
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect observation of o-benzyne formation in photochemical hexadehydro-Diels–Alder (hν-HDDA) reactions†
Xiaonan
Ma
ab,
Jan
Maier
 c,
Michael
Wenzel
a,
Alexandra
Friedrich
c,
Andreas
Steffen
cd,
Todd B.
Marder
*c,
Roland
Mitrić
*a and
Tobias
Brixner
*a
c,
Michael
Wenzel
a,
Alexandra
Friedrich
c,
Andreas
Steffen
cd,
Todd B.
Marder
*c,
Roland
Mitrić
*a and
Tobias
Brixner
*a
aInstitut für Physikalische und Theoretische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: xiaonanma@tju.edu.cn; brixner@uni-wuerzburg.de; roland.mitric@uni-wuerzburg.de
bInstitute of Molecular Plus, Tianjin University, No. 92 Weijin Road, Nankai District, 300072 Tianjin, China
cInstitut für Anorganische Chemie, Institute for Sustainable Chemistry & Catalysis with Boron, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: todd.marder@uni-wuerzburg.de
dInstitut für Anorganische Chemie, Technische Universität Dortmund, Otto-Hahn-Str.6, 44227 Dortmund, Germany
First published on 14th August 2020
Abstract
Reactive ortho-benzyne derivatives are believed to be the initial products of liquid-phase [4 + 2]-cycloadditions between a 1,3-diyne and an alkyne via what is known as a hexadehydro-Diels–Alder (HDDA) reaction. The UV/VIS spectroscopic observation of o-benzyne derivatives and their photochemical dynamics in solution, however, have not been reported previously. Herein, we report direct UV/VIS spectroscopic evidence for the existence of an o-benzyne in solution, and establish the dynamics of its formation in a photoinduced reaction. For this purpose, we investigated a bis-diyne compound using femtosecond transient absorption spectroscopy in the ultraviolet/visible region. In the first step, we observe excited-state isomerization on a sub-10 ps time scale. For identification of the o-benzyne species formed within 50–70 ps, and the corresponding photochemical hexadehydro-Diels–Alder (hν-HDDA) reactions, we employed two intermolecular trapping strategies. In the first case, the o-benzyne was trapped by a second bis-diyne, i.e., self-trapping. The self-trapping products were then identified in the transient absorption experiments by comparing their spectral features to those of the isolated products. In the second case, we used perylene for trapping and reconstructed the spectrum of the trapping product by removing the contribution of irrelevant species from the experimentally observed spectra. Taken together, the UV/VIS spectroscopic data provide a consistent picture for o-benzyne derivatives in solution as the products of photo-initiated HDDA reactions, and we deduce the time scales for their formation.
Introduction
After the discovery of a [4 + 2]-cycloaddition reaction between a conjugated diene and an alkene by Diels and Alder in 1928,1 it took almost 70 years until Johnson2–6 and Ueda7–11 explored the [4 + 2]-cycloaddition between a diyne and an alkyne. This latter reaction is now known as a “hexadehydro-Diels–Alder” (HDDA) reaction so named by the Hoye group in 2012 as a logical extension of the dehydro-Diels–Alder reaction.12 Instead of forming cyclohexene derivatives, the HDDA reaction13–16 is believed to produce a highly reactive o-benzyne intermediate, and is of much current interest due to its unusual reactivity/mechanism17–27 and numerous applications in synthetic chemistry.28–35 A series of reports within the last three years,36–43 examining the HDDA reaction from a more mechanistic viewpoint, demonstrate the immense versatility of HDDA-generated o-benzynes, including reaction with an alkyne forming a highly reactive benzocyclobutadiene21 or reaction with electron-deficient thioamides giving stabilized ammonium ylides in a new type of [3 + 2]-cycloaddition.24 These and other examples emphasize the importance of HDDA-generated o-benzynes for the discovery of new reaction pathways.Although the HDDA reaction is becoming increasingly useful in synthesis, intriguing questions remain. Does o-benzyne really form as an intermediate during the HDDA reaction? Can we confirm the existence of o-benzyne derivatives as initial products of the HDDA reaction in solution? In reported investigations on the HDDA reaction, o-benzyne was inferred indirectly by “backtracking” its structure from the products resulting from trapping reactions, i.e., a secondary product of the HDDA reaction.14–16 A trapping reaction of unique interest is the double hydrogen transfer, first reported in 2009 by Tsui and Sterenberg.44 This transfer of two vicinal hydrogen atoms from solvents was further investigated in more detail by Hoye et al.17 correlating the reactivity of different solvents with the degree of eclipsing of two adjacent C–H bonds among their low-energy conformers. A subsequent promising trapping reaction is that of perylene with traditional45,46 and HDDA-generated47o-benzyne derivatives, generating new polycyclic aromatic hydrocarbons. However, trapping experiments do not provide a direct observation of the proposed o-benzyne intermediate, and UV/VIS spectroscopic evidence for o-benzynes in solution remains elusive.
The formation, characterization, and application of benzynes has fascinated chemists for more than 90 years due to their Kekulé-type diradical structure, special role in combustion and astrochemistry, and great potential in synthetic chemistry.48–53o-Benzyne was first proposed as an intermediate in 1927 by Bachmann and Clarke,54 and IR,55–61 microwave,62–66 NMR,67,68 and photoelectron69–74 spectra of o-benzynes have been reported in the last half century. A recent report,75 however, explains that the NMR data68 of an o-benzyne generated inside of a hemicarcerand do not belong to an o-benzyne inside the molecular cage, but instead, the signals result from a species formed from a reaction of the o-benzyne with the molecular cage. This had been one of the very few reports of the spectroscopic identifications of an o-benzyne in “solution” at room temperature. There are only few investigations of electronic absorption spectra in the ultraviolet/visible (UV/VIS) range, in the gas phase or in low-temperature matrices.76–82 To the best of our knowledge, the electronic absorption spectrum, i.e., the UV/VIS spectrum, of o-benzyne in solution, has not been reported.
In addition to the thermal HDDA reaction discussed above, the photochemical HDDA (hν-HDDA) reaction has also been reported recently, initiated by UV excitation at low temperature (−70 °C). The reaction cascade and corresponding trapping product were found to be identical to those formed thermally.83 Extending the previous work which employed continuous-wave illumination, we show herein that using time-resolved excitation and transient absorption spectroscopy makes the direct detection of o-benzynes possible, and allows us to deduce their formation dynamics. In our study, we employed ultrafast spectroscopy that has proven to be a powerful tool by which to elucidate chemical reaction mechanisms, identifying the product as well as the pathways leading to it.
Thus, we performed ultrafast pump–probe (transient absorption) spectroscopy in the UV/VIS region and accurate quantum chemical calculations to explain the observed transient spectra in order to investigate the excited-state dynamics and photochemical reaction of bis-diyne compound Me-BD-Me (Fig. 1, top left), where Me is –CH3 and the bis-diyne (BD) is –4-C6H4–(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)2–(CH2)3–(CC)2–4-C6H4–. We combine transient absorption measurements with intermolecular trapping strategies (Fig. 1, bottom) in tetrahydrofuran (THF) solution (path 1), using self-trapping (paths 2 and 3), and the reaction with perylene (path 4). Thus, we obtained spectroscopic evidence for the existence of the o-benzyne hν-HDDA cascade and recorded the highly plausible transient UV/VIS absorption spectrum of an o-benzyne species in solution for the first time.
C)2–(CH2)3–(CC)2–4-C6H4–. We combine transient absorption measurements with intermolecular trapping strategies (Fig. 1, bottom) in tetrahydrofuran (THF) solution (path 1), using self-trapping (paths 2 and 3), and the reaction with perylene (path 4). Thus, we obtained spectroscopic evidence for the existence of the o-benzyne hν-HDDA cascade and recorded the highly plausible transient UV/VIS absorption spectrum of an o-benzyne species in solution for the first time.
 | ||
| Fig. 1 Compounds and reactivity scheme. Chemical structure of the bis-diyne investigated, 1,11-bis(p-tolyl)undeca-1,3,8,10-tetrayne (Me-BD-Me, top left) with Me = –CH3, BD = –4-C6H4–(CC)2–(CH2)3–(CC)2–4-C6H4–. We also show the hν-HDDA reaction-generated o-benzyne species (Me-BZ-Me, top center) and the following trapping reactions with tetrahydrofuran (bottom left, path 1), a second Me-BD-Me (self-trapping, bottom center, path 2 and 3), and perylene (bottom right, path 4). | ||
Results and discussion
Transient absorption experiment
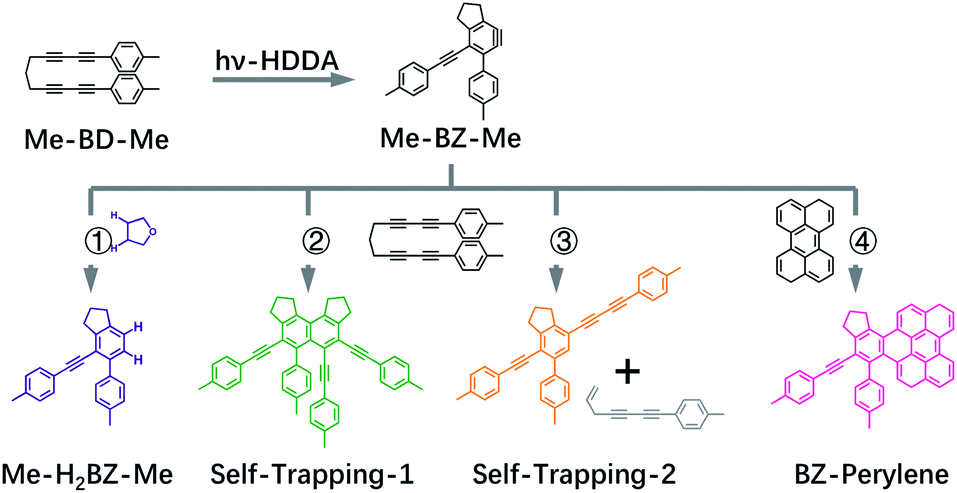
The transient UV/VIS absorption spectra of Me-BD-Me in THF following UV excitation at a central wavelength of λ = 295 nm are shown in Fig. 2, which display the optical density difference spectra (ΔOD) in the 320–670 nm regime. The transient absorption spectra of Me-BD-Me in CHCl3 and CH3CN are displayed in Fig. S1† with similar manner. | ||
| Fig. 2 Transient UV/VIS absorption spectra in 320–670 nm regime. Data are shown for Me-BD-Me dissolved in THF upon photoexcitation at 295 nm in the delay ranges of (a) 500 fs to 15 ps, (b) 15 ps to 300 ps, and (c) 300 ps to 3.8 ns. | ||
The transient absorption signal is dominated by a cascaded sequence of positive peaks extending from the VIS to the near-UV range with dynamic changes observed from sub-picosecond to several-nanosecond time scales. The positive peaks can be attributed to excited-state absorption of initially populated states as well as the ground-state and excited-state absorption resulting from intermediates or final photoproducts, to be assigned below. No negative ground-state bleach from the initial bis-diyne is observed in the recorded spectral window as the measured static UV/VIS spectrum of Me-BD-Me in THF (Fig. S2†) indicates its electronic absorption to be mostly in the deep-UV (<300 nm) range. The transient absorption spectra of Me-BD-Me in THF, CHCl3, and CH3CN are qualitatively similar, and three spectral bands are observed in all three cases. At early pump–probe time delays (Fig. 2a, S1a and d†), the pronounced positive band at 350–450 nm (subsequently referred to as band 1) appears immediately upon excitation and loses amplitude on a several-picosecond time scale (see below for a global fit and quantitative data). Band 1 of Me-BD-Me in different solvents shows multi-peak character (Fig. S3†). Taking Me-BD-Me in THF as an example (Fig. 2a), two pronounced peaks (2.98 eV/416 nm and 3.44 eV/360 nm) with comparable intensities were observed at 0.5 ps delay for Me-BD-Me, while an additional shoulder (3.14 eV/395 nm) of the 2.98 eV/416 nm peak is also observed. Subsequently, the 2.98 eV/416 nm peak rapidly decays together with the shoulder, while the 3.44 eV/360 nm peak undergoes red-shifting as a new band grows at 3.34 eV/371 nm (Fig. 2b). Similar spectral character and behaviors can also be observed for Me-BD-Me in both CHCl3 and CH3CN. Considering the fast decay behavior, we tentatively attribute the multi-peak band 1 to the excited-state absorption (ESA) band of the initially populated S1 state of Me-BD-Me. The observed decay of band 1 within hundreds of picoseconds might correspond to the electronic deactivation (S1 → S0) or the following photochemical transformation to bleach the S1 state. During the peak red-shifting process in the range of 50 to 100 ps delay, the 416 nm peak of the ESA band also decays and disappears with a similar time scale, which confirms that the red-shifting is a result of the decay of ESA and the growing of a new band at 371 nm. We performed TD-DFT calculations84–87 on the structurally optimized S1 state of Me-BD-Me in THF solution with the CAM-B3LYP88 functional and def2-TZVP89,90 basis set, which predicted two vertical transitions with pronounced oscillator strength, i.e. S1 → S2 (1.31 eV/946 nm) and S1 → S6 (2.73 eV/454 nm). The latter is reasonably consistent with the experimentally observed peak at 2.98 eV/416 nm, while the S1 → S2 transition is beyond our probe wavelength range. Note that the calculated (454 nm) and experimentally observed wavelength (416 nm) show a discrepancy of 2.98 eV (416 nm) − 2.73 eV (454 nm) = 0.25 eV, which is still in the expected confidence range of DFT calculations of electronic transition energies that are conventionally associated with errors of ±0.3 eV. A clear rise in the transient spectra can be observed at the red edge (Fig. 2a, b, S1a, b, d and e†), which shows a decay process similar to band 1, and implies the existence of an extra positive band in the near-infrared regime. Such an observation is consistent with the calculated S1 → S2 ESA band. The peaks at 3.14 eV/395 nm and 3.44 eV/360 nm observed in band 1 may originate from vibrational progressions or excitations to higher states.
The disappearance of band 1 indicates excited-state deactivation that subsequently leads to the rise of a strong and narrow band (band 2, peak at 3.35 eV/370 nm) in the 350–400 nm region (Fig. 2b, S1b and e†) that builds up within hundreds of picoseconds. Band 2 is slightly red-shifted from the 3.44 eV/360 nm peak of band 1; apparently bands 1 and 2 are strongly overlapped with each other in the 350–450 nm regime. It is difficult to conclude whether band 2 is newly formed but overlapped with band 1, or band 2 is subsequently transformed from the 350–400 nm part of band 1, which we attempt to clarify by the quantitative target analysis in the following sections. We postulate that this corresponds to the photochemical generation of a reactive intermediate formed from the excited bis-diyne, namely the o-benzyne (Me-BZ-Me), discussed further and confirmed below. The TD-DFT calculations also predicted the vertical excitation energy of o-benzyne Me-BZ-Me to be 3.33 eV/372 nm (S0 → S1), which is consistent with the experimentally observed energy of band 2, i.e. 3.35 eV/370 nm.
In the following step (Fig. 2c, S1c and f†), band 2 slowly decays with the concurrent rise of a new band (band 3) at even shorter wavelengths (320–370 nm), visible as a shoulder of band 2. Several peaks can be observed within band 3, which may originate from vibrational progressions. The sequential nature of the photoreaction sequence (band 1 → band 2 → band 3) is nicely observed by the occurrence of isosbestic points at 393 nm (Fig. 2a, b, S1a and b†), 368 nm (Fig. 2c) for Me-BD-Me in THF and CHCl3, and at 388 nm (Fig. S1d and e†) and 356 nm (Fig. S1f†) for Me-BD-Me in CH3CN.
We performed a target analysis of the transient UV/VIS absorption spectra to confirm the sequential process of reactive steps, obtain time constants, and assign the associated species. Singular-value decomposition indicated the presence of four linearly independent components (species I, II, III, IV) above the experimental noise level, and three time constants [τ1(I → II), τ2(II → III), τ3(III → IV)] describing their temporal evolution. The target analysis, therefore, employed the assumption of a sequential kinetic model consisting of three steps (I → II → III → IV) with any possible decay of species IV occurring beyond the maximum time delay and, thus, not observable. The set of species-associated spectra (SAS) obtained and corresponding fitted time constants of this analysis, as well as others further discussed below, are shown in Fig. 3a and summarized in Table 1. The quality of the fit is shown in Fig. S4† with time traces at selected probe wavelengths, while the target analysis fitted concentration evolution of each transient species of Me-BD-Me and perylene in CHCl3 are displayed in Fig. S5.†
 | ||
| Fig. 3 Species-associated spectra (SAS) obtained from target analysis of transient UV/VIS spectra. Data are shown for Me-BD-Me in (a) THF, (b) a 50/50 mixture of THF and 1,4-dioxane, (c) 1,4-dioxane, (d) CHCl3, and (e) CH3CN. The sequential model (I → II → III → IV) was applied for the target analysis to estimate the SAS and corresponding time constants which are listed in Table 1. See text for detailed assignments. The scale bar in each subplot corresponds to an absorbance change of 5 mOD. | ||
| BDa in THFb | BDa in THFb/Dc | BDa in Dc | BD in CH3CN | |
|---|---|---|---|---|
| a BD: bis-diyne, i.e., Me-BD-Me. b THF: tetrahydrofuran. c D: 1,4-dioxane. d P: perylene. e The transient absorption spectra of perylene were fitted with a three-step model (I → II → III→), wherein the third time constant indicates the decay of species III, i.e. τ3 (III→), instead of τ3 (III → IV). See Fig. S5 for details. | ||||
| τ 1(I → II)/ps | 3.17 ± 0.28 | 2.77 ± 0.31 | 2.62 ± 0.24 | 3.03 ± 0.32 |
| τ 2(II → III)/ps | 48.5 ± 5.0 | 50.5 ± 4.7 | 51.3 ± 4.5 | 63.2 ± 5.9 |
| τ 3(III → IV)/ns | 4.03 ± 0.78 | 4.15 ± 0.76 | 4.64 ± 0.82 | 2.47 ± 0.30 |
As shown in Fig. 3a, d and e (red and orange curves), the target analysis successfully isolated the bi-exponential decay (I → II→) of band 1 observed in the transient absorption spectra (Fig. 2). Such a bi-exponential decay of an ESA band can be explained by a typical non-radiative decay of the excited state via a pronounced potential energy barrier,91–93i.e., the initially populated excited state (species I) relaxes [τ1(I → II)] within the S1 potential energy surface until it reaches a local minimum (species II). Then, the trapped wave packet must overcome an energy barrier [τ2(II → III)] to reach a conical intersection (CI) associated with further photochemical conversion.
Regarding process I → II, we firstly exclude two potential assignments that might in principle show up on a several picoseconds time scale in the early stages of excited-state evolution following optical excitation: (1) vibrational relaxation in the excited state, i.e., the wave packet escaping from the initially populated Franck–Condon area, can be excluded as we observed neither red-shift nor spectral narrowing during the I → II process; (2) we also exclude ultrafast solvation, because solvation is usually highly dependent on solvent polarity whereas, in our case, the fitted time constants of I → II show no significant change upon varying the solvent, from high-polarity acetonitrile (ε = 35.95) to low-polarity 1,4-dioxane (ε = 2.21). Next, the DFT/TD-DFT calculations indicate that both the ground (S0) and excited state (S1) of Me-BD-Me have two conformers with comparable energies: the “open” conformation in which the two –CC–CC– units are relatively far from each other and the “closed” conformation in which the two –CC–CC– units are close to each other (see Fig. S6†). The “open” conformation was shown to be the structure of ground-state Me-BD-Me in the solid state by single-crystal X-ray diffraction,94 and our DFT calculations on Me-BD-Me also indicate a preference for the open conformation with ΔG(Open) − ΔG(Closed) = −3.3 kJ mol−1 in favor of the open conformer. However, it is logical to assume that the “closed” conformation leads to an increased HDDA reaction probability between the two –CC–CC– units in the excited state because the two units have to approach each other for the reaction to occur. Thus, we tentatively attribute the process I → II to the excited-state isomerization of Me-BD-Me from “open” to “closed” conformation or other structural alteration for assisting the subsequent HDDA reaction, although this particular assignment cannot be proved unambiguously with the available data. As the electronic absorption spectrum is normally insensitive to such structural conformational changes, the SAS of species I and II are similar to one another and differ only by their associated time scales. These time constants of several picoseconds are consistent with the typical time scales of intramolecular excited-state isomerization.95,96 We note that the TD-DFT calculations are not accurate enough (at a ∼0.3 eV confidence level) to predict the small differences between the transient absorption spectra of species I and II. The isomerization process includes no drastic changes in geometry and the electronic states should be able to follow the geometry adiabatically. Larger differences in the spectrum might be expected if the geometry change during the isomerization gave rise to new electronic couplings. This does not seem to be the case, however, according to the experimental evidence and the calculations.
The process II → III produces pronounced absorption bands centered at 370 nm with formation time constants of 48.5 ps (in THF), 70.2 ps (in CHCl3) and 63.2 ps (in CH3CN), respectively, visible in the green curves in Fig. 3a, d and e. Species III then slowly converts [τ3(III → IV) = 3–5 ns] to species IV, visible as a new band with a vibrational progression at shorter wavelengths (left peak(s) in blue curves of Fig. 3). While we assign species I and II to be the initially populated S1 state in the “open” form and its relaxed “closed” conformer, respectively, the assignment of species III and IV requires more consideration. As we mentioned above, the vertical excitation energy of o-benzyne (Me-BZ-Me) is 3.33 eV (S0 → S1), which is consistent with the experimentally observed energy of band 2 (3.35 eV). However, such consistency alone is not sufficient to assign band 2 to the benzyne species. Considering the fact that an experimental electronic spectrum of benzyne species in solution has not been reported previously, direct comparison with other experimental data is difficult. Therefore, we turned to an indirect procedure by performing a series of intra and intermolecular trapping experiments on benzyne that, together with the consistent DFT description, makes the assignment possible, which is discussed in the following sections.
If we assume the observed sequence (I → II → III → IV) to be the hν-HDDA reaction, species III, with a strong absorption band (band 2) at ∼370 nm, might belong to the o-benzyne derivative (denoted as Me-BZ-Me) generated. Then, species IV (band 3) can be assigned to secondary products of reactions of the o-benzyne with abundant possibilities, highly depending on the chemical environments. In order to identify the photoproducts, and assign the transient absorption features, we carried out different time-resolved intermolecular trapping experiments as described in the following paragraphs. The strategies were designed to confirm the existence of the transient o-benzyne spectrum in our transient absorption observations. Our objective is the assignment of the slowly rising band 3 observed in transient absorption spectra (see Fig. 2c, S1c and f†) of Me-BD-Me in THF, CHCl3, and CH3CN, i.e., species IV from the target analysis (see Fig. 3).
Double-hydrogen transfer
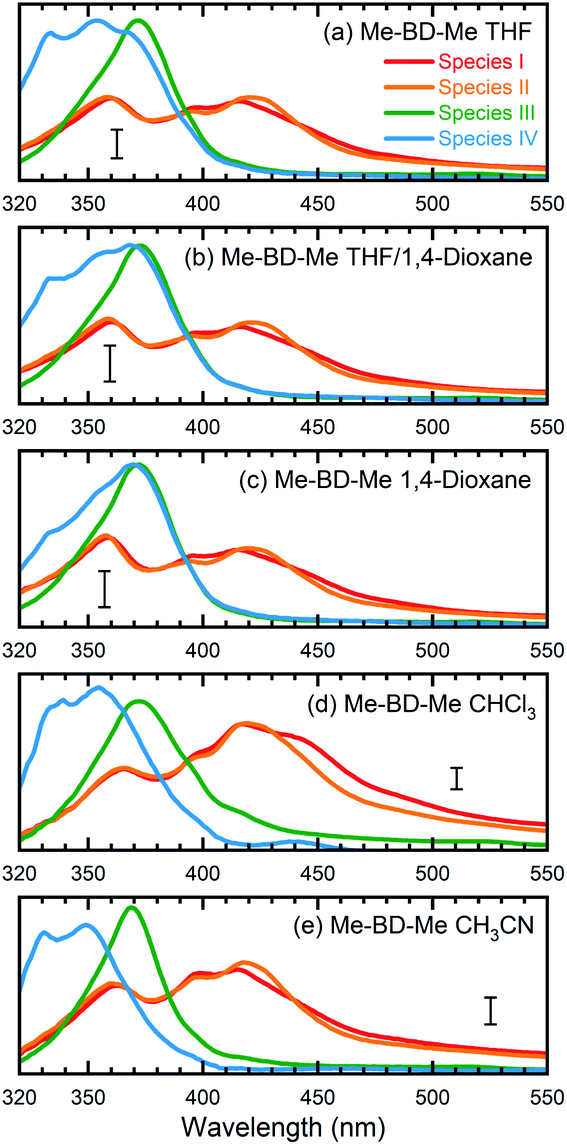
As a first possibility, we consider the scenario that double-hydrogen transfer from an alkane solvent generates the benzene species Me-H2BZ-Me (Fig. 1, path 1). This is plausible because THF was reported to be a relatively good 2H donor (with a yield of ∼60%) for reaction with o-benzyne.17 Thus, we carried out transient absorption measurements on Me-BD-Me in 1,4-dioxane and a THF/1,4-dioxane mixture because the 2H transfer yield is almost zero in 1,4-dioxane.17The results of transient absorption measurements on Me-BD-Me are shown for a 3.8 ns delay in Fig. 4a for THF (red), a 50/50 mixture of THF and 1,4-dioxane (pink), and pure 1,4-dioxane solvent (light pink), and the results of the corresponding target analysis are displayed in Fig. 3b for the 50/50 mixture, and in Fig. 3c for pure 1,4-dioxane. It is clear that band 3 builds substantially even in 1,4-dioxane, although it is weaker than in THF. Thus, it can be tentatively excluded that band 3 is dominated by the Me-H2BZ-Me species. In order to provide more direct evidence, chemical synthesis and purification of Me-H2BZ-Me was conducted together with comprehensive structural characterization (ESI Note 2†). The measured absorption spectrum of Me-H2BZ-Me (Fig. 4b, navy line) is located mostly at wavelengths shorter than 320 nm, i.e., outside the probe window of our transient absorption measurements. Therefore, although the double-hydrogen transfer is well-known as an important secondary reaction following benzyne formation in solution, any Me-H2BZ-Me contribution to band 3 must be very small even if the species is formed. The Me-H2BZ-Me species can be safely excluded as a potential assignment of the slowly formed band 3 following band 2 at 370 nm.
 | ||
| Fig. 4 Comparison between transient absorption spectra of Me-BD-Me and spectra of chemically isolated trapping products. (a and c) Transient absorption spectra in the 320–500 nm region at 300 ps (gray filled) and 3800 ps (colored solid lines) delay of Me-BD-Me in THF, THF/1,4-dioxane, 1,4-dioxane (a) and CHCl3 (c). (b and d) Static absorption spectra of chemically isolated Me-H2BZ-Me (navy line) and of two self-trapping species (orange and green lines) in THF (b) and CHCl3 (d). Several dashed lines are used for assisting the comparison. See Fig. 1 for structural formulae, and text for details. | ||
Intermolecular self-trapping by bis-diyne
With Me-H2BZ-Me excluded to be responsible for transient absorption band 3, we performed chemical separation of a thermal reaction of Me-BD-Me. Two dominating and stable products were separated using flash chromatography. The structural characterization confirmed a product with a dimeric structure, i.e., a naphthalene derivative,21,94 denoted Self-trapping-1 with its structure illustrated in Fig. 1 (path 2, green). In addition, an indane product (Self-trapping-2, Fig. 1, path 3, orange) and a fragment (Fig. 1, path 3 gray) were also isolated and identified.94 Although the isolated products arose from a thermal reaction, it has been reported that the HDDA products arising from both the thermally and photochemically generated o-benzyne intermediate are the same.83 Thus, it was assumed that the electronic state of the o-benzyne intermediate is the same in both cases. Fig. 4b and d show the absorption spectra in THF and CHCl3, respectively, of the two products Self-trapping-1 (orange) and Self-trapping-2 (green). It can be seen that these spectra match the transient spectral features of band 3 for both THF and CHCl3 (vertical dashed lines in Fig. 4). This correspondence indicates that HDDA-generated o-benzyne is probably self-trapped by an extra aryl butadiyne moiety of Me-BD-Me (for a plausible reaction mechanism see ref. 94). With the most plausible assignment of band 3 thus in place, band 2 at ∼370 nm can be concluded to be the o-benzyne species.In addition to the direct interpretation of the transient spectra in Fig. 4, we compare the steady-state absorption spectra of the isolated products with the SAS from target analysis in Fig. S10.† It can be seen that the SAS of species IV in THF and CHCl3 are consistent with spectra of the isolated products, i.e., Self-trapping-1 (at ∼335 nm) and Self-trapping-2 (at ∼354 nm and ∼368 nm). The trapping experiment thus provides evidence that the observed band 3 can be tentatively attributed to the self-trapping product of o-benzyne with Me-BD-Me itself, and that, in turn, band 2 probably belongs to the prior o-benzyne intermediate from which the self-trapping species emerges.
Intermolecular trapping by perylene
An alternative strategy for trapping o-benzyne is its reaction with species containing –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– bonds such as in a π-conjugated system reported recently (without time resolution).12,15,26,47,83 We also made use of this reaction, but again employing femtosecond time-resolved techniques. The trapping product is expected to absorb in the >400 nm wavelength region where the transient absorption of the reactant bis-diynes is sufficiently weak to allow the largely unhindered observation of the trapping products. Among the reported options, perylene, with its absorption in the 400–450 nm region,47 is ideal for our application. In such cases, we have to consider that the intermolecular trapping process occurs as a bimolecular reaction with diffusion-controlled kinetics.97–100 To promote the diffusion and enable us to observe the trapping product within our maximum delay range, we employed low-viscosity CHCl3 as a solvent for the perylene trapping experiments. Meanwhile, according to our target analysis, Me-BD-Me shows the slowest self-trapping reaction in CHCl3 compared with reactions in THF and CH3CN (see Table 1). Thus, utilizing CHCl3 minimizes the interference of the self-trapping reaction in the delay range employed. Although perylene can also be excited directly at our pump wavelength (295 nm), the remaining ground-state concentration should be sufficient for trapping o-benzyne, as the molar extinction coefficient of perylene at 295 nm is small (Fig. S11†).
C– bonds such as in a π-conjugated system reported recently (without time resolution).12,15,26,47,83 We also made use of this reaction, but again employing femtosecond time-resolved techniques. The trapping product is expected to absorb in the >400 nm wavelength region where the transient absorption of the reactant bis-diynes is sufficiently weak to allow the largely unhindered observation of the trapping products. Among the reported options, perylene, with its absorption in the 400–450 nm region,47 is ideal for our application. In such cases, we have to consider that the intermolecular trapping process occurs as a bimolecular reaction with diffusion-controlled kinetics.97–100 To promote the diffusion and enable us to observe the trapping product within our maximum delay range, we employed low-viscosity CHCl3 as a solvent for the perylene trapping experiments. Meanwhile, according to our target analysis, Me-BD-Me shows the slowest self-trapping reaction in CHCl3 compared with reactions in THF and CH3CN (see Table 1). Thus, utilizing CHCl3 minimizes the interference of the self-trapping reaction in the delay range employed. Although perylene can also be excited directly at our pump wavelength (295 nm), the remaining ground-state concentration should be sufficient for trapping o-benzyne, as the molar extinction coefficient of perylene at 295 nm is small (Fig. S11†).
We carried out transient absorption measurements on Me-BD-Me and on Me-BD-Me/perylene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 concentration ratio), referred to as “trapping mixture”, both in CHCl3 solvent under otherwise similar conditions as described in the sections above. Again, we performed a target analysis using a sequential model on the temporal–spectral maps measured, and the resulting SAS are displayed in Fig. 5a (trapping mixture) and Fig. 5b (pure Me-BD-Me). For comparison, we also measured the transient absorption spectra of pure perylene in CHCl3 under identical concentration and excitation conditions, such as excitation wavelength and power, which produced the transient absorption spectra shown in Fig. S12a and b,† and the target analysis results shown in Fig. 5, S12c and d.†
:5 concentration ratio), referred to as “trapping mixture”, both in CHCl3 solvent under otherwise similar conditions as described in the sections above. Again, we performed a target analysis using a sequential model on the temporal–spectral maps measured, and the resulting SAS are displayed in Fig. 5a (trapping mixture) and Fig. 5b (pure Me-BD-Me). For comparison, we also measured the transient absorption spectra of pure perylene in CHCl3 under identical concentration and excitation conditions, such as excitation wavelength and power, which produced the transient absorption spectra shown in Fig. S12a and b,† and the target analysis results shown in Fig. 5, S12c and d.†
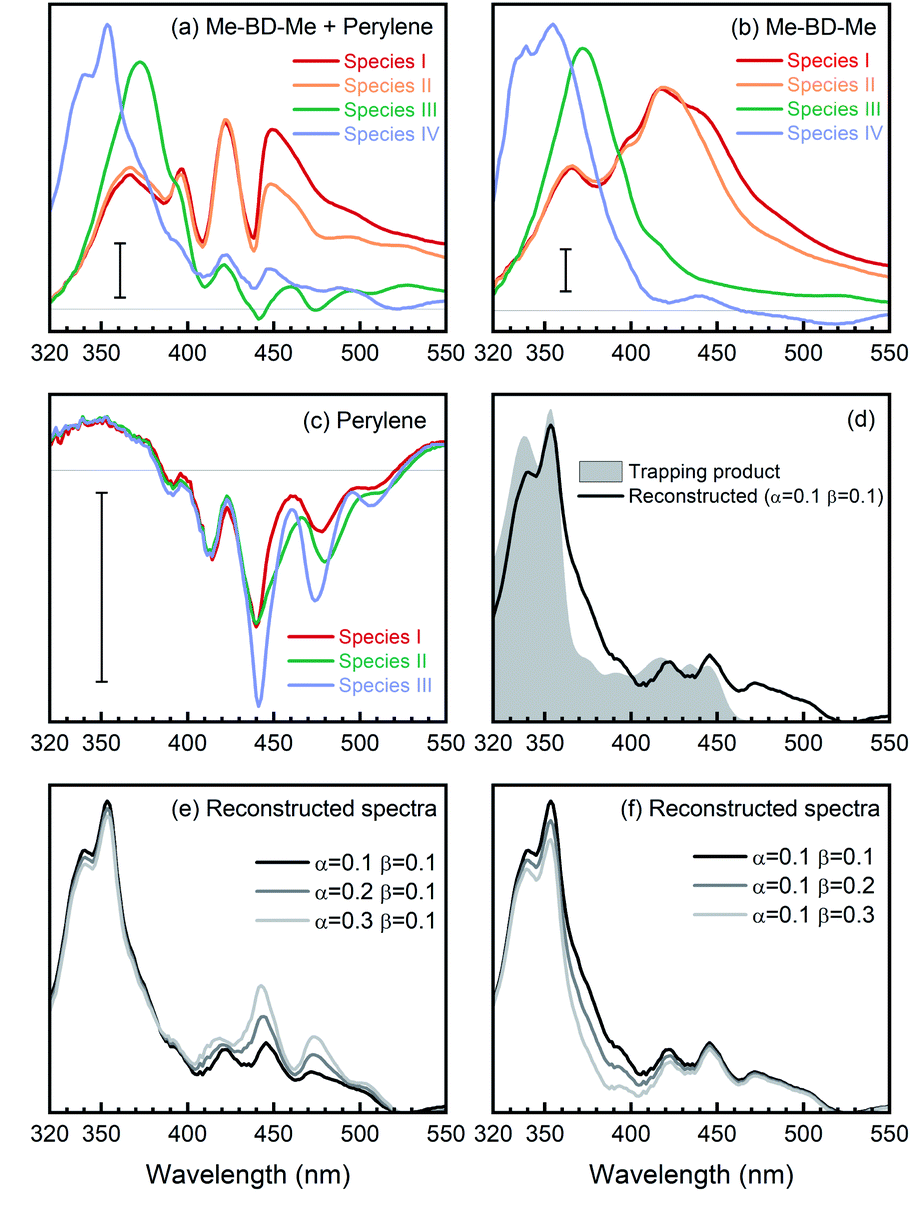 | ||
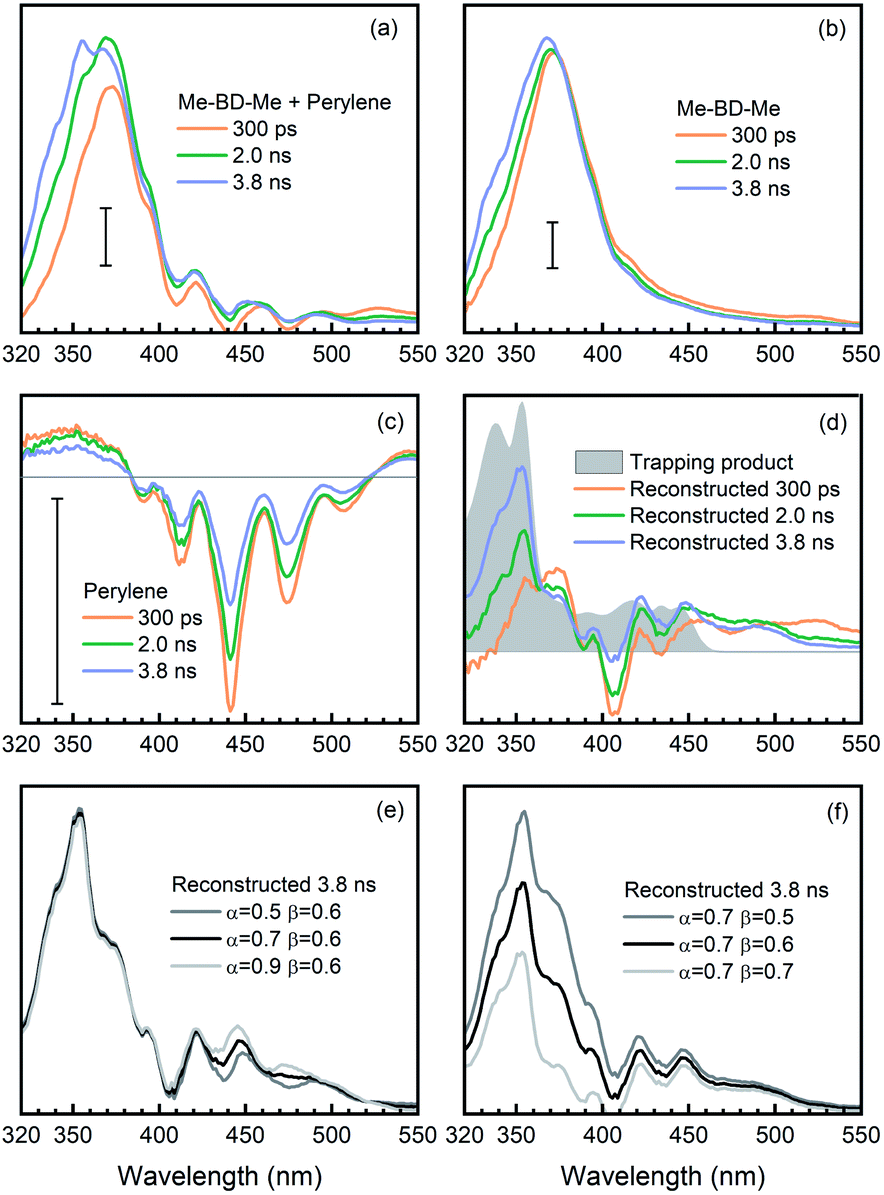
| Fig. 5 Reconstruction of o-benzyne trapping with perylene in CHCl3 solvent using SAS. (a) SAS from target analysis of transient absorption of Me-BD-Me/perylene mixture [SM(N,λ)], wherein N stands for the numbering of species from the target analysis. (b) SAS of pure Me-BD-Me [SB(N,λ)]. (c) SAS of pure perylene [SP(N,λ)]. (d) Reconstructed spectrum of trapping product [ST(λ), pink line] calculated by ST(λ) = SM(4,λ) − αSP(3,λ) − βSB(3,λ), and steady-state absorption spectrum of chemically isolated trapping product (gray filled). (e and f) Reconstructed spectra of trapping products using different weighting parameters α (e) and β (f). The scale bars correspond to an absorbance change of 5 mOD. | ||
The transient spectra of Me-BD-Me in CHCl3 display features similar to the ones in THF or 1,4-dioxane, while UV-excited perylene contributes multi-peak structures originating from ground-state bleach and stimulated emission in the 380–520 nm wavelength range (Fig. S12†) which persist until the maximum delay time (3.8 ns). Such a structural signature also appears in the transient absorption spectra of the trapping mixture (Fig. S13†), i.e., they are superimposed on the weak signals of the expected trapping products. Upon UV photolysis, both bis-diyne (Me-BD-Me) and perylene can be excited to their respective S1 states. Hence, as listed in Table 2, several components might contribute to the transient absorption of the trapping mixture after several nanoseconds of electronic deactivation and reaction: the excited state of perylene, hν-HDDA-generated o-benzyne, o-benzyne-perylene trapping product, and self-trapping products which are, however, expected to be of minor importance. In order to extract the desired trapping signal from the mixture of all possible contributions, we employed two spectral reconstruction strategies to remove the perylene and o-benzyne contributions as explained further below. As the most reliable reference, we isolated the o-benzyne-perylene trapping product (ESI Note 3†) and measured its absorption spectrum (see Fig. 5d). Although quantitative dynamics of bimolecular reactions in solution have been extensively reported for other species, the precondition for these studies is that only one of the reactants can be excited by the pump pulses.101–106 Thus, we limit our analysis to a qualitative discussion due to the existence of both excited bis-diynes and excited perylene.
| Species | Representative SAS | Transient spectra |
|---|---|---|
| Mixture | Mixture SAS4, SM(4,λ) | Mixture, AM(λ,t) |
| Perylene | Perylene SAS3, SP(3,λ) | Perylene, AP(λ,t) |
| o-Benzyne | Me-BD-Me SAS3, SB(3,λ) | Me-BD-Me, AB(λ,t) |
| Trapping product | Reconstructed, ST(λ) | Reconstructed, AT(λ,t) |
| Self-trapping products | Of minor importance | |
Target analysis with a sequential model (I → II → III → IV) successfully reproduced the transient absorption spectra of bis-diynes and their corresponding mixture with perylene in CHCl3, as can be inferred by the quality of the fit (Fig. S16†). For the first strategy, we removed the contribution of unreacted o-benzyne and perylene from the SAS of the trapping mixture and, thus, “manually” reconstructed the “spectral signature” of the trapping product. As summarized in Table 2, the SAS4 of the mixture [SM(4,λ)] arises from unreacted perylene [corresponds to SAS3 of perylene, SP(3,λ)], unreacted o-benzyne [SAS3 of Me-BD-Me, SB(3,λ)], trapping product [ST(λ)], and ignorable self-trapping products. With proper proportionality constants, the spectrum of the trapping product can thus be expressed as ST(λ) ≈ SM(4,λ) − αSP(3,λ) − βSB(3,λ). Considering the highly complicated nature of the system, accurate estimation of factors α and β is challenging. In the first step, the factor α = 0.1 was fitted as it minimizes the spectral modulation in the range of 380–520 nm. The factor β = 0.1 was subsequently used to reach a convincing spectral shape without an unreasonable negative signal. The resulting reconstructed spectrum of the trapping product from transient absorption (Fig. 5d, black) is consistent with the spectrum of the chemically isolated product (gray). Nevertheless, an additional band can be observed in the longer wavelength regime (460–510 nm), which cannot be attributed to the simple 1:1 trapping product, but might belong to a double (2:1) adduct structure with extended conjugation.47 Meanwhile, although values of the factors α and β are determined with some uncertainty, we found that alternations of α (Fig. 5e) or β (Fig. 5f) do not substantially change the shape of reconstructed spectra. Therefore, we assign the reconstructed spectral feature to the expected trapping product.
In addition, as a second method to obtain the trapping product signature, we removed the contribution of pure perylene from the transient spectra of the trapping mixture and, thus, “manually” reconstructed the time-resolved spectra of the trapping product during its formation. This was done by subtracting the transient absorption spectra of pure perylene [AP(λ,t), Fig. 6c] and bis-diyne [AB(λ,t), Fig. 6b], with suitable proportionality constants, from the spectra of the trapping mixture [AM(λ,t), Fig. 6a] at selected delay times, AT(λ,t) ≈ AM(λ,t) − αAP(λ,t) − βAB(λ,t), to obtain the spectral contribution of the trapping product [AT(λ,t)].
 | ||
| Fig. 6 Reconstruction of o-benzyne trapping with perylene in CHCl3 solvent by using transient absorption spectra at selected delay times (300 ps, 2.0 ns and 3.8 ns). (a) Time-resolved transient absorption spectra of Me-BD-Me/perylene mixture [AM(λ,t)]. (b) Time-resolved transient absorption spectra of Me-BD-Me [AB(λ,t)]. (c) Time-resolved transient absorption spectra of perylene [AP(λ,t)]. (d) Reconstructed transient absorption spectra of the trapping product [AT(λ,t), colored lines] calculated by AT(λ,t) = AM(λ,t) − αAP(λ,t) − βAB(λ,t), and the absorption spectrum of the chemically isolated trapping product (gray filled). (e and f) Reconstructed transient spectra of trapping products at 3.8 ns delay time by using different weighted parameters α (e) and β (f). The scale bars correspond to an absorbance change of 5 mOD. | ||
Again, the factor α was optimized to α = 0.6, so that the modulation of perylene in the reconstructed spectra can be minimized in the 380–520 nm regime. Meanwhile, as illustrated in Fig. 6d (gray filled) the trapping products have no absorption in the spectral region >550 nm, i.e., AT(λ,t) ≈ 0, and the factor for the o-benzyne contribution was optimized to β = 0.6 so as to make the reconstructed spectra [AM(λ,t) − αAP(λ,t) − βAB(λ,t)] as close as possible to the spectral baseline (ΔA = 0). The reconstructed spectra at selected delay times are shown in Fig. 6d (colored lines) in comparison with the isolated spectrum (gray). One can clearly see the rise of a positive band at 400–510 nm from 0.3 ns to 3.8 ns time delay, which is very similar in spectral range and shape to the reconstructed SAS (see Fig. 5d, black line) obtained from target analysis. Analogously to the discussion above, the precise values of the factors α (Fig. 6e) or β (Fig. 6f) do not affect significantly the spectral feature of reconstructed spectra at 3.8 ns delay time. This provides further evidence that indeed the trapping product of HDDA-generated o-benzyne with perylene was observed in our experiments.
Considering both trapping experiments together, we have successfully observed and confirmed the existence of trapping products of o-benzyne intermediates. By backtracking the reaction sequence, we believe that the most plausible assignment for the strong transient absorption band centered at 372 nm is the hν-HDDA reaction-produced o-benzyne intermediate, which arises from excited-state bis-diyne (Me-BD-Me, 400–450 nm) with a time constant of ∼50 ps, and decays on a nanosecond time scale via self-trapping by an additional bis-diyne or by perylene trapping.
Lastly, the thermal-HDDA reaction was reported to occur via a stepwise(-like) mechanism with a diradical intermediate,26 which is a reaction from the electronic ground state in contrast to the photoreaction we describe in the present manuscript (see a comparative illustration of reaction pathways in Fig. 7). The hν-HDDA reaction relies on the UV excitation to populate the S1 state directly, from which the wave packet might decay via a S1/S0 conical intersection leading to a highly efficient reaction.
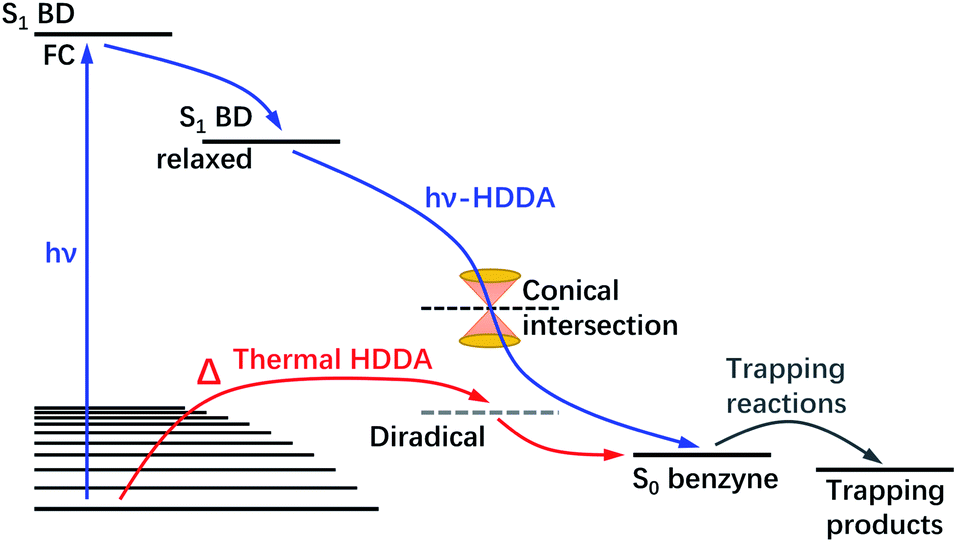 | ||
| Fig. 7 Schematic illustration of the mechanism of the photochemical (hν) and thermal HDDA reactions. The hν-HDDA mechanism (blue path) is revealed by the present work, while the thermal HDDA mechanism was reported by Wang et al.26 and included here for comparison. | ||
Conclusions
In this work, the UV-induced photochemistry of the bis-diyne compound Me-BD-Me, where Me is –CH3 and BD is –4-C6H4–(CC)2–(CH2)3–(CC)2–4-C6H4–, was measured by femtosecond transient absorption spectroscopy in the UV/VIS region and in a 100 fs to 3.8 ns time range. We deduced multisequential kinetics for which, in the first step, the UV-populated first excited singlet state of Me-BD-Me isomerizes from an “open” to a “closed” conformation [τ1(I → II) = 2–5 ps]. The following processes are the formation [τ2(II → III) = 50–70 ps] of an intermediate and its slow decay [τ3(III → IV) > 3 ns]. By employing intermolecular trapping strategies via trapping reactions with a second Me-BD-Me molecule, as well as reaction with perylene, we assigned the process II → III, observed experimentally, to be the formation of the o-benzyne intermediate in the hν-HDDA reaction. Use of the trapping strategies allowed us to circumvent the general insensitivity of UV/VIS transient absorption spectroscopy to chemical structure. The HDDA-generated o-benzyne species from Me-BD-Me displayed an absorption at ∼372 nm. To the best of our knowledge, this is the first report which spectroscopically confirms the existence of an o-benzyne species in solution as a product of the hν-HDDA reaction. The HDDA-generated o-benzyne has seen rapidly increased general interest and application in organic syntheses during the last eight years.15 A wide variety of substrates have been investigated for reaction with HDDA-generated o-benzynes. They range from small molecules such as furan107 to highly complex natural products.20
Experimental section
Sample preparation
Me-BD-Me was synthesized according to the literature.94 The general information on chemical synthesis and isolation is summarized in ESI Note 1.† In all transient absorption spectroscopic experiments, samples were dissolved in spectroscopic-grade solvents (Sigma-Aldrich) as received. The static absorption spectra for sample preparation were measured with a JASCO V-670 UV/VIS spectrometer.Spectroscopic measurements
The details of the ultrafast transient absorption setup have been described elsewhere.108,109 Briefly, a commercial 1 kHz Ti:sapphire laser system (Solstice, Spectra-Physics) delivered 120 fs pulses centered at 800 nm. The ∼100 fs excitation pulses (λpump = 295 nm) were derived from the second-harmonic output of a commercial nonlinear optical parametric amplifier (TOPAS White, Light Conversion). Approximately 0.5–1.0 μJ pulse energy was measured at the sample position. The broadband UV/VIS probe pulses were generated by focusing a small portion of the Ti:sapphire laser fundamental into a linearly moving CaF2 window, resulting in a white-light spectrum between 320 nm and 670 nm. Pump and probe beams were spatially overlapped in a flow cuvette with a sample thickness of 200 μm which, at the chosen sample concentration, led to an optical density of 0.5 at 295 nm. The polarizations of the pump and probe beams were set to the magic angle of 54.7°. A cross correlation of ∼120 fs between pump and probe pulses was achieved. The pump–probe delay time was varied up to 3.8 ns by delaying the probe beam with a mechanical translation stage (M-IMS600, Newport). Every second pump pulse was blocked by a chopper driven at 500 Hz. After passing the sample, the probe pulses were dispersed in a spectrometer (Acton SP2500i, Princeton Instruments) and detected shot-to-shot by a CCD camera (Pixis 2K, Princeton Instruments). During the transient absorption measurements, the sample solution was circulated through a 0.1 mm thick flow-cell (48/UTWA2/Q/0.1, Starna GmbH) by using a micro-gear pump (mzr-4605, HNP Mikrosysteme). We conducted a control experiment to verify the reliability of our transient absorption measurements when dealing with irreversible photochemical reactions (Fig. S17†). We measured the transient absorption spectra at 350 ps (orange line) and then repeated the measurement (blue line) after constant illumination, for 1 h, with the 295 nm pump beam and, as can be seen in Fig. S17,† we observed no significant alteration of the transient absorption spectra, i.e., any irreversible photodegradation of the sample does not create any uncertainty in the transient spectra. The transient data were evaluated via target analysis110 with the software package Glotaran based on the R-package TIMP.111Theoretical calculations
All TD-DFT calculations were executed with the Gaussian 09 program package,112 using the CAM-B3LYP functional combined with the def2-TZVP basis set. Oscillator strengths among excited states were calculated using the wavefunction analyzer program Multiwfn.113 Dispersion effects were incorporated by the use of Grimme's dispersion correction with Becke–Johnson damping114 and solvation was treated with the polarizable continuum model.115Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. B. M., R. M. and T. B. gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (Research Training School GRK 2112). Financial support by the European Research Council (ERC) Consolidator Grant “DYNAMO” (No. 646737) is gratefully acknowledged by M. W., and T. B. M. thanks the Julius-Maximilians-Universität Würzburg for support.References
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- A. Z. Bradley and R. P. Johnson, J. Am. Chem. Soc., 1997, 119, 9917–9918 CrossRef CAS.
- A. Ajaz, A. Z. Bradley, R. C. Burrell, W. H. H. Li, K. J. Daoust, L. B. Bovee, K. J. DiRico and R. P. Johnson, J. Org. Chem., 2011, 76, 9320–9328 CrossRef CAS PubMed.
- K. J. Cahill, A. Ajaz and R. P. Johnson, Aust. J. Chem., 2010, 63, 1007–1012 CrossRef CAS.
- R. P. Johnson, J. Phys. Org. Chem., 2010, 23, 283–292 CAS.
- S. L. Skraba-Joiner, R. P. Johnson and J. Agarwal, J. Org. Chem., 2015, 80, 11779–11787 CrossRef CAS PubMed.
- K. Miyawaki, R. Suzuki, T. Kawano and I. Ueda, Tetrahedron Lett., 1997, 38, 3943–3946 CrossRef CAS.
- I. Ueda, Y. Sakurai, T. Kawano, Y. Wada and M. Futai, Tetrahedron Lett., 1999, 40, 319–322 CrossRef CAS.
- K. Miyawaki, T. Kawano and I. Ueda, Tetrahedron Lett., 1998, 39, 6923–6926 CrossRef CAS.
- K. Miyawaki, T. Kawano and I. Ueda, Tetrahedron Lett., 2000, 41, 1447–1451 CrossRef CAS.
- T. Kawano, H. Inai, K. Miyawaki and I. Ueda, Tetrahedron Lett., 2005, 46, 1233–1236 CrossRef CAS.
- T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208–212 CrossRef CAS PubMed.
- R. W. Hoffmann and K. Suzuki, Angew. Chem., Int. Ed., 2013, 52, 2655–2656 CrossRef CAS PubMed.
- C. Holden and M. F. Greaney, Angew. Chem., Int. Ed., 2014, 53, 5746–5749 CrossRef CAS PubMed.
- O. J. Diamond and T. B. Marder, Org. Chem. Front., 2017, 4, 891–910 RSC.
- W. Li, L. Zhou and J. Zhang, Chem.–Eur. J., 2016, 22, 1558–1571 CrossRef CAS PubMed.
- D. Niu, P. H. Willoughby, B. P. Woods, B. Baire and T. R. Hoye, Nature, 2013, 501, 531–534 CrossRef CAS PubMed.
- T. R. Hoye, B. Baire and T. Wang, Chem. Sci., 2014, 5, 545–550 RSC.
- Y. Liang, X. Hong, P. Yu and K. N. Houk, Org. Lett., 2014, 16, 5702–5705 CrossRef CAS PubMed.
- S. P. Ross and T. R. Hoye, Nat. Chem., 2017, 9, 523–530 CrossRef CAS PubMed.
- X. Xiao, B. P. Woods, W. Xiu and T. R. Hoye, Angew. Chem., Int. Ed., 2018, 57, 9901–9905 CrossRef CAS PubMed.
- Y. Wang and T. R. Hoye, Org. Lett., 2018, 20, 88–91 CrossRef CAS PubMed.
- X. Xiao and T. R. Hoye, Nat. Chem., 2018, 10, 838–844 CrossRef CAS PubMed.
- J. Zhang, A. C. S. Page, V. Palani, J. Chen and T. R. Hoye, Org. Lett., 2018, 20, 5550–5553 CrossRef CAS PubMed.
- P. H. Willoughby, D. Niu, T. Wang, M. K. Haj, C. J. Cramer and T. R. Hoye, J. Am. Chem. Soc., 2014, 136, 13657–13665 CrossRef CAS PubMed.
- T. Wang, D. Niu and T. R. Hoye, J. Am. Chem. Soc., 2016, 138, 7832–7835 CrossRef CAS PubMed.
- T. Wang, R. R. Naredla, S. K. Thompson and T. R. Hoye, Nature, 2016, 532, 484–488 CrossRef CAS PubMed.
- R. Karmakar and D. Lee, Org. Lett., 2016, 18, 6105–6107 CrossRef CAS PubMed.
- T. Wang, C. Oswood and T. Hoye, Synlett, 2017, 28, 2933–2935 CrossRef CAS.
- Q. Hu, L. Li, F. Yin, H. Zhang, Y. Hu, B. Liu and Y. Hu, RSC Adv., 2017, 7, 49810–49816 RSC.
- X. Meng, S. Lv, D. Cheng, Q. Hu, J. Ma, B. Liu and Y. Hu, Chem.–Eur. J., 2017, 23, 6264–6271 CrossRef CAS PubMed.
- Y. Hu, J. Ma, L. Li, Q. Hu, S. Lv, B. Liu and S. Wang, Chem. Commun., 2017, 53, 1542–1545 RSC.
- S. Yoshida, K. Shimizu, K. Uchida, Y. Hazama, K. Igawa, K. Tomooka and T. Hosoya, Chem.–Eur. J., 2017, 23, 15332–15335 CrossRef CAS PubMed.
- S. P. Ross and T. R. Hoye, Org. Lett., 2018, 20, 100–103 CrossRef CAS PubMed.
- M. P. Smela and T. R. Hoye, Org. Lett., 2018, 20, 5502–5505 CrossRef CAS PubMed.
- S. P. Ross, B. Baire and T. R. Hoye, Org. Lett., 2017, 19, 5705–5708 CrossRef CAS PubMed.
- S. Ghorai and D. Lee, Tetrahedron, 2017, 73, 4062–4069 CrossRef CAS.
- H. Shen, X. Xiao, M. K. Haj, P. H. Willoughby and T. R. Hoye, J. Am. Chem. Soc., 2018, 140, 15616–15620 CrossRef CAS PubMed.
- X. Xiao, T. Wang, F. Xu and T. R. Hoye, Angew. Chem., Int. Ed., 2018, 130, 16802–16806 CrossRef.
- Y. Wang, L. Zheng and T. R. Hoye, Org. Lett., 2018, 20, 7145–7148 CrossRef CAS PubMed.
- S. K. Thompson and T. R. Hoye, J. Am. Chem. Soc., 2019, 141, 19575–19580 CrossRef CAS PubMed.
- S. Ghorai and D. Lee, Org. Lett., 2019, 21, 7390–7393 CrossRef CAS PubMed.
- M. Chen, C. Q. He and K. N. Houk, J. Org. Chem., 2019, 84, 1959–1963 CrossRef CAS PubMed.
- J. A. Tsui and B. T. Sterenberg, Organometallics, 2009, 28, 4906–4908 CrossRef CAS.
- E. H. Fort and L. T. Scott, Tetrahedron Lett., 2011, 52, 2051–2053 CrossRef CAS.
- B. Schuler, S. Collazos, L. Gross, G. Meyer, D. Pérez, E. Guitián and D. Peña, Angew. Chem., Int. Ed., 2014, 53, 9004–9006 CrossRef CAS PubMed.
- F. Xu, X. Xiao and T. R. Hoye, Org. Lett., 2016, 18, 5636–5639 CrossRef CAS PubMed.
- H. H. Wenk, M. Winkler and W. Sander, Angew. Chem., Int. Ed., 2003, 42, 502–528 CrossRef CAS PubMed.
- C. Wentrup, Aust. J. Chem., 2010, 63, 979–986 CrossRef CAS.
- T. Kitamura, Aust. J. Chem., 2010, 63, 987–1001 CrossRef CAS.
- H. Pellissier and M. Santelli, Tetrahedron, 2003, 59, 701–730 CrossRef CAS.
- C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766–3778 CrossRef CAS PubMed.
- P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef CAS PubMed.
- W. E. Bachmann and H. T. Clarke, J. Am. Chem. Soc., 1927, 49, 2089–2098 CrossRef CAS.
- J. G. Radziszewski, J. Waluk, P. Kaszynski and J. Spanget-Larsen, J. Phys. Chem. A, 2002, 106, 6730–6737 CrossRef CAS.
- J. G. Radziszewski, B. A. Hess and R. Zahradnik, J. Am. Chem. Soc., 1992, 114, 52–57 CrossRef CAS.
- H.-H. Nam and G. E. Leroi, J. Mol. Struct., 1987, 157, 301–304 CrossRef CAS.
- M. J. S. Dewar, G. P. Ford and H. S. Rzepa, J. Mol. Struct., 1979, 51, 275–279 CrossRef CAS.
- O. L. Chapman, C. C. Chang, J. Kolc, N. R. Rosenquist and H. Tomioka, J. Am. Chem. Soc., 1975, 97, 6586–6588 CrossRef CAS.
- O. L. Chapman, K. Mattes, C. L. McIntosh, J. Pacansky, G. V. Calder and G. Orr, J. Am. Chem. Soc., 1973, 95, 6134–6135 CrossRef CAS.
- X. Zhang, A. T. Maccarone, M. R. Nimlos, S. Kato, V. M. Bierbaum, G. B. Ellison, B. Ruscic, A. C. Simmonett, W. D. Allen and H. F. Schaefer, J. Chem. Phys., 2007, 126, 044312 CrossRef PubMed.
- R. D. Brown, P. D. Godfrey and M. Rodler, J. Am. Chem. Soc., 1986, 108, 1296–1297 CrossRef CAS.
- P. D. Godfrey, Aust. J. Chem., 2010, 63, 1061 CrossRef CAS.
- S. G. Kukolich, C. Tanjaroon, M. C. McCarthy and P. Thaddeus, J. Chem. Phys., 2003, 119, 4353–4359 CrossRef CAS.
- S. G. Kukolich, M. C. McCarthy and P. Thaddeus, J. Phys. Chem. A, 2004, 108, 2645–2651 CrossRef CAS.
- E. G. Robertson, P. D. Godfrey and D. McNaughton, J. Mol. Spectrosc., 2003, 217, 123–126 CrossRef CAS.
- A. M. Orendt, J. C. Facelli, J. G. Radziszewski, W. J. Horton, D. M. Grant and J. Michl, J. Am. Chem. Soc., 1996, 118, 846–852 CrossRef CAS.
- R. Warmuth, Angew. Chem., Int. Ed. Engl., 1997, 36, 1347–1350 CrossRef CAS.
- M. J. S. Dewar and T.-P. Tien, J. Chem. Soc., Chem. Commun., 1985, 1243 RSC.
- J. G. Günther Simon, H. Specht and A. Schweig, Chem. Phys. Lett., 1992, 200, 459–464 CrossRef.
- X. Zhang and P. Chen, J. Am. Chem. Soc., 1992, 114, 3147–3148 CrossRef CAS.
- N. H. Werstiuk, C. D. Roy and J. Ma, Can. J. Chem., 1995, 73, 146–149 CrossRef CAS.
- D. Kaiser, E. Reusch, P. Hemberger, A. Bodi, E. Welz, B. Engels and I. Fischer, Phys. Chem. Chem. Phys., 2018, 20, 3988–3996 RSC.
- F. Hirsch, E. Reusch, P. Constantinidis, I. Fischer, S. Bakels, A. M. Rijs and P. Hemberger, J. Phys. Chem. A, 2018, 122, 9563–9571 CrossRef CAS PubMed.
- A. C. Castro, A. Romero-Rivera, S. Osuna, K. N. Houk and M. Swart, Chem.–Eur. J., 2020, 26, 2626–2634 CrossRef CAS PubMed.
- A. Schweig, N. Münzel, H. Meyer and A. Heidenreich, Struct. Chem., 1990, 1, 89–100 CrossRef CAS.
- R. S. Berry, G. N. Spokes and M. Stiles, J. Am. Chem. Soc., 1962, 84, 3570–3577 CrossRef CAS.
- J. Fulara, A. Nagy, K. Filipkowski, V. S. Thimmakondu, J. F. Stanton and J. P. Maier, J. Phys. Chem. A, 2013, 117, 13605–13615 CrossRef CAS PubMed.
- J. G. G. Simon, A. Schweig, Y. Xie and H. F. Schaefer, Chem. Phys. Lett., 1992, 200, 631–634 CrossRef CAS.
- J. Kolc, Tetrahedron Lett., 1972, 13, 5321–5324 CrossRef.
- N. Münzel and A. Schweig, Chem. Phys. Lett., 1988, 147, 192–194 CrossRef.
- J. G. G. Simon, N. Münzel and A. Schweig, Chem. Phys. Lett., 1990, 170, 187–192 CrossRef CAS.
- F. Xu, X. Xiao and T. R. Hoye, J. Am. Chem. Soc., 2017, 139, 8400–8403 CrossRef CAS PubMed.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. E. Casida, in Recent Advances in Density Functional Methods, World Scientific Publishing Europe Ltd., London, 1995, vol. 1, pp. 155–192 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. Nenov, W. J. Schreier, F. O. Koller, M. Braun, R. de Vivie-Riedle, W. Zinth and I. Pugliesi, J. Phys. Chem. A, 2012, 116, 10518–10528 CrossRef CAS PubMed.
- H. Sotome, T. Nagasaka, K. Une, S. Morikawa, T. Katayama, S. Kobatake, M. Irie and H. Miyasaka, J. Am. Chem. Soc., 2017, 139, 17159–17167 CrossRef CAS PubMed.
- C. L. Ward and C. G. Elles, J. Phys. Chem. Lett., 2012, 3, 2995–3000 CrossRef CAS PubMed.
- J. Maier, M. Deutsch, J. Merz, Q. Ye, O. Diamond, M.-T. Schilling, A. Friedrich, B. Engels and T. B. Marder, Chem.–Eur. J., 2020 DOI:10.1002/chem.202002511 , accepted article.
- J. Buback, M. Kullmann, F. Langhojer, P. Nuernberger, R. Schmidt, F. Würthner and T. Brixner, J. Am. Chem. Soc., 2010, 132, 16510–16519 CrossRef CAS PubMed.
- J. Dasgupta, R. R. Frontiera, K. C. Taylor, J. C. Lagarias and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1784–1789 CrossRef CAS PubMed.
- A. J. Orr-Ewing, Annu. Rev. Phys. Chem., 2015, 66, 119–141 CrossRef CAS PubMed.
- A. J. Orr-Ewing, Chem. Soc. Rev., 2017, 46, 7597–7614 RSC.
- T. R. Waite, J. Chem. Phys., 1958, 28, 103–106 CrossRef CAS.
- T. R. Waite, J. Chem. Phys., 1960, 32, 21–23 CrossRef CAS.
- S. J. Greaves, R. A. Rose, T. A. A. Oliver, D. R. Glowacki, M. N. R. Ashfold, J. N. Harvey, I. P. Clark, G. M. Greetham, A. W. Parker, M. Towrie and A. J. Orr-Ewing, Science, 2011, 331, 1423–1426 CrossRef CAS PubMed.
- D. R. Glowacki, R. A. Rose, S. J. Greaves, A. J. Orr-Ewing and J. N. Harvey, Nat. Chem., 2011, 3, 850–855 CrossRef CAS PubMed.
- D. Koyama, P. Coulter, M. P. Grubb, G. M. Greetham, I. P. Clark and A. J. Orr-Ewing, J. Phys. Chem. A, 2015, 119, 12924–12934 CrossRef CAS PubMed.
- D. Koyama, P. M. Donaldson and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2017, 19, 12981–12991 RSC.
- D. Koyama and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2016, 18, 12115–12127 RSC.
- P. Coulter, M. P. Grubb, D. Koyama, I. V. Sazanovich, G. M. Greetham and A. J. Orr-Ewing, J. Phys. Chem. A, 2015, 119, 12911–12923 CrossRef CAS PubMed.
- S. Nobusue, H. Yamane, H. Miyoshi and Y. Tobe, Org. Lett., 2014, 16, 1940–1943 CrossRef CAS PubMed.
- S. Schott, L. Ress, J. Hrušák, P. Nuernberger and T. Brixner, Phys. Chem. Chem. Phys., 2016, 18, 33287–33302 RSC.
- X. Ma, M. Wenzel, H.-C. Schmitt, M. Flock, E. Reusch, R. Mitrić, I. Fischer and T. Brixner, Phys. Chem. Chem. Phys., 2018, 20, 15434–15444 RSC.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2017, 18, 1–46 Search PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. Hratchian, A. Izmaylov, J. Bloino, G. Zheng, J. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. Peralta, F. Ogliaro, M. Bearpark, J. Heyd, E. Brothers, K. Kudin, V. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. Burant, S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. Millam, M. Klene, J. Knox, J. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. Stratmann, O. Yazyev, A. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. Martin, K. Morokuma, V. Zakrzewski, G. Voth, P. Salvador, J. Dannenberg, S. Dapprich, A. Daniels, O. Farkas, J. Foresman, J. Ortiz, J. Cioslowski and D. Fox, Gaussian 09 Revis. D01, Gaussian Inc., Wallingford CT Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03184d |
| This journal is © The Royal Society of Chemistry 2020 |