
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic enantioselective allene–anhydride approach to β,γ-unsaturated enones bearing an α-all-carbon-quarternary center†
Yuan
Yuan
a,
Xue
Zhang
 *b,
Hui
Qian
*a and
Shengming
Ma
*ab
*b,
Hui
Qian
*a and
Shengming
Ma
*ab
aResearch Center for Molecular Recognition and Synthesis, Department of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P. R. China. E-mail: qian_hui@fudan.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu,, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn
First published on 22nd July 2020
Abstract
A protocol of highly regio- and enantioselective copper-catalyzed hydroacylation of the non-terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in 1,1-disubstituted terminal allenes with anhydrides has been developed. Both aromatic and aliphatic carboxylic anhydrides are applicable to the efficient construction of all carbon quarternary centers connected with a versatile CC bond and a useful ketone functionality. The synthetic potentials of the enantioenriched products have also been demonstrated. Density functional theory (DFT) calculations were performed to explain the steric outcome of the products: the hydroacylation proceeds through a six-membered transition state and the ligand-substrate steric interactions account for the observed enantioselectivity although the chiral ligand is far away from the to-be-genetated chiral center.
C bond in 1,1-disubstituted terminal allenes with anhydrides has been developed. Both aromatic and aliphatic carboxylic anhydrides are applicable to the efficient construction of all carbon quarternary centers connected with a versatile CC bond and a useful ketone functionality. The synthetic potentials of the enantioenriched products have also been demonstrated. Density functional theory (DFT) calculations were performed to explain the steric outcome of the products: the hydroacylation proceeds through a six-membered transition state and the ligand-substrate steric interactions account for the observed enantioselectivity although the chiral ligand is far away from the to-be-genetated chiral center.
Introduction
Carboxylic anhydrides are readily available, stable, easy-to-handle, and widely used as the acyl source to afford ketones by treatment with organomagnesium, lithium, or zinc reagents (Scheme 1A-a).1 However, the challenge lies in the formation of tertiary alcohols as by-products via over-addition to the in situ formed ketones.2 Miura,3a Krische,3b (Scheme 1A-b), Buchwald (Scheme 1A-c)4 and their coworkers reported such transition metal-catalyzed reactions of aryl anhydrides with in situ generated organometallic reagents from hydrometalation of styrenes with different hydride donors offering exclusive approaches from carboxylic anhydrides to ketones. On the other hand, attention has been paid to the corresponding reactions involving allenes5,6 with anhydrides: in 2015, Fujihara and Tsuji reported the Pd-catalyzed formal hydroacylation of mono-substituted terminal allenes with aryl carboxylic anhydrides to afford α,β-unsaturated enones (Scheme 1A-d).7 In 2018, Tsuji and coworkers reported a copper-catalyzed boroacylation of 1,1-disubstituted allenes8 with carboxylic anhydrides, which provide an access to racemic β-boryl-β,γ-unsaturated ketones bearing an all-carbon quarternary center in good to high yields (Scheme 1A-e).9–11 We herein wish to present the first example of highly regio- and enantioselective hydrocupration of 1,1-disubstituted allenes followed by trapping with both aryl and alkyl carboxylic anhydrides to afford optically active β,γ-unsaturated enones bearing an α-all-carbon-quarternary stereocenter, which are challenging to construct (Scheme 1B).12–16 It should be mentioned that the formation of regioisomeric α,β-unsaturated ketones was not observed.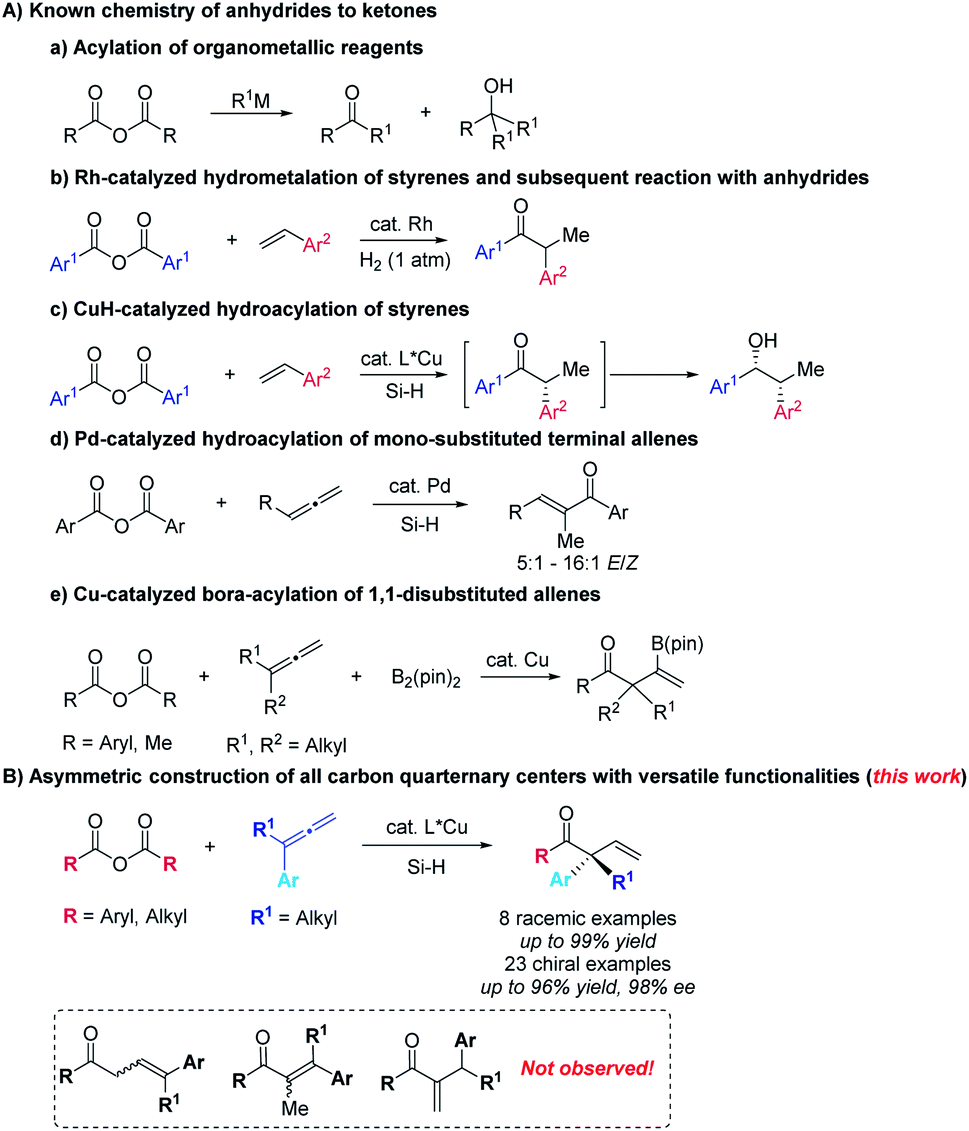 | ||
| Scheme 1 Acylations with carboxylic anhydrides as acyl source. | ||
Results and discussion
Initially, we studied the reaction of Cu(OAc)2-catalyzed hydroacylation of 1,1-disubstituted allene 1a17 with carboxylic acid anhydride 2avia brief screening of non-chiral ligands and solvents (Table 1). Commercially available bidentate ligands such as DPPM, DPPE, DPPP, DPPF, and BINAP were examined and we were delighted to find DPPE could afford 31% yield of the desired β,γ-unsaturated enone rac-3aa (entries 1–5). PCy3 led to full recovery of starting material 1a (entry 6). Studies on the effect of solvents with DPPE as the ligand indicated that the reactions in dioxane, dichloromethane, and toluene afforded the expected product rac-3aa in low yields (entries 8–10) and the best solvent is THF. After further optimization, it was observed that the reaction utilizing 0.5 mmol anhydride 2a, 2.0 equiv. of allene 1a, 2.5 mol% of Cu(OAc)2, 2.5 mol% of DPPE, and 3.0 equiv. of Me(MeO)2SiH in THF at room temperature for 6.0 h could afford rac-3aa in 66% yield (entry 12).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | NMR yield of rac-3aa (%) | Recovery of 1a (%) |
| a Reaction conditions: allene 1a (0.1 mmol), anhydride 2a (1.5 equiv.), Cu(OAc)2 (2.5 mol%), ligand (2.5 mol%), and Me(MeO)2SiH (3.0 equiv.) in solvent (1.0 mL) at room temperature for 6 h. b PCy3 (5.0 mol%) was used. c Allene 1a (0.5 mmol) and 2a (1.5 equiv.) were used. d Allene 1a (2.0 equiv.) and 2a (0.5 mmol) were used. | ||||
| 1 | DPPM | THF | 0 | 100 |
| 2 | DPPE | THF | 31 | 63 |
| 3 | DPPP | THF | 0 | 100 |
| 4 | DPPF | THF | 8 | 84 |
| 5 | BINAP | THF | 21 | 75 |
| 6b | PCy3 | THF | 0 | 100 |
| 7 | DPPE | Cyclohexane | 0 | 100 |
| 8 | DPPE | Dioxane | 15 | 78 |
| 9 | DPPE | DCM | 10 | 79 |
| 10 | DPPE | Toluene | 24 | 62 |
| 11c | DPPE | THF | 49 | 4 |
| 12d | DPPE | THF | 66 | — |

|
||||

The scope of this reaction was then demonstrated and the typical results were shown in Table 2. Racemic β,γ-unsaturated enones rac-3 were obtained in good to excellent yields and the reaction is amenable to both terminal 1-electron-withdrawing or electron-donating group substituted aryl-1-alkylallenes and differently substituted aryl anhydrides (m-Me, p-F, p-Cl, and p-t-Bu).

Encouraged by these results, we turned our efforts to develop the enantioselective reaction by using 3-phenyl-nona-1,2-diene (1a) and benzoic anhydride (2a) as the model substrates again (Table 3). To our delight, when (R)-BINAP was used as the chiral ligand, the desired product 3aa was obtained in 74% NMR yield with 79% ee. Biaryl-based (R)-MeO-Biphep, (R)-C3-Tunephos, (R)-Garphos, (S)-Synphos, and (R)-Segphos could afford the desired product 3aa in good yield with 78–87% ee. Non-rigid (R,R)-BDPP could also give a moderate enantio-selectivity while the spiro-ligand (R)-SDP was not working for this reaction. Finally, we had identified successfully (R,R)-BPE as a promising ligand, affording (R)-3aa in 34% NMR yield with 91% ee.
| a Reaction conditions: 1a (0.1 mmol), 2a (1.5 equiv.), Cu(OAc)2 (5 mol%), ligand (5 mol%), and Me(MeO)2SiH (4.0 equiv.) in THF (1.0 mL) at 40 °C for 12 h. Yield was determined by 1H NMR analysis using dibromomethane as the internal standard. |
|---|

|
When the reaction was conducted at room temperature, both the yield and ee value increased (Table 4, entry 1). Shortening the reaction time to 5 h improved the yield to 92% with 96% ee (Table 4, entry 2). Three equivalents of Me(MeO)2SiH were still efficient for this process (Table 4, entry 3). However, recovery of 1a was observed when further decreasing the loading of silane (Table 4, entries 4 & 5). When the reaction was conducted on 1.0 mmol scale for 6 h, the loading of Cu(OAc)2 and (R,R)-BPE could be reduced to 2.5 mol% affording (R)-3aa in 86% NMR yield with 95% ee, which was chosen as the optimal reaction conditions (Table 4, entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | x | t (h) | Recovery of 1ab (%) | NMR yield of 3aab (%) | ee (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), Cu(OAc)2 (5 mol%), (R,R)-BPE (5 mol%), and Me(MeO)2SiH in THF (1.0 mL) at room temperature unless otherwise noted. b Determined by 1H NMR analysis using MeNO2 as the internal standard. c The reaction was conducted using Cu(OAc)2 (2.5 mol%) and (R,R)-BPE (2.5 mol%). d The reaction was conducted on 1.0 mmol scale. | |||||
| 1 | 4.0 | 12 | — | 68 | 93 |
| 2 | 4.0 | 5 | — | 92 | 96 |
| 3 | 3.0 | 5 | — | 89 | 94 |
| 4 | 2.0 | 5 | 15 | 82 | 94 |
| 5 | 1.0 | 5 | 50 | 50 | 94 |
| 6c,d | 3.0 | 6 | — | 86 | 95 |
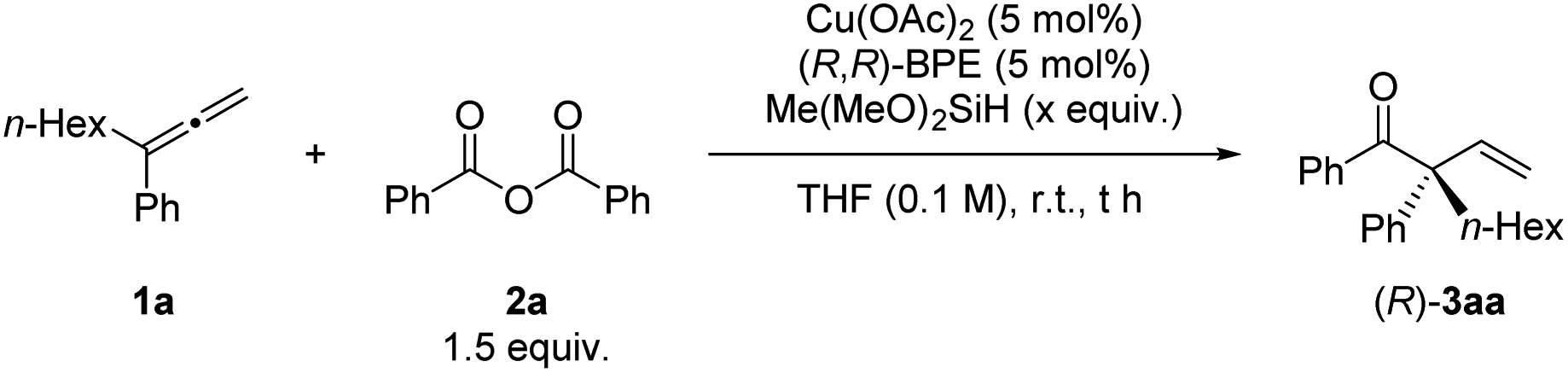
With the optimized conditions in hand, we next examined the scope of this enantioselective reaction. As shown in Table 5, a wide range of terminal 1,1-disubstituted allenes bearing one aryl group and one alkyl group reacted smoothly with 2a to form the corresponding β,γ-unsaturated enone products (R)-3 with excellent enantioselectivity. The absolute configuration of these products has been established via X-ray crystallography of (R)-3ea. Allenes with the aryl group (Ar) bearing different substituents, including alkoxy group and halides, are all suitable substrates ((R)-3fa to (R)-3ka). Furthermore, the substrate scope of carboxylic anhydrides with allene 1a was examed. Both electron-rich and electron-deficient aryl carboxylic anhydrides gave the products in high to excellent yields and ees ((R)-3ab to (R)-3ag). It is worth mentioning that alkyl carboxylic acid anhydrides also worked well under the optimized conditions to afford the desired products with excellent enantioselectivity ((R)-3ah–3aj). When (S,S)-BPE was used as the ligand, the enantiomers could be obtained ((S)-3af, (S)-3ai, and (S)-3aj). In all these reactions, only the more substituted CC bond in allene reacted formally.
| a Reaction conditions: allene 1 (1.0 mmol), anhydride 2 (1.5 equiv.), Cu(OAc)2 (2.5 mol%), (R,R)-BPE (2.5 mol%) and Me(MeO)2SiH (3.0 equiv.) in THF (10.0 mL) at room temperature for 6 h unless otherwise noted. b Allene 1 (1.5 equiv.), anhydride 2a (1.0 mmol) and Me(MeO)2SiH (1.5 equiv.) were used instead. c Allene 1a (2.0 equiv.), anhydride 2 (1.0 mmol) were used instead. d Allene 1a (1.5 equiv.), anhydride 2 (0.5 mmol), and (S,S)-BPE (2.5 mol%) were used instead. |
|---|

|
The practicality of this reaction has been demonstrated by executing a 10.0 mmol scale reaction, affording 2.4974 g of (R)-3ka in 79% yield with 96% ee (Scheme 2a). The highly functionalized products could be easily transformed to other useful molecules (Scheme 2b): after treatment with hydroxylamine hydrochloride,18 the β,γ-unsaturated enone could be transformed to β,γ-unsaturated ketoxime (R,Z)-4, which was reported to be very useful and convenient starting material for the synthesis of isoxazoline.18,19 The aryl–Br bond could undergo Suzuki coupling to afford (R)-5 in 90% yield with 96% ee. The carbon–carbon double bond may undergo aldehyde-selective Wacker-type oxidation and hydrogenation to form the corresponding products (R)-6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and (R)-7 in excellent enantiomeric excess.
20 and (R)-7 in excellent enantiomeric excess.
 | ||
| Scheme 2 Gram scale synthesis of (R)-3ka and its synthetic potentials. | ||
A plausible catalytic cycle was proposed (Scheme 3): hydrocupration of the terminal CC bond in allene 1d with the in situ formed copper-hydride species I would generate the allyl copper specie E-II. Subsequent nucleophilic addition of E-II with anhydride 2a would afford the copper species IIIvia the six-membered ring chair-like transition state (TS). The β-O elimination of III would eventually regenerate the CO bond affording enantioenriched (R)-3da and the Cu-OBz specie IV, which would react with Me(MeO)2SiH to regenerate copper-hydride I to finish the catalytic cycle.
 | ||
| Scheme 3 The proposed mechanism. | ||
It should be noted that the chiral ligand is far away from the to-be-generated chiral center as shown in the six-membered ring chair-like transition state (TS). To pursue a further understanding of the observed enantioselectivity of the current reaction, DFT calculations were performed at the M06/6-311+G(d,p)-SDD/SMD(THF)//B3LYP/6-31G(d)-LANL2DZ level, using allene 1d and benzoic anhydride 2a as the model substrates (see computational methods in the ESI for detailed information†). The reported energies are Gibbs energies that incorporate the effect of the THF solvent.
The reaction proceeds initially from the hydrocupration of allene 1d with the in situ generated copper-hydride as shown in Scheme 4. Both terminal and internal double bonds of allene 1d could participate in the hydrocupration, thus, four competing transition structures associated with the hydrocupration step are obtained, which are denoted as TS1_a–d, separately (see the ESI for other less favorable transition structures†). The activation barriers required for TS1_a–d are 18.3, 17.0, 19.9 and 18.8 kcal mol−1 respectively, indicating that this hydrocupration step favours terminal CC bond over internal CC bond in allene 1d.
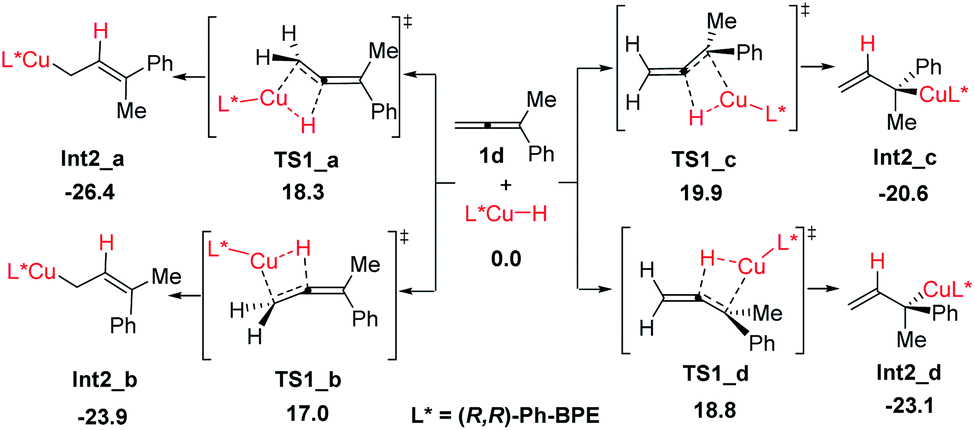 | ||
| Scheme 4 Four competing transition structures associated with the hydro-cupration step. All energies are in kcal mol−1 with respect of CuH and allene (1d). | ||
Four allylcoppers Int2_a–d could be provided irreversibly from the hydrocupration, among which the kinetically less favorable E-isomer of the terminal allylic copper Int2_a is the thermodynamically most stable. Further calculation reveals that Int2_a–d may isomerize to each other via 1,3-Cu shift (Scheme 5), and these interconversions are rather facial with barriers of less than 8 kcal mol−1.
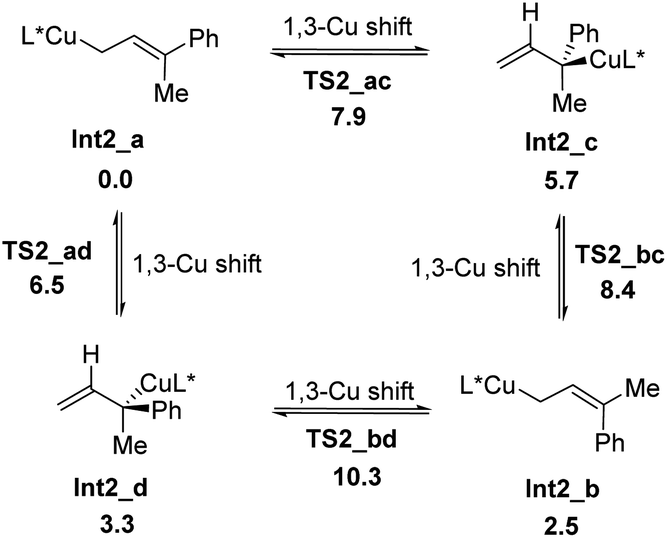 | ||
| Scheme 5 The isomerization of Int2_a–d. All energies are in kcal mol−1 with respect of Int2_a. | ||
The subsequent addition of the benzoic anhydride to terminal allylic copper Int2_a or Int2_b could proceed via the six-membered cyclic chair-like transition states. The benzoic anhydride 2a approaches from both faces of the prochiral center of Int2_a and Int2_b, leading to four six-membered transition states, which are denoted as TS3_a1, TS3_a2, TS3_b1 and TS3_b2, separately (Scheme 6a) (The benzoic anhydride 2a participates with Re-face in these four transition states (see the ESI for the other four less favorable transition structures with Si-face 2a participation†)). Geometry analysis of the six-membered transition structures indicates that TS3 should be a late transition state (see the ESI for the detail†).
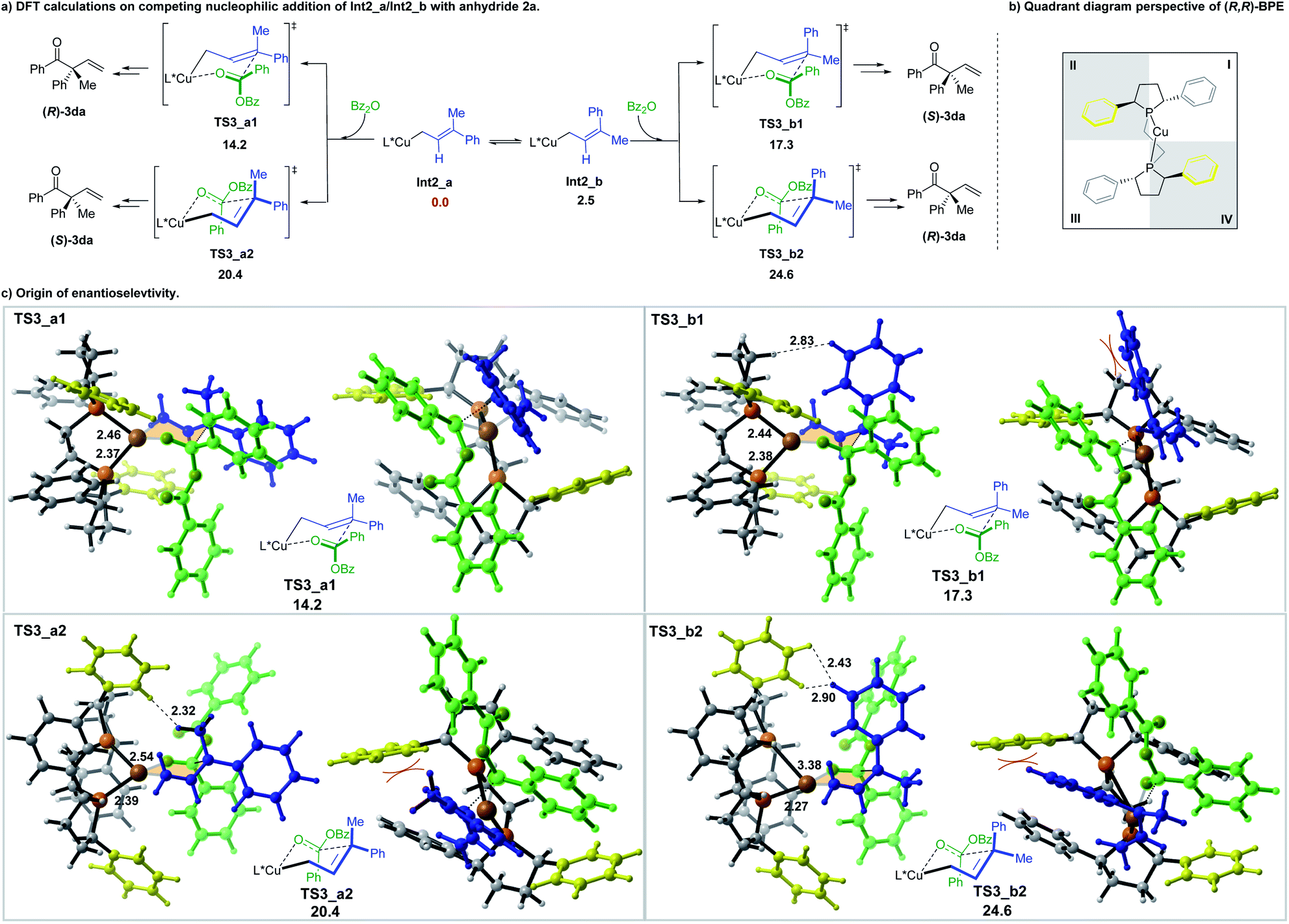 | ||
| Scheme 6 The origin of enatioselectivity of the nucleophilic addition of 2a to Int2_a and Int2_b. All energies are in kcal mol−1 with respect of Int2_a. Bond lengths are given in angstroms. The right side drawings in (c) are the views in quadrant as shown in (b). | ||
Among the four transition states, a Re-face approach of 2a to the E-allyl intermediate Int2_a (TS3_a1, 14.2 kcal mol−1), which leads to the formation of (R)-3da, is found to be the most favorable one, thus, accounting for the domination of the R-product observed in the experiment (Scheme 6).
The origin of the enantioselectivity can be visualized in the quadrant diagrams. Scheme 6b shows a quadrant diagram perspective of the (R,R)-BPE ligand, while Scheme 6c are the illustration of the four six-membered transition structures. In Scheme 6b, the phenyl groups in quadrants II and IV extend forward, which is in the same direction with the coordinated copper center, thus making these two locations more hindered than the other two. In the transition structures of TS3_a1 and TS3_a2, which both originated from the E-allyl intermediate Int2_a, the smaller substituent (Me) locates at the pseudoaxial position of the chair-like six-member ring. The allyl moiety of TS3_a1 is placed in the less hindered quadrant I. However, the methyl substituent of TS3_a2 suffers an unfavourable steric repulsion with ligand phenyl group in quadrant II (H⋯H distance of only 2.32 Å), thus interpreting the preference of TS3_a1 over TS3_a2 by 6.2 kcal mol−1. Alternatively, in TS3_b1 and TS3_b2, associated with the Z-allyl intermediate Int2_b, the pseudoaxial occupation of the large phenyl group destabilizes both structures. The steric hindrance between the phenyl group and the carbon chain in phospha-five-membered-ring of ligand makes TS3_b1 less favorable by 3.1 kcal mol−1 in energy than TS3_a1. Moreover, severe steric interaction between the pseudoaxial phenyl group and the ligand phenyl group in quadrant II further destabilizes TS3_b2, makes it the least stable one (10.4 kcal mol−1 higher in energy than TS3_a1). To reduce the steric repulsion, one phosphine is obliged to partly dissociate with the metal center, leading to the increasing of one of the Cu-P distance to 3.38 Å.
Conclusions
In summary, we have developed a highly efficient regio- and enantio-selective method for the construction of all carbon quarternary stereocenter linked with a CC bond and a carbonyl group from 1,1-disubstituted allenes and carboxylic anhydrides. The current reaction represents the first copper-catalyzed enantioselective hydroacylation of allenes with carboxylic anhydrides. Enabled by the mild reaction conditions, the reactions proceed efficiently with good functional group compatibility. Synthetic transformations to other quarternary-carbon-center-containing molecules have also been established. Based on DFT calculations, the origin of the enantioselectivity has also been rationalized.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (Grant No. 21690063 for S. Ma and Grant No. 21801041 for H. Qian) are greatly appreciated. We thank Mr Yizhan Zhai in this group for reproducing the results of rac-3ae presented in Table 2, and Mr Yulong Song in this group for reproducing the results of (R)-3ea and (R)-3aj in Table 5.Notes and references
-
R. C. Larock, A. V. Dubrovskiy and N. A. Markina, in Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 3rd edn, 2018, pp. 1–95 Search PubMed
.
- Y. Miki, H. Hachiken and I. Yoshikawa, Heterocycles, 1997, 6, 1143 CrossRef
-
(a) K. Kokubo, M. Miura and M. Nomura, Organometallics, 1995, 14, 4521 CrossRef CAS
- J. S. Bandar, E. Ascic and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 5821 CrossRef CAS PubMed
- For selected book and reviews on synthesis of allenes, see:
(a)
N. Krause and A. S. K. Hashimi, in Modern Allene Chemistry, Wiley-VCH, Weinheim, 2004, vol. 1 CrossRef
- For selected book and reviews on transformation of allenes, see:
(a)
N. Krause and A. S. K. Hashimi, in Modern Allene Chemistry, Wiley-VCH, Weinheim, 2004, vol. 2 CrossRef
- T. Fujihara, T. Hosomi, C. Cong, T. Hosoki, J. Terao and Y. Tsuji, Tetrahedron, 2015, 71, 4570 CrossRef CAS
- For selected reviews on hydrometalation of allenes, see:
(a) A. P. Pulis, K. Yeung and D. J. Procter, Chem. Sci., 2017, 8, 5240 RSC
- A. Sawada, T. Fujihara and Y. Tsuji, Adv. Synth. Catal., 2018, 360, 2621 CrossRef CAS
- For non-enantioselective hydrocupration of allenes, see:
(a) Y. Tani, K. Kuga, T. Fujihara, J. Terao and Y. Tsuji, Chem. Commun., 2015, 51, 13020 RSC
- For non-enantioselective borylcupration of allenes, see:
(a) J. Rae, K. Yeung, J. J. W. McDouall and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 1102 CrossRef CAS PubMed
- For enantioslelective hydrocupration of allenes, trapping with ketones, see:
(a) E. Y. Tsai, R. Y. Liu, Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 2007 CrossRef CAS PubMed
- For enantioslelective borylcupration of allenes, trapping with imines, see:
(a) H. Jang, F. Romiti, S. Torker and A. H. Hoveyda, Nat. Chem., 2017, 9, 1269 CrossRef CAS PubMed
- For selected reviews on construction of quarternary carbon centers, see:
(a) Y. Li and S. Xu, Chem.–Eur. J., 2018, 24, 16218 CrossRef CAS PubMed
- For selected reviews on construction of quarternary carbon centers in acyclic systems, see:
(a) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564 CrossRef CAS PubMed
- For selected examples on asymmetric synthesis of α-all-carbon-quarternary β,γ-unsaturated carbonyl compounds, see:
(a) P. A. Evans, S. Oliver and J. Chae, J. Am. Chem. Soc., 2012, 134, 19314 CrossRef CAS PubMed
- J. Li, C. Zhou, C. Fu and S. Ma, Tetrahedron, 2009, 65, 3695 CrossRef CAS
- F. Chen, F.-F. Zhu, M. Zhang, R.-H. Liu, W. Yu and B. Han, Org. Lett., 2017, 19, 3255 CrossRef CAS PubMed
-
(a) M.-K. Zhu, J.-F. Zhao and T.-P. Loh, J. Am. Chem. Soc., 2010, 132, 6284 CrossRef CAS PubMed
- K. E. Kim, J. Li, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 13179 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1939837. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03227a |
| This journal is © The Royal Society of Chemistry 2020 |