
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-and temperature induced phase transitions, piezochromism, NLC behaviour and pressure controlled Jahn–Teller switching in a Cu-based framework†
Charles J.
McMonagle
a,
Priyanka
Comar
a,
Gary S.
Nichol
 a,
David R.
Allan
b,
Jesús
González
c,
José A.
Barreda-Argüeso
c,
Fernando
Rodríguez
c,
Rafael
Valiente
d,
Gemma F.
Turner
e,
Euan K.
Brechin
*a and
Stephen A.
Moggach
*e
a,
David R.
Allan
b,
Jesús
González
c,
José A.
Barreda-Argüeso
c,
Fernando
Rodríguez
c,
Rafael
Valiente
d,
Gemma F.
Turner
e,
Euan K.
Brechin
*a and
Stephen A.
Moggach
*e
aSchool of Chemistry and Centre for Science at Extreme Conditions, The University of Edinburgh, King's Buildings, David Brewster Road, Edinburgh, Scotland EH9 3FJ, UK
bDiamond Light Source, Harwell Campus, Didcot, OX11 ODE, UK
cMALTA Team, CITIMAC, Facultad de Ciencias, University of Cantabria, 39005 Santander, Spain
dApplied Physics Dept, Facultad de Cienicas, University of Cantabria – IDIVAL, 39005 Santander, Spain
eSchool of Molecular Sciences and Centre for Microscopy, Characterisation and Analysis, University of Western Australia, 35 Stirling Highway, Crawley, Perth, 6005, Western Australia, Australia. E-mail: stephen.moggach@uwa.edu.au
First published on 30th July 2020
Abstract
In situ single-crystal diffraction and spectroscopic techniques have been used to study a previously unreported Cu-framework bis[1-(4-pyridyl)butane-1,3-dione]copper(II) (CuPyr-I). CuPyr-I was found to exhibit high-pressure and low-temperature phase transitions, piezochromism, negative linear compressibility, and a pressure induced Jahn–Teller switch, where the switching pressure was hydrostatic media dependent.
High-pressure crystallographic experiments over the last 25 years or so have proved to be a unique tool in probing the mechanical properties of the organic solid state,1 metal-complexes, and 2D/3D coordination compounds.2 In particular, high-pressure techniques have been used to study an array of mechanical and chemical properties of crystals, such as changes in electrical and thermal conductivity,3 pressure-induced melting,4 solubility,5 amorphisation,6 post-synthetic modification,7 and chemical reactions such as polymerisation,8 cycloaddition9 and nanoparticle formation.10 Previous high-pressure experiments on porous metal-organic framework (MOF) materials have shown that on loading a diamond anvil cell (DAC) with a single-crystal or polycrystalline powder, the hydrostatic medium that surrounds the sample (to ensure hydrostatic conditions) can be forced inside the pores on increasing pressure, causing the pore and sample volume to increase with applied pressure.11 This technique has also been used to determine the position of CH4 and CO2 molecules inside the small pores of a Sc-based MOF at room temperature using a laboratory X-ray diffractometer, and has proved useful in experimentally determining the maximum size of guest molecules that can penetrate into a pore.12
On direct compression of more dense frameworks, negative linear compressibility (NLC) has also been observed, which results in an expansion of one or more of the unit cell dimensions with an overall contraction in volume. Such changes in the compressibility behaviour of metal-containing framework materials is usually as a result of common structural motifs which rotate or bend in order to accommodate increases in length along particular crystallographic directions.13 Changes in coordination environment can also be induced at pressure, as metal–ligand bonds are more susceptible to compression than covalent bonds.2a In previous high pressure studies on metal complexes or coordination compounds, in which the metal ion has an asymmetric octahedral environment caused by Jahn–Teller (JT) distortions for example (such as those observed in Cu2+ and Mn3+ complexes), the application of pressure can result in compression of the JT axis, and can even be switched to lie along another bonding direction within the octahedron.14 Such distortions often result in piezochromism, often observed within a single crystal.15
Here, we present a high-pressure crystallographic study on a novel and unreported Cu-framework bis[1-(4-pyridyl)butane-1,3-dione]copper(II) (hereafter referred to as CuPyr-I). On application of pressure, CuPyr-I is highly unusual in that it demonstrates several of these phenomena within the same framework, including a single-crystal to single-crystal phase transition, a switching of the JT axis that depends on the hydrostatic medium used to compress the crystal, piezochromism and NLC behaviour. To date, we are unaware of any other material which exhibits all of these phenomena, with the first ever reported hydrostatic media ‘tuneable’ JT-switching.
Under ambient temperature and pressure CuPyr-I crystallises in the rhombohedral space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (a/b = 26.5936(31) Å, c = 7.7475(9) Å). Each Cu-centre is coordinated to four 1-(4-pyridyl)butane-1,3-dione linkers, two of these ligands are bound through the dione O-atoms, with the final two bonding through the N-atom of the pyridine ring to form a 3D polymer. The crystal structure of CuPyr-I is composed of an interpenetration of these 3D polymers to form one-dimensional porous channels (∼2 Å in diameter) that run along the c-axis direction (Fig. 1).
(a/b = 26.5936(31) Å, c = 7.7475(9) Å). Each Cu-centre is coordinated to four 1-(4-pyridyl)butane-1,3-dione linkers, two of these ligands are bound through the dione O-atoms, with the final two bonding through the N-atom of the pyridine ring to form a 3D polymer. The crystal structure of CuPyr-I is composed of an interpenetration of these 3D polymers to form one-dimensional porous channels (∼2 Å in diameter) that run along the c-axis direction (Fig. 1).
 | ||
| Fig. 1 Ball and stick model showing the coordination environment around the Cu2+ ion in CuPyr-I, and 3D-pore structure as viewed along the c-axis direction. The yellow sphere represents the available pore-space. Colour scheme is red: oxygen, blue: nitrogen, black: carbon, white: hydrogen and cyan: copper. The Cu2+ octahedron is illustrated in green. | ||
On increasing pressure from 0.07 GPa to 1.56 GPa using Fluorinert FC-70 (a mixture of large perfluorinated hydrocarbons) as a hydrostatic medium, compression of the framework occurs, resulting in a 9.89% reduction in volume, while the a/b-axes and c-axis are reduced by 4.46% and 1.25% respectively (Fig. 2 (blue triangles) and Table S1†).
 | ||
| Fig. 2 a/b and c-axes as a function of pressure in a hydrostatic medium of FC-70 (blue triangles) and MeOH (red/black circles). The vertical line indicates the transition from CuPyr-I (red circles) to CuPyr-II (black circles) above 2.15 GPa. Errors in cell-lengths are smaller than the symbols plotted. | ||
On increasing pressure to 1.84 GPa, the framework became amorphous, though this is unsurprising as the hydrostatic limit for FC-70 is ∼2 GPa, and compression of frameworks in non-hydrostatic conditions usually results in amorphisation.16 On increasing pressure to 1.56 GPa, the three-symmetry independent Cu–O/N bond lengths to the ligand were monitored (Fig. 3 and Table S5†). Under ambient pressure conditions, the two Cu–N1 pyridine bonds are longer than the four Cu–O1/O2 dione bonds, typical for an elongated JT distorted Cu2+ complex. However, on increasing pressure the direction of the JT axis gradually changed from Cu–N1 to the Cu–O1 bond (the dione oxygen in the 3-position), becoming equidistant at ∼0.57 GPa. By 1.56 GPa, the lengths of the Cu–N1 and Cu–O1 bonds had steadily reduced and increased by 12.3% and 8.9%, respectively. Throughout this the Cu–O2 bond remained essentially unchanged.
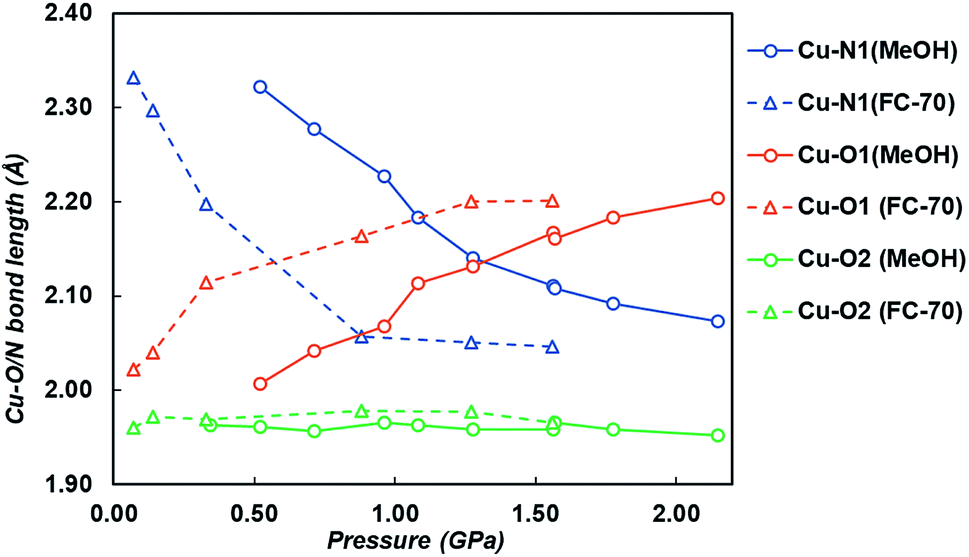 | ||
| Fig. 3 Cu–O1 (orange), Cu–N1 (blue) and Cu–O2 (green) bond lengths on increasing pressure in both FC-70 (triangles and dashed lines) and MeOH (circles and solid lines). | ||
Pressure induced JT switching has been observed in other systems, including a Mn12 single-molecule magnet cluster that re-orientates the JT axis on one of the Mn centres at 2.5 GPa.14a A similar transition was also observed in [CuF2(H2O)2(pyz)] (pyz = pyrazine) and Rb2CuCl4(H2O)2,15 where the JT axis was reoriented from the Cu–N bond to the perpendicular Cu–O bond, though this occurs during a crystallographic phase transition at 1.8 GPa.18 Here, in CuPyr-I, no phase transition takes place, and unusually the JT switching appears to occur progressively on increasing pressure with no phase transition.14b
Using methanol (MeOH) as the hydrostatic medium, CuPyr-I was compressed in two separate experiments, from 0.52 GPa to 5.28 GPa using synchrotron radiation, and from 0.34 GPa to 2.95 GPa using a laboratory X-ray diffractometer. On increasing pressure to 2.15 GPa, the a/b and c-axes compressed by 6.22% and 0.39% respectively (Fig. 2, Tables S2 and S3†). On increasing pressure from 2.15 GPa to 2.78 GPa, CuPyr-I underwent a single-crystal to single-crystal isosymmetric phase transition to a previously unobserved phase (hereafter referred to as CuPyr-II).
The transition to CuPyr-II resulted in a doubling of the a/b-axes, whilst the c-axis remained essentially unchanged. On increasing the pressure further, the a/b-axes continued to be compressed, whilst the c-axis increased in length, exhibiting negative linear compressibility (NLC) until the sample became amorphous at 5.28 GPa. The diffraction data were of poor quality after the phase transition, and only the connectivity of the CuPyr-II phase could be determined at 3.34 GPa. Above 3.34 GPa, only unit cell dimensions could be extracted. The occurrence of positive linear compressibility (PLC) followed by NLC is unusual in a framework material, and we could find only a few examples in the literature where this occurs.19
During the NLC, the c-axis expanded by 1.46%, to give a compressibility of KNLC = −5.3 (0.8) TPa−1 (Δp = 2.23–4.90 GPa). KNLC is calculated using the relationship K = −1/l(∂l/∂p)T, where l is the length of the axis and (∂l/∂p)T is the length change in pressure at constant temperature.20 The value of KNLC here is rather small compared to the massive NLC behaviour observed in the low pressure phase of Ag3[Co(CN)6]9 (KNLC = −76(9) TPa−1, Δp = 0–0.19 GPa) or the flexible MOF MIL-53(Al) (KNLC = −28 TPa−1, Δp = 0–3 GPa) for example,17b and is much more comparable to the dense Zn formate MOF [NH4][Zn(HCOO)3] (−1.8(8) TPa−1 (Δp = 0–0.94 GPa)).21 Because of the quality of the data, the exact nature, or reason for the NLC in CuPyr-II is unknown, although we aim to investigate this in the future.
On increasing pressure using MeOH, the JT axis was again supressed on compression, with the Cu–N1 bond reducing in length by 0.288 Å (12%) between 0.34 and 2.15 GPa, while the Cu–O1 bond length increased by 0.216 Å (11%). The pressure at which Cu–N1 and Cu–O1 became equidistant was 1.28 GPa, measuring 2.140(5) Å and 2.131(6) Å respectively (Fig. 3 and Table S6†). Across the entire pressure range, little to no compression or expansion was observed in the Cu–O2 bond in the 1-position of the dione in CuPyr-I, the same trend observed when compressed in FC-70. The JT switching pressure in MeOH however was 0.71 GPa higher than observed by direct compression in FC-70 (0.57 GPa). This, to our knowledge, is the first time that pressure induced JT switching has been observed to be hydrostatic media dependent.
Changes to the Cu–N and Cu–O bond lengths were supported by high-pressure Raman spectroscopy of CuPyr-I, using MeOH as the hydrostatic medium (Fig. S10†). Gradual growth of a shoulder on a band at ∼700 cm−1 during compression is tentatively assigned to the Cu2+ coordination environment shifting from elongated to compressed JT distorted geometry. The shouldered peak becomes split above 2 GPa, after which the isosymmetric phase transition occurs.
The gradual JT switch is thought to be principally responsible for reversible piezochromism in single crystals of CuPyr-I, which change in colour from green to dark red under applied pressure (Fig. 4b, S1 and S2†). UV-visible spectroscopy confirms a bariometric blue-shift in the absorption peak at ∼700 nm assigned to d–d electronic transitions, and a red-shift of the tentatively assigned ligand-to-metal charge-transfer (LMCT) edge around 450 nm during the elongated to compressed switch (Fig. 4a and S8†), accounting for this colour change. The red-shift is observed during compression in both Fluorinert® FC-70 and MeOH hydrostatic media, with a slightly suppressed shift measured in the latter due to filling of the framework pores (Table S2†). Geometric switching at the metal centre leads to electronic stabilisation of the Cu2+ ion, as electrons transfer from higher energy dx2−y2 (Cu–O) orbitals to the lower energy dz2 (Cu–N) state (Fig. S7†), evidenced by the blue-shift of the d–d intraconfigurational band as the dz2 (Cu–N) is progressively mixing with dx2−y2 (Cu–O) increasing its energy with respect to the lower energy dxy and dxz,yz levels becoming the highest energy level at the nearly compressed rhombic geometry. On the other hand, the redshift in the hesitantly assigned O2− to Cu2+ LMCT band below 450 nm is ascribed to increase of the Cu–O bond distance and a likely bandwidth broadening with pressure both yielding a pressure redshift of the absorption band gap edge (Fig. 4a).
 | ||
| Fig. 4 (a) UV-visible spectroscopy of CuPyr-I during compression in Fluorinert® FC-70 showing a gradual BLUE-shift in the d–d intraconfigurational band (∼700 nm) and a gradual red-shift of the absorption band assigned to LMCT (∼450 nm) with increasing pressure. (b) Gradual pressure-induced Jahn–Teller switch of the Cu2+ octahedral coordination environment in CuPyr-1 from tetragonal elongated (left, green) to rhombic compressed (right, red), causing piezochromism. Atom colouring follows previous figures. | ||
Compression of the coordination bonds was not the only distortion to take place in CuPyr-I, with the Cu-octahedra also twisting with respect to the 1-(4-pyridyl)butane-1,3-dione linkers on increasing pressure. Twisting of the Cu-octahedra in CuPyr-I with respect to the dione section of the linker could be quantified by measuring both the ∠N1Cu1O2C4 and the ∠N1Cu1O1C2 torsion angles from the X-ray data, which in MeOH gradually decrease and increase by 12.2° and 7.3°, respectively, to 2.15 GPa (Table S8†). In FC-70, ∠N1Cu1O2C4 and ∠N1Cu1O1C2 decrease and increase by 5.4° and 2.8°, respectively, to 1.56 GPa. On increasing pressure to 1.57 GPa in a hydrostatic medium of MeOH, a difference of ∼5° for both angles was observed compared to FC-70 at 1.56 GPa. Twisting about the octahedra allows compression of the channels to take place in a ‘screw’ like fashion and has been observed in other porous materials with channel structures.22 The overall effect is to reduce the pore volume, and decrease the size of the channels (Tables S2 and S3†). Using MeOH as a hydrostatic medium therefore appears to reduce this effect by decreasing the compressibility of the framework.
It was not possible to determine the pressure dependence in other longer-chain alcohols, including ethanol (EtOH) and isopropanol (IPA), due to cracking of the crystal upon loading into the diamond anvil cell (Fig. S1†). We believe this is a result of these longer chain alcohols acting as reducing agents, as indicated by the loss in colour of the crystals.
To ascertain the origin of the hydrostatic media-induced change in the JT switching pressure and unit cell compressibility, the pore size and content were monitored as a function of pressure. A dried crystal of CuPyr-I was collected at ambient pressure and temperature in order to compare to the high-pressure data and is included in the ESI.† The pore volume and electron density were estimated and modelled respectively using the SQUEEZE algorithm within PLATON (Tables S1–S3†).23 CuPyr-I under ambient pressure conditions has three symmetry equivalent channels per unit cell with a total volume of ∼1152 Å3 containing diethyl ether (2.5 wt%) trapped in the pores during the synthesis of the framework, confirmed by TGA analysis (Fig. S6†).
On surrounding the crystal with FC-70, direct compression of the framework occurred. The pore content remained almost constant during compression up to 0.88 GPa, inferring no change in the pore contents. On increasing pressure further to 1.56 GPa, an increase in the calculated electron density was observed (23%), though the data here were of depreciating quality and less reliable. During compression of CuPyr-I in MeOH to 0.52 GPa, the pore volume and electron density in the channels increased by 4.5% and 54%, respectively, reflecting ingress of MeOH into the pores. The electron density in the channels continued to increase to a maximum of 0.466e− A−3 by 0.96 GPa, although the pore volume began to decrease at this pressure. The uptake of MeOH into the pores therefore results in the marked decrease in compressibility, as noted above.
Previous high-pressure experiments on porous MOFs have resulted in similar behaviour on application of pressure, with the uptake of the media significantly decreasing the compressibility of the framework.24 However, using different hydrostatic media to control the JT switch in any material is, to the best of our knowledge, previously unreported. On increasing pressure above 0.96 GPa, the electron density in the pores decreases, and coincides with a steady reduction in volume of the unit cell. Both an initial increase and then subsequent decrease in uptake of hydrostatic media is common in high-pressure studies of MOFs, and has been seen several times, for example in HKUST-1 (ref. 24c) and MOF-5.24a The ingress of MeOH into the pores on initially increasing pressure to 0.52 GPa is also reflected in a twisting of the octahedra, in-particular the ∠N1Cu1O2C4 angle decreases by 5.8° in MeOH, whereas on compression in FC-70, little to no change is observed in the ∠N1Cu1O2C4 angle to 1.56 GPa. These angles represent a twisting of the dione backbone, which we speculate must interact with the MeOH molecules which penetrate into the framework.
Upon compression in n-pentane, the lightest alkane that is a liquid at ambient temperature, we see different behaviour to that in MeOH or FC-70. Poor data quality permitted only the extraction of unit cell parameters but from this it can be seen that CuPyr-I has undergone the transition to CuPyr-II by 0.77 GPa. This is a significantly lower pressure than is required to induce the phase transition in MeOH (ca. 2.15 GPa). We speculate that this difference in pressure is caused by the n-pentane entering the channels at a lower pressure than MeOH due to the hydrophobic nature of the channels. This can be overcome by MeOH but not until substantially higher pressures, as seen in other MOFs that contain hydrophobic pores.25
On undergoing the transition to CuPyr-II at 2.78 GPa the unit cell volume quadruples, resulting in three symmetry independent channels (12 per unit cell), with the % pore volume continuing to decrease (Table S4†). Additionally, the reflections become much broader, significantly depreciating the data quality. Nevertheless, changes in metal–ligand bond lengths and general packing features can be extracted. In particular, the transition to CuPyr-II results in two independent Cu-centres, with six independent Cu–N/O bond distances per Cu. Each exhibits a continuation of the trend seen in CuPyr-I, with the Cu–O bonds (equivalent to the Cu–O1 bond in CuPyr-I) remaining longer than the JT suppressed Cu–N bonds. However, the transition to CuPyr-II results in both an increase and decrease in three of the four Cu–N and Cu–O bonds respectively, compared to CuPyr-I at 2.15 GPa (Table S6†). The net result is a framework which contains a Cu-centre where the coordination bonds are more equidistant, while the JT axis becomes much more prominent in the other Cu-centre, with the Cu–O dione bond continuing to increase in length. The data for CuPyr-II depreciates rapidly after the phase transition, and more work would be required to study the effect of the anisotropic compression of the JT axis in CuPyr-II on increasing pressure further.
It is difficult to determine the mechanism behind the NLC behaviour observed upon compression of CuPyr-II because the phase transition results in a significant reduction in data quality. Further work will be carried out computationally in order to elucidate the structural mechanism that gives rise to the PLC followed by NLC. However, we propose this effect is inherent to this framework and the ingress of MeOH molecules into the channels allows the retention of crystallinity to allow this behaviour to be observed crystallographically.
In order to determine whether the JT switch could be induced by decreasing temperature and remove any effect the ingress of hydrostatic media has into the pores on the JT switch, variable temperature X-ray diffraction measurements were undertaken on a powder and single-crystal sample. On cooling below 175 K and 150 K in a powder and single-crystal sample respectively, a phase transition was observed, however, this was to a completely different triclinic phase, hereafter referred to as CuPyr-III. The transition here appears to occur when the disordered diethyl ether becomes ordered in the pores, confirmed by determination of the structure by single-crystal X-ray diffraction, where the diethylether could be modelled inside the pore-channel (see ESI Sections 7 & 8 for details†).
In conclusion, we have presented a compression study on the newly synthesised Cu-based porous framework bis[1-(4-pyridyl)butane-1,3-dione]copper(II), referred to as CuPyr, compressed in FC-70 to 1.56 GPa and MeOH to 4.90 GPa. In both FC-70 and MeOH hydrostatic media, the JT axis, which extends along the Cu–N pyridyl bond, steadily compresses and then switches to lie along one of the Cu–O dione bonds. Compression in MeOH results in ingress of the medium into the framework pores, which increases the JT switching pressure to 1.47 GPa, compared with 0.64 GPa during compression in Fluorinert® FC-70. Interaction of stored MeOH with the host framework prompts twisting of the ligand backbone, which is not observed in the absence of adsorbed guest. Suppression of the JT axis is accompanied by a piezochromic colour change in the single crystals from green to dark red, as confirmed by crystallographic and spectroscopic measurements. Increasing the applied pressure to at least 2.15 GPa causes the framework to undergo an isosymmetric phase transition to a previously unobserved phase, characterised by a doubling of the a/b axes. Between 2.15 GPa and 4.90 GPa, NLC behaviour is observed.
This is to the best of our knowledge the first time a phase transition, NLC, piezochromic and pressure induced JT switching behaviour have been observed within the same material. We have also reported for the first time a pressure induced JT axis switch which is hydrostatic media dependent. In further analysis of this system, we intend to study the magnetic properties under ambient and high pressure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments. EKB thanks the EPSRC for funding grant EP/N01331X/1. The support by the Spanish Ministerio de Economía, Industria y Competitividad (PGC2018-101464-B-I00), and INNVAL 18/28 is also acknowledged.References
- (a) S. A. Moggach, S. Parsons and P. A. Wood, Crystallogr. Rev., 2008, 14, 143–184 CrossRef CAS; (b) E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 916–917 CrossRef CAS.
- (a) S. A. Moggach and S. Parsons, in Spectroscopic Properties of Inorganic and Organometallic Compounds: Volume 40, ed J. Yarwood, R. Douthwaite and S. B. Duckett, The Royal Society of Chemistry, 2009, vol. 40, pp. 324–354 Search PubMed; (b) I. E. Collings and A. L. Goodwin, J. Appl. Phys., 2019, 126, 181101–181113 CrossRef.
- (a) K. Ohta, Y. Kuwayama, K. Hirose, K. Shimizu and Y. Ohishi, Nature, 2016, 534, 95–98 CrossRef CAS PubMed; (b) P. Saha, A. Mazumder and G. D. Mukherjee, Geosci. Front., 2020 Search PubMed.
- R. N. Widmer, G. I. Lampronti, S. Anzellini, G. Romain, S. Farsang, C. Zhou, A. M. Belenguer, C. W. Wilson, H. Palmer, A. K. Kleppe, M. T. Wharmby, X. Yu, S. M. Cohen, S. G. Telfer, S. A. T. Redfern, F.-X. Coudert, S. G. MacLeod and T. D. Bennett, Nat. Mater., 2019, 18, 370–376 CrossRef CAS PubMed.
- E. Patyk-Kazmierczak, M. R. Warren, D. R. Allan and A. Katrusiak, Phys. Chem. Chem. Phys., 2017, 19, 9086–9091 RSC.
- D. Umeyama, S. Horike, C. Tassel, H. Kageyama, Y. Higo, K. Hagi, N. Ogiwara and S. Kitagawa, APL Mater., 2014, 2, 124401 CrossRef.
- S. C. McKellar, J. Sotelo, J. P. S. Mowat, P. A. Wright and S. A. Moggach, CrystEngComm, 2016, 18, 1273–1276 RSC.
- B. F. Johnston, W. G. Marshall, S. Parsons, A. J. Urquhart and I. D. H. Oswald, J. Phys. B: At., Mol. Opt. Phys., 2014, 118, 4044–4051 CAS.
- V. D. Kiselev, Int. J. Chem. Kinet., 2013, 45, 613–622 CrossRef CAS.
- H. Yan, F. Yang, D. Pan, Y. Lin, J. N. Hohman, D. Solis-Ibarra, F. H. Li, J. E. P. Dahl, R. M. K. Carlson, B. A. Tkachcenko, A. A. Fokin, P. R. Schreiner, G. Galli, W. L. Mao, Z.-X. Shen and N. A. Melosh, Nature, 2018, 554, 505–510 CrossRef CAS PubMed.
- (a) S. C. McKellar and S. A. Moggach, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 587–607 CrossRef CAS PubMed; (b) R. Monteagudo-Olivan, L. Paseta, G. Potier, P. Lopez-Ram-de-Viu and J. Coronas, Eur. J. Inorg. Chem., 2018, 2019, 29–36 CrossRef.
- J. Sotelo, C. H. Woodall, D. R. Allan, E. Gregoryanz, R. T. Howie, K. V. Kamenev, M. R. Probert, P. A. Wright and S. A. Moggach, Angew. Chem., Int. Ed., 2015, 54, 13332–13336 CrossRef CAS PubMed.
- (a) A. B. Cairns and A. L. Goodwin, Phys. Chem. Chem. Phys., 2015, 17, 20449–20465 RSC; (b) Y. Yan, A. E. O'Connor, G. Kanthasamy, G. Atkinson, D. R. Allan, A. J. Blake and M. Schroder, J. Am. Chem. Soc., 2018, 130, 3952–3958 CrossRef PubMed.
- (a) P. Parois, S. A. Moggach, J. Sanchez-Benitez, K. V. Kamenev, A. R. Lennie, J. E. Warren, E. K. Brechin, S. Parsons and M. Murrie, Chem. Commun., 2010, 46, 1881–1883 RSC; (b) A. Prescimone, C. Morien, D. Allan, J. A. Schlueter, S. W. Tozer, J. L. Manson, S. Parsons, E. K. Brechin and S. Hill, Angew. Chem., Int. Ed., 2012, 51, 7490–7494 CrossRef CAS PubMed; (c) H. L. B. Bostrom, I. E. Collings, A. B. Cairns, C. P. Romao and A. L. Goodwin, Dalton Trans., 2019, 48, 1647–1655 RSC.
- F. Aguado, F. Rodriguez, R. Valiente, J. P. Itie and P. Munsch, Phys. Rev. B, 2004, 70, 214104 CrossRef.
- M. Andrzejewski, N. Casati and A. Katrusiak, Dalton Trans., 2017, 46, 14795–14803 RSC.
- (a) A. U. Ortiz, A. Boutin, A. H. Fuchs and F.-X. Coudert, J. Phys. Chem. Lett., 2013, 4, 1861–1865 CrossRef CAS PubMed; (b) T. D. Bennett, P. J. Saines, D. A. Keen, J.-C. Tan and A. K. Cheetham, Chem.–Eur. J., 2013, 19, 7049–7055 CrossRef CAS PubMed.
- A. Lanza, C. Fiolka, M. Fisch, N. Casati, M. Skoulatos, C. Ruegg, K. W. Kramer and P. Macchi, Chem. Commun., 2014, 50, 14504–14507 RSC.
- (a) H. H. M. Yeung, R. Kilmurray, C. L. Hobday, S. C. McKellar, A. K. Cheetham, D. R. Allan and S. A. Moggach, Phys. Chem. Chem. Phys., 2017, 19, 3544–3549 RSC; (b) D. V. Korabel'nikov and Y. N. Zhuravlev, Phys. Chem. Chem. Phys., 2016, 18, 33126–33133 RSC.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- W. Li, M. R. Probert, M. Kosa, T. D. Bennett, A. Thirumurugan, R. P. Burwood, M. Parinello, J. A. K. Howard and A. K. Cheetham, J. Am. Chem. Soc., 2012, 134, 11940–11943 CrossRef CAS PubMed.
- S. A. Moggach, C. H. Gorbitz and J. E. Warren, CrystEngComm, 2010, 12, 2322–2324 RSC.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- (a) A. J. Graham, D. R. Allan, A. Muszkiewicz, C. A. Morrison and S. A. Moggach, Angew. Chem., Int. Ed., 2011, 50, 11138–11141 CrossRef CAS PubMed; (b) A. J. Graham, A. M. Banu, T. Duren, A. Greenaway, S. C. McKellar, J. P. S. Mowat, K. Ward, P. A. Wright and S. A. Moggach, J. Am. Chem. Soc., 2014, 136, 8606–8613 CrossRef CAS PubMed; (c) A. J. Graham, J.-C. Tan, D. R. Allan and S. A. Moggach, Chem. Commun., 2012, 48, 1535–1537 RSC.
- S. C. McKellar, A. J. Graham, D. R. Allan, M. I. H. Mohideen, R. E. Morris and S. A. Moggach, Nanoscale, 2014, 6, 4163–4173 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1564952, 1564954–1564972, 2008843. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03229h |
| This journal is © The Royal Society of Chemistry 2020 |