 11603 CrossRef CAS; (b) B. Borthakur, B. Silvi, R. Dewhurst and A. K. Phukan, J. Comput. Chem., 2016, 37, 1484 CrossRef CAS.
- (a) M. Asay, B. Donnadieu, A. Baceiredo, M. Soleilhavoup and G. Bertrand, Inorg. Chem., 2008, 47, 3949 CrossRef CAS PubMed; (b) J. Kobayashi, S.-y. Nakafuji, A. Yatabe and T. Kawashima, Chem. Commun., 2008, 6233 RSC.
- H. Steinert, C. Schwarz, A. Kroll and V. H. Gessner, Molecules, 2020, 25, 796 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754 CrossRef CAS PubMed.
- M. Asay, S. Inoue and M. Driess, Angew. Chem., Int. Ed., 2011, 50, 9589 CrossRef CAS PubMed.
- A. Schmidpeter, H.-P. Schrödel and J. Knizek, Chem. Ber., 1998, 9, 103 CAS.
- I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2016, 128, 16375 CrossRef.
- I. Alvarado-Beltran, A. Rosas-Sánchez, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10481 CrossRef CAS PubMed.
- J. Berthe, J. M. Garcia, E. Ocando, T. Kato, N. Saffon-Merceron, A. De Cózar, F. P. Cossío and A. Baceiredo, J. Am. Chem. Soc., 2011, 133, 15930 CrossRef CAS PubMed.
- (a) N. Del Rio, A. Baceiredo, N. Saffon-Merceron, D. Hahizume, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 4753 CrossRef CAS PubMed; (b) A. Rosas-Sánchez, I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, S. Massou, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10549 CrossRef PubMed.
- A. Rosas-Sánchez, I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, S. Massou, D. Hashizume, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 16132 CrossRef.
- N. Del Rio, M. Lopez-Reyes, A. Baceiredo, N. Saffon-Merceron, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 1365 CrossRef CAS PubMed.
- C. Mohapatra, L. Scharf, T. Scherpf, B. Mallick, K.-S. Feichtner, C. Schwarz and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 7459 CrossRef CAS PubMed.
- (a) A. Schmidpeter, H. Noeth, G. Jochem, H.-P. Schroedel and K. Karaghiosoff, Chem. Ber., 1995, 128, 379 CrossRef CAS; (b) A. Schmidpeter, G. Jochem, C. Klinger, C. Robl and H. Nöth, J. Organomet. Chem., 1997, 529, 87 CrossRef CAS; (c) H.-P. Schroedel, A. Schmidpeter and H. Nöth, Heteroat. Chem., 1996, 7, 355 CrossRef CAS; (d) G. Jochem, F. Breitsameter, A. Schier and A. Schmidpeter, Heteroat. Chem., 1996, 7, 239 CrossRef CAS; (e) A. Schmidpeter and G. Jochem, Tetrahedron Lett., 1992, 33, 471 CrossRef CAS.
- R. Hoffmann, L. Radom, J. A. Pople, P. v. R. Schleyer, W. J. Hehre and L. Salem, J. Am. Chem. Soc., 1972, 94, 6221 CrossRef CAS.
- D. Gudat, Coord. Chem. Rev., 1997, 163, 71 CrossRef CAS.
- F. Breitsameter, A. Schmidpeter and H. Nöth, Chem.–Eur. J., 2000, 6, 3531 CrossRef CAS.
- (a) A. Schmidpeter and M. Thiele, Angew. Chem., Int. Ed. Engl., 1991, 30, 308 CrossRef; (b) D. Gudat, E. Nieger and M. Schrott, Chem. Ber., 1995, 128, 259 CrossRef CAS.
- (a) T. Scherpf, C. Schwarz, L. T. Scharf, J.-A. Zur, A. Helbig and V. H. Gessner, Angew. Chem., Int. Ed., 2018, 57, 12859 CrossRef CAS PubMed; (b) C. Schwarz, T. Scherpf, I. Rodstein, J. Weismann, K. -S. Feichtner and V. H. Gessner, ChemistryOpen, 2019, 8, 621 CrossRef CAS.
- K. Issleib and R. Lindner, Justus Liebigs Ann. Chem., 1966, 699, 40 CrossRef CAS.
- For ylide-substituted arsenic and antimony compounds, see: (a) H. Schmidbaur and W. Tronich, Chem. Ber., 1968, 101, 3545 CrossRef CAS; (b) F. Breitsameter, H.-P. Schrödel, A. Schmidpeter, H. Nöth and S. Rojas-Lima, Z. Anorg. Allg. Chem., 1999, 625, 1293 CrossRef CAS; (c) H. Grützmacher and H. Pitzkow, Chem. Ber., 1989, 122, 1417 CrossRef.
- (a) P. Weber, T. Scherpf, I. Rodstein, D. Lichte, L. T. Scharf, L. J. Gooßen and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 3203 CrossRef CAS PubMed; (b) X.-Q. Hu, D. Lichte, I. Rodstein, P. Weber, A.-K. Seitz, T. Scherpf, V. H. Gessner and L. J. Gooßen, Org. Lett., 2019, 21(18), 7558 CrossRef CAS PubMed; (c) C. Schwarz, J. Handelmann, D. M. Baier, A. Ouissa and V. H. Gessner, Catal. Sci. Technol., 2019, 9, 6808 RSC; (d) L. T. Scharf, I. Rodstein, M. Schmidt, T. Scherpf and V. H. Gessner, ACS Catal., 2020, 10, 999 CrossRef CAS PubMed; (e) J. Tappen, I. Rodstein, K. McGuire, A. Großjohann, J. Löffler, T. Scherpf and V. H. Gessner, Chem.–Eur. J., 2020, 26, 4281 CrossRef CAS PubMed; (f) T. Scherpf, H. Steinert, A. Großjohann, K. Dilchert, J. Tappen, I. Rodstein and V. H. Gessner, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202008866.
- (a) N. R. Paisley, M. W. Lui, R. McDonald and E. Rivard, Dalton Trans., 2016, 45, 9860 RSC; (b) M. W. Lui, O. Shynkaruk, M. S. Oakley, R. Sinelnikov, R. McDonald, M. J. Ferguson, M. Klobukowski and E. Rivard, Dalton Trans., 2017, 46, 5946 RSC; (c) J. Ruiz, M. E. G. Mosquera, G. García, F. Marquínez and V. Riera, Angew. Chem., Int. Ed., 2005, 44, 102 CrossRef CAS; (d) A. Dumrath, X.-F. Wu, H. Neumann, A. Spannenberg, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8988 CrossRef CAS PubMed.
- M. A. Wünsche, P. Mehlmann, T. Witteler, F. Bus, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857 CrossRef PubMed.
- (a) S. Ullrich, B. Kovačević, X. Xie and J. Sundermeyer, Angew. Chem., Int. Ed., 2019, 58, 10335 CrossRef CAS PubMed. For an earlier report: (b) H. Schmidbaur and G. Jonas, Chem. Ber., 1968, 101, 1271–1285 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |