
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-mediated peptide arylation selective for the N-terminus†
Mary K.
Miller‡
 ,
Haopei
Wang‡
,
Kengo
Hanaya
,
Olivia
Zhang
,
Alex
Berlaga
and
Zachary T.
Ball
*
,
Haopei
Wang‡
,
Kengo
Hanaya
,
Olivia
Zhang
,
Alex
Berlaga
and
Zachary T.
Ball
*
Department of Chemistry, Rice University, Houston, TX 77005, USA. E-mail: zb1@rice.edu
First published on 14th September 2020
Abstract
Polypeptides present remarkable selectivity challenges for chemical methods. Amino groups are ubiquitous in polypeptide structure, yet few paradigms exist for reactivity and selectivity in arylation of amine groups. This communication describes the utilization of boronic acid reagents bearing certain o-electron withdrawing groups for copper-mediated amine arylation of the N-terminus under mild conditions and primarily aqueous solvent. The method adds to the toolkit of boronic acid reagents for polypeptide modification under mild conditions in water that shows complete selectivity for the N-terminus in the presence of lysine side chains.
Introduction
Methods for selective modification of polypeptides and proteins allow access to drug conjugates, imaging probes, and hybrid materials.1,2 At the same time, polypeptides represent a stringent test of chemists' ability to control reactivity and chemoselectivity in a diverse, polyfunctional environment. The amino (–NH2) group is a common bioconjugation site, found primarily at the N-terminus and at lysine side chains.Cross-coupling reactivity, typically mediated by transition metal complexes, have become important tools for N–C bond formation, and they represent an interesting approach to selective modification of peptides and proteins.3–7 Chemoselective cysteine coupling has been achieved with a variety of transition metal-mediated approaches,8–11 and tyrosine is selectively modified with π-allyl intermediates.12 In contrast, cross-coupling at amine groups in polypeptides are limited to a few special cases, including arylpalladium reagents that mediate arylation of lysine side chains in non-aqueous solvents13 and arylation of individual amino acid derivatives with Cu14–16 or Pd,17,18 typically at elevated temperatures under non-aqueous conditions. Perhaps surprisingly, cross-coupling at amino groups (N-terminus and/or lysine) under physiologically relevant aqueous conditions remains an unsolved problem, and selectivity questions for the common case of multiple amino groups are largely unaddressed. While copper is well known to bind N-terminal motifs,19–21 these coordination complexes have not been utilized as intermediates in productive bond-forming catalysis, and indeed binding to the amino terminus is typically employed as a protecting group to block reactivity. Herein, we describe arylation with remarkable selectivity for the amino terminus, without competing reactivity at lysine. Simple copper(II) salts mediate the reaction with arylboronic acid reagents under mild conditions in aqueous buffer.
During investigations into boronic acid couplings with backbone amide N–H bonds,3,4,22 we observed a switch in chemoselectivity with certain ortho-substituted arylboronic acids. With sulfonamide-substituted reagent 1, we observed no reactivity at amide N–H bonds, but instead observed highly chemoselective arylation of the N-terminal amino group (Fig. 1a and b). We decided to investigate this unique selectivity, mindful that N-terminal cross-coupling could serve as a useful complement to alternative N-terminal modification methods—such as aldehyde condensation,23–29 pH-controlled acylation,30–33 N-terminal oxidation,34 and diazo transfer33—and to other emerging concepts for amine-selective bioconjugation.35
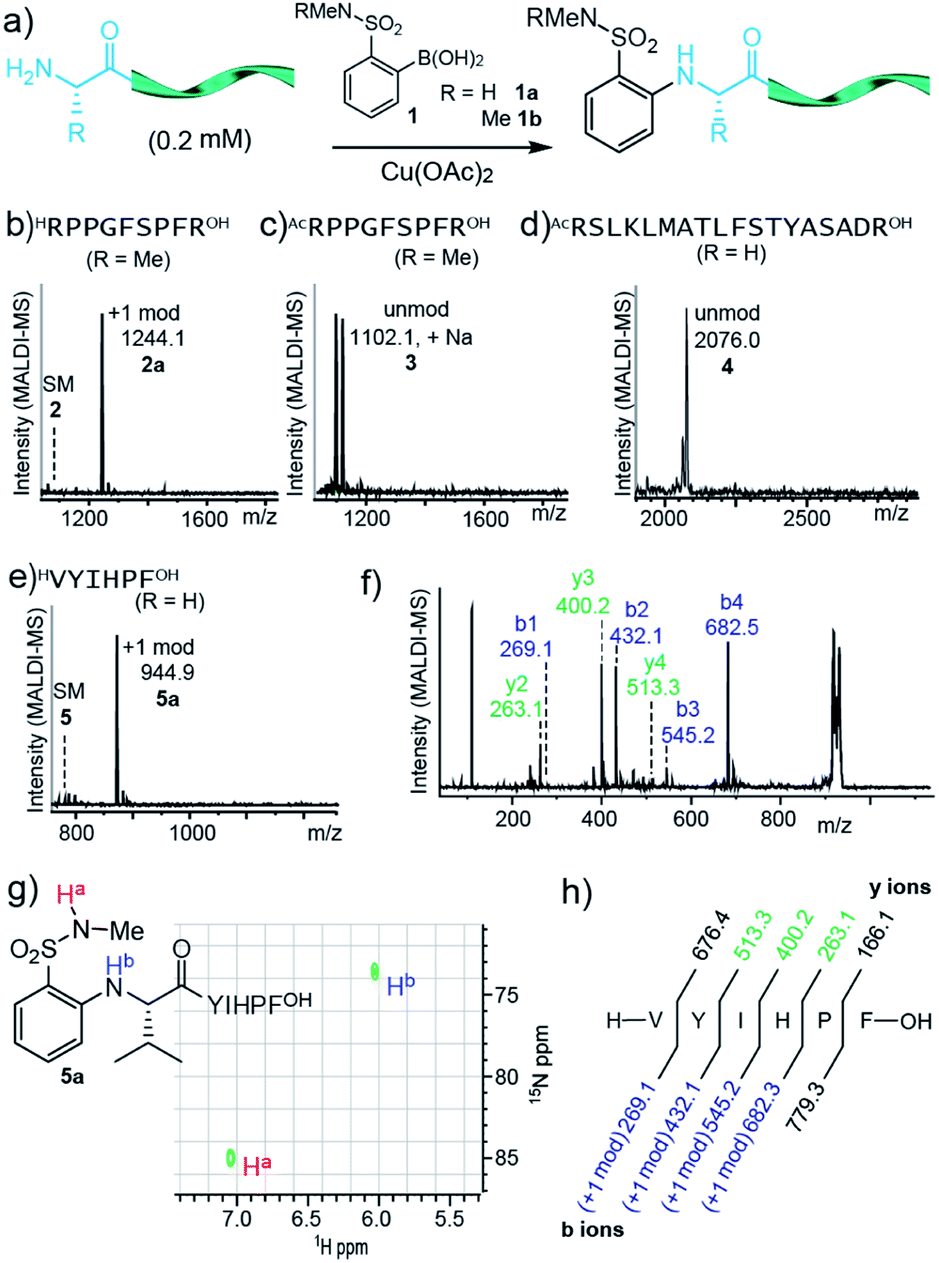 | ||
| Fig. 1 (a) Modification of peptides 2–5 with boronic acids 1a and 1b. Conditions: peptide (0.2 mM), boronic acid 1a/b (2 mM) and Cu(OAc)2 (0.1 mM) in NMM buffer (10 mM, pH 9.0) with 30% TFE at 37 °C for 18 h. For 5, HEPES buffer (10 mM, pH = 7.0) with 20% acetonitrile was employed. (b–e) MALDI-MS spectrum (crude) for reaction of peptide 2–5. (f) MS/MS spectrum of product 5a. (g) 1H–15N HSQC NMR spectrum of product 5a. (h) Sequence and fragmentation ladder of product 5a. Observed b and y ions are indicated. | ||
Results and discussion
Remarkably, primary amine groups of lysine side chain were completely unreactive (Fig. 1d), and an N-terminally acetylated analog of a reactive sequence resulted in no modification, consistent with reaction at an amino group. MALDI-MS/MS (Fig. S27 and S28†) verified N-terminal reactivity in these cases. The structure of modified peptide 5a was confirmed by MALDI-MS/MS (Fig. 1f and h) and 15N HSQC experiments (Fig. 1g), and MALDI-MS/MS also established N-terminal selectivity for peptides with multiple amino groups.Encouraged by these observations, we set out to understand the scope of the reaction conditions using a model peptide, 5 (Table 1). Yields were determined by HPLC analysis with an internal standard (see ESI and Fig. S3† for details). For reactions with peptide 5, pH = 7.0 was best; increasing or decreasing pH resulted in decreased yields (entry 1, 3–4). Pleasantly, the reaction was scalable, and the arylated peptide (entry 2) was isolated in 68% yield, similar to that observed by HPLC on small scale under identical conditions.
|
|
||||
|---|---|---|---|---|
| Entry | pH | Buffer | Cosolvent | Yielda (%) |
| a Yield calculated by RP-HPLC. b 5 mg scale reaction. c Isolated yield. | ||||
| 1 | 6.0 | NMM | 30% TFE | 15 |
| 2 | 7.0 | NMM | 30% TFE | 66b (68)c |
| 3 | 8.0 | NMM | 30% TFE | 33 |
| 4 | 9.0 | NMM | 30% TFE | 18 |
| 5 | 7.0 | NMM | None | 53 |
| 6 | 7.0 | NMM | 30% DMSO | 70 |
| 7 | 7.0 | NMM | 30% MeCN | 87 |
| 8 | 7.0 | NMM | 40% MeCN | 44 |
| 9 | 7.0 | Tris | 20% MeCN | 11 |
| 10 | 7.0 | HEPES | 20% MeCN | 97 |
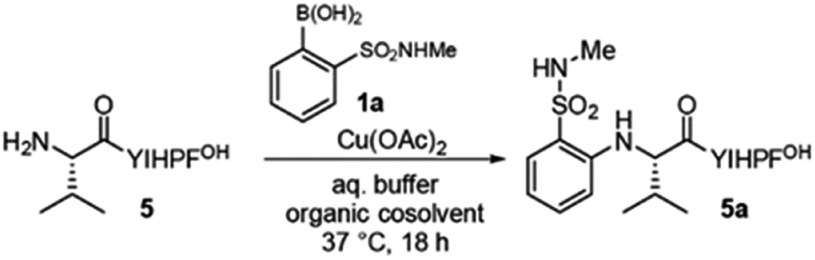
While the reaction can be performed under strictly aqueous conditions (entry 5), the addition of an organic cosolvent improved yields (entry 7). The nature of the buffer impacts reaction efficiency. The yield increased to near-quantitative levels (97%) for reactions in HEPES buffer (entry 10), while Tris, which contains a primary amine, was a poor buffer choice (entry 9).
Examining the scope of boronic acids revealed a structure–reactivity relationship (Fig. 2). Arylation products with peptide 2 were observed only with select electron-withdrawing ortho substituents: sulfonamide group (1b–c), sulfone group (1d) and halogen groups (1e–i). This trend is quite different from that observed with metal-catalyzed reactions of boronic acids with backbone amide3 or cysteine8 side chains. Additional substitution at distal positions (1c, 1g, 1i) is tolerated. The successful coupling with 1i, for example, introduces an arylbromide handle for later elaboration. The reaction has a strict requirement for ortho substitution (1l). Surprisingly, no product was observed with an ortho-nitro group, despite the excellent reactivity of this compound in both cysteine8 and amide N–H5 arylation. These observations implicate a substantially different reactivity type in the present N-terminal reactivity.36,37
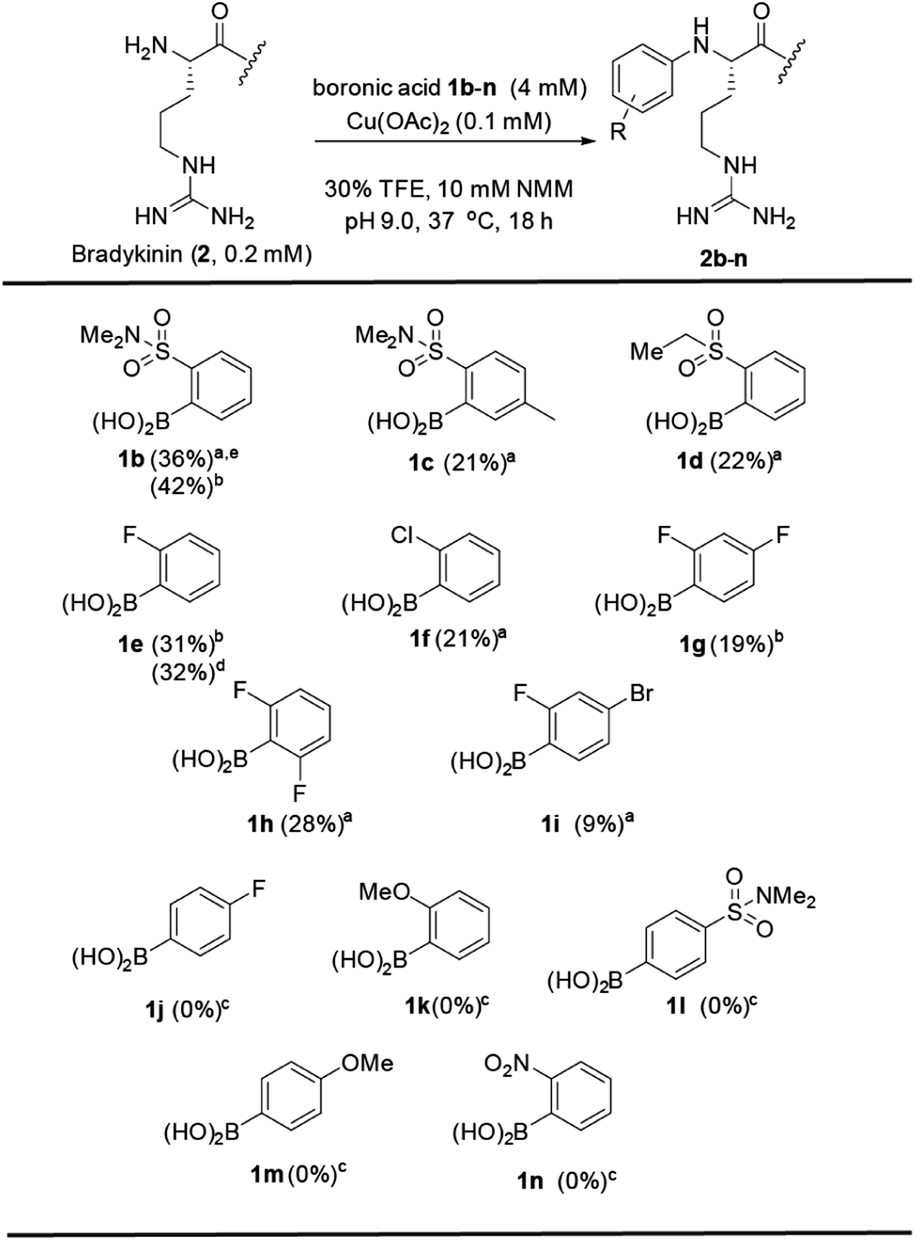 | ||
| Fig. 2 Scope of the boronic acid reagents. Conditions: Bradykinin (2) (0.2 mM), boronic acid 1b–n (4 mM), and Cu(OAc)2 (0.1 mM) in NMM buffer (10 mM, pH = 9.0) at 37 °C for 18 h. aRP-HPLC yield determined using internal standards. bIsolated yield on a 10 mg scale. cYield determined by ESI-MS. dYield determined using peak area of a known concentration of isolated product. eBoronic acid was added as seven aliquots over 25 h. | ||
To determine the tolerance of the reaction for different N-terminal residues, we synthesized variants at the N-terminal residue (5–13, Table 2). The reaction tolerates a wide variety of N-terminal residues (entries 1–5, 7–11), including bulky residues (tryptophan, valine, and leucine), charged residues (arginine, aspartate) and glycine. Proline, which contains a secondary amine (entry 6), was not tolerated. In all cases, only a single product is observed. HPLC analysis shows no evidence of coupling at sites other than the N-terminus, or of any side products, even for peptides with potentially reactive N-terminal side chains (i.e.4, 8) (see Fig. S6–S13†). The modest yields observed in a few cases (i.e.8, 12) are the result of incomplete conversion or, more commonly, partial starting material decomposition into unknown species.
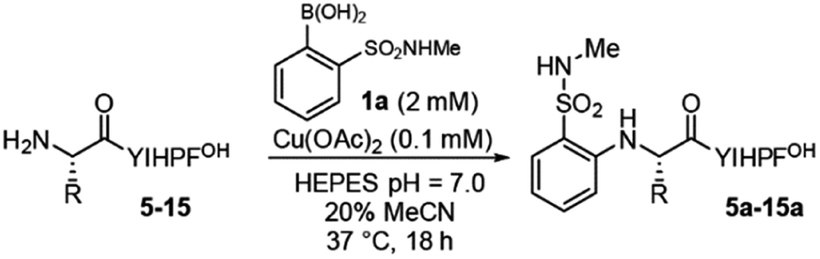
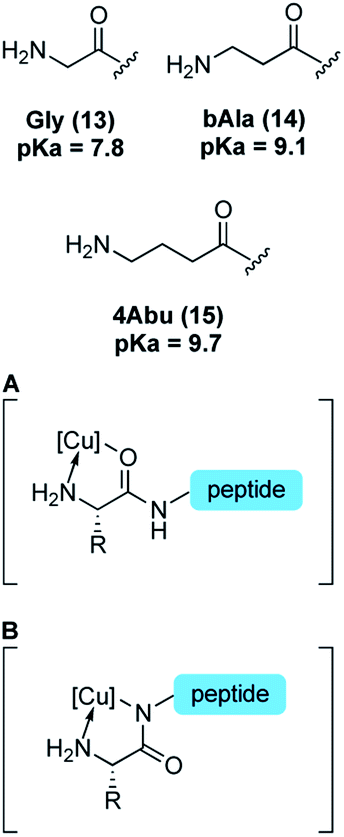
Reactivity at N-terminal amino groups in the presence of lysine side-chain amines is an interesting chemoselectivity. We hypothesized that the origin of chemoselectivity could be either pKa differences38 or the intermediacy of a chelation complex (Table 2, at right) of the N-terminal amine, not possible at lysine side chains. To probe this, we examined peptides with unnatural amino acids that would require larger ring-chelate structures: β-alanine (bAla) (entry 10) and γ-aminobutyric acid (4Abu) (entry 11). Relative to glycine, these peptide variants have a higher pKa (Table 2).39 We found that the bAla peptide, capable of forming a 6-membered ring chelate, retained reactivity, while the 4Abu variant—which would require at least a 7-membered ring intermediate—was unreactive. The chelation-driven selectivity model seems most in accord with these results, although additional study is warranted. In this context, it is worth noting that the reaction does not exhibit the hallmark increasing reactivity with increasing pH that is typically observed in amine functionalization governed by pKa.
Reactivity studies also shed light on the nature of a putative κ2 copper binding. While copper binding to N-terminal sequences is well studied,19–21 canonical structures typically adopt N-bound amidate structures (Table 2, B). In N-terminal arylation reported here, peptides with proline as the second amino acid, which have no amide N–H and thus cannot adopt ATCUN-like amidate structures, nonetheless are competent reaction partners (Table 3, entries 1–2). This observation would seem to indicate that neutral, O-bound proximal amide groups (Table 2, A) are competent species in catalysis.
We next examined reactivity of other peptide sequences (Table 3). A number of naturally occurring peptide sequences were amenable to this reaction. A 21-mer peptide indicates that the reaction tolerates quite lengthy sequences (entry 4), hinting at potential use in more demanding bioconjugation challenges.
The alkyne group is one of the most useful and general handles for manipulation of biomolecules, and we were gratified to find that boronic acids containing an alkyne handle retained efficient reactivity. A sulfonamide-linked alkyne boronic acid was an efficient reagent for N-terminal arylation (Fig. 3a).
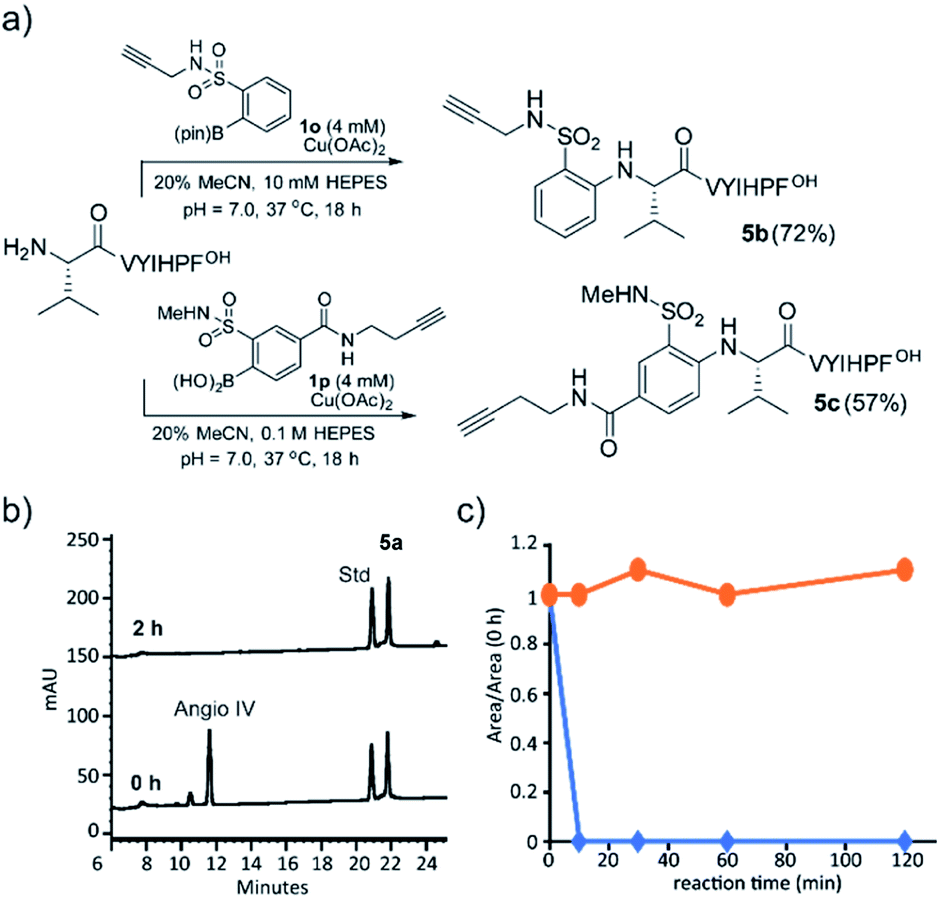 | ||
| Fig. 3 (a) Modification of peptide 5 with boronate ester 1o and 1p. Yield calculated by RP-HPLC. (b) RP-HPLC analysis of the enzymatic degradation of angiotensin IV (5) and arylated angiotensin IV (5a) with aminopeptidase I. Conditions: 100 μM 5, 40 μM 5a, 0.08 μg mL−1 aminopeptidase I, and 20 μM Co(OAc)2 in NaOAc buffer (5 mM, pH = 6.0) at 95 °C. Internal standard 19 for quantification. (c) Peptidase activity: time course for aminopeptidease I cleavage of arylated angiotensin IV (5a, orange) and angiotensin IV (5, blue). | ||
Arylation significantly alters the N-terminal charge state, since the product aniline is expected to be uncharged under physiological conditions. We decided to investigate whether the charge and structural perturbation afforded by the N-arylation engendered peptide stability towards enzymatic degradation. Using aminopeptidase I, an enzyme that liberates the N-terminal residue from peptides and proteins, we followed the reaction of both angiotensin IV 5 and its arylated analog 5a with Pfu aminopeptidase I and found that after 10 minutes 5 was completely consumed (Fig. 3b) while the 5a remained stable even after 2 h incubation (Fig. 3c).
Conclusion
In conclusion, copper(II) salts together with boronic acids bearing ortho-sulfonamide groups induce N–H arylation that is specific for the N-terminus. The reaction proceeds under neutral conditions in water and allows arylation of a wide variety of N-terminal residues. The reactivity is indicative of a new selectivity paradigm for copper-catalyzed amine functionalization that relies on local structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Quinn Kleerekoper for assistance with NMR studies. Funding for this research was provided by the NSF under award number CHE-1904865 and by the the Robert A. Welch Foundation Research Grant C-1680.References
- N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 Search PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 Search PubMed.
- J. Ohata, M. B. Minus, M. E. Abernathy and Z. T. Ball, J. Am. Chem. Soc., 2016, 138, 7472–7475 Search PubMed.
- J. Ohata, Y. Zeng, L. Segatori and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 4015–4019 Search PubMed.
- K. Hanaya, M. K. Miller and Z. T. Ball, Org. Lett., 2019, 21, 2445–2448 Search PubMed.
- J. Ohata, S. C. Martin and Z. T. Ball, Angew. Chem., Int. Ed., 2019, 58, 6176–6199 Search PubMed.
- J. Ruiz-Rodríguez, F. Albericio and R. Lavilla, Chem. Eur. J., 2010, 16, 1124–1127 Search PubMed.
- K. Hanaya, J. Ohata, M. K. Miller, A. E. Mangubat-Medina, M. J. Swierczynski, D. C. Yang, R. M. Rosenthal, B. V. Popp and Z. T. Ball, Chem. Commun., 2019, 55, 2841–2844 Search PubMed.
- K. K.-Y. Kung, H.-M. Ko, J.-F. Cui, H.-C. Chong, Y.-C. Leung and M.-K. Wong, Chem. Commun., 2014, 50, 11899–11902 Search PubMed.
- M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2018, 140, 7065–7069 Search PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 Search PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 Search PubMed.
- H. G. Lee, G. Lautrette, B. L. Pentelute and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 3177–3181 Search PubMed.
- D. Ma, Y. Zhang, J. Yao, S. Wu and F. Tao, J. Am. Chem. Soc., 1998, 120, 12459–12467 Search PubMed.
- Z. Lu and R. J. Twieg, Tetrahedron Lett., 2005, 46, 2997–3001 Search PubMed.
- N. Narendar and S. Velmathi, Tetrahedron Lett., 2009, 50, 5159–5161 Search PubMed.
- F. Ma, X. Xie, L. Ding, J. Gao and Z. Zhang, Tetrahedron, 2011, 67, 9405–9410 Search PubMed.
- S. M. King and S. L. Buchwald, Org. Lett., 2016, 18, 4128–4131 Search PubMed.
- Y. Jin, M. A. Lewis, N. H. Gokhale, E. C. Long and J. A. Cowan, J. Am. Chem. Soc., 2007, 129, 8353–8361 Search PubMed.
- Y.-A. Choi, J. O. Keem, C. Y. Kim, H. R. Yoon, W. D. Heo, B. H. Chung and Y. Jung, Chem. Sci., 2015, 6, 1301–1307 Search PubMed.
- L. W. Donaldson, N. R. Skrynnikov, W.-Y. Choy, D. R. Muhandiram, B. Sarkar, J. D. Forman-Kay and L. E. Kay, J. Am. Chem. Soc., 2001, 123, 9843–9847 Search PubMed.
- A. E. Mangubat-Medina, S. C. Martin, K. Hanaya and Z. T. Ball, J. Am. Chem. Soc., 2018, 140, 8401–8404 Search PubMed.
- H. B. F. Dixon, J. Protein Chem., 1984, 3, 99–108 Search PubMed.
- C. Cennamo, Naturwissenschaften, 1954, 41, 39 Search PubMed.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 Search PubMed.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C.-H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 Search PubMed.
- M. Zhang, X. Zhang, J. Li, Q. Guo and Q. Xiao, Chin. J. Chem., 2011, 29, 1715–1720 Search PubMed.
- D. Chen, M. M. Disotuar, X. Xiong, Y. Wang and D. H.-C. Chou, Chem. Sci., 2017, 8, 2717–2722 Search PubMed.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 Search PubMed.
- E. Reid, Nature, 1951, 168, 955 Search PubMed.
- G. Gaudriault and J.-P. Vincent, Peptides, 1992, 13, 1187–1192 Search PubMed.
- I. Sélo, L. Négroni, C. Créminon, J. Grassi and J. M. Wal, J. Immunol. Methods, 1996, 199, 127–138 Search PubMed.
- S. Schoffelen, M. B. van Eldijk, B. Rooijakkers, R. Raijmakers, A. J. R. Heck and J. C. M. van Hest, Chem. Sci., 2011, 2, 701–705 Search PubMed.
- A. C. Obermeyer, J. B. Jarman and M. B. Francis, J. Am. Chem. Soc., 2014, 136, 9572–9579 Search PubMed.
- Y. E. Sim, O. Nwajiobi, S. Mahesh, R. D. Cohen, M. Y. Reibarkh and M. Raj, Chem. Sci., 2019, 11, 53–61 Search PubMed.
- M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 Search PubMed.
- J. C. Vantourout, H. N. Miras, A. Isidro-Llobet, S. Sproules and A. J. B. Watson, J. Am. Chem. Soc., 2017, 139, 4769–4779 Search PubMed.
- T. J. Sereda, C. T. Mant, A. M. Quinn and R. S. Hodges, J. Chromatogr. A, 1993, 646, 17–30 Search PubMed.
- H. K. Hall, J. Am. Chem. Soc., 1957, 79, 5441–5444 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02933e |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |