
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular-level insight in supported olefin metathesis catalysts by combining surface organometallic chemistry, high throughput experimentation, and data analysis†
Jordan
De Jesus Silva
 a,
Marco A. B.
Ferreira
bc,
Alexey
Fedorov
*ad,
Matthew S.
Sigman
*b and
Christophe
Copéret
*a
a,
Marco A. B.
Ferreira
bc,
Alexey
Fedorov
*ad,
Matthew S.
Sigman
*b and
Christophe
Copéret
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1 5, CH 8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch; fedoroal@ethz.ch
bDepartment of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, Utah 84112, USA. E-mail: matt.sigman@utah.edu
cCentre for Excellence for Research in Sustainable Chemistry (CERSusChem), Department of Chemistry, Federal University of São Carlos – UFSCar, Rodovia Washington Luís, Km 235, SP-310, São Carlos, São Paulo 13565-905, Brazil
dDepartment of Mechanical and Process Engineering, ETH Zürich, Leonhardstrasse 21, CH 8092 Zürich, Switzerland
First published on 10th June 2020
Abstract
A combination of high-throughput experimentation (HTE), surface organometallic chemistry (SOMC) and statistical data analysis provided the platform to analyze in situ silica-grafted Mo imido alkylidene catalysts based on a library of 35 phenols. Overall, these tools allowed for the identification of σ-donor electronic effects and dispersive interactions and as key drivers in a prototypical metathesis reaction, homodimerization of 1-nonene. Univariate and multivariate correlation analysis confirmed the categorization of the catalytic data into two groups, depending on the presence of aryl groups in ortho position of the phenol ligand. The initial activity (TOFin) was predominantly correlated to the σ-donor ability of the aryloxy ligands, while the overall catalytic performance (TON1 h) was mainly dependent on attractive dispersive interactions with the used phenol ligands featuring aryl ortho substituents and, in sharp contrast, repulsive dispersive interactions with phenol free of aryl ortho substituents. This work outlines a fast and efficient workflow of gaining molecular-level insight into supported metathesis catalysts and highlights σ-donor ability and noncovalent interactions as crucial properties for designing active d0 supported metathesis catalysts.
Introduction
Research in academic and industrial laboratories over the last several decades has produced impressive advances in the field of alkene metathesis.1–6 This research has helped establishing detailed structure–activity relationships (SAR) for well-defined Mo-, W- and Ru-based molecular catalysts,7,8 and thereby aided in the rational development of catalytic systems with improved activity, selectivity and stability.9–11 Within this, high-throughput experimentation (HTE) can accelerate building robust SAR as it allows for a rapid and systematic acquisition of data on large libraries of compounds and formulations enabling the identification of catalysts.12–17 Utilization of robotized HTE methods for data acquisition is particularly advantageous because they allow obtaining reproducible data sets that are statistically significant to uncover robust and specific ligand properties in catalytic processes. We have recently integrated HTE with statistical analysis tools, inspired by methods of physical organic chemistry, that allow correlating various experimental and calculated steric or electronic ligand descriptors to performance indicators.11,18 These reaction outputs include turnover numbers and frequencies (TONs and TOFs, respectively) as well as selectivity and stability.8,10,18–25As an example of exploiting this methodology, we investigated the selective ethenolysis of cyclic olefins that relied on evaluating 29 well-defined Ru metathesis catalysts via HTE tools interfaced with statistical modeling. This effort ultimately provided a rational for the relative performance of catalysts, wherein the importance of π-backbonding and the size of the supporting NHC ligand for the selective formation of α,ω-dienes was revealed (Fig. 1).7 We also recently reported, using a similar methodology, the importance of noncovalent interactions (NCI) in controlling the activity and the stability of Schrock-type metathesis catalysts.8 Of particular note, the catalytic performance could be categorized by the type of phenols used to initiate the catalytic processes, wherein attractive non-covalent interactions (NCIs) were found to predominantly impact performance of catalysts that contained simple phenols devoid of ortho-aryl substituents. While powerful, this methodology has so far been rarely applied to the development and understanding of heterogeneous metathesis catalysts.26,27
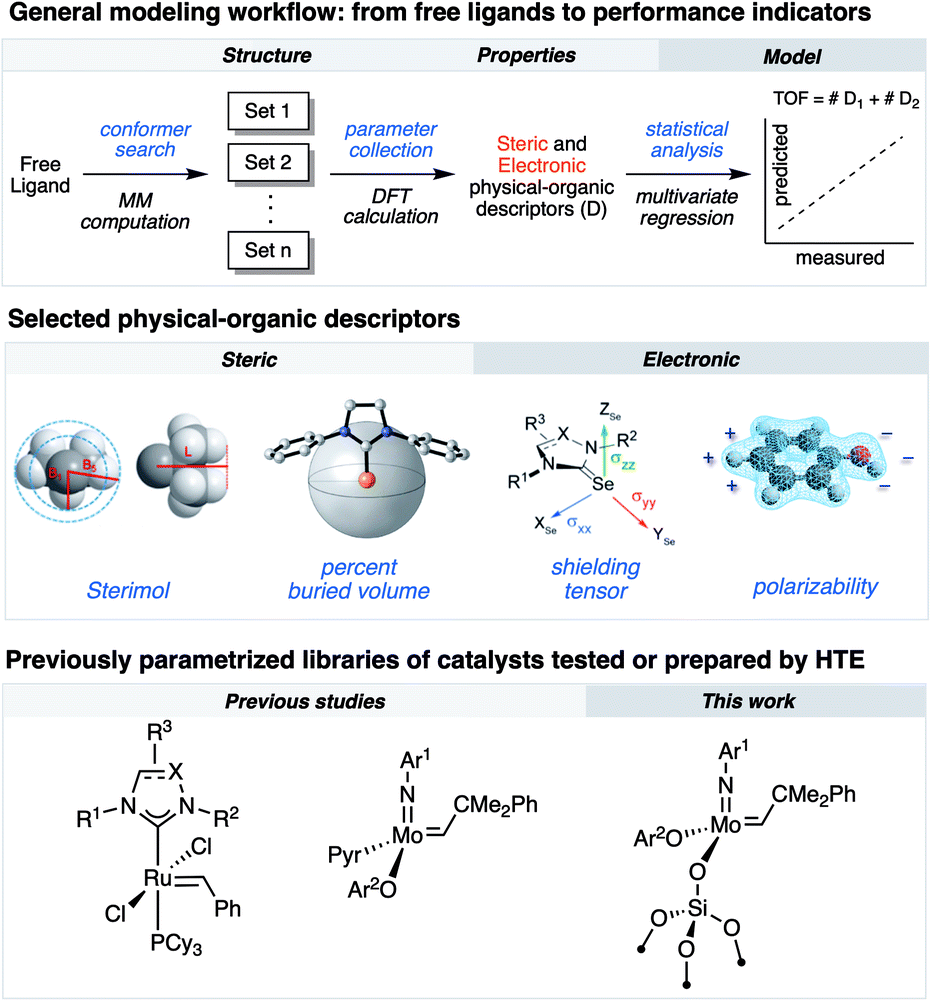 | ||
| Fig. 1 The concept of integrating HTE with statistical modeling. | ||
In parallel, surface organometallic chemistry (SOMC) has been established as a powerful approach to generate well-defined heterogeneous catalysts where the ligand effects can be probed.9 In this approach, the surface is exploited as a ligand to anchor (covalently graft) molecular catalysts. One of the most prominent examples of SOMC is the development of silica-supported catalysts, wherein surface silanols are used to graft the molecular complex via protonolysis of an anionic ligand of the molecular precursor.4,28 Besides the classical advantage of supported catalysts (ease of separation and recycling), this approach exploits surface site isolation to avoid bimolecular deactivation pathways, thereby increasing the stability of the corresponding well-defined supported catalysts compared to their homogenous analogues. In addition, these supported catalysts often feature activities exceeding those of their molecular counterparts.29–34
Herein, we demonstrate that combining HTE-SOMC13 with data analysis aiming at the correlation of molecular properties is a powerful approach to understand the catalytic performance of silica-supported metathesis catalysts at the molecular level, using the homodimerization of 1-nonene as a prototypical reaction. Within this study, by applying multivariate statistical modeling, we reveal that NCIs, which are typically associated with molecular catalyst, also govern the catalytic activity of heterogeneous, silica-supported Schrock-type catalysts.
Results and discussion
Testing in situ grafted Mo metathesis catalysts in the homodimerization of 1-nonene
To initiate this study, in situ formulations of a range of catalysts were prepared using 35 phenols with two precursor bis-pyrrolido Mo alkylidene complexes (2,5-(Me)2-Pyr)2Mo(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NAr)(CHCMe2Ph), where Ar = 2,6-(i-Pr)2-Ph (Mo-1) and 2,6-(Me)2-Ph (Mo-2), and silica partially dehydroxylated at 700 °C (SiO2-700) using HTE automation tools. The phenol library was designed on the basis of our previous studies.8 The formulations were prepared using, 2
NAr)(CHCMe2Ph), where Ar = 2,6-(i-Pr)2-Ph (Mo-1) and 2,6-(Me)2-Ph (Mo-2), and silica partially dehydroxylated at 700 °C (SiO2-700) using HTE automation tools. The phenol library was designed on the basis of our previous studies.8 The formulations were prepared using, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 molar ratio of ArOH, Mo-1/2 and
:1:2 molar ratio of ArOH, Mo-1/2 and ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiOH sites of SiO2-700 support, respectively, in order to complete the ligand exchange and surface grafting, targeting in situ synthesis of monoaryloxide surface-grafted species (Fig. 2A and see also S2A†). Specifically, bis-pyrrolides Mo-1 or Mo-2 (1 equiv.) were contacted with each phenol (2 equiv.) in toluene for 5 minutes prior to adding the resultant solution to SiO2-700 (2 equiv. of the surface SiOH), which was followed by keeping each reaction mixture for 3 h at 27 °C. We have recently shown using in situ1H NMR experiments that reacting Mo-1 in a 1:2 ratio with various ArOH used in this work typically leads to the formation of a single new alkylidene resonance.8 We reasoned that irrespective of the initial identity of the molecular alkylidene species present in solution (i.e. mono-aryloxide pyrrolide (MAP) or bisaryloxide species), the grafting reaction with SiO2-700 will lead to the monografted aryloxy Mo species (Fig. 2A), owing to the known exclusive exchange of the pyrrolide ligand in preference to aryloxide ligand during grafting of MAP complexes.35 In all cases, as the grafting reaction proceeds, the solution becomes colourless while the silica-supports becomes coloured. Prior to the catalytic test, all materials were washed to remove possible physisorbed molecular species on the silica material (see ESI† for details). Subsequently, a solution of 1-nonene in toluene was added to each in situ grafted material (0.1 mol% catalyst loading assuming quantitative grafting). All these steps were performed by an automated liquid handling robotic system operated inside an inert (N2) atmosphere glovebox. The reaction mixtures were agitated at 27 °C in open vials while GC aliquots were automatically withdrawn for analysis after ca. 6, 16, 39, 72, 135, 258 and 501 minutes, giving conversion of 1-nonene (X), selectivity to hexadec-8-ene (SC16 and SC16 (E/Z)), and respective TONs and TOFs that are reported based on the yield hexadec-8-ene. Complete catalytic data is presented in the ESI (Tables S1, S2, Fig. S3–S5 and S12–S83).† Robustness tests were performed in triplicates with new batches of 1-nonene, exhibiting good reproducibility (Tables S3 and S4†). In the discussion below, we focus on two selected activity indicators, TOFin and TON1 h (data points collected after ca. 6 and 72 min, respectively). TOFin reflects the initial activity of the catalyst formulation. Given that formulations on average reach X1 h > 40% after 72 min but no formulation reaches full conversion at this time point, the TON1 h indicator provides information about catalyst stability (Fig. 2, see ESI† for such plots using results with Mo-2).
SiOH sites of SiO2-700 support, respectively, in order to complete the ligand exchange and surface grafting, targeting in situ synthesis of monoaryloxide surface-grafted species (Fig. 2A and see also S2A†). Specifically, bis-pyrrolides Mo-1 or Mo-2 (1 equiv.) were contacted with each phenol (2 equiv.) in toluene for 5 minutes prior to adding the resultant solution to SiO2-700 (2 equiv. of the surface SiOH), which was followed by keeping each reaction mixture for 3 h at 27 °C. We have recently shown using in situ1H NMR experiments that reacting Mo-1 in a 1:2 ratio with various ArOH used in this work typically leads to the formation of a single new alkylidene resonance.8 We reasoned that irrespective of the initial identity of the molecular alkylidene species present in solution (i.e. mono-aryloxide pyrrolide (MAP) or bisaryloxide species), the grafting reaction with SiO2-700 will lead to the monografted aryloxy Mo species (Fig. 2A), owing to the known exclusive exchange of the pyrrolide ligand in preference to aryloxide ligand during grafting of MAP complexes.35 In all cases, as the grafting reaction proceeds, the solution becomes colourless while the silica-supports becomes coloured. Prior to the catalytic test, all materials were washed to remove possible physisorbed molecular species on the silica material (see ESI† for details). Subsequently, a solution of 1-nonene in toluene was added to each in situ grafted material (0.1 mol% catalyst loading assuming quantitative grafting). All these steps were performed by an automated liquid handling robotic system operated inside an inert (N2) atmosphere glovebox. The reaction mixtures were agitated at 27 °C in open vials while GC aliquots were automatically withdrawn for analysis after ca. 6, 16, 39, 72, 135, 258 and 501 minutes, giving conversion of 1-nonene (X), selectivity to hexadec-8-ene (SC16 and SC16 (E/Z)), and respective TONs and TOFs that are reported based on the yield hexadec-8-ene. Complete catalytic data is presented in the ESI (Tables S1, S2, Fig. S3–S5 and S12–S83).† Robustness tests were performed in triplicates with new batches of 1-nonene, exhibiting good reproducibility (Tables S3 and S4†). In the discussion below, we focus on two selected activity indicators, TOFin and TON1 h (data points collected after ca. 6 and 72 min, respectively). TOFin reflects the initial activity of the catalyst formulation. Given that formulations on average reach X1 h > 40% after 72 min but no formulation reaches full conversion at this time point, the TON1 h indicator provides information about catalyst stability (Fig. 2, see ESI† for such plots using results with Mo-2).
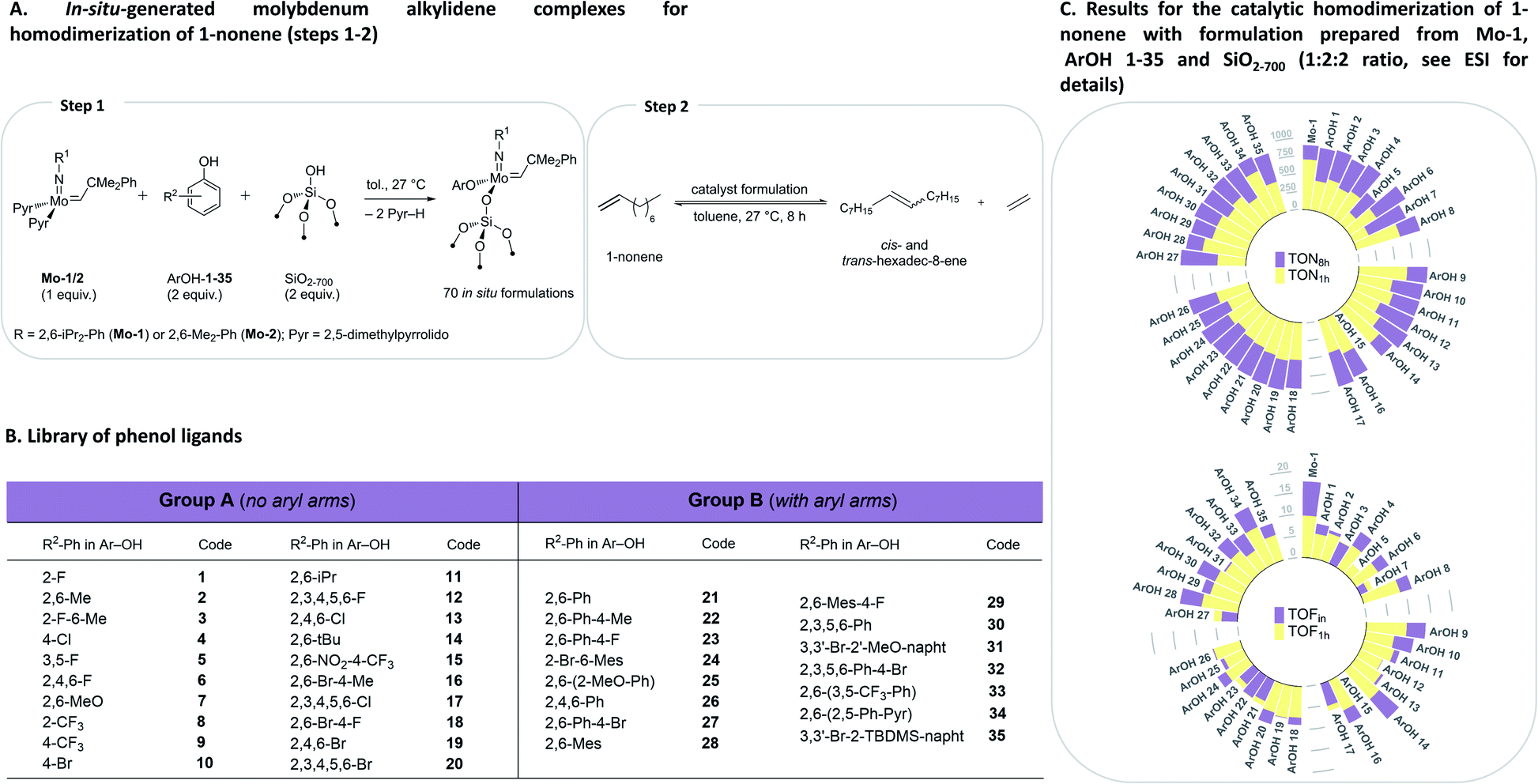 | ||
| Fig. 2 Design of the HTE study (A and B) and catalytic results (C) for in situ grafted formulations with Mo-1 with ArOH 1–35. | ||
Control experiments performed using longer premixing of various selected phenols and Mo-1 prior to contacting with SiO2-700 (i.e. 180 vs. 5 min, 2:1:2 molar ratio, respectively, Table S5†) show no notable differences in catalytic results beyond experimental error, which suggest formation of the same grafted species irrespective of premixing time. This indicates that even if the starting unreacted bis-pyrrolide complex Mo-1 might graft onto SiO2-700 faster than the pyrrolide ligand exchanges with ArOH, the latter ligand exchange can also proceed on the grafted Mo-1/SiO2-700 species. To confirm this, we have contacted well-characterized Mo-1/SiO2-700 material described previously15 with 2 equiv. of ArOH-2 or 13 and followed the reaction by in situ1H NMR spectroscopy. Quantifications of released 2,5-dimethylpyrrole in solution shows that with ArOH-2 or 13, the exchange proceeds quantitatively within 3 hours. However, the exchange reaction is accompanied by a partial de-grafting (7 and 14% for ArOH-2 or 13, respectively, Table S6†) as indicated by the alkylidene signal of bisaryloxide alkylidene species in solution. Thus, we conclude that the exchange of the 2,5-dimethylpyrrolide ligand for the aryloxide ligand also proceeds in the grafted Mo-1/SiO2-700 species, which leads to the target grafted aryloxide species in these in situ prepared formulations. Because the washing step implemented in the in situ grafting protocol removes soluble molecular alkylidene species, the measured catalytic activity discussed below is predominantly due to the grafted aryloxide surface species (Fig. 2A).
Comparison between formulations with Mo-1 and Mo-2 for TOFin or TON1 h reveals that in situ catalysts derived from the smaller 2,6-dimethylphenylimido ligand (Mo-2) exhibit significantly reduced TOFin and TON1 h (Fig. 2, S4 and S5†). This trend is consistent with our previous results on inferior activity of homogeneous formulations derived from Mo-2 in the self-metathesis of 1-nonene.8 Here, we observe that phenols without aryl groups in ortho positions (Fig. 2B, Group A) in general lead to lower activities (Fig. S4†). However, phenols with pendant aryls (Group B) yield similarly activities irrespective of the size of the imido moiety. In what follows, for brevity we concentrate the discussion on the results obtained with Mo-1.
In situ grafting of Mo-1 onto SiO2-700 leads to a formulation (Mo-1/SiO2-700, mono-siloxide pyrrolide species) featuring TOFin = 17.7 min−1 and TON1 h = 700, which is notable as unsupported Mo-1 is nearly inactive. Grafting of Mo-1 in the presence of phenols 1–35 gives formulations with TOFin and TON1 h values generally lower than those of Mo-1/SiO2-700 or respective molecular formulations. However, almost every grafted formulation reaches conversions exceeding 95% after 8 h with an average SC16 selectivity for all 35 ligands at 97% and 94% after 1 h and 8 h, respectively. Comparison of E/Z8 h, TOFin, TON1 h, and TON8 h between in situ prepared silica-supported and respective molecular formulations reveal that while the initial rate of the in situ grafted formulations is reduced relative to molecular formulations, the deactivation is generally retarded for grafted catalysts, as assessed by the narrow range of TON1 h around approximately 480 (Fig. S9†). This is presumably due to the suppression of bimolecular deactivation pathways for site-isolated grafted metathesis catalysts.31–34
TOFin and TON1 h values for formulations based on Mo-1 are correlated with R2 = 0.73, which suggests similar deactivation pathways/relative rates for most ligands (Fig. S7†). For all formulations, the (E/Z) ratios for the SC16 (E) isomer increase as the reaction progresses, approaching the thermodynamic ratio SC16 (E/Z)8 h = 5.25 (84:16 trans:cis product). No highly Z-selective catalyst formulations were formed using SiO2-700 as a support, in contrast to what was observed previously with molecular systems where ArOH-28, 33, 34 and 35 gave Z-selective formulations (Fig. S9†).8,36–40 The highest Z-selectivity of ca. 40% was found for the grafted formulation derived from Mo-1 and ArOH-5; this selectivity, however, was stable during the catalytic test.
Univariate modeling
In order to compare differences between the trends reported for homogeneous systems derived from Mo-1 and the grafted aryloxides, we used the same set of molecular descriptors of the phenolic ligands as in our previous study.8 We started by classifying the TON1 h catalytic data according to the nature of substituents on the phenol ligands to those without and with aryl groups in ortho positions of ArOH (Groups A and B in Fig. 2, respectively). This was accomplished using the Sterimol parameter Lsum, which quantifies steric volume along the axes of both ortho substitutes in aryloxide ligands.41 Parameter Lsum indicates different catalytic regimes for the respective subsets of ligands in the grafted formulations and is in line with our previous findings on respective molecular analogues (Fig. S8†).8Analysis of outliers and control experiments
Analysis of the univariate correlations also uncovered systematic outliers that were excluded for further analysis. As in our previous study, (2,6-NO2-4-CF3)-PhOH 15 yielded an inactive formulation, likely owing to protonation of the alkylidene.8 Phenols with an increasingly large size such as ArOH-14, ArOH-28 or ArOH-34, were identified as slow exchangers in our previous study,8 and provided materials with TOFin similar to that of the control catalyst, Mo-1/SiO2-700. This suggests that the exchange has likely proceeded only to a low extent, resulting in the same grafted species in Mo-1/SiO2-700 and with formulations containing phenols ArOH-14, 28, 34 (Fig. 2C and Table S1†).Formulations with low TOF are observed for phenols with ortho-methoxy substituents (ArOH-7 and 25), likely due to coordination of this group to Mo and blocking the olefin coordination site.8 With that said, TON8 h values reached by these formulations are similar to formulations with other tested phenols. This can be explained by the generally improved stability of grafted catalysts. Interestingly, three particular outliers were identified (Fig. S8†), involving the fluorine-bearing ligands 3,5-F-PhOH, 2-CF3-PhOH and 4-CF3-PhOH (ArOH-5, 8 and 9, respectively). Formulations with ArOH-5 show lower activity than other Group A ligands, whereas ArOH-8 and ArOH-9 display high activity, reminiscent of grafted Mo-1 without addition of an ArOH ligand. We speculate that fluorine interactions with the silica surface are at the origin of these observations. In particular, the exchange of the aryloxide ligand between Mo-1/SiO2-700 and ArOH-9 is hindered, proceeding to only 18% after 3 h according to in situ1H NMR experiment (Table S6†), and in contrast to what was observed for ArOH-2 or 13 discussed above. However, while a quantitative exchange is observed between Mo-1/SiO2-700 and ArOH-5, low activity of this formulation is likely due to the fluorine–silica interaction. As described above, ArOH-5 provides the most Z-selective catalyst among all tested formulations.
Correlations of ortho-isosteric ligands
To further examine the robustness of the acquired experimental data and the electronic impact of aryloxides, univariate inter-correlations within the data set were analyzed. Therefore, sub-classes of phenols bearing the same ortho substituents were selected following insight from our earlier work:8 phenols with the 2,6-dibromo and 2,6-diphenyl ligands (ArOH-16, 18, 19, 20 from Group A, and ArOH-21, 22, 23, 26, 27, 30, 32 from Group B). Analysis of 2,6-Br ligands (Fig. 3A) identifies a good correlation of TOFin and the dipole moment μ, with a commensurate increase in TOFin as the permanent charge separation within the ligand is enhanced. This is possibly related to σ-donor abilities of the ligand. In contrast, an excellent correlation is observed between TON1 h and the antibonding σ*(C–O) NBO energies (Eσ*(C–O), R2 = 0.92, Fig. 3A), marked by an increase in turnover number with increasing electron density on the pendant aryl substituents. This reflects the σ-donation due to inductive effects of the substituents on the phenol ring. Analogous analysis of the 2,6-Ph series of phenols reveals that increasing the polarizability enhances the rates and turnover numbers, as defined by a correlation between TOFin and TON1 h with Pol parameter (R2 = 0.88 and 0.66, respectively, Fig. 3B).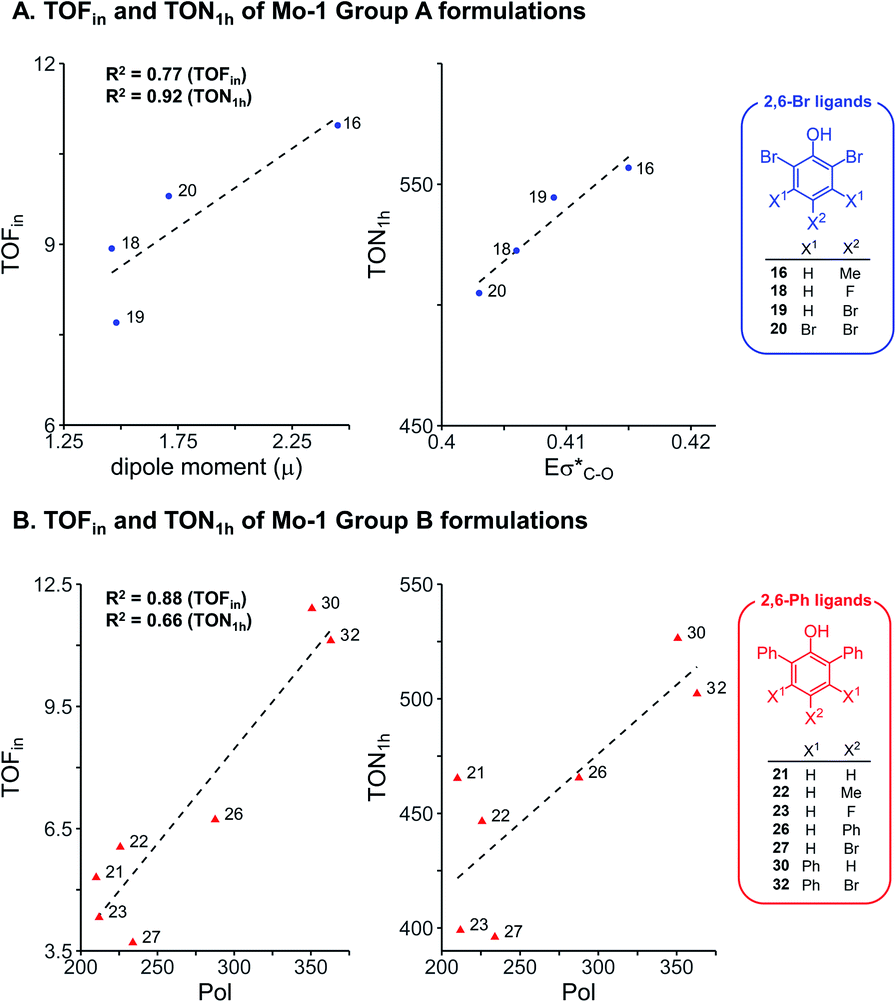 | ||
| Fig. 3 Univariate correlation of electronic descriptors for selected formulations using Mo-1 and ligands without (A) and with (B) ortho aryl substituents. | ||
Overall, this data suggests that electronic effects (as reflected in μ and Eσ*(C–O) parameters) are key factors for Group A ligands. Group B on the other hand, is impacted by the polarizability Pol, a descriptor with hybrid character expected for ligands of larger size, for which attractive interactions with the silica surface could potentially be important.42
Multivariate regression analysis
The assessment of cooperative effects on the catalytic performance was investigated though multivariate linear regression analysis on the TOFin and TON1 h responses for Group A and B (Fig. 4, Table S7 and 8†). The consistency of the models is probed with internal-validation techniques (leave-one-out (LOO) and k-fold methods), yielding good scores for all cases consistent with a well-validated model.7–9 The trained models from normalized descriptors gave coefficients that revealed the significance of each of represented effects.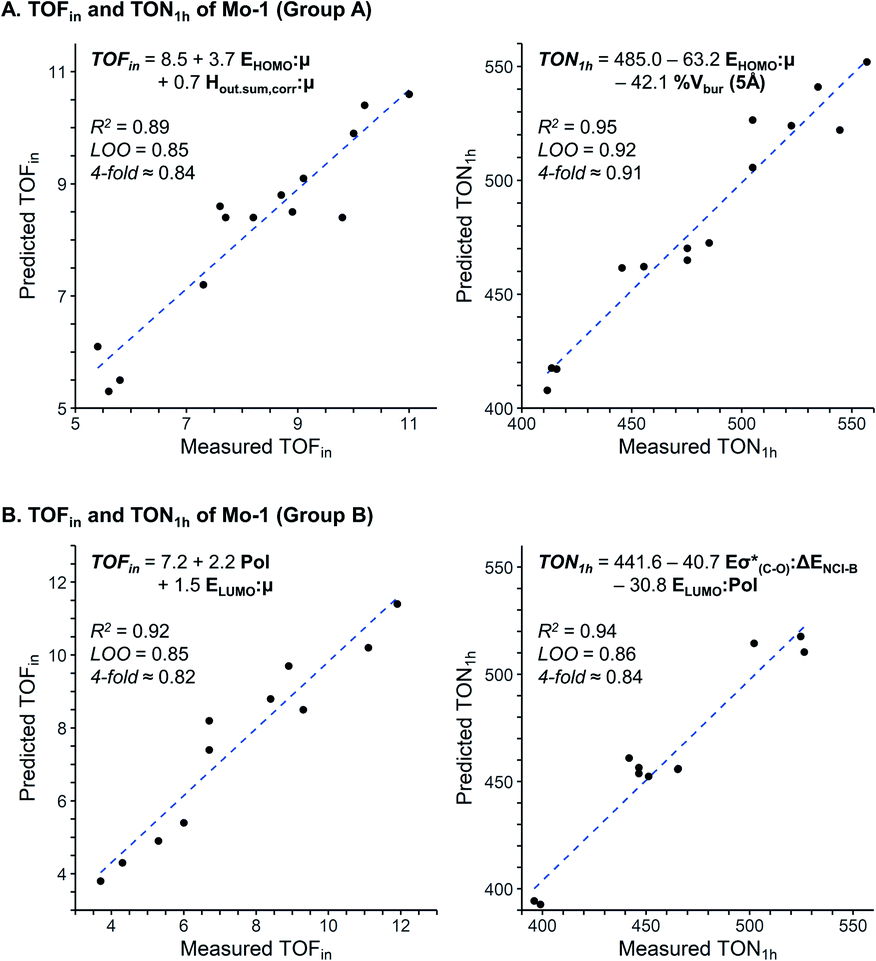 | ||
| Fig. 4 Multivariate linear regression model to predict TOFin and TON1 h for Group A (A) and Group B (B). | ||
The multivariate models obtained for the Group A feature steric and electronic hybrid interactions terms for TOFin and TON1 h (Fig. 4A and B). Consistent with our previous results on molecular catalysts,8 the electronic effect is dominant for both TOFin and TON1 h. The first descriptor, that includes μ and HOMOphenol, has the highest significance in both models. This interaction term describes the σ-donor ability of the aryloxy ligand; it is expected that stronger σ-donor ligands (less negative interaction term) increase the activity of the complex by increasing the electronic dissymmetry at the metal centre and their higher trans-influence with respect to the weaker σ-donating surface siloxy ligand.43–45 The hybrid stereoelectronic descriptor used for modeling TOFin, an interaction term of μ and Hout,sum,corr, reflects the perturbation of the permanent dipole by the electron density of the pendant substituents on the phenolic ligands. This steric descriptor captures the increase in catalytic activity by the repulsive NCI exerted by ortho pendant substituents. Notably, the steric effects gain in importance with increasing reaction times as indicated by the inclusion of % Vbur (5 Å) in the TON1 h model.
Consequently, statistical modeling was also employed for Group B. The correlations found for the TOFin and TON1 h responses showcase a strong significance of the polarizability Pol of the ligands, as indicated by the large coefficient (Fig. 4B). The polarizability could possibly be an effect of the silica surface (attractive interaction of the surface with aryl moiety).46 In the model for TOFin, the polarizability appears as a single term, accompanied by an interaction term (LUMOphenol and μ), that can be viewed again as the σ-donor ability of the phenol oxygen. This is in line with the previous empirical observation that stronger σ-donors produced higher catalytic activity, as illustrated by the electron-rich 2,6-Ph ligands of Group B (Fig. 3).
Evaluation of the TON1 h response shows that the polarizability appears in an interaction term (Pol and LUMOphenol), together with a second stereoelectronic descriptor (Eσ*(C–O) and ΔENCI-B). The LUMO and Eσ*(C–O) essentially describe related phenomena due to the perturbation of the electron density by pendant substituents with varying electronic properties. The non-covalent interaction term ΔENCI-B not only modulates the decrease of activity with the increase of size of ligand, but in concert with the polarizability, highlights the importance of dispersive forces in enhancing catalytic performance.
Conclusions
In summary, we developed a practical protocol based on high-throughput experimentation combined with surface organometallic chemistry that allows generating and testing the catalytic performance of large libraries of in situ grafted Mo imido alkylidene metathesis catalyst, based on 35 phenol ligands and two precursor Mo bis-pyrrolido alkylidene complexes. Control experiments indicated that grafted aryloxide Mo alkylidene surface species were formed. Using statistical data analysis, we identified σ-donation ability of the ligands and dispersive forces to be essential in promoting catalytic activity. Univariate modeling allowed distinguishing two groups of phenoxy ligands, either without aryl arms in ortho position (Group A) or with aryl arms in ortho position (Group B). This finding on supported catalysts is reminiscent of what is observed in the corresponding libraries of molecular catalysts, indicating that catalysts prepared by SOMC retained a molecular character. In comparison to their molecular counterparts, all grafted catalysts display lower initial rates (as evaluated by TOFin), but higher stability as seen by a narrow range of TON1 h approximately 480. After 8 h of reaction, 44 from 70 grafted catalyst formulations reached conversion of 1-nonene exceeding 90% (as opposed to only 31 formulations for molecular in situ prepared formulations). This is likely due to higher stability of grafted species, as site-isolated metathesis catalysts do not suffer from bimolecular deactivation pathways.47 Overall the initial rates for both groups are dominated by the σ-donor ability of the aryloxy ligands, supporting the view that electronic dissymmetry at the metal centre improves the activity of Schrock-type metathesis catalysts by facilitating coordination of the olefin substrate as well as the retrocyclization. However, as reaction times increase, opposite trends in the catalytic performance (as evaluated by TON1 h) arise with an increase of the ortho pendant substituent size. While for Group A the increase in steric bulk (described by % Vbur (5 Å)) is associated with lower turnover numbers, Group B displays an increase in catalytic performance with increasing steric bulk, likely owing to non-covalent interactions of the aryl moieties with the silica surface. This work showcases how molecular aspects of heterogeneous catalysts prepared via an HTE-SOMC approach can be evaluated via statistical methods. We confirmed that grafting enhances the stability of metathesis catalysts and corroborated that promoting electronic dissymmetry at the metal centre by modulating the σ-donor ability of the aryloxy ligands increases activity. Furthermore, we highlighted the importance of dispersive interaction of ligands, aryl alcohols or silica support, in enhancing the catalyst activity in grafted d0 metathesis catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Scientific Equipment Program of ETH Zürich and the SNSF (R'Equip grant 206021_150709/1) for financial support of the high throughput catalyst screening facility (HTE@ETH). M. A. B. F. thanks Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, 17/13306-1, 15/08541-6, and 14/50249-8) and GSK for financial support. J. D. J. S. was supported by the National Research Fund, Luxembourg (AFR Individual PhD Grant 12516655). M. S. S. thanks the National Science Foundation (CHE-1763436) for funding. The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged. The authors thank XiMo Hungary Ltd for donation of Mo-1,2 and selected phenol ligands.Notes and references
- R. H. Grubbs, Handbook of Metathesis, Wiley-VCH, Weinheim, 2014 Search PubMed.
- C. S. Higman, J. A. M. Lummiss and D. E. Fogg, Angew. Chem., Int. Ed., 2016, 55, 3552 CrossRef CAS PubMed.
- E. S. Sattely, S. J. Meek, S. J. Malcolmson, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 943–953 CrossRef CAS PubMed.
- C. Copéret, F. Allouche, K. W. Chan, M. P. D. Conley, F. Murielle, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398 CrossRef PubMed.
- A. H. Hoveyda, J. Org. Chem., 2014, 79, 4763 CrossRef CAS PubMed.
- M. J. Benedikter, F. Ziegler, J. Groos, P. M. Hauser, R. Schowner and M. R. Buchmeiser, Coord. Chem. Rev., 2020, 415, 213315 CrossRef CAS.
- P. S. Engl, C. B. Santiago, C. P. Gordon, W. C. Liao, A. Fedorov, C. Copeŕet, M. S. Sigman and A. Togni, J. Am. Chem. Soc., 2017, 139, 13117 CrossRef CAS PubMed.
- M. A. B. Ferreira, J. De Jesus Silva, S. Grosslight, A. Fedorov, M. S. Sigman and C. Copéret, J. Am. Chem. Soc., 2019, 141, 10788–10800 CrossRef CAS.
- V. Mougel, C. B. Santiago, P. A. Zhizhko, E. N. Bess, J. Varga, G. Frater, M. S. Sigman and C. Copeŕet, J. Am. Chem. Soc., 2015, 137, 6699 CrossRef CAS PubMed.
- J. Y. Guo, Y. Minko, C. B. Santiago and M. S. Sigman, ACS Catal., 2017, 7, 4144 CrossRef CAS.
- C. B. Santiago, J. Y. Guo and M. S. Sigman, Chem. Sci., 2018, 9, 2398 RSC.
- S. T. Alvaro Gordillo, C. Futter, M. L. Lejkowski, E. Prasetyo, T. Luis, A. Rupflin, T. Emmert and S. A. Schunk, High-Throughput Experimentation in Catalysis and Materials Science, Wiley-VCH, Weinheim, 2014 Search PubMed.
- R. Arancon, M. Saab, A. Morvan, A. Bonduelle-Skrzypczak, A.-L. Taleb, A.-S. Gay, C. Legens, O. Ersen, K. Searles, V. Mougel, A. Fedorov, C. Copéret and P. Raybaud, J. Phys. Chem. C, 2019, 123, 24659–24669 CrossRef CAS.
- A. Fedorov, H.-J. Liu, H.-K. Lo and C. Copéret, J. Am. Chem. Soc., 2016, 138, 16502–16507 CrossRef CAS PubMed.
- P. A. Zhizhko, V. Mougel, J. De Jesus Silva and C. Copéret, Helv. Chim. Acta, 2018, 101, e1700302 CrossRef.
- S. Monfette, J. M. Blacquiere and D. E. Fogg, Organometallics, 2011, 30, 36–42 CrossRef CAS.
- P. Wyrębek, P. Małecki, A. Sytniczuk, W. Kośnik, A. Gawin, J. Kostrzewa, A. Kajetanowicz and K. Grela, ACS Omega, 2018, 3, 18481–18488 CrossRef PubMed.
- F. D. Toste, M. S. Sigman and S. J. Miller, Acc. Chem. Res., 2017, 50, 609 CrossRef CAS PubMed.
- C. B. Santiago, A. Milo and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 13424 CrossRef CAS PubMed.
- C. A. Hollingsworth, P. G. Seybold and C. M. Hadad, Int. J. Quantum Chem., 2002, 90, 1396 CrossRef CAS.
- A. Verloop and E. J. Ariens, Drug Design, 1976 Search PubMed.
- A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322 CrossRef CAS.
- M. Orlandi, J. A. S. Coelho, M. J. Hilton, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 6803 CrossRef CAS PubMed.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637 CrossRef CAS PubMed.
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274 CrossRef CAS PubMed.
- M. Stoyanova, U. Rodemerck, U. Bentrup, U. Dingerdissen, D. Linke, R. W. Mayer, H. G. J. Lansink Rotgerink and T. Tacke, Appl. Catal., A, 2008, 340, 242–249 CrossRef CAS.
- D. R. Romer, V. J. Sussman, K. Burdett, Y. Chen and K. J. Miller, ACS Comb. Sci., 2014, 16, 551–557 CrossRef CAS PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- N. Rendón, F. Blanc and C. Copéret, Coord. Chem. Rev., 2009, 253, 2015–2020 CrossRef.
- C. Copéret, Dalton Trans., 2007, 5498–5504 RSC.
- J. Robbins, G. C. Bazan, J. S. Murdzek, M. B. O'Regan and R. R. Schrock, Organometallics, 1991, 10, 2902–2907 CrossRef CAS.
- L. P. H. Lopez and R. R. Schrock, J. Am. Chem. Soc., 2004, 126, 9526–9527 CrossRef CAS PubMed.
- A. Sinha, L. P. H. Lopez, R. R. Schrock, A. S. Hock and P. Müller, Organometallics, 2006, 25, 1412–1423 CrossRef CAS.
- L. P. H. Lopez, R. R. Schrock and P. Müller, Organometallics, 2006, 25, 1978–1986 CrossRef CAS.
- N. Rendón, R. Berthoud, F. Blanc, D. Gajan, T. Maishal, J.-M. Basset, C. Copéret, A. Lesage, L. Emsley, S. C. Marinescu, R. Singh and R. R. Schrock, Chem.–Eur. J., 2009, 15, 5083–5089 CrossRef PubMed.
- M. J. Koh, T. T. Nguyen, J. K. Lam, S. Torker, J. Hyvl, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 542, 80 CrossRef CAS PubMed.
- S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 471, 461–466 CrossRef CAS.
- M. M. Flook, L. C. H. Gerber, G. T. Debelouchina and R. R. Schrock, Macromolecules, 2010, 43, 7515–7522 CrossRef CAS PubMed.
- A. J. Jiang, Y. Zhao, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 16630 CrossRef CAS PubMed.
- M. M. Flook, A. J. Jiang, R. R. Schrock, P. Müller and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7962–7963 CrossRef CAS PubMed.
- A. V. Brethomé, S. P. Fletcher and R. S. Paton, ACS Catal., 2019, 9, 2313–2323 CrossRef.
- F. Blanc, J.-M. Basset, C. Copéret, A. Sinha, Z. J. Tonzetich, R. R. Schrock, X. Solans-Monfort, E. Clot, O. Eisenstein, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2008, 130, 5886–5900 CrossRef CAS PubMed.
- V. Mougel and C. Copéret, Chem. Sci., 2014, 5, 2475 RSC.
- A. Poater, X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, J. Am. Chem. Soc., 2007, 129, 8207–8216 CrossRef CAS PubMed.
- X. Solans-Monfort, C. Copeŕet and O. Eisenstein, Organometallics, 2012, 31, 6812 CrossRef CAS.
- B. Rhers, A. Salameh, A. Baudouin, E. A. Quadrelli, M. Taoufik, C. Copéret, F. Lefebvre, J.-M. Basset, X. Solans-Monfort, O. Eisenstein, W. W. Lukens, L. P. H. Lopez, A. Sinha and R. R. Schrock, Organometallics, 2006, 25, 3554–3557 CrossRef CAS.
- F. Blanc, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage, L. Emsley, A. Sinha and R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 1216–1220 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02594a |
| This journal is © The Royal Society of Chemistry 2020 |