
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCellular uptake and targeting of low dispersity, dual emissive, segmented block copolymer nanofibers†
Steven T. G.
Street‡§
 ab,
Yunxiang
He‡
a,
Xu-Hui
Jin
ac,
Lorna
Hodgson
d,
Paul
Verkade
d and
Ian
Manners
*ab
ab,
Yunxiang
He‡
a,
Xu-Hui
Jin
ac,
Lorna
Hodgson
d,
Paul
Verkade
d and
Ian
Manners
*ab
aSchool of Chemistry, University of Bristol, Bristol BS8 1TS, UK
bDepartment of Chemistry, University of Victoria, Victoria, BC V8W 3V6, Canada. E-mail: imanners@uvic.ca
cSchool of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing, China
dSchool of Biochemistry, University of Bristol, Bristol BS8 1TD, UK
First published on 8th July 2020
Abstract
Polymer-based nanoparticles show substantial promise in the treatment and diagnosis of cancer and other diseases. Herein we report an exploration of the cellular uptake of tailored, low dispersity segmented 1D nanoparticles which were prepared from an amphiphilic block copolymer, poly(dihexylfluorene)-b-poly(ethyleneglycol) (PDHF13-b-PEG227), with a crystallizable PDHF core-forming block and a ‘stealth’ PEG corona-forming block with different end-group functionalities. Segmented C–B–A–B–C pentablock 1D nanofibers with varied spatially-defined coronal chemistries and a selected length (95 nm) were prepared using the living crystallization-driven self-assembly (CDSA) seeded-growth method. As the blue fluorescence of PDHF is often subject to environment-related quenching, a far-red BODIPY (BD) fluorophore was attached to the PEG end-group of the coronal B segments to provide additional tracking capability. Folic acid (FA) was also incorporated as a targeting group in the terminal C segments. These dual-emissive pentablock nanofibers exhibited uptake into >97% of folate receptor positive HeLa cells by flow cytometry. In the absence of FA, no significant uptake was detected and nanofibers with either FA or BD coronal groups showed no significant toxicity. Correlative light and electron microscopy (CLEM) studies revealed receptor-mediated endocytosis as an uptake pathway, with subsequent localization to the perinuclear region. A significant proportion of the nanofibers also appeared to interact with the cell membrane in an end-on fashion, which was coupled with fluorescence quenching of the PDHF core. These results provide new insights into the cellular uptake of polymer-based nanofibers and suggest their potential use in targeted therapies and diagnostics.
Introduction
Nanoparticle-mediated therapeutics show considerable promise in the diagnosis and treatment of a plethora of diseases that affect human health, especially cancer.1,2 The delivery of cargo such as drugs, proteins, imaging agents and nucleic acids to specific locations inside cells in target tissue in the human body however remains a major challenge,3 despite their considerable potential. The ideal nanoparticle delivery system therefore has several requirements such as biocompatibility,4 high specificity,5 and a high loading capacity,6 maximizing efficacy whilst remaining as cost effective as possible. In practical terms, this means maintaining a modular, versatile design whilst simultaneously exhibiting precise control over nanoparticle size, shape, rigidity, and surface chemistry.7–11 To this end, 1D nanomaterials have attracted substantial recent attention, with a wide range of potential advantages evidenced over considerably more well-studied spherical systems such as improved circulation,12 retention,13,14 adhesion,15,16 specificity,17 and cell uptake.18 The 1D shape has also been shown to enable unique endocytosis mechanisms19 involving improved membrane wrapping20 and reduced macrophage uptake (which is length dependent)21,22 thereby offering the promise of enhanced active targeting capabilities.One of the most well-studied active targeting agents is Folic Acid (FA), with several FA conjugates in clinical trials.23 FA is the substrate for folate receptors such as FRα, which are overexpressed in numerous types of cancer, and have represented a significant target for the delivery of tailored therapeutics.23–27 A variety of anisotropic nanoparticles has been functionalized with folic acid, such as polyacrylic acid-b-polystyrene spherical and cylindrical micelles,28 gold nanorods,29 and coordination-complex nanotubes,30 with anisotropic particles displaying features such as increased uptake over spheres,28 and disassembly upon cellular internalization.30 Other polymeric systems that have used FA as a targeting agent include spherical micelles and star-shaped polymers based on PLA31,32 and spherical micelles based on PCL.33,34
Recently, a seeded-growth approach termed ‘living’ crystallization-driven self-assembly (CDSA), has been developed which allows access to a wide range of morphologically pure, low dispersity 1D (and also 2D) core–shell nanomaterials.35–58 Briefly, amphiphilic block copolymers (‘unimers’) with a crystallizable core-forming block are dissolved in a ‘common’ solvent which is compatible with both blocks and the resulting solution is then mixed with a ‘selective’ solvent which solvates only one block (the corona-forming block). These conditions yield polydisperse fiber-like micelles with an insoluble, solvophobic crystalline-core and a solubilizing solvophilic corona, via a self-nucleation mechanism. Sonication of the resulting polydisperse fiber-like micelles causes fragmentation, yielding small ‘seed’ micelles. Further addition of unimer to the seed micelles leads to epitaxial growth and low dispersity micelles with a length controlled by the unimer to seed ratio in a manner analogous to a living covalent polymerization of molecular monomers.37 This process is uniquely suited towards the generation of nanoparticles that are otherwise hard to access, such as uniform 1D nanofibers,38–44 (and also 2D platelets)45,46,48 as well as more complex assemblies.50 For example, ‘living’ CDSA can also be used to generate hierarchical nanomaterials, such as segmented nanofibers with spatially-defined functionalizable regions,36,38,39,49–52 as well as random- and gradient co-micelles by the sequential or simultaneous seeded growth of different block copolymers with distinct coronal chemistries.52 Significantly, the ability to tailor surface chemistry in individual regions allows for modular functionalization.53,54
Despite the substantial recent progress made with self-assembled nanomaterials formed via ‘living’ CDSA, the majority of systems described so far have involved the use of organic rather than aqueous media, limiting their potential for biomedical applications. Only a few examples exist of the use of ‘living’ CDSA to prepare low dispersity fibers of controlled length which can be dispersed in water. Several are based on a crystallizable polyferrocenylsilane (PFS) core.38,42 The Dove and O′Reilly groups have reported the first example of ‘living’ CDSA in aqueous media based on a biocompatible and biodegradable polycaprolactone (PCL) core-forming block, thereby accessing fibers with lengths of up to 800 nm.44 In addition, we recently reported the ‘living’ CDSA of a biocompatible polycarbonate core-forming block, with morphologically pure 1D block co-micelles accessed with lengths up to ∼1.6 μm and which are colloidally stable in aqueous media.39 Despite these advances, the application of functional nanoparticles produced via CDSA to biomedicine remains a nascent field, requiring much further development in areas such as scalability, incorporation of functionality, and biological activity.
Block copolymers with a crystallizable π-conjugated core have also been shown to undergo ‘living’ CDSA.55–58 One such class of π-conjugated materials is polyfluorenes (PFs),59 which exhibit strong luminescence, making them excellent candidates for chemo/biosensors, diagnostics and imaging agents.60–62 Whilst most work has focused on optoelectronic properties, some studies have explored biological applications.63–66 The majority of studies have focused on the use of a conjugated PF backbone, with charged side chains that provide aqueous stability. The resultant polymers self-assemble into spherical nanoparticles, with FA either covalently linked to the polymer, leading to selective cell uptake67 or electrostatically bound, leading to FA dependent fluorescence quenching of PF.68 Numerous examples also exist of PF containing cationic π-conjugated polymers for nucleic acid binding and the detection of pathogens65 whilst π-conjugated polythiophenes have also been used for the delivery of nucleic acids to cells.69
Associated with the limited development of 1D fiber-like micelles prepared by ‘living’ CDSA that are dispersible in aqueous media,38,39,42,44 a paucity of biological data currently exists, with currently available studies largely limited to cytotoxicity experiments. Whilst polydisperse worm-like micelles with amorphous cores have been investigated,70 fundamental questions remain regarding the in vivo and in vitro effects of low dispersity polymer nanofibers in which the core is crystalline and more rigid. It is noteworthy that, to date, there have only been limited studies on the effect of 1D fiber length on cell uptake,7–11,18,71–73 with no reports on the behavior of polymer-based nanofibers. Furthermore, there are very few examples of easily functionalizable fiber-like micelles that are dispersible in water. Fiber-like micelles with a π-conjugated PF core offer bright fluorescence of potential interest for imaging, tracking nanoparticles inside cells, and sensing. Herein we describe the preparation of length-controlled, low dispersity 1D PDHF-b-PEG (PDHF = poly(di(n-hexyl)fluorene), PEG = poly(ethyleneglycol)) nanofibers in aqueous media, and studies of their functionalization, cytotoxicity, cellular targeting, uptake, and localization.
Results
Preparation of colloidally stable dual emissive PDHF triblock and pentablock nanofibers in water
The PF block copolymer PDHF-b-PEG was selected in cellular uptake studies because PEG is known as a ‘stealth’ polymer and can provide biocompatibility as well as aqueous colloidal stability. First, alkyne-terminated PDHF13 homopolymer was prepared (Mn = 4400 g mol−1, DP = 13, determined via MALDI-TOF MS, ĐM = 1.22, determined by GPC, Fig. S1†) via Grignard metathesis (GRIM) polymerization using a previously described procedure (Scheme S1†).74 Heterobifunctional PEG was synthesized by mono-tosylation of HO–PEG249–OH (Fig. S2†) and, after substitution of the tosylate for azide, the resulting mixture of HO–PEG249–OH and HO–PEG249–N3 was used without further purification in the Huisgen 1,3-dipolar cycloaddition ‘click’ coupling with the alkyne-terminated PDHF13 according to the previously reported method.74 Excess HO–PEG249–OH was removed via precipitation to yield PDHF13-b-PEG227 (Fig. 1A, Mn = 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1, ĐM = 1.12 determined by GPC, block ratio determined by 1H-NMR, Fig. S3 and S4†). While the π-conjugated PDHF core of the PDHF13-b-PEG227 micelles is inherently fluorescent, in biological systems the blue emission is subject to fluorescence quenching upon interaction with a variety of species,68,75 and will also compete with background cell autofluorescence. Thus, to supplement the results from PDHF fluorescence, we also introduced a far-red fluorophore to allow for an additional tracking capability. The far-red BODIPY630/650-X (BD) fluorophore (λex = 630 nm, λem = 650 nm, excitation/emission in superscript) was selected to attach to the PEG terminus (Fig. 1A). This was achieved by using the terminal hydroxyl group of the PEG block for further chemical functionalization (Scheme S1†). Condensation of PDHF13-b-PEG227 with Boc-β-alanine, followed by Boc deprotection yielded a terminal amine residue, which was then further modified with the BODIPY630/650-XN-hydroxysuccinimide (NHS) ester to yield an end-functionalized, dual-emissive PDHF13-b-PEG227 polymer, termed PDHF13-b-PEG227–BD (Fig. 1A). The ability to functionalize the corona chain end with an amine also allowed us to employ amide coupling chemistry to attach targeting moieties for the active uptake of nanofibers into a target environment or cell type via receptor-mediated endocytosis. To this end, we first selected FA as our targeting group of choice as it is well established that folate receptors are over-expressed in many different types of cancer, with several treatments involving folate targeting undergoing clinical evaluation such as vintafolide.23 Thus, we adapted the chemistry developed for attaching the BD dye to the PEG chain terminus, instead attaching an N-hydroxysuccinimide activated FA derivative (Scheme S1†). This yielded FA functionalized PDHF13-b-PEG227, termed PDHF13-b-PEG227–FA (Fig. 1A).
900 g mol−1, ĐM = 1.12 determined by GPC, block ratio determined by 1H-NMR, Fig. S3 and S4†). While the π-conjugated PDHF core of the PDHF13-b-PEG227 micelles is inherently fluorescent, in biological systems the blue emission is subject to fluorescence quenching upon interaction with a variety of species,68,75 and will also compete with background cell autofluorescence. Thus, to supplement the results from PDHF fluorescence, we also introduced a far-red fluorophore to allow for an additional tracking capability. The far-red BODIPY630/650-X (BD) fluorophore (λex = 630 nm, λem = 650 nm, excitation/emission in superscript) was selected to attach to the PEG terminus (Fig. 1A). This was achieved by using the terminal hydroxyl group of the PEG block for further chemical functionalization (Scheme S1†). Condensation of PDHF13-b-PEG227 with Boc-β-alanine, followed by Boc deprotection yielded a terminal amine residue, which was then further modified with the BODIPY630/650-XN-hydroxysuccinimide (NHS) ester to yield an end-functionalized, dual-emissive PDHF13-b-PEG227 polymer, termed PDHF13-b-PEG227–BD (Fig. 1A). The ability to functionalize the corona chain end with an amine also allowed us to employ amide coupling chemistry to attach targeting moieties for the active uptake of nanofibers into a target environment or cell type via receptor-mediated endocytosis. To this end, we first selected FA as our targeting group of choice as it is well established that folate receptors are over-expressed in many different types of cancer, with several treatments involving folate targeting undergoing clinical evaluation such as vintafolide.23 Thus, we adapted the chemistry developed for attaching the BD dye to the PEG chain terminus, instead attaching an N-hydroxysuccinimide activated FA derivative (Scheme S1†). This yielded FA functionalized PDHF13-b-PEG227, termed PDHF13-b-PEG227–FA (Fig. 1A).
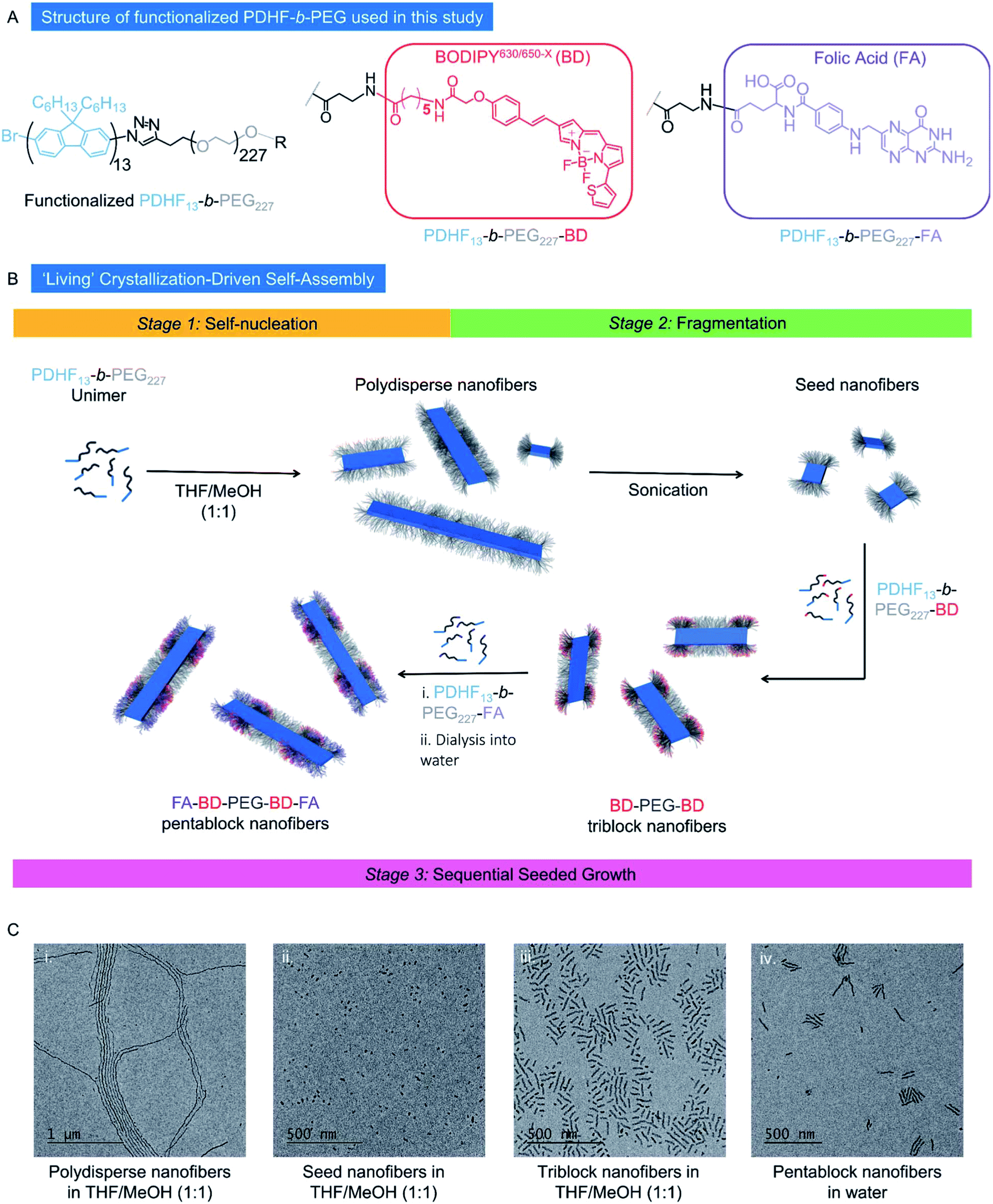 | ||
| Fig. 1 (A) Structure of functionalized PDHF-b-PEG diblock copolymers used in this study. (B) Schematic representation of the preparation of low dispersity PDHF13-b-PEG227 nanofibers produced through ‘living’ CDSA. (C) Transmission electron microscopy (TEM) images of (i) polydisperse nanofibers. (ii) Seed nanofibers (Ln: 21 nm, Lw/Ln: 1.07, σL = 8 nm). (iii) Triblock nanofibers in MeOH/THF (1:1, Ln = 56 nm, Lw/Ln = 1.09, σL = 18 nm), and (iv) pentablock nanofibers in H2O (Ln = 95 nm, Lw/Ln = 1.17, σL = 39 nm, Wn = 13 nm, Ww/Wn = 1.02, σW = 2 nm). Samples were prepared at 0.5 mg mL−1. | ||
According to previous studies, dimensions of ca. 10–100 nm represent the most desirable size range for nanoparticles to be used as drug delivery vectors, as this leads to optimum circulation in the bloodstream. Objects within this size regime are sufficiently large to avoid renal and lymphatic clearance, yet sufficiently small to avoid opsonization.3,76 Therefore, we aimed to prepare low dispersity 1D fiber-like micelles of PDHF-b-PEG with lengths of ≤ca. 100 nm in this study. By comparison, assuming a chain-extended structure for the PDHF segment as previously found,74 the core cross-section of the PDHF14-b-PEG227 1D fibers is 65 nm2 (Wn × Hn = 13 × 5 nm, where Wn and Hn are the number average width and height respectively). The ability to prepare segmented nanofibers via living CDSA should also yield advantages for the optimal presentation of targeting groups (such as FA) and cargo (such as BODIPY630/650-X) as well as facilitating modular nanoparticle construction. To avoid complications with the self-assembly process, unfunctionalized PDHF13-b-PEG227 was used to form the initial seed micelles, after which the PDHF13-b-PEG227–BD and/or PDHF13-b-PEG227–FA unimers were added sequentially to create segmented nanofibers. To facilitate optimum cellular uptake, the segmented nanofibers were designed to possess terminal PDHF13-b-PEG227–FA blocks, as previous work has revealed that receptor mediated endocytosis of 1D nanomaterials occurs primarily through association of the nanoparticle tip with the cell membrane.77
The nanofibers were prepared via the seeded-growth method (Fig. 1B). Briefly, polydisperse PDHF13-b-PEG227 nanofibers were formed by the addition of MeOH to a solution of PDHF13-b-PEG227 in THF and aged for 24 h (Fig. S5A†). Seed nanofibers (Ln = 21 nm, Lw/Ln = 1.07, σ = 8 nm) were prepared by sonication of the resultant polydisperse nanofibers for 3 h at 22 °C in MeOH/THF (1:1, Fig. S5B and C†). Addition of PDHF13-b-PEG227–BD unimer to the seed nanofibers in MeOH/THF yielded intermediate (PDHF13-b-PEG227–BD)-m-(PDHF13-b-PEG227–OH)-m-(PDHF13-b-PEG227–BD) dual-emissive B–A–B triblock nanofibers (Ln = 56 nm, Lw/Ln = 1.09, σ = 18 nm, Fig. 1C and S6A†). Further addition of PDHF13-b-PEG227–FA unimer to the triblock nanofibers yielded (PDHF13-b-PEG227–FA)-m-(PDHF13-b-PEG227–BD)-m-(PDHF13-b-PEG227–OH)-m-(PDHF13-b-PEG227–BD)-m-(PDHF13-b-PEG227–FA) C–B–A–B–C pentablock nanofibers (Ln = 117 nm, Lw/Ln = 1.05, σ = 25 nm, termed FA–BD–PEG–BD–FA pentablock nanofibers), bearing both fluorescence and active targeting capabilities (Fig. 1C and S6B†). The FA–BD–PEG–BD–FA pentablock nanofibers consisted of a 21 nm central segment with a PEG corona derived from the seed, 18 nm inner BD functionalized blocks, and 31 nm outer FA functionalized blocks, and were transferred into aqueous media for future experiments by dialysis from MeOH/THF (1:1), with a very small amount of fragmentation observed based on the slight increase in dispersity and reduction in average length as measured by TEM (Ln = 95 nm, Lw/Ln = 1.17, σ of length (σL) = 39 nm, Fig. S6C–E,†Wn = 13 nm, Ww/Wn = 1.02, σ of width (σW) = 2 nm, Fig. S7A†).
To assess the effects of both FA and BD on cell uptake, triblock comicelles containing solely BD or FA functionalization were required as controls. To ensure that results were comparable to those obtained for pentablock nanofibers with both BD and FA decorated segments, and to ensure that nanofiber length was not a variable affecting results, we aimed to produce nanofibers with lengths comparable to the FA–BD–PEG–BD–FA pentablock nanofibers prepared previously. Addition of PDHF13-b-PEG227–BD unimer to seed nanofibers (Ln: 21 nm, Lw/Ln: 1.07, σL = 8 nm) yielded (PDHF13-b-PEG227–BD)-m-(PDHF13-b-PEG227–OH)-m-(PDHF13-b-PEG227–BD) dual-emissive B–A–B triblock co-micelles termed BD–PEG–BD triblock nanofibers (Ln = 113 nm, Lw/Ln = 1.10, σL = 36 nm, Fig. 2A and B and S8A†). BD–PEG–BD triblock nanofibers were transferred into aqueous media by dialysis from MeOH/THF (1:1), with a very small amount of fragmentation observed (Ln = 85 nm, Lw/Ln = 1.19, σL = 38 nm, Wn = 11 nm, Ww/Wn = 1.02, σW = 2 nm, Fig. 2C, S7B and S8B and C†). In a similar manner, (PDHF13-b-PEG227–FA)-m-(PDHF13-b-PEG227–OH)-m-(PDHF13-b-PEG227–FA) triblock co-micelles (termed FA–PEG–FA triblock nanofibers) were prepared from PDHF13-b-PEG227 seed micelles (Ln = 42 nm, Lw/Ln = 1.07, σL = 12 nm, Fig. S9A†), with an average length of 105 nm (Lw/Ln = 1.05, σL = 24 nm) before dialysis in THF/MeOH (1:1, Fig. 2D and E) and an average length of 90 nm (Lw/Ln = 1.11, σL = 30 nm, Wn = 12 nm, Ww/Wn = 1.02, σW = 2 nm) after dialysis into water (Fig. 2F, S7C and S9B–E†).
 | ||
| Fig. 2 (A) Schematic representation and (B and C) TEM micrographs of BD–PEG–BD triblock nanofibers in (B) THF/MeOH (1:1, Ln = 113 nm, Lw/Ln = 1.10, σL = 36 nm) and (C) water (Ln = 85 nm, Lw/Ln = 1.19, σL = 38 nm, Wn = 11 nm, Ww/Wn = 1.02, σW = 2 nm). (D) Schematic representation and (E and F) TEM micrographs of FA–PEG–FA triblock nanofibers in (E) THF/MeOH (1:1, Ln = 105 nm, Lw/Ln = 1.05, σL = 12 nm) and (F) water (Ln = 90 nm, Lw/Ln = 1.11, σL = 30 nm, Wn = 12 nm, Ww/Wn = 1.02, σW = 2 nm). Samples were prepared at 0.5 mg mL−1. | ||
UV/vis absorption and fluorescence profiles of PDHF13-b-PEG227 nanofibers
The absorption and fluorescence profiles of BD–PEG–BD and FA–PEG–FA triblock nanofibers, and FA–BD–PEG–BD–FA pentablock nanofibers in water and PBS were investigated prior to cellular experiments (Fig. S10–S14†). The absorbance and fluorescence excitation profile for all nanofibers exhibited a λmax of 375 nm, closely matching previously reported spectra for PDHF in organic solvents.74,78 The emission profiles of BD–PEG–BD triblock nanofibers and FA–BD–PEG–BD–FA pentablock nanofibers both exhibited a peak at 650 nm, which corresponds to the BD dye (Fig. S10B and C†). The excitation profile for the emission of BD at 650 nm matched the excitation of PDHF, indicative of Förster resonance energy transfer (FRET) between the π-conjugated PDHF core and the BD dye. As FRET interactions are very sensitive to distance,79 the results indicate that the BD dye is in close proximity (within ca. 10 nm) to the PDHF core in water and is presumably located near the core–corona interface. Confocal Laser Scanning Microscopy (CLSM) and fluorescence measurements in PBS and cell media revealed that both PDHF and BD can be tracked in complex media for use in cell uptake studies (Fig. 3, S13 and S14†). | ||
| Fig. 3 CLSM images of the dual-emissive FA–BD–PEG–BD–FA pentablock nanofibers (100 μg mL−1) in MEM cellular medium with 10% FBS. (A) Blue channel from PDHF core (λex = 405 nm, λem = 415–478 nm); (B) red channel from BD fluorescence (λex = 633 nm, λem = 640–700 nm); and (C) overlay of (A) and (B) showing correlation of red and blue fluorescence. Scale bars correspond to 2 μm. | ||
Cellular uptake of BD–PEG–BD triblock nanofibers
We sought to utilize the dual-emissive nature of BD–PEG–BD triblock nanofibers to investigate if untargeted PDHF nanofibers were capable of cellular uptake. Investigations began by incubating the same low dispersity 85 nm BD–PEG–BD triblock nanofibers (Lw/Ln = 1.19, σL = 38 nm) with HeLa cells for 1 h at a concentration of 50 μg mL−1. After incubation, the cells were fixed, and the nucleus was stained with DAPI (4′,6-diamidino-2-phenylindole), the F-actin was stained with Alexa Fluor 488 Phalloidin, and the cells were imaged using CLSM. The results (Fig. S15†) revealed that limited intracellular fluorescence was observed upon excitation for the BD fluorophore (λex = 633 nm, λem = 640–700 nm) for cells incubated with BD labeled nanofibers over the fluorescence arising from control cells which had not been exposed to any nanofibers. These results, which implied that BD–PEG–BD triblock nanofibers are not internalized by cells, were confirmed by live cell imaging (Fig. S16A–F†), where incubation of the BD–PEG–BD triblock nanofibers with HeLa cells for 45 minutes at a concentration of 50 μg mL−1 also led to similar results, with no observable emission from either PDHF or BD. Finally, similar live cell experiments were conducted with BD–PEG–BD triblock nanofibers where the supernatant was left in suspension over the cells for 1 h before imaging. The results from this experiment (Fig. S16G–I†) revealed that the fluorescence from the BD fluorophore was located extracellularly, confirming the successful visualization of the BD labeled nanofibers in the presence of cells. Taken together, these results indicate that BD–PEG–BD triblock nanofibers with a neutral PEG corona alone are incapable of being internalized by the cells studied, and that the introduction of active targeting (in the form of FA) is required to enable cellular internalization to take place.Cellular uptake of folic acid-decorated dual-emissive PDHF-b-PEG nanofibers
In order to investigate whether the addition of FA to PDHF13-b-PEG227 nanofibers facilitates cellular uptake, dual-emissive FA–BD–PEG–BD–FA pentablock nanofibers (10 μg mL−1, Ln = 95 nm, Lw/Ln = 1.17, σL = 39 nm) were incubated with HeLa cells, and imaged via live cell CLSM. After 30 minutes incubation, significant uptake of FA–BD–PEG–BD–FA pentablock nanofibers was observed (Fig. 4A–D). While negligible fluorescence was observed in the blue channel for PDHF, there was significant fluorescence observed from BD. The punctate fluorescence appeared to be within the cell throughout the cytosol, concentrated around the perinuclear region, whilst little uptake was observed in a central region, presumably the nucleus. The observed fluorescence around the perinuclear region may correspond to nanofibers that are located around the nuclear membrane. Further experiments with cells where the nucleus was labelled with DAPI, and the F-actin labelled with Alexa Fluor 488-Phalloidin (Fig. 4E–H) confirmed that little fluorescence is found within the nucleus, implying that FA–BD–PEG–BD–FA pentablock nanofibers are unable to localize in that region. Examination of z-stack data of both fixed and live cells (Fig. S17†) revealed that the punctate fluorescence was located within the cell, rather than on the surface, indicating that the nanofibers are internalized inside the cell and are not attached to the exterior of the plasma membrane. Fluorescence quenching of the PDHF core of the nanofibers was also observed (see ESI Page S3†). | ||
| Fig. 4 (A–D) CLSM maximum intensity projections of live HeLa cells after 30 minutes exposure to FA–BD–PEG–BD–FA pentablock nanofibers (10 μg mL−1, Ln = 95 nm, Lw/Ln = 1.17, σL = 39 nm). (A) Blue channel from PDHF core (λex = 405 nm, λem = 415–478 nm). (B) Brightfield transmitted light channel. (C) Red channel from BD fluorescence (λex = 633 nm, λem = 640–700 nm). (D) Overlay of images (A–C). (E–H) CLSM maximum intensity projections of fixed HeLa cells after 1 h exposure to the dual-emissive FA–BD–PEG–BD–FA pentablock nanofibers (50 μg mL−1). (E) Nucleus stained with DAPI. (F) F-Actin stained with Alexa Fluor 488 Phalloidin. (G) BD fluorescence from FA–BD–PEG–BD–FA pentablock nanofibers. (H) Overlay of images (E–G). All scale bars correspond to 20 μm. | ||
Next, to quantitatively probe the cellular uptake of FA–BD–PEG–BD–FA pentablock nanofibers and compare this to BD–PEG–BD triblock nanofibers lacking FA, we undertook flow cytometry experiments with HeLa cells (Fig. 5 and Table S1†). After 45 minutes of incubation with BD labelled nanofibers either bearing FA (Ln = 95 nm, Lw/Ln = 1.17, σL = 39 nm) or lacking FA (Ln = 85 nm, Lw/Ln = 1.19, σL = 38 nm), cells were detached with Accutase® and counted via flow cytometry. After gating sequentially for cells, single cells, and live cells, the remaining cells were gated for either BD fluorescence (λex = 633 nm, λem = 660/20 nm) or PDHF fluorescence (λex = 405 nm, λem = 450/50 nm). Results for 85 nm BD–PEG–BD triblock nanofibers indicated that they were not uptaken by the cells, as BD fluorescence was equal to that of control HeLa cells that had not been exposed to any nanofibers. In contrast, results for 95 nm FA–BD–PEG–BD–FA pentablock nanofibers revealed a shift in BD fluorescence, with >99% of the cells counted (>10000) in every repeat displaying a significant increase in BD fluorescence (Fig. 5H). The increase in BD fluorescence over control HeLa cells observed in ∼100% of the cells counted implies that the addition of FA to the periphery of the terminal segments of the corona of the nanofibers successfully facilitates cellular uptake. Furthermore, because nanofibers without FA do not undergo internalization, the uptake should be entirely dependent on the presence (or absence) of folate receptors on the target cell, opening up the possibility of using PDHF nanofibers for the targeted imaging or delivery of therapeutics for diseases such as cancer. Analysis of the median fluorescence intensity of each cell (Fig. 5G) revealed that for fibers without FA, median fluorescence was comparable to control HeLa cells (149% of control) whereas fibers with FA exhibited a ca. 1660% increase in fluorescence intensity. The large increase in median fluorescence intensity for fibers with FA is further evidence for their uptake into cells. Nanoparticle uptake can also be measured by changes in side scattering from flow cytometry,80 however no significant differences in side scattering were observed for any of the experiments conducted on these nanofibers (Fig. 5I).
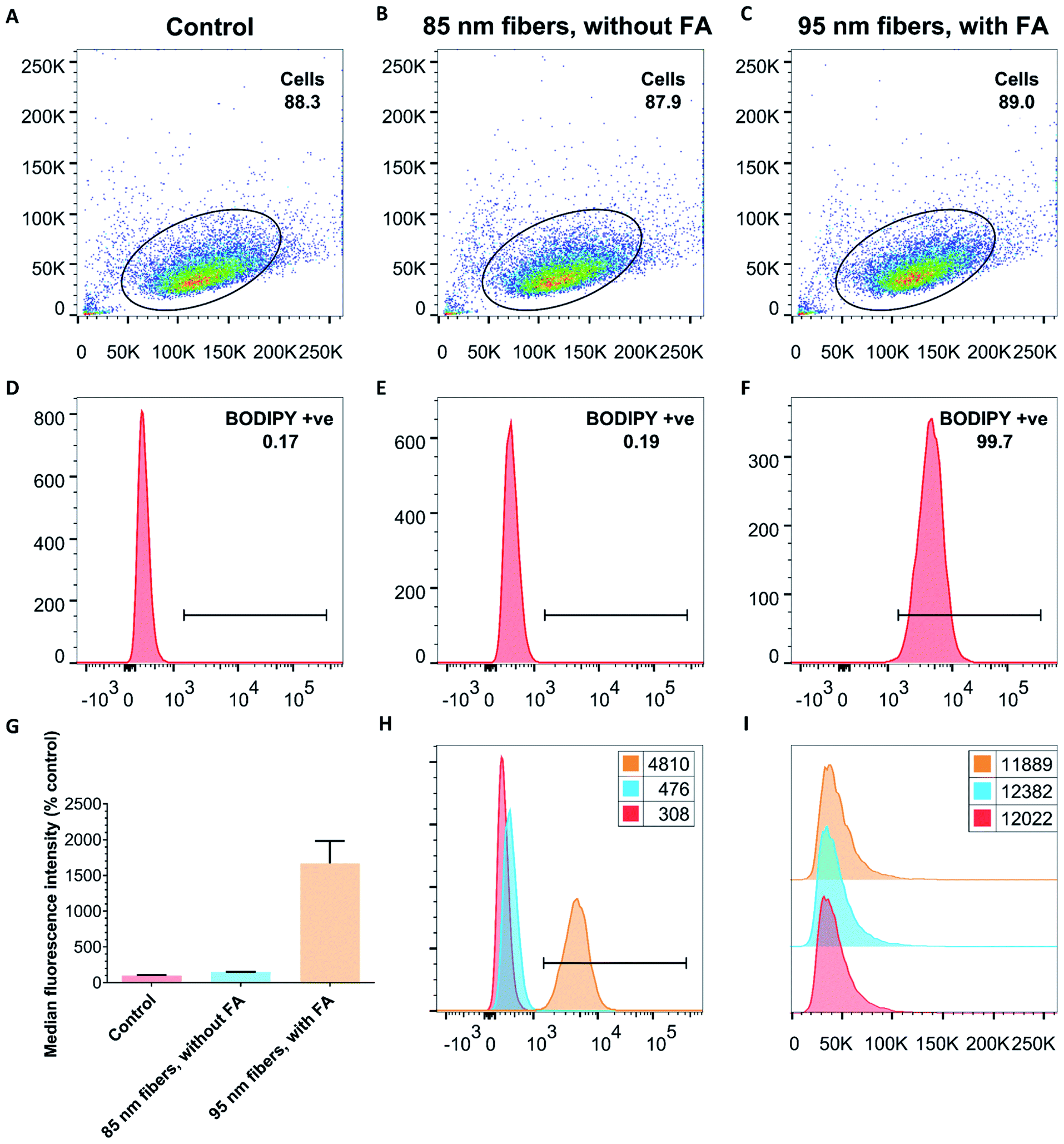 | ||
| Fig. 5 Representative flow cytometry data from experiments with either control cells (no nanofibers added, (A and D) or with dual-emissive PDHF triblock (without FA, (B and E)) and pentablock (with FA, (C and F)) nanofibers. (A–C) Double scatter plots of side scattering (y-axis) versus forward scattering (x-axis), and (D–F) fluorescence intensity distributions (in logarithmic scale on x-axis) versus cell count (y-axis) following BD fluorescence (λex = 633 nm, λem = 660/20 nm) of HeLa cells after 45 minutes exposure to either nothing (control, (A and D)), or 50 μg mL−1 of BD labelled nanofibers either without FA (B and E), or with FA (C and F). (G) Normalized (relative to % control) median fluorescence intensity of BD fluorescence (expressed as mean of all live cells, error is σ). Cells exposed to fibers without FA have median fluorescence comparable to control cells, whilst cells exposed to fibers with FA exhibit a 1660% increase in fluorescence intensity per cell. (H) Overlay of BD fluorescence intensity histograms from (D–F) (x-axis) vs. cell count (y-axis) illustrating that FA is essential for the uptake of PDHF13-b-PEG227 nanofibers. (I) Histogram of side scattering (x-axis) vs. cell count (y-axis). No changes were observed for any sample. (G–I) Control is red, 85 nm triblock nanofibers without FA are light blue, and 95 nm pentablock nanofibers with FA are yellow. Numbers in the legends refer to the geometric mean. For more information, see Table S1, Fig. S18 and S19.† | ||
Investigations into uptake pathway and intracellular localization
Intrigued by the differences between PDHF and BD fluorescence, we attempted to further investigate the cellular uptake pathway of FA–BD–PEG–BD–FA pentablock nanofibers via correlated CLSM and electron microscopy (CLEM) on cells cooled to 4 °C. At this temperature, active transport mechanisms are considerably slowed down. Thus, if internalization still occurs it is likely to proceed through temperature-independent invagination, whereas if the nanofibers are only bound to the outer cell membrane then uptake is likely to occur through one of the many active transport mechanisms. Considering that the FA–BD–PEG–BD–FA pentablock nanofibers discussed here are decorated with FA, one might assume that uptake occurs through receptor-mediated endocytosis, as reported for other FA decorated nanoparticles.23–26,65,67,81–86Our initial experiments involved the addition of FA–BD–PEG–BD–FA pentablock nanofibers (50 μg mL−1, Ln = 95 nm, Lw/Ln = 1.17, σL = 39 nm) to HeLa cells expressing GRASP65-GFP (that contain GFP labelled Golgi apparatus as a reference)87 on ice. After 10 minutes of incubation (which should allow for association between FA residues and folate receptors on the cell surface), cells were imaged via CLSM. Results (Fig. 6A–D) indicated that BD fluorescence was observed partially inside cells as well as around the cell membrane (e.g.Fig. 6C), which was interesting as uptake of FA–BD–PEG–BD–FA pentablock nanofibers at 4 °C was unexpected, potentially pointing to two different uptake mechanisms operating.
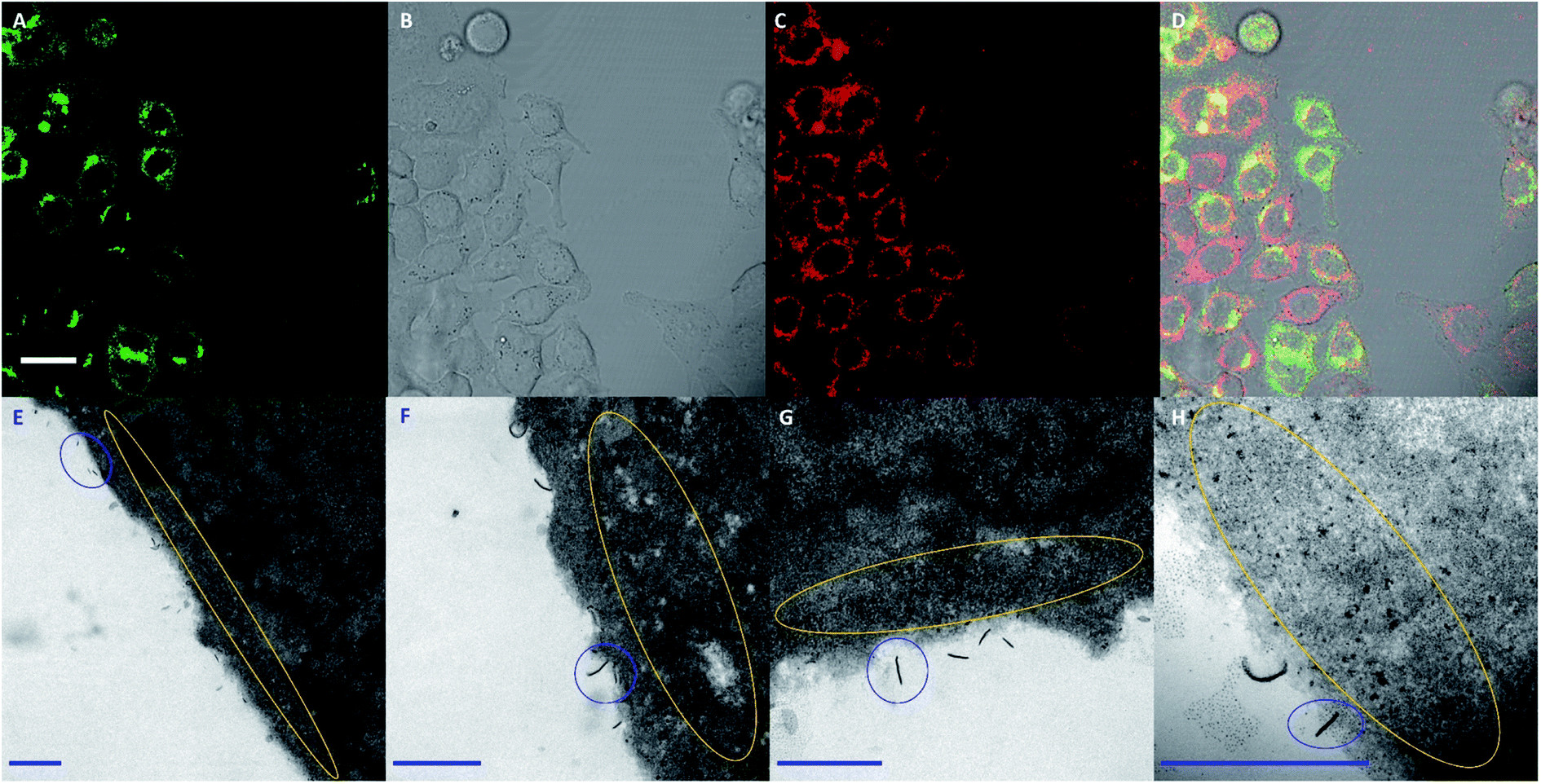 | ||
| Fig. 6 (A–D) CLSM images of HeLa cells expressing GRASP65-GFP after 10 minutes incubation at 4 °C with FA–BD–PEG–BD–FA pentablock nanofibers (50 μg mL−1, Ln = 95 nm, Lw/Ln = 1.17, σL = 39 nm). (A) Fluorescence from GRASP65-GFP labelled Golgi apparatus. (B) Brightfield transmitted light channel. (C) Red channel from BD fluorescence (λex = 633 nm, λem = 640–700 nm). (D) Overlay of images (A–C). (E–H) Representative transmission electron microscopy images of the same cells, after processing. Highlighted in blue are examples of FA–BD–PEG–BD–FA pentablock nanofibers bound to the membrane, with a higher than expected number exhibiting end-on interactions with the cell membrane (e.g. in (H)). Highlighted in yellow are the electron-dense fragments observed around the periphery of the cells, which we hypothesize are nanofiber fragments. Scale bars for (A–D) correspond to 20 μm, while (E–H) correspond to 500 nm. | ||
TEM analysis of 70 nm slices of the cells revealed individual nanofibers interacting with the cell membrane, as well as smaller electron-dense anisotropic particles proximal to the cell membrane (Fig. 6E and H, S20 and S21†). Analysis of the lengths and widths of these electron-dense anisotropic particles (Fig. S22†) revealed a Ln of 19 nm (Lw/Ln = 1.11, σL = 6 nm) and a Wn of 10 nm (Lw/Ln = 1.14, σW = 4 nm), values that are comparable to the dimensions expected for the PDHF core.74 We hypothesize that the anisotropic electron-dense particles observed may correspond to fragments of PDHF13-b-PEG227 nanofibers, and they will be referred to as ‘fragments’ henceforth. It is important to note that whilst the fragments observed closely match the BD fluorescence in CLSM data, their small size and shape (10–20 nm) also closely match those of other natural cellular structures such as ribosomes, glycogen granules, and nucleosomes, preventing definitive assignment via TEM. Further evidence for the fragmentation of nanofibers upon cellular internalization was provided by TEM micrographs where a lower contrast ‘corona’ (Fig. S21,† red circle) was observed around the nanofibers, which appeared to be associated with cleavage (Fig. S21,† green circle).
Further analysis of cell slices of a single cell imaged via CLEM revealed both intact FA–BD–PEG–BD–FA pentablock nanofibers and fragments throughout the cell, concentrated around the perinuclear region (Fig. 7 and S23†). Intact nanofibers were observed inside endosomal-like vesicles (Fig. 7, circled yellow and Fig. S23†), alongside fragments (Fig. 7, circled green), which may correspond to late endosomes.88 Free intact nanofibers (Fig. 7, circled red) and free fragments (Fig. 7, circled purple) were also both observed inside the cytosol. The presence of intact nanofibers inside endosomes is consistent with receptor-mediated endocytosis being an active uptake pathway for these FA decorated nanofibers. The presence of intact nanofibers and fragments in the cytosol, as well as the enrichment of fragments inside endosomes raises questions about the endosomal escape of the materials, as well as other potentially active endocytosis mechanisms that may be operating. Transmembrane penetration by passive diffusion of nanoparticles has been reported,89 and may be a second internalization pathway in operation for these nanofibers, given the uptake detected at 4 °C via CLSM. Statistical analysis of the lengths of the intact nanofibers that were observable inside a single cell revealed 446 individual nanofiber-like objects (excluding fragments), with lengths averaging 115 nm (Lw/Ln = 1.12, σL = 40 nm, Fig. S24 and Table S2†). This length is very similar to that for FA–BD–PEG–BD–FA pentablock nanofibers before cellular experiments (95 nm, Lw/Ln = 1.17, σL = 39 nm), providing further evidence for their identity. The fibers and fragments observed via TEM correlate with the intracellular localization of BD fluorescence observed via CLSM for CLEM experiments (Fig. 7B), and suggest that FA–BD–PEG–BD–FA pentablock nanofibers interact with the cell membrane, leading to receptor-mediated endocytosis, and nanofiber fragmentation. Dalhaimer et al.90 also observed fragmentation of multimicrometer long polymeric worm-like micelles upon cellular uptake, although the resulting fragments were still up to ca. 500 nm in length. The number of intact anisotropic particles observed (446) also provides a rough indication for how many nanofibers may be uptaken by an individual cell. If nanofiber fragmentation is taken into consideration, this number is likely to be higher.
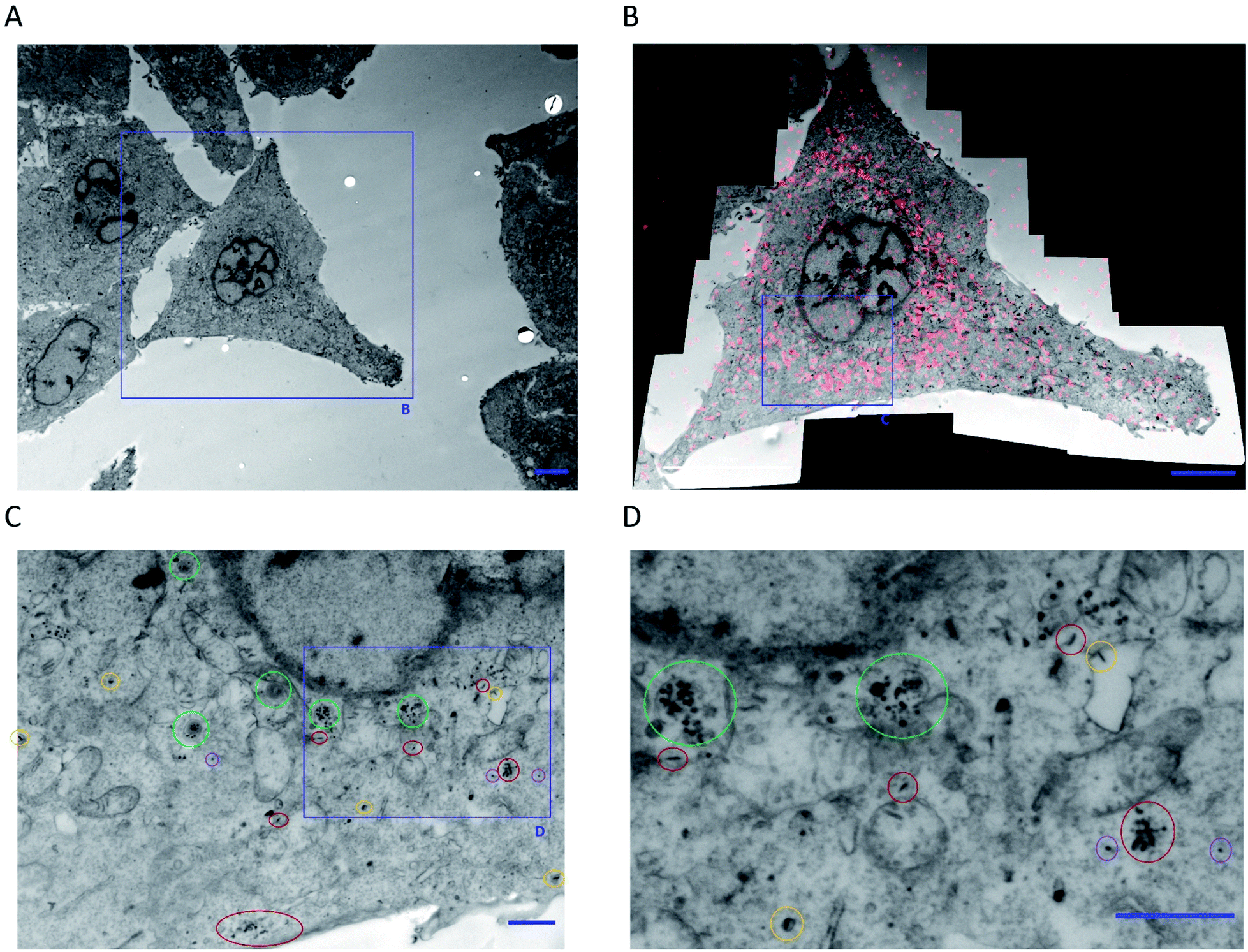 | ||
| Fig. 7 (A) Low magnification TEM micrograph of HeLa cells expressing GRASP65-GFP following 10 minutes incubation at 4 °C with FA–BD–PEG–BD–FA pentablock nanofibers (50 μg mL−1, Ln = 95 nm, Lw/Ln = 1.17, σL = 39 nm). (B) CLEM overlay of a high-resolution composite TEM micrograph (16 individual micrographs) and a CLSM z-slice image (following BD fluorescence) of the cell highlighted in (A). The overlay correlates BD fluorescence with the nanofibers and possible fragments observed via TEM. (C and D) Magnification of part of the perinuclear region of (B), highlighting intact nanofibers inside endosomes (circled yellow), fragments inside endosomes (circled green), free intact nanofibers (circled red), and free fragments (circled purple). Scale bars correspond to 5 μm for (A) and (B), and 1 μm for (C) and (D). For more information, see Fig. S23 in the ESI.† | ||
Observations of membrane-bound FA–BD–PEG–BD–FA pentablock nanofibers during experiments at 4 °C revealed a larger than expected fraction of nanofibers interacting with the cell membrane through end-on interactions (several examples are circled in blue in Fig. 6E and H, and further cases in Fig. S20†). This end on interaction mode should be statistically of low frequency if particle anisotropy had no effect on membrane binding. Thus, these images suggest that FA–BD–PEG–BD–FA pentablock nanofibers favor interaction with the cell membrane through an end-on binding mode. Analysis of the entry angle observed between FA–BD–PEG–BD–FA pentablock nanofibers and the cell membrane observed via TEM revealed two distributions, centered around 90° and 165° respectively (Fig. 8 and S25†). These correspond to nanofibers which are either ‘end-on’ (∼90°, Fig. S25A†) or ‘side-on’ (∼165°, Fig. S25C†). Such modes have been investigated previously, both theoretically (for generic rod-like nanoparticles),19,20 and experimentally (for carbon nanotubes),77 and have been reported to be facilitated through increased membrane wrapping of 1D materials, owing to the effects of stiffness, length, and aspect ratio on the uptake mechanism.91–94
 | ||
| Fig. 8 (A) Histogram of the entry angle of FA–BD–PEG–BD–FA pentablock nanofibers to the cellular membrane (N = 104, blue) and cumulative frequency of observed entry angles (orange). (B) First derivative of the cumulative frequency of the observed nanofiber entry angle with cells, revealing two distributions, centered around 90° and 165° respectively. These correspond to ‘end-on’ (90°, perpendicular) and ‘side-on’ (∼180°, parallel) binding modes with the cellular membrane. (C) Schematic representation of the two major proposed association methods: ‘end-on’ (A) and ‘side-on’ (B). Transition between the two states may be possible, with ‘end-on’ nanofibers undergoing cellular internalization. Measurement of entry angle was conducted using Fiji software (ImageJ) with examples in Fig. S25.† | ||
Analogous CLEM experiments where HeLa cells expressing GRASP65-GFP were incubated with FA–BD–PEG–BD–FA pentablock nanofibers for 90 minutes at 22 °C (after association at 4 °C) revealed electron rich fragments and intact nanofibers located around the perinuclear region via TEM micrographs of the resulting cell slices (Fig. S26 and S27†), which correlated with the fluorescence observed via CLSM when data is overlaid (Fig. S26B and C†). The localization of these intact nanofibers and objects via TEM correlates with the fluorescence observed from BD around the perinuclear region, and supports the hypothesis that the anisotropic particles correspond to FA–BD–PEG–BD–FA pentablock nanofiber fragments. Finally, to confirm that these results were not due to interference from the presence of the BD dye, FA–PEG–FA triblock nanofibers (100 μg mL−1 or 500 μg mL−1, Ln = 90 nm, Lw/Ln = 1.11, σL = 30 nm) were incubated with HeLa cells for 5 minutes and 75 minutes respectively, prepared as before, and imaged via TEM (Fig. S28 and S29†). Whilst some intact nanofibers were observed in TEM micrographs of HeLa cells exposed to 100 μg mL−1 of FA–PEG–FA triblock nanofibers for 5 minutes (Fig. S28,† circled red), fragments and clusters were also observed throughout the cell (Fig. S28,† circled blue), including inside endosomes/lysosomes (Fig. S28,† circled green). Results from HeLa cells exposed to 500 μg mL−1 of FA–PEG–FA triblock nanofibers for 75 minutes also revealed intact nanofibers and fragments throughout the cell, as well as inside endosomes/lysosomes (Fig. S29†). There appeared to be an increase in the number of fragments present, which is in accordance with the higher concentration of FA–PEG–FA triblock nanofibers leading to a higher number of fragments inside the cell. Analysis of the small electron-rich fragments present in HeLa cells exposed to 100 μg mL−1 of FA–PEG–FA triblock nanofibers for 5 minutes revealed a length of 26 nm (Ln = 26 nm, Lw/Ln = 1.09, σL = 8 nm) and a width of 12 nm (Wn = 12 nm, Ww/Wn = 1.04, σW = 3 nm, Fig. S30†), in close agreement with the measured width of FA–PEG–FA triblock nanofibers (Wn = 12 nm, Ww/Wn = 1.02, σW = 2 nm).
In summary, our results indicate that FA functionalized PDHF13-b-PEG227 nanofibers appear to interact with the cell membrane at either a 90° (perpendicular) or 165° (parallel) angle of contact, with perpendicular fibers appearing to undergo cellular internalization whilst parallel fibers are either not internalized, or shift to a perpendicular orientation before entering the cell.77 Upon cellular internalization, we hypothesize that some of the nanofibers fragment into ∼20 nm long particles. Both intact nanofibers and fragments were observed inside cells, as well as intact nanofibers inside endosomes, indicating receptor-mediated endocytosis is one active uptake mechanism, but passive diffusion may also be operational. Localization primarily occurs to the perinuclear region however nanofibers and fragments were observed throughout the cell.
Discussion
Precision functional, modular PDHF nanofibers for biomedical applications
In this work we have demonstrated the formation of 95 nm pentablock co-micelles with an average segment length of only 19 nm, which is close to the lower limit for the lengths of 1D nanomaterials produced via living CDSA to date. This represents the highest density of segments produced in a polymer nanofiber to date. In principle, the highly modular nature of the synthetic route to end-group modification of the PDHF-b-PEG polymer allows for a diverse range of targeting groups, imaging agents, and cargo such as drugs to be incorporated into the nanofibers. As a proof of concept, we have taken FA; one of the most well-studied ligands for targeted drug delivery to cancer cells. We have produced two sets of PDHF13-b-PEG227 nanofibers: those with FA, and those lacking FA. Both nanofibers have a neutral PEG corona which should confer the nanofibers with ‘stealth’ properties. Overall, these results show that complex nanomaterials can be prepared using living CDSA on a length-scale appropriate for biological applications and provide a method for precisely tailoring surface chemistry in small nanoparticles.Cellular uptake of untargeted vs. targeted PDHF-b-PEG nanofibers
Initial CLSM experiments on PDHF13-b-PEG227 nanofibers lacking FA revealed that, while no discernable cytotoxicity was detected (see ESI Page S2, Fig. S31, S32 and Tables S3–S6, see ESI† for results and discussion), no cellular uptake was observed over a 1 h period either. This result was reinforced by flow cytometry experiments, which revealed basal levels of BD emission, on a par with untreated control cells. One plausible explanation is that a longer time period is required before significant uptake will be observed, as the internalization of neutral PEG-coated gold nanorods was observed to occur over a 24 h period,95 though uptake after 24 h was only 2% of the total added. CLSM indicated that FA-mediated nanofiber uptake occurs within 30 minutes, leading primarily to localization in the perinuclear region. Flow cytometry allowed for a comparison of the uptake efficiency of targeted vs. untargeted nanofibers, with >99% of HeLa cells exhibiting uptake of FA decorated nanofibers, versus <1% of HeLa cells for those lacking FA.Intracellular fate of folic acid decorated PDHF-b-PEG nanofibers
Analysis of TEM images of FA–BD–PEG–BD–FA pentablock nanofibers in the region of the cellular membrane of HeLa cells at 4 °C revealed a larger than expected number of nanofibers that interact with the cell membrane in an ‘end-on’ (perpendicular) and ‘side-on’ (parallel) fashion. The experimental results obtained here are consistent with theoretical and experimental studies by Shi,77 and Möller21et al., where cellular internalization of rod-like nanoparticles appears to occur firstly via association with the tip of the fiber, followed by rotation to a 90° (perpendicular) angle of contact that is driven by a relaxation in elastic energy in the cell membrane. Our results concur with those of Shi et al., where only nanofibers with high angles of contact are observed undergoing cellular internalization. The observation of nanofibers with ‘end-on’ membrane interactions and curved, flexible tails (Fig. S20†) supports the proposed transition from ‘side-on’ to ‘end-on’ before internalization. ‘End-on’ internalization is presumably further driven by the segmented block-like structure of the nanofibers, where the FA targeting group is located solely at the fiber ends, facilitating cellular uptake.CLEM studies of HeLa cells incubated with FA–BD–PEG–BD–FA pentablock nanofibers at 4 °C revealed intact nanofibers as well as small, high contrast ‘fragments’ in the immediate region around the cellular membrane and throughout the cell via TEM, which correlated with BD fluorescence observed in CLSM. Some of the nanofibers were found within endosomes, indicating that receptor-mediated endocytosis is an active uptake mechanism, consistent with other FA containing nanomaterials.23,25,26 The observation of intact nanofibers and fragments outside of endosomes via TEM, and the presence of intracellular BD fluorescence at 4 °C via CLSM indicates that another uptake mechanism may also be present, such as passive diffusion. CLEM and TEM experiments involving nanofibers both with and without the BD dye reveal similar intact nanofibers and high contrast fragments observed within the cell and around the nuclear membrane. Taken together, we hypothesize that fragmentation/disassembly of the nanofibers occurs upon cellular internalization, with subsequent localization primarily in the perinuclear region. Nanofiber fragmentation would also fit with the observed fluorescence quenching of the PDHF core (Fig. S33, see ESI† for results and discussion), as it could be imagined that the forces driving nanofiber cleavage might involve interaction of the π-conjugated core with species capable of causing fluorescence quenching. Fragmentation of the PDHF nanofibers upon cellular internalization would also be consistent with the behavior of FA-functionalized coordination complex nanotubes observed by Wang et al.,30 and of PEG-b-PCL filomicelles by Geng et al.12 raising the possibility that this may be a more general consequence of the cellular internalization of 1D nanomaterials with specific properties.96 Nanofiber fragmentation currently remains a hypothesis, however, as further studies are required to probe and confirm this phenomenon. As many questions remain regarding the cellular internalization and localization of FA targeted PDHF-b-PEG nanofibers, future work will focus on probing this process in more detail.
Summary
Using the living CDSA approach, we have developed colloidally stable, hydrophilic segmented 1D nanofibers with a crystalline π-conjugated PDHF core, a ‘stealth’ PEG corona, and spatially confined functionality. Segmented pentablock nanofibers of length 95 nm were prepared through a seeded-growth process, which possess the highest density of different corona-forming blocks in a segmented nanofiber to date. The development of nanofibers with length control, and the ability to easily present different functional groups in a modular, controlled fashion over length scales relevant to biomedical applications represents a potentially significant advance. In the absence of targeting groups, the nanofibers were not capable of being internalized by HeLa cells after 1 h, however cell uptake was detected by CLSM and flow cytometry within 30 minutes for nanofibers functionalized in the terminal segment with FA, which binds to folate receptors that are overexpressed in cancer cells such as the HeLa cell line examined here. Nanofibers without FA were uptaken into <1% of HeLa cells, in contrast with > 97% uptake of FA decorated nanofibers. The lack of cellular uptake for nanofibers without FA implies that nanofibers bearing this moiety may act as a targeted diagnostic, preferentially undergoing internalization into cells that express folate receptors, such as those in tumors. FA decorated nanofibers were observed to undergo internalization into HeLa cells at 4 °C, with some observed in the cellular membrane and others inside the cell. A significant number of membrane-bound nanofibers were observed to interact with the cell membrane in either an ‘end-on’ or ‘side-on’ fashion. Only ‘end-on’ fibers were observed to undergo internalization, providing experimental evidence for the unique uptake mechanism of high aspect ratio 1D nanomaterials. Small, high contrast, anisotropic particles (∼10 × 20 nm) were observed inside HeLa cells proximal to the cell membrane, leading us to hypothesize that FA–BD–PEG–BD–FA pentablock nanofibers may undergo fragmentation upon cellular internalization at 4 °C. Analogous experiments at room temperature revealed similar particles throughout the cell, but concentrated around the perinuclear region. If the small, high contrast particles observed do correspond to nanofiber fragments, this would point towards a unique uptake and disassembly mechanism for this type of 1D material. Intact nanofibers were also observed throughout the cell, with examples of both nanofibers and fragments free in the cytosol, as well as inside endosomes. Examples of intact nanofibers inside endosomes indicate that receptor-mediated endocytosis is an active uptake mechanism for the FA decorated nanofibers.Overall, these results indicate that the nanofibers are capable of active targeting towards different cell lines, with minimal cellular uptake observed for those lacking an active targeting group. If nanofiber fragmentation upon cellular internalization is confirmed, this could allow for the benefits of targeted 1D nanomaterials in vivo, whilst releasing smaller particles after cellular internalization that could have other, additional benefits (e.g. nuclear localization if fragment size can be tuned to <5 nm). These results also provide new insights into the cellular uptake of low dispersity 1D nanoparticles, revealing the potential for π-conjugated PDHF nanofibers to act as fluorescence turn-off sensors for cells rich in folate receptors. This work also provides valuable information on the uptake mechanism for anisotropic 1D polymer nanoparticles. Finally, the study indicates that the ability of ‘living’ CDSA to generate anisotropic polymer nanoparticles with near uniform dimensions and a segmented structure should facilitate further investigations of nanoparticle uptake into cells, and where features such as fiber length, width, stiffness, and the spatial location and choice of targeting groups are varied. Analogous 1D nanoparticles also have the potential to deliver therapeutic cargoes, with relevant studies currently in progress.
Author contributions
Y. He, S. Street and I. Manners conceived the project. X. Jin synthesized PDHF homopolymer. Y. He conceived and conducted the synthesis, self-assembly, and characterization of all polymer nanofibers. S. Street conceived and conducted all other experiments and analyzed the data. The manuscript was written by S. Street and I. Manners, with input from Y. He. P. Verkade conceived and conducted CLEM studies with S. Street. L. Hodgson assisted with CLEM studies and discussions. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Street thanks the EPSRC (UK) for a DTP Doctoral Prize Fellowship (EP/N509619/1). I. Manners thanks NSERC (Canada) for an NSERC Discovery Grant, the Canadian Government for a Canada 150 Research Chair, the University of Victoria for start-up funds and the Canada Foundation for Innovation (CFI), and the British Columbia Knowledge Development Fund (BCKDF) for equipment and instrumentational support. Y. He thanks the EPSRC funded Bristol Chemical Synthesis CDT for funding. L. Hodgson thanks the BBSRC (UK) for a strategic LoLa award (BB/M002969/1). The authors wish to acknowledge the assistance of Dr Andrew Herman and Lorena Sueiro Ballesteros and the University of Bristol Faculty of Biomedical Sciences Flow Cytometry Facility with flow cytometry experiments, BrisSynBio (BB/L01386X/1) for use of cell culture facilities, and Prof. Cornelia Bohne for use of the QM40 fluorimeter. S. Street and Y. He also thank the Bristol Chemistry Electron Microscopy Unit for the use of TEM facilities. All the authors would like to thank Jon Lane for providing the GRASP65-GFP HeLa cell line, Prof. M. C. Galan for providing WI-38 cells, and Holly Baum, George Banting, and Dek Woolfson for useful discussions (supported by BBSRC grants BB/L010518/1 and BB/L01386X/1). We are grateful for the support by the EM and LM units of the Wolfson Bioimaging Facility (BBSRC grant BB/L014181/1). The authors also wish to thank the reviewers for their insightful comments and suggestions.References
- S. Hua, M. B. C. de Matos, J. M. Metselaar and G. Storm, Front. Pharmacol., 2018, 9, 790 CrossRef PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- S. Naahidi, M. Jafari, F. Edalat, K. Raymond, A. Khademhosseini and P. Chen, J. Controlled Release, 2013, 166, 182–194 CrossRef CAS PubMed.
- A. D. Friedman, S. E. Claypool and R. Liu, Curr. Pharm. Des., 2013, 19, 6315–6329 CrossRef CAS PubMed.
- S. Shen, Y. Wu, Y. Liu and D. Wu, Int. J. Nanomed., 2017, 12, 4085–4109 CrossRef CAS PubMed.
- J. M. Williford, J. L. Santos, R. Shyam and H. Q. Mao, Biomater. Sci., 2015, 3, 894–907 RSC.
- C. Kinnear, T. L. Moore, L. Rodriguez-Lorenzo, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2017, 117, 11476–11521 CrossRef CAS PubMed.
- S. Kaga, N. P. Truong, L. Esser, D. Senyschyn, A. Sanyal, R. Sanyal, J. F. Quinn, T. P. Davis, L. M. Kaminskas and M. R. Whittaker, Biomacromolecules, 2017, 18, 3963–3970 CrossRef CAS PubMed.
- A. Banerjee, J. Qi, R. Gogoi, J. Wong and S. Mitragotri, J. Controlled Release, 2016, 238, 176–185 CrossRef CAS PubMed.
- R. Toy, P. M. Peiris, K. B. Ghaghada and E. Karathanasis, Nanomedicine, 2014, 9, 121–134 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- M. A. Bruckman, L. N. Randolph, A. VanMeter, S. Hern, A. J. Shoffstall, R. E. Taurog and N. F. Steinmetz, Virology, 2014, 449, 163–173 CrossRef CAS PubMed.
- Y. Zhao, Y. Wang, F. Ran, Y. Cui, C. Liu, Q. Zhao, Y. Gao, D. Wang and S. Wang, Sci. Rep., 2017, 7, 4131 CrossRef PubMed.
- N. Doshi, B. Prabhakarpandian, A. Rea-Ramsey, K. Pant, S. Sundaram and S. Mitragotri, J. Controlled Release, 2010, 146, 196–200 CrossRef CAS PubMed.
- P. Kolhar, A. C. Anselmo, V. Gupta, K. Pant, B. Prabhakarpandian, E. Ruoslahti and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10753–10758 CrossRef CAS PubMed.
- S. Barua, J. W. Yoo, P. Kolhar, A. Wakankar, Y. R. Gokarn and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3270–3275 CrossRef CAS PubMed.
- X. Huang, X. Teng, D. Chen, F. Tang and J. He, Biomaterials, 2010, 31, 438–448 CrossRef CAS PubMed.
- S. Dasgupta, T. Auth and G. Gompper, Nano Lett., 2014, 14, 687–693 CrossRef CAS PubMed.
- R. Vácha, F. J. Martinez-Veracoechea and D. Frenkel, Nano Lett., 2011, 11, 5391–5395 CrossRef PubMed.
- J. Möller, T. Luehmann, H. Hall and V. Vogel, Nano Lett., 2012, 12, 2901–2905 CrossRef PubMed.
- J. A. Champion and S. Mitragotri, Pharm. Res., 2009, 26, 244–249 CrossRef CAS PubMed.
- M. Fernández, F. Javaid and V. Chudasama, Chem. Sci., 2018, 9, 790–810 RSC.
- M. H. Han, H. Zheng, Y. F. Guo, Y. H. Wang, X. Y. Qi and X. T. Wang, RSC Adv., 2016, 6, 45664–45672 RSC.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef CAS PubMed.
- B. Stella, S. Arpicco, M. T. Peracchia, D. Desmaële, J. Hoebeke, M. Renoir, J. D'Angelo, L. Cattel and P. Couvreur, J. Pharm. Sci., 2000, 89, 1452–1464 CrossRef CAS PubMed.
- B. Bahrami, M. Mohammadnia-Afrouzi, P. Bakhshaei, Y. Yazdani, G. Ghalamfarsa, M. Yousefi, S. Sadreddini, F. Jadidi-Niaragh and M. Hojjat-Farsangi, Tumor Biol., 2015, 36, 5727–5742 CrossRef CAS PubMed.
- K. Zhang, R. Rossin, A. Hagooly, Z. Chen, M. J. Welch and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7578–7583 CrossRef CAS PubMed.
- A. Mehdizadeh, S. Pandesh, A. Shakeri-Zadeh, S. K. Kamrava, M. Habib-Agahi, M. Farhadi, M. Pishghadam, A. Ahmadi, S. Arami and Y. Fedutik, Lasers Med. Sci., 2014, 29, 939–948 CrossRef PubMed.
- Y. Wang, C. Zhang, H. Li, G. Zhu, S. S. Bao, S. Wei, L. M. Zheng, M. Ren and Z. Xu, J. Mater. Chem. B, 2015, 3, 296–305 RSC.
- J. Zhu, Z. Zhou, C. Yang, D. Kong, Y. Wan and Z. Wang, J. Biomed. Mater. Res., Part A, 2011, 97A, 498–508 CrossRef CAS PubMed.
- Z. Hami, M. Amini, M. Ghazi-Khansari, S. M. Rezayat and K. Gilani, DARU J. Pharm. Sci., 2014, 22, 30 CrossRef PubMed.
- W. C. Wu, C. M. Huang and P. W. Liao, React. Funct. Polym., 2014, 81, 82–90 CrossRef CAS.
- X. Guo, C. Shi, J. Wang, S. Di and S. Zhou, Biomaterials, 2013, 34, 4544–4554 CrossRef CAS PubMed.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS PubMed.
- A. Nazemi, C. E. Boott, D. J. Lunn, J. Gwyther, D. W. Hayward, R. M. Richardson, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2016, 138, 4484–4493 CrossRef CAS PubMed.
- J. R. Finnegan, X. He, S. T. G. Street, J. D. Garcia-Hernandez, D. W. Hayward, R. L. Harniman, R. M. Richardson, G. R. Whittell and I. Manners, J. Am. Chem. Soc., 2018, 140, 17127–17140 CrossRef CAS PubMed.
- L. Sun, N. Petzetakis, A. Pitto-Barry, T. L. Schiller, N. Kirby, D. J. Keddie, B. J. Boyd, R. K. O'Reilly and A. P. Dove, Macromolecules, 2013, 46, 9074–9082 CrossRef CAS.
- J. Schöbel, M. Karg, D. Rosenbach, G. Krauss, A. Greiner and H. Schmalz, Macromolecules, 2016, 49, 2761–2771 CrossRef.
- Q. Yu, M. G. Roberts, S. Pearce, A. M. Oliver, H. Zhou, C. Allen, I. Manners and M. A. Winnik, Macromolecules, 2019, 52, 5231–5244 CrossRef CAS.
- Y. He, J. C. Eloi, R. L. Harniman, R. M. Richardson, G. R. Whittell, R. T. Mathers, A. P. Dove, R. K. O'Reilly and I. Manners, J. Am. Chem. Soc., 2019, 141, 19088–19098 CrossRef CAS PubMed.
- M. C. Arno, M. Inam, Z. Coe, G. Cambridge, L. J. Macdougall, R. Keogh, A. P. Dove and R. K. O'Reilly, J. Am. Chem. Soc., 2017, 139, 16980–16985 CrossRef CAS.
- Z. Li, Y. Zhang, L. Wu, W. Yu, T. R. Wilks, A. P. Dove, H. M. Ding, R. K. O'Reilly, G. Chen and M. Jiang, ACS Macro Lett., 2019, 8, 596–602 CrossRef CAS.
- Z. M. Hudson, C. E. Boott, M. E. Robinson, P. A. Rupar, M. A. Winnik and I. Manners, Nat. Chem., 2014, 6, 893–898 CrossRef CAS PubMed.
- B. Fan, R. Y. Wang, X. Y. Wang, J. T. Xu, B. Y. Du and Z. Q. Fan, Macromolecules, 2017, 50, 2006–2015 CrossRef CAS.
- S. Pearce, X. He, M. S. Hsiao, R. L. Harniman, L. R. MacFarlane and I. Manners, Macromolecules, 2019, 52, 6068–6079 CrossRef CAS.
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329–1332 CrossRef CAS PubMed.
- S. Ganda and M. H. Stenzel, Prog. Polym. Sci., 2019, 101, 101195 CrossRef.
- J. Xu, H. Zhou, Q. Yu, G. Guerin, I. Manners and M. A. Winnik, Chem. Sci., 2019, 10, 2280–2284 RSC.
- J. R. Finnegan, D. J. Lunn, O. E. C. Gould, Z. M. Hudson, G. R. Whittell, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2014, 136, 13835–13844 CrossRef CAS PubMed.
- Z. M. Hudson, D. J. Lunn, M. A. Winnik and I. Manners, Nat. Commun., 2014, 5, 3372 CrossRef PubMed.
- D. J. Lunn, O. E. C. Gould, G. R. Whittell, D. P. Armstrong, K. P. Mineart, M. A. Winnik, R. J. Spontak, P. G. Pringle and I. Manners, Nat. Commun., 2016, 7, 12371 CrossRef PubMed.
- S. Shin, F. Menk, Y. Kim, J. Lim, K. Char, R. Zentel and T. L. Choi, J. Am. Chem. Soc., 2018, 140, 6088–6094 CrossRef CAS PubMed.
- J. Gwyther, J. B. Gilroy, P. A. Rupar, D. J. Lunn, E. Kynaston, S. K. Patra, G. R. Whittell, M. A. Winnik and I. Manners, Chem.–Eur. J., 2013, 19, 9186–9197 CrossRef CAS PubMed.
- M. Wang, Y. Zhu, L. Han, R. Qi and F. He, Polym. Chem., 2020, 11, 61–67 RSC.
- D. Tao, Z. Wang, X. Huang, M. Tian, G. Lu, I. Manners, M. A. Winnik and C. Feng, Angew. Chem., Int. Ed., 2020, 59, 8232–8239 CrossRef CAS PubMed.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- C. Duan, K. Zhang, C. Zhong, F. Huang and Y. Cao, Chem. Soc. Rev., 2013, 42, 9071–9104 RSC.
- B. Liu and G. C. Bazan, Conjugated Polyelectrolytes: Fundamentals and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Berlin, 2013 Search PubMed.
- W. Wu, G. C. Bazan and B. Liu, Chem, 2017, 2, 760–790 CAS.
- M. Sun, B. Sun, Y. Liu, Q. D. Shen and S. Jiang, Sci. Rep., 2016, 6, 22368 CrossRef CAS PubMed.
- Q. Zhao, C. Zhang, S. Liu, Y. Liu, K. Y. Zhang, X. Zhou, J. Jiang, W. Xu, T. Yang and W. Huang, Sci. Rep., 2015, 5, 16420 CrossRef CAS PubMed.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed.
- K. Deng, X. Zhao, F. Liu, J. Peng, C. Meng, Y. Huang, L. Ma, C. Chang and H. Wei, ACS Biomater. Sci. Eng., 2019, 5, 3419–3428 CrossRef CAS.
- K. Y. Pu, K. Li and B. Liu, Adv. Funct. Mater., 2010, 20, 2770–2777 CrossRef CAS.
- S. Roy, A. Gunukula, B. Ghosh and C. Chakraborty, Sensors & Actuators, B Chemical, 2019, 291, 337–344 CrossRef CAS.
- C. Zhang, J. Ji, X. Shi, X. Zheng, X. Wang and F. Feng, ACS Appl. Mater. Interfaces, 2018, 10, 4519–4529 CrossRef CAS PubMed.
- N. S. Oltra, P. Nair and D. E. Discher, Annu. Rev. Chem. Biomol. Eng., 2014, 5, 281–299 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- Z. Li, L. Sun, Y. Zhang, A. P. Dove, R. K. O'Reilly and G. Chen, ACS Macro Lett., 2016, 5, 1059–1064 CrossRef CAS PubMed.
- M. Müllner, S. J. Dodds, T. H. Nguyen, D. Senyschyn, C. J. H. Porter, B. J. Boyd and F. Caruso, ACS Nano, 2015, 9, 1294–1304 CrossRef PubMed.
- X. H. Jin, M. B. Price, J. R. Finnegan, C. E. Boott, J. M. Richter, A. Rao, S. M. Menke, R. H. Friend, G. R. Whittell and I. Manners, Science, 2018, 360, 897–900 CrossRef CAS PubMed.
- Y. Zhang, B. Liu and Y. Cao, Chem.–Asian J., 2008, 3, 739–745 CrossRef CAS PubMed.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS PubMed.
- X. Shi, A. Von Dem Bussche, R. H. Hurt, A. B. Kane and H. Gao, Nat. Nanotechnol., 2011, 6, 714–719 CrossRef CAS PubMed.
- P. Najechalski, Y. Morel, O. Stéphan and P. L. Baldeck, Chem. Phys. Lett., 2001, 343, 44–48 CrossRef CAS.
- L. Stryer and R. P. Haugland, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 719–726 CrossRef CAS PubMed.
- A. Salvati, I. Nelissen, A. Haase, C. Åberg, S. Moya, A. Jacobs, F. Alnasser, T. Bewersdorff, S. Deville, A. Luch and K. A. Dawson, NanoImpact, 2018, 9, 42–50 CrossRef.
- S. Dhar, Z. Liu, J. Thomale, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 11467–11476 CrossRef CAS PubMed.
- A. W. York, F. Huang and C. L. McCormick, Biomacromolecules, 2010, 11, 505–514 CrossRef CAS PubMed.
- D. Cao, S. Tian, H. Huang, J. Chen and S. Pan, Mol. Pharm., 2015, 12, 240–252 CrossRef CAS PubMed.
- A. W. York, Y. Zhang, A. C. Holley, Y. Guo, F. Huang and C. L. McCormick, Biomacromolecules, 2009, 10, 936–943 CrossRef CAS PubMed.
- D. Goren, A. T. Horowitz, D. Tzemach, M. Tarshish, S. Zalipsky and A. Gabizon, Clin. Cancer Res., 2000, 6, 1949–1957 CAS.
- T. Yoshida, N. Oide, T. Sakamoto, S. Yotsumoto, Y. Negishi, S. Tsuchiya and Y. Aramaki, J. Controlled Release, 2006, 111, 325–332 CrossRef CAS PubMed.
- J. D. Lane, J. Lucocq, J. Pryde, F. A. Barr, P. G. Woodman, V. J. Allan and M. Lowe, J. Cell Biol., 2002, 156, 495–509 CrossRef CAS PubMed.
- I. G. Ganley, K. Carroll, L. Bittova and S. Pfeffer, Mol. Biol. Cell, 2004, 15, 5420–5430 CrossRef CAS PubMed.
- S. Snipstad, S. Westrøm, Y. Mørch, M. Afadzi, A. K. O. Åslund and C. de Lange Davies, Cancer Nanotechnol., 2014, 5, 8 CrossRef PubMed.
- P. Dalhaimer, A. J. Engler, R. Parthasarathy and D. E. Discher, Biomacromolecules, 2004, 5, 1714–1719 CrossRef CAS PubMed.
- Z. P. Xu, M. Niebert, K. Porazik, T. L. Walker, H. M. Cooper, A. P. J. Middelberg, P. P. Gray, P. F. Bartlett and G. Q. Lu, J. Controlled Release, 2008, 130, 86–94 CrossRef CAS PubMed.
- J. Zhao, H. Lu, Y. Yao, S. Ganda and M. H. Stenzel, J. Mater. Chem. B, 2018, 6, 4223–4231 RSC.
- X. Liu, F. Wu, Y. Tian, M. Wu, Q. Zhou, S. Jiang and Z. Niu, Sci. Rep., 2016, 6, 24567 CrossRef CAS PubMed.
- S. Zhang, H. Gao and G. Bao, ACS Nano, 2015, 9, 8655–8671 CrossRef CAS PubMed.
- X. Xie, J. Liao, X. Shao, Q. Li and Y. Lin, Sci. Rep., 2017, 7, 3827 CrossRef PubMed.
- J. Zhao and M. H. Stenzel, Polym. Chem., 2018, 9, 259–272 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02593c |
| ‡ S. T. G. S. and Y. H. contributed equally to this work. |
| § Present address: Department of Chemistry, University of Victoria, Victoria, BC V8W 3V6, Canada. |
| This journal is © The Royal Society of Chemistry 2020 |