
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissipative self-assembly, competition and inhibition in a self-reproducing protocell model†
Elias A. J.
Post
 and
Stephen P.
Fletcher
*
and
Stephen P.
Fletcher
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: stephen.fletcher@chem.ox.ac.uk
First published on 12th August 2020
Abstract
The bottom-up synthesis of artificial, life-like systems promises to enable the study of emergent properties distinctive to life. Here, we report protocell systems generated from phase-separated building blocks. Vesicle protocells self-reproduce through a phase-transfer mechanism, catalysing their own formation. Dissipative self-assembly by the protocells is achieved when a hydrolysis step to destroy the surfactant is introduced. Competition between micelle and vesicle based replicators for a common feedstock shows that environmental conditions can control what species predominates: under basic conditions vesicles predominate, but in a neutral medium micelles are selected for via a mechanism which inhibits vesicle formation. Finally, the protocells enable orthogonal reactivity by catalysing in situ formation of an amphiphilic organocatalyst, which after incorporation into the vesicle bilayer enantioselectively forms a secondary product.
Introduction
Virchow stated that all cells come from cells,1 rejecting the idea that living systems could arise from non-living material. It is still not clear how simple life forms can arise and reproduce in the absence of biological machinery, but the bottom-up synthesis of protocells promises to provide new insight into living systems.2–4Compartmentalized self-reproducing chemical systems have been used as model protocells to study how inanimate components may transition to living systems.5–7 Yet compartmentalization has received less attention than genetics or metabolism in the emergence of life. A protocell membrane has the crucial function of providing a protective boundary that concentrates reactants, and provides spatial confinement for reactions and genetic material.8,9 It has been argued that membrane compartments are essential for genetic and metabolic processes to manifest and evolve. In the Lipid World10 model, assemblies of simple, reproducing amphiphiles could form rudimentary systems with life-like properties in the absence of genetic material and metabolic functionalities.11,12
Study of protocellular systems has shown that fatty acid vesicles can grow and divide under certain stimuli.13–16 Lipids capable of fully autonomous self-reproduction have also been reported,17–21 and are capable of complex functions including linking the self-reproduction of giant vesicles to the amplification of encapsulated DNA.22
Synthetic protocells comprised of self-reproducing lipid systems are generally driven by movement toward thermodynamic equilibrium. This is fundamentally different to the natural self-assembly processes these systems aim to mimic, which require the continuous consumption of energy to maintain structure and function.23,24 Key to creating more life-like systems is dissipative self-assembly by the protocells which has been achieved using an external enzyme25 or by consuming an internal substrate26 or fuel.27 We have reported self-reproduction of micellar lipid aggregates28,29 and how autocatalytic micelle formation can become transient using thermodynamically unstable lipids that are destroyed at time-scales similar to their formation,30,31 enabling kinetically controlled selection from a pool of replicators.32
Recently there has been much experimental progress in mimicking individual features of life such as self-reproduction, compartmentalization, emergent function, adaptation in response to signals, formation of non-equilibrium structures and metabolism. Systems that integrate multiple such life-like properties into a functioning, dynamic whole, would represent a major advancement in the field.33
Here, self-reproducing phospholipid vesicles are generated from phase-separated reagents via a Cu-catalyzed alkyne–azide cycloaddition (CuAAC). This coupling reaction has previously been employed as a biomimetic method of driving de novo membrane assembly.20,34,35 The system reported here can self-reproduce while maintaining a dissipative population of vesicles, out-compete micelles under certain conditions, and features orthogonal reactivity through in situ generation of an organocatalyst (Fig. 1). Supramolecular phase transfer processes cause the lipids to self-reproduce by accelerating their own formation, and dissipative self-assembly by vesicles is achieved using conditions where the phospholipid is first created and then destroyed by hydrolysis. When vesicle forming protocell building blocks compete for a common feedstock with micelle forming building blocks, rudimentary selection processes are observed. Under pH-neutral conditions only the formation of micelles was observed due to inhibition of vesicle formation by the micelles (Fig. 1b), while under basic conditions the formation of vesicles was favoured over micellar aggregates (Fig. 1c). The vesicle protocells enable secondary reactivity by catalysing in situ formation of amphiphilic organocatalysts which become embedded in the vesicle bilayer and allow the protocell to act as an enantioselective nanoreactor. Overall, by combining minimalistic building blocks, a protocell model is generated that shows several emergent life-like properties.
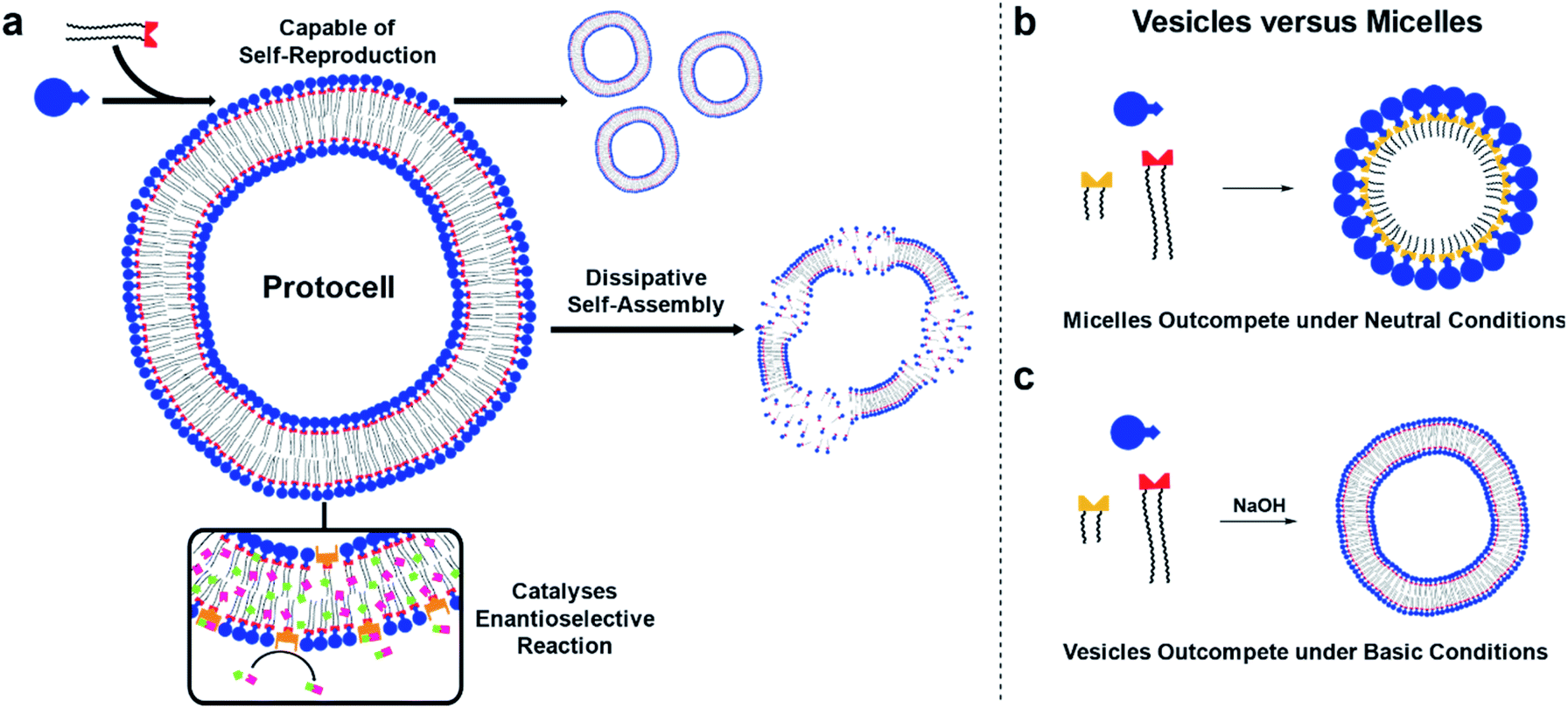 | ||
| Fig. 1 A protocell model featuring several emergent functionalities. (a) Schematic representation of a protocell that is capable of self-reproduction, dissipative self-assembly, and can catalyse an orthogonal enantioselective reaction. Competition experiments between two replicators select for micellar aggregates under neutral conditions (b) and vesicular aggregates under basic conditions (c). | ||
Results and discussion
A self-reproducing protocell model
The protocell systems here operate through a physical autocatalysis mechanism.28,29,36,37 Initially, hydrophobic azide 1 that constitutes the organic phase reacts slowly at the interface with hydrophilic phosphocholine 2 which is dissolved in water to form phospholipid 3via a CuAAC reaction with a hydrophobic copper–ligand complex (step i, Fig. 2a). Once a critical concentration of surfactant is reached, self-assembly by 3 into vesicles occurs (step ii). These vesicles then act as phase-transfer catalysts by taking 1 and the hydrophobic Cu-complex up from the organic phase into their bilayer (step iii) and facilitate intermolecular interaction with 2, thereby accelerating the formation of 3 closing the autocatalytic cycle (step iv).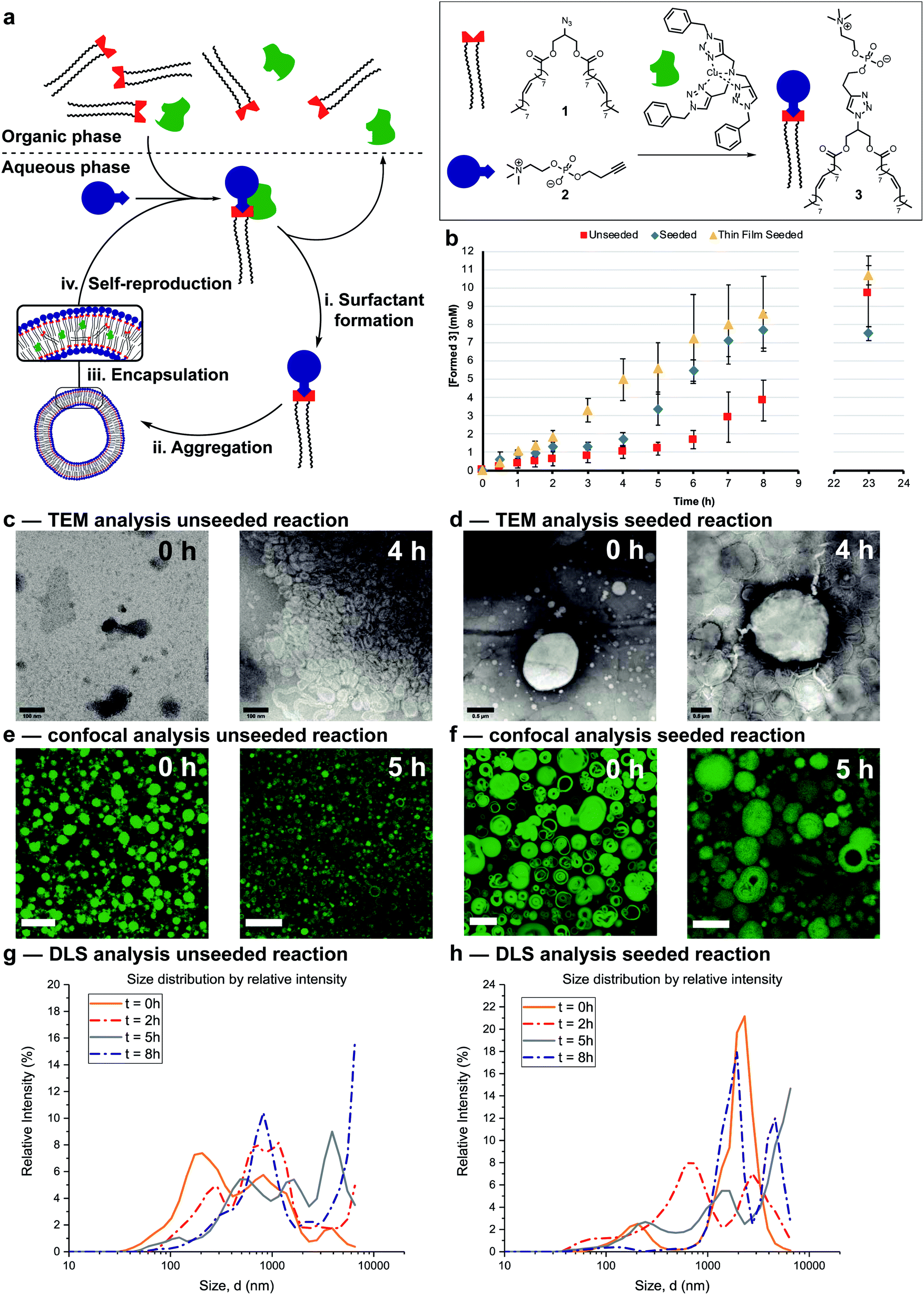 | ||
| Fig. 2 Autocatalytic formation, and characterization of vesicles. (a) Schematic representation of the mechanism of self-reproducing vesicles. (b) Kinetic results showing autocatalytic formation of 3 over time under unseeded (red squares), seeded (blue diamonds) or seeded through thin-film hydration (yellow triangles) conditions. Reactions were performed in the presence of 2 (20 mM, 1 eq.), 1 (1.56 eq.), CuSO4·5H2O (0.1 eq.) and monitored by UPLC-analysis. Points are the mean of three independent experiments and the error bars are the standard deviation. (c and d) TEM images at different time points of aliquots of the unseeded reaction and seeded reaction respectively (negatively stained with 2% uranyl acetate solution). Scale bar, 100 nm and 0.5 μm respectively. (e and f) Confocal images at different time points of aliquots of the unseeded reaction and seeded reaction respectively (stained with 5 μM coumarin 6 solution). Scale bar, 10 μm. (g and h) Size distribution by intensity of the aggregates in aliquots of the unseeded reaction and seeded reaction respectively at different time points measured by DLS. | ||
The supramolecular properties of 3 were investigated by a variety of analytical techniques. Fluorimetry measurements indicate that 3 has a critical aggregation constant (CAC) of 0.68 μM (Fig. S1†). Dynamic Light Scattering (DLS) measurements of 3 at different concentrations indicated an increase in diameter of the vesicles at higher concentrations, from 300 nm at 40 μM, to micron sized vesicles at 4 mM (Fig. S3†). When the same samples were stirred, a decrease in size of the aggregates was observed, possibly due to shearing forces. Absorption measurements showed an increase in turbidity for samples higher in concentration of 3 (Fig. S11†). TEM analysis showed the formation of spherical aggregates largely in the range of 1–30 nm, but up to 180 nm at 4 mM (Fig. S12†). Confocal microscopy of 3 at different concentrations shows that multilamellar vesicles increase in size and density with increasing concentration (Fig. S16–S24†).
Reaction conditions were tuned so that the kinetics of the formation of 3 was at convenient time scales. Kinetic experiments reveal a lag period followed by a subsequent period with a faster rate for the formation of 3 (Fig. 2b, red squares). Seeding the reaction at t = 0 with 3 at a concentration (4 mM) above the CAC reduces the length of the lag period and increases the rate of product formation (blue diamonds). These observations suggest a physical autocatalytic mechanism, however the lag period was not completely eliminated as it often is in seeded micellar autocatalytic systems.28–31,36 When the unseeded reaction was performed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/tert-butanol mixture to facilitate phase mixing, the lag period disappeared and complete conversion was observed in 1.5 hours (Fig. S37†), supporting the idea that phase behaviour is responsible for the lag period and non-linear kinetics.
:1 water/tert-butanol mixture to facilitate phase mixing, the lag period disappeared and complete conversion was observed in 1.5 hours (Fig. S37†), supporting the idea that phase behaviour is responsible for the lag period and non-linear kinetics.
The supramolecular properties of the aggregates were studied throughout the reaction. When aliquots of the unseeded reaction were studied by TEM no aggregates were observed at t = 0 only salt crystals of the negative stain were present, but increasingly larger aggregates were visible at later time points (Fig. 2c and S14† for additional time points). Confocal microscopy only showed stained oil droplets of 1 at t = 0, and largely unilaminar <5 micron vesicles at later times (Fig. 2e and S26† for additional time points). DLS analysis showed a growing population of micron sized vesicles when analysed based on intensity of scattering (Fig. 2g). The majority of vesicles stay in the 30–100 nm range independent of reaction time (Fig. S6†), perhaps due to the shearing forces introduced by stirring the reaction.
In seeded reactions, aggregates were already observed at t = 0 by both TEM and confocal analysis (Fig. 2d, f, S15 and S27† for additional time points). Aggregates with sizes of >20 micron were observed by confocal microscopy, where some vesicles were multilamellar to such a degree that they appeared to be completely filled with membrane leaving no visible aqueous interior (Fig. S27† for additional time points). DLS analysis by intensity showed the presence of a growing population of aggregates in the 1–10 micron range (Fig. 2h) but when analysed by number the majority was again in the 20–100 nm range independent of reaction time (Fig. S7†).
Overall the supramolecular aggregates of 3 formed in situ differ from those formed by seeding the reaction with premade 3. Aggregates formed in situ remain smaller and more unilaminar whereas the population of seeded aggregates remains large and multilamellar throughout the reaction without significant formation of small, unilaminar vesicles. Multilamellar vesicles might affect self-reproduction, as catalysts or reagents sequestered in inner layers could be physically separated and unable to participate in forming 3, and inner layers might not be available to transfer material between phases. This may explain the lag period observed for seeded reactions (Fig. 2b). To test this, giant unilaminar vesicles (GUV) of 3 were prepared through the hydration of a thin film38–40 as opposed to simple dissolution. Confocal analysis showed unilaminar aggregates (Fig. S25†) and DLS indicated a decrease in size compared to normal dissolution of 3 (Fig. S4†). When the reaction was seeded with these GUVs no lag period and a higher autocatalytic efficiency was observed (Fig. 2b, yellow triangles). Confocal micrographs of aliquots showed that there are still some unilaminar vesicles present at later time points (Fig. S28†), while DLS indicated that the majority of aggregates are 20 nm in size (Fig. S8†). We conclude that variations in supramolecular composition of the vesicles heavily influences the efficiency of the autocatalyst and the kinetics of the overall reaction.
Dissipative self-assembly
Mimicking dissipative self-assembly processes occurring in nature that rely on continuous consumption of chemical energy enable complex functions in synthetic systems such as self-healing in supramolecular materials41 and oscillations in reaction networks.42 A self-reproducing system may exhibit dissipative self-assembly through the use of a transient replicator.30,31 An autocatalyst that is under pressure of degradation but is also continuously regenerated will be unable to reach thermodynamic equilibrium, enabling study of the system in a non-equilibrium state.Introducing a hydrolysis step by adding base makes 3 transient. Phase separation prevents significant hydrolysis of 1 at reaction time scales as judged by 1H NMR spectroscopic analysis. The hydrolysis of one or two ester moieties in 3 gives 4 or 5 respectively, as well as oleate as a side product (Fig. 3a). Significant hydrolysis should lead to destruction of the monomers, the supramolecular aggregates, and the system's ability to reproduce. To fulfil the second requirement for dissipative self-reproduction the building blocks to regenerate the transient replicator have to be continuously available. This was achieved through continuous addition of hydrophobic (Fig. 3b, syringe i) and hydrophilic (syringe ii) starting materials and removal of waste products (syringe iii) from the reaction using a syringe pump set up. These components together form a continuously stirred-tank reactor (CSTR) which prevents the system from fully equilibrating.
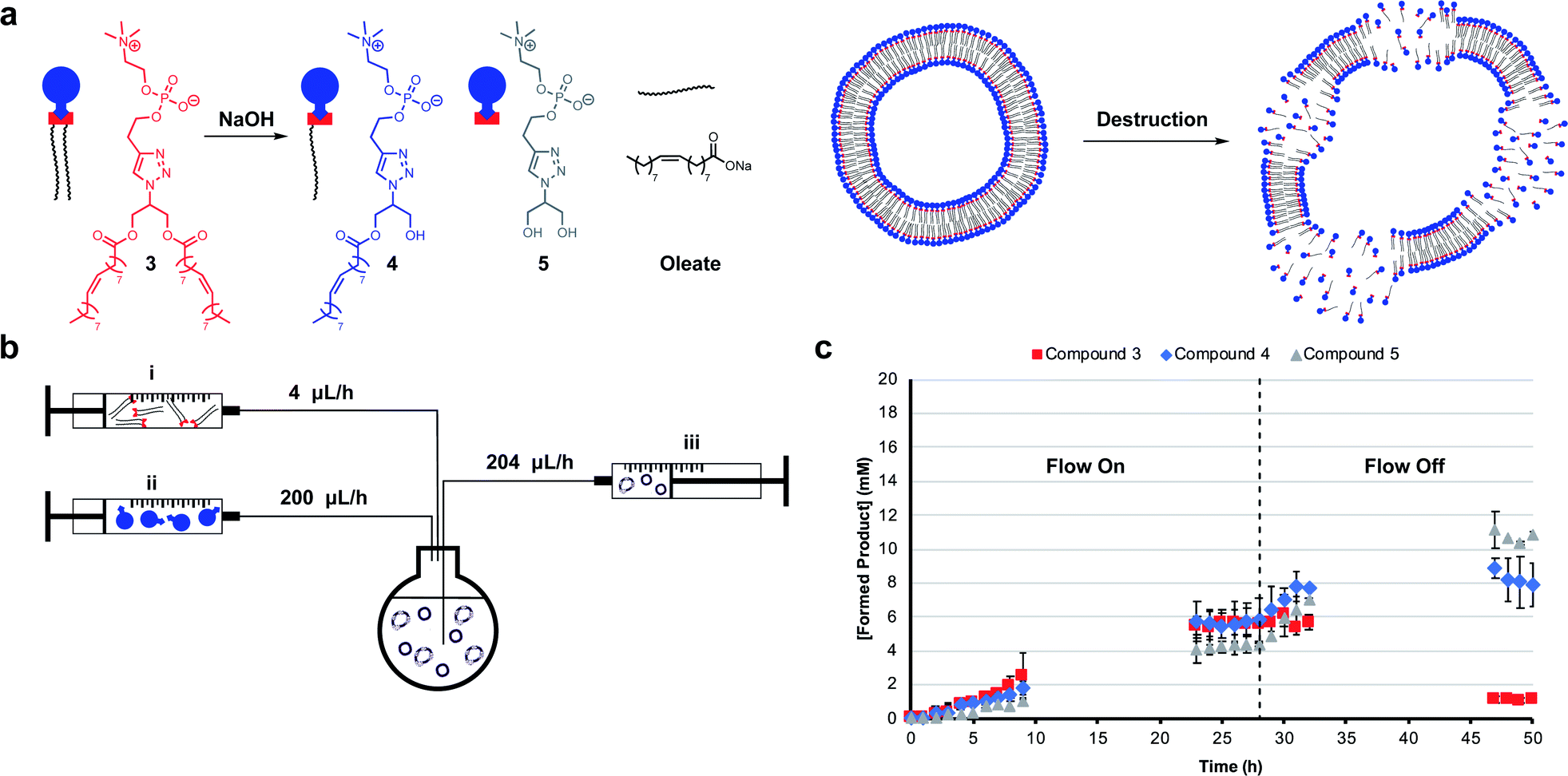 | ||
| Fig. 3 Dissipative self-assembly through transient phospholipid formation. (a) Schematic representation of the destruction of vesicle forming compound 3 to hydrolysis products 4 and 5. (b) Schematic representation of the continuously stirred-tank reactor (CSTR) setup. (c) Kinetic results showing a stable non-equilibrium state of metastable 3 with 100 mM NaOH while a flow through the CSTR is maintained followed by degradation when the flow is turned off. Reactions were performed in the presence of 2 (20 mM, 1 eq.), 1 (1.56 eq.), CuSO4·5H2O (0.4 eq.), NaOH (100 mM) and monitored by UPLC-analysis. Points are the mean of two independent experiments and the error bars are the standard deviation. | ||
The parameters used in kinetic experiments had to be reoptimized as hydrolysing conditions significantly slowed the formation of 3, and the loading of the copper catalyst was quadrupled (Fig. S38†). The concentration of base was fine tuned to control the degree of hydrolysis. DLS analysis showed a decrease in aggregate size at higher concentrations of NaOH (Fig. S9†). Slower reaction rates and more hydrolysis was observed going from 0 mM to 50 mM NaOH (Fig. S39–S41†). However, at these concentrations due to a lack of base, no significant further hydrolysis of 3 was observed when the flow was turned off and equilibrium composition was reached. At 100 mM NaOH a steady-state of 3 was observed at t = 23 h (Fig. 3c) which was stable for the next 6 hours while the flow was maintained. Turning off the flow brought the system to thermodynamic equilibrium by further degradation to 4 and 5, with almost complete destruction of 3 after 47 hours.‡
DLS analysis indicated the presence of <100 nm aggregates throughout the reaction (Fig. S10†). These sizes are below the detection limit of conventional confocal microscopy and can therefore not be resolved with this technique. Confocal analysis of the reaction in Fig. 3c showed a mixture of amorphous material with vesicles at earlier time points (<8 h), whereas aliquots taken after 24 hours showed only oil droplets indicating decomposition of larger vesicles (Fig. S29†). Overall, dissipative self-assembly into vesicles was achieved through transient formation of 3 by coupling autocatalytic surfactant formation to base induced degradation.
The winner of competition experiments depends on the reaction conditions
Living systems are often under external pressure leading to selection. The graded autocatalysis replication domain (GARD) model predicts that environmental changes can lead to selection between heterogeneous amphiphile assemblies of different composition resembling a form of evolution.10,12,43 Here we developed a rudimentary test of the GARD model using two competing amphiphiles, one that should form vesicles and one based on micelles.A micellar replicator was generated by coupling azide 6 to 2 to yield 7 (Fig. 4a), differing from 3 only in the length of the hydrophobic chains. Due to these shorter chains 7 assembles into micelles as opposed to vesicles44 with a diameter of 10 nm and a critical micelle concentration (CMC) of 27.6 μM, where aggregate size is independent of concentration, as judged by fluorimetry, DLS and TEM (Fig. S2, S5 and S13†).
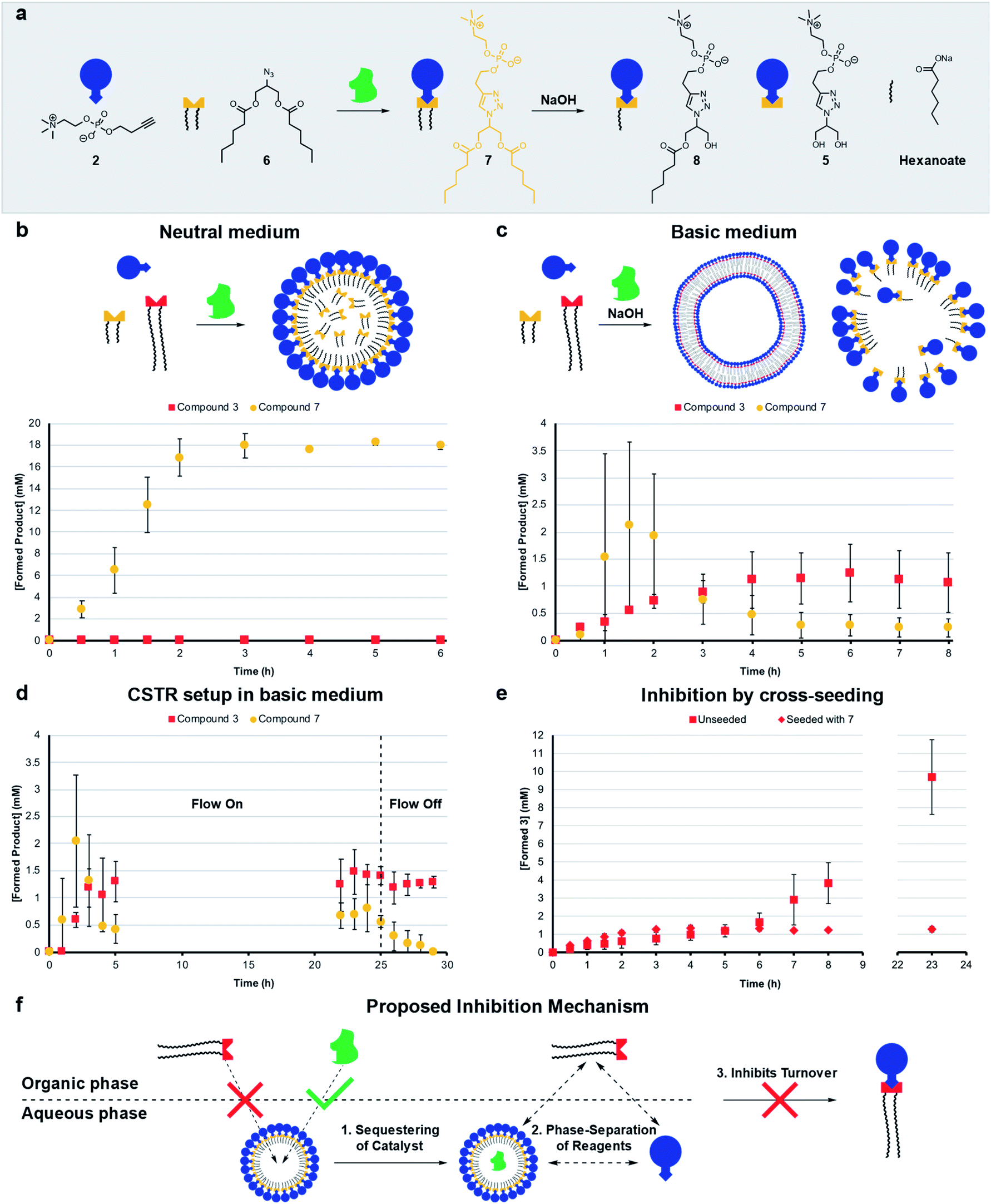 | ||
| Fig. 4 Competition experiments between a self-reproducing protocell and a micellar replicator. (a) Representation of micelle forming 7 being produced from azide 6 and 2 and destroyed by hydrolysis to 5, 8 and hexanoate. (b) Competition between two azides (1 (1.56 eq.) and 6 (1.56 eq.)) with alkyne 2 (20 mM, 1 eq.) and CuSO4·5H2O (0.4 eq.) to yield 3 and 7 respectively under neutral conditions. (c) Competition between 1 and 6 to give 3 and 7 in the presence of 50 mM NaOH (analogous conditions to b). (d) Competition between azides 1 and 6 to give 3 and 7 with 50 mM NaOH in a CSTR setup (analogous conditions to b). The flow is turned off after 25 hours. (e) Comparison of unseeded (Fig. 2b) with cross-seeding experiment between azide 1 (1.56 eq.) and alkyne 2 (20 mM, 1 eq.) with CuSO4·5H2O (0.1 eq.) to give 3 seeded with 7 (4 mM). All reactions monitored by UPLC-analysis. Points are the mean of three independent experiments and the error bars are the standard deviation. (f) Proposed mechanism by which micelles of 7 inhibit the formation of 3. | ||
The kinetic data we obtained is consistent with physical autocatalysis.36 The formation of 7 is fast (Fig. S43†), likely due to the increased solubility of shorter tailed 6 facilitating interfacial reactions, consistent with trends seen in other systems.28,32 Reducing the stirring rate to 400 rpm revealed an ∼1 h lag period that could be eliminated by seeding with 7 (4 mM) or by performing the reaction under homogeneous conditions (Fig. S37 and S44†).
Competition between azide 1 and 6 for alkyne 2 under neutral conditions (Fig. 4b) gives only product 7. However, in competition experiments where 50 mM NaOH is present, both 7 and 3 are observed, and 3 reaches a higher relative concentration at completion (Fig. 4c, zoomed in for clarity and see Fig. S45† for hydrolysis products). In a CSTR setup with base, a steady-state reached after 5 hours was maintained for a further 20 hours. Under these conditions the concentration of 3 is higher than 7 (Fig. 4d, zoomed in for clarity and S46†). When the flow was turned off 7 completely degraded whereas 3 is stable.
Cross-seeding experiments were investigated to probe the effect of competing surfactants on reproduction. Kinetics for the formation of 7 are the same when seeded with 3 or 7, so with 7 there appears to be no difference in rate of formation with either micelle or vesicle forming surfactants (c.f. Fig. S44 and S47†). Remarkably, formation of long tailed 3 stopped (at 1 mM) after two hours when the reaction was cross-seeded with 7, although the lag period was eliminated (c.fFig. 2b and 4e). Addition of 7 inhibits the reaction, which is consistent with the data in Fig. 4b where only 7 is generated in the absence of base. However, in the presence of base (Fig. 4c), 7 does not inhibit the reaction, probably as it is continuously destroyed by hydrolysis. 3 is much less labile (see Fig. S48†), leading to a higher concentration of 3 than 7 under those conditions.
Mechanisms of selection and inhibition
The micelles were expected to have different reproducing properties to the vesicles. Micelles are dynamic and rapidly exchange micellar components with the solvent relative to phospholipid vesicles.4,45 Micelles also have no inner layers of membrane whereas multilamellar vesicles have bilayers impermeable to polar compounds that subsequently cannot reach any inner layers of membrane.46 Phospholipid vesicles were anticipated to be more stable to hydrolysis than micelles. Sutherland showed acylation and hydrolysis cycling allowed selection in longer-chain acylglycerol-phosphates,47 which self-assembled into vesicles more stable towards hydrolysis than short chain compounds. Boekhoven observed a selection mechanism where shorter-tailed anhydrides are hydrolysed preferentially over longer-tailed anhydrides which phase-separated into oil droplets stabilising them to hydrolysis.48Hydrolysis of the aggregates was probed by stirring 20 mM of 3 or 7 in 50 mM NaOH which resulted in limited degradation of 3 whereas 7 was completely hydrolysed in 15 minutes (Fig. S48 and S49†). Repeating the experiment for 3 in a 1:1 water/tert-butanol mixture gave almost complete hydrolysis to 5 in 1 hour (Fig. S50†). When 3 or 7 are individually generated in situ in the presence of 50 mM NaOH a similar trend is observed where 7 largely degrades and 3 predominates (Fig. S38 and S51†) indicating that 3 is indeed more stable than 7 to hydrolysis. However, in competition experiments under basic conditions 3 reaches a higher concentration than 7 (Fig. 4c), which is not simply due to relative stability as 3 does not form in competition experiments under neutral conditions.
Diffusion ordered spectroscopy (DOSY) experiments were performed to probe how micelle forming 7 inhibits the formation of 3. The observed diffusion rates for the systems components at different concentrations (Table S1†) are consistent with hydrophobic components being solubilized by surfactant aggregates, and hydrophilic components not being associated with micelles of 7. Strikingly, however, the diffusion rates suggest that 7 is not capable of effectively solubilizing 1. At lower concentrations of 7 (4 mM) no uptake of 1 is observed (Fig. 4f, step 1). Whereas, 6 and the Cu–ligand complex are solubilized as shown by comparing D-values of these components and 7. We suggest that the reason micelles of 7 do not take up 1, is the large size of 1 which might disrupt the packing of 7 into micelles.
A second factor contributing to the inhibition of 3 might be sequestering of the copper complex into the aqueous phase by the micelles (Fig. 4f, step 2), which readily take up the catalyst (Table S1,† entry M). This effectively phase separates the catalyst and 1, which is restricted to the organic phase and prevents the formation of 3 (step 3). Overall, three hours are required for the system to reach an equilibrium composition where formation is fully inhibited, meanwhile a background reaction is operating possibly accelerated by 7 through lowering of interfacial tension. Repeating the cross-seeding experiment, but in a 1:1 water/tert-butanol mixture gave full conversion in 45 minutes (Fig. S52†), showing phase separation and self-assembly are required for the inhibition mechanism to operate.
A mixture of 3 and 7 studied by confocal microscopy shows that the morphology of the aggregates is no longer limited to spherical shapes – tubular structures and fused vesicles are also observed (Fig. S30†). Analysis of the hydrolysing competition reaction (Fig. 4c) by confocal microscopy showed clusters of large (up to 40 micron) oil droplets at t = 1 h, likely emulsified by the small amounts of 3 and 7 present,49 while later time points showed clusters of vesicles combined with amorphous material (Fig. S31†).
Downstream orthogonal reactivity
Life depends on interconnected reactions that together constitute metabolism.50 A network of reactions was introduced to this system by linking protocell formation to the in situ generation of a catalytically active surfactant which can be used as an organocatalyst51–54 to promote an enantioselective aldol reaction between phase separated substrates to form an orthogonal product.Proline-derived catalyst 10 was generated in situ through CuAAC of alkyne 9 and 1 in the presence of protocells (Fig. 5). Organocatalyst 10 catalyses an enantioselective reaction between nitrobenzaldehyde 11 and cyclohexane 12 to give 13. Control reactions showed that precatalyst 9 is not able to catalyse formation of 13, likely due to substrate phase separation (Fig. 5a and d, entry 1). When 9 is combined with amphiphile 3 still no product is observed even though 11 and 12 are now solubilized (Fig. 5b and d, entry 2). We assume this lack of reactivity derives from the inability of 9 to penetrate the lipid bilayer. Surprisingly, attempts to generate 10in situ from 1 and 9 were unsuccessful and also no aldol products were observed (entry 3). This is ascribed to phase separation between 1 and 9 preventing 10 from forming. Preformed catalyst 10, added as a solid, also does not yield 13 likely due to low the solubility of 10 in water. However, a thin-film of 10 generated to cover the entire surface of the flask could give 13 (entry 4, 39% conversion, 89:11 anti:syn ratio, 80% ee of anti). When 10 is combined with 3 to generate a protocell assembly where the organocatalyst is embedded in the bilayer, an effective nanoreactor is obtained, which generates 13 faster and with higher anti/syn- and enantioselectivity (Fig. 5c and d, entry 5). Doubling the loading of aldol reagents and catalyst 10 relative to 3 reduces the reaction time and increases the ee to 95% (entry 6). No degradation, precipitation or phase separation was observed while monitoring these reactions.
 | ||
| Fig. 5 Enantioselective catalysis with a self-reproducing protocell nanoreactor. (a) Schematic representation of entry 1 in table (d) where 9 is phase separated from 11 and 12, and no aldol reaction occurs. (b) Schematic representation of entry 2 where 9 is phase separated from 11 and 12 which are encapsulated in the bilayer of 3, and no aldol reaction occurs. (c) Schematic representation of entry 5 where 10 is embedded in the bilayer of 3 with 11 and 12 enabling the formation 13. (d) Table showing the experimental results of aldol experiments. All reactions were carried out at room temperature in the presence of 11 (13.3 mM) and 12 (66.6 mM) in 1 mL of H2O while stirring at 1000 rpm for 10 days. aDetermined by 1H-NMR spectroscopy of crude reaction mixture. bDetermined by SFC on a chiral non-racemic stationary phase. cCuSO4 (8 mM), TBTA (8 mM) and sodium ascorbate (24 mM) were also present in 5 mL of H2O. dConversion observed when the reaction was performed in a tBuOH:H2O (1:1) mixture. e11 (26.6 mM), 12 (133.2 mM) and 6 days of reaction time. | ||
The entire system was generated in situ from 1, 2 and 9 to yield 3 and 10 (entry 7). In this case, formation of 13 was observed (c.f. entry 3). The protocell appears to not only enhance the efficiency of the aldol reaction, but also facilitates the formation of the active catalyst 10. However, under these conditions the observed anti/syn ratio and ee are reduced. Interestingly, when the experiments described in entry 5 and 7 were repeated in a 1:1 water/tert-butanol mixture (conversion value shown in brackets), under which conditions no self-assembly into vesicles occurs, no formation of 13 was observed at the time scale of the reaction even though 10 is formed. We attribute this effect to the importance of compartmentalization of the reaction which can increase the local concentration of reactants and accelerate reaction times. Overall, a reaction network was developed where the protocell facilitates synthesis of an organocatalyst, which together with the vesicle catalyses the enantioselective formation of an orthogonal product.
Conclusions
We describe self-assembling, self-reproducing protocell models generated from simple, phase-separated chemical building blocks. Running the system under hydrolysing conditions introduced a destructive step to the autocatalytic systems and rendered the surfactants transient, enabling dissipative self-assembly. Competition between the building blocks of vesicular and micellar replicators for a common feedstock allows pH to act as a selection pressure and control which species predominates. Under basic conditions vesicular surfactants are favoured because their aggregates are less prone to hydrolysis, but at neutral pH micelle forming structures are favoured. We have also discovered an unusual selection mechanism whereby micelles can inhibit the formation of vesicle forming surfactants by effectively phase separating the reactants needed to form vesicles. A network of linked reactions was created through protocell enabled formation of a bilayer imbedded catalyst which subsequently catalyses the formation of enantioenriched secondary products. Overall, this protocell model incorporates several emergent properties into functioning, dynamic artificial cells, and shows that even a simple bottom-up synthetic molecular system can display complex, life-like behaviour.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dhanya Babu, Dr Dipak Samanta and Prof. Nathalie Katsonis (University of Twente) for access to their facilities and their support in the supramolecular characterisation of the protocells. We are grateful to Dr Andrew Bissette and Dr Elena Palmieri for constructive feedback on the manuscript. The ERC (Consolidator Grant, Autocat, 681491) is gratefully acknowledged for financial support.Notes and references
- R. L. C. Virchow, Cellular Pathology, John Churchill, London, special edn, 1859 Search PubMed.
- C. Xu, S. Hu and X. Chen, Mater. Today, 2016, 19, 516–532 CrossRef CAS.
- P. A. Beales, B. Ciani and S. Mann, Interface Focus, 2018, 8, 20180046 CrossRef.
- J. C. Blain and J. W. Szostak, Annu. Rev. Biochem., 2014, 83, 615–640 CrossRef CAS PubMed.
- R. V. Solé, A. Munteanu, C. Rodriguez-Caso and J. Macía, Philos. Trans. R. Soc., B, 2007, 362, 1727–1739 CrossRef.
- N. Amy Yewdall, A. F. Mason and J. C. M. van Hest, Interface Focus, 2018, 8, 20180023 CrossRef.
- J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387–390 CrossRef CAS PubMed.
- J. Lombard, P. López-García and D. Moreira, Nat. Rev. Microbiol., 2012, 10, 507–515 CrossRef CAS PubMed.
- B. C. Buddingh and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef CAS PubMed.
- D. Segré, D. Ben-Eli, D. W. Deamer and D. Lancet, Origins Life Evol. Biospheres, 2001, 31, 119–145 CrossRef.
- A. Kahana, P. Schmitt-Kopplin and D. Lancet, Astrobiology, 2019, 19, 1–16 CrossRef PubMed.
- D. Lancet, R. Zidovetzki and O. Markovitch, J. R. Soc., Interface, 2018, 15, 20180159 CrossRef PubMed.
- T. F. Zhu and J. W. Szostak, J. Am. Chem. Soc., 2009, 131, 5705–5713 CrossRef CAS.
- M. M. Hanczyc, Science, 2003, 302, 618–622 CrossRef CAS PubMed.
- K. Adamala and J. W. Szostak, Nat. Chem., 2013, 5, 495–501 CrossRef CAS PubMed.
- I. A. Chen, R. W. Roberts and J. W. Szostak, Science, 2004, 305, 1474–1476 CrossRef CAS PubMed.
- P. Walde, R. Wick, M. Fresta, A. Mangone and P. L. Luisi, J. Am. Chem. Soc., 1994, 116, 11649–11654 CrossRef CAS.
- P. Walde, P. A. Monnard, M. Wessicken, A. Goto and P. L. Luisi, J. Am. Chem. Soc., 1994, 116, 7541–7547 CrossRef CAS.
- K. Takakura, T. Toyota and T. Sugawara, J. Am. Chem. Soc., 2003, 125, 8134–8140 CrossRef CAS.
- M. D. Hardy, J. Yang, J. Selimkhanov, C. M. Cole, L. S. Tsimring and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8187–8192 CrossRef CAS PubMed.
- A. J. Bissette, B. Odell and S. P. Fletcher, Nat. Commun., 2014, 5, 4607 CrossRef CAS.
- K. Kurihara, M. Tamura, K. Shohda, T. Toyota, K. Suzuki and T. Sugawara, Nat. Chem., 2011, 3, 775–781 CrossRef CAS PubMed.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed.
- R. D. Astumian, Nat. Commun., 2019, 10, 3837 CrossRef PubMed.
- S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed.
- P. Solís Muñana, G. Ragazzon, J. Dupont, C. Z. J. Ren, L. J. Prins and J. L. Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 16469–16474 CrossRef.
- C. Wanzke, A. Jussupow, F. Kohler, H. Dietz, V. R. I. Kaila and J. Boekhoven, ChemSystemsChem, 2020, 2, 1–7 CrossRef.
- E. A. J. Post, A. J. Bissette and S. P. Fletcher, Chem. Commun., 2018, 54, 8777–8780 RSC.
- E. A. J. Post and S. P. Fletcher, J. Org. Chem., 2019, 84, 2741–2755 CrossRef CAS.
- I. Colomer, S. M. Morrow and S. P. Fletcher, Nat. Commun., 2018, 9, 2239 CrossRef.
- S. M. Morrow, I. Colomer and S. P. Fletcher, Nat. Commun., 2019, 10, 1011 CrossRef PubMed.
- I. Colomer, A. Borissov and S. P. Fletcher, Nat. Commun., 2020, 11, 176 CrossRef CAS.
- D. Kroiss, G. Ashkenasy, A. B. Braunschweig, T. Tuttle and R. V. Ulijn, Chem, 2019, 5, 1917–1920 CAS.
- I. Budin and N. K. Devaraj, J. Am. Chem. Soc., 2012, 134, 751–753 CrossRef CAS PubMed.
- M. D. Hardy, D. Konetski, C. N. Bowman and N. K. Devaraj, Org. Biomol. Chem., 2016, 14, 5555–5558 RSC.
- P. A. Bachmann, P. L. Luisi and J. Lang, Nature, 1992, 357, 57–59 CrossRef CAS.
- A. J. Bissette, B. Odell and S. P. Fletcher, Nat. Commun., 2014, 5, 4607 CrossRef CAS.
- A. Jesorka, N. Stepanyants, H. Zhang, B. Ortmen, B. Hakonen and O. Orwar, Nat. Protoc., 2011, 6, 791–805 CrossRef CAS.
- M. Karlsson, K. Sott, M. Davidson, A.-S. Cans, P. Linderholm, D. Chiu and O. Orwar, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11573–11578 CrossRef CAS PubMed.
- M. Karlsson, K. Nolkrantz, M. J. Davidson, A. Strömberg, F. Ryttsén, B. Åkerman and O. Orwar, Anal. Chem., 2000, 72, 5857–5862 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- S. N. Semenov, L. J. Kraft, A. Ainla, M. Zhao, M. Baghbanzadeh, V. E. Campbell, K. Kang, J. M. Fox and G. M. Whitesides, Nature, 2016, 537, 656–660 CrossRef CAS.
- D. Segré, D. Ben-Eli and D. Lancet, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4112–4117 CrossRef.
- C. Tanford, The hydrophobic effect: formation of micelles and biological membranes, Wiley-Interscience, New York, 1980 Search PubMed.
- R. M. Garavito and S. Ferguson-Miller, J. Biol. Chem., 2001, 276, 32403–32406 CrossRef CAS.
- I. A. Chen and P. Walde, Cold Spring Harbor Perspect. Biol., 2010, 2, a002170 Search PubMed.
- C. Bonfio, C. Caumes, C. D. Duffy, B. H. Patel, C. Percivalle, M. Tsanakopoulou and J. D. Sutherland, J. Am. Chem. Soc., 2019, 141, 3934–3939 CrossRef CAS.
- M. Tena-Solsona, C. Wanzke, B. Riess, A. R. Bausch and J. Boekhoven, Nat. Commun., 2018, 9, 2044 CrossRef PubMed.
- M. J. Rosen and J. T. Kunjappu, Surfactants and Interfacial Phenomena, John Wiley & Sons, Inc., Hoboken, 4th edn, 2012 Search PubMed.
- J. C. Letelier, M. L. Cárdenas and A. Cornish-Bowden, J. Theor. Biol., 2011, 286, 100–113 CrossRef PubMed.
- Y. Hayashi, S. Aratake, T. Okano, J. Takahashi, T. Sumiya and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 5527–5529 CrossRef CAS.
- J. Huang, X. Zhang and D. W. Armstrong, Angew. Chem., Int. Ed., 2007, 46, 9073–9077 CrossRef CAS PubMed.
- M. C. M. van Oers, W. S. Veldmate, J. C. M. van Hest and F. P. J. T. Rutjes, Polym. Chem., 2015, 6, 5358–5361 RSC.
- B. H. Lipshutz and S. Ghorai, Org. Lett., 2012, 14, 422–425 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02768e |
| ‡ A control experiment under homogeneous conditions gave complete conversion to 5 within 5 hours and no change in composition was observed when the flow was turned off (Fig. S42). Phase-separation and self-assembly appear to be required for the non-equilibrium kinetics. |
| This journal is © The Royal Society of Chemistry 2020 |