DOI:
10.1039/D0SC02540B
(Edge Article)
Chem. Sci., 2020,
11, 8025-8035
The crystal engineering of radiation-sensitive diacetylene cocrystals and salts†
Received
5th May 2020
, Accepted 18th July 2020
First published on 20th July 2020
Abstract
In this work we develop photoreactive cocrystals/salts of a commercially-important diacetylene, 10,12-pentacosadiynoic acid (PCDA, 1) and report the first X-ray crystal structures of PCDA based systems. The topochemical reactivity of the system is modified depending on the coformer used and correlates with the structural parameters. Crystallisation of 1 with 4,4′-azopyridine (2), 4,4′-bipyridyl (3), and trans-1,2-bis(4-pyridyl)ethylene (4) results in unreactive 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystals or a salt in the case of 4,4′-bipiperidine (5). However, salt formation with morpholine (6), diethylamine (7), and n-butylamine (8), results in highly photoreactive salts 12·7 and 1·8 whose reactivity can be explained using topochemical criteria. The salt 1·6 is also highly photoreactive and is compared to a model morpholinium butanoate salt. Resonance Raman spectroscopy reveals structural details of the photopolymer including its conformational disorder in comparison to less photoactive alkali metal salts and the extent of solid state conversion can be monitored by CP-MAS NMR spectroscopy. We also report an unusual catalysis in which amine evaporation from photopolymerised PCDA ammonium salts effectively acts as a catalyst for polymerisation of PCDA itself. The new photoreactive salts exhibit more reactivity but decreased conjugation compared to the commercial lithium salt and are of considerable practical potential in terms of tunable colours and greater range in UV, X-ray, and γ-ray dosimetry applications.
:1 cocrystals or a salt in the case of 4,4′-bipiperidine (5). However, salt formation with morpholine (6), diethylamine (7), and n-butylamine (8), results in highly photoreactive salts 12·7 and 1·8 whose reactivity can be explained using topochemical criteria. The salt 1·6 is also highly photoreactive and is compared to a model morpholinium butanoate salt. Resonance Raman spectroscopy reveals structural details of the photopolymer including its conformational disorder in comparison to less photoactive alkali metal salts and the extent of solid state conversion can be monitored by CP-MAS NMR spectroscopy. We also report an unusual catalysis in which amine evaporation from photopolymerised PCDA ammonium salts effectively acts as a catalyst for polymerisation of PCDA itself. The new photoreactive salts exhibit more reactivity but decreased conjugation compared to the commercial lithium salt and are of considerable practical potential in terms of tunable colours and greater range in UV, X-ray, and γ-ray dosimetry applications.
Introduction
The solid state 1,4-addition polymerisation of diacetylenes can be initiated by radiation and heat, and results in a conjugated ene–yne polymeric chain. The reaction is thought to only occur if the topochemical parameters of the diacetylene packing are optimal. The first report of topochemical reactivity in the solid state was in an alkene system described by Schmidt in 1964 who suggested that carbon double bonds must be separated by a maximum distance of 4.2 Å for successful polymerisation.1 In 1969, Wegner reported the first example of diacetylene polymerisation in the solid state,2,3 while 15 years later, Enkelmann proposed strict criteria for diacetylene reactivity, whereby adjacent diacetylene monomers will react when the reactive groups are separated by a C1–C4′ contact distance (d) of ≤3.8 Å, a translational period repeat spacing (r) of ≤4.9 Å, along with an orientation angle (θ) to the crystal axis at an optimum value of 45° (Scheme 1).4–6 The diacetylene polymerisation parameters highlight the importance of molecular organisation in the topochemical reaction.7,8
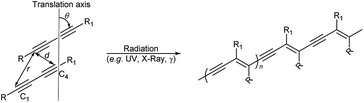 |
| | Scheme 1 The topochemical parameters for diacetylene 1,4-addition polymerisation requires the tilt angle (θ) of the monomers to be 45°, the C1–C4′ distance (r) to be ≤3.8 Å, and the translational repeat distance (d) to be ≤4.9 Å to yield a polydiacetylene. | |
The monomer-to-polymer transition is clearly observed by a colour change from colourless to blue, due to the rearrangement of the diacetylene monomers to give an ene–yne chromophore. The blue colour is due to π–π* transitions in the ordered, conjugated chain with the reorganisation of the chains controlling the degree of diacetylene polymerisation.9 Additional external stimuli on the polymerised diacetylene (polydiacetylene) such as extended heating,10–12 pH change,10,13–16 treatment with organic solvents,10,17–21 mechanical stress,22,23 and ligand–receptor interactions,24,25 can cause the polydiacetylenes to exhibit a range of colours from blue, to red, to yellow.15 These chromic changes can be explained by a conformational rearrangement within the polydiacetylene assembly which disrupts the conjugated backbone, causing reduced overlap of the π orbitals, resulting in a widening of the HOMO–LUMO energy gap and hence the polydiacetylene absorbing light at a higher energy.12,26,27 The commercially important diacetylene, 10,12-pentacosadiynoic acid (PCDA, 1), is used to provide a colourimetric change in practical chemosensors,28–33 biosensors,24,34–36 and dosimeters.37–40 Although PCDA is somewhat photoreactive, further tuning of its photoresponse is of considerable interest, especially for radiation dosimetry applications. Covalent modification offers a viable strategy to PCDA analogues with a tuned photoresponse.41–43 However, since the solid state reactivity of dialkynes depends on their crystal packing arrangement, a simpler strategy is to address the dialkyne reactivity through modification of non-covalent interactions by cocrystal or salt formation.44–47 Whether a cocrystal or salt will form depends on the difference in pKa of the two components. For a cocrystal, the ΔpKa must be <2–3log units, while salt formation is expected for a greater difference.48,49 Cocrystals of carboxylic acids can be prepared using the robust hydrogen-bonded COOH⋯Npyridine heterosynthon, while salts can be based on ammonium complexes of more basic amines.50–54 In this work we explore the relationship between structure and photochemistry for PCDA (1) with three different pyridine-containing coformers 4,4′-azopyridine (2), 4,4′-bipyridyl (3), and trans-1,2-bis(4-pyridyl)ethylene (4) and compare the photoreactivity of the resulting cocrystals with aliphatic amine salts of 4,4′-bipiperidine (5), morpholine (6) diethylamine (7), and n-butylamine (8) (Scheme 2). To date no X-ray structure information has been reported for PCDA or related photoactive surfactant-like molecules despite its commercial importance and hence no structure–reactivity relationship has been elucidated.
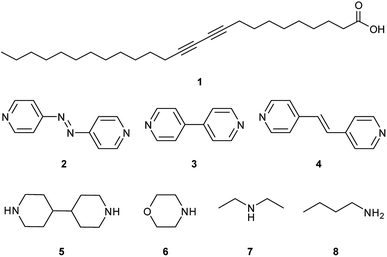 |
| | Scheme 2 Structures of PCDA (1), with coformers 4,4′-azopyridine (2), 4,4′-bipyridyl (3), trans-1,2-bis(4-pyridyl)ethylene (4), and salt formers 4,4′-bipiperidine (5), morpholine (6), diethylamine (7) and n-butylamine (8). | |
Results and discussion
The X-ray crystal structure of PCDA is unknown and the preparation of single crystals of such surfactant-like compounds is generally regarded as difficult. However, preliminary powder X-ray diffraction (PXRD) analysis indicated that compound 1 is crystalline with a lamellar structure (ESI Fig. S1†). Slow evaporation of an acetone solution of PCDA gave high a quality sample suitable for analysis at the I19 beamline at the Diamond Light Source, Oxfordshire. Some decay of photosensitive 1 in the high-intensity synchrotron X-ray beam was noted, however, a structure determination was successfully carried out. The X-ray structure of 1 revealed a centrosymmetric structure (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group) based on the well-known OH⋯O R22(8) ring hydrogen-bonded carboxylic acid dimer synthon55,56 (Fig. 1). The structure is based on a head-to-head bilayer arrangement of molecules and the aliphatic substituents at either end of the dialkyne unit adopt an anti-conformation. The crystallographic c-axis of 46.647(3) Å is long, which is also apparent in the PXRD pattern, which shows a lamellar progression of low-angle peaks from 1.9–17.1° 2θ, all correlating to reflections in the (00l) plane (Fig. S1†).57 The structure conforms to the topochemical postulate for photoreactivity with a translational repeat distance of ≤4.9 Å, at 4.574(1) Å, along with a tilt angle of 44.7° that very close to the desired value of 45°. The inter-alkyne C1–C4′ distance between adjacent molecules of 1 is 3.712(1) Å, which is close to the upper limit of the topochemical postulate for alkyne reactivity (a maximum distance of 3.8 Å).3 These values are consistent with the limited observed solid state photoreactivity of 1 which, while it gradually turns visually blue upon exposure to UV and X-ray, the actual photoconversion as monitored by CP-MAS 13C NMR spectroscopy displays no alkene peaks, implying less than 1% even upon prolonged exposure (Fig. S2–S5†). Therefore, the question is raised as to whether the topochemistry of 1 can be engineered to provide an altered and tunable radiation response of use in radiation dosimetry by incorporating 1 into a cocrystal or a salt. The lithium salt of PCDA has been reported in this context in the patent literature,58 however, no X-ray crystal structure has been obtained to understand the photoreactivity of the salt. As a result, organic coformers potentially offer a structural insight to photoreactivity, along with improved versatility and more facile processing because of enhanced solubility.
space group) based on the well-known OH⋯O R22(8) ring hydrogen-bonded carboxylic acid dimer synthon55,56 (Fig. 1). The structure is based on a head-to-head bilayer arrangement of molecules and the aliphatic substituents at either end of the dialkyne unit adopt an anti-conformation. The crystallographic c-axis of 46.647(3) Å is long, which is also apparent in the PXRD pattern, which shows a lamellar progression of low-angle peaks from 1.9–17.1° 2θ, all correlating to reflections in the (00l) plane (Fig. S1†).57 The structure conforms to the topochemical postulate for photoreactivity with a translational repeat distance of ≤4.9 Å, at 4.574(1) Å, along with a tilt angle of 44.7° that very close to the desired value of 45°. The inter-alkyne C1–C4′ distance between adjacent molecules of 1 is 3.712(1) Å, which is close to the upper limit of the topochemical postulate for alkyne reactivity (a maximum distance of 3.8 Å).3 These values are consistent with the limited observed solid state photoreactivity of 1 which, while it gradually turns visually blue upon exposure to UV and X-ray, the actual photoconversion as monitored by CP-MAS 13C NMR spectroscopy displays no alkene peaks, implying less than 1% even upon prolonged exposure (Fig. S2–S5†). Therefore, the question is raised as to whether the topochemistry of 1 can be engineered to provide an altered and tunable radiation response of use in radiation dosimetry by incorporating 1 into a cocrystal or a salt. The lithium salt of PCDA has been reported in this context in the patent literature,58 however, no X-ray crystal structure has been obtained to understand the photoreactivity of the salt. As a result, organic coformers potentially offer a structural insight to photoreactivity, along with improved versatility and more facile processing because of enhanced solubility.
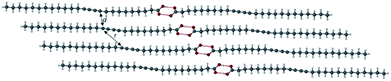 |
| | Fig. 1 The X-ray structure of 1 showing the carboxylic acid dimer (O⋯O distance of 2.6581(9) Å) and the anti-conformation of the diacetylene substituents. The C1–C4′ distance and intermolecular repeat distance are denoted by d and r, respectively. | |
PCDA cocrystals
Grinding PCDA (1) and 4,4′-azopyridine (2) in a Retsch MM 200 mixer mill for 1 hour in a 2:1 ratio, respectively (which reflects the single hydrogen bond donor group of 1 and the two hydrogen bond acceptor groups of 2), gave a powder of cocrystal 12·2, which was characterised by PXRD and used for seeding the cocrystallisation of 1 and 2 in acetone. No other stoichiometries were attempted. After the evaporation of solvent at room temperature for one week, plate-shaped crystals of 12·2 formed and were analysed by single crystal X-ray diffraction (SC-XRD). The structure of 12·2 reveals a 2:1 stoichiometry with the diacetylene substituents in anti-conformation, analogous to the structure of 1, with OH⋯N hydrogen bonds from the carboxylic acid protons of 1 to the pyridyl nitrogen atoms of 2 (Fig. 2a). The O⋯N distance of 2.677(4) Å is consistent with a strong, carboxylic acid OH⋯pyridyl hydrogen bond. The carboxylic acid proton was located experimentally in the X-ray structure and is situated on the oxygen atom of the carboxylic acid, ruling out the possibility of salt formation. The unit cell of 12·2 has a shorter crystallographic c-axis of 39.920(2) Å compared to 1 itself (by a considerable 6.87 Å) implying a more slanted orientation of the lamellar structure (Fig. 2b). Compared to 1, the cocrystal 12·2 also has a significantly shorter inter-alkyne C1–C4′ distance of 3.633(1) Å, however, the tilt angle of 1 in the cocrystal is greater than the optimum value at 48.4°, along with a translational repeat distance outside of the desired range for topochemical reactivity at 5.354(1) Å.
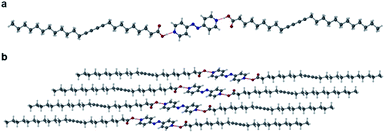 |
| | Fig. 2 (a) The X-ray structure of 12·2 showing the OH⋯N hydrogen bond (O⋯N distance of 2.677(4)) with the diacetylene substituents in an anti-conformation. (b) The packing diagram of 12·2 in the (100) crystallographic plane. | |
Along with SC-XRD, 12·2 was characterised by Differential Scanning Calorimetry (DSC) which displayed a melting onset temperature of 57 °C (Fig. S6†), which is lower than the melting temperatures of the individual components (62 °C for 1 (Fig. S7†) and 96 °C for 2 (ref. 59)) implying relatively weak interactions. The Fourier-transform infrared (FTIR) spectrum displays a hydrogen-bonded carbonyl stretching band at 1695 cm−1, compared to 1692 cm−1 in pure 1 implying slightly weaker hydrogen bonding (Fig. S8†). The cocrystal displays significant anisotropic thermal expansion along the c-axis which increases from 39.33 Å to 40.99 Å between 120 and 273 K. The differences in the unit cell made the calculated and experimental PXRD data difficult to compare, although it is clear that the single crystal studied is representative of the bulk material (Fig. S9†).
Two further cocrystals 12·3 and 12·4 were synthesised from 4,4′-bipyridyl and trans-1,2-bis(4-pyridyl)ethylene, respectively, by grinding the coformers with PCDA in a 2:1 ratio for 45 minutes in a mixer mill, to yield the cocrystal in powder form. Samples were characterised by PXRD and then used in seeded crystallisations in acetone. These experiments gave plates of 12·3 and 12·4 after the evaporation of solvent at room temperature for one week. A single crystal of 12·3 was analysed at the I19 beamline at the Diamond Light Source at 100 K, while crystals of 12·4 were analysed on a Bruker D8 Venture diffractometer at 120 K. The two materials are isostructural and crystallise in the monoclinic space group P21/c. The X-ray structures of cocrystals 12·3 and 12·4 consist of hydrogen bonds between the carboxylic acid hydrogen atom of 1 and the pyridyl nitrogen atom of the coformer at an O⋯N distance of 2.652(4) Å in 12·3 (Fig. 3a) and 2.6579(17) Å in 12·4 (Fig. 3b). Interestingly the dialkyne moieties in both structures adopt a syn-conformation, in contrast to the anti-conformation in 1 and 12·2 indicating that subtle modification of conformer can have a significant effect on crystal packing mode. The X-ray structures of 12·3 and 12·4 and their packing diagrams are shown in Fig. 3c and d. The ethylene bond of 12·4 is disordered over two positions. The C1–C4′ inter-alkyne distances between adjacent PCDA molecules in both cocrystals are both within the topochemical postulate at distances of 3.730(1) Å and 3.726(2) Å, respectively, although they are longer than those found in 1 and 12·2, because of the syn conformation of the dialkyne fragments. However, the tilt angle of PCDA in the cocrystals and the translational repeat distance for 12·3 is 47.4° and 5.442(1) Å, respectively, with similar values for 12·4 with a tilt angle of 1 in the cocrystal at 47.3° and a repeat distance of 5.449(2) Å. Therefore, neither of the isostructural cocrystals are expected to be photoreactive, as only one out of three of the parameters are within the topochemical postulate. The syn conformation of the dialkyne substituents allows an interdigitated, bilayer packing arrangement which translates to the much longer crystallographic c axes which encompass four folded molecules in the cocrystals of 3 and 4 as opposed to two extended molecules in 12·2.
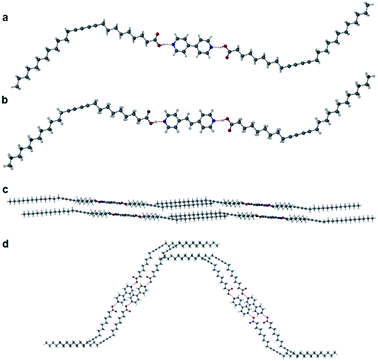 |
| | Fig. 3 (a) The X-ray structure of 12·3 with components joined by an OH⋯N hydrogen bond (O⋯N distance of 2.6448(9) Å) showing the syn-conformation of the dialkyne substituents. (b) The X-ray structure of 12·4 with OH⋯N hydrogen bonds (O⋯N distance of 2.6579(17) Å) and the diacetylene substituents in a syn-conformation (disorder omitted). Packing diagram of 12·3 in the (c) (010) and (d) (001) crystallographic planes. | |
The DSC thermogram of 12·3 displays a melt onset endotherm of 73 °C (Fig. S10†) (compared to the coformer melt temperatures for 1 and 3 of 62 °C and 114 °C,60 respectively), while 12·4 exhibits a melting onset temperature of 72 °C (Fig. S11†), compared to 150 °C for 4.61 The similar melting temperatures for the two cocrystals are expected due to the isostructural nature of these materials. The FTIR spectra for these cocrystals display a hydrogen-bonded carbonyl stretch at 1683 cm−1 and 1688 cm−1 respectively, compared to 1692 cm−1 in pure 1, implying slightly stronger hydrogen bonding (Fig. S12 and S13†). In a similar way to 12·2, cocrystals 12·3 and 12·4 show considerable anisotropic thermal expansion on warming (see Table S1†). This makes the calculated PXRD patterns appear somewhat different to the room temperature experimental patterns (Fig. S14 and S15†).
PCDA salts
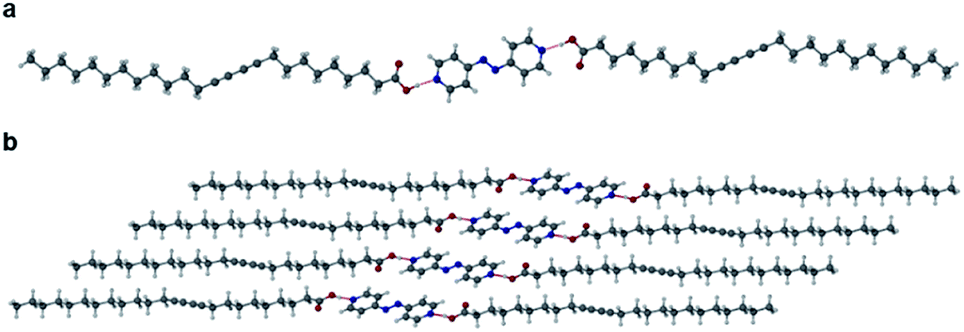
Cocrystals of PCDA with bifunctional coformers 2–4 appear to give structures that are unlikely to be photoreactive based on their topochemical metrics. As a result, we examined both mono- and bifunctional coformers with higher basicity intended to deprotonate the PCDA acid functionality and hence alter the hydrogen bonding pattern and change the consequent stacking of the PCDA units. Salt formation was undertaken with a bifunctional diamine (5), a cyclic amine (6), a linear secondary amine (7), and a linear terminal amine (8). PCDA and compounds 5–8 were mechanochemically ground in a mixer mill to give a range of new salt materials as indicated by FTIR analysis. The carboxylate asymmetric carbonyl stretching modes proved to be at lower wavenumbers than in the free acid (1, 1692 cm−1) with a carbonyl stretch at 1653 cm−1 in 12·5 (Fig. S16†) and 1·6 (Fig. S17†), 1627 cm−1 in 12·7 (Fig. S18†), and 1649 cm−1 in 1·8 (Fig. S19†), suggesting stronger hydrogen bonding in the salts than the cocrystals and a delocalised carboxylate anion structure. The X-ray structure of 12·5 reveals a salt with two anions of 1 and a dication of double protonated 5 in a 2:1 stoichiometry, respectively, consisting of NH⋯O hydrogen bonds from the amine hydrogen atom of 5 and the oxygen atom of 1, at an N⋯O distance of 2.717(1) Å (Fig. 4). The salt 12·5 crystallises with the same symmetry as 1 and 12·2 in the space group P, with the crystallographic c-axis at the shortest observed so far at 23.0041(15) Å. The C1–C4′ inter-alkyne distance between adjacent molecules of 1 is 3.760(2) Å, which is within the topochemical postulate for the reactivity of diacetylenes (≤3.8 Å), however, the tilt angle of 1 in the salt cocrystal is below the desired value (45°) at 24.1°, and the translational repeat distance of 5.577(2) Å is outside the maximum distance for this parameter (≤4.9 Å) again suggesting limited photoreactivity.
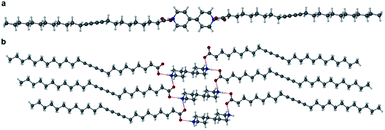 |
| | Fig. 4 The X-ray structure of the salt cocrystal 12·5 in the (a) (100) and (b) (001) crystallographic planes. | |
The morpholinium salt 1·6 was crystallised by the slow evaporation of acetone at room temperature, however, due to poor crystal quality after repeated crystallisation attempts, no SC-XRD analysis of 1·6 could be undertaken. To model the interactions between the two components, the synthesis of the butanoic acid (BuA) salt of 6 was attempted. Large single crystals of BuA·6 formed from equimolar amounts of reagents in a sealed flask allowed to stand overnight. The X-ray structure reveals a salt with a butanoate anion and protonated morpholinium cation (Fig. 5). The structure involves two unique NH⋯O hydrogen bonding interactions with N⋯O distances of 2.673(1) Å and 2.732(1) Å. Based on the similar pKa of 1 and butanoic acid it is possible that 1·6 is also a salt with similar head-group structure, although the relevance of this model system to the PCDA analogue is otherwise limited.
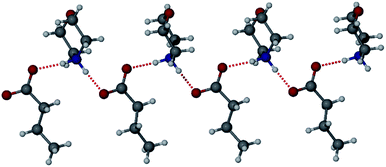 |
| | Fig. 5 The X-ray structure of BuA·6 showing two different hydrogen bonding interactions. | |
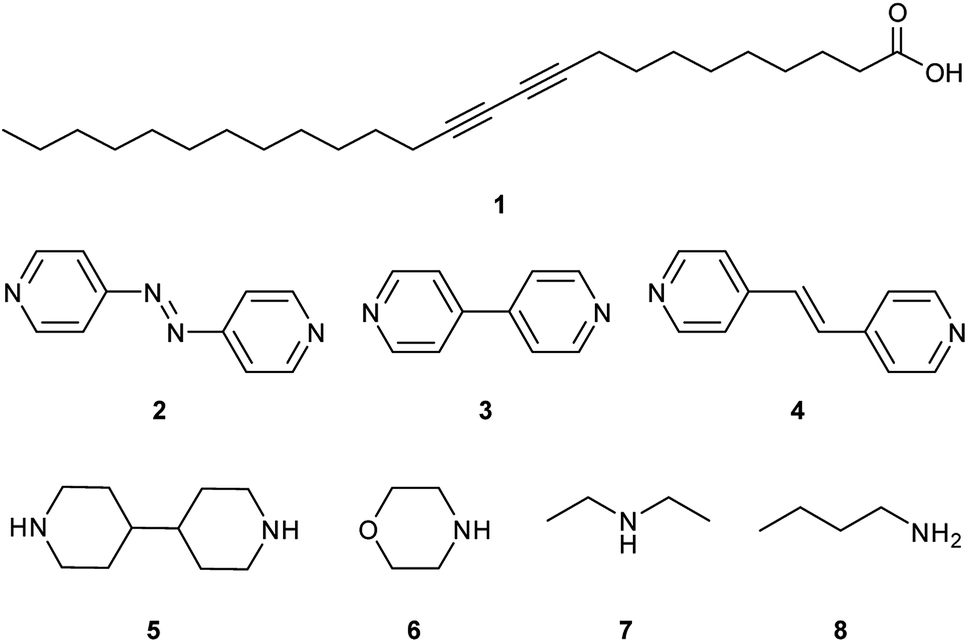
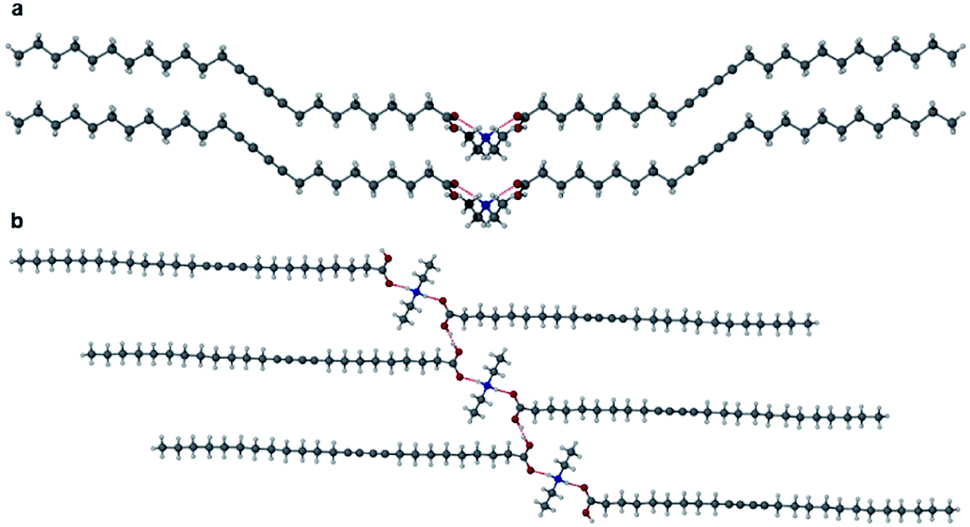
Salts of PCDA with diethylamine (7) and n-butylamine (8) crystallised by slow evaporation of acetone solutions at room temperature. Surprisingly the crystals are highly coloured purple and blue, respectively, consistent with facile photopolymerisation (Fig. 6). However, the X-ray structure determinations reveal salts of unpolymerised PCDA and hence the colouration is likely to be a surface effect. Indeed, cutting a single crystal in half revealed a colourless inner core. The structure of the diethylammonium salt proved to be a salt cocrystal that also includes a neutral molecule of 1 (Fig. 7), with formula 12·7. The butylammonium compound is a 1:1 salt of formula 1·8. The structure adopts a stacked bilayer arrangement (Fig. 8). In 12·7, hydrogen bonding occurs from the ammonium NH hydrogen atoms to the carbonyl oxygen of 1, with an N⋯O distance of 2.737(1) Å. The carboxylic acid group of the neutral PCDA hydrogen bonds to the carboxylate functionality on the PCDA anion with a very short O⋯O distance of 2.444(1) Å (the additional hydrogen atom present between PCDA and the PCDA anion is disordered). In the 1:1 salt 1·8, there are three different hydrogen bond interactions form from the NH3+ cation to the carboxylate oxygen atoms of the PCDA anion, with NH⋯O distances of 2.671(1) Å, 2.725(1) Å, and 2.784(1) Å. The 12·7 structure also has a large c-axis of 57.520(4) Å, which is the longest c-axis of all the structures studied reflecting the linear, parallel arrangement of the PCDA components. Salts 12·7 and 1·8 have similar C1–C4′ inter-alkyne distances of 3.776(2) Å and 3.779(1) Å, respectively, with tilt angles of 41.9° and 43.7°, and translational repeat distances of 4.644(3) Å and 4.593(1) Å. For these two salts, all three values are well within the optimum values of the topochemical postulate, and they are therefore are expected to show significant photoreactivity, consistent with the spontaneous surface colouration of the crystals.
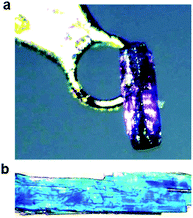 |
| | Fig. 6 Photographs of (a) the purple crystal of 12·7 and (b) the blue crystals of 1·8, taken before X-ray irradiation. | |
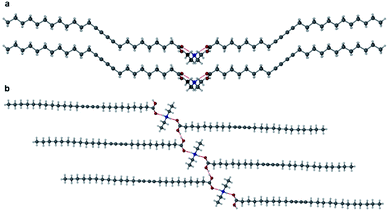 |
| | Fig. 7 The X-ray structure of 12·7 in the (a) (100) and (b) (010) crystallographic planes. | |
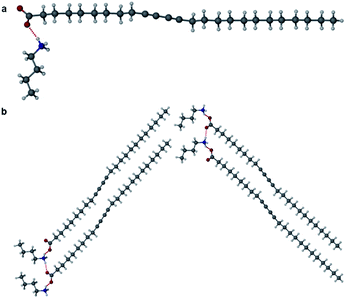 |
| | Fig. 8 (a) The X-ray structure of 1·8 (b) in the (100) crystallographic plane. | |
DSC analyses of the PCDA salts of 5–8 reveal melt onset endotherms of 107 °C for 12·5 (Fig. S20†) (compared to 62 °C and 170 °C (ref. 62) for the parent components 1 and 5, respectively). This relatively high value likely reflects the fact that proton transfer has occurred as well as the higher melting point of the bipiperidine coformer. The morpholinium salt 1·6 has a low melting onset of 44 °C (Fig. S21†) consistent with the fact that morpholine is a liquid at room temperature (it boils at 128 °C).63 In the same way as 1·6, the DSC thermogram of 12·7 exhibits a melt onset endotherm of 46 °C (Fig. S22†), in comparison to the boiling temperature of 55 °C for 7,64 while 1·8 displays a melt onset endotherm at 57 °C (Fig. S23†), with the salt former 8 boiling at 77 °C.65
Cocrystal and salt response to UV and X-rays
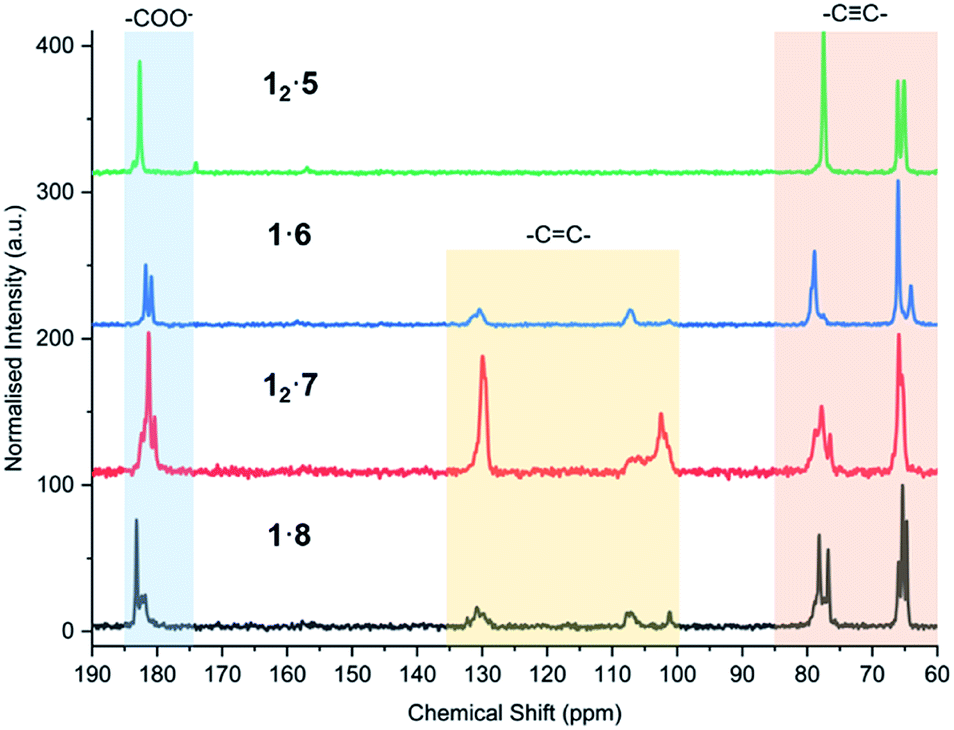
The powder of each cocrystal and salt was placed on filter paper in a dark box and exposed to a 6-Watt handheld UV light at 254 nm for varying durations (Fig. 9). It is known that the azobenzene coformer 2 itself undergoes photoisomerisation to the cis form when irradiated at 365 nm (ref. 66) and so 12·2 was also irradiated at this wavelength in order to probe photoresponse of the coformer component within the cocrystal. While PCDA powder itself gradually darkens from a white to deep blue upon irradiation, all of the cocrystals with coformers 2–4 do not change colour despite the close proximity of the dialkyne functionalities, which are within the distance specified by the topochemical postulate. However, the tilt angles of 1 in the cocrystals, and the translational repeat distances of the cocrystals are outside of the desired values. The irradiated cocrystals were analysed by PXRD, solid-state CP-MAS 13C NMR spectroscopy, and FTIR spectroscopy. This data confirmed that the cocrystal samples do not undergo photopolymerisation after irradiation (Fig. S24–S32†). However, after one hour and one day, 12·2 and 12·4 after one day, display additional peaks in the PXRD patterns and solid-state NMR spectra that are attributable to 1. This suggests that irradiation decomposes the cocrystals to liberate free 1, particularly in the case of the azobenzene complex 12·2. This behaviour is attributed to the photoisomerisation of coformer 2 resulting in degradation of the cocrystal. The PXRD pattern and solid-state NMR spectra also imply a less-crystalline material at one hour in 12·2 and show the presence of crystalline 1 after one day of irradiation. In addition, the solid-state NMR spectroscopic data revealed that even the deep blue colour of the irradiated sample of PCDA is a surface effect and the material undergoes <1% photopolymerization, implying that the radiation is not penetrating the bulk of the sample. The bipiperidinium salt 12·5 is also not photoresponsive (Fig. S33 and S34†), consistent with the unfavourable translational repeat distance observed in the X-ray structure. From these results, bifunctional salt/coformers in general seem to give rise to a slightly offset packing mode that consistently results in an unfavourable repeat distance and tilt angle and hence essentially no photosensitivity. In contrast, the salts of monofunctional ammonium cations are all highly photoactive. Significant visual colour change occurs after just five minutes of irradiation for the salts 1·6, 12·7 and 1·8 (Fig. 9). Signals assigned to photopolymerised material are clearly visible by CP-MAS 13C NMR spectroscopy (Fig. 10 and S35–S37†). Salt 12·7 shows the greatest sensitivity towards UV radiation by CP-MAS 13C NMR spectroscopy with the most significant change occurring in the alkene region (100–140 ppm) of the spectrum corresponding to the ene–yne photopolymer functionality (Fig. 10). However, even in these systems the conversion is slow, and the sharpness of the NMR resonances imply a relatively low degree of oligomerisation. This kind of slow reactivity reflects the solid-state nature of the process resulting in poor radiation penetration into the bulk of the sample. However, this gradual response is desirable in dosimetry applications making these materials of considerable interest. The significant photoreactivity of 12·7 and 1·8 is consistent with the crystal packing revealed by their structures, which both show parameters within the range specified by the topochemical postulate. While structural data is not available for the morpholinium salt, it seems likely that this too is within the topochemical postulate range. The topochemical parameters for each compound are summarised in Table 1.
 |
| | Fig. 9 The coformer 1 and cocrystals 12·2, 12·3 and 12·4, and salts 12·5, 1·6, 12·7, 1·8 and the lithium salt (Li salt) before and after UV irradiation at 254 nm for different durations. The 12·2a sample was irradiated with UV light at 365 nm while 254 nm radiation was used for the 12·2b sample. The initial colour changes to UV radiation show that only 1 and the salt samples visibly change colour with prolonged irradiation, with 12·7 being the most radiation sensitive to the naked eye and experimentally. | |
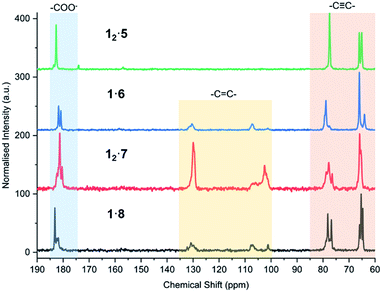 |
| | Fig. 10 CP-MAS 13C NMR spectra of 12·5, 1·6, 12·7, and 1·8 after 7 days of UV irradiation at 254 nm, highlighting the alkyne carbons (60–85 ppm), alkene carbons of the photoreactive salts (100–135 ppm), and carboxylate environment of each salt (175–185 ppm). | |
Table 1 Topochemical parameters for PCDA cocrystals and salts structurally characterised in this work. The tilt angle (θ) is calculated from the orientation of 1 in the cocrystal and salt cocrystal samples
| Compound |
d/Å |
r/Å |
θ/° |
|
1
|
3.712(1) |
4.574(1) |
44.7 |
|
12·2
|
3.633(1) |
5.354(1) |
48.4 |
|
12·3
|
3.730(1) |
5.442(1) |
47.4 |
|
12·4
|
3.726(2) |
5.449(2) |
47.3 |
|
12·5
|
3.760(2) |
5.577(2) |
24.1 |
|
12·7
|
3.776(2) |
4.644(3) |
41.9 |
|
1·8
|
3.779(1) |
4.593(1) |
43.7 |
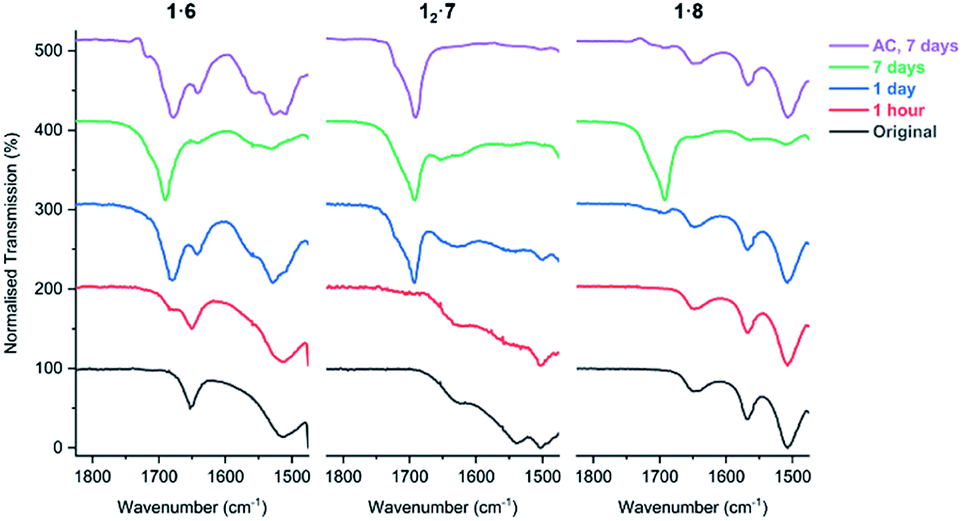
FTIR analysis of the salt cocrystals after irradiation shows that salts 1·6, 12·7, and 1·8 begin to lose their volatile coformers after prolonged UV exposure and revert to free carboxylic acids. This is evidenced by the decrease in intensity of the carboxylate asymmetric stretch band νasymm(CO2) of the salt (1653 cm−1 in 1·6, 1627 cm−1 in 12·7, and 1649 cm−1 in 1·8) and the emergence of a free acid peak at 1692 cm−1 close to the value of PCDA as the sample is irradiated (Fig. 11). The effect is highly pronounced for the morpholinium salt 1·6 which reverts to free acid after just one hour while 7 and 8 begin to separate from their respective salts after one day of irradiation. The resulting carboxylic acid is a mixture of free PCDA and photopolymer. These findings are also supported by PXRD analysis of the irradiated salts (Fig. S38–S40†). Interestingly, given the very limited photoreactivity of PCDA itself, salt formation followed by removal of the amine in this way gives an interesting route to the free acid photopolymer and hence transient amine complexation effectively catalyses the photopolymerisation reaction of PCDA itself. The salts were also exposed to ambient conditions for seven days to investigate whether the amine is lost from the salts without irradiation. The FTIR of salts 1·6 and 1·8 display changes consistent with UV irradiation for one day, while salt 12·7 displays total loss of amine from the salt under ambient conditions after seven days (Fig. 11).
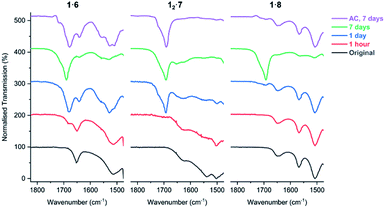 |
| | Fig. 11 The FTIR spectra of salts 1·6, 12·7, and 1·8 in the range of 1475–1825 cm−1 showing the carboxylate environment of the salts with volatile amines as the samples are irradiated with UV light (254 nm), and exposure to ambient conditions (AC) for seven days. | |
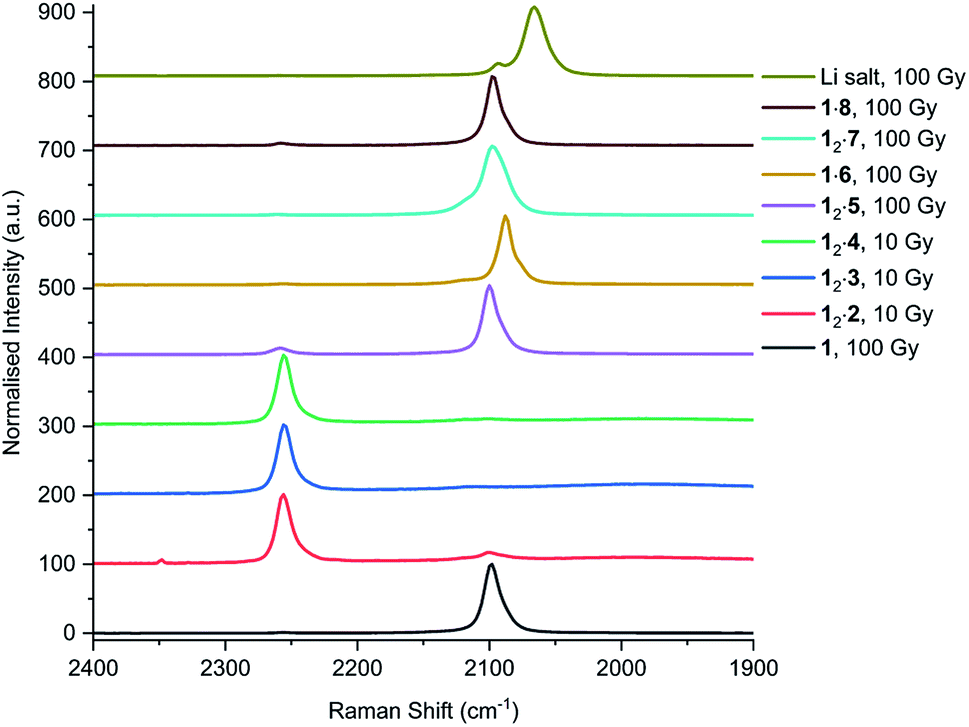
In addition to UV irradiation, the effect of X-ray on PCDA and its derivatives was also analysed. Free PCDA (1) was irradiated with 100 Gy of X-ray radiation and analysed by Raman spectroscopy (Fig. 12). This revealed a clear ene–yne polymer alkyne band at 2098.8 cm−1 with a small residual dialkyne band at 2253.3 cm−1 (Fig. S41†). The enhanced appearance of the 2098.8 cm−1 band despite the very limited photoreactivity of free 1 is a reflection of a pre-resonance Raman effect since the excitation wavelength of the laser 785 nm overlaps with the absorption band of the photopolymer, resulting in significant enhancement of the chromophore Raman bands. This is consistent with the visual observation of some blue colouration despite the 13C CP MAS-NMR data that indicate a very low degree of bulk conversion (Fig. S4†). In contrast, when all three cocrystals with coformers 2–4 were irradiated with 10 Gy of X-ray radiation they showed very little photoreactivity, as evidenced by the low intensity peaks in the conjugated ene–yne region (approx. 2100 cm−1)67 that exists both before and after irradiation (Fig. S42–S44†). Cocrystal 12·2 has a small ene–yne band present at 2100.4 cm−1 compared to the other two cocrystals likely arising from small amounts of 1 photopolymer present as a contaminant in the starting PCDA. Similarly to the cocrystals, salt 12·5 displays a band at 2258.4 cm−1 assigned to unreacted dialkyne even after 100 Gy of X-ray irradiation which further reinforces that the salt is photostable (Fig. S45†). The small ene–yne photopolymer band at 2100.3 cm−1 is likely to arise from small amounts of photopolymerised PCDA impurities. On the other hand, salt 1·6 shows impressive sensitivity to X-ray radiation as indicated by the presence of the significant ene–yne band at 2088.1 cm−1 (Fig. S46†). This band is significantly red-shifted compared to photopolymerised PCDA, indicating a more planar, conjugated conformation of the chromophore. This is in contrast to 1 alone which exhibits torsional strain on the π-bonds of the chromophore when irradiated.68 Salt 1·6 also shows significantly more visual colour change upon irradiation compared to 1 alone. It is likely that the increased hydrogen bonding in the salt brings the monomers of 1 in a closer spatial arrangement and hence makes it more photosensitive. After 100 Gy of X-ray irradiation, salt 12·7 also displays a prominent photopolymer alkyne band at 2097.7 cm−1 with minimal residual dialkyne signal (Fig. S4†). The pre-resonance Raman effect is very evident in 12·7 and can be seen in the exaggeration of the ene–yne band in Fig. 12 relative to the dialkyne band in the region of 2250 cm−1. Solid-state NMR results indicate about 53% polymerisation (Fig. S47†), however the Raman signal for the colourless monomer is almost invisible. Interestingly in the Raman spectrum of 12·7, the breadth of the ene–yne band, and the presence of an additional alkene peak at 1500 cm−1 at slightly higher wavenumber than the typical major 1445 cm−1 alkene band indicates multiple conformations of the polymerised material, and implies some structural differences in the resulting chromophore suggesting that multiple conformations of the polymerised salt exist. For 1·8, Raman analysis of the 100 Gy X-ray irradiated sample shows that the salt gradually photopolymerises and has a similar radiation sensitivity as 1 alone, with an ene–yne band at 2098.1 cm−1 (Fig. S48†). Additionally, for the photosensitive salts, the C–H wagging progressions arising between 1300 cm−1 and 1150 cm−1 from the polymer side chains of 1 in the salts change with irradiation to suggest a changed conformational structure when compared to the lithium salt (Fig. 13). The change of the side chain conformation is due to difference in phase angles of coupled oscillations between methylene groups. These differences in C–H wagging progressions can be used as an additional conformational tool for detecting the presence of a PCDA polymer. Close examination of the differences in frequency within the wagging mode progressions may also indicate stresses on the side chains due to their close approach to each other as the polymer is formed. Interestingly, the positions of the ene–yne alkyne bands in the irradiated diethylammonium and butylammonium salts of around 2100 cm−1 contrast sharply with the value of 2066.3 cm−1 obtained for commercial lithium PCDA. This significantly red-shifted value implies a much more planar ‘ordered’ chromophore in the lithium salt and hence while the commercial material exists in an ordered ‘blue state’ the use of the organic salt-formers give a less ordered ‘red state’ photopolymer. The value of 2088 cm−1 for the morpholinium salt is somewhere in between and implies that the polymer ordering and hence, potentially, colour may be tunable.
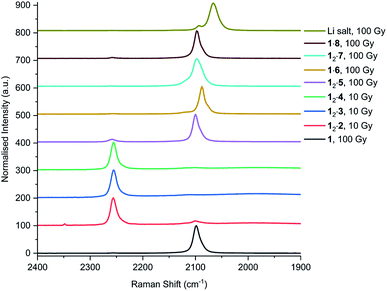 |
| | Fig. 12 Raman spectra of 1, and cocrystal and salts after X-ray irradiation. The peaks at approx. 2260 cm−1 correlate to the alkyne band of monomeric 1, while the internal alkyne of the photopolymer is displayed at approx. 2100 cm−1. | |
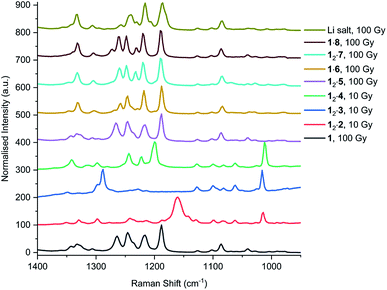 |
| | Fig. 13 The methylene wagging vibrations of 1 and each cocrystal and salt after X-ray irradiation, analysed by Raman spectroscopy. | |
In an attempt to assess the degree of polymerisation, the irradiated samples were analysed by two different mass spectrometry techniques, matrix-assisted laser desorption/ionisation (MALDI) and atmospheric solid analysis probe (ASAP). However, neither method was able to fully quantify the amount of polymer present in the samples nor the molecular weight distribution. MALDI did not generate any signals assignable to photopolymerised material. In contrast, the ASAP technique did show peaks assigned to PCDA monomer, dimer, and trimer, however, the distribution of these signals was the same before and after UV irradiation for one day for PCDA itself and even for 12·7 UV irradiated for 14 days. It is likely that higher molecular weight oligomers are present given the broadness of the CP-MAS NMR spectra, therefore, these are not volatile enough to be detected by mass spectrometry in this way. Similarly, the poor solubility of the polymerised materials made them unsuitable for gel permeation chromatography.
Conclusion
Three PCDA cocrystals 12·2, 12·3, and 12·4 have been prepared which were engineered to exhibit the common carboxylic acid⋯pyridine supramolecular synthon. The lamellar structures of these surfactant-like compounds exhibit two very different packing modes with either a syn or anti conformation of the dialkyne substituents and display considerable thermal expansion in the c-axis direction upon warming. The cocrystal with the shortest dialkyne distance of 3.631(6) Å is 12·2, which is shorter than in PCDA itself and all cocrystals have inter-alkyne distances within the topochemical postulate. However, they all exhibit very little response to UV and X-ray radiation, because the translational repeat distances are greater than the maximum value. Hence, cocrystallisation appears to significantly stabilise PCDA in the solid state. This observation is somewhat surprising in the light of the photoreactivity of related pyridyl-containing cocrystals of di- and trialkynes.45,69 In contrast, salts of monofunctional amines gave highly photoreactive materials. For example, the morpholinium salt 1·6 changes from pink-to-blue-to-black in under 10 minutes of UV irradiation. Similarly the salt cocrystal with diethylamine 12·7 and the butylammonium salt 1·8 are both highly radiation sensitive and undergo an impressive lilac-to-black colour change with under 5 minutes of UV irradiation for 12·7 and a pink-to-navy colour change for 1·8, in powder form. Their X-ray crystal structures indicate that these salts adhere to the topochemical postulate. These radiation-sensitive salts are of considerable commercial interest for the development of radiochromic films and due to their different sensitivities to radiation, can be applied to different radiation dose ranges, and therapeutic areas. Preliminary indications suggest that they outperform the commercial lithium salt in terms of photosensitivity, while Raman spectroscopy shows that the photopolymers are relatively disordered with ene–yne bands in the range 2088–2100 cm−1, potentially allowing access to a range of colours. The novel feature of amine evaporation over time means that transient ammonium salt formation with a volatile amine effectively catalyses the solid-state photopolymerisation of the relatively unreactive PCDA.
Experimental
General
10,12-Pentacosadynoic acid (1) was supplied by Ashland LLC, with all other reagents and solvents purchased from standard commercial sources and used without further purification. IR spectra were measured with a PerkinElmer 100 FT-IR spectrometer with an uATR attachment. Raman spectra were collected on a PerkinElmer Ramanstation 400F with 5–10 accumulations of 10–60 second scans, using an excitation laser with a wavelength of 785 nm. Solid-state NMR spectra were recorded at 100.63 MHz using a Bruker Avance III HD spectrometer and a 4 mm magic-angle spinning probe. Spectra were obtained using cross-polarisation with a 20 s recycle delay with 7 ms contact time at ambient probe temperature (approx. 25 °C) at a sample spin rate of 10 kHz with 400 repetitions. Spectral referencing was with respect to an external sample of neat tetramethylsilane. Differential scanning calorimetry thermograms were recorded using a PerkinElmer 8500 calorimeter, calibrated using an indium standard, with samples accurately weighed (±0.01 mg) into standard aluminium pans.
Mass spectra by the ASAP technique were recorded using a LCT Premier XE mass spectrometer (Waters Ltd) heating approximately 1 mg of powder isothermally at 350 °C. Single crystal X-ray data for the cocrystals and salts 12·2, 12·4, 12·5, BuA·6, and 12·7 were collected at 120.0(2) K on a Bruker D8 Venture diffractometer (Photon 100 CMOS detector, IuS-microsource focusing mirrors) equipped with Cryostream (Oxford Cryostreams) open-flow nitrogen cryostat and using Mo Kα and Cu Kα radiation with wavelengths of 0.71073 Å and 1.54178 Å, respectively. The single crystal data for 1·8 was collected at 120.0 K on an XCalibur Agilent, Sapphire3 diffractometer equipped with Cryostream 700 nitrogen cryostat and using Mo Kα radiation with a wavelength of 0.71073 Å. Single crystal data for 1 and 12·3 were collected at 100.0(2) K at I19 beamline (Dectris Pilatus 2M pixel-array photon-counting detector, undulator, graphite monochromator, λ = 0.6889 Å) at the Diamond Light Source, Oxfordshire. All structures were solved using direct methods and refined by full-matrix least squares on F2 for all data using SHELXL50 and OLEX2 software.51 All non-hydrogen atoms were refined with anisotropic displacement parameters. CH hydrogen atoms were placed in calculated positions, assigned an isotropic displacement factor that is a multiple of the parent carbon atom and allowed to ride. H atoms attached to oxygen atoms were located on the difference map when possible or placed in calculated positions. X-ray powder diffraction patterns were performed on glass slides, using a Bruker AXS D8 Advance diffractometer, with a Lynxeye Soller PSD Detector, using Cu Kα radiation at a wavelength of 1.5406 Å. CCDC deposition numbers 2000830–2000837.† The powdered cocrystal and salt samples were placed on filter paper in a dark box and exposed to a 6-Watt handheld UV light at 254 nm. Compound 12·2 was also irradiated at 365 nm. The powdered samples were mixed at regular intervals to ensure an even exposure of the bulk to irradiation.
Synthesis of PCDA, cocrystals, and salts
10,12-Pentacosadiynoic acid (1).
Analysis calc. of C25H42O2: C 80.16, H 11.30%, found: C 80.05, H 11.18%; FTIR (ν/cm−1): 2956, 2918, 2848, 1692, 1467, 1460, 1417, 1291, 1264, 932, 722. Colourless needle crystals of high quality were grown from the slow evaporation of acetone at room temperature for one week from a failed cocrystallisation experiment with pyrazine. Crystal data: M = 374.58 g mol−1, 0.12 × 0.04 × 0.01 mm3, triclinic, space group P (no. 2), a = 4.5738(3) Å, b = 5.3909(3) Å, c = 46.647(3) Å, α = 88.6499(15)°, β = 88.5073(14)°, γ = 81.4017(14)°, V = 1136.64(12) Å3, Z = 2, Dc = 1.094 g cm−3, F000 = 416.0, synchrotron radiation, λ = 0.6889 Å, T = 100 K, 2θmax = 58.994°, 21023 reflections collected, 6893 unique (Rint = 0.0574). Final GooF = 1.035, R1 = 0.0640, wR2 = 0.1772, R indices based on 6893 reflections with I ≥ 2σ(I) (refinement on F2), 249 parameters, 0 restraints, μ = 0.063 mm−1.
PCDA 4,4′-azopyridine cocrystal (12·2).
Cocrystal 12·2 was prepared by grinding 1 (0.15 g, 0.40 mmol) and 2 (0.037 g, 0.20 mmol) in a 2:1 ratio, respectively, in a Retsch MM 200 mixer mill for 1 hour at a frequency of 20 s−1 (yield 0.10 g, 0.22 mmol, 55%). The resulting powder was used as a seed (ca. 0.005 g, 0.011 mmol) for the cocrystallisation of 1 (0.025 g, 0.067 mmol) and 2 (0.006 g, 0.033 mmol) in acetone (2 mL). After brief sonication, the solution was left to crystallise by slow evaporation at room temperature, which yielded large colourless plate crystals. Analysis for C30H46N2O2 calc.: C 77.19, H 9.94, N 6.01%, found: C 77.19, H 9.93, N 5.32%; FTIR (ν/cm−1): 2936, 2919, 2850, 1695, 1598, 1470, 1410, 1253, 1214, 1184, 1011, 848, 719, 573. Crystal data: M = 466.69 g mol−1, 0.32 × 0.09 × 0.02 mm3, triclinic, space group P (no. 2), a = 5.3544(3) Å, b = 6.8239(4) Å, c = 39.920(2) Å, α = 87.742(4)°, β = 88.869(4)°, γ = 75.291(4)°, V = 1409.64(14) Å3, Z = 2, Dc = 1.100 g cm−3, F000 = 512.0, Cu Kα radiation, λ = 1.54178 Å, T = 120 K, 2θmax = 137.98°, 33397 reflections collected, 5160 unique (Rint = 0.1445). Final GooF = 1.090, R1 = 0.0994, wR2 = 0.1798, R indices based on 5160 reflections with I ≥ 2σ(I) (refinement on F2), 312 parameters, 0 restraints, μ = 0.522 mm−1.
PCDA 4,4′-bipyridyl cocrystal (12·3).
Cocrystal 12·3 was prepared by grinding 1 (0.15 g, 0.40 mmol) and 3 (0.032 g, 0.20 mmol) in a Retsch MM 200 mixer mill for 45 minutes at a frequency of 20 s−1 (yield = 0.091 g, 0.20 mmol, 50%). The resulting powder was used as a seed (ca. 0.005 g, 0.011 mmol) for the cocrystallisation of 1 (0.025 g, 0.067 mmol) and 3 (0.053 g, 0.034 mmol) in acetone (2 mL). After brief sonication, the solution was left to crystallise by slow evaporation at room temperature, which yielded small colourless plate crystals. Analysis calc. of C30H46N2O2: C 79.60, H 10.24, N 3.09%, found: C 79.24, H 10.20, N 3.12%; FTIR (ν/cm−1): 2919, 2850, 1683, 1600, 1471, 1408, 1365, 1325, 1287, 1253, 1212, 1187, 1071, 1000, 821, 718, 625. Crystal data: M = 452.68 g mol−1, 0.12 × 0.04 × 0.03 mm3, monoclinic, space group P21/c (no. 14), a = 5.4415(2) Å, b = 8.9535(4) Å, c = 55.673(3) Å, β = 90.8823(10)°, V = 2712.1(2) Å3, Z = 4, Dc = 1.109 g cm−3, F000 = 996.0, synchrotron radiation, λ = 0.6889 Å, T = 100 K, 2θmax = 57.994°, 47696 reflections collected, 7923 unique (Rint = 0.0663). Final GooF = 1.069, R1 = 0.0529, wR2 = 0.1510, R indices based on 7923 reflections with I ≥ 2σ(I) (refinement on F2), 303 parameters, 0 restraints, μ = 0.064 mm−1.
PCDA trans-1,2-bis(4-pyridine)ethylene cocrystal (12·4).
Cocrystal 12·4 was prepared by grinding 1 (0.15 g, 0.40 mmol) and 4 (0.039 g, 0.21 mmol) in a Retsch MM 200 mixer mill for 45 minutes at a frequency of 20 s−1 (yield = 0.12 g, 0.26 mmol, 65%). The resulting powder was used as a seed (ca. 0.005 g, 0.011 mmol) for the cocrystallisation of 1 (0.025 g, 0.067 mmol) and 4 (0.0061 g, 0.033 mmol) in acetone (2 mL). After brief sonication, the solution was left to crystallise by slow evaporation at room temperature and yielded small colourless block crystals. Analysis calc. of C31H47NO2: C 79.95, H 10.17, N 3.01%, found: C 79.84, H 10.14, N 2.86%; FTIR (ν/cm−1): 2919, 2850, 1688, 1604, 1471, 1414, 1325, 1252, 1235, 1213, 1183, 1100, 974, 826, 718, 553. Crystal data: M = 465.69 g mol−1, 0.36 × 0.31 × 0.23 mm3, monoclinic, space group P21/c (no. 14), a = 5.4494(3) Å, b = 8.9235(5) Å, c = 57.441(3) Å, β = 92.643(2)°, V = 2790.2(3) Å3, Z = 4, Dc = 1.109 g cm−3, F000 = 1024.0, Mo Kα radiation, λ = 0.71073 Å, T = 120 K, 2θmax = 56.98°, 34945 reflections collected, 6537 unique (Rint = 0.0635). Final GooF = 1.034, R1 = 0.0560, wR2 = 0.1073, R indices based on 6537 reflections with I ≥ 2σ(I) (refinement on F2), 321 parameters, 1 restraint, μ = 0.067 mm−1.
PCDA 4,4′-bipiperidine salt (12·5).
Salt 12·5 was prepared by grinding 1 (0.2 g, 0.53 mmol) and 5 (0.045 g, 0.27 mmol) in a 2:1 ratio in a Retsch MM 200 mixer mill for 90 minutes at a frequency of 20 s−1 (yield = 0.22 g, 0.40 mmol, 89%). The resulting powder (0.030 g) was combined with ethanol (2 mL) and briefly sonicated and left to crystallise by slow evaporation at room temperature. Colourless plate crystals formed after 2 weeks. Analysis calc. of C60H104N2O4: C 78.55, H 11.43, N 3.05%, found: C 77.94, H 11.36, N 2.83%; FTIR (ν/cm−1): 2917, 2849, 1692, 1644, 1527, 1467, 1418, 1305, 1266, 1231, 1099, 1011, 921, 868, 807, 719, 639. Crystal data: M = 917.45 g mol−1, 0.767 × 0.314 × 0.1 mm3, triclinic, space group P (no. 2), a = 5.5770(4) Å, b = 11.8339(8) Å, c = 23.0041(15) Å, α = 100.670(2)°, β = 96.096(2)°, γ = 103.007(2)°, V = 1436.25(17) Å3, Z = 1, Dc = 1.061 g cm−3, F000 = 510.0, Mo Kα radiation, λ = 0.71073 Å, T = 120 K, 2θmax = 65°, 36532 reflections collected, 10400 unique (Rint = 0.0455). Final GooF = 1.019, R1 = 0.0497, wR2 = 0.1263, R indices based on 10400 reflections with I ≥ 2σ(I) (refinement on F2), 312 parameters, 0 restraints, μ = 0.064 mm−1.
PCDA morpholine salt (1·6).
Salt 1·6 was prepared by grinding 1 (0.2 g, 0.53 mmol) and 6 (0.046 mL, 0.53 mmol) in a Retsch MM 200 mixer mill for 45 minutes at a frequency of 20 s−1 (yield = 0.24 g, 0.52 mmol, 98%). The resulting powder of 1·6 (0.030 g) was combined with acetone (3 mL) and briefly sonicated and left to crystallise by slow evaporation at room temperature. Colourless plate crystals formed after 2 weeks, however, crystallisations with and without seeding yielded poor-quality crystals that did not diffract sufficiently to allow single crystal structure determination. Analysis calc. of C29H51NO3: C 75.45, H 11.14, N 3.03%, found C 75.18, H 11.08, N 2.80%; FTIR (ν/cm−1): 2917, 2850, 1652, 1515, 1474, 1406, 1297, 1108, 879, 728, 616.
Morpholinium butanoate salt (BuA·6).
Salt 1·6 was prepared by combining butanoic acid (0.05 mL, 0.55 mmol) and 6 (0.047 mL, 0.55 mmol) to give a yellow oil and was left to precipitate slowly overnight in a sealed round-bottom flask to yield large colourless plate crystals (yield = 0.088 g, 0.5 mmol, 91%). Analysis calc. of C8H17NO3: C 54.84, H 9.78, N 7.99%, found: C 54.38, H 10.06, N 7.70%; FTIR (ν/cm−1): 2961, 2871, 1711, 1545, 1456, 1394, 1379, 1303, 1243, 1195, 1107, 1049, 997, 878, 829, 766, 615. Crystal data: M = 175.22 g mol−1, 0.44 × 0.25 × 0.21 mm3, monoclinic, space group C2/c (no. 15), a = 20.0926(14) Å, b = 8.0678(6) Å, c = 11.6061(8) Å, β = 97.064(3)°, V = 1867.1(2) Å3, Z = 8, Dc = 1.247 g cm−3, F000 = 768.0, Mo Kα radiation, λ = 0.71073 Å, T = 120 K, 2θmax = 58.994°, 13471 reflections collected, 2587 unique (Rint = 0.0313). Final GooF = 1.023, R1 = 0.0356, wR2 = 0.0931, R indices based on 2587 reflections with I ≥ 2σ(I) (refinement on F2), 177 parameters, 0 restraints, μ = 0.094 mm−1.
PCDA diethylamine salt cocrystal (12·7).
Salt cocrystal 12·7 was prepared by grinding 1 (0.2 g, 0.53 mmol) and 7 (0.055 mL, 0.53 mmol) in a Retsch MM 200 mixer mill for 45 minutes at a frequency of 20 s−1 (yield = 0.22 g, 0.5 mmol, 94%). The resulting powder of 12·7 (0.030 g) was combined with acetone (3 mL) and briefly sonicated and left to crystallise by slow evaporation at room temperature. Purple plate crystals formed after 2 weeks to reveal a 2:1 (PCDA:diethylamine) stoichiometry with the formula (C25H42O2)·(C25H41O2)−·(C4H12N+). Analysis calc. for 12·7: C 78.87, H 11.64, N 1.70%, found: C 77.47, H 11.61, N 1.70%; FTIR (ν/cm−1): 2923, 2846, 1627, 1461, 1423, 1384, 1068, 1010, 955, 854, 811, 724, 592. Crystal data: M = 822.3 g mol−1, 0.5 × 0.12 × 0.1 mm3, monoclinic, space group P2/n (no. 13), a = 9.5968(6) Å, b = 4.6441(3) Å, c = 57.520(4) Å, β = 92.590(2)°,V = 2561.0(3) Å3, Z = 2, Dc = 1.066 g cm−3, F000 = 916.0, Mo Kα radiation, λ = 0.71073 Å, T = 120 K, 2θmax = 55.996°, 30938 reflections collected, 6051 unique (Rint = 0.0502). Final GooF = 1.049, R1 = 0.0561, wR2 = 0.1217, R indices based on 6051 reflections with I ≥ 2σ(I) (refinement on F2), 274 parameters, 0 restraints, μ = 0.065 mm−1.
PCDA n-butylamine salt (1·8).
Salt 1·8 was prepared by grinding 1 (0.2 g, 0.53 mmol) and 8 (0.053 mL, 0.53 mmol) in a Retsch MM 200 mixer mill for 45 minutes at a frequency of 20 s−1 (yield = 0.23 g, 0.5 mmol, 96%). The resulting powder of 1·8 (0.030 g) was combined with acetone (2 mL) and briefly sonicated. A powder seed (ca. 0.004 g, 0.0089 mmol) was added to the sample and left to crystallise by slow evaporation at room temperature. Blue block crystals formed after one month. Analysis calc. of C29H53NO2: C 77.79, H 11.93, N 3.13%, found: C 77.50, H 11.84, N 3.08%; FTIR (ν/cm−1): 2919, 2848, 2675, 2594, 2183, 1650, 1567, 1508, 1461, 1411, 1334, 1309, 1272, 1239, 1201, 1115, 1095, 1056, 1028, 932, 921, 750, 722, 650. Crystal data: M = 447.72 g mol−1, 0.36 × 0.08 × 0.05 mm3, monoclinic, space group P21 (no. 4), a = 4.5934(6) Å, b = 56.597(9) Å, c = 5.5096(6) Å, β = 99.130(10)°, V = 1414.2(3) Å3, Z = 2, Dc = 1.051 g cm−3, F000 = 500.0, Mo Kα radiation, λ = 0.71073 Å, T = 120 K, 2θmax = 53.97°, 14179 reflections collected, 5663 unique (Rint = 0.1264). Final GooF = 1.026, R1 = 0.1156, wR2 = 0.2163, R indices based on 5663 reflections with I ≥ 2σ(I) (refinement on F2), 293 parameters, 22 restraints, μ = 0.064 mm−1.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Ashland LLC and the Engineering and Physical Sciences Research Council for studentship funding. We also thank the Diamond Light Source for an award of instrument time on the Station I19 (MT 16117 and CY222240) and the instrument scientists for their support and patience. Additionally, we thank Mr W. Douglas Carswell for his assistance with DSC measurements, Dr David Parker for his help and expertise with mass spectrometry experiments, and Dr Andrei Batsanov for collection data of compound 12·5.
Notes and references
- F. L. Hirshfeld and G. M. J. Schmidt, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 2181–2190 CrossRef.
- G. Wegner, Z. Naturforsch., B: J. Chem. Sci., 1969, 24, 824–832 CAS.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CAS.
- V. Enkelmann, Adv. Polym. Sci., 1984, 63, 91–136 CrossRef CAS.
- J. W. Lauher, F. W. Fowler and N. S. Goroff, Acc. Chem. Res., 2008, 41, 1215–1229 CrossRef CAS PubMed.
- S. C. Wang, Y. Li, H. Liu, J. P. Li, T. S. Li, Y. J. Wu, S. Okada and H. Nakanishi, Org. Biomol. Chem., 2015, 13, 5467–5474 RSC.
- R. H. Baughman, J. Appl. Phys., 1972, 43, 4362–4370 CrossRef CAS.
- R. H. Baughman and K. C. Yee, J. Polym. Sci., Part D: Macromol. Rev., 1978, 13, 219–239 CAS.
- A. Mueller and D. F. O'Brien, Chem. Rev., 2002, 102, 727–757 CrossRef CAS PubMed.
- N. Charoenthai, T. Pattanatornchai, S. Wacharasindhu, M. Sukwattanasinitt and R. Traiphol, J. Colloid Interface Sci., 2011, 360, 565–573 CrossRef CAS PubMed.
- D. J. Ahn, E. H. Chae, G. S. Lee, H. Y. Shim, T. E. Chang, K. D. Ahn and J. M. Kim, J. Am. Chem. Soc., 2003, 125, 8976–8977 CrossRef CAS PubMed.
- C. Khanantong, N. Charoenthai, F. Kielar, N. Traiphol and R. Traiphol, Colloids Surf., A, 2019, 561, 226–235 CrossRef CAS.
- H. Jeon, J. Lee, M. H. Kim and J. Yoon, Macromol. Rapid Commun., 2012, 33, 972–976 CrossRef CAS PubMed.
- G. S. Lee, S. J. Hyun and D. J. Ahn, Macromol. Res., 2018, 26, 566–570 CrossRef CAS.
- K. Yoo, S. Kim, N. Han, G. E. Kim, M. J. Shin, J. S. Shin and M. Kim, Dyes Pigm., 2018, 149, 242–245 CrossRef CAS.
- J. P. Yapor, A. Alharby, C. Gentry-Weeks, M. M. Reynolds, A. Alam and Y. V. Li, ACS Omega, 2017, 2, 7334–7342 CrossRef CAS PubMed.
- T. Pattanatornchai, N. Charoenthai, S. Wacharasindhu, M. Sukwattanasinitt and R. Traiphol, J. Colloid Interface Sci., 2013, 391, 45–53 CrossRef CAS PubMed.
- B. Yoon, S. Lee and J. M. Kim, Chem. Soc. Rev., 2009, 38, 1958–1968 RSC.
- M. J. Shin and J. D. Kim, Langmuir, 2016, 32, 882–888 CrossRef CAS PubMed.
- R. Kassis, M. Bassil and A. Al Choueiry, Mater. Res. Express, 2019, 6, 1–7 Search PubMed.
- M. J. Shin and J. S. Shin, J. Appl. Polym. Sci., 2019, 136, 1–7 Search PubMed.
- R. W. Carpick, D. Y. Sasaki, M. S. Marcus, M. A. Eriksson and A. R. Burns, J. Phys.: Condens. Matter, 2004, 16, 679–697 CrossRef.
- H. S. Peng, X. M. Sun, F. J. Cai, X. L. Chen, Y. C. Zhu, G. P. Liao, D. Y. Chen, Q. W. Li, Y. F. Lu, Y. T. Zhu and Q. X. Jia, Nat. Nanotechnol., 2009, 4, 738–741 CrossRef CAS PubMed.
- J. L. Deng, Z. H. Sheng, K. Zhou, M. X. Duan, C. Y. Yu and L. Jiang, Bioconjugate Chem., 2009, 20, 533–537 CrossRef CAS PubMed.
- D. J. Ahn and J. M. Kim, Acc. Chem. Res., 2008, 41, 805–816 CrossRef CAS PubMed.
- J. Pang, L. Yang, B. F. McCaughey, H. Peng, H. S. Ashbaugh, C. J. Brinker and Y. Lu, J. Phys. Chem. B, 2006, 110, 7221–7225 CrossRef CAS PubMed.
- C. Khanantong, N. Charoenthai, S. Wacharasindhu, M. Sukwattanasinitt, N. Traiphol and R. Traiphol, J. Ind. Eng. Chem., 2018, 58, 258–265 CrossRef CAS.
- R. Kassis, M. Bassil and A. Al Choueiry, Mater. Res. Express, 2019, 6, 1–7 Search PubMed.
- Y. L. Su, React. Funct. Polym., 2006, 66, 967–973 CrossRef CAS.
- Y. J. Gwon, C. Kim and T. S. Lee, Sens. Actuators, B, 2019, 281, 343–349 CrossRef CAS.
- D. E. Wang, X. H. Gao, G. B. Li, T. Xue, H. Yang and H. Y. Xu, Sens. Actuators, B, 2019, 289, 85–92 CrossRef CAS.
- S. Ali, F. Ahmed and A. Khatri, Mehran Univ. Res. J. Eng. Technol., 2016, 35, 287–292 CAS.
- M. Kim, Y. J. Shin, S. W. Hwang, M. J. Shin and J. S. Shin, J. Appl. Polym. Sci., 2018, 135, 5 Search PubMed.
- J. P. Jeong, E. Cho, D. Yun, T. Kim, I. S. Lee and S. Jung, Polymers, 2017, 9, 9 Search PubMed.
- J. Luo, K. Y. Fu, H. Y. Dong and D. Y. Chen, Chin. J. Chem. Phys., 2016, 29, 749–753 CrossRef CAS.
- A. Kamphan, C. J. Gong, K. Maiti, S. Sur, R. Traiphol and D. P. Arya, RSC Adv., 2017, 7, 41435–41443 RSC.
- P. Sun, Y. C. Fu, J. Hu, N. Hao, W. Huang and B. Jiang, Radiat. Meas., 2016, 85, 116–125 CrossRef CAS.
- S. Devic, N. Tomic and D. Lewis, Phys. Med., 2016, 32, 541–556 CrossRef PubMed.
- A. Rink, D. F. Lewis, S. Varma, I. A. Vitkin and D. A. Jaffray, J. Med. Phys., 2008, 35, 4545–4555 CrossRef CAS PubMed.
- Y. S. Soliman, A. A. Abdel-Fattah, A. A. Hamed and A. M. M. Bayomi, Radiat. Phys. Chem., 2018, 144, 56–62 CrossRef CAS.
- K. Fahsi, J. Deschamps, K. Chougrani, L. Viau, B. Boury, A. Vioux, A. van der Lee and S. G. Dutremez, CrystEngComm, 2013, 15, 4261–4279 RSC.
- H. Matsuzawa, S. Okada, A. Sarkar, H. Matsuda and H. Nakanishi, Polym. J., 2001, 33, 182–189 CrossRef CAS.
- B. C. Roy and S. Mallik, Org. Lett., 2001, 3, 1877–1879 CrossRef CAS PubMed.
- S. M. Curtis, N. Le, F. W. Fowler and J. W. Lauher, Cryst. Growth Des., 2005, 5, 2313–2321 CrossRef CAS.
- J. J. Kane, R. F. Liao, J. W. Lauher and F. W. Fowler, J. Am. Chem. Soc., 1995, 117, 12003–12004 CrossRef CAS.
- N. S. Goroff, S. M. Curtis, J. A. Webb, F. W. Fowler and J. W. Lauher, Org. Lett., 2005, 7, 1891–1893 CrossRef CAS PubMed.
- L. Luo, C. Wilhelm, A. W. Sun, C. P. Grey, J. W. Lauher and N. S. Goroff, J. Am. Chem. Soc., 2008, 130, 7702–7709 CrossRef CAS PubMed.
- W.-Q. Tong and G. Whitesell, Pharm. Dev. Technol., 1998, 3, 215–223 CrossRef CAS PubMed.
- D. J. Berry and J. W. Steed, Adv. Drug Delivery Rev., 2017, 117, 3–24 CrossRef CAS PubMed.
- M. Khan, V. Enkelmann and G. Brunklaus, Cryst. Growth Des., 2009, 9, 2354–2362 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- T. R. Shattock, K. K. Arora, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2008, 8, 4533–4545 CrossRef CAS.
- T. K. Adalder, R. Sankolli and P. Dastidar, Cryst. Growth Des., 2012, 12, 2533–2542 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- F. H. Allen, W. D. S. Motherwell, P. R. Raithby, G. P. Shields and R. Taylor, New J. Chem., 1999, 23, 25–34 RSC.
- M. Okaniwa, Y. Oaki, S. Kaneko, K. Ishida, H. Maki and H. Imai, Chem. Mater., 2015, 27, 2627–2632 CrossRef CAS.
-
J. Anyumba, D. F. Lewis, H.-Y. Shih, X. Yu, Europe Pat., EP1614002A4, 2004 Search PubMed.
- C. B. Aakeroy, S. Panikkattu, B. DeHaven and J. Desper, CrystEngComm, 2013, 15, 463–470 RSC.
- A. O. Surov, A. A. Simagina, N. G. Manin, L. G. Kuzmina, A. V. Churakov and G. L. Perlovich, Cryst. Growth Des., 2015, 15, 228–238 CrossRef CAS.
- K. Tsaggeos, N. Masiera, A. Niwicka, V. Dokorou, M. G. Siskos, S. Skoulika and A. Michaelides, Cryst. Growth Des., 2012, 12, 2187–2194 CrossRef CAS.
-
Sigma-Aldrich, 4,4′-Bipiperidine, https://www.sigmaaldrich.com/catalog/product/aldrich/705845?lang=en%26region=GB, accessed 27/01/2020, 2020.
- D. Vedal, O. H. Ellestad, P. Klaboe and G. Hagen, Spectrochim. Acta, Part A, 1976, 32, 877–890 CrossRef.
- A. Aucejo, S. Loras, R. Munoz, J. Wisniak and H. Segura, J. Chem. Eng. Data, 1997, 42, 1201–1207 CrossRef CAS.
- M. Dominguez, S. Martin, H. Artigas, M. C. Lopez and F. M. Royo, J. Chem. Eng. Data, 2002, 47, 405–410 CrossRef CAS.
- M. Zhu and L. Yu, J. Therm. Anal. Calorim., 2018, 132, 463–469 CrossRef CAS.
- Y. Y. Xu, S. Y. Fu, F. Y. Liu, H. Y. Yu and J. G. Gao, Soft Matter, 2018, 14, 8044–8050 RSC.
- M. Wenzel and G. H. Atkinson, J. Am. Chem. Soc., 1989, 111, 6123–6127 CrossRef CAS.
- J. Xiao, M. Yang, J. W. Lauher and F. W. Fowler, Angew. Chem., Int. Ed., 2000, 39, 2216–2219 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: XRPD, IR, Raman, DSC, SS NMR and unit cell data. CCDC 2000830–2000837. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02540b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Dmitry S.
Yufit
a,
David C.
Apperley
a,
Larry
Senak
b,
Osama M.
Musa
b,
David K.
Hood
b and
Jonathan W.
Steed
a,
Dmitry S.
Yufit
a,
David C.
Apperley
a,
Larry
Senak
b,
Osama M.
Musa
b,
David K.
Hood
b and
Jonathan W.
Steed