
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heteroleptic actinocenes: a thorium(IV)–cyclobutadienyl–cyclooctatetraenyl–di-potassium-cyclooctatetraenyl complex†
Josef T.
Boronski
 ,
Ashley J.
Wooles
and
Stephen T.
Liddle
*
,
Ashley J.
Wooles
and
Stephen T.
Liddle
*
Department of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: steve.liddle@manchester.ac.uk
First published on 10th June 2020
Abstract
Despite the vast array of ηn-carbocyclic C5–8 complexes reported for actinides, cyclobutadienyl (C4) remain exceedingly rare, being restricted to six uranium examples. Here, overcoming the inherent challenges of installing highly reducing C4-ligands onto actinides when using polar starting materials such as halides, we report that reaction of [Th(η8-C8H8)2] with [K2{C4(SiMe3)4}] gives [{Th(η4-C4[SiMe3]4)(μ-η8-C8H8)(μ-η2-C8H8)(K[C6H5Me]2)}2{K(C6H5Me)}{K}] (1), a new type of heteroleptic actinocene. Quantum chemical calculations suggest that the thorium ion engages in π- and δ-bonding to the η4-cyclobutadienyl and η8-cyclooctatetraenyl ligands, respectively. Furthermore, the coordination sphere of this bent thorocene analogue is supplemented by an η2-cyclooctatetraenyl interaction, which calculations suggest is composed of σ- and π-symmetry donations from in-plane in- and out-of-phase C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C 2p-orbital combinations to vacant thorium 6d orbitals. The characterisation data are consistent with this being a metal–alkene-type interaction that is integral to the bent structure and stability of this complex.
C 2p-orbital combinations to vacant thorium 6d orbitals. The characterisation data are consistent with this being a metal–alkene-type interaction that is integral to the bent structure and stability of this complex.
Introduction
The synthesis of uranocene, [U(η8-C8H8)2], the first actinocene, was reported in 1968, and its cousin thorocene, [Th(η8-C8H8)2], emerged in 1969.1 Uranocene was a landmark discovery, not only in the field of f-element chemistry, but organometallic chemistry as a whole, and it inspired research into the degree of 5f/6d orbital participation in metal–ligand bonding that is still burgeoning today.2 Furthermore, due to the steric and electronic versatility of cyclooctatetraenyl dianions, which provide four-fold symmetry bonding combinations with 5f/6d orbitals, this ligand class has been used widely in organoactinide chemistry,2d,3 with uranium- and thorium–cyclooctatetraenyl complexes being found to exhibit uncommon bonding, oxidation state, and ligand motifs.4Despite advances in actinide science enabled by cyclooctatetraenyl ligands, the closely-related, but far smaller, dianionic cyclobutadienyl ligand, which also provides up to four-fold symmetry bonding combinations to metals, has, thus far, remained barely investigated. This possibly reflects the paucity of suitable starting materials and the proclivity of cyclobutadienyls to decompose via reductive, protonolysis, or cyclometallation routes when in the coordination sphere of polar f-elements. Indeed, although transition metal cyclobutadienyl chemistry was established in the 1960s,5 the first f-element-cyclobutadienyl complex, an inverted sandwich tetraphenylcyclobutadienyl diuranium(IV) species, was reported in 2013.6,7 Very recently, the uranium(IV)–cyclobutadienyl half sandwich pianostool complex [U{C4(SiMe3)4}(BH4)3][Li(THF)4] (A) was reported,8 and shortly after that four uranium(IV)–cyclobutadienyl complexes [U{C4(SiMe3)4}(BH4)3][Na(12-crown-4)],9 [U{C4(SiMe3)4}(BH4)2(μ-BH4){K(THF)2}]2,9 [U(BH4){C4(SiMe3)4}{κ3-C4H(SiMe3)3-κ-(CH2SiMe2)}][Na(tBuOMe)3.6(THF)0.4],9 and [{U(C4[SiMe3]4)(μ-I)2}3{μ3-O}][Mg(THF)6] were disclosed.10 These six uranium–cyclobutadienyl complexes constitute all actinide–cyclobutadienyl chemistry to date, in stark contrast to the plethora of reported f-element ηn-carbocyclic C5–9 complexes.11 Thus, a thorium–cyclobutadienyl complex of any kind is conspicuous by its absence, but realising such a target would provide comparisons between uranium and thorium and provide further insight into actinide–cyclobutadienyl bonding.
Herein, we report the synthesis of the first thorium–cyclobutadienyl complex, which, containing cyclobutadienyl and cyclooctatetraenyl ligands, is an unprecedented heteroleptic actinocene analogue. The thorium ion engages in π- and δ-bonding to the cyclobutadienyl and cyclooctatetraenyl ligands, respectively, and the coordination sphere of this bent thorocene analogue is supplemented by an η2-cyclooctatetraenyl interaction from co-complexed dipotassium-cyclooctatetraenyl. Quantum chemical studies reveal that this thorium–η2-cyclooctatetraenyl interaction is composed of σ- and π-symmetry alkene-like donation from in-plane in- and out-of-phase CC 2p-orbital combinations to formally vacant thorium 6d orbitals.
Results and discussion
Synthetic considerations
We previously reported that reaction of [Li2{C4(SiMe3)4}(THF)2]12 with UCl4 led to reduction of uranium(IV) to intractable mixtures of uranium(III)-containing products.8 However, thorium(IV) has a far greater reduction potential (Eθ Th(IV) → Th(III) −3.7 V) than that of uranium(IV) (Eθ U(IV) → U(III) −0.6 V).13 Nevertheless, addition of a solution of [K2{C4(SiMe3)4}]14 to [ThCl4(THF)3.5]15 resulted in oxidation of dianionic {C4(SiMe3)4}2− to the neutral cyclobutadiene {C4(SiMe3)4},16 as confirmed by multinuclear NMR spectroscopy, and deposition of an insoluble dark grey precipitate presumed to be KCl and colloidal thorium.17 This underscores the strongly reducing nature of {C4(SiMe3)4}2− and led us to conclude that polar actinide halides are unsuitable starting materials for our purposes. Advancing an alternate strategy, it was reasoned that less polar thorium(IV)–carbocyclic complexes would be less prone to the undesirable redox chemistry observed for actinide-halides. On hard-soft acid-base theory grounds, [Th(η8-C8H8)2] was selected as a starting material1,18 since we anticipated that displacement of the soft, charge-diffuse 10π-{C8H8}2− dianion (1.25e− per carbon) by the hard, charge-concentrated 6π-{C4(SiMe3)4}2− dianion (1.5e− per carbon) would be favourable for the hard thorium(IV) ion.Condensation of THF onto a cold (−196 °C) solid mixture of [Th(η8-C8H8)2] and [K2{C4(SiMe3)4}], followed by thawing and heating for two hours, led to the formation of a bright orange solution. After work-up, recrystallisation from toluene afforded bright orange crystals, the colour of which assures the presence of thorium(IV), formulated as the co-complex [{Th(η4-C4[SiMe3]4)(μ-η8-C8H8)(μ-η2-C8H8)(K[C6H5Me]2)}2{K(C6H5Me)}{K}] (1) in 78% isolated yield (Scheme 1).17 Interestingly, 1 does not appear to react further if an additional equivalent of [K2{C4(SiMe3)4}] is added. The coordinated toluene molecules in 1 do not de-coordinate when 1 is placed under dynamic vacuum, but they do exchange with benzene when 1 is dissolved in this solvent.
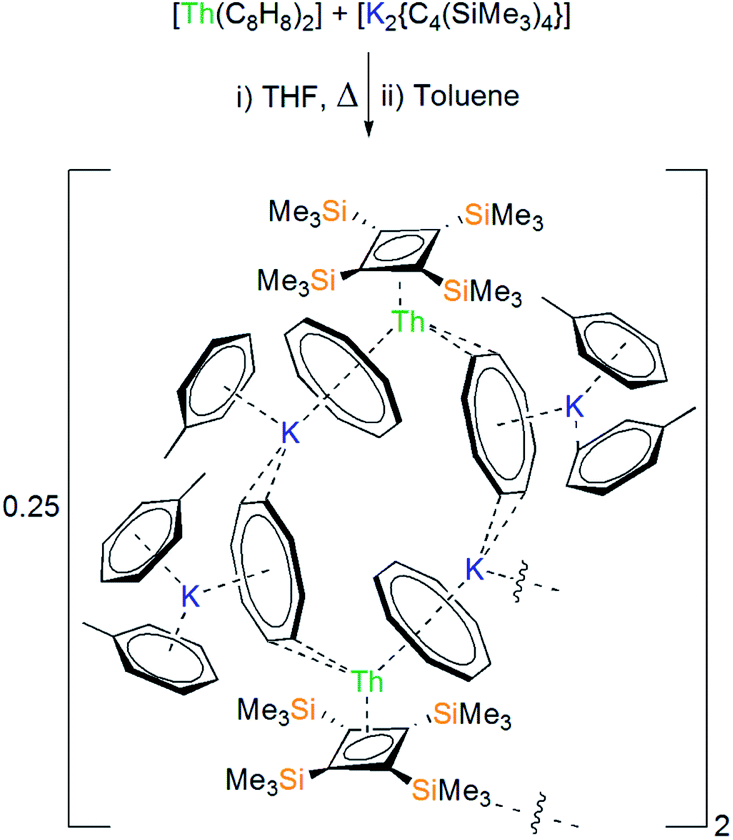 | ||
| Scheme 1 Synthesis of 1 from [Th(η8-C8H8)2] and [K2{C4(SiMe3)4}]. | ||
Crystallographic characterisation
The solid-state structure of 1 was determined by X-ray diffraction (Fig. 1). Complex 1 crystallises with two [Th{η4-C4(SiMe3)4}(μ-η8-C8H8)(μ-η2-C8H8)(K)2] units in the crystallographic asymmetric unit, rendered inequivalent to each other by the nature of K-coordination environments: K4-(η6-C6H5Me)(η8-C8H8)(η2-C8H8); K3-(η6-C6H5Me)(η1-C6H5Me)(η8-C8H8); K2-(η6-C6H5Me)(η6-C6H5Me)(η8-C8H8); K1-(η8-C8H8)(η2-C8H8)(κ1-MeSiMe2). The latter interactions double-up the asymmetric unit resulting in a tetrathorium aggregate overall.17 In more detail, the salient structural features of each thorium-containing unit are η4-coordination of the {C4(SiMe3)4}2− dianion and two {C8H8}2− ligands which are η8-and η2-coordinated, the latter of which is highly unusual for a {C8H8}2− ligand. Thus, the coordination geometry at each thorium ion resembles that of a bent C4/C8-metallocene with a coordinated alkene, the latter being a rare interaction for lanthanides19 and unknown in actinide chemistry, with the closest example for actinides being the reduced 1,2-ethanediide [{(η8-C8H4[SiPri3]2)(η5-C5Me5)U}2(μ-η2-η2-C2H4)].20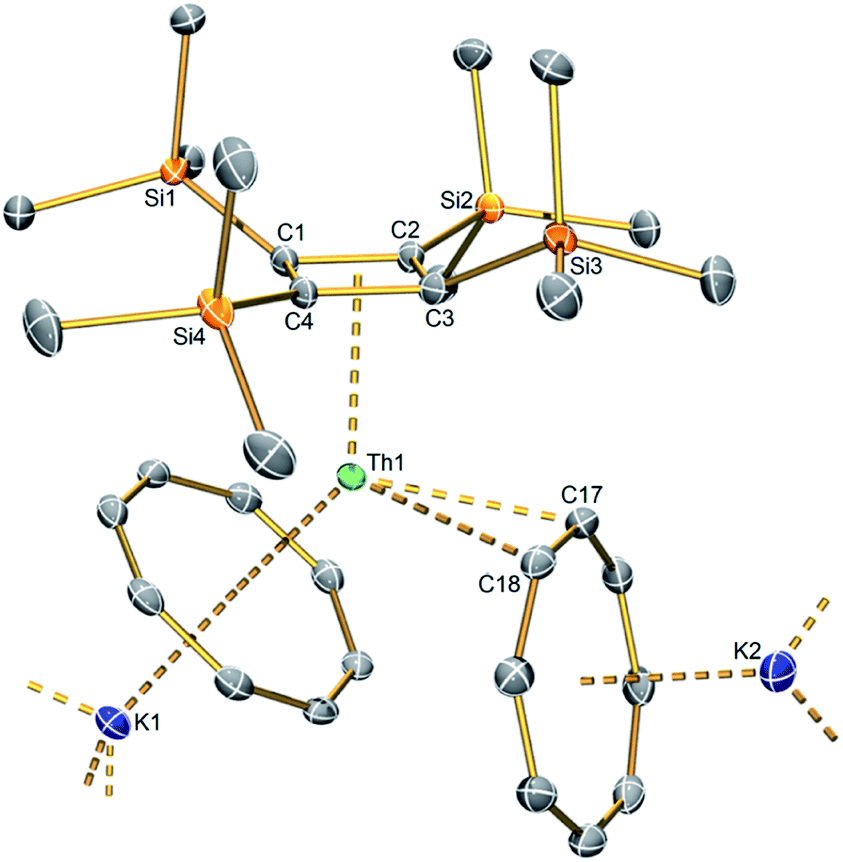 | ||
| Fig. 1 Solid state structure of the heteroleptic thorocene unit in 1 at 100 K with displacement ellipsoids set to 40%. Hydrogen atoms, the five coordinated toluene, and lattice toluene molecules omitted for clarity. Only one of the two [Th{η4-C4(SiMe3)4}(μ-η8-C8H8)(μ-η2-C8H8)(K)2] fragments from the asymmetric unit is shown; they are both very similar to each other, differing principally only in the varied multi-hapto coordination of toluene solvent and cyclooctatetraene and agostic-type trimethylsilyl interactions. | ||
The average Th–C(η4-C4(SiMe3)4) distances are 2.651(9) and 2.649(9) Å for Th1 and Th2, respectively; ∼0.14 Å greater than analogous value found for A (2.513(17) Å), though when considering the 3σ-criterion this difference falls to 0.06 Å.8 The latter is close to the 0.05 Å difference in the ionic six-coordinate radii of thorium(IV) (0.94 Å) and uranium(IV) (0.89 Å).21 Interestingly, the Th–C(η4-C4(SiMe3)4) distances span quite different ranges at Th1 and Th2, being 2.627(3)–2.672(3) and 2.557(3)–2.739(3) Å, respectively, which overall is a larger range of variance than A (2.477(11)–2.549(12) Å).8 These structural differences may reflect more polar Th–C bonding compared the U–C bonding in A, but may also reflect that the Th1 thorocene unit is more restricted within the tetrathorium motif than the Th2 thorocene unit and the coordination environments of A and 1 are rather different. However, for a firmer comparison, the Th–C(η5-C5Me5) distances in the bent thorium–pentamethylcyclopentadienyl–cyclooctatetraenyl metallocene [{(η8-C8H8)(η5-C5Me5)Th}2(μ-η3-η3-C8H8)] (B) range from 2.813(6)–2.864(6) Å, and average 2.843(8) Å.22 This is evidently greater than the average Th–C(η4-C4(SiMe3)4) distance in 1, reflecting the formal dianionic charge on the cyclobutadienyl ligand in 1, versus the mono anionic charge of the pentamethylcyclopentadienyl ligand in B. The average cyclobutadienyl C4-ring C–C distances in 1 are identical at 1.481(8) Å, and statistically indistinguishable from the analogous metric for A (1.488(17) Å).8–10
The displacement of the cyclobutadienyl silyl substituents in 1, out of the plane of the C4-ring away from the respective thorium centres, is notable due to their asymmetry. Opposing silyl groups are displaced by similar distances to each other, but each pair is quite different to the other. At Th1, Si1/Si3 and Si2/Si4 pairs are displaced by 0.839(9)/0.830(12) and 0.233(9)/0.224(8) Å, respectively. At Th2, a larger range is found, with Si5/Si7 and Si6/Si8 displaced by 0.097(9)/0.297(8) and 0.678(6)/1.097(10) Å, respectively. In A, silyl displacements are also observed, but over a narrower range (0.452(3)–0.566(3) Å).8 This most likely can be attributed to the presence of three symmetrically coordinated κ3-borohydrides at uranium in A whereas in 1 the (η8-C8H8)(η2-C8H8) arrangement is asymmetric.
The average Th–C(η8-C8H8) distances are typical of such interactions, at 2.787(12) and 2.765(17) Å for Th1 and Th2, respectively,1,4e,22 and are unsurprisingly longer than the average Th–C(η4-C4(SiMe3)4) distances within 1. For Th1 and Th2, the average Th–C(η2-C8H8) distances are 2.741(10) and 2.788(9) Å, respectively. Interestingly, the η2-C8H8 interaction appears to induce a lengthening of the η2-C–C distances (1.441(4) and 1.427(4) Å) compared to the rest of the C8-rings (1.397(4)–1.418(4) and 1.398(4)–1.414(4) Å, respectively); whilst there is some overlap in these distances by the 3σ-criterion it is certainly the case that the range for the η2-C–C distances sits at the top end of these C–C bond distances, hinting at depletion of C–C bond electron density.
There are few examples of structurally characterised metal-η2–cyclooctatetraenyl interactions,22,23 but they usually involve small, very polarising metals such as lithium and magnesium. The normal trend for cyclooctatetraenyl bonding is that the hapticity increases with increasing metal size,24 but when lower η values are observed they are overwhelmingly η4 butadienyl-like or η3 allyl-like. However, 1 exhibits thorium, one of the larger metals in the periodic table with the η2 alkene-like coordination mode. The C–C distances of the C8H8 unit are not obviously perturbed by η2-coordination to lanthanum in the complex [(μ-η8:η2-C8H8)La{η5-C5H3(SiMe3)2}(μ-η8:η8-C8H8)La{η5-C5H3(SiMe3)2}2][K(18-crown-6)],25 but would appear to be in the case of 1, when η2-coordinated to thorium.
The Cnt(η8-C8H8)-Th-Cnt(η4-C4(SiMe3)4) (Cnt = centroid of the Cn-ring) angles within 1 deviate significantly from linearity, at 139.3° for Th1 and 136.9° for Th2. These values are similar to the Cnt-Th-Cnt angle of 144.2° for the bent thorocene complex [Th(η8-C8H8)2(Me4phen)] (Me4phen = 3,4,7,8-tetramethyl-1,10-phenanthroline) and B (132.11°).4e,22 The C8⋯K distances are unexceptional.26
Spectroscopic analyses
The 1H NMR spectrum of 1 in C6D6 exhibits a sharp resonance (full width half maximum = 1.98 Hz) at 0.74 ppm, corresponding to the trimethylsilyl substituents of the cyclobutadienyl ligands. Additionally, two equally broad resonances from the cyclooctatetraenyl hydrogen atoms are exhibited at 5.71 and 6.47 ppm, with full width half maxima values of 26.5 and 28.7 Hz, respectively. This indicates that two distinct cyclooctatetraenyl environments are present in solution, a situation which is also observed for the crude post-reaction product dissolved in THF (Fig. S8†). Unfortunately, variable-temperature and DOSY experiments (arenes or THF) were not possible due to 1 precipitating on standing or cooling or showing signs of decomposing upon heating for prolonged periods. It seems likely that the more downfield resonance corresponds to the {η8-C8H8}2− dianion,18,22 since this is close to the value of 6.50 ppm for thorocene. The upfield resonance is more in the region associated with neutral tetraolefinic C8H8, and if the η2-coordination of the [C8H8]2− dianion to thorium is maintained in solution, this would be expected to result in C–C π-bond relocalisation to some extent,27 as suggested by the structural data for 1. We note that the upfield resonance is shifted slightly from [K2(C8H8)] (5.76 ppm),28 which is not unexpected given 1 is a co-complex.The optical spectrum of 1 in toluene is dominated by a broad charge transfer band, which spans from ∼18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 into the UV (Fig. S11†). This is consistent with the intense colour of 1 in solution and solid phases, and is modelled well by TD-DFT calculations (vide infra).
000 cm−1 into the UV (Fig. S11†). This is consistent with the intense colour of 1 in solution and solid phases, and is modelled well by TD-DFT calculations (vide infra).
Computational analyses
In order to further understand the nature of the bent, heteroleptic thorocene unit in 1 we undertook DFT calculations.17 With the whole tetrathorium assembly of 1 being unrealistic to compute, we focussed on a discrete [Th{η4-C4(SiMe3)4}(η8-C8H8)] unit. However, geometry optimisation converges to a less bent structure in the gas phase, with a computed Cnt(η8-C8H8)-Th-Cnt(η4-C4(SiMe3)4) angle of 160.5°. A single point energy calculation on the crystallographic coordinates returns a total bond energy that is ∼332 kcal mol−1 (3.6%) destabilised compared to the geometry optimised total bond energy. This clearly represents the upper bound at most, since the single point energy coordinates have not been allowed to relax in any way in this scenario, but is an instructive parameter since it suggests that the Th-η2-C8H8 interaction brings significant stabilisation to 1 since its inclusion clearly compensates for the energetic penalty of bending the [Th{η4-C4(SiMe3)4}(η8-C8H8)] unit to accommodate it.To provide a benchmark, we first describe pertinent aspects of the electronic structure of the [Th{η4-C4(SiMe3)4}(η8-C8H8)] (1′) unit from the single point energy calculation on the crystallographic coordinates derived from 1 (Fig. 2a–d). The HOMO and HOMO−1 of 1′ are two quasi-degenerate (−4.119 and −4.218 eV, respectively) thorium–cyclobutadienyl π-bonds each composed of 22:78% Th:C character, deriving from the ψ2 and ψ3 π-symmetry molecular orbitals of cyclobutadienyl and thorium (6d:5f – 69:31%). The HOMO−2 and HOMO−3 are two quasi-degenerate (−5.036 and −5.093 eV) thorium–cyclooctatetraenyl δ-bonds each composed of 19:81% Th:C character, deriving from the ψ4 and ψ5 δ-symmetry molecular orbitals of cyclooctatetraenyl and thorium orbitals (6d:5f – 92:8%). Whilst the thorium percentage character contributions to the molecular orbitals of 1′ are quite similar for both ring types, and thorium utilises mainly 6d-orbital character in its bonding generally, substantially more 5f character is involved with the smaller cyclobutadienyl ring compared to the more expansive cyclooctatetraenyl ring; this may reflect the more angular character of 5f-compared to 6d-orbitals.2b,6 When compared to A,8 the thorium contributions to the thorium–cyclobutadienyl π-bonds in 1′ are ∼8% lower than for uranium (U:C 30:70%) but, whilst the thorium 5f contribution is ca. half that found in A (6d:5f – 33:67%) it is surprisingly high for thorium.
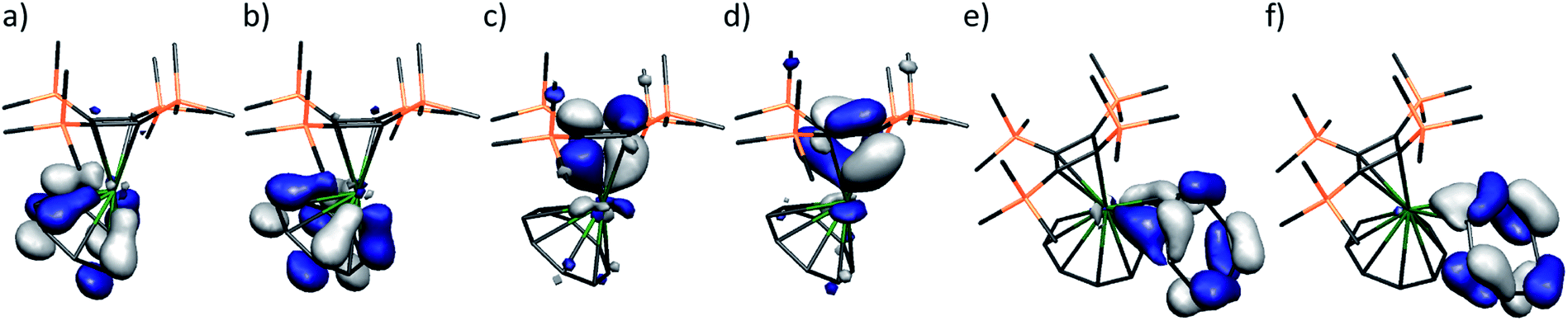 | ||
| Fig. 2 Frontier molecular orbitals of computational models 1′ and 1′′. (a) HOMO−3 δ-bond (164, −5.093 eV) of 1′, (b) HOMO−2 δ-bond (165, −5.036 eV) of 1′, (c) HOMO−1 π-bond (166, −4.218 eV) of 1′, (d) HOMO π-bond (167, −4.119 eV) of 1′, (e) HOMO−1 π-bond (195, 3.018 eV) of 1′′, (f) HOMO σ-bond (196, 3.145 eV) of 1′′. The positive energies of the HOMO and HOMO−1 of 1′′ reflect the formal 2− charge applied to the system, but the electrons are clearly not detached. Hydrogen atoms are omitted for clarity. | ||
The Th–C Mayer bond orders for 1′ average 0.50 and 0.36 for the Th–C(η4-C4(SiMe3)4) and Th–C(η8-C8H8) interactions, respectively, and sum to 2.0 and 2.84, respectively. The C–C bond orders are 1.0 and 1.2 for the cyclobutadienyl and cyclooctatetraenyl rings, and the computed charges of +1.9 (Th), −1.7 (C4), and −2.3 (C8) perhaps suggest a greater engagement of the cyclobutadienyl ligand to thorium than the cyclooctatetraenyl ring, in-line with the Mayer bond orders.
Since DFT calculations produce an orbital-based bonding picture, we examined the bonding topology in 1′ with the QTAIM method. This reveals computed average ρ/∇2ρ/H (energy)/ε (ellipticity) 3,−1 bond critical point values for the Th–C(η4-C4(SiMe3)4) and Th–C(η8-C8H8) interactions in 1′ of 0.05/0.08/−0.02/0.13 and 0.03/0.08/−0.02/0.49; these data in gross terms compare well to computed data for [U(η5-C5H5)4],29 though for the cyclobutadienyl component they indicate slightly weaker Th–C interactions than in A.8
With the bonding within 1' benchmarked, we now consider the bonding of [Th{η4-C4(SiMe3)4}(η8-C8H8)(η2-C8H8)]2− (1′′). To enable meaningful comparisons to be made between 1′ and 1′′ this analysis is on a single point energy calculation on crystallographic coordinates, though the analysis is anyway qualitative due to the necessity to introduce the formal 2− charge for 1′′ since inclusion of charge-balancing potassium ions and toluene solvent in a realistic model was impracticable given the pseudo-periodic nature of 1. The most obvious effect of binding of the η2-C8H8 ligand to thorium is that the Th–C(η4-C4(SiMe3)4) and Th–C(η8-C8H8) Mayer bond orders decrease to 0.33 and 0.29, respectively. In this context the Th–C(η2-C8H8) average Mayer bond order of 0.35 is significant. Inspection of the molecular orbital manifold reveals that in 1′′ the thorium–cyclobutadienyl and –cyclooctatetraenyl bonding interactions are far more mixed than in 1', across HOMOs −2 to −5. The HOMO and HOMO−1 account for the Th-η2-C8H8 interaction (Fig. 2d and e). With respect to the coordinated CC unit 2p π-symmetry orbitals, the HOMO is an in-phase in-plane σ-donation, whereas HOMO−1 is an out-of-phase in-plane π-donation. These two orbitals correspond to combinations from the doubly occupied ψ4 and ψ5 δ-symmetry molecular orbitals of cyclooctatetraenyl into vacant thorium 6d orbitals.
QTAIM analysis finds average ρ/∇2ρ/H (energy)/ε (ellipticity) 3,−1 bond critical point values for the two Th–C(η2-C8H8) interactions of 0.04/0.08/−0.02/0.31; these are in between the Th–C(η4-C4(SiMe3)4) and Th–C(η8-C8H8) values for 1′. When taking all these data together, it is clear that the Th–C(η2-C8H8) interaction is significant.
The calculations on 1′′ provide a basis to probe the experimental spectroscopic data of 1. The principal absorbances in the 18000–24000 cm−1 region from a TD-DFT calculation17 on 1′′ correspond to transitions into the LUMO, which is the ψ4 δ-symmetry molecular orbital of cyclobutadienyl, from the thorium–cyclobutadienyl and –cyclooctatetraenyl bonding molecular orbitals. The transition that gives 1 its striking orange colour is LMCT at 24270 cm−1 from the η2-coordinated cyclooctatetraenyl ligand to vacant thorium 6d orbitals. As expected, TD-DFT calculations on [Th{η4-C4(SiMe3)4}(η8-C8H8)] (crystallographic or geometry optimised coordinates), where the additional η2-(C8H8)2− dianion ligand is omitted, reveals transitions principally involving ψ2/3 transitions to ψ4 for the Th–C4 interactions in the 20000–23000 cm−1 region, then above 30000 cm−1 transitions involve Th–C8 (δ) to Th–C4 (δ) transitions, but the 23000–30000 cm−1 region is essentially void of transitions, with any oscillator strengths being 2–4 orders of magnitude lower in intensity.
Summary and conclusions
To conclude, although the {C4(SiMe3)4}2− dianion is too reducing for polar thorium tetrachloride, reacting to produce intractable precipitates and the neutral cyclobutadiene {C4(SiMe3)4}, we find that less polar thorocene is an effective starting material from which to prepare a thorium–cyclobutadienyl complex, which is notable since actinocenes are usually regarded as thermodynamic sinks. Within this new type of thorocene co-complex is the unprecedented heteroleptic thorocene dianion unit [Th{η4-C4(SiMe3)4}(η8-C8H8)(η2-C8H8)]2−, which exhibits an exceedingly unusual thorium–η2-C8H8 alkene-type interaction that is composed of σ- and π-symmetry alkene-like donation from in-plane in- and out-of-phase CC p-orbital combinations to formally vacant thorium 6d orbitals. Complementary spectroscopic and computational characterisation data provide evidence which suggests that LMCT from the (η2-C8H8)2− ligand to vacant thorium 6d orbitals accounts for the vivid orange colour of 1. The combination of cyclobutadienyl and cyclooctatetraenyl ligands bound to one thorium showcases the flexible nature of thorium–ligand bonding, not only in terms of π vs. δ bonding to carbocyclic rings of different sizes, but also the surprising use of 5f-as well as 6d-orbital character to support the bonding to small aromatic rings. Whilst the general picture of thorium–ligand bonding being more polar than uranium is reflected in our calculations, they also reveal surprisingly high levels of thorium 5f orbital character in the bonding, which is not entirely in-line with the traditional of thorium and uranium displaying 6d vs. 5f orbital character, respectively.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the UK EPSRC (grants EP/M027015/1 and EP/P001386/1), ERC (grant CoG612724), and the University of Manchester for generous funding and support. We thank Dr Ralph Adams (University of Manchester) for informative discussions regarding the NMR data of 1.Notes and references
- (a) A. Streitwieser and U. Müller-Westerhoff, J. Am. Chem. Soc., 1968, 90, 7364 CrossRef CAS; (b) A. Streitwieser and N. Yoshida, J. Am. Chem. Soc., 1969, 91, 7528 CrossRef CAS; (c) A. Avdeef, K. N. Raymond, K. O. Hodgson and A. Zalkin, Inorg. Chem., 1972, 11, 1083 CrossRef CAS; (d) J. Goffart, J. F. B. Gilbert, B. Kanellakopulos and G. Duyckaerts, Inorg. Nucl. Chem. Lett., 1972, 8, 403 CrossRef CAS; (e) D. F. Starks and A. Streitwieser, J. Am. Chem. Soc., 1973, 95, 3423 CrossRef CAS.
- (a) D. Seyferth, Organometallics, 2004, 23, 3562 CrossRef CAS; (b) N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407 CrossRef CAS PubMed; (c) M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394 CrossRef CAS; (d) S. T. Liddle, Coord. Chem. Rev., 2015, 293–294, 211 CrossRef CAS.
- (a) M. Ephritikhine, Dalton Trans., 2006, 2501 RSC; (b) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137 CrossRef CAS PubMed; (c) H. S. La Pierre and K. Meyer, Prog. Inorg. Chem., 2014, 58, 303 CAS; (d) S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604 CrossRef CAS PubMed; (e) O. Walter, Chem.–Eur. J., 2019, 25, 2927 CAS.
- (a) J. S. Parry, F. G. N. Cloke, S. J. Coles and M. B. Hursthouse, J. Am. Chem. Soc., 1999, 121, 6867 CrossRef CAS; (b) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829 CrossRef CAS PubMed; (c) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602 CrossRef CAS PubMed; (d) A. S. Frey, F. G. N. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitken, J. Am. Chem. Soc., 2008, 130, 13816 CrossRef CAS PubMed; (e) J.-C. Berthet, P. thuery, N. Garin, J.-P. Dognon, T. Cantat and M. Ephritikhine, J. Am. Chem. Soc., 2013, 135, 10003 CrossRef CAS PubMed; (f) S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor, R. L. Martin, D. K. Shuh and T. Tyliszczak, Chem. Sci., 2014, 5, 351 RSC; (g) N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624 RSC.
- (a) R. P. Dodge and V. Schomaker, Nature, 1960, 186, 798 CrossRef CAS; (b) A. Efraty, Chem. Rev., 1977, 77, 691 CrossRef CAS; (c) S. Y. Chu and R. Hoffmann, J. Phys. Chem., 1982, 86, 1289 CrossRef CAS.
- D. Patel, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Commun., 2013, 4, 2323 CrossRef PubMed.
- D. Patel, F. Moro, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 10388 CrossRef CAS PubMed.
- J. T. Boronski, L. R. Doyle, J. A. Seed, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2020, 59, 295 CrossRef CAS PubMed.
- N. Tsoureas, A. Mansikkamäki and R. A. Layfield, Chem. Commun., 2020, 56, 944 RSC.
- J. T. Boronski, L. R. Doyle, A. J. Wooles, J. A. Seed and S. T. Liddle, Organometallics, 2020, 39, 1824 CrossRef CAS.
- Representative reviews: (a) S. Arndt and J. Okuda, Chem. Rev., 2002, 102, 1953 CrossRef CAS PubMed; (b) M. N. Bochkarev, Chem. Rev., 2002, 102, 2089 CrossRef CAS PubMed; (c) W. J. Evans and B. L. Davis, Chem. Rev., 2002, 102, 2199 Search PubMed; (d) W. J. Evans, Inorg. Chem., 2007, 46, 3435 CrossRef CAS PubMed; (e) Organometallic and Coordination Chemistry of the Actinides, ed. T. E. Albrecht–Schmitt, Springer, Berlin, Heidelberg, 2008 Search PubMed; (f) F. T. Edelmann, Coord. Chem. Rev., 2009, 256, 1151 CrossRef; (g) F. T. Edelmann, Coord. Chem. Rev., 2010, 23–24, 2641 Search PubMed; (h) F. T. Edelmann, Coord. Chem. Rev., 2012, 261, 73 CrossRef; (i) F. T. Edelmann, Coord. Chem. Rev., 2013, 284, 124 CrossRef; (j) M. Ephritikhine, Organometallics, 2013, 32, 2464 CrossRef CAS; (k) F. T. Edelmann, Coord. Chem. Rev., 2014, 306, 346 CrossRef; (l) F. T. Edelmann, Coord. Chem. Rev., 2015, 318, 29 CrossRef; (m) W. J. Evans, Organometallics, 2016, 35, 3088 CrossRef CAS; (n) F. T. Edelmann, Coord. Chem. Rev., 2016, 338, 27 CrossRef; (o) F. T. Edelmann, Coord. Chem. Rev., 2017, 370, 129 CrossRef; (p) M. Xémard, S. Zimmer, M. Cordier, V. Goudy, L. Ricard, C. Clavaguéra and G. Nocton, J. Am. Chem. Soc., 2018, 140, 14433 CrossRef PubMed; (q) L. Münzfeld, C. Schoo, S. Bestgen, E. Moreno-Pineda, R. Köppe, M. Ruben and P. W. Roesky, Nat. Commun., 2019, 10, 3135 CrossRef PubMed.
- (a) A. Sekiguchi, T. Matsuo and H. Watanabe, J. Am. Chem. Soc., 2000, 122, 5652 CrossRef CAS; (b) A. Sekiguchi, T. Matsuo, H. Watanabe and M. Nakamoto, Russ. Chem. Bull., 2004, 53, 1109 CrossRef CAS.
- (a) L. J. Nugent, R. D. Baybarz, J. L. Burnett and J. L. Ryan, J. Phys. Chem., 1973, 77, 1528 CrossRef CAS; (b) C. J. Inman and F. G. N. Cloke, Dalton Trans., 2019, 48, 10782 RSC.
- B. M. Day, F.-S. Gui, S. R. Giblin, A. Sekiguchi, A. Mansikkamäki and R. A. Layfield, Chem.–Eur. J., 2018, 24, 16779 CrossRef CAS PubMed.
- T. Cantat, B. L. Scotta and J. L. Kiplinger, Chem. Commun., 2010, 46, 919 RSC.
- A. Sekiguchi, M. Tanaka, T. Matsuo and H. Watanabe, Angew. Chem., Int. Ed., 2000, 40, 1675 CrossRef.
- See the ESI† for full details.
- A. Hervé, P. Thuéry, M. Ephritikhine and J.-C. Berthet, Organometallics, 2014, 33, 2088 CrossRef.
- (a) C. J. Burns and R. A. Andersen, J. Am. Chem. Soc., 1987, 109, 915 CrossRef CAS; (b) C. Eaborn, P. B. Hitchcock, K. Izod, Z.-R. Lu and J. D. Smith, Organometallics, 1996, 15, 4783 CrossRef CAS.
- N. Tsoureas, L. Maron, A. F. R. Kilpatrick, R. A. Layfield and F. G. N. Cloke, J. Am. Chem. Soc., 2020, 142, 89 CrossRef CAS PubMed.
- G. R. Choppin, Radiochim. Acta, 1983, 32, 43 CAS.
- (a) W. J. Evans, G. W. Nyce and J. W. Ziller, Angew. Chem., Int. Ed., 2000, 39, 240 CrossRef CAS; (b) W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568 CrossRef CAS.
- (a) T. G. Wetzel, S. Dehnen and P. W. Roesky, Organometallics, 1999, 18, 3835 CrossRef CAS; (b) V. Lorenz, A. Edelmann, S. Blaurock, F. Freise and F. T. Edelmann, Organometallics, 2007, 26, 6681 CrossRef CAS; (c) C. Meermann, K. Ohno, K. W. Tornroos, K. Mashima and R. Anwander, Eur. J. Inorg. Chem., 2009, 76 CrossRef CAS; (d) S. J. Bonyhady, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., Int. Ed., 2009, 48, 2973 CrossRef CAS PubMed; (e) M. Jeletic, P.-H. Lin, J. J. Le Roy, I. Korobkov, S. I. Gorelsky and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 19286 CrossRef CAS PubMed; (f) J. Rausch, P. Liebing, V. Lorenz, L. Hilfert, J. Friedrich and F. T. Edelmann, Angew. Chem., Int. Ed., 2017, 56, 7238 CrossRef CAS PubMed.
- C. Elschenbroich, Organometallics, Wiley-VCH, Weinheim, Germany, 3rd edn, 2005 Search PubMed.
- C. T. Palumbo, L. E. Darago, M. T. Dumas, J. W. Ziller, J. R. Long and W. J. Evans, Organometallics, 2018, 37, 3322 CrossRef CAS.
- J.-C. Berthet, P. Thuéry and M. Ephritikhine, C. R. Chim., 2014, 17, 526 CrossRef CAS.
- (a) S. M. Hubig, S. V. Lindeman and J. K. Kochi, Coord. Chem. Rev., 2000, 200–202, 831 CrossRef CAS; (b) R. H. Mitchell, Chem. Rev., 2001, 101, 1301 CrossRef CAS.
- W. J. Evans, D. J. Wink, A. L. Wayda and D. A. Little, J. Org. Chem., 1981, 46, 3925 CrossRef CAS.
- M. J. Tassell and N. Kaltsoyannis, Dalton Trans., 2010, 39, 6719 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, structural, spectroscopic, and computational details. CCDC 1992448 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02479a |
| This journal is © The Royal Society of Chemistry 2020 |