
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled synthesis of organic two-dimensional nanostructures via reaction-driven, cooperative supramolecular polymerization†
Shikha
Dhiman‡
 ,
Rita
Ghosh‡§
,
Souvik
Sarkar
and
Subi J.
George
*
,
Rita
Ghosh‡§
,
Souvik
Sarkar
and
Subi J.
George
*
Supramolecular Chemistry Laboratory, New Chemistry Unit, School of Advanced Materials (SAMat), Jawaharlal Nehru Centre of Advanced Scientific Research (JNCASR), Jakkur, Bangalore 560064, India. E-mail: george@jncasr.ac.in; subijg@gmail.com
First published on 21st July 2020
Abstract
The bottom-up approach of supramolecular polymerization is an effective synthetic method for functional organic nanostructures. However, the uncontrolled growth and polydisperse structural outcome often lead to low functional efficiency. Thus, precise control over the structural characteristics of supramolecular polymers is the current scientific hurdle. Research so far has tended to focus on systems with inherent kinetic control by the presence of metastable state monomers either through conformational molecular design or by exploring pathway complexity. The need of the hour is to create generic strategies for dormant states of monomers that can be extended to different molecules and various structural organizations and dimensions. Here we venture to demonstrate chemical reaction-driven cooperative supramolecular polymerization as an alternative strategy for the controlled synthesis of organic two-dimensional nanostructures. In our approach, the dynamic imine bond is exploited to convert a non-assembling dormant monomer to an activated amphiphilic structure in a kinetically controlled manner. The chemical reaction governed retarded nucleation–elongation growth provides control over dispersity and size.
Introduction
Organic two-dimensional (2D) nanostructures1 composed of semiconducting π-conjugated molecules are important for various optoelectronic applications.2,3 The bottom-up approach of self-assembly has been proven to be an effective synthetic method for the synthesis of defect-free 2D-nanosheets by the well-ordered organization of organic π-conjugated amphiphiles.4 The structural control over the size and dispersity of these 2D organic nanostructures is crucial for their functional outcome.3 However, the controlled growth of organic monomers in two-dimensions has been a challenge and is seldom reported.5 In a pioneering study, crystallization-driven self-assembly6 (CDSA) has been employed to control the micellar organization of block copolymer amphiphiles to construct precision 2D nanostructures.7In this context, the structural control of one-dimensional organic nanostructures has been recently realized by living supramolecular polymerization (LSP).8 The strategy involves kinetic control over the nucleation or initiation phase during the nucleation-growth supramolecular polymerization to prevent spontaneous growth.9 In a unique strategy, a conformationally inactive metastable monomeric state10 was utilized for initiator-mediated chain-growth11 supramolecular polymerization. Alternatively, the most common design involves the kinetic trapping of monomers in non-equilibrium metastable aggregated states12 by exploiting the pathway complexity13 to realize seed-induced LSP. Sugiyasu and co-workers recently extended the seeded approach for the synthesis of 2D supramolecular polymers with a controlled area and narrow dispersity.14
In an attempt to generalize the design of LSP and to make it independent of the monomer structure, we have established the concept of dormant monomers in fuel or chemical reaction-driven LSP.15 In this bioinspired approach, the kinetics of the reaction, which can be modulated by external molecular cues, determine the rate of supramolecular polymerization.16,17 This strategy comprises a dormant monomer which, unlike the above-mentioned metastable dormant state, due to its thermodynamic stability, does not undergo self-assembly on its own (Scheme 1a). Hence, an external cue, an activator, is essential to in situ convert the non-assembling dormant monomer to an assembling active monomer which undergoes kinetically controlled, cooperative supramolecular polymerization.16 As a consequence, the activation step (kA) would be the rate-determining step (R.D.S.) and hence both the extent and kinetics of the self-assembly process can be directly modulated by the concentration of the activator. This was demonstrated recently by our group to attain complete control over the structural and temporal features of 1D supramolecular polymers.17 So far the fuel-driven strategy has only been applied to 1-dimensional (1D) nanostructures. However, controlled synthesis of 2D nanostructures has immense importance in organic materials, as mentioned above.
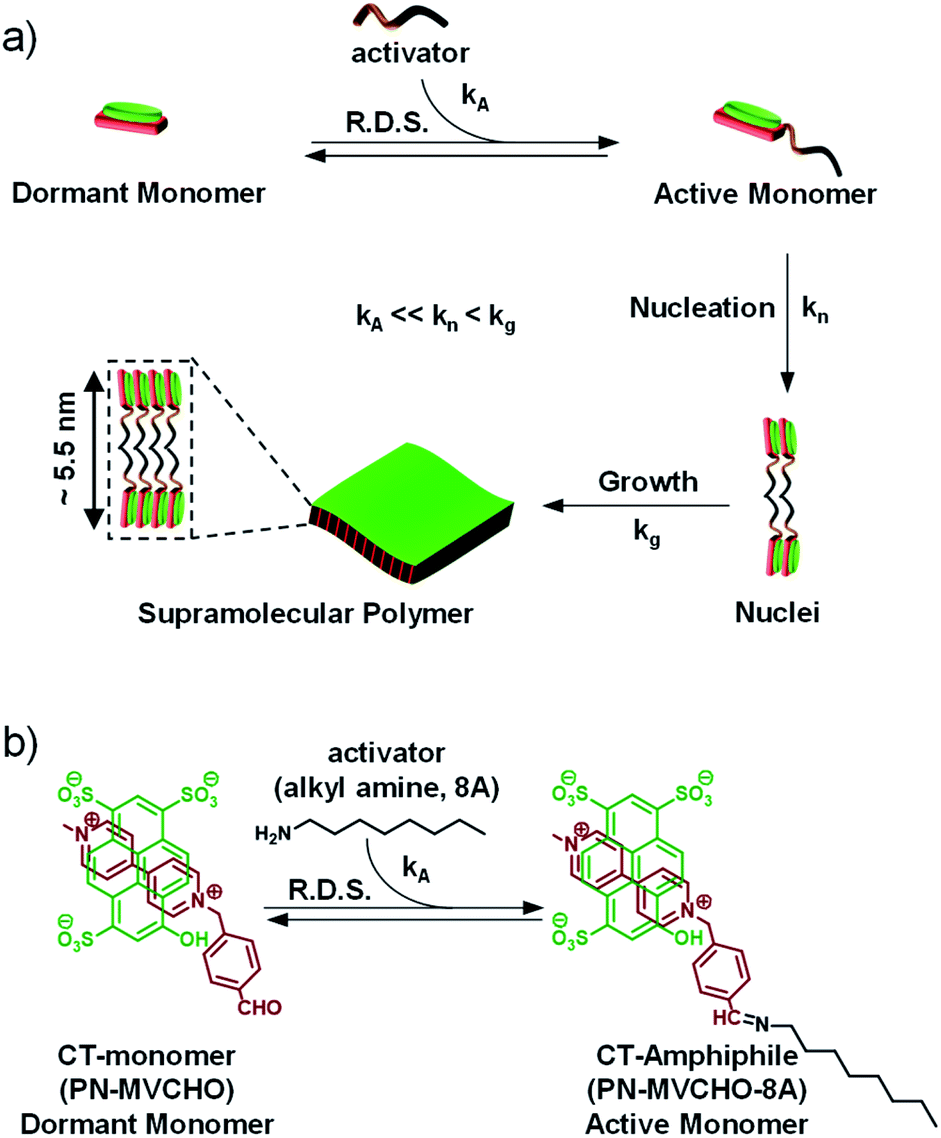 | ||
| Scheme 1 (a) Schematic illustration of the chemical reaction (imine bond)-driven activation of a dormant monomer (charge-transfer (CT)-monomer) to an active monomer (CT-amphiphile) by an activator which further undergoes nucleation-growth into bilayer sheets. (b) Chemical structure and reaction for the activation of a CT-complex composed of a pyranine (PN) donor and methyl viologen benzaldehyde (MVCHO) acceptor to the CT-amphiphile by octyl amine (8A). kA = rate of activation, kn = rate of nucleation, kg = rate of growth/elongation and R.D.S. = rate determining step. | ||
Herein we report the generic nature of the chemical reaction-driven cooperative supramolecular approach for the controlled synthesis of 2D nanostructures from organic semiconducting monomers, which can be modulated by variation of the concentration of molecular cues for a controlled dispersity and size.
Design strategy
Charge-transfer (CT) supramolecular amphiphiles, with donor (D) and acceptor (A) π-stacked components, were extensively studied by Zhang's group and by our group independently to create semiconducting alternating supramolecular polymers in various dimensions for their optoelectronic and stimuli-responsive applications.18 However, due to spontaneous self-assembly (Scheme 1, kn and kg are high), they lack controlled dimensions, and thus there is high scope to increase their functional efficiency. For this, kinetic regulation (kA ≪ kn) over the supramolecular polymerization process is required that can retard the nucleation process.19 However, the strategy of kinetic trapping of monomers via an off-nucleation pathway or conformational metastable state cannot be applied in these systems as most of the CT supramolecular monomers are highly dynamic and fall spontaneously into thermodynamic equilibrium. Therefore, in our approach to realizing kinetically controlled supramolecular polymerization, we designed a dormant monomer state comprising an oppositely charged D and A that form the CT-complex (PN-MVCHO).Since the CT-complex, PN-MVCHO (dormant CT-monomer), is charged, it remains well-dispersed in aqueous solution as a discrete, non-covalent monomer and lacks any propensity to aggregate (Scheme 1). Moreover, the aldehyde group appended to the acceptor acts as the reactive center for the nucleophilic reaction of amine to form the dynamic imine bond.20 As a consequence, the imine reaction orchestrates the conversion of the dormant CT-monomers to the active monomeric state (PN-MVCHO-8A, kA, Scheme 1), with a non-covalent amphiphile structure, in a temporal manner that governs the kinetically controlled supramolecular polymerization process and hence is the rate-determining step (R.D.S.). Further, the kinetics and extent of this chemical reaction-driven supramolecular polymerization can be modulated by the change in reactivity (by changing the pH) or the concentration of the substrates, amine (8A) in the present case.
Results and discussion
Packing factor calculation for two-dimensional supramolecular assembly
We aim to construct a two-dimensional supramolecular assembly of non-covalent CT-amphiphilic monomers, with controlled dispersity and size. Amphiphilic systems are highly designable with a predictable morphological outcome. Hence, to predict the nanostructure morphology, we have calculated the packing factor of the supramolecular CT-amphiphile using the Tanford equation.21 In our previous study, we employed the CT-amphiphile with coronene tetra potassium salt (CS) as the donor component and the imine adduct of octyl amine (8A) and methyl viologen benzaldehyde (MVCHO) as the acceptor component.17 The resultant supramolecular CT-amphiphile (CS-MVCHO-8A) with a head size of 7.5 Å and a tail size of 11.62 Å yielded a packing factor of 0.47 corresponding to a cylindrical micellar arrangement (see the ESI†). The calculation is in agreement with the experimentally observed one-dimensional nanostructures with micellar packing of the amphiphilic CT-monomers.17On the other hand, a two-dimensional (2D) bilayer organization demands a packing factor of 0.9–1.0 according to the Tanford equation. To increase the packing factor, the interfacial area and head size of the amphiphile should be decreased to half that for the CS-MVCHO-8A, i.e. ∼3.75 Å. Keeping this in mind, in the present study, we have replaced the CS with a smaller pyranine donor molecule, PN, and used the same acceptor component. As a result, the head group size decreases, and a resultant packing factor of 0.91 is obtained. Hence we envisage that the bilayer assembly of non-covalent PN-MVCHO-8A amphiphilic monomers would yield bilayer 2D sheets.
Imine reaction-driven 2D supramolecular polymerization
We aim to control the structural properties of 2D assemblies via kinetically controlled growth, mediated by the chemical reaction, herein the imine reaction. The imine reaction-driven kinetics of self-assembly would depend on three factors (Scheme 1). Firstly, the reactivity of the amine to convert the CT-complex to the CT-amphiphile via the imine reaction. Secondly, the thermodynamic stability of the end product, herein CT self-assembly, shifts the dynamic reaction equilibrium towards the aggregated state and thus amine is further consumed. The third is the concentration of amine that influences both the above-stated factors.Before the self-assembly study, we investigated the formation and stoichiometry of the D and A components in the inactive CT-monomer. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of PN (donor) and MVCHO (acceptor) in the CT-monomer was calculated from the titration curve where a charge-transfer induced quenching of PN emission (λex = 350 nm) and appearance of a new CT-absorption band around 550–650 nm in a 5 × 10−4 M aqueous solution are observed (Fig. S1a and S2†). The concentration dependent absorption study of the CT complex shows a linear change in the CT absorption band, i.e. it follows the Lambert–Beer law in the concentration regime of 1 × 10−3 to 8 × 10−5 M (Fig. S3†). This suggests that the molecule does not undergo any assembly–disassembly phenomenon and a mere change of its concentration occurs. Further absence of scattering at higher wavelength confirms that the non-covalent CT-monomer is highly soluble in water and has no propensity to self-assemble in an alkaline solution.
:1 stoichiometry of PN (donor) and MVCHO (acceptor) in the CT-monomer was calculated from the titration curve where a charge-transfer induced quenching of PN emission (λex = 350 nm) and appearance of a new CT-absorption band around 550–650 nm in a 5 × 10−4 M aqueous solution are observed (Fig. S1a and S2†). The concentration dependent absorption study of the CT complex shows a linear change in the CT absorption band, i.e. it follows the Lambert–Beer law in the concentration regime of 1 × 10−3 to 8 × 10−5 M (Fig. S3†). This suggests that the molecule does not undergo any assembly–disassembly phenomenon and a mere change of its concentration occurs. Further absence of scattering at higher wavelength confirms that the non-covalent CT-monomer is highly soluble in water and has no propensity to self-assemble in an alkaline solution.
Next, we investigated the imine-driven supramolecular polymerization to a controlled extent. Since CT-interaction is known to get enhanced with self-assembly, we believed that the conversion of the CT-monomer to the CT-amphiphile and subsequent self-assembly can be probed by the increase in the CT-absorption band and a further quenching of PN emission (Fig. S4†). To investigate this, 1 equivalent (eq.) of octyl amine (8A) was added to an aqueous solution of 5 × 10−4 M PN-MVCHO at pH 11.5, and the spectral properties were probed after 2 hours of incubation. As the reactivity of amine determines the kinetics of the imine reaction, we kept the pH alkaline at ca. 11.5, which ensures that the deprotonated form of 8A exhibits high nucleophilicity. After the incubation time, an enhancement in the CT-absorption band along with a further quenching in PN emission was observed for PN-MVCHO-8A in comparison with the inactive CT monomer, PN-MVCHO, solution (Fig. S4†). The formation of self-assembled structures in solution is reiterated by the increase in size from 220 to 620 nm observed by dynamic light scattering (DLS) measurements (Fig. S5†).
The strengthening of the CT-interaction on amphiphilicity-driven self-assembly was further proven by the increase in the association constant (Ka) between the donor and acceptor from 1.7 × 105 M for the CT-monomer (PN-MVCHO) to 3.6 × 105 M for the CT-amphiphile (PN-MVCHO-8A) by fitting their emission titration curves to a 1:1 binding model (Fig. S1†). As the CT strength is also determined by the distance between the D and A components, the amphiphilicity-driven self-assembly further strengthens the existing CT-interaction in monomers.
To confirm that the observed self-assembly is indeed driven by the imine formation, 1H NMR was performed to quantify the imine reaction by monitoring the consumption of MVCHO through the protons of its aldehyde functional group. The 1H NMR of PN-MVCHO in D2O at pH 11.5 with 1 eq. of an internal standard of sodium acetate was first recorded (Fig. S6a and b†). To the same solution, 1 eq. of 8A in D2O was added and after 2 hours of incubation NMR was recorded. The 1H NMR spectrum shows complete disappearance of the aldehyde peak and broadening of other peaks suggesting 100% imine formation to promote self-assembly (Fig. S6a†). This depicts the interdependency of imine-driven self-assembly and the shifts of the dynamic imine reaction equilibrium due to the thermodynamic stability of CT self-assembly.17
As discussed earlier, most of the CT-based assemblies reported in the literature are highly dynamic and fall instantaneously to the thermodynamic minimum and thus lack any kinetic control over their supramolecular polymerization process.18 Similarly, the CT formation is very fast in the present system and we envisage that the kinetics of the formation of imine should be the rate determining step in the growth process (Scheme 1). To investigate this, the change in absorbance on the formation of CT extended assembly from inactive CT-monomers was probed during the growth process. For this, a solution of 5 × 10−4 M PN-MVCHO at pH 11.5 was taken and to it 1 eq. of 8A was added and the time-dependent absorbance changes at 670 nm (characteristic of CT) were monitored. The kinetic experiment revealed a non-linear increase in absorbance saturating at ∼2000 seconds (Fig. 1a). The changes in the absorbance could be due to the combined effect of an increase in the CT absorption and the increased scattering upon the self-assembly process. However, the kinetics monitored via light scattering which exclusively probes the growth process matches well with absorbance kinetics, suggesting that spectroscopic changes indeed probe the growth process. As the inactive CT-monomer has no inherent kinetics of growth, the kinetics observed is attributed to the rate of formation of imine.
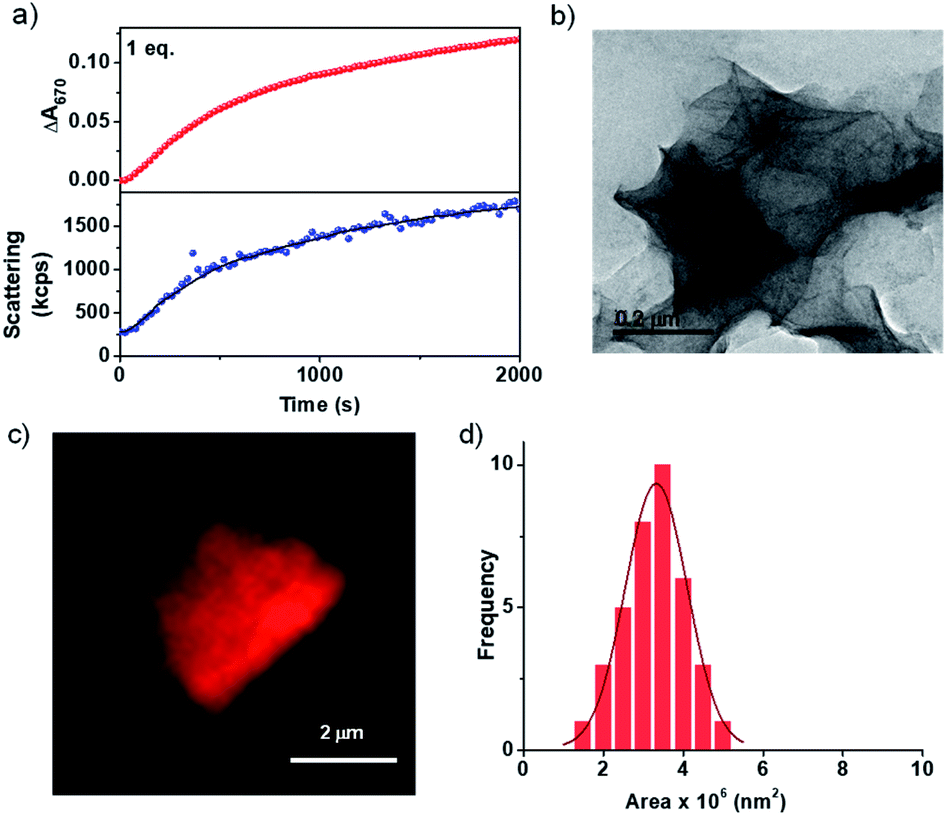 | ||
| Fig. 1 (a) Probing the growth of the PN-MVCHO-8A assembly via the time-dependent changes in absorbance at 670 nm and via scattering changes (monitored via DLS) showing a similar trend in growth with 1.0 eq. 8A. (b) TEM image and (c) confocal image (Nile red encapsulated in the hydrophobic bilayer) of the PN-MVCHO-8A assembly showing the formation of 2D sheets. [Nile red] = 1 × 10−6 M. (d) Area distribution profile obtained from confocal microscopy images depicting a controlled PDI of 1.06. [PN] = [MVCHO] = [8A] = 5 × 10−4 M, pH = 11.5. | ||
Thus the spectroscopic probing of the growth process suggests a kinetically controlled supramolecular polymerization by imine-reaction triggered self-assembly. Hence we next investigated its effect on the structural characteristics of the supramolecular polymer thus obtained. The morphological outcome of PN-MVCHO-8A self-assembly was investigated through transmission electron microscopy (TEM) which displayed the formed 2D sheets with >1 μm size (Fig. 1b and S7†) is consistent with the prediction from the packing factor. Atomic Force Microscopy (AFM) also showed thin sheets of a 5.5 nm height, revealing a non-interdigitated bilayer packing of the supramolecular amphiphile, PN-MVCHO-8A, along the thickness of the sheets, as calculated from the energy minimized structure (Fig. S8†). Zeta potential measurements suggest a negatively charged surface (−37 mV) of the sheets due to the dominance of anionic groups of PN-MVCHO (trianionic PNversus dicationic MVCHO) on the surface, which helps to obtain the single-layered sheets due to electrostatic repulsion (Fig. S9†). To confirm the bilayer packing, a polarity responsive hydrophobic dye, Nile red, was used to stain the sheets. Confocal fluorescence microscopy images show the formation of 2D sheets in solution imaged using Nile red dye encapsulation in the hydrophobic pockets of the nanostructures, which also allowed the visualization of 2D nanostructures in the solution phase without any solvent drying effects (Fig. 1c). Statistical analyses of these fluorescent 2D nanostructures revealed an average surface area of 3.3 × 106 nm2 with a narrow polydispersity index (PDI) of 1.06 (Fig. 1d and S10†). Thus it is evident that the imine reaction drives the conversion of the dormant monomer to the active monomer that undergoes kinetically controlled supramolecular polymerization into well-defined 2D sheets with a bilayer organization.
To further investigate the primary factor which is the effect of reactivity of the amine on the kinetics of self-assembly, the nucleophilicity of the amines is altered. Amines with high nucleophilicity are expected to undergo fast imine formation whereas protonated amines would not have any propensity towards imine formation. To scrutinize imine-assisted supramolecular polymerization, first, pH-dependence on the extent of imine formation is studied by 1H NMR spectroscopy. The 1H NMR spectra of PN-MVCHO with and without 8A were measured at pH 7 and 10 (Fig. S6c and d†). Integration with respect to the internal standard sodium acetate revealed 8% and 37% aldehyde consumption, respectively, in the presence of 8A (Fig. S6e†). Thus, at lower pH, the extent of imine formation is low due to lower nucleophilicity of the amine. This influence of pH on the chemical reaction also directly influences the rate and extent of self-assembly as elucidated by the absorption kinetic profiles. At pH 7, no growth occurs whereas at pH 10 there is sigmoidal growth but the rate and extent of self-assembly are too low compared to those at pH 11.5 (keeping the concentration of all other components constant) as evident from the absorption kinetics (Fig. S11†).
Hence, this proves that our strategy of chemical reaction-driven supramolecular polymerization provides a tool to modulate the extent of self-assembly and its kinetics by altering the reactivity of the amine which could be controlled by an external cue, pH in this case.
Chemical reaction-driven, cooperative supramolecular polymerization
An important characteristic of chemical reaction-driven self-assembly is the possible modulation of the rate of the reaction and hence the growth by variation of the external cues such as pH (vide supra) or concentration of substrates. Here we explore the effect of the change in the initial concentration of amine on the rate of the imine reaction and subsequent growth of the supramolecular polymer as well as the extent of self-assembly (Fig. 2). In order to explore this, the amount of 8A added to 5 × 10−4 M PN-MVCHO at pH 11.5 was varied from 0.2 eq. to 1.0 eq. and the corresponding absorbance changes at 670 nm were monitored for the growth process. The kinetics show a significant deceleration as the concentration of 8A was decreased (Fig. 2b–d and S12†). A remarkable sigmoidal growth was observed depicting a cooperative mechanism of growth, which is confirmed by the good fit of the growth kinetics to Watzky–Finke's two-state nucleation-growth model (Fig. S13†). The nucleation rate constant (kn) calculated for 0.3 eq. of 8A was 2.3 × 10−5 s−1 in comparison to the 5.6 × 10−4 s−1 for 0.4 eq. of 8A (Fig. 2b–d). Further, the growth kinetics acquired from the absorbance changes show a direct correlation with scattering confirming the changes monitored by absorbance are indeed due to growth (Fig. 2b and c).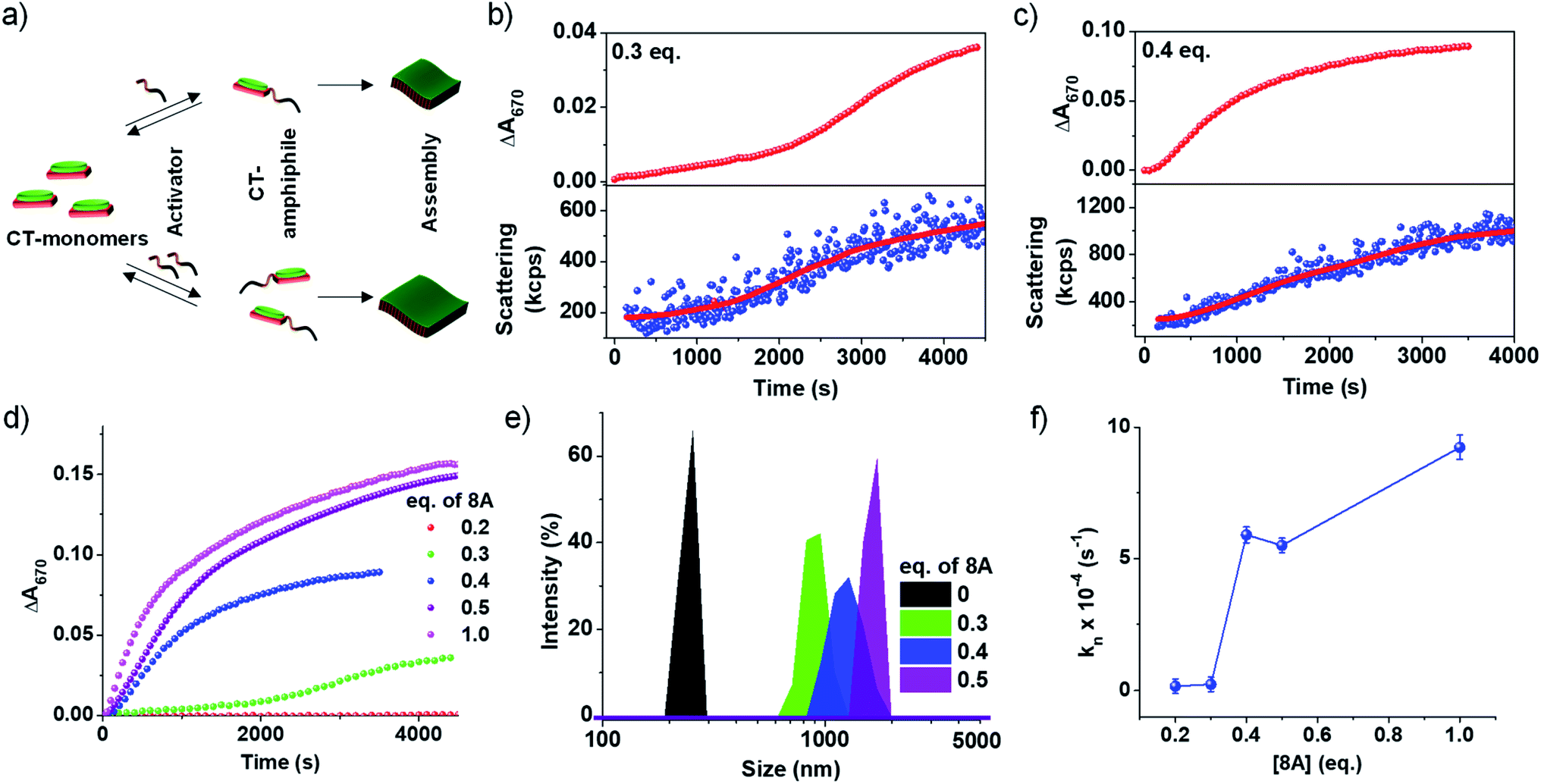 | ||
| Fig. 2 Dependence of amine concentration [8A] on reaction driven 2D self-assembly of PN-MVCHO-8A. (a) Schematic representation of reaction-driven 2D self-assembly and its dependence on the concentration of amine (8A) used. Time-dependent changes in absorbance at 670 nm and scattering changes showing the effect of change in the concentration of 8A on the growth kinetics of the supramolecular polymer with (b) 0.3 eq. and (c) 0.4 eq. of 8A. (d) Time-dependent changes in absorbance at 670 nm with different concentrations of 8A. (e) DLS data showing the concentration of 8A dependent size of kinetically grown CT supramolecular polymers. (f) Effect of the nucleation rate (kn) on 8A concentration. [PN] = [MVCHO] = 5 × 10−4 M, pH = 11.5. | ||
In the case of 1 eq. of 8A, non-sigmoidal kinetics with a half time (t50) of 485 seconds with no lag phase was observed (Fig. 2d). However, lowering the concentration of amine to 0.3 eq. of 8A resulted in sigmoidal growth with a t50 of 1700 seconds and a lag phase of ca. 165 seconds (Fig. 2d and S14†). Final intensity % size data show a dependence of size on the concentration of amine. For 0.3 eq. a size of 950 ± 130 nm, for 0.4 eq., 1280 ± 160 nm and for 0.5 eq., 1425 ± 120 nm were obtained indicating that a higher concentration of amine results in bigger assemblies (Fig. 2e and S7b†). As DLS assumes a spherical morphology and the present study deals with 2-dimensional assembly, DLS is only employed to provide a trend (qualitative analysis) of the size difference and we do not emphasize the exact numerical values (quantitative analysis).
To further investigate the growth process, the kinetic parameters obtained for 0.2–0.4 eq. were calculated as ≥0.5 eq. of 8A do not exhibit any clear lag phase owing to fast kinetics (Fig. 2d and S13†). The kinetic parameters show an increase in the maximum rate (vmax) and a decrease in the halftime (t50) and lag phase (tlag) with an increase in the concentration of the activator (8A) (Fig. S14†). Further, the fitting of the kinetics to the Finke–Watzky model shows an increase in the nucleation rate (kn) on increasing the 8A concentration (Fig. 2f and S13†). Since the kinetics of growth are determined by the reaction kinetics and monomer-assembly equilibrium, at low 8A concentration, the reaction is slow and hence the growth is less. At a higher 8A concentration, the reaction kinetics become fast and further autocatalyzed by the aggregation process. This results in the non-linear effect in the nucleation rate on increasing the 8A concentration (Fig. 2f).
Hence, the concentration of molecular cues, herein amine, can be utilized to directly affect the rate of imine formation and hence the growth of the supramolecular polymer which is not possible in other cooperative supramolecular assemblies where either solvent composition or temperature modulation is required to influence the energy landscape of the supramolecular polymerization process. This bioinspired strategy for chemical reaction-driven supramolecular polymerization is analogous to fuel-driven cooperative supramolecular polymerization in biological systems like actin.16
Chemical reaction-driven seeded supramolecular polymerization
The use of imine chemistry provides a remarkable control over the kinetics of self-assembly via a chemical reaction-driven strategy. If the supramolecular polymerization undergoes a nucleation–elongation mechanism of growth and the nucleation step is well-retarded via slow kinetic control, it can exhibit seeded growth characteristics with a new batch of monomers. The in situ activated monomers in the present study can either grow independently via a homogeneous nucleation mechanism or grow on an existing assembly (referred to as a seed/nucleus) via heterogeneous nucleation. Amongst the two, the former occurs in a non-spontaneous nucleation process whereas the latter can spontaneously occur. Thus, activated monomers tend to choose the second pathway which is termed seeded supramolecular polymerization and which has been recently exploited to gain structural control of supramolecular polymers (Fig. 3). Further seeded supramolecular polymerization is a ubiquitous process in fuel-driven supramolecular assemblies in the biological realm.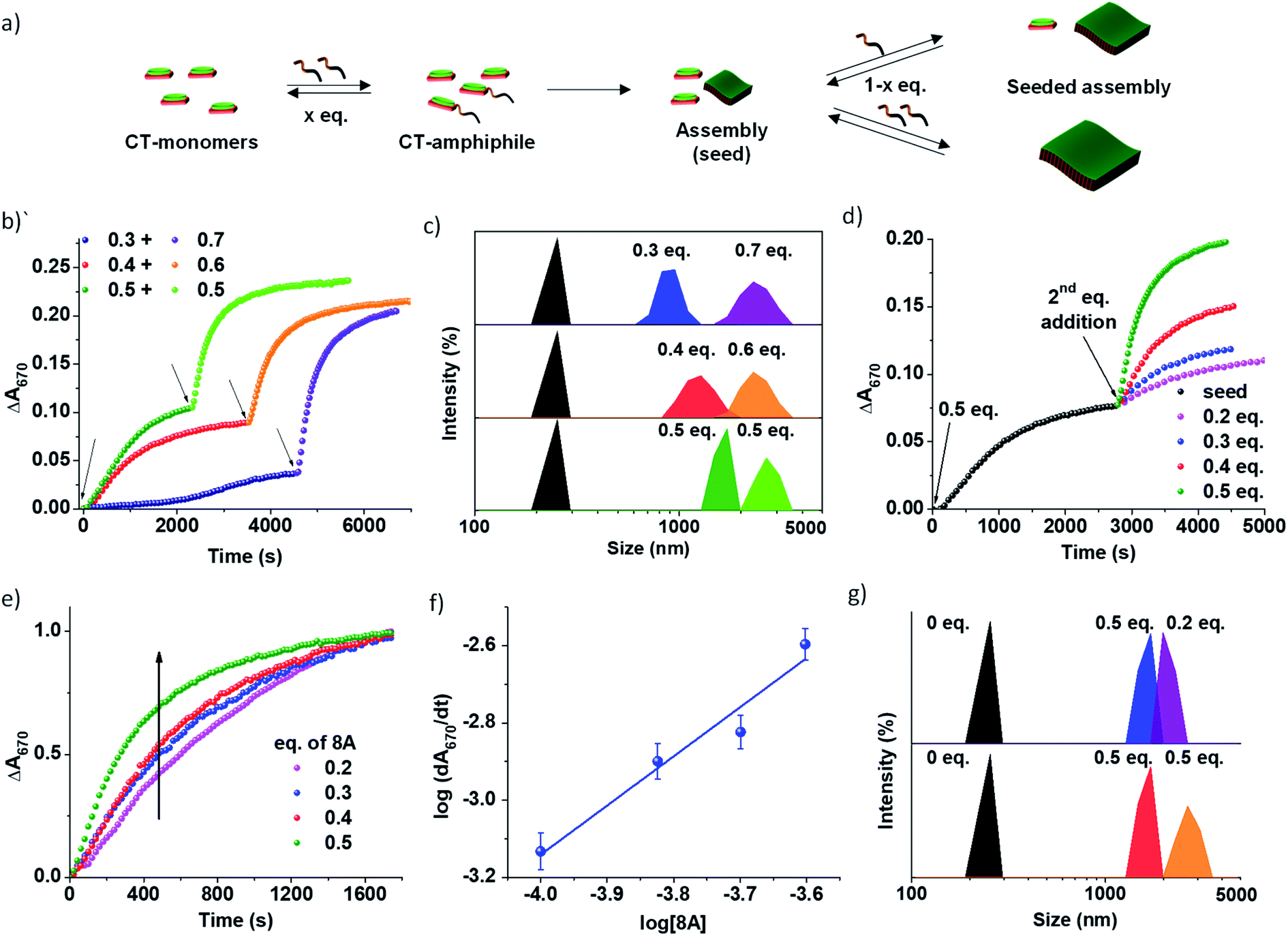 | ||
| Fig. 3 Imine reaction driven seeded 2D self-assembly of PN-MVCHO-8A. (a) Schematic representation of seeded supramolecular assembly with addition of 8A in batches. Temporal change in absorbance at 670 nm showing the initial sigmoidal kinetics for unseeded growth with subsequent non-sigmoidal kinetics on seeding (b) by adding 8A in batches with the seed + monomer concentration kept constant with 1 eq. of 8A and (d) constant seed (0.5 eq. 8A) and variable monomer concentration (0.2–0.5 eq. 8A). (c and g) DLS data showing a gradual increase in the size of the supramolecular assembly on seeding with the second batch of amine corresponding to the experiments in (b) and (d), respectively. (e) Normalized absorbance changes at 670 with time for seeding at a constant seed concentration depicting direct dependence of the rate of growth on the concentration of 8A. (f) log–log plot of the rate of seeded growth with the concentration of active monomers (eq. of 8A) at a constant seed concentration of 0.5 eq. [PN] = [MVCHO] = 5 × 10−4 M, pH = 11.5. | ||
Since the PN-MVCHO-8A system exhibits sigmoidal growth indicating a nucleation–elongation mechanism, we next investigate its seeding behavior. To investigate the seeded nature of reaction-driven self-assembly, we have introduced amines in batches (Fig. 3a) into a solution containing pre-grown assemblies and an excess of inactive dormant monomers, instead of a conventional seeding process where the seed is added to monomers trapped in a metastable state.5 This method would ensure the in situ generation of active monomers in solution by the imine-reaction without undergoing any concentration dependent changes in the reaction equilibrium. Hence, we performed kinetically controlled growth by adding amine in batches where its final concentration after the last addition is kept constant at 1.0 eq. For this, to a solution of 5 × 10−4 M PN-MVCHO, 0.3 eq. of 8A was added and the time-dependent absorbance changes at 670 nm were monitored (Fig. 3a and b). Sigmoidal growth saturating at ∼4560 seconds was observed with a nucleation rate of 2.3 × 10−5 s−1 (Fig. 3b). Addition of the remaining 0.7 eq. of 8A to the same solution resulted in non-sigmoidal growth with a rate of 2.3 × 10−3 s−1 confirming the seeded growth of the in situ generated active monomers on the existing supramolecular polymer rather than formation of their independent nuclei (Fig. 3b).
Moreover, a similar trend was displayed when sequential additions of 0.4 + 0.6 eq. of 8A and 0.5 + 0.5 eq. of 8A were attempted. The DLS intensity % data suggested a gradual increase in size with the addition of the second batch of 8A (Fig. 3c). In the case of 0.3 eq. of 8A, an increase in size from 950 ± 400 nm to 2255 ± 130 nm is observed with the addition of 0.7 eq. of 8A. Batch wise addition of 0.4 + 0.6 eq. of 8A results in an increase in size from 1280 ± 365 nm to 2300 ± 160 nm. From batch-wise addition of 0.5 eq. of 8A, the first addition gives a 1430 ± 300 nm size and the second seeding gives a 2670 ± 120 nm size (Fig. 3b and S15a†). It is observed that the increase in the extent of growth with the second batch of addition is proportional to the concentration of amine introduced, is consistent with seeded assembly (Fig. 3b).8
Würthner and coworkers suggested that multicycle seeding is a mandatory requirement for the seeded supramolecular polymerization to exhibit living characteristics.8 Hence, to explore the possibility of multicycle seeding, we added 8A in three batches of 0.3 eq. each (Fig. S15b†). Similar to previous cases, the first growth was sigmoidal, whereas the second and third were non-sigmoidal with rates of 1.08 × 10−3 s−1 and 1.04 × 10−3 s−1, respectively, reiterating seeded growth over multiple cycles of fuel addition (Fig. S15b†).
All the growth curves were non-sigmoidal depicting seeded kinetics. The increase in eq. of 8A from 0.2 to 0.5 eq. resulted in an enhanced rate of seeded growth from 7 × 10−4 s−1 to 2.5 × 10−3 s−1 (Fig. S16†). Also, the amount of amine directly determines the extent of self-assembly, which is evident from the seeding experiments with a fixed concentration of the seeds and with varying concentrations of amine 8A for the second batch of addition (Fig. 3d). Interestingly, the plot of the log of the rate of seeded growth versus the log of the concentration of active monomers (eq. of 8A) follows a linear increase which indicates the first-order rate of supramolecular polymerization with respect to amine and which along with non-sigmoidal growth validates a reaction-driven seeded growth (Fig. 3f). Further, the DLS intensity % data show a higher increase in size with higher eq. of the second batch of 8A (Fig. 3g). Hence, the present system demonstrates chemical reaction-modulated kinetically controlled 2D supramolecular polymerization with seeded growth.
Controlled dispersity and size via seeded supramolecular polymerization
Kinetically controlled seeded growth aims to provide control over dispersity and size. Thus, it is necessary to scrutinize the dispersity and size obtained after the kinetic growth (Fig. 3). Although DLS gives a relative change in the size of aggregates upon the seeding process, it cannot be used as a probe to determine the polydispersity index (PDI) and the size of two-dimensional assemblies. Consequently, to probe the structural aspects of the two-dimensional nanostructures in solution, we have used confocal fluorescence microscopy. Since the nanosheets in their native state are non-fluorescent due to their CT character, an external polarity sensitive probe, Nile red, is used for their visualization as mentioned before, which exhibits a turn-on fluorescence when encapsulated in the bilayers of PN-MVCHO-8A sheets.To further investigate the seeding characteristics of the PN-MVCHO-8A assembly, we added 8A in two batches of 0.5 + 0.5 eq. in the presence of Nile red and attempted size analysis as in the previous case. Firstly, 0.5 eq. of 8A was added and an average sheet area of 1.7 ± 0.18 × 106 nm2 with an aspect ratio of 1.10 and polydispersity index of 1.10 is observed (Fig. 4 and S17†). This is a smaller size compared to that obtained with 1 eq. confirming the amine-dependent degree of polymerization. Interestingly, on the addition of the second batch of 0.5 eq. of 8A, an increase in size from 1.7 ± 0.18 × 106 nm2 to 3.5 ± 0.05 × 106 nm2, with approximately two times the area, is observed. However, PDI remained narrow (∼1.10) with a negligible difference in the aspect ratio i.e. 1.13 (Fig. 4 and S18†). This substantiates that the imine bond directed kinetically controlled growth results in two-dimensional seeding of the CT monomers in a living manner (Scheme 1).
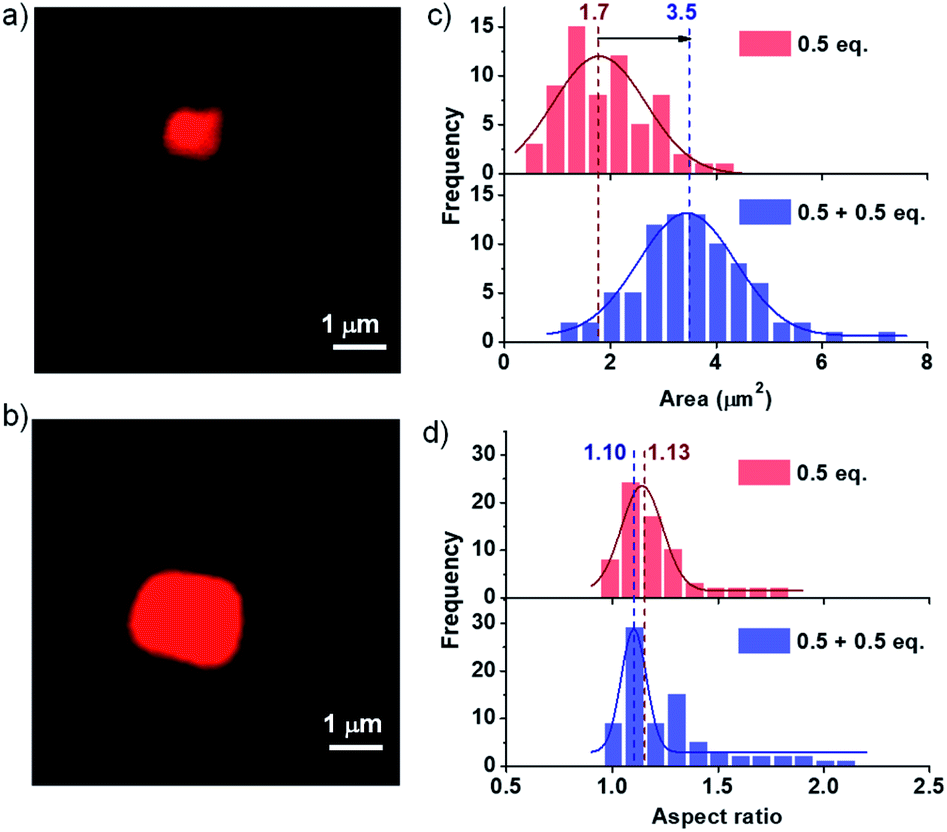 | ||
| Fig. 4 Confocal microscopy images. (a) 0.5 eq. unseeded and (b) 0.5 eq. seeded on 0.5 eq. unseeded supramolecular assembly. The distribution profiles of (c) area and (d) aspect ratio obtained from confocal microscopy images depicting a controlled PDI and aspect ratio. [PN] = [MVCHO] = 5 × 10−4 M, pH = 11.5, [Nile red] = 1 μM. | ||
In order to get unequivocal proof of 2D seeded growth, Nile red was introduced only during the seeding process. Subsequent visualization confocal microscopy images revealed a frame-like structure with an inner unseeded dark region and outer fluorescent seeded region of PN-MVCHO-8A (Fig. S19†). Thus, indeed the chemical reaction-driven cooperative supramolecular polymerization provides an opportunity for kinetically controlled growth and seeded supramolecular polymerization for controlled size and dispersity. The difference in growth kinetics of previously studied CS-MVCHO-8A and PN-MVCHO-8A in the current work under the same reaction conditions indicated that self-assembly autocatalyzes the formation of imine bonds and hence plays an important role in determining the kinetics (Fig. S20†).
Conclusion
In conclusion, we demonstrated chemical reaction-driven controlled synthesis of 2D organic nanostructures composed of supramolecular CT-monomers. The study depicts the importance of chemical reaction-driven cooperative supramolecular polymerization as a generic strategy to achieve two fold-control over the growth process: first, the retarded nucleation step by the in situ kinetically controlled generation of active monomers for narrow dispersity, and secondly, the seeded supramolecular polymerization for a controlled size and aspect ratio. Moreover, the extent of supramolecular polymerization and the rate of growth could be controlled by external cues such as the concentration and reactivity of the substrate. We envisage that this strategy can be extended for precision supramolecular nanostructures in all dimensions, without the kinetic trapping of highly dynamic monomers in their metastable states. The strategy of dynamic covalent chemistry of imine can be applied to any amphiphilicity driven supramolecular polymerization and hence has a high substrate scope. Finally, a CT supramolecular assembly is highly programmable in terms of its dimensions by changing the size of the donor, acceptor, or alkyl tail which is not so predictable in other molecular assemblies. The aforementioned strategy also paves the way to construct radial organic heterostructures with fine microstructure control using multicomponent seeded supramolecular polymerization.22Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. C. N. R. Rao, FRS for his support and guidance. We would like to thank the JNCASR and the Department of Science and Technology, Government of India for financial support. Also, the funding from the Sheikh Saqr Laboratory (SSL), JNCASR is acknowledged. S. J. G. acknowledges the funding received from Nanomission, DST (SR/NM/TP-25/2016) and the SwarnaJayanti Fellowship Award (DST/SJF/CSA-01/2016-2017).Notes and references
- X. Zhuang, Y. Mai, D. Wu, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 403–427 CrossRef CAS PubMed; C. E. Boott, A. Nazemi and I. Manners, Angew. Chem., Int. Ed., 2015, 54, 13876–13894 CrossRef PubMed; B. Shen, Y. Kim and M. Lee, Adv. Mater., 2020, 1905669 CrossRef PubMed; F. Ishiwari, Y. Shoji and T. Fukushima, Chem. Sci., 2018, 9, 2028 RSC; G. Wu, P. Verwilst, K. Liu, M. Smet, C. F. J. Faul and X. Zhang, Chem. Sci., 2013, 4, 4486–4493 RSC.
- S. K. Park, J. H. Kim and S. Y. Park, Adv. Mater., 2018, 30, 1704759 CrossRef CAS; F. Yang, S. Cheng, X. Zhang, X. Ren, R. Li, H. Dong and W. Hu, Adv. Mater., 2018, 30, 1702415 CrossRef PubMed; S. Ghosh, D. S. Philips, A. Saeki and A. Ajayaghosh, Adv. Mater., 2017, 29, 1605408 CrossRef PubMed; A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2005, 3245–3258 RSC.
- A. Jain and S. J. George, Mater. Today, 2015, 18, 206–214 CrossRef CAS.
- E. Lee, J. K. Kim and M. Lee, Angew. Chem., Int. Ed., 2009, 48, 3657–3660 CrossRef CAS PubMed; X. Liu, X. Zhou, B. Shen, Y. Kim, H. Wang, W. Pan, J. Kim and M. Lee, J. Am. Chem. Soc., 2020, 142(4), 1904–1910 CrossRef PubMed; Z. Chen, Y.-T. Chan, D. Miyajima, T. Kajitani, A. Kosaka, T. Fukushima, J. M. Lobez and T. Aida, Nat. Commun., 2016, 7, 13640 CrossRef PubMed; Z. Lin, J. Sun, Y. Zhou, Y. Wang, H. Xu, X. Yang, H. Su, H. Cui, T. Aida, W. Zhang and S. Z. D. Cheng, J. Am. Chem. Soc., 2017, 139, 5883–5889 CrossRef; D. Sahoo, M. Peterca, E. Aqad, B. E. Partridge, P. A. Heiney, R. Graf, H. W. Spiess, X. Zeng and V. Percec, J. Am. Chem. Soc., 2016, 138, 14798–14807 CrossRef; Y. Kim, S. Shin, T. Kim, D. Lee, C. Seok and M. Lee, Angew. Chem., Int. Ed., 2013, 52, 6426–6429 CrossRef PubMed; J. del Barrio, J. Liu, R. A. Brady, C. S. Y. Tan, S. Chiodini, M. Ricci, R. Fernández-Leiro, C.-J. Tsai, P. Vasileiadi, L. Di Michele, D. Lairez, C. Toprakcioglu and O. A. Scherman, J. Am. Chem. Soc., 2019, 141, 14021–14025 CrossRef PubMed; B. Narayan, S. P. Senanayak, A. Jain, K. S. Narayan and S. J. George, Adv. Funct. Mater., 2013, 23, 3053–3060 CrossRef; K. Liu, Y. Yao, C. Wang, Y. Liu, Z. Li and X. Zhang, Chem.–Eur. J., 2012, 18, 8622–8628 CrossRef.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed; N. Sasaki, J. Yuan, T. Fukui, M. Takeuchi and K. Sugiyasu, Chem.–Eur. J., 2020, 26, 7840–7846 CrossRef; D. S. Pal and S. Ghosh, Chem.–Eur. J., 2018, 24, 8519–8523 CrossRef.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566 CrossRef CAS; S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570–10577 CrossRef PubMed; X. He, M.-S. Hsiao, C. E. Boott, R. L. Harniman, A. Nazemi, X. Li, M. A. Winnik and I. Manners, Nat. Mater., 2017, 16, 481 CrossRef PubMed.
- Z. M. Hudson, C. E. Boott, M. E. Robinson, P. A. Rupar, M. A. Winnik and I. Manners, Nat. Chem., 2014, 6, 893–898 CrossRef CAS PubMed; X. He, Y. He, M. S. Hsiao, R. L. Harniman, S. Pearce, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2017, 139, 9221–9228 CrossRef PubMed.
- D. van der Zwaag, T. F. A. de Greef and E. W. Meijer, Angew. Chem., Int. Ed., 2015, 54, 8334–8336 CrossRef CAS PubMed; M. Wehner and F. Würthner, Nat. Rev. Chem., 2019, 4, 38–53 CrossRef; R. D. Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241–242 CrossRef PubMed; A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn., 2018, 91, 687–699 CrossRef CAS; T. Fukui, N. Sasaki, M. Takeuchi and K. Sugiyasu, Chem. Sci., 2019, 10, 6770–6776 RSC; J. Matern, Y. Dorca, L. Sánchez and G. Fernandez, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef PubMed; G. Ghosh, P. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757–6769 RSC; M. Hartlieb, E. D. H. Mansfield and S. Perrier, Polym. Chem., 2014, 11, 1083–1110 RSC.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed; S. Ogi, V. Stepanenko, J. Thein and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670–678 CrossRef PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed; P. A. Korevaar, T. F. A. de Greef and E. W. Meijer, Chem. Mater., 2014, 26, 576–586 CrossRef; J. Matern, K. K. Kartha, L. Sánchez and G. Fernandez, Chem. Sci., 2020, 11, 6780–6788 RSC.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed.
- S. Dhiman, A. Sarkar and S. J. George, RSC Adv., 2018, 8, 18913–18925 RSC; S. Dhiman and S. J. George, in Advances in the Chemistry and Physics of Materials, ed. S. J. George, C. Narayana and C. N. R. Rao, World Scientific, 2019, pp. 123–147 Search PubMed.
- A. Mishra, D. B. Korlepara, M. Kumar, A. Jain, N. Jonnalagadda, K. K. Bejagam, S. Balasubramanian and S. J. George, Nat. Commun., 2018, 9, 1295 CrossRef CAS PubMed; A. Mishra, D. B. Korlepara, S. Balasubramanian and S. J. George, Chem. Commun., 2020, 56, 1505–1508 RSC; L. J. Iglesias, A. Sorrenti, A. Sato, P. A. Dunne and T. M. Hermans, Chem. Commun., 2016, 52, 9009–9012 RSC.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS PubMed.
- A. A. Sagade, K. V. Rao, U. Mogera, S. J. George, A. Datta and G. U. Kulkarni, Adv. Mater., 2013, 25, 559–564 CrossRef CAS PubMed; X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94–101 RSC; C. Wang, Y. Guo, Y. Wang, H. Xu, R. Wang and X. Zhang, Angew. Chem., Int. Ed., 2009, 48, 8962–8965 CrossRef PubMed; K. Liu, C. Wang, Z. Li and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 4952–4956 CrossRef PubMed; M. Kumar, K. V. Rao and S. J. George, Phys. Chem. Chem. Phys., 2014, 16, 1300–1303 RSC; K. Jalani, S. Dhiman, A. Jain and S. J. George, Chem. Sci., 2017, 8, 6030–6036 RSC; B. V. V. S. P. Kumar, K. V. Rao, T. Soumya, S. J. George and M. Eswaramoorthy, J. Am. Chem. Soc., 2013, 135, 10902–10905 CrossRef PubMed; A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef PubMed.
- B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2018, 140, 534–537 CrossRef CAS PubMed; H. Frisch, E.-C. Fritz, F. Stricker, L. Schmüser, D. Spitzer, T. Weidner, B. J. Ravoo and P. Besenius, Angew. Chem., Int. Ed., 2016, 55, 7242–7246 CrossRef PubMed; J. P. Wojciechowski, A. D. Martin and P. Thordarson, J. Am. Chem. Soc., 2018, 140, 2869–2874 CrossRef PubMed.
- R. Nguyen, L. Allouche, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2009, 48, 1093–1096 CrossRef CAS PubMed; Y. Kang, K. Liu and X. Zhang, Langmuir, 2014, 30, 5989–6001 CrossRef PubMed; C. B. Minkenberg, L. Florusse, R. Eelkema, G. J. M. Koper and J. H. van Esch, J. Am. Chem. Soc., 2009, 131, 11274–11275 CrossRef PubMed; C. Wang, G. Wang, Z. Wang and X. Zhang, Chem.–Eur. J., 2011, 17, 3322–3325 CrossRef PubMed.
- C. Tanford, J. Phys. Chem., 1972, 76, 3020–3024 CrossRef CAS.
- A. Sarkar, R. Sasmal, C. Empereur-Mot, D. Bochicchio, S. V. K. Kompella, K. Sharma, S. Dhiman, S. Balasubramanian, S. S. Agasti, G. M. Pavan and S. J. George, J. Am. Chem. Soc., 2020, 142, 7606–7617 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02670k |
| ‡ Both the authors contributed equally. |
| § Current affiliation: Department of Chemistry, St. Joseph's College (Autonomous), Bangalore, India. |
| This journal is © The Royal Society of Chemistry 2020 |