
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances of group 14 dimetallenes and dimetallynes in bond activation and catalysis
Franziska
Hanusch
 ,
Lisa
Groll
and
Shigeyoshi
Inoue
*
,
Lisa
Groll
and
Shigeyoshi
Inoue
*
Department of Chemistry, Catalysis Research Center and Institute of Silicon Chemistry, Technische Universität München, Lichtenbergstrasse 4, Garching bei München, 85748, Germany. E-mail: s.inoue@tum.de
First published on 3rd August 2020
Abstract
Since the first heavy alkene analogues of germanium and tin were isolated in 1976, followed by West's disilene in 1981, the chemistry of stable group 14 dimetallenes and dimetallynes has advanced immensely. Recent developments in this field veered the focus from the isolation of novel bonding motifs to mimicking transition metals in their ability to activate small molecules and perform catalysis. The potential of these homonuclear multiply bonded compounds has been demonstrated numerous times in the activation of H2, NH3, CO2 and other small molecules. Hereby, the strong relationship between structure and reactivity warrants close attention towards rational ligand design. This minireview provides an overview on recent developments in regard to bond activation with group 14 dimetallenes and dimetallynes with the perspective of potential catalytic applications of these compounds.
 Franziska Hanusch | Franziska Hanusch graduated from the Department of Chemistry at the Technische Universität München with a M.Sc. in 2014 under the guidance of Prof. Florian Kraus. After a research fellowship at the Ruhr-Universität-Bochum with Prof. Roland Fischer, she joined the group of Prof. Shigeyoshi Inoue as a PhD student in 2016. She is currently working on bis-NHI-stabilized low-valent silicon compounds, their transition metal complexes and reactivity towards small molecules. |
 Lisa Groll | Lisa Groll received her M.Sc. degree from the Technische Universität München in 2019 under the supervision of Prof. Klaus Köhler. After that, she started her doctoral work in the research group of Prof. Shigeyoshi Inoue. There she is pursuing her research interests in the isolation of low-coordinate germanium and tin compounds with the goal of small molecule activation and catalysis. |
 Shigeyoshi Inoue | Shigeyoshi Inoue studied at the University of Tsukuba and carried out his doctoral studies under the supervision of Prof. Akira Sekiguchi, obtaining his PhD in 2008. As a Humboldt grantee and a JSPS grantee, he spent the academic years 2008–2010 at the Technische Universität Berlin in the group of Prof. Matthias Drieß. In 2010, he established an independent research group within the framework of the Sofja Kovalevskaja program at the Technische Universität, Berlin. Since 2015 he has been on the faculty at the Technische Universität München (TUM). His current research interests focus on the synthesis, characterization, and reactivity investigation of compounds containing low-valent main group elements with unusual structures and unique electronic properties, with the goal of finding novel applications in synthesis and catalysis. A particular emphasis is placed on low-valent silicon and aluminium compounds. |
Introduction
Much of the early advances in heavier group 14 chemistry stem from the vast richness of organic compounds and the motivation of mimicking those with the heavier congeners of carbon. Due to their ubiquity in hydrocarbons and simultaneous scarcity in heavier homologs, double and triple bonds are highly sought-after bonding motifs in Si, Ge, Sn and Pb chemistry. In the infancy of this chemistry, isolation of heavy group 14 multiple bonds proved to be a challenging endeavor. The consensus went even so far that the so-called “double bond rule”, according to which heavy homologues of carbon (and heavy p block elements in general) cannot form stable multiple bonds, was widely accepted.1–3 This rule was, however, unequivocally disproved with the first heavy alkene analogues of germanium and tin by Lappert in 1976, followed by West's disilene as well as Brook's silaethene and Yoshifuji's diphosphene only 5 years later.4–8 With this, the search for novel, previously labeled “non-existent”, compounds sparked off. But it took almost two decades until the first heavy acetylene analogue, a diplumbyne9 by Power, was realized, followed by the completion of the series in subsequent years.10–12 With the isolation of more and more stable compounds, the motivation for novel bonding motifs was expanded by the incentive to mimic transition metals in their ability to activate small molecules and perform catalysis.13 Up until recently, this kind of reactivity was thought to be reserved almost exclusively for d-block elements. Low valent main-group species do, however, allow comparison to open-shell transition-metal complexes due to possession of donor/acceptor valence orbitals with small energy gaps. Dimetallenes and dimetallynes combine the reactivity of electron rich multiple bonds and the unique electronic properties of low valent group 14 compounds. Moreover, as dinuclear complexes they could possibly benefit from cooperative effects of two tetrel atoms. This combination makes them promising candidates for the activation of enthalpically strong small molecules. Power's seminal review in 2010 highlighted at first the great potential of main-group compounds in that regard.13 A challenge, nonetheless, is still the flexible interchange between several oxidation states displayed by d-block complexes. Group 14 multiply bonded species tend to form very stable oxidative addition products, making reductive elimination, the next key step in a typical catalytic cycle for transition metals, very difficult.14,15 There is, however, significant progress being made in that regard, so much so that recently the first catalytic cyclotrimerization of terminal alkynes by a digermyne could be realized.16 This minireview aims to provide insight into the recent developments towards small molecule activation, bond activation and catalytic applications of group 14 compounds with homonuclear multiple bonds, i.e. heavy alkene and alkyne analogues as well as interconnected bismetallylenes.Bonding nature
Dimetallenes and dimetallynes are compounds of the form R2E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ER2 and RE
ER2 and RE![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ER, where E is a heavier group 14 element (E = Si, Ge, Sn, Pb). In their formula, they are the direct descendants of alkenes and alkynes. However, they manifest not as planar E2H4 (like ethylene) or linear E2H2 (like acetylene), but as a trans-bent structure. The reason for this drastic structural difference stems from an increased size difference of the valence orbitals leading to a less effective hybridization of the heavier elements.17,18 To illustrate the resulting deviation in bonding, parent group 14 multiple bonds can be seen as an interaction of two monomeric ‘H2E:’ or ‘HE:·’ units, respectively. While methylene (H2C:) is a ground state triplet (ΔEST = −14.0 kcal mol−1) and therefore is capable of forming a classical planar double bond (Fig. 1a), heavier tetrylenes (H2E:) almost exclusively occupy a singlet ground state (ΔEST = 16.7 kcal mol−1 (Si), 21.8 kcal mol−1 (Ge), 24.8 kcal mol−1 (Sn), 34.8 kcal mol−1 (Pb)).19 In triply bonded compounds in consequence, alkynes possess sp-hybridized carbon atoms in the quartet ground state that result in three covalent bonds and a linear structure (Fig. 2a). In dimetallynes the doublet ‘HE:·’ moieties are not capable of forming covalent triple bonds. In both cases, significant Pauli repulsion between the occupied orbitals or the excitation into the singlet or quartet state, respectively, would need to be overcome in order to form a standard E–E multiple bond.13,20–23 Therefore a double donor–acceptor interaction is energetically favored in which each partner of the multiple bond donates from their lone pair of electrons into the vacant p orbital of the other (Fig. 1b and 2b).
ER, where E is a heavier group 14 element (E = Si, Ge, Sn, Pb). In their formula, they are the direct descendants of alkenes and alkynes. However, they manifest not as planar E2H4 (like ethylene) or linear E2H2 (like acetylene), but as a trans-bent structure. The reason for this drastic structural difference stems from an increased size difference of the valence orbitals leading to a less effective hybridization of the heavier elements.17,18 To illustrate the resulting deviation in bonding, parent group 14 multiple bonds can be seen as an interaction of two monomeric ‘H2E:’ or ‘HE:·’ units, respectively. While methylene (H2C:) is a ground state triplet (ΔEST = −14.0 kcal mol−1) and therefore is capable of forming a classical planar double bond (Fig. 1a), heavier tetrylenes (H2E:) almost exclusively occupy a singlet ground state (ΔEST = 16.7 kcal mol−1 (Si), 21.8 kcal mol−1 (Ge), 24.8 kcal mol−1 (Sn), 34.8 kcal mol−1 (Pb)).19 In triply bonded compounds in consequence, alkynes possess sp-hybridized carbon atoms in the quartet ground state that result in three covalent bonds and a linear structure (Fig. 2a). In dimetallynes the doublet ‘HE:·’ moieties are not capable of forming covalent triple bonds. In both cases, significant Pauli repulsion between the occupied orbitals or the excitation into the singlet or quartet state, respectively, would need to be overcome in order to form a standard E–E multiple bond.13,20–23 Therefore a double donor–acceptor interaction is energetically favored in which each partner of the multiple bond donates from their lone pair of electrons into the vacant p orbital of the other (Fig. 1b and 2b).
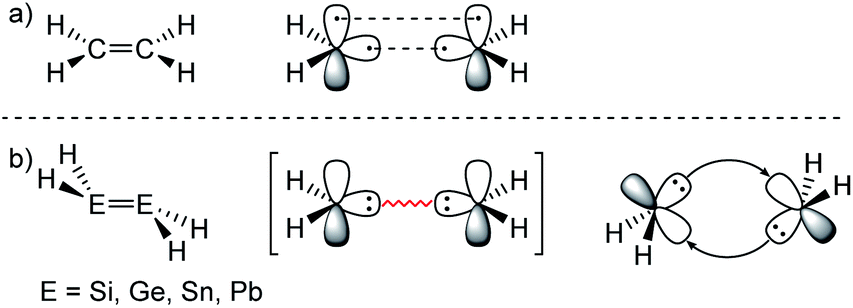 | ||
| Fig. 1 (a) Ethylene with a covalent bonding description. (b) CGMT model of dimetallenes with a bonding description. | ||
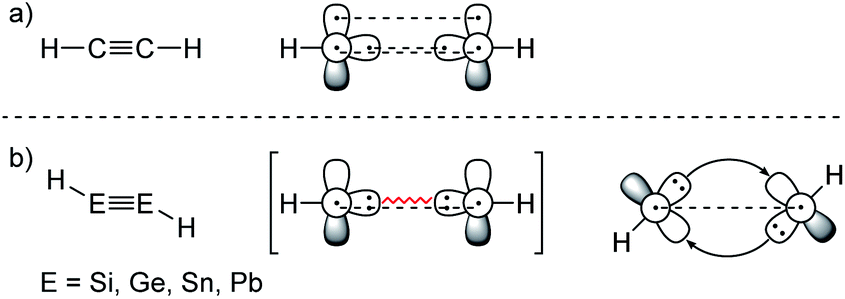 | ||
| Fig. 2 (a) Acetylene with a covalent bonding description. (b) CGMT model of dimetallynes with a bonding description. | ||
The Carter–Goddard–Malrieu–Trinquier (CGMT) model that describes the bonding of main-group multiple bonds was initially proposed for Lappert's distannene and is probably the most intuitive approach, since the principle of a classical multiple bond is retained.5,21,22 The increasing deviation from a planar double bond of dimetallenes with the descend down group 14 can be interpreted by an increasing stability of the singlet ground state, leading to more pronounced trans-bending and weakening of the EE bond.24,25 For dimetallynes, the same effect can be seen in the orbital scheme in Fig. 3. It can be used to describe the severeness of the bending and shows the orbital mixing of HEEH in linear, trans-bent and H:E–E:H structures in the right-angled form according to the Jahn–Teller distortion model. The mixing of σ and π orbitals of suitable symmetry leads to distorted orbitals and therefore to the observed morphed structures.17,26,27
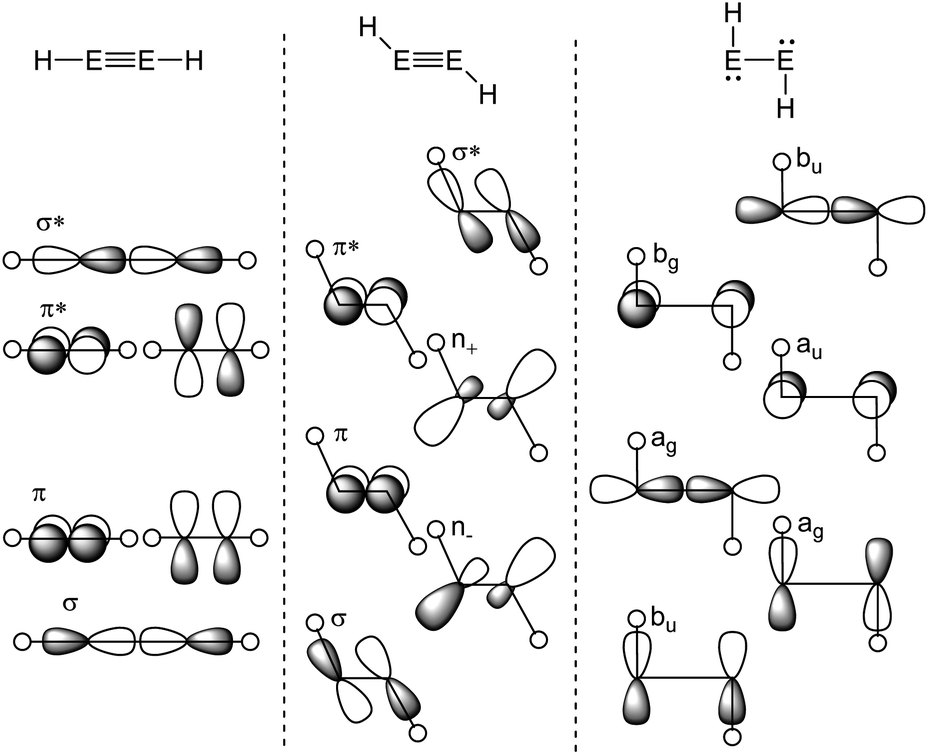 | ||
| Fig. 3 Orbital scheme of the second order Jahn–Teller distortion in HEEH (E = Si, Ge, Sn, Pb).17,28,29 | ||
As in the case of dimetallenes, the donor–acceptor type interactions in dimetallynes show fading stability when going down the group (Fig. 4). The trend starts with disilynes, that show strong triple bond character with short Si–Si bonds (Si2H2, Si–Sicalc. = 2.1 Å31) and relatively big H–Si–Si angles (Si2H2calc. ∼ 125°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31),32,33 and ends with diplumbynes that are almost exclusively represented in the form of bisplumbylenes – singly bonded compounds with two lone pairs of electrons, having long Pb–Pb distances (Pb2H2, Pb–Pbcalc. = 3.2 Å34) and high trans-bent angles near 90°.34 In between, the bonding characteristics for Ge- and Sn-compounds are hard to be defined precisely and therefore need to be discussed in every case using theoretical considerations and structural characteristics.26,30,35 Therefore, different resonance structures with varying weighting contribute to the bonding motifs of dimetallynes (Fig. 4). Beyond the pure understanding of bonding characters, the described effects influence strongly the reactivity that is observed and expected for heavier group 14 multiply bonded compounds that make them so attractive for small molecule activation. Like all multiple bonds, the E–E bonds also show high electron densities that result in a pronounced nucleophilicity. Nevertheless, unlike organic bonds, donor–acceptor type interactions in dimetallenes and dimetallynes allow further reactivities due to lower E–E bond energies and two lone pairs of electrons that are embedded into the multiple bond (see Fig. 1b and 2b). Like in multiply bonded transition-metal M–M and M–C compounds,36–38 cycloaddition is an often-observed reaction for dimetallenes and dimetallynes.16,39–53 Moreover, when going down the group these effects get even stronger, with more exposed lone pairs and decreasing E–E bond energies. For dimetallynes, singlet diradical character can be seen as a mesomeric structure and in reaction, respectively. In a recent study Britt and Power could indeed show the dissociation of the distannyne Ar′SnSnAr′ (5b) into Sn(I)-radicals :SṅAr′ in toluene solution that could be detected via EPR spectroscopy. These findings also strongly support the concept of charge-shift bonds being present in dimetallynes.54 Not ending at this point, dimetallenes and dimetallynes are dinuclear complexes, in which two E atoms build the reactive core within the complex. Taking nature as a benchmark for efficient and low energy consuming activation and conversion of relatively inert small molecules, one strategy that could be mimicked by chemists is the use of two or more metal centers within the active site of a catalyst. These catalysts benefit from cooperative and synergetic effects between the metal centers that lower activation barriers and improve selectivity.55 In dimetallenes and dimetallynes cooperative effects can take place between two neighboring tetrel elements. Due to the multiple bond character, addition of small molecules would not necessarily break the E–E bond and with that bond still intact, conversion, insertion, or addition of another molecule is rendered possible. Additionally, the release of a product molecule could be accompanied by restoration of the multiple bond. All these steps are crucial for performing small molecule activation to achieve full catalytic cycles and applications.
31),32,33 and ends with diplumbynes that are almost exclusively represented in the form of bisplumbylenes – singly bonded compounds with two lone pairs of electrons, having long Pb–Pb distances (Pb2H2, Pb–Pbcalc. = 3.2 Å34) and high trans-bent angles near 90°.34 In between, the bonding characteristics for Ge- and Sn-compounds are hard to be defined precisely and therefore need to be discussed in every case using theoretical considerations and structural characteristics.26,30,35 Therefore, different resonance structures with varying weighting contribute to the bonding motifs of dimetallynes (Fig. 4). Beyond the pure understanding of bonding characters, the described effects influence strongly the reactivity that is observed and expected for heavier group 14 multiply bonded compounds that make them so attractive for small molecule activation. Like all multiple bonds, the E–E bonds also show high electron densities that result in a pronounced nucleophilicity. Nevertheless, unlike organic bonds, donor–acceptor type interactions in dimetallenes and dimetallynes allow further reactivities due to lower E–E bond energies and two lone pairs of electrons that are embedded into the multiple bond (see Fig. 1b and 2b). Like in multiply bonded transition-metal M–M and M–C compounds,36–38 cycloaddition is an often-observed reaction for dimetallenes and dimetallynes.16,39–53 Moreover, when going down the group these effects get even stronger, with more exposed lone pairs and decreasing E–E bond energies. For dimetallynes, singlet diradical character can be seen as a mesomeric structure and in reaction, respectively. In a recent study Britt and Power could indeed show the dissociation of the distannyne Ar′SnSnAr′ (5b) into Sn(I)-radicals :SṅAr′ in toluene solution that could be detected via EPR spectroscopy. These findings also strongly support the concept of charge-shift bonds being present in dimetallynes.54 Not ending at this point, dimetallenes and dimetallynes are dinuclear complexes, in which two E atoms build the reactive core within the complex. Taking nature as a benchmark for efficient and low energy consuming activation and conversion of relatively inert small molecules, one strategy that could be mimicked by chemists is the use of two or more metal centers within the active site of a catalyst. These catalysts benefit from cooperative and synergetic effects between the metal centers that lower activation barriers and improve selectivity.55 In dimetallenes and dimetallynes cooperative effects can take place between two neighboring tetrel elements. Due to the multiple bond character, addition of small molecules would not necessarily break the E–E bond and with that bond still intact, conversion, insertion, or addition of another molecule is rendered possible. Additionally, the release of a product molecule could be accompanied by restoration of the multiple bond. All these steps are crucial for performing small molecule activation to achieve full catalytic cycles and applications.
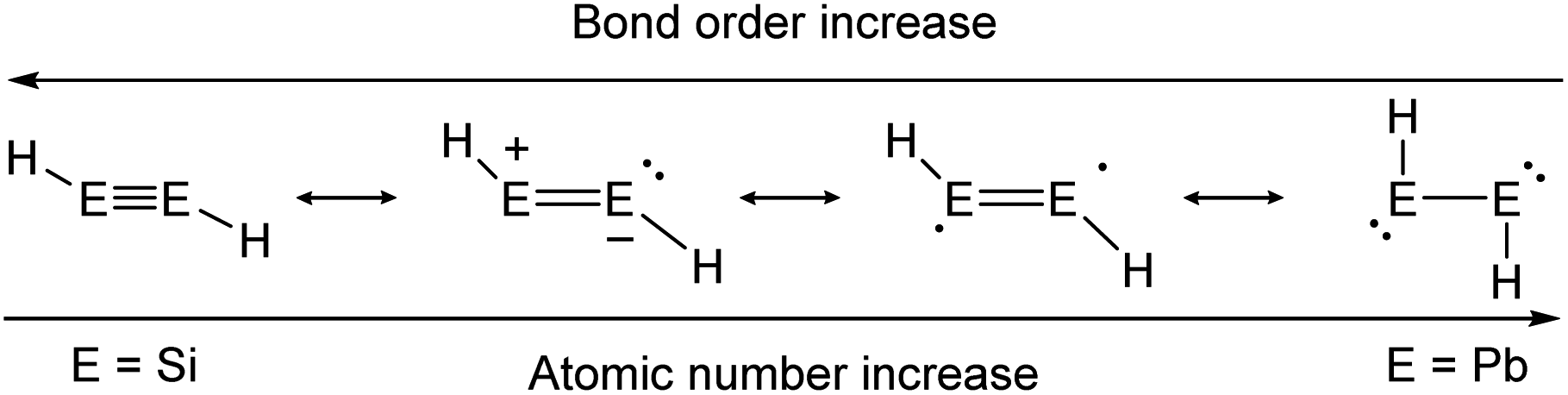 | ||
| Fig. 4 Trends in bonding of heavier triple bonds, illustrated by mesomeric structures of HEEH (E = Si, Ge, Sn, Pb).26,30 | ||
Synthesis and ligand design
The synthesis of E–E multiple bonds has proven to be quite diverse. Scheme 1 and 2, however, show the main synthesis pathways for dimetallenes and dimetallynes, respectively. Reductive dehalogenation of di- (R2EX2) or trihalometallanes (REX3), dimetallanes (X2REERX2), dimetallenes (XREERX), or metallylenes (XRE:) using reductive agents (red.) such as KC8,45,47,56–58 metal naphthalenides,59,60 and [(MesNacnac)Mg]2 (MesNacnac = (MesNCMe)2CH)61 is the central motif in isolation of these compounds.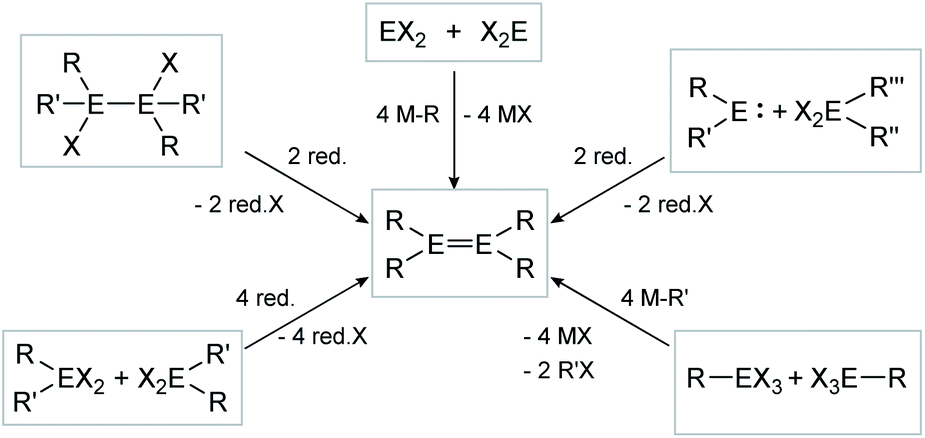 | ||
| Scheme 1 Possible synthesis routes for the isolation of dimetallenes. E = Si, Ge, Sn, Pb; R = stabilizing ligand; X = halogen; red. = reductive agent. | ||
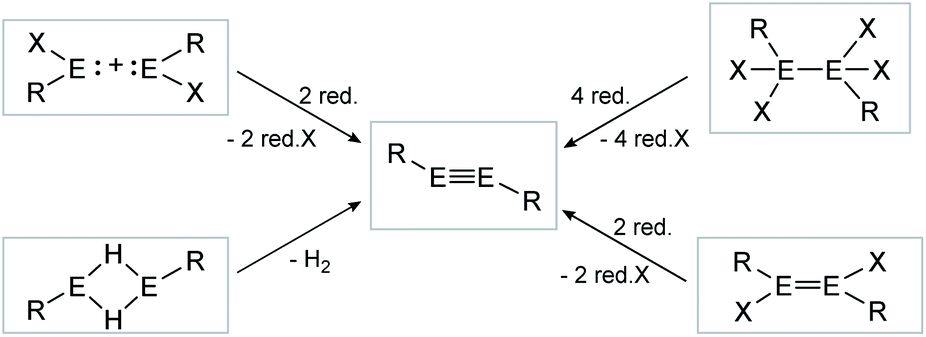 | ||
| Scheme 2 Possible synthesis routes for the isolation of dimetallynes. E = Si, Ge, Sn, Pb; R = stabilizing ligand; X = halogen; red. = reductive agent. Note: Hydrogen elimination route (bottom left) is only known for E = Sn, Pb. | ||
Consecutive dimerization takes place for monomeric starting materials. Ligands are mainly applied before reduction but could also be applied throughout the reduction process. Disilene 12 (Fig. 8), for example, is synthesized from N-heterocyclic imine (NHI)-stabilized NHI–SiBr3 and KSi(SiMe3)3, transferring the silyl ligand.62 In dimetallyne chemistry the elimination of H2 is also known to form triply bonded tin56 and lead9,29,63 compounds, starting from halogenated XRE via an X/H exchange using hydride sources like LiAlH4 or DIBAL-H. Since it can be seen as the reverse of H2 activation, this synthetic route towards dimetallynes of tin and lead is given particular attention in this contribution although it is not yet applied for all heavier group 14 elements.
The strength and overall nature of multiply bonded group 14 compounds are not to be carved in stone, rather they can be greatly influenced by the applied ligands.64,65 The steric demand of the ligand system is crucial to achieve kinetic stabilization of E–E multiple bonds.31,66 If this stabilization is not provided, thermodynamic driving forces could lead to favored di- and oligomerization or isomerization as predicted by quantum chemical calculations.
Looking at the plausible structures of Si2H2 (Fig. 5, A–C), for example, reveals no minimum for A, but a non-stable minimum for B. Isomerization of B, as a consequence of a deficient kinetic stabilization, would lead to structure C or bridged structures that are thermodynamically more stable; with double H-bridging being a global minimum in this system.31
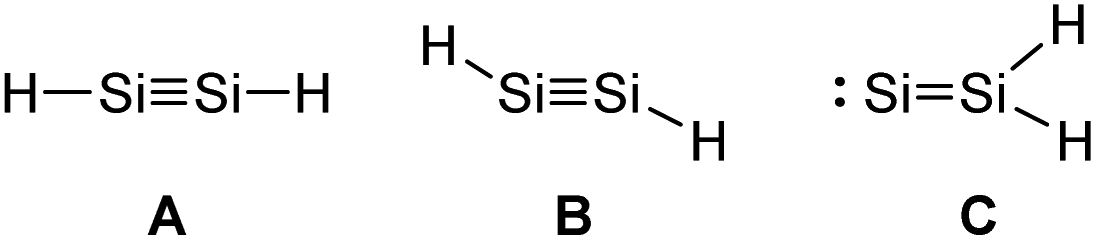 | ||
| Fig. 5 Three plausible isomers of parent disilyne Si2H2 with A: linear, B: trans-bent, or C: SiSiH2 structure.31 | ||
That said, recent accounts have highlighted the importance of London dispersion force (LDF) energies that contribute via dispersion forces between hydrocarbon groups.67,68 While these parts of van der Waals forces have only a minor influence on themselves, they add up in large ligands to contributions that have an observable impact on the stability of kinetically stabilized complexes.25,69Fig. 6 shows selected examples of dimetallenes and dimetallynes that are stabilized by sterically encumbered aryl ligands that achieve primarily kinetic stabilization.
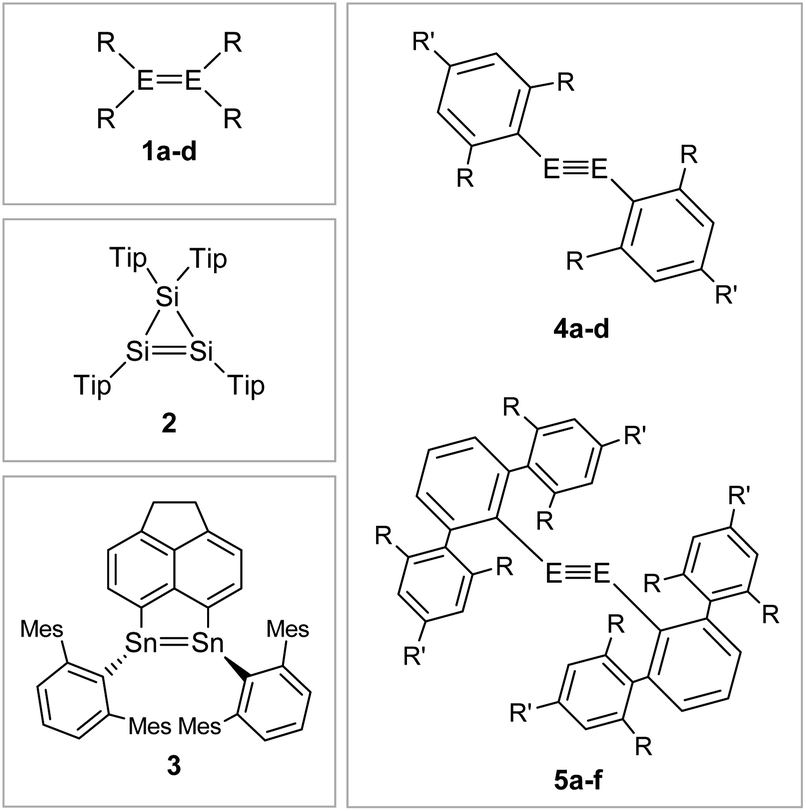 | ||
| Fig. 6 Selected examples of aryl-type stabilized dimetallenes and dimetallynes. 1: 1a6: E = Si, R = Mes (Mes = 2,4,6-trimethylphenyl) 1b70,71: E = Ge, R = Mes 1c72: E = Si, R = Tip (Tip = 2,4,6-triisopropylphenyl) 1d73: E = Ge, R = 2,5-tBu2C6H3; 2;743;754: 4a60: E = Si, Bbt: R = CH(SiMe3)2 R′ = C(SiMe3)34b76: E = Ge, Bbt: R = CH(SiMe3)2 R′ = C(SiMe3)34c47: E = Si, Tbb: R = CH(SiMe3)2 R′ = t-Bu 4d45: E = Ge, Tbb: R = CH(SiMe3)2 R′ = t-Bu; 5: 5a12: E = Ge, Ar′: R = i-Pr R′ = H 5b11: E = Sn, Ar′: R = i-Pr R′ = H 5c29: E = Pb, Ar′: R = i-Pr R′ = H 5d77,78: E = Ge, Ar*: R = i-Pr R′ = i-Pr 5e56: E = Sn, Ar*: R = i-Pr R′ = i-Pr 5f9: E = Pb, Ar*: R = i-Pr R′ = i-Pr. | ||
If one goes beyond kinetic stabilization and also brings in thermodynamic influence on the system, silyl ligands like Sit-Bu3, Sii-Pr2Me, and Sii-Pr(CH(SiMe3)2)2 strengthen and shorten the E–E bond in dimetallenes and dimetallynes by decreasing the singlet–triplet gap or doublet–quartet gap of the element centers, due to stabilization of the excited singlet and doublet states, respectively.62,64,79,80Fig. 7 shows examples of silyl stabilized dimetallenes and dimetallynes.
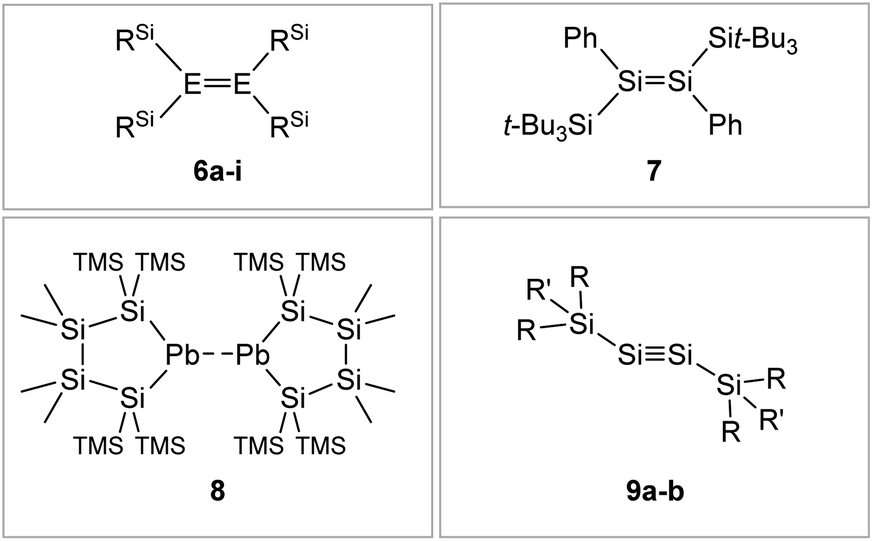 | ||
| Fig. 7 Selected examples of silyl-type stabilized dimetallenes and dimetallynes. 680–84: 6a: E = Si, RSi = i-Pr2MeSi 6b: E = Si, RSi = t-BuMe2Si 6c: E = Si, RSi = i-Pr3Si 6d: E = Si, RSi = t-Bu2MeSi 6e: E = Ge, RSi = i-Pr2MeSi 6f: E = Ge, RSi = t-BuMe2Si 6g: E = Ge, RSi = i-Pr3Si 6h: E = Ge, RSi = t-Bu2MeSi 6i: E = Sn, RSi = t-Bu2MeSi; 785; 869: TMS = SiMe3; 9: 9a10: Si1: R = CH(SiMe3)2 R′ = i-Pr 9b59: Si2: R = Sit-Bu3 R′ = Me. | ||
Boryl ligands such as 9-BBN or B(NArCH)2, in contrast (Fig. 8), incorporate electron accepting properties into the ligand sphere, due to the vacant p orbital of the boron center. These ligands contribute π-acceptor and σ-donor capabilities, which can for example lead to π-conjugation between the disilene double bond and the boron in compound 11.57,86–88
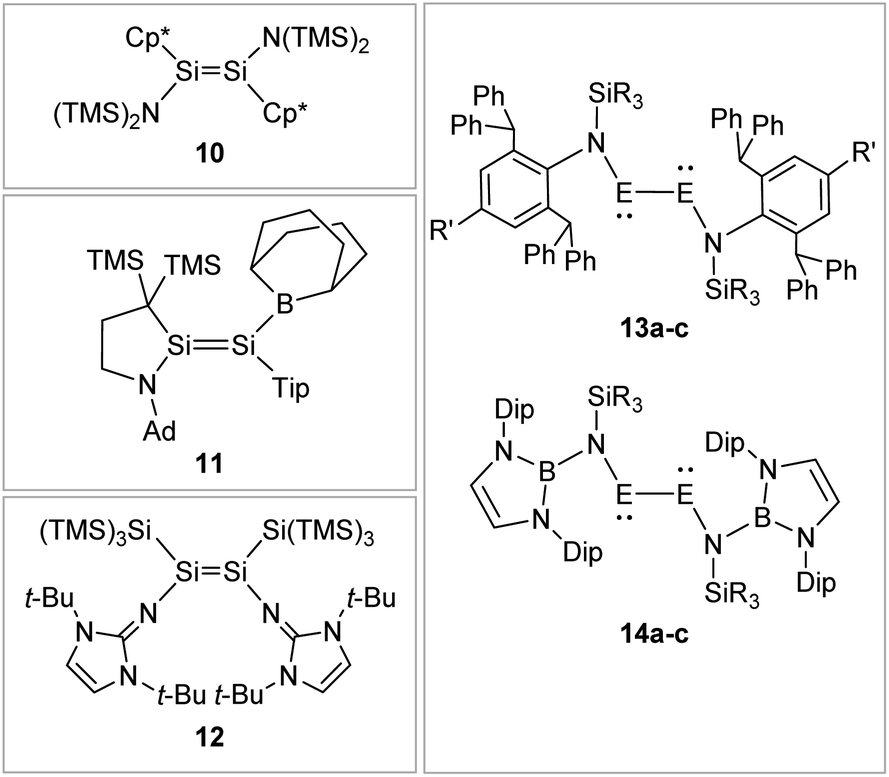 | ||
| Fig. 8 Selected examples of N-type stabilized dimetallenes and dimetallynes. 1089,90: Cp* = η1-Me5C5; 1157: TMS = SiMe3, Ad = 1-adamantyl, Tip = 2,4,6-triisopropylphenyl; 1262,91; 13: 13a61: E = Ge, N1: R = Me R′ = Me 13b92: E = Ge, N2: R = i-Pr R′ = i-Pr 13c93: E = Sn, N2: R = i-Pr R′ = i-Pr; 1494: Dip = 2,6-diisopropylphenyl 14a: E = Ge, R = Me 14b: E = Ge, R = Ph 14c: E = Sn, R = Ph. | ||
N-Donor ligands like amino, amide or NHI95 do have a diverse influence on E–E multiple bonds. While in dimetallenes they result in a pronounced multiple bond character like for 10, in combination with silyl ligand Si(SiMe3)3, the NHI ligand in 12 causes a stretching, twisting, and trans-bending of the double bond (Fig. 8). A highly twisted and trans-bent EE bond can lead to destabilization of the HOMO orbital due to reduced pπ–pπ orbital overlap, resulting in a decreased HOMO–LUMO energy gap. In the case of amido ligands, (SiR3)ArN (dimetallynes 13) and (SiR3)BoN (dimetallynes 14, Bo = boryl) reduce the multiple bond character of the resulting digermyne and distannyne tremendously, and this results in a very high bismetallylene character (Fig. 8). Utilization of additional intramolecular donors and chelating ligands has resulted in the isolation of dimetallynes in the form of interconnected bismetallylenes, as displayed in examples 15–18 in Fig. 9.
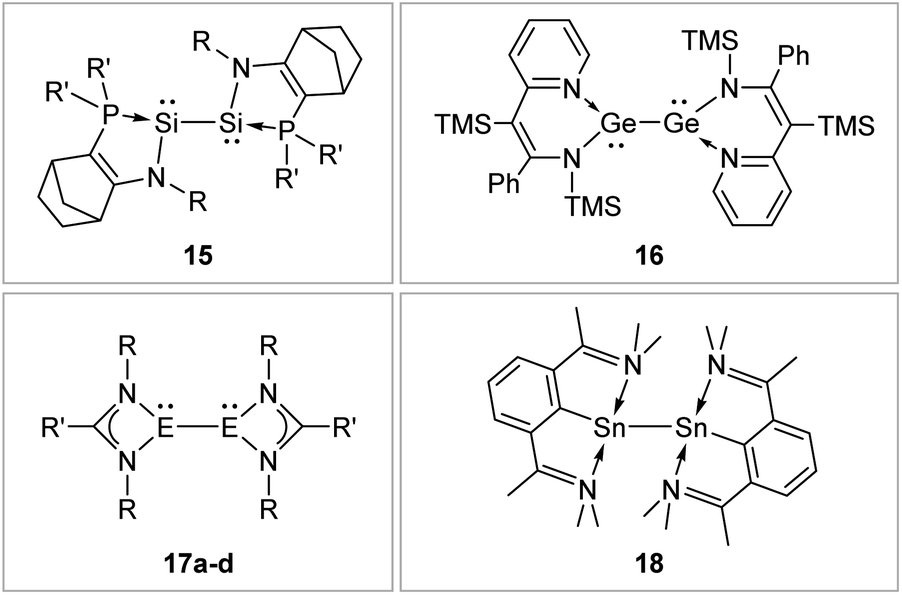 | ||
| Fig. 9 Selected examples of bismetallylenes, stabilized by chelating donor ligands. 1596: R = Dip (Dip = 2,6-i-Pr2C6H3) R′ = t-Bu; 1697: TMS = SiMe3; 17: 17a98: E = Si, R = t-Bu R′ = Ph 17b99: E = Si, R = Dip R′ = 4-t-BuC6H417c100: E = Ge, R = Dip R′ = t-Bu 17d100: E = Ge, R = Dip R′ = Ni–Pr2; 18.101 | ||
Small molecule activations of dimetallenes and dimetallynes/bismetallylenes
While the number of stable dimetallenes and dimetallynes steadily expands, accounts of reactivity towards small molecules and other bond activation reactions are relatively scarce. To date, bond activation via insertion into the multiple bond or various addition reactions with small molecules have been achieved a number of times. What seems to be challenging is firstly, the activation of a molecule without breaking the E–E bond altogether and secondly, achieving reversibility of the reaction, i.e. regeneration of the multiple bond as a key step for any future catalytic application. To our knowledge, there are no examples of catalytic reactions performed by a group 14 dimetallene to date and bond activation is mostly reserved for disilenes, whereas dimetallynes and bismetallylenes show a broader range of reactivity that peaks in the first complete catalytic cycle found for the Tbb-substituted digermyne 4d in 2018.16 The adjacent reactivity of monomeric tetrylene compounds, low valent hydrides and other low valent group 14 species have been discussed in a number of reviews,14,15,102,103 but are beyond the scope of this account. Although there is noteworthy progress in bond activation of functional groups as well,52,104–109 in the following paragraphs only the activation of classic small molecules such as H2, NH3, CO2, CO, alkenes and alkynes are discussed in further detail.H2 activation
The activation of hydrogen is an important preliminary experiment on the way to industrially relevant catalytic chemical reactions like hydrogenations of unsaturated hydrocarbons, to name only one.H2 activation was achieved by only a few disilenes up till now, with the first account being the selective formation of the trans-hydrogenation product 19 under mild conditions starting from NHI-stabilized disilene 12 (Scheme 3).62 DFT calculations suggest a concerted mechanism which is enabled by the relatively easy rotation along the double bond due to the highly twisted and trans-bent structure and the staggered ligands. Additionally, in 2018 Iwamoto reported that the treatment of 1-amino-2-boryldisilene 11 with dihydrogen at room temperature resulted in concomitant formation of trihydridodisilane 20 and hydroborane (Scheme 4) via the initial cleavage of the Si–B bond as a first step.57,87 Since theoretical calculations suggested an overpowering effect of the π-accepting boryl moiety on the polarity and HOMO–LUMO gap (and therefore reactivity) of the SiSi bond, similar disilenes, each with either only the boryl or amino ligand attached respectively, were also treated with H2. It turned out that the interaction of the low lying empty 2p boron orbital with the π*(SiSi) orbital is essential for the reactivity. While the monoboryldisilene 2,2,5,5-(SiMe3)4–C4H4SiSi(Bo)Tip (Bo = 9-borabicyclo[3.3.1]-nonane) (11a) reacts with H2 in a similar manner as 11, the monoaminodisilene N-(Ad)-5,5-(SiMe3)2C3H4SiSi(i-Pr)Tip (11b) is not capable of activating dihydrogen even at elevated temperatures. Interestingly, the conversion of H2 with 11a takes significantly longer compared to 11, suggesting that the amine moiety does contribute marginally to the reactivity of the compound despite an almost negligible destabilization of the HOMO energy in 11 according to computational studies.87
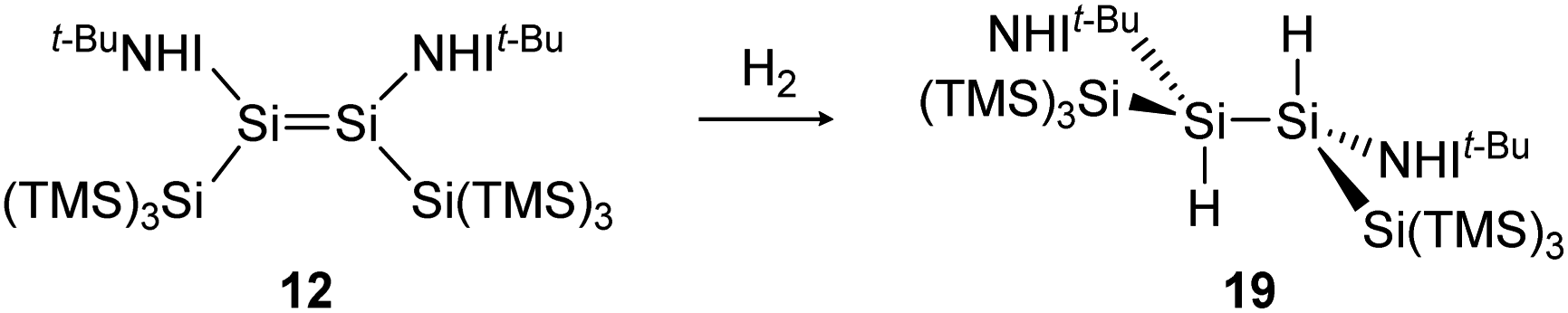 | ||
| Scheme 3 Selective trans addition of H2 by iminodisilene 12. TMS = SiMe3.62 | ||
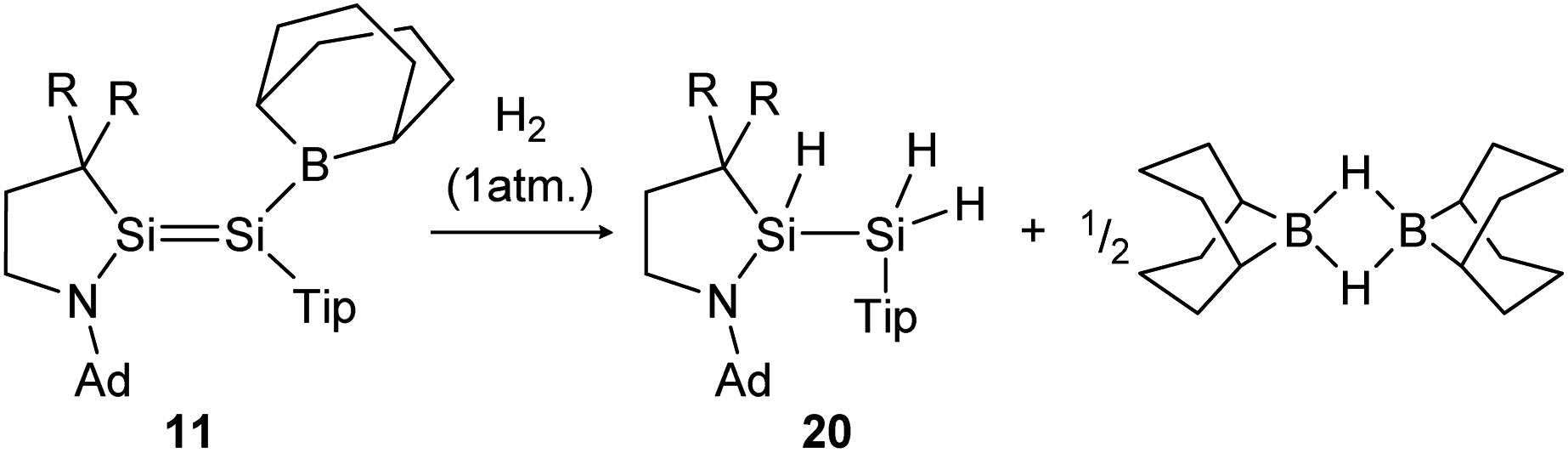 | ||
| Scheme 4 Cleavage of two hydrogen molecules under formation of trihydridodisilane and hydroborane by 1-amino-2-boryldisilene 11.57 R = SiMe3. | ||
Amongst activation products, the hydrogen binding modes have an impact on the hydrogen-transfer ability of those complexes, if a catalytic cycle is intended. In this respect, dimetallynes show a variety of hydrogen binding modes that – unfortunately – lack reliable selectivity in most cases. However, the number of H2 activating dimetallynes and bismetallylenes is limited to a few examples featuring aryl and amide stabilization. In the first report from 2005, Power treated 5a (Ar′GeGeAr′) with H2 at room temperature and atmospheric pressure (Scheme 5).110 In a product mixture they found the digermene 21, digermane 22 and germane 23, shown in Scheme 5. This account presents the first example of H2 activation by using a molecular main-group species without a catalyst that is estimated to be due to a singlet biradical character of the ground state of 5a. The heavier analogue 5b (Ar′SnSnAr′) was shown in 2018 to activate dihydrogen in a reversible form (Scheme 5, bottom) to produce hydrogen bridged 24.111 The addition of H2 proceeds at room temperature, whereas it is released again by heating to 80 °C. A tremendous connection between ligand nature and size and reactivity could be shown by reacting slightly bigger distannyne 5g with H2 under ambient conditions. In this case the reaction brings forth the non-bridged hydrogenation product 25.111 For amide ligands H2-activation has been proven possible for bisgermylene 13a and 13b and bisstannylene 13c each with high selectivity.61,92,93 While 13a reacts quantitatively with 1 bar of H2 at ambient temperature to hydridogermane-germylene N1GeGeH2N1 (27), the slightly modified ligand N2 in 13b leads, under similar conditions, to the hydrido-digermene N2HGeGeHN2 (26) (Scheme 6, top). Attempts to form di- or tri-hydrogenation products by applying harsher conditions failed in both cases. In order to understand the differences in the reaction mechanism that result in the formation of the two diverging products, theoretical reaction profiles were explored, leading to the conclusion that NHGeGeHN is formed in both reactions, followed by a rearrangement in the case of 27 due to a low activation barrier and higher thermal stability.113–115 In 2014, the reaction of 13c with H2 was explored, yielding the third product motif N2Sn(μ-H)2SnN2 (28) (Scheme 6, bottom).
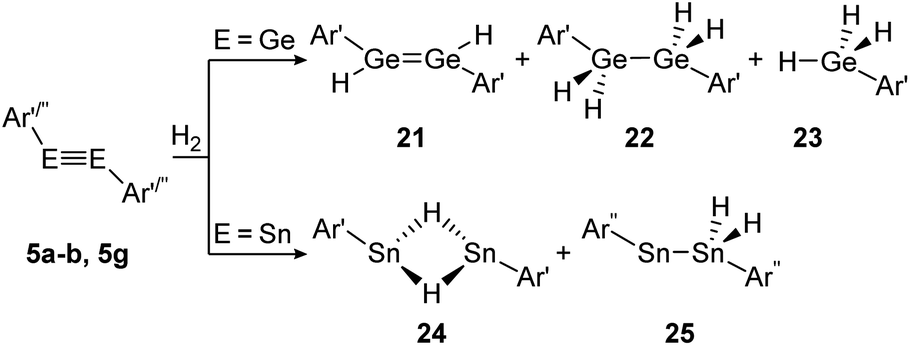 | ||
| Scheme 5 Reactivity of digermyne 5a (Ar′ = C6H3-2,6-(C6H3-2,6-i-Pr2)2) and distannynes 5b and 5g (Ar′′ = C6H-2,6-(C6H2-2,4,6-i-Pr3)2-3,5-i-Pr2) with H2.110–112 | ||
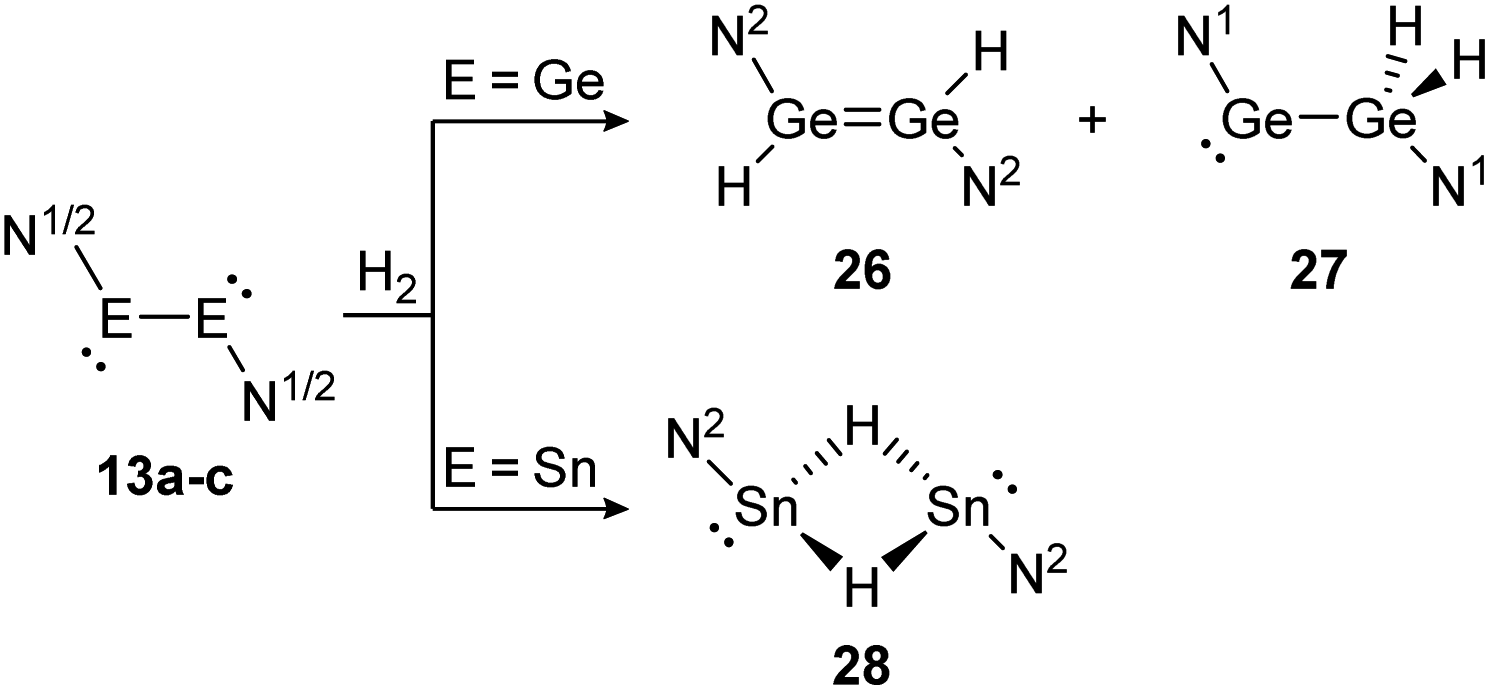 | ||
| Scheme 6 Reactivity of bisgermylenes 13a and 13b and bisstannylene 13c with H2. N1 = N(Ar1) (SiMe3) (Ar1 = 2,6-(CHPh2)2-4-MeC6H2), N2 = N(Ar2) (Sii-Pr3) (Ar2 = 2,6-(CHPh2)2-4-i-PrC6H2).61,92,93 | ||
NH3 activation
The splitting of N–H bonds is especially interesting in regard to the activation of ammonia in order to facilitate transfer reactions to olefins or arenes. Breaking this strong bond is still challenging, even for transition-metal complexes. Still, some group 14 dimetallene compounds have shown the ability to perform hydroamination reactions.In the first example, a tetrasilabuta-1,3-diene (1f), each double bond reacts with ammonia in a 1,2-addition reaction, yielding six-membered ring compound 30. This is somewhat surprising, since the simpler Tipp2SiSiTipp2 congener (1c) with only one SiSi double bond, is inert towards NH3, indicating the increased reactivity of the conjugated double bonds (Scheme 7).116
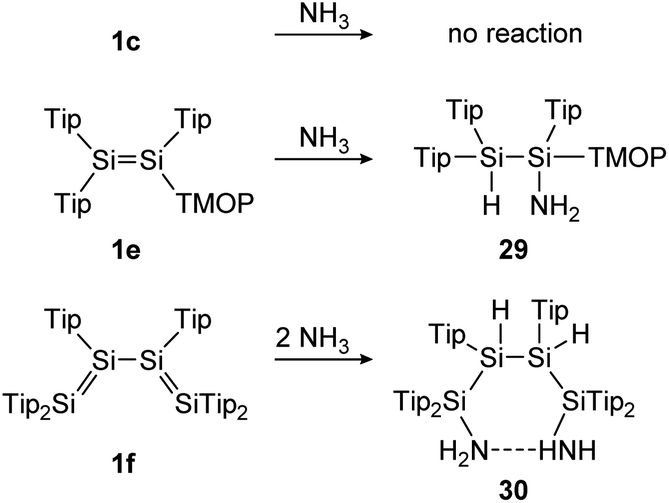 | ||
| Scheme 7 Reactivity of various Tip substituted disilenes 1e,1171c, and 1f116 towards NH3. Tip = 2,4,6-triisopropylphenyl; TMOP = 2,4,6-trimethoxyphenyl. | ||
Similarly, Scheschkewitz could demonstrate an increased reactivity of the SiSi bond by substituting one ligand moiety with a TMOP ligand (1e), resulting in a prominent polarization of the SiSi bond and enabling a regioselective 1,2-addition of ammonia to addition product 29 (Scheme 7).117
In a similar manner to the 1,2-addition of hydrogen to the iminodisilene 12, an analogous trans-addition product with NH3 was reported more recently.91 The ammonia reaction to 31 progresses at a much lower temperature (−78 °C) which can be explained by the significantly lower energy barrier of the anti-addition of NH3 (7.7 kcal mol−1) compared to that of H2 (15.6 kcal mol−1). When the reaction is performed at room temperature, the formal oxidative addition product 32 of the corresponding silylene with NH3 is observed (Scheme 8).
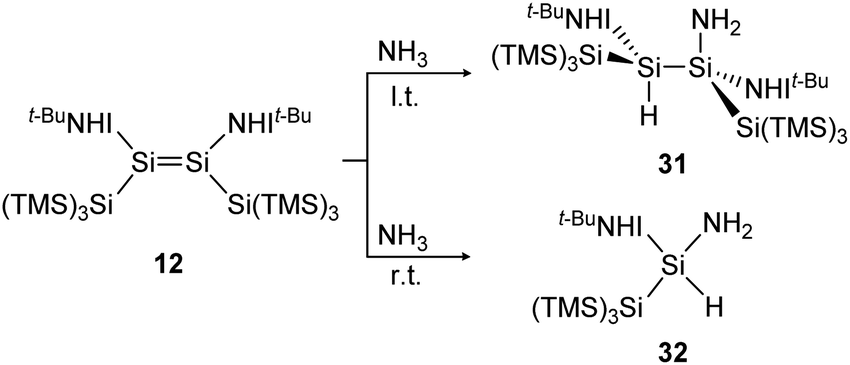 | ||
| Scheme 8 Reaction of NH3 with iminodisilene 12 at l.t. = −78 °C or r.t. = room temperature. TMS = SiMe3.91 | ||
CO2 activation
The activation and reduction of CO2 has attracted much attention over the last few decades, because it could make it possible to use this abundant gas, which is a critically discussed greenhouse gas, and transform it into value-added chemicals such as carbon monoxide or higher hydrocarbons.118 In main-group chemistry the number of examples of CO2 activations has increased over the years but is still limited compared to other reactivities.119 Up till now, the aforementioned iminodisilene (12) and the supersilyl substituted disilene, (Sit-Bu3)PhSiSiPh(Sit-Bu3) (7) showed reactivity towards the molecule.
Treatment of 12 with CO2, again has different outcomes depending on the reaction temperature. Either selective adduct 33 is formed at −78 °C or a mixture of products including the novel five-membered silacycle 34, which could be identified by stepwise and partial oxidation of the double bond with N2O as well as O2 and comparison of NMR data (Scheme 9).91 The disilene 7 activates CO2 in a [2 + 2] cycloaddition reaction as shown in Scheme 10 to obtain 35.85
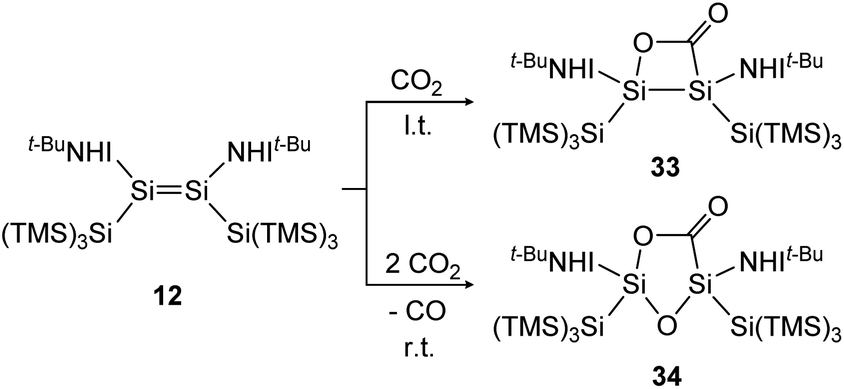 | ||
| Scheme 9 Activation of CO2 by iminodisilene 12 at l.t. = −78 °C or r.t. = room temperature. TMS = SiMe3.91 | ||
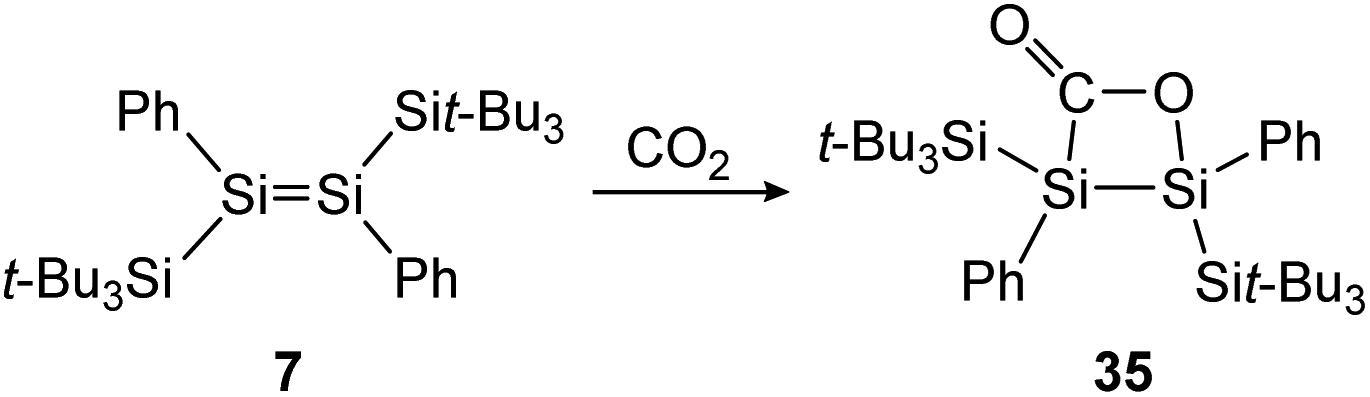 | ||
| Scheme 10 [2 + 2] cycloaddition of CO2 to supersilyl substituted disilene 7.85 | ||
For classical dimetallynes no activation of CO2 is known, however, examples involving interconnected bismetallylenes are reported. Upon exposure of a toluene solution of 13a to CO2 at −70 °C the reaction proceeds via the intermediate N1GeO2CGeN1 (36′) and ends with the bisgermylene oxide N1GeOGeN1 (36) as shown in Scheme 11.109 Analysis of the head space gas reveals the release of CO. Theoretical analysis of the reaction profile suggests a three step mechanism, in which CO2 connects through a side-on approach to one or both germanium centers. Via Ge–Ge bond rupture and rearrangement, intermediate 36′ is formed, which yields product 36 after elimination of one CO molecule.114 The analogue reaction with CS2 afforded N1GeSGeN1, respectively.
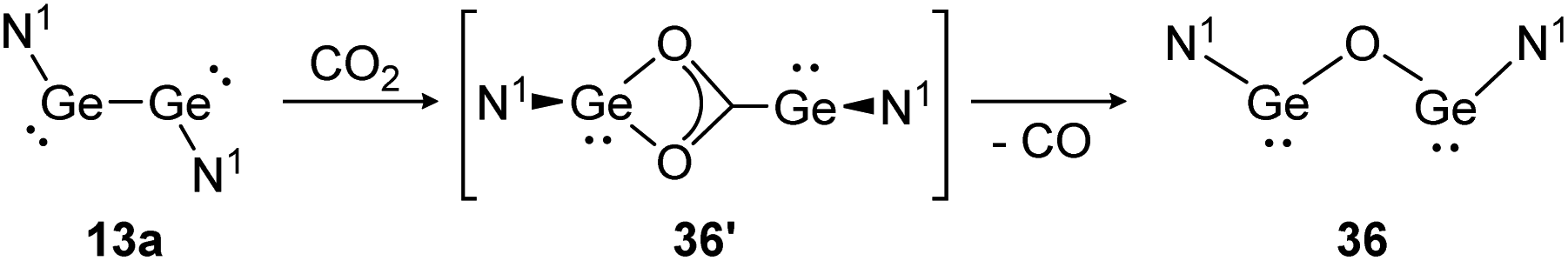 | ||
| Scheme 11 Reactivity of bisgermylene 13a with CO2. N1 = N(Ar1)(SiMe3) (Ar1 = 2,6-(CHPh2)2-4-MeC6H2).109 | ||
Another deoxygenation of CO2 was reported by Baceiredo and Kato in 2011.96 In detail, three molecules of CO2 are deoxygenated concomitant with the formation of three Si–O–Si units upon Si–Si bond rupture. In a subsequent step the carbonate unit is formed via insertion of one additional CO2 molecule into a previously built Si–O bond, resulting in product 37 (Scheme 12). The high reactivity of the bissilylene is clearly indicated by the rapid reaction with CO2 that is completed upon mixing of the reactants.
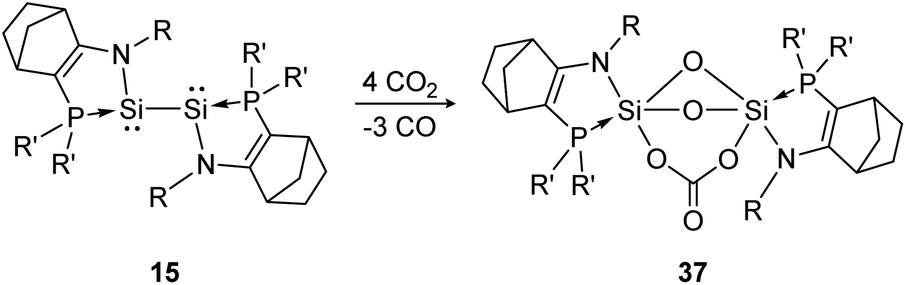 | ||
| Scheme 12 Reactivity of bissilylene 15 with CO2 (R = Dip, R′ = t-Bu).96 | ||
Apart from the singly bonded and lone pair exposed structure of the bissilylene, a peculiar ligand effect also acts on the central Si-atoms, where the phosphine moiety induces a strong π donor–acceptor character of the SiI centers.
CO activation
The activation and bond cleavage of CO is in the focus of synthetic chemists mainly for the possible utilization of CO as a C1 building block as well as application in C–C bond formation in Fischer–Tropsch processes.120 In group 14 chemistry, activations of CO are scarce but have been achieved for low valent silicon and germanium complexes, for example.121,122 In dimetallene and dimetallyne chemistry this reactivity was mainly pioneered by the group of Scheschkewitz, who already reported in 2015 about CO cleavage and C–C bond formation using a Tip substituted disilenide (1g) as shown in Scheme 13.123 Proposed reaction intermediates include the twofold addition of CO to the disilenide forming a silylenoid and a dimerization product. Overall, two molecules of disilenide are needed to activate four molecules of CO within the reaction product 38.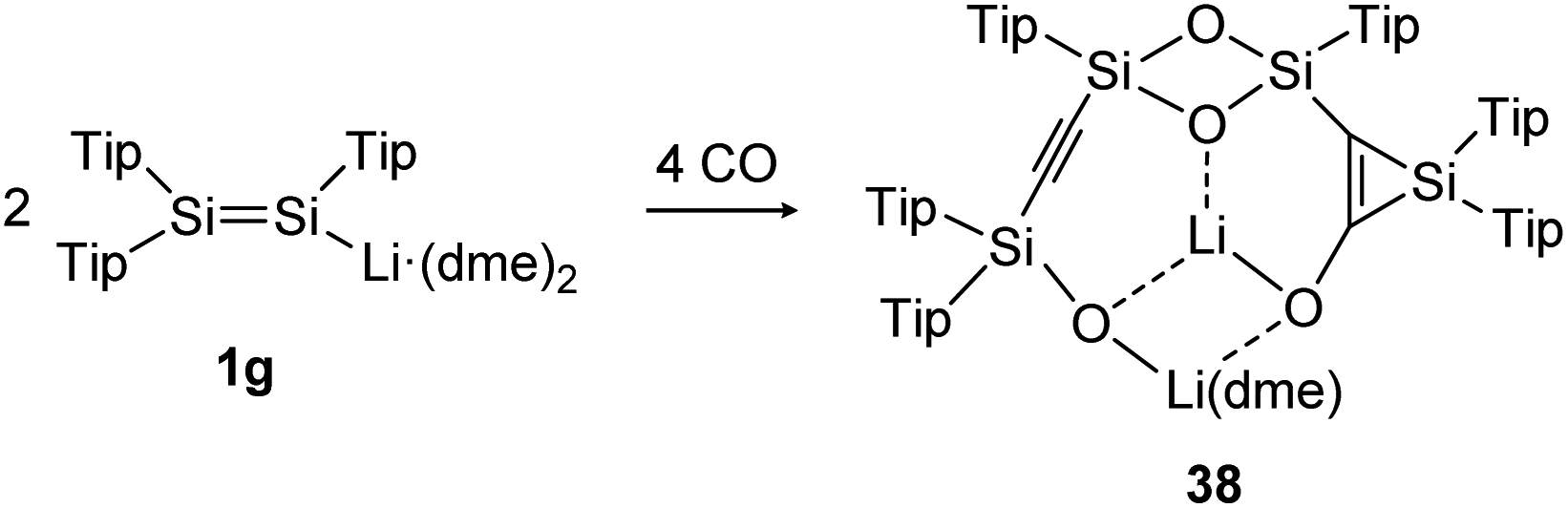 | ||
| Scheme 13 Reactivity of disilenide 1g with CO. Tip = 2,4,6-triisopropylphenyl, dme = dimethoxyethane.123 | ||
The same group could find a reactivity of a functionalized cyclic disilene – called cyclotrisilene (2) – towards CO. While in 2013 the 1:1 reaction product could only be isolated as dimer 39 (Scheme 14, top),124 one year later the monomer 40 could be trapped using Et2O·B(C6F5)3 (Scheme 14, bottom).125
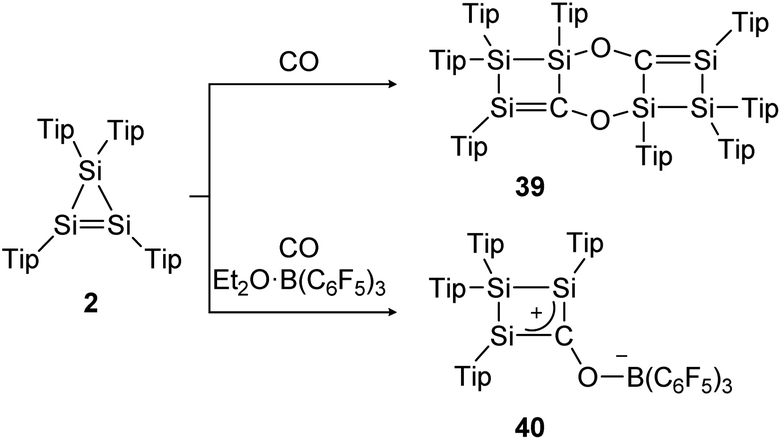 | ||
| Scheme 14 Reactivity of cyclotrisilene 2 with CO and additional Et2O·B(C6F5)3. Tip = 2,4,6-triisopropylphenyl.124,125 | ||
N2O activation
Nitrous oxide is one of the classic small molecules, however, its activation is easy enough for N2O to find application as a mild oxidant in synthetic chemistry. Utilization of this readily available industrial waste gas as a mono oxygen transfer agent offers the advantage of producing only greenhouse-neutral and non-toxic N2 as a by-product.126Reaction of iminodisilene 12 with N2O produces three-membered ring oxadisilirane 41 in cis confirmation (Scheme 15). When exposed to oxygen, in contrast, a dioxadisiletane with a Si2O2-central 4-membered ring is formed via the trans analogue of 41 as an intermediate.
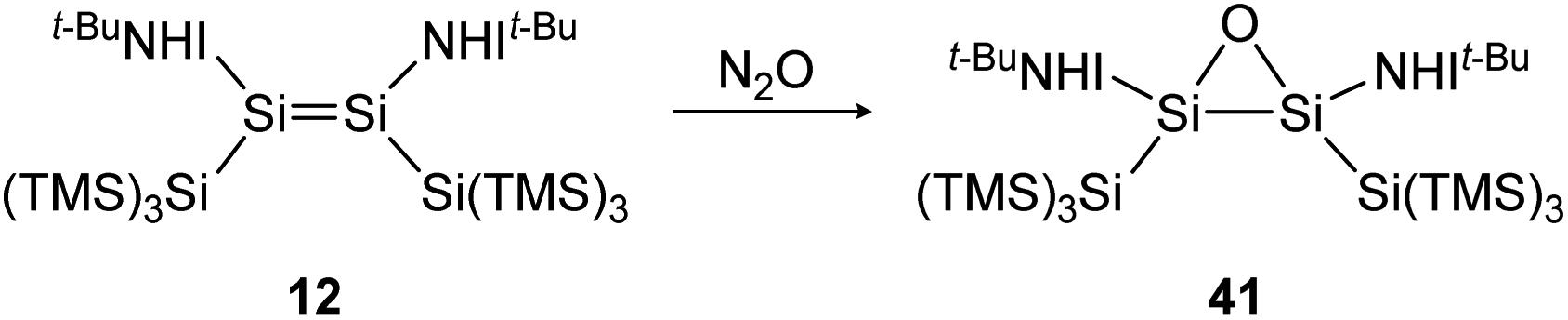 | ||
| Scheme 15 Formation of oxadisilirane via the reaction of 12 with N2O. TMS = SiMe3.91 | ||
The first example of a disilene that produces both the cis (42b) and trans (42a) isomers of a dioxadisiletane upon reaction with N2O is trans-[(TMS)2N(η1-Me5C5)SiSi(η1-Me5C5)N(TMS)2] (10), displayed in Scheme 16.127 DFT calculations revealed the two isomers to be isoenergetic, with the cis counterpart 42b being slightly higher in energy by 3.3 kcal mol−1.
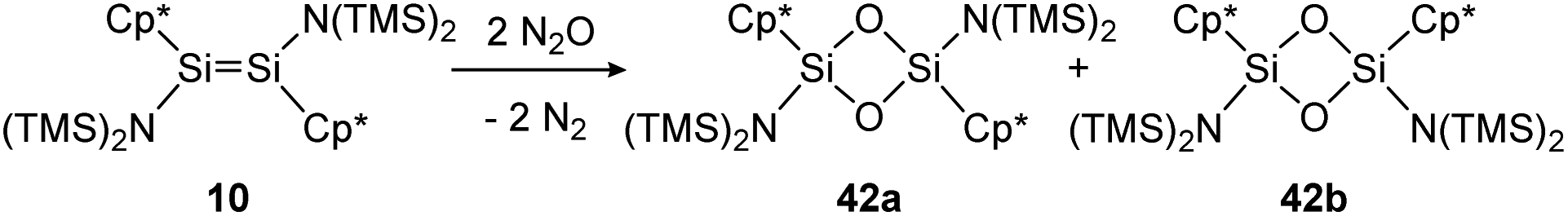 | ||
| Scheme 16 Reaction of trans-disilene 10 with N2O results in formation of both cis- (42b) and trans-dioxadisiletane (42a). TMS = SiMe3.127 | ||
Depending on the attached ligands, three reaction pathways of the addition of N2O to a SiSi bond likely take place according to a recent theoretical study.128 The first one, starting with dissociation of the double bond into silylene monomers followed by a nucleophilic attack of N2O and concluding with the dimerization of the resulted silanone intermediates, affords cis- and trans-products. This pathway is favored by substituents without a lone pair of electrons, leading to a more unstable disilene and therefore facilitating the subsequent attack of N2O. Alternatively, N2O reacts directly with the double bond either as a nucleophile or an electrophile. Nucleophilic N2O leads to an end-on attack whereas electrophilic N2O interacts with the disilene in a side-on fashion.128 Within dimetallyne chemistry the activation of N2O is reported with several outcomes. For bisgermylene 13a, the reaction proceeds with a simple oxygen transfer that breaks and inserts into the Ge–Ge bond (Scheme 17).109 In this regard, it is an alternative synthetic pathway to yield the CO2 activation product 36.
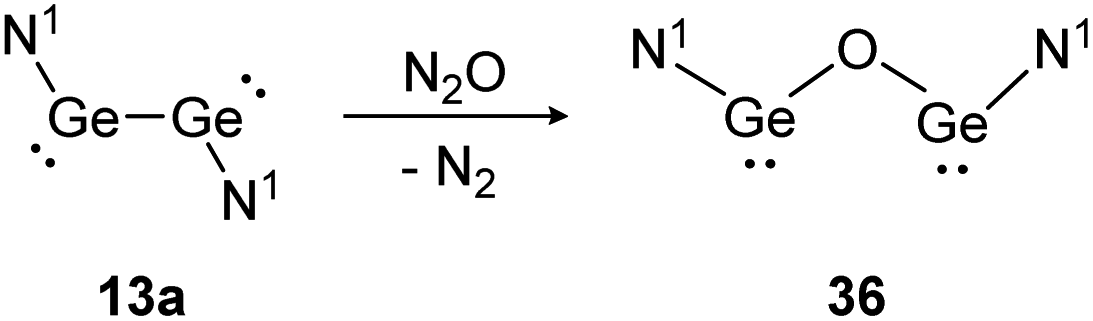 | ||
| Scheme 17 Reactivity of bisgermylene 13a with N2O. N1 = N(Ar1)(SiMe3) (Ar1 = 2,6-(CHPh2)2-4-MeC6H2).109 | ||
In contrast, reaction of N2O with digermyne 5a that bears significantly higher multiple bond character produces a Ge2O3-ring with two exocyclic hydroxo groups (43) (Scheme 18).129 Theoretical calculations suggest that the reaction proceeds via a radical mechanism that forms a peroxo bridge, followed by subsequent deoxygenation of additional N2O molecules. Formation of the terminal hydroxo groups in 43 could stem from dehydrogenation of solvent molecules.
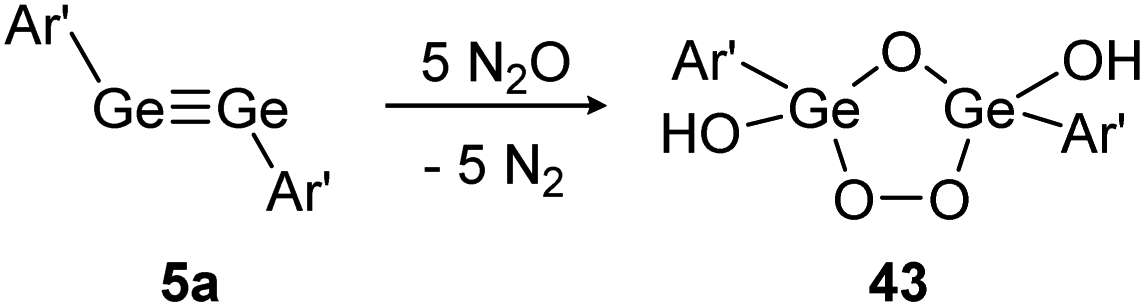 | ||
| Scheme 18 Reactivity of digermyne 5a with N2O. Ar′ = C6H3-2,6-(C6H3-2,6-i-Pr2)2.129 | ||
Base-stabilized bissilylene 17a consumes three equivalents of N2O at ambient temperature and forms the eight-membered Si4–O4-ring compound 44 upon dimerization of two bissilanone moieties (44′′) induced by the polar SiO double bonds (Scheme 19). The reaction mechanism is proposed to proceed via primary insertion of an oxygen atom into the Si–Si bond (44′) and subsequent oxygenation to 44′′. Each bissilylene unit consist of a Si(μ-O)2Si core. Reaction with t-BuNCO affords the same product (44).
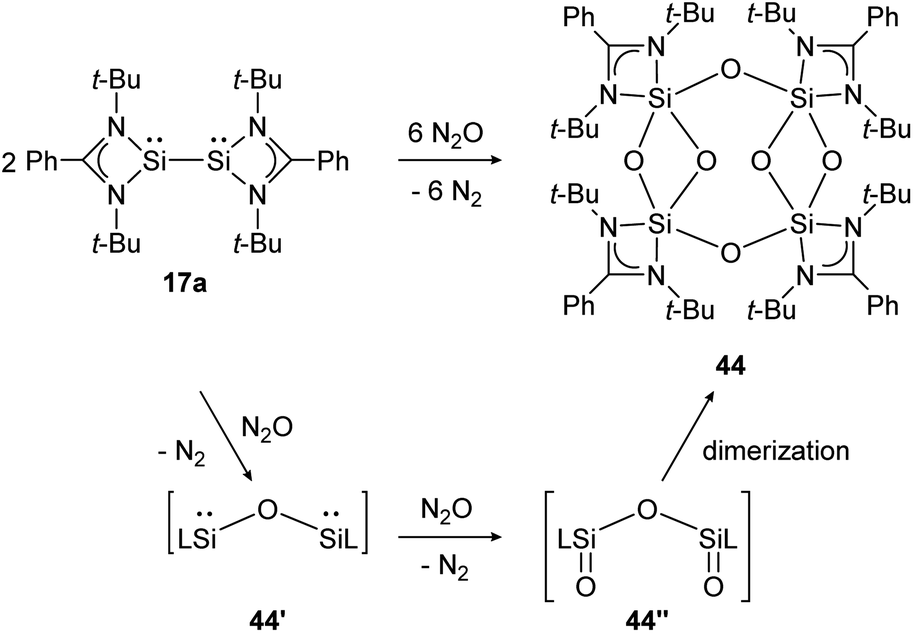 | ||
| Scheme 19 Reactivity of bissilylene 17a with N2O.130 | ||
Cycloaddition of alkenes and alkynes
Cycloadditions of dimetallenes with unsaturated hydrocarbons and other multiple bonds are often observed. In particular, the reactivity of disilenes and digermenes with alkenes, alkynes, CN, CO and other heteroatom multiple bonds has been reported, resulting in otherwise difficult to obtain ring compounds.104,105,131–135 For the reactions of disilenes with terminal alkenes and alkynes, a radical pathway with a stepwise mechanism can be proposed.134,136 Baines showed, for example, that the reaction of tetramesityldisilene (1a) with trans-d-styrene resulted in a 7:3 diastereomeric mixture of the cis and trans [2 + 2] cycloaddition products 45 (Scheme 20), indicating a bond rotation of the intermediate 45′, yielding the cis isomer.137
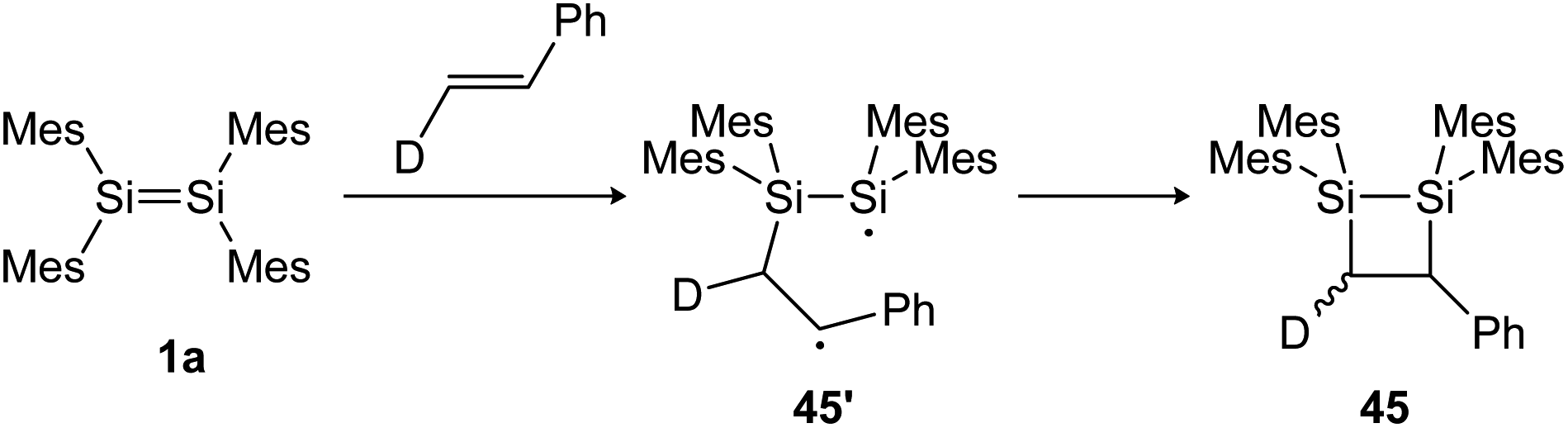 | ||
| Scheme 20 [2 + 2] cycloaddition of tetramesityldisilene 1a and trans-d-styrene gives a diastereomeric product mixture. Mes = 2,4,6-trimethylphenyl.137 | ||
Similarly, the cycloaddition reaction of tetramesityldigermene (1b) with a cyclopropylcarbinyl probe suggests the formation of several intermediate diradicals (46′, 46′′) resulting in various cyclization products of which 46a and 46b are shown (Scheme 21).134
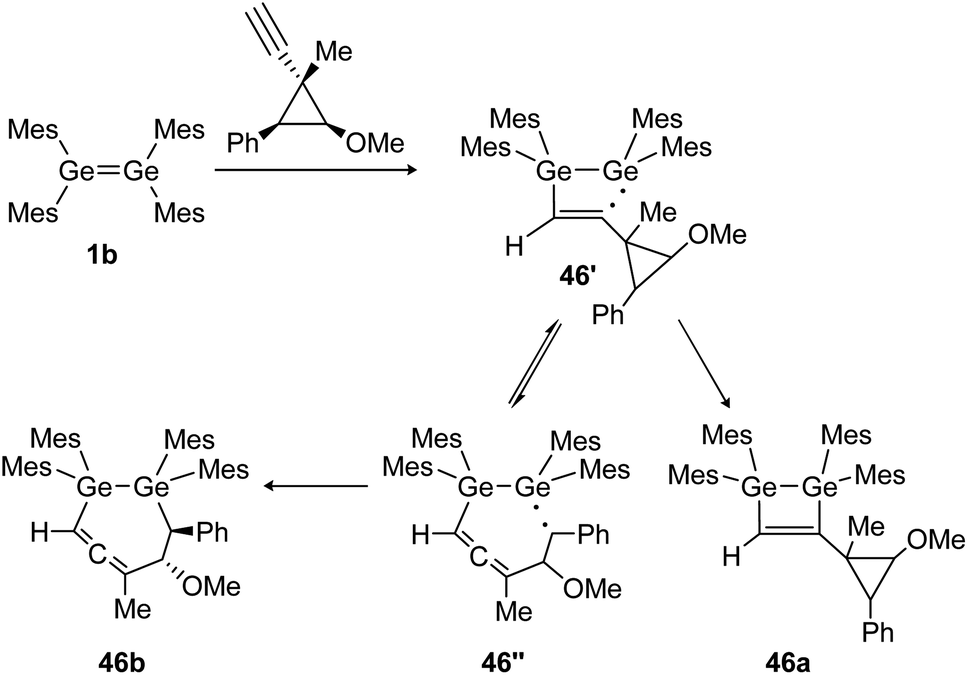 | ||
| Scheme 21 Reaction of tetramesityldigermene with a cyclopropyl alkyne. Mes = 2,4,6-trimethylphenyl.134 | ||
Moreover, two linker-bridged distannenes showed the ability to activate terminal alkynes at ambient temperature. The reaction of 3 with trimethylsilylacetylene and phenylacetylene occurs via a formal [2 + 2] cycloaddition reaction. The gradual reverse reaction of the isolated product 47 could be observed in a C6D6 solution, confirmed by NMR experiments (Scheme 22, top).75 Other distannenes like 3a with similar backbones (Scheme 22, bottom) are also able to perform cycloaddition with alkynes to exemplified product 48, but show no reversibility or only with some alkynes with no obvious correlation with the respective ligand structures. In all cases, the SnSn bond lengths do not change significantly upon cycloaddition (from 2.729–2.784 Å before cycloaddition to 2.744–2.767 Å after).75 Cycloaddition reactions of dimetallenes are often not reversible and the reactivity of stable distannenes with terminal alkenes has only been observed a handful of times. To our knowledge, the only example of a reversible cycloaddition to a digermene is the [2 + 2]-cycloaddition of acetonitrile, which can be reversed at elevated temperatures.104 Dimetallynes – like many multiply bonded compounds – are also prone to cycloaddition reactions. Therefore, a broad variety of examples are known today.
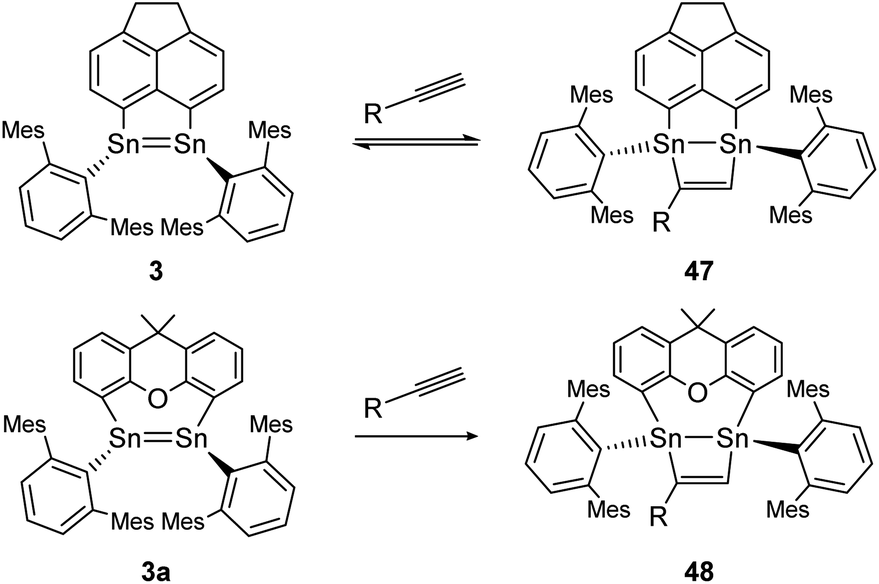 | ||
| Scheme 22 [2 + 2] cycloaddition of terminal alkynes to linker bridged distannenes 3 and 3a. Mes = 2,4,6-trimethylphenyl; 3: R = SiMe3 or Ph; 3a: R = Ph.75 | ||
Multiple studies have shown that the cycloaddition of olefins follows a certain pathway.43,46,48 Within two addition and rearrangement reactions dimetallynes are able to form 4-membered cycloadducts and upon addition of an extra alkene molecule, 3-membered cycloadducts also (Scheme 23). The product distribution of those reactions depends on reaction conditions as well as on the central group 14 element and the choice of ligand.
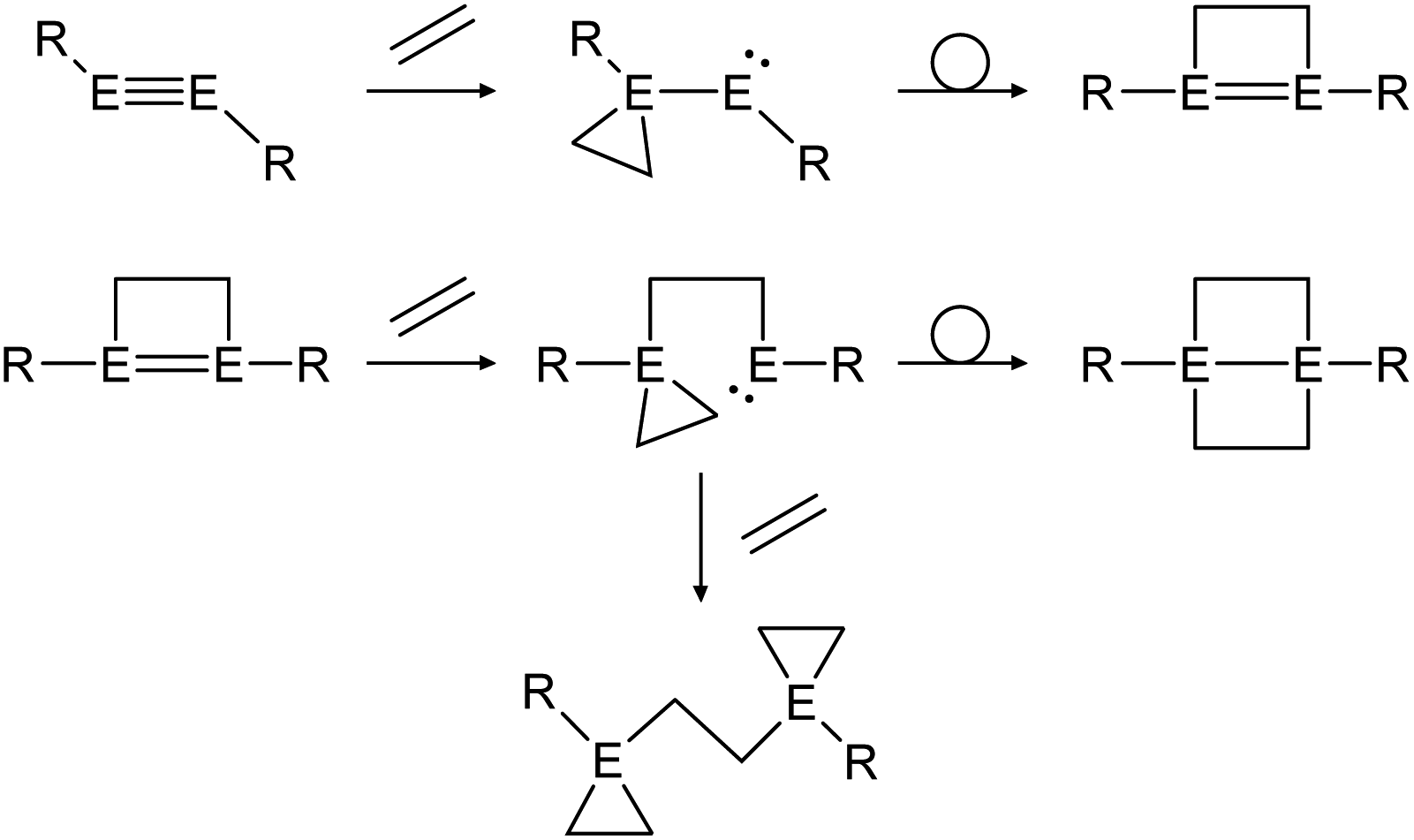 | ||
| Scheme 23 General reaction pathway for cycloaddition reactions of dimetallynes with olefins exemplified by ethylene. E = Si, Ge, Sn; R = stabilizing ligand. | ||
In a similar fashion, reactions with alkynes show a common reaction mechanism.47,49,51 Upon addition of two alkyne molecules either 1,2-ditetrel-benzene or 1,4-ditetrel-benzene are isolated as stable products. In case of a 1,4-ditetrel-benzene a third alkyne can be added resulting in 1,4-ditetrelbicyclo-[2.2.2]octa-2,5,7-triene derivatives (Scheme 24).
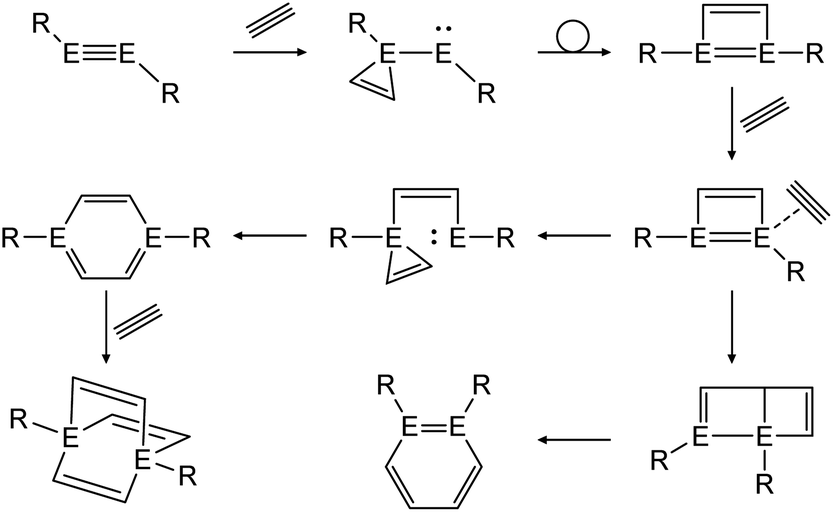 | ||
| Scheme 24 General reaction pathway for cycloaddition reactions of dimetallynes with alkynes exemplified by acetylene. E = Si, Ge, Sn; R = stabilizing ligand. | ||
A highly regarded report about olefin cycloaddition on tetrel triple bonds was published by Power in 2009.138 It describes the reversible ethylene addition to distannyne 5b that proceeds readily at room temperature to form the cycloadduct 49. Under reduced pressure both ethylene molecules dissociate and distannyne 5b is recovered. Theoretical calculations point out a rare combination of electronic, steric and thermodynamic effects that result in considerable weak Sn–C cycloadduct bonds. Noteworthily, the reactivity with ethylene is shared by the sterically more protected distannyne AriPr8SnSnAriPr8 (AriPr8 = C6H-2,6-(C6H2-2,4,6-i-Pr3)2-3,5-i-Pr2), whereas the reversible addition of norbornadiene (nbd) to 50 can be obtained exclusively by using 5b (Scheme 25).
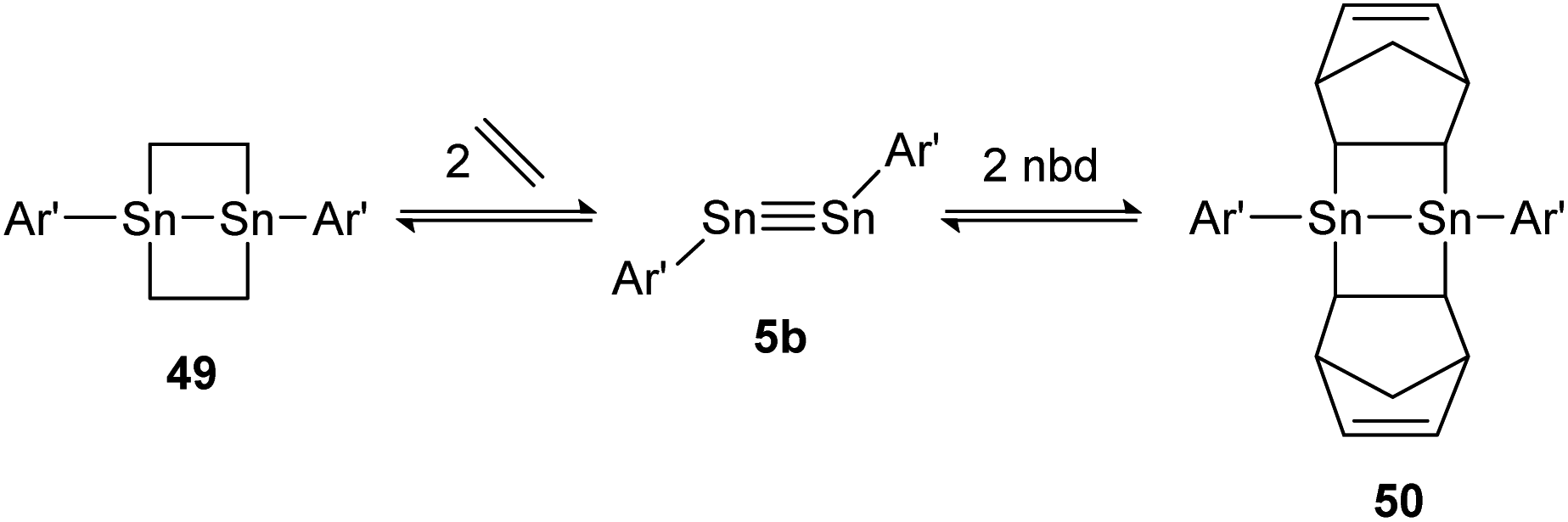 | ||
| Scheme 25 Reversible cycloaddition of ethylene to distannyne 5b. Ar′ = C6H3-2,6-(C6H3-2,6-i-Pr2)2, nbd = norbornadiene.138 | ||
In 2002 Power had already accomplished activation of the alkene 2,3-dimethyl-1,3-butadiene with digermyne 5d.139 Upon addition of three diene molecules the previously unknown ‘bridged’ reaction product 51 was furnished. Two years later, the same group reported on a cycloaddition of 5a (Ar′GeGeAr′) with SiMe3CCH and diphenylacetylene (Scheme 26, right).39 While for diphenylacetylene only one addition takes place forming product 52, in case of the sterically less demanding trimethylsilylacetylene two molecules can be added and form the postulated diradical intermediate 53′. A third cycloaddition process is initiated by one of the ligand's Dip-rings, finally forming the rather unusual product 53 (Scheme 26, bottom).
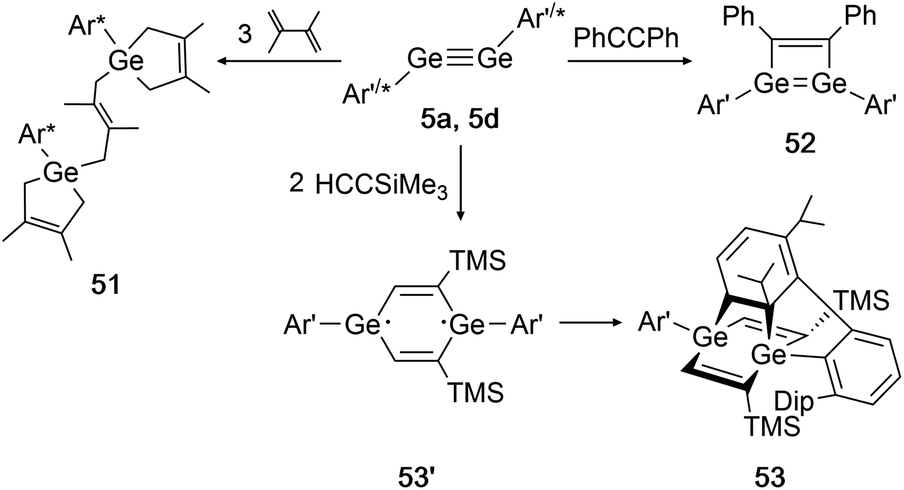 | ||
| Scheme 26 Cycloaddition reactions of digermyne 5a with diphenylacetylene, trimethylsilylacetylene, and digermyne 5d with 2,3-dimethyl-1,3-butadiene. Ar′ = C6H3-2,6-(C6H3-2,6-i-Pr2)2, Ar* = C6H3-2,6-(C6H2-i-Pr3)2, TMS = SiMe3.39,52,139 | ||
In 2007 Sekiguchi could find the first examples of cycloaddition on disilyne 9a (Si1SiSiSi1, Si1 = Sii-Pr[CH(SiMe3)2]2) when reacting it with either 2-butene or phenylacetylene (Scheme 27).43 For the reaction with 2-butene, a significant time dependence could be revealed. While after 30 minutes the reaction affords 55 with the methyl groups in cis formation, after 24 hours 55 could be obtained in trans formation solely. In the course of this investigation, Sekiguchi could also find a working synthetic procedure for the isolation of stable 1,2-disilabenzene 54a and 54b that are formed in the ratio of 54a:54b = 2:3.
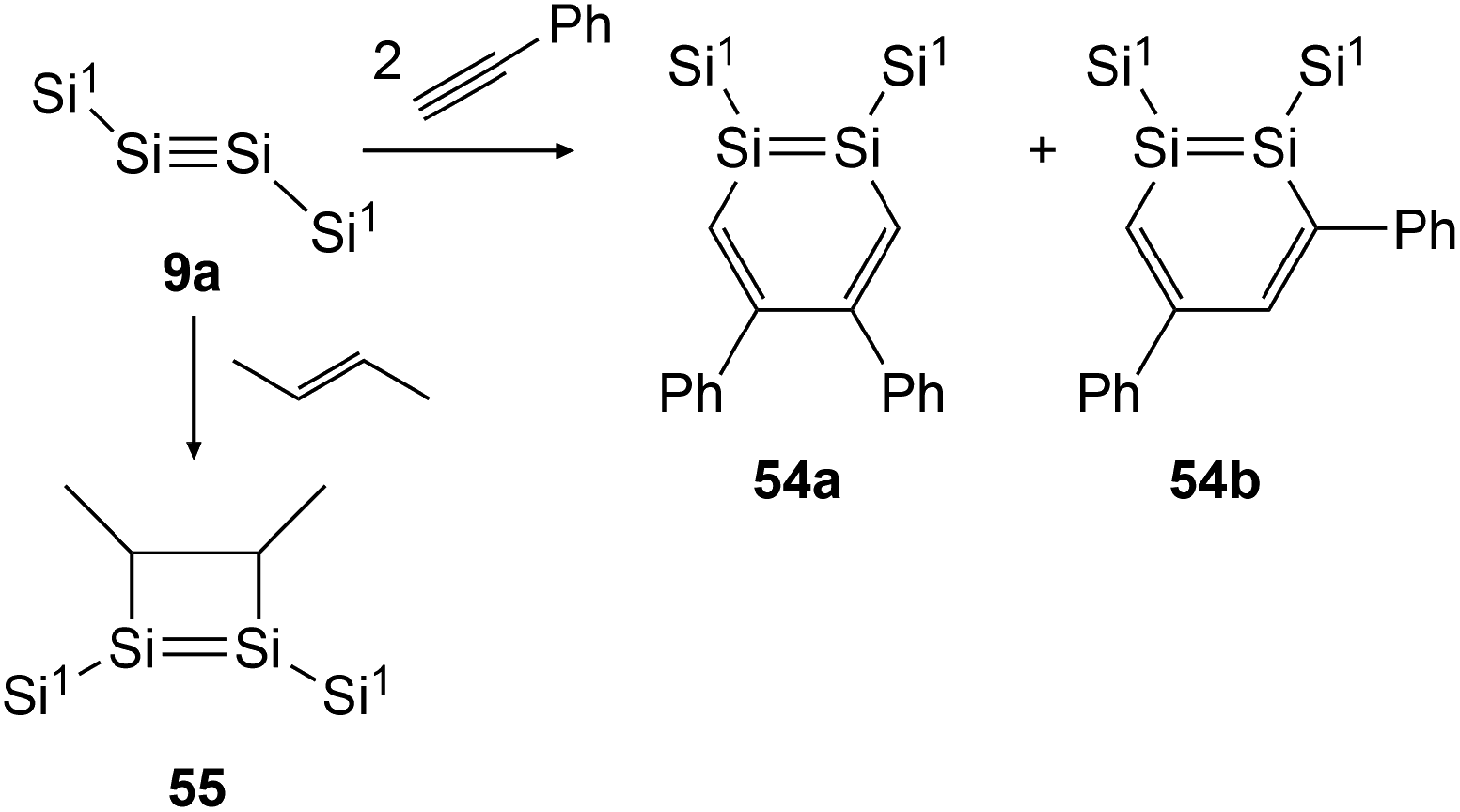 | ||
| Scheme 27 Cycloaddition reactions of disilyne 9a with phenylacetylene and 2-butene. Si1 = Sii-Pr[CH(SiMe3)2]2.43,50 | ||
In the following years the same strategy has led to several examples of stable ditetrelbenzenes and a rich follow up chemistry. In 2010, Tokitoh investigated the cycloaddition of olefins and alkynes to disilyne BbtSiSiBbt (4a).41,42 Depending on the steric demand of the olefins, the reactions result in the 3-membered cycloadduct 56 for ethylene and the [2 + 2] addition product 58 for cyclohexene (Scheme 28, left). In the addition of 2,3-dimethyl-1,3-butadiene to the disilyne, only one addition takes place affording the unusual product 57 as anti-tricyclo[3.1.0.02,4]1,2-disilahexane (Scheme 28, top right). As mentioned before, using a similar synthetic pathway to that used by Sekiguchi, Bbt substituted 1,2-disilabenzene derivative 59 can be furnished for acetylene, phenylacetylene and trimethylsilylacetylene (Scheme 28, bottom right).
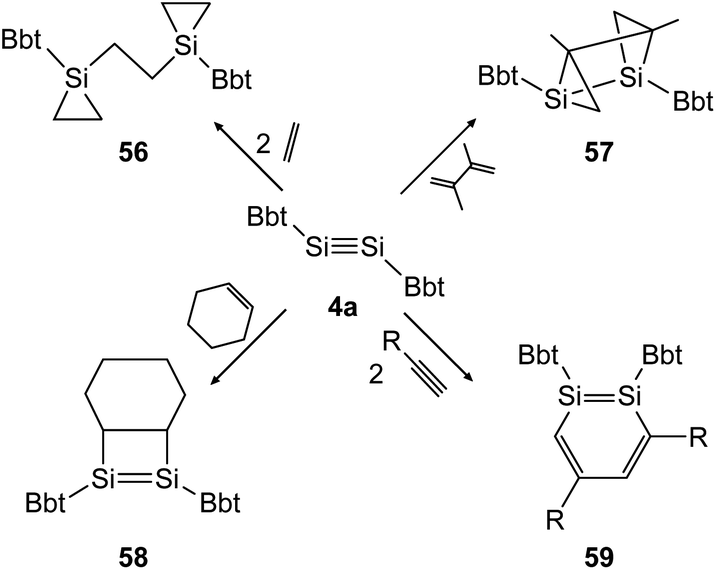 | ||
| Scheme 28 Cycloaddition reactions of disilyne 4a with ethylene, cyclohexene, acetylene derivatives (R = H, Ph, SiMe3), and 2,3-dimethyl-1,3-butadiene.41,42 | ||
Similar reactivity was found for digermyne BbtGeGeBbt (4b) upon reaction with ethylene and terminal olefins (Scheme 29).46,48 Depending on the applied conditions, both stable addition products, 3-membered and 4-membered cycloadduct 60 and 61 can be isolated separately. Theoretical calculations suggest that 1,4-digermabicyclo[2.2.0]hexane (61) is the thermodynamically favored product whereas bis(germiranyl)ethane (60) is stabilized kinetically. Using substituted terminal olefins, products of the form of 62 were obtained exclusively.
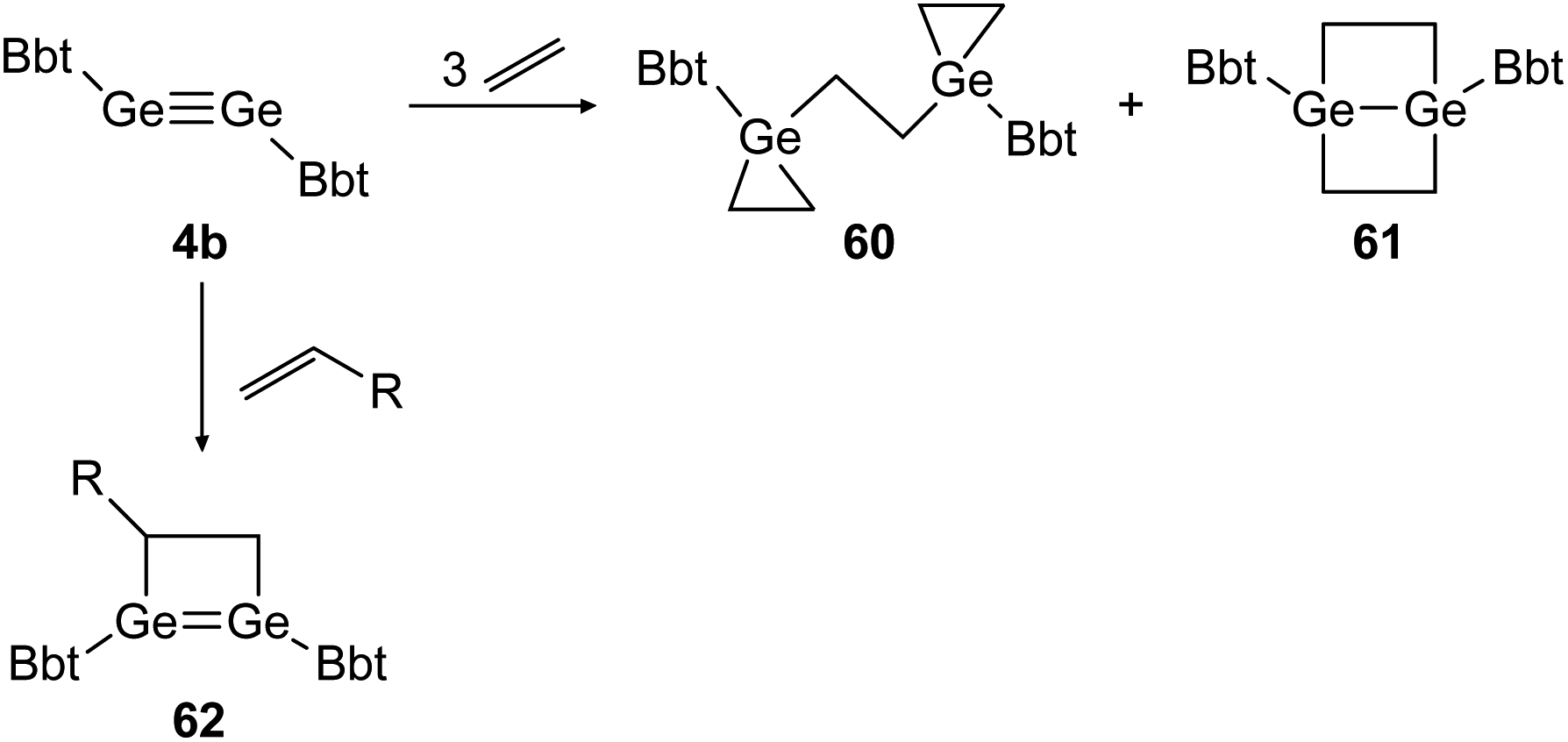 | ||
| Scheme 29 Cycloaddition reactions of digermyne 4b with ethylene and terminal olefins R = n-C4H9, Ph.46,48 | ||
In 2018 Sasamori could uncover the reactivity of disilyne 4c (TbbSiSiTbb) with organic triple bonds (Scheme 30) that gave rise to the 1,2-disilabenzene 65 and 1,4-disilabenzene 63 depending on the size of the substrate.47 While reaction with terminal acetylene derivatives afforded 1,2-disilabenzene 65 within 10 minutes selectively, the slightly bigger 3-hexyne first formed 1,4-disilabenzene 63.49 Remarkably, a transformation of 63 can be induced photochemically, yielding disilabenzvalene derivative 64 (Scheme 30).
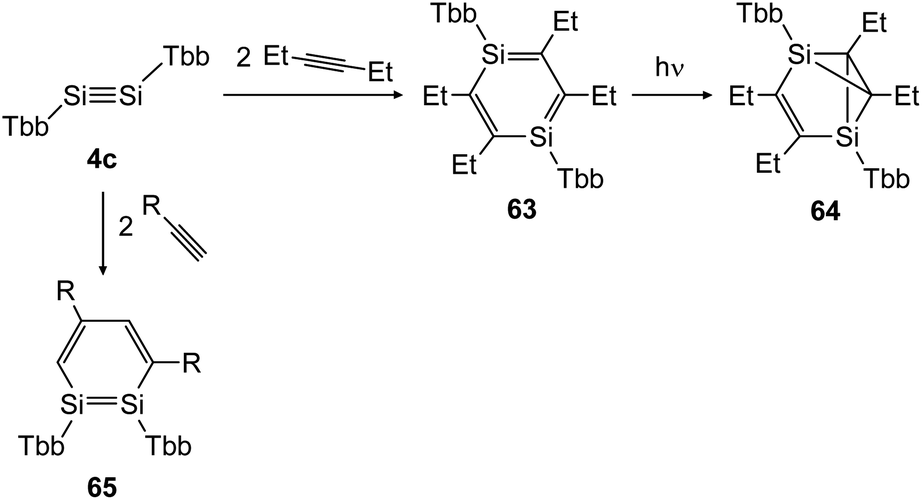 | ||
| Scheme 30 Cycloaddition reactions of disilyne 4c with mono and disubstituted acetylene derivatives R = H, Ph.47,49 | ||
Apart from dimetallynes with high multiple bond character, amide stabilized bisgermylene N1GeGeN1 (13a) shows a standard [2 + 2] cycloaddition reaction for ethylene and cod that results in 66 and 68 (Scheme 31, left) but proceeds via fracture of the Ge–Ge bond in case of nbd and methylacetylene, yielding 67 and 69 (Scheme 31, right).40,53 Theoretical investigation points out an initial [2 + 1] addition to one metal atom and consecutive rearrangement to the cyclic species 66 in case of ethylene. In contrast, for addition of nbd and methylacetylene, rotation around the carbon–carbon bond and thereafter, Ge–Ge bond rupture takes place to yield compounds 67 and 69, respectively.
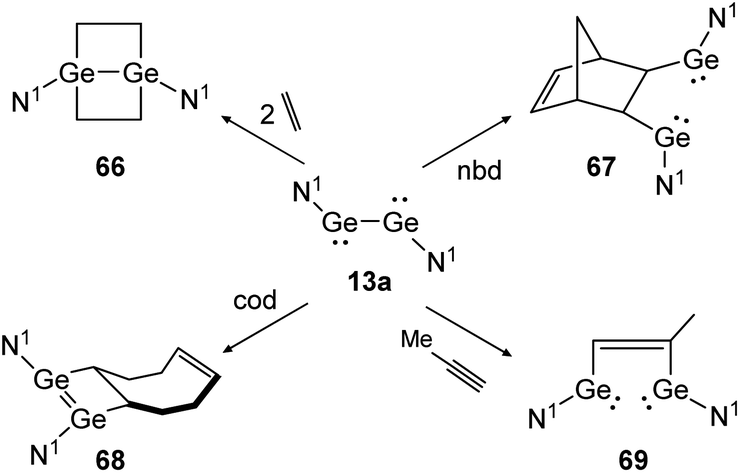 | ||
| Scheme 31 Cycloaddition reactions of bisgermylene 13a with ethylene, cyclooctadiene (cod), methylacetylene, and norbornadiene. N1 = N(Ar1) (SiMe3) (Ar1 = 2,6-(CHPh2)2-4-MeC6H2).40,53 | ||
The same reaction motif can be found for base stabilized bissilylene 17a upon addition of substituted acetylenes (Scheme 32).140 This study shows, in a remarkable way, how the product structure depends on the steric demand of the utilized alkyne. For phenylacetylene, a [1 + 2] addition and insertion with Si–Si bond rupture leads to cis-1,2-disilylenylethene 70. Using the sterically more demanding diphenylacetylene, a singlet delocalized biradicaloid (71) has been synthesized that is stabilized through the donor ligand as well as delocalization within the Si2C4-ring.
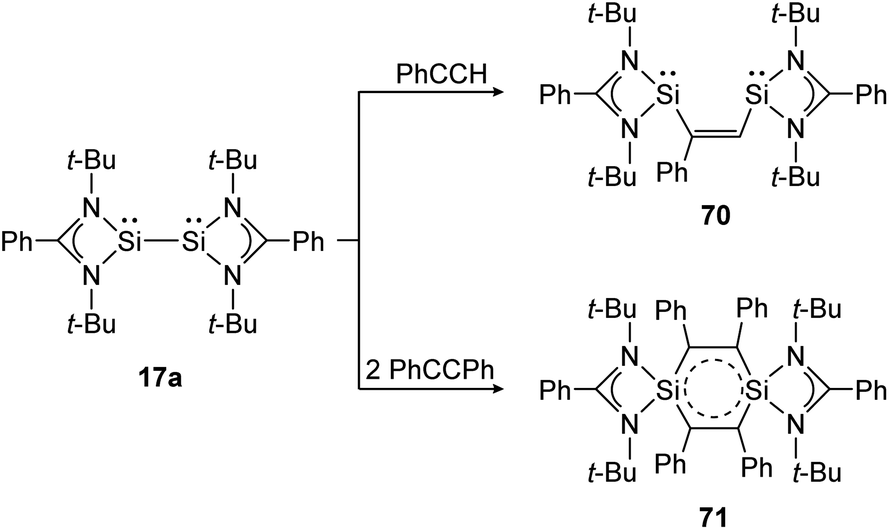 | ||
| Scheme 32 Cycloaddition reactions of bissilylene 17a with phenylacetylene and diphenylacetylene.140 | ||
Catalytic application
Recently, Sasamori and Tokitoh reported on a cyclotrimerization of terminal alkynes (Reppe reaction) using TbbGeGeTbb (4d) as a pre-catalyst.16,45 In this reaction route two equivalents of PhCCH are added to the digermyne and form 3,5-diphenyldigermabenzene (72) within three minutes. 72 acts as the catalyst, to which three more molecules of PhCCH are added during the catalytic cycle to obtain intermediate products 73, 74, and 75 (Scheme 33) and to release 1,2,4-triphenylbenzene from 75b with high selectivity. A range of arylic substrates was tested. The high regioselectivity remains with all tested terminal alkynes and challenges even transition-metal catalysts in its performance. The mechanism of the reaction suggests that the two Ge atoms act in a cooperative fashion during the redox processes between Ge(II) and Ge(IV).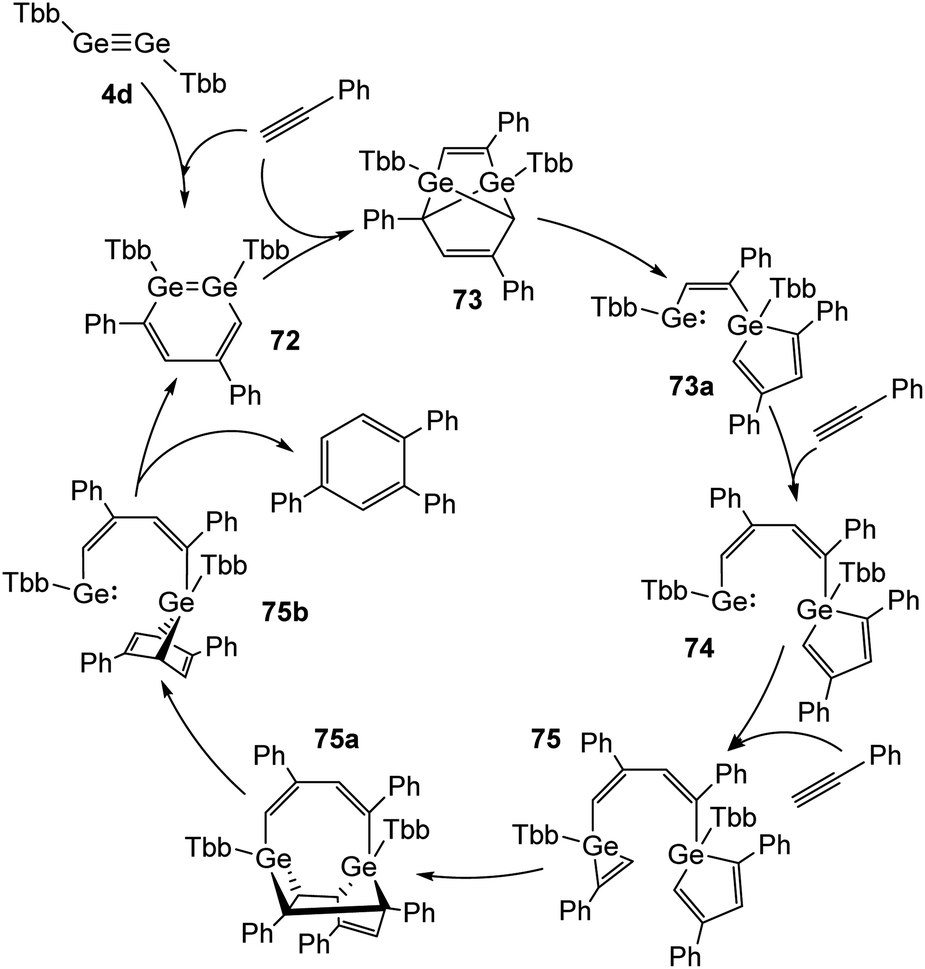 | ||
| Scheme 33 Full catalytic cycle for the cyclotrimerization of phenylacetylene using digermyne 4d.16 | ||
Conclusions
In 2010, Power's seminal review highlighted the great potential of main-group compounds in regard to mimicking transition-metal behavior for the first time. Since then, the growing field of group 14 dimetallenes and dimetallynes has been more and more appreciated and their potential for bond activation and catalysis, increasingly recognized. The influence of ligand properties and central group 14 atoms on the structure and electronic nature of these multiple bonds is better understood. This allows rational design of new compounds with the intention to optimize and decrease HOMO–LUMO gaps and fine-tune the reactivity towards target molecules. Multiple accounts could show, for example, the importance of strongly donating ligands, such as sterically encumbered NHIs or silanides and certain boryl moieties, in enabling activation of H2, NH3, CO2 and other industrially relevant building blocks. A persistent challenge in activating small molecules remains the reversibility of the reaction, i.e. reductive elimination, as the key step in most catalytic reactions to close the cycle and recover the catalyst. There is, however, an increasing number of accounts that show reversibility. For example, the activation of terminal alkynes with a number of bridged distannenes, the reaction of acetonitrile with tetramesityldigermene and the addition of H2 to the distannyne Ar′SnSnAr′ have been reported. Probably the most notable stride in the reactivity of dimetallenes and dimetallynes is the catalytic cyclotrimerization of terminal alkynes with TbbGeGeTbb. This highly regioselective process can certainly compete with transition metal catalysts and represents an interesting new concept of cooperative interactions of two tetrel centers in discrete oxidation states.There is no doubt that the chemistry of group 14 multiple bonds will continue to flourish and open up new alternatives to scarce, expensive and often more toxic transition-metal catalysts. Whilst germanium and tin are more promising to achieve transition-metal mimicking conversions, silicon is so abundant and environmentally benign that it has the highest potential to offer ecological and economic incentives for future application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are exceptionally grateful to the WACKER Chemie AG, the European Research Council (SILION 63794) and the Deutsche Forschungsgemeinschaft (In 234/7-1) for financial support.Notes and references
- L. E. Gusel'nikov and N. S. Nametkin, Chem. Rev., 1979, 79, 529 CrossRef.
- P. Jutzi, Angew. Chem., Int. Ed. Engl., 1975, 14, 232 CrossRef.
- R. West, Angew. Chem., Int. Ed. Engl., 1987, 26, 1201 CrossRef.
- P. J. Davidson, D. H. Harris and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1976, 2268 RSC.
- D. E. Goldberg, D. H. Harris, M. F. Lappert and K. M. Thomas, J. Chem. Soc., Chem. Commun., 1976, 261 RSC.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343 CrossRef CAS PubMed.
- A. G. Brook, F. Abdesaken, B. Gutekunst, G. Gutekunst and R. K. Kallury, J. Chem. Soc., Chem. Commun., 1981, 191 RSC.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587 CrossRef CAS.
- L. Pu, B. Twamley and P. P. Power, J. Am. Chem. Soc., 2000, 122, 3524 CrossRef CAS.
- A. Sekiguchi, R. Kinjo and M. Ichinohe, Science, 2004, 305, 1755 CrossRef CAS PubMed.
- A. D. Phillips, R. J. Wright, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2002, 124, 5930 CrossRef CAS PubMed.
- M. Stender, A. D. Phillips, R. J. Wright and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 1785 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213 CrossRef CAS.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef CAS PubMed.
- T. Sugahara, J.-D. Guo, T. Sasamori, S. Nagase and N. Tokitoh, Angew. Chem., Int. Ed., 2018, 57, 3499 CrossRef CAS PubMed.
- W. Kutzelnigg, Angew. Chem., Int. Ed. Engl., 1984, 23, 272 CrossRef.
- M. Lein, A. Krapp and G. Frenking, J. Am. Chem. Soc., 2005, 127, 6290 CrossRef CAS PubMed.
- G. Trinquier, J. Am. Chem. Soc., 1990, 112, 2130 CrossRef CAS.
- G. Trinquier, J. P. Malrieu and P. Riviere, J. Am. Chem. Soc., 1982, 104, 4529 CrossRef CAS.
- E. A. Carter and W. A. Goddard, J. Phys. Chem., 1986, 90, 998 CrossRef CAS.
- G. Trinquier and J. P. Malrieu, J. Am. Chem. Soc., 1987, 109, 5303 CrossRef CAS.
- S. Nagase, K. Kobayashi and N. Takagi, J. Organomet. Chem., 2000, 611, 264 CrossRef CAS.
- H. B. Wedler, P. Wendelboe and P. P. Power, Organometallics, 2018, 37, 2929 CrossRef CAS.
- P. P. Power, Chem. Rev., 1999, 99, 3463 CrossRef CAS PubMed.
- Y. Jung, M. Brynda, P. P. Power and M. Head-Gordon, J. Am. Chem. Soc., 2006, 128, 7185 CrossRef CAS PubMed.
- H. B. Wedler, P. Wendelboe, D. J. Tantillo and P. P. Power, Dalton Trans., 2020, 49, 5175 RSC.
- P. P. Power, Organometallics, 2007, 26, 4362 CrossRef CAS.
- J. D. Queen, M. Bursch, J. Seibert, L. R. Maurer, B. D. Ellis, J. C. Fettinger, S. Grimme and P. P. Power, J. Am. Chem. Soc., 2019, 141, 14370 CrossRef CAS PubMed.
- W. Kurlancheek, Y. Jung and M. Head-Gordon, Dalton Trans., 2008, 4428 RSC.
- K. Kobayashi and S. Nagase, Organometallics, 1997, 16, 2489 CrossRef CAS.
- V. Kravchenko, R. Kinjo, A. Sekiguchi, M. Ichinohe, R. West, Y. S. Balazs, A. Schmidt, M. Karni and Y. Apeloig, J. Am. Chem. Soc., 2006, 128, 14472 CrossRef CAS PubMed.
- M. Takahashi and Y. Kawazoe, Organometallics, 2008, 27, 4829 CrossRef CAS.
- Y. Chen, M. Hartmann, M. Diedenhofen and G. Frenking, Angew. Chem., Int. Ed., 2001, 40, 2051 CrossRef PubMed.
- N. Takagi and S. Nagase, Organometallics, 2001, 20, 5498 CrossRef CAS.
- D. Astruc, in Organometallic Chemistry and Catalysis, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, p. 197 Search PubMed.
- D. J. Cardin, B. Cetinkaya and M. F. Lappert, Chem. Rev., 1972, 72, 545 CrossRef CAS.
- R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592 CrossRef CAS PubMed.
- C. Cui, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 126, 5062 CrossRef CAS PubMed.
- T. J. Hadlington, J. Li, M. Hermann, A. Davey, G. Frenking and C. Jones, Organometallics, 2015, 34, 3175 CrossRef CAS.
- J. S. Han, T. Sasamori, Y. Mizuhata and N. Tokitoh, J. Am. Chem. Soc., 2010, 132, 2546 CrossRef CAS PubMed.
- J. S. Han, T. Sasamori, Y. Mizuhata and N. Tokitoh, Dalton Trans., 2010, 39, 9238 RSC.
- R. Kinjo, M. Ichinohe, A. Sekiguchi, N. Takagi, M. Sumimoto and S. Nagase, J. Am. Chem. Soc., 2007, 129, 7766 CrossRef CAS PubMed.
- T. Koike, S. Honda, S. Ishida and T. Iwamoto, Organometallics, 2020 DOI:10.1021/acs.organomet.9b00828.
- T. Sasamori, T. Sugahara, T. Agou, J.-D. Guo, S. Nagase, R. Streubel and N. Tokitoh, Organometallics, 2015, 34, 2106 CrossRef CAS.
- T. Sasamori, T. Sugahara, T. Agou, K. Sugamata, J.-D. Guo, S. Nagase and N. Tokitoh, Chem. Sci., 2015, 6, 5526 RSC.
- T. Sugahara, J.-D. Guo, D. Hashizume, T. Sasamori, S. Nagase and N. Tokitoh, Dalton Trans., 2018, 47, 13318 RSC.
- T. Sugahara, J.-D. Guo, T. Sasamori, S. Nagase and N. Tokitoh, Chem. Commun., 2018, 54, 519 RSC.
- T. Sugahara, T. Sasamori and N. Tokitoh, Dalton Trans., 2019, 48, 9053 RSC.
- K. Takeuchi, M. Ichinohe, A. Sekiguchi, J.-D. Guo and S. Nagase, Organometallics, 2009, 28, 2658 CrossRef CAS.
- N. Y. Tashkandi, L. C. Pavelka, C. A. Caputo, P. D. Boyle, P. P. Power and K. M. Baines, Dalton Trans., 2016, 45, 7226 RSC.
- X. Wang, Y. Peng, Z. Zhu, J. C. Fettinger, P. P. Power, J. Guo and S. Nagase, Angew. Chem., Int. Ed., 2010, 49, 4593 CrossRef CAS PubMed.
- L. Zhao, C. Jones and G. Frenking, Chem.–Eur. J., 2015, 21, 12405 CrossRef CAS PubMed.
- T. Y. Lai, L. Tao, R. D. Britt and P. P. Power, J. Am. Chem. Soc., 2019, 141, 12527 CrossRef CAS PubMed.
- P. Buchwalter, J. Rose and P. Braunstein, Chem. Rev., 2015, 115, 28 CrossRef CAS PubMed.
- L. G. Perla, J. M. Kulenkampff, J. C. Fettinger and P. P. Power, Organometallics, 2018, 37, 4048 CrossRef CAS.
- T. Kosai and T. Iwamoto, J. Am. Chem. Soc., 2017, 139, 18146 CrossRef CAS PubMed.
- Y. Murata, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2010, 132, 16768 CrossRef CAS PubMed.
- N. Wiberg, S. K. Vasisht, G. Fischer and P. Mayer, Z. Anorg. Allg. Chem., 2004, 630, 1823 CrossRef CAS.
- T. Sasamori, K. Hironaka, Y. Sugiyama, N. Takagi, S. Nagase, Y. Hosoi, Y. Furukawa and N. Tokitoh, J. Am. Chem. Soc., 2008, 130, 13856 CrossRef CAS PubMed.
- J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622 CrossRef CAS PubMed.
- D. Wendel, T. Szilvási, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 9156 CrossRef CAS PubMed.
- J. Schneider, C. P. Sindlinger, K. Eichele, H. Schubert and L. Wesemann, J. Am. Chem. Soc., 2017, 139, 6542 CrossRef CAS PubMed.
- M. Karni and Y. Apeloig, J. Am. Chem. Soc., 1990, 112, 8589 CrossRef CAS.
- C. Liang and L. C. Allen, J. Am. Chem. Soc., 1990, 112, 1039 CrossRef CAS.
- K. Kobayashi, N. Takagi and S. Nagase, Organometallics, 2001, 20, 234 CrossRef CAS.
- P. P. Power, Organometallics, 2020 DOI:10.1021/acs.organomet.0c00200.
- J.-D. Guo, D. J. Liptrot, S. Nagase and P. P. Power, Chem. Sci., 2015, 6, 6235 RSC.
- H. Arp, J. Baumgartner, C. Marschner, P. Zark and T. Müller, J. Am. Chem. Soc., 2012, 134, 6409 CrossRef CAS PubMed.
- A. Wataru, T. Takeshi and S. Akira, Chem. Lett., 1987, 16, 317 CrossRef.
- K. L. Hurni, P. A. Rupar, N. C. Payne and K. M. Baines, Organometallics, 2007, 26, 5569 CrossRef CAS.
- W. Hamao, T. Ken, F. Norio, K. Motohiko, G. Midori and N. Yoichiro, Chem. Lett., 1987, 16, 1341 CrossRef.
- B. Pampuch, W. Saak and M. Weidenbruch, J. Organomet. Chem., 2006, 691, 3540 CrossRef CAS.
- K. Leszczyńska, K. Abersfelder, A. Mix, B. Neumann, H.-G. Stammler, M. J. Cowley, P. Jutzi and D. Scheschkewitz, Angew. Chem., Int. Ed., 2012, 51, 6785 CrossRef PubMed.
- J. Schneider, J. Henning, J. Edrich, H. Schubert and L. Wesemann, Inorg. Chem., 2015, 54, 6020 CrossRef CAS PubMed.
- Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023 CrossRef CAS PubMed.
- Y. Peng, R. C. Fischer, W. A. Merrill, J. Fischer, L. Pu, B. D. Ellis, J. C. Fettinger, R. H. Herber and P. P. Power, Chem. Sci., 2010, 1, 461 RSC.
- M. Stender, L. Pu and P. P. Power, Organometallics, 2001, 20, 1820 CrossRef CAS.
- V. Y. Lee, K. McNiece, Y. Ito, A. Sekiguchi, N. Geinik and J. Y. Becker, Heteroat. Chem., 2014, 25, 313 CrossRef CAS.
- M. Kira, T. Maruyama, C. Kabuto, K. Ebata and H. Sakurai, Angew. Chem., Int. Ed. Engl., 1994, 33, 1489 CrossRef.
- A. Sekiguchi, S. Inoue, M. Ichinohe and Y. Arai, J. Am. Chem. Soc., 2004, 126, 9626 CrossRef CAS PubMed.
- M. Kira, T. Iwamoto, T. Maruyama, C. Kabuto and H. Sakurai, Organometallics, 1996, 15, 3767 CrossRef CAS.
- V. Y. Lee, K. McNeice, Y. Ito and A. Sekiguchi, Chem. Commun., 2011, 47, 3272 RSC.
- V. Y. Lee, T. Fukawa, M. Nakamoto, A. Sekiguchi, B. L. Tumanskii, M. Karni and Y. Apeloig, J. Am. Chem. Soc., 2006, 128, 11643 CrossRef CAS PubMed.
- N. Wiberg, W. Niedermayer, K. Polborn and P. Mayer, Chem.–Eur. J., 2002, 8, 2730 CrossRef CAS.
- I. Shigeyoshi, I. Masaaki and S. Akira, Chem. Lett., 2008, 37, 1044 CrossRef.
- T. Kosai and T. Iwamoto, Chem.–Eur. J., 2018, 24, 7774 CrossRef CAS PubMed.
- Z. Liu, J. Zhang, H. Yang and C. Cui, Organometallics, 2020 DOI:10.1021/acs.organomet.0c00148.
- P. Jutzi, A. Mix, B. Rummel, W. W. Schoeller, B. Neumann and H.-G. Stammler, Science, 2004, 305, 849 CrossRef CAS PubMed.
- P. Jutzi, A. Mix, B. Neumann, B. Rummel, W. W. Schoeller, H.-G. Stammler and A. B. Rozhenko, J. Am. Chem. Soc., 2009, 131, 12137 CrossRef CAS PubMed.
- D. Wendel, T. Szilvási, D. Henschel, P. J. Altmann, C. Jandl, S. Inoue and B. Rieger, Angew. Chem., Int. Ed., 2018, 57, 14575 CrossRef CAS PubMed.
- T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199 CrossRef CAS PubMed.
- T. J. Hadlington and C. Jones, Chem. Commun., 2014, 50, 2321 RSC.
- J. A. Kelly, M. Juckel, T. J. Hadlington, I. Fernández, G. Frenking and C. Jones, Chem.–Eur. J., 2019, 25, 2773 CAS.
- T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327 RSC.
- D. Gau, R. Rodriguez, T. Kato, N. Saffon-Merceron, A. de Cózar, F. P. Cossío and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 1092 CrossRef CAS PubMed.
- W.-P. Leung, W.-K. Chiu and T. C. W. Mak, Organometallics, 2014, 33, 225 CrossRef CAS.
- S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536 CrossRef CAS PubMed.
- C. Jones, S. J. Bonyhady, N. Holzmann, G. Frenking and A. Stasch, Inorg. Chem., 2011, 50, 12315 CrossRef CAS PubMed.
- S. P. Green, C. Jones, P. C. Junk, K.-A. Lippert and A. Stasch, Chem. Commun., 2006, 3978 RSC.
- R. Jambor, B. Kašná, K. N. Kirschner, M. Schürmann and K. Jurkschat, Angew. Chem., Int. Ed., 2008, 47, 1650 CrossRef CAS PubMed.
- T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176 RSC.
- E. Rivard, Chem. Soc. Rev., 2016, 45, 989 RSC.
- J. A. Hardwick and K. M. Baines, Angew. Chem., Int. Ed., 2015, 54, 6600 CrossRef CAS PubMed.
- J. A. Hardwick and K. M. Baines, Chem.–Eur. J., 2015, 21, 2480 CrossRef CAS PubMed.
- X. Wang, C. Ni, Z. Zhu, J. C. Fettinger and P. P. Power, Inorg. Chem., 2009, 48, 2464 CrossRef CAS PubMed.
- C. Cui, M. M. Olmstead, J. C. Fettinger, G. H. Spikes and P. P. Power, J. Am. Chem. Soc., 2005, 127, 17530 CrossRef CAS PubMed.
- G. H. Spikes, Y. Peng, J. C. Fettinger, J. Steiner and P. P. Power, Chem. Commun., 2005, 6041 RSC.
- J. Li, M. Hermann, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 8611 CrossRef CAS PubMed.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232 CrossRef CAS PubMed.
- Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard and P. P. Power, Chem. Commun., 2008, 6042 RSC.
- S. Wang, T. J. Sherbow, L. A. Berben and P. P. Power, J. Am. Chem. Soc., 2018, 140, 590 CrossRef CAS PubMed.
- M. Hermann, C. Goedecke, C. Jones and G. Frenking, Organometallics, 2013, 32, 6666 CrossRef CAS.
- M. Hermann, C. Jones and G. Frenking, Inorg. Chem., 2014, 53, 6482 CrossRef CAS PubMed.
- L. Zhao, F. Huang, G. Lu, Z.-X. Wang and P. v. R. Schleyer, J. Am. Chem. Soc., 2012, 134, 8856 CrossRef CAS PubMed.
- S. Boomgaarden, W. Saak, M. Weidenbruch and H. Marsmann, Z. Anorg. Allg. Chem., 2001, 627, 349 CrossRef CAS.
- A. Meltzer, M. Majumdar, A. J. P. White, V. Huch and D. Scheschkewitz, Organometallics, 2013, 32, 6844 CrossRef CAS.
- X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27 CrossRef CAS.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707 RSC.
- A. A. Adesina, Appl. Catal., A, 1996, 138, 345 CrossRef CAS.
- A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. Á. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808 CrossRef CAS PubMed.
- X. Wang, Z. Zhu, Y. Peng, H. Lei, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2009, 131, 6912 CrossRef CAS PubMed.
- M. Majumdar, I. Omlor, C. B. Yildiz, A. Azizoglu, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed. Engl., 2015, 54, 8746 CrossRef CAS PubMed.
- M. J. Cowley, Y. Ohmori, V. Huch, M. Ichinohe, A. Sekiguchi and D. Scheschkewitz, Angew. Chem., Int. Ed. Engl., 2013, 52, 13247 CrossRef CAS PubMed.
- M. J. Cowley, V. Huch and D. Scheschkewitz, Chem.–Eur. J., 2014, 20, 9221 CrossRef CAS PubMed.
- T. D. Rapson and H. Dacres, TrAC, Trends Anal. Chem., 2014, 54, 65 CrossRef CAS.
- S. Khan, R. Michel, D. Koley, H. W. Roesky and D. Stalke, Inorg. Chem., 2011, 50, 10878 CrossRef CAS PubMed.
- B. Maity and D. Koley, J. Phys. Chem. A, 2017, 121, 401 CrossRef CAS PubMed.
- P. P. Power, Appl. Organomet. Chem., 2005, 19, 488 CrossRef CAS.
- S. S. Sen, G. Tavčar, H. W. Roesky, D. Kratzert, J. Hey and D. Stalke, Organometallics, 2010, 29, 2343 CrossRef CAS.
- O. Renji and R. West, in Adv. Organomet. Chem., ed. F. Gordon, A. Stone and R. West, Academic Press, 1996, vol. 39, p. 231 Search PubMed.
- M. Kira and T. Iwamoto, in Adv. Organomet. Chem., ed. R. West and A. F. Hill, Academic Press, 2006, vol. 54, p. 73 Search PubMed.
- O. M. Nefedov, M. P. Egorov, A. M. Gal'minas, S. P. Kolesnikov, A. Krebs and J. Berndt, J. Organomet. Chem., 1986, 301, C21 CrossRef CAS.
- K. L. Hurni and K. M. Baines, Chem. Commun., 2011, 47, 8382 RSC.
- S. A. Batcheller and S. Masamune, Tetrahedron Lett., 1988, 29, 3383 CrossRef CAS.
- S. E. Gottschling, K. K. Milnes, M. C. Jennings and K. M. Baines, Organometallics, 2005, 24, 3811 CrossRef CAS.
- C. E. Dixon and K. M. Baines, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 124, 123 Search PubMed.
- Y. Peng, B. D. Ellis, X. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668 CrossRef CAS PubMed.
- M. Stender, A. D. Phillips and P. P. Power, Chem. Commun., 2002, 1312 RSC.
- H.-X. Yeong, H.-W. Xi, K. H. Lim and C.-W. So, Chem.–Eur. J., 2010, 16, 12956 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |