
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical synthesis and biological activity of peptides incorporating an ether bridge as a surrogate for a disulfide bond†
Rui
Zhao‡
ab,
Pan
Shi‡
a,
Junyou
Chen‡
b,
Shuaishuai
Sun
b,
Jingnan
Chen
b,
Jibin
Cui
b,
Fangming
Wu
f,
Gemin
Fang
 e,
Changlin
Tian
a,
Jing
Shi
*a,
Donald
Bierer
d,
Lei
Liu
c and
Yi-Ming
Li
*b
e,
Changlin
Tian
a,
Jing
Shi
*a,
Donald
Bierer
d,
Lei
Liu
c and
Yi-Ming
Li
*b
aHefei National Laboratory of Physical Sciences at Microscale, Department of Chemistry, School of Life Sciences, University of Science and Technology of China, Hefei, Anhui 230009, China. E-mail: shijing@ustc.edu.cn
bSchool of Food and Biological Engineering, Hefei University of Technology, Hefei, Anhui 230009, China. E-mail: ymli@hfut.edu.cn
cDepartment of Chemistry, Tsinghua University, Beijing 100084, China
dDepartment of Medicinal Chemistry, Bayer AG, Aprather Weg 18A, 42096 Wuppertal, Germany
eSchool of Life Science, Institute of Physical Science and Information Technology, Anhui University, Hefei 230601, China
fHigh Magnetic Field Laboratory, Chinese Academy of Sciences, Hefei 230031, China
First published on 8th July 2020
Abstract
Disulfide bridges contribute to the definition and rigidity of polypeptides, but they are inherently unstable in reducing environments and in the presence of isomerases and nucleophiles. Strategies to address these deficiencies, ideally without significantly perturbing the structure of the polypeptide, would be of great interest. One possible surrogate for the disulfide bridge is a simple thioether, but these are susceptible to oxidation. We report the introduction of an ether linkage into the biologically active, disulfide-rich peptides oxytocin, tachyplesin I, and conotoxin α-ImI, using an ether-containing diaminodiacid as the key building block, obtained by the stereoselective ring-opening addition reaction of an aziridine skeleton with a hydroxy group. NMR studies indicated that the derivatives with an ether surrogate bridge exhibited very small change of their three-dimensional structures. The analogs obtained using this novel substitution strategy were found to be more stable than the original peptide in oxidative and reductive conditions; without a loss of bioactivity. This strategy is therefore proposed as a practical and versatile solution to the stability problems associated with cysteine-rich peptides.
Introduction
Disulfide-rich polypeptides have emerged as promising drug candidates due to their excellent metabolic stability, potent activity and high target selectivity.1 These favorable characteristics are in part due to the disulfide bonds themselves, which support the definition of the structure and influence the topological orientation of the pharmacophore, thereby imparting stability and selectivity to the peptides.2 However, disulfide bonds are inherently unstable in reducing environments and in the presence of isomerases and nucleophiles, resulting in structural rearrangement of the entire peptide and a complete loss of biological activity.3 Accordingly, the replacement of this basic structural element with a more stable, non-reducing isostere would be highly desirable; and thioether,4 diselenide bridge,5 triazole,6 and hydrocarbon bridge7 surrogates have all been proposed. In practice, however, drawbacks are associated with all of these disulfide bond surrogates. First, the rigidity and planarity of triazole and dicarba (olefin or hydrocarbon) bridges generally precludes their proper accommodation into the parent structures of peptides, changing their overall shape, properties, and activity.8 For example, some triazole-containing insulin analogs were poorly active due to structural distortions introduced into their backbone by the triazole moiety.8a Furthermore, although thioether and selenoether bridges are similar in structure and reactivity to the native disulfides, they are also prone to oxidation, which can be problematic – some thioether-substituted conotoxin α-ImI derivatives have previously been revealed to be easily oxidized to sulfoxide, leading to a loss of activity.4aAnother possible disulfide bridge isostere is an ether bridge, which in principle has several advantages over those previously mentioned. For example, ether bridges are inherently more stable than thioether bridges, are expected to be readily accommodated by the structure of the peptide, being neither rigid nor planar, and not susceptible to racemization during solid phase peptide synthesis (SPPS). However, the formation of ether bonds is difficult, especially in the context of side chain cyclization, and usually requires forcing conditions not compatible with delicate peptide functionality.9
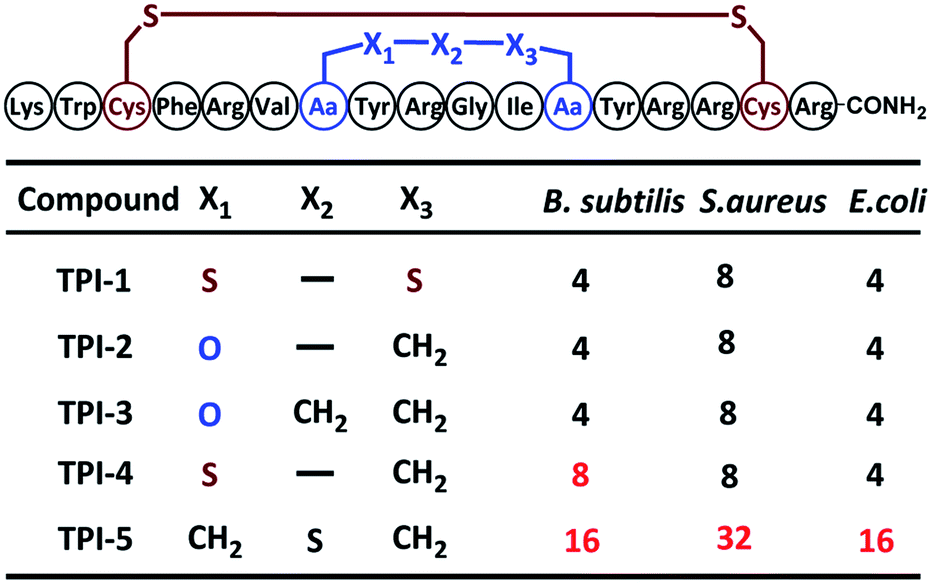
Herein, we disclose a new and practical strategy for the synthesis of cysteine-rich peptides incorporating an ether linkage as a disulfide-bond mimic using an orthogonally protected, ether-containing diaminodiacid, obtained by the stereoselective ring-opening addition reaction of an aziridine with a hydroxy group (Scheme 1).10 The utility of this strategy was exemplified in syntheses of the ether analogs of oxytocin; the bacteriostatic peptide tachyplesin I (TPI-1), which was shown to be more stable than that its wild-type (WT) counterpart under reducing conditions but just as bioactive; and an ether-substituted conotoxin α-ImI analog, which was significantly more stable in oxidative environments compared to conotoxin α-ImI analogs incorporating thioether bonds.
 | ||
| Scheme 1 Diaminodiacid strategy enabled the development of the first example of cysteine-rich peptide exploiting the ether-bond as disulfide-bond mimic. These ether-containing peptide mimics exhibit significant stability to both reduction and oxidation conditions. | ||
Results and discussion
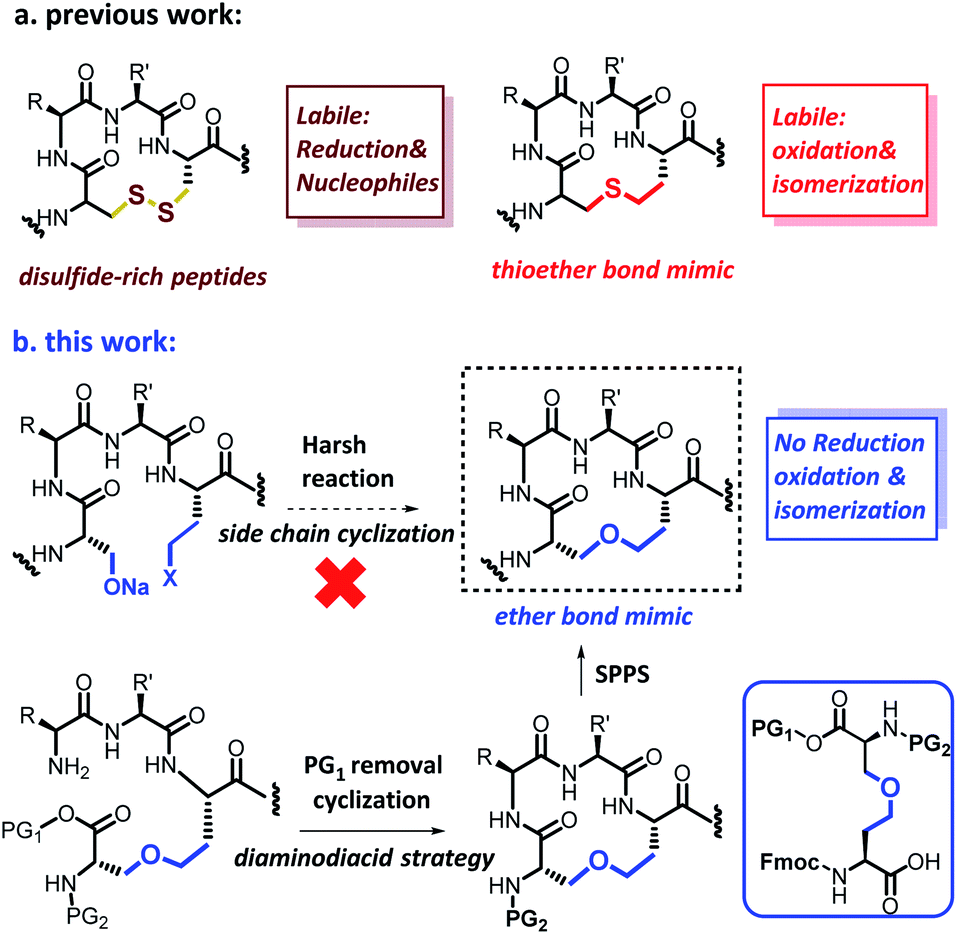
Although ether bridge possibly has advantages over previously mentioned surrogates, the replacement of a disulfide moiety with an ether is not straightforward. The major reason for this is that whereas a disulfide bridge can be readily formed by the gentle oxidation of two proximal thiols, the reaction conditions used to construct ether bonds are relatively harsh; for example, the Williamson ether synthesis requires high temperatures, strongly basic sodium alkoxides, and prolonged reaction times.9 To avoid having to expose a fully-formed peptide to such forcing conditions, we envisioned pre-preparation of a diaminodiacid unit containing an ether bond, followed by its incorporation into a polypeptide by conventional SPPS. We expected that under Lewis acid catalysis, selective β-position ring-opening addition of azacyclopropane derivatives through the side chain hydroxyl groups of homoserine derivatives will construct the key skeleton of the ether-containing diaminodiacid.10 Vederas et al. used a similar reaction to a synthesize a natural product DAP analog which was crucial for bacterial cell wall layer formation.10dFirst, we synthesized aziridine derivative 1b by cyclization of 1a using methanesulfonyl chloride and triethylamine. To activate the aziridine for regioselective ring opening at the β-position, we chose the electron-withdrawing p-nitrobenzyloxycarbonyl group (pNZ) for nitrogen protection.11 After removal of the Trt group, pNZ-Cl was added to obtain segment 1c. We further obtained Fmoc-homoserine-tBu 2a as previously reported,4j and then successfully constructed the key intermediate 3a containing an ether bond by reacting 2a and 1c in the presence of catalytic BF3·OEt2 (yield 30%). Finally, the ether-containing diaminodiacid product 4a was obtained by removing the tBu group with TFA (overall yield 3.1%, 7 steps from L-serine) (Scheme 2). Because the ether bond is slightly shorter than the disulfide bond, we further prepared a side-chain extended ether-containing diaminodiacid 4b by coupling Fmoc-homohomoserine-tBu (2b) with 1c, to test the influence of linkage length on peptide bio-activity (Scheme S5†).
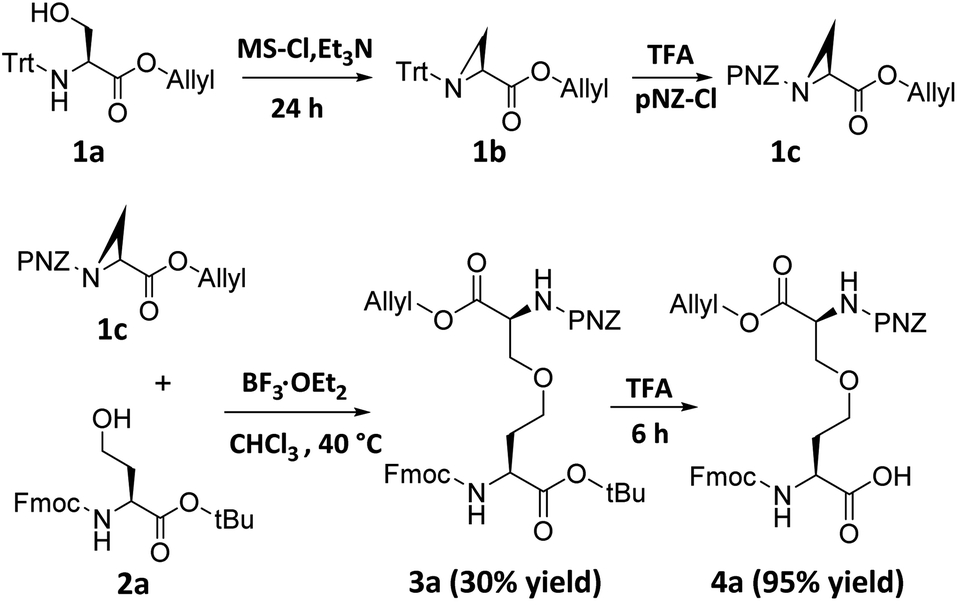 | ||
| Scheme 2 Chemical synthesis of ether-containing diaminodiacids 4a. | ||
With ether-containing diaminodiacids in hand, we first tested their compatibility with Fmoc-based SPPS by synthesizing ether-containing analogs of oxytocin, a peptide hormone with a plethora of both physiological and psychological biological functions.12 The synthesis of the ether-containing analog of oxytocin was attempted first. Gly, Leu and Pro were sequentially coupled to Rink amide AM resin to give 5. Then, 4a was condensed to the α-amino of the pendant Pro of 5 to yield 6. Finally, the Tyr–Ile–Gln–Asn tetrapeptide was added on to the 4a moiety of 6. After removal of the allyl protecting group, the side-chain carboxyl group of 4a and N-terminal amino group of Tyr were efficiently cyclized using PyAOP/NMM. Then, the pNZ group was removed and finally the oxytocin analog 9 was obtained after cleavage from the resin using TFA (Fig. 1a). HPLC analysis of the purified products gave rise to a single peak, and 10 was also identified by LC-MS as an oxytocin derivative containing an ether bond (Fig. 1b), HPLC analysis of purified 10 showed a single product peak, and it was identified by LC-MS as an oxytocin derivative containing an ether bond (Fig. 1b), proving that the new diaminodiacid can be readily used in SPPS. The circular dichroism (CD) spectrum confirmed that 10 had a secondary structure, very similar to that of its native counterpart (Fig. 1c).4e To verify the three-dimensional structure of diaminodiacid-replaced analogue, solution NMR experiments including DQF-COSY, TOCSY, NOESY and ROESY were carried out to determine the structure of 10. With a total of 52 NOE distance restraints (including 3 long-range NOEs) and 6 dihedral restraints, 20 lowest energy structures selected from 100 calculated structures were observed with an all heavy atom RMSD (residue 1–8) of 1.476 Å (Fig. 1d and ESI Table S1†). The ROESY cross peaks and NMR structure showed that 10 incorporated the correct Cys1–Cys6 ether bond connectivity (Fig. 1d). The overall solution NMR structure of 10 is a turning loop structure, almost identical to the native oxytocin. In our proton assignment, similar chemical shifts for Cα–H atoms were observed for native oxytocin4h and 10, except for the replaced Cys residues (Fig. S11†), thus indicating that replacement of the Cys1–Cys6 disulfide bond with an ether bond had little influence on the tertiary structure of Oxytocin.
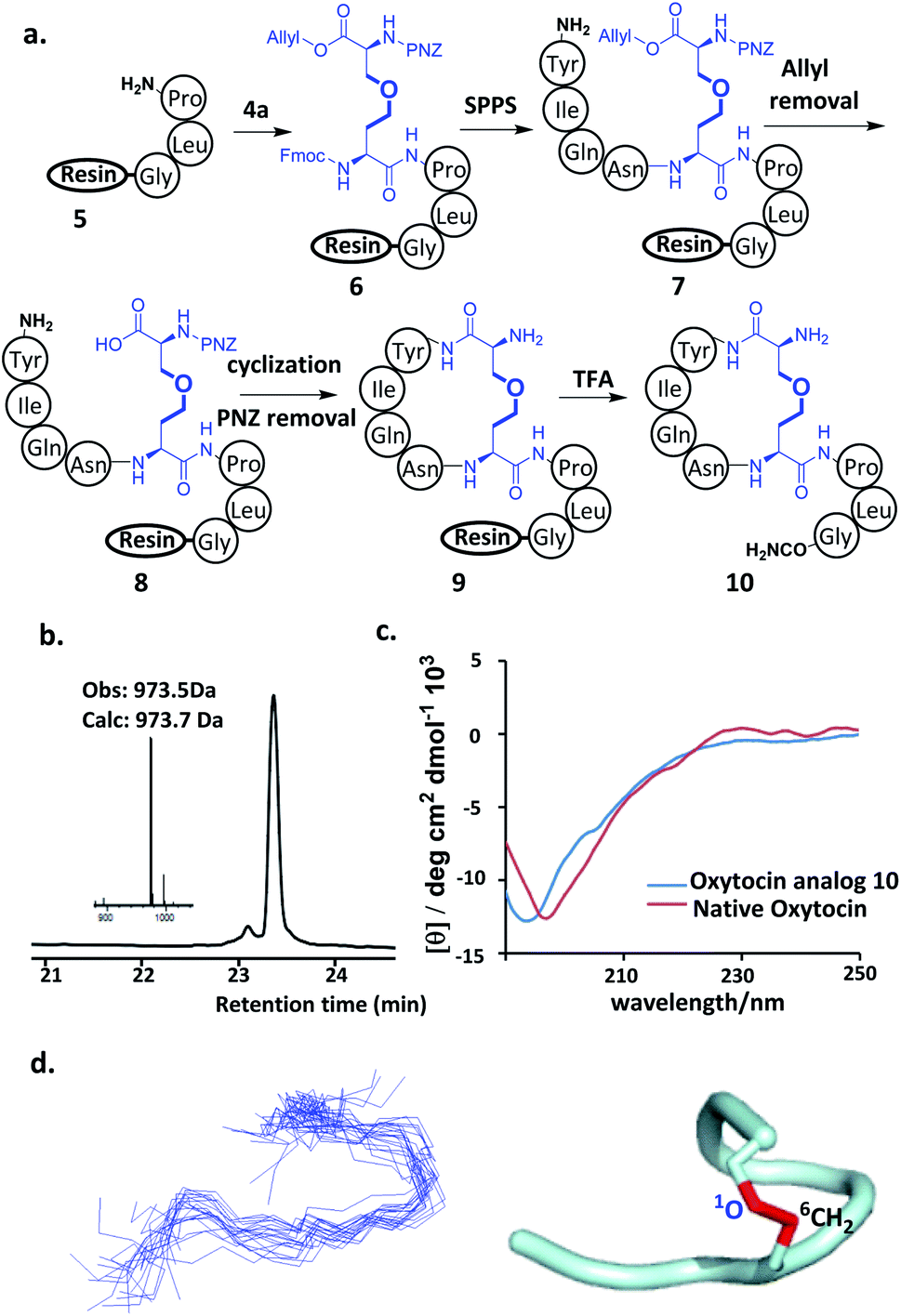 | ||
| Fig. 1 (a). Synthetic route for the preparation of oxytocin analog using diaminodiacid 4a. (b) HPLC traces and ESI-MS analysis of purified oxytocin analog. (c) CD spectra of oxytocin analogue in water. (d) Backbone ensembles of 20 lowest-energy NMR structures of oxytocin analog 10 (left) and a ribbon representation of 10 (right). The C–O bridge is shown in red. | ||
Next, our attention turned to tachyplesin I (TPI-1). TPI-1 adopts a β-sheet conformation stabilized by two cross-linked disulfide bonds,13 and the folding and activity of its analogs are sensitive to the nature of the disulfide bond surrogates. For example, when the Cys7–Cys12 disulfide bond was replaced with a thioether bond, the bio-activity of the resulting TPI-1 analog was significantly reduced compared to TPI-1.4b Our synthesis of a TPI-1 derivative incorporating an ether bond (TPI-2) in place of the Cys7–Cys12 disulfide bond is depicted in Fig. 2a. A linear peptide 11 containing 4a was first assembled on the resin. Removal of the allyl and Fmoc groups of 11 gave peptide 12. Cyclization using PyAOP and NMM followed by removal of the pNZ protecting group gave 13. After condensation of the hexapeptide Lys–Trp–Cys–Phe–Arg–Val with 13, the resulting peptide was cleaved from the resin and purified by HPLC to give the full-length peptide 14, which was refolded under neutral conditions (33% acetonitrile, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 GSSG/GSH, pH 7.5). HPLC and ESI-MS analysis demonstrated the purity and correct molecular weight of the ether bond substituted TPI-2 (Fig. 2b). For comparison, wildtype (WT) TPI-1 and TPI-3 incorporating a side-chain containing an extra methylene group were obtained under the same conditions. Using previous reported thioether-containing diaminodiacids, we further synthesized two other TPI-1 derivatives incorporating thioether linkages of different lengths (TPI-4 and TPI-5) (Fig. 2d). The CD spectra of TPI-1 and its ether bond analogs were measured; WT TPI-1 was found to give rise to a positive peak at 196 nm and a negative peak at 218 nm, while the spectra of its ether bond analogs showed a weak negative absorption at 218 nm (Fig. 2c). This suggests the β-sheet structure of TPI-2 and TPI-3 to be less ordered than that of TPI-1, consistent with the previous results of thioether derivatives.4b We further conducted solution NMR spectroscopy (including DQF-COSY, TOCSY, NOESY, 13C-HSQC and 15N-HSQC) for TPI-2. The determined solution structures revealed that a total of 136 NOE distance restraints (including 8 long-range NOEs) and 22 dihedral restraints, 20 lowest energy structures selected from 100 calculated structures were observed with an all heavy atom RMSD (residue 5–14) of 1.317 Å (ESI Table S2 and Fig. S15†). The NOESY cross peaks and NMR structure showed that TPI-2 incorporated the correct Cys7–Cys12 ether bond connectivity (Fig. 2d). Furthermore, we also observed the similar chemical shifts for Cα–H of TPI-2 (The residue Lys1 subject to fast protein exchange) compared with native TPI-14b (Fig. S13†), except for those corresponding to the two replaced Cys residues, thus confirming that TPI-2 maintained a three dimensional structure similar to the TPI-1.
:1 GSSG/GSH, pH 7.5). HPLC and ESI-MS analysis demonstrated the purity and correct molecular weight of the ether bond substituted TPI-2 (Fig. 2b). For comparison, wildtype (WT) TPI-1 and TPI-3 incorporating a side-chain containing an extra methylene group were obtained under the same conditions. Using previous reported thioether-containing diaminodiacids, we further synthesized two other TPI-1 derivatives incorporating thioether linkages of different lengths (TPI-4 and TPI-5) (Fig. 2d). The CD spectra of TPI-1 and its ether bond analogs were measured; WT TPI-1 was found to give rise to a positive peak at 196 nm and a negative peak at 218 nm, while the spectra of its ether bond analogs showed a weak negative absorption at 218 nm (Fig. 2c). This suggests the β-sheet structure of TPI-2 and TPI-3 to be less ordered than that of TPI-1, consistent with the previous results of thioether derivatives.4b We further conducted solution NMR spectroscopy (including DQF-COSY, TOCSY, NOESY, 13C-HSQC and 15N-HSQC) for TPI-2. The determined solution structures revealed that a total of 136 NOE distance restraints (including 8 long-range NOEs) and 22 dihedral restraints, 20 lowest energy structures selected from 100 calculated structures were observed with an all heavy atom RMSD (residue 5–14) of 1.317 Å (ESI Table S2 and Fig. S15†). The NOESY cross peaks and NMR structure showed that TPI-2 incorporated the correct Cys7–Cys12 ether bond connectivity (Fig. 2d). Furthermore, we also observed the similar chemical shifts for Cα–H of TPI-2 (The residue Lys1 subject to fast protein exchange) compared with native TPI-14b (Fig. S13†), except for those corresponding to the two replaced Cys residues, thus confirming that TPI-2 maintained a three dimensional structure similar to the TPI-1.
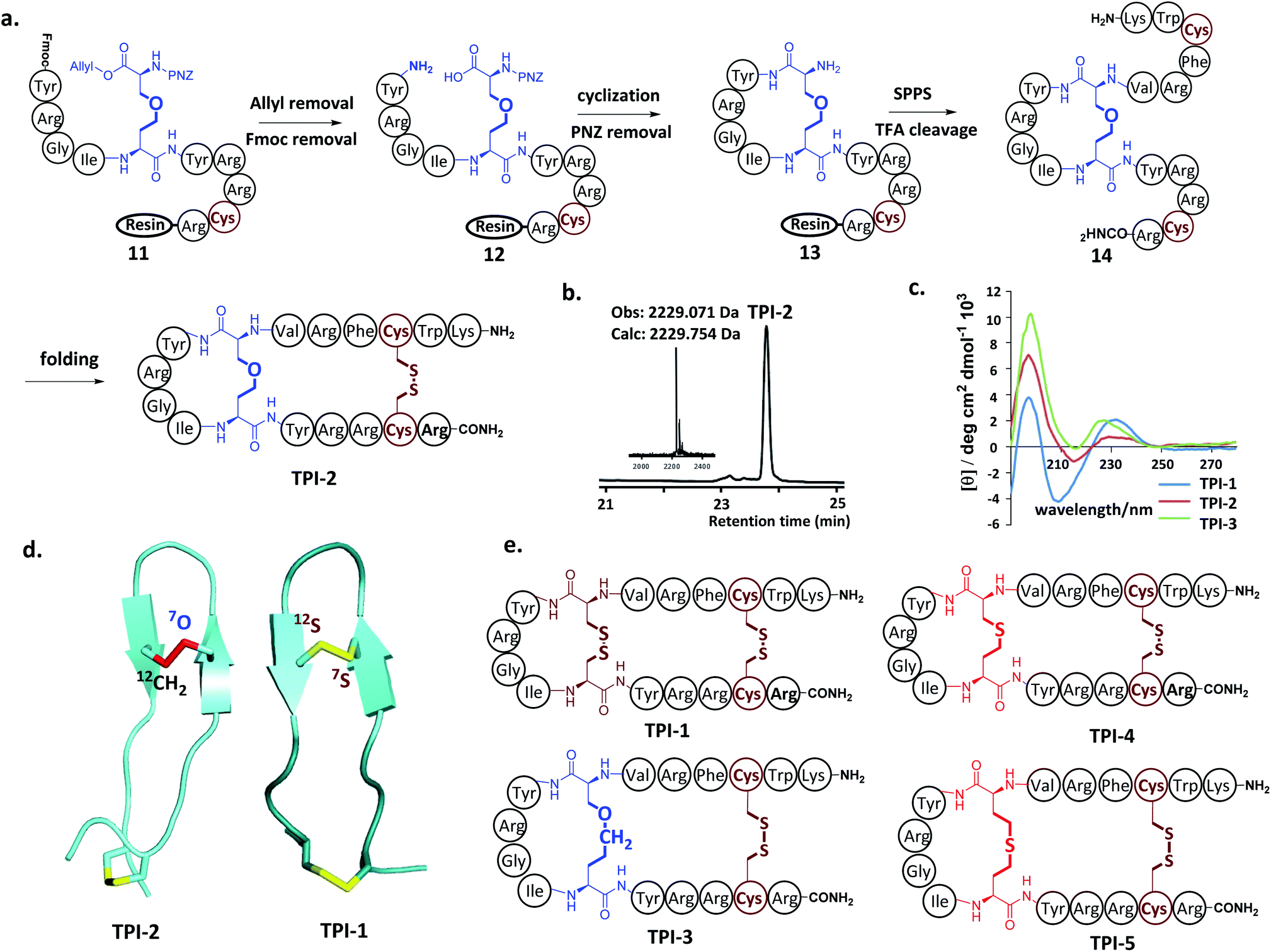 | ||
| Fig. 2 (a) Solid-phase synthetic route for the preparation of ether-containing TPI-2 using diaminodiacid 4a. (b) HPLC traces and ESI-MS analysis of purified refolded TPI-2. (c) CD spectra of TPI-1, TPI-2 and TPI-3 in water. (d) NMR structure of TPI-1 (right) and ether-containing analogue TPI-2 (left). Disulfides are shown in yellow and the ether bond in red. (e) The structure of side-chain extended ether-containing TPI-3 and different lengths of thioether linkage substituted TPI-1 derivatives TPI-4 and TPI-5. | ||
Antimicrobial assays were then used to ascertain the biological activity of the TPI analogs. Three bacterial strains, including two Gram-positive bacteria (Bacillus subtilis and Staphylococcus epidermidis) and one Gram-negative bacteria (Escherichia coli) were used. The three strains were cultured to 108 CFU at 37 °C using LB medium; and the minimal inhibitory concentration (MIC) of the TPI derivatives tested using the standard two-fold dilution method.14 Each experiment was repeated three times. The ether-containing TPI-2 and TPI-3 had identical antibacterial activity to WT TPI-1, exerting significant antibacterial activity against both Gram-positive and Gram-negative bacteria at low concentrations. TPI-4 had a slightly inferior MIC value for Bacillus subtilis compared to TPI-1, whereas the antibacterial activity of TPI-5 incorporating a side-chain containing an extended thioether bond was dramatically decreased, for both Gram-positive and Gram-negative bacteria (Table 1). Collectively, these results indicate that ether bond substitution may exert less of an effect on the activity of disulfide-rich polypeptides than thioether bonds, especially in peptides sensitive to structural alterations such as TPI-1.
|
We also studied the stability of these peptides in a reducing environment. After one hour in aqueous dithiothreitol (DTT, 250 μm) solution, 80% TPI-1 had been completely reduced to linear peptide, whereas TPI-2 exhibited less than 40% reduction (Fig. S9†). Thus, the replacement of the disulfide functionality with an ether improves the stability of the parent peptide, as expected.
Although thioether crosslinks are among the most popular disulfide surrogates, they remain susceptible to sulfoxide formation under oxidative conditions, which may influence biological activity. Conotoxin α-ImI was selected as the basis for a study to examine whether ether-bond substitutions are more stable than thioethers in an oxidative environment.4a Conotoxin α-ImI analogs α-ImI-1 and α-ImI-2 incorporating different surrogates for the Cys2–Cys7 disulfide bond (Fig. 3) were readily prepared using 4a and thioether-containing diaminodiacids, respectively. HPLC and MS analysis found that about 30% α-ImI-2 was oxidized to sulfoxide after 4 hours under 0.1% hydrogen peroxide. When the oxidation process was extended to 12 hours, almost all the α-ImI-2 was oxidized. Meanwhile, no oxidation by-products of α-ImI-1 were observed after 12 hours under the same oxidation conditions (Fig. 3). These results indicate that the ether-substituted peptide is more stable under oxidative conditions than the thioether-containing analog. Therefore, the ether-bond substitution strategy developed herein is expected to replace the thioether bond strategy for disulfide-rich peptides that are sensitive to oxidative conditions.
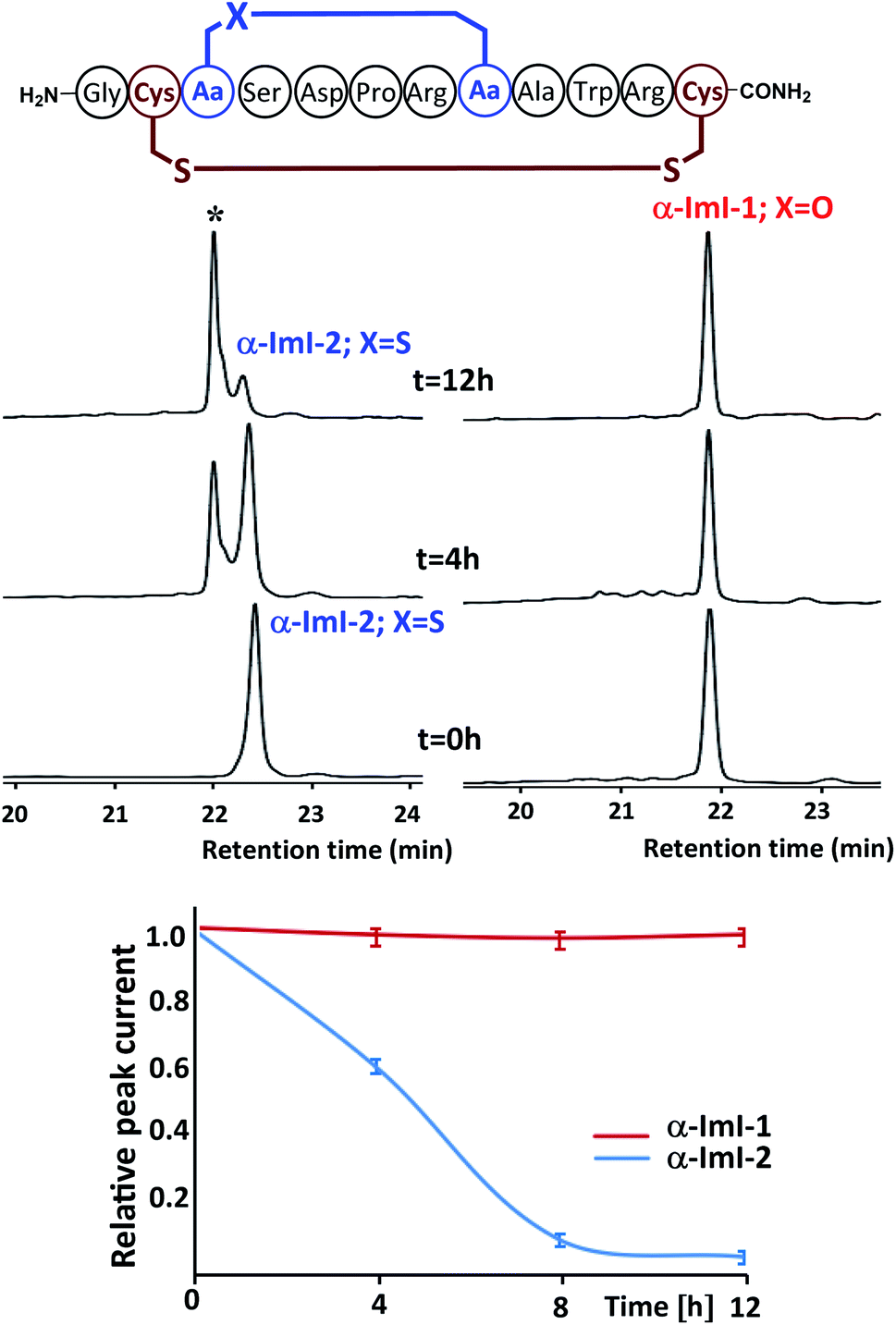 | ||
| Fig. 3 Oxidative stability studies on α-ImI-1 and α-ImI-2 under 0.1% hydrogen peroxide. After 12 hours, almost all α-ImI-2 is oxidized, whereas no oxidation by-products of α-ImI-1 were observed. * refers to α-ImI-2 in which thioether is oxidized to sulfoxide. | ||
Conclusions
A selection of cysteine-rich peptides incorporating an ether-bond as a disulfide surrogate have been efficiently synthesized using a strategy based on diaminodiacid building blocks, obtained by the stereoselective ring-opening addition reaction of an aziridine skeleton with a hydroxy group. NMR studies suggested that these ether-containing derivatives exhibited very small change of their tertiary structures. The ether analog of the bacteriostatic peptide TPI-2 was found to be more stable under reducing conditions than wildtype TPI-1, but equally bioactive. Also, the ether-containing conotoxin analog was more stable in an oxidative environment compared to an analog incorporating a thioether. Further studies to explore the scope of this strategy for the improvement of the properties of cysteine-rich peptides are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFA0505400 and 2017YFA0505200), the National Natural Science Foundation of China (No. 91753205, 21877024, 21977089, 81621002, 31971152 and 21621003), the Fundamental Research Funds for the Central Universities (JZ2019HGPB0105). Bayer AG would like to acknowledge Professor Dr Ulrich Koert from the Philipps University of Marburg for useful scientific discussions.Notes and references
- (a) W. J. Wedemeyer, E. Welker and M. Narayan, Biochemistry, 2000, 39(15), 4207–4216 CrossRef CAS PubMed; (b) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed; (c) M. Góngora-Benítez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef PubMed.
- (a) J. Gehrmann, P. F. Alewood and D. J. Craik, J. Mol. Biol., 1998, 278, 401–415 CrossRef CAS PubMed; (b) V. Lavergne, R. J. Taft and P. F. Alewood, Curr. Top. Med. Chem., 2012, 12, 1514–1533 CrossRef CAS.
- (a) C. J. Armishaw, N. L. Daly, S. T. Nevin, D. J. Adams, D. J. Craik and P. F. Alewood, J. Biol. Chem., 2006, 281, 14136–14143 CrossRef CAS PubMed; (b) J.-Y. Chang, B.-Y. Lu and L. Li, Anal. Biochem., 2005, 342, 78–85 CrossRef CAS PubMed; (c) K. Hu, W. Xiong, C. J. Sun, C. Wang, J. X. Li, F. Yin, Y. X. Jiang, M.-R. Zhang, Z. Li, X. W. Wang and Z. G. Li, CCS Chem., 2020, 2, 42–51 CrossRef.
- (a) Z. Dekan, I. Vetter, N. L. Daly, D. J. Craik, R. J. Lewis and P. F. Alewood, J. Am. Chem. Soc., 2011, 133, 15866–15869 CrossRef CAS PubMed; (b) H. K. Cui, Y. Guo, F. L. Feng, H. N. Chang, Y. J. Wang, F. M. Wu, C. Tian and L. Liu, Angew. Chem., Int. Ed., 2013, 52, 9558–9562 CrossRef CAS PubMed; (c) Y. Guo, D. M. Sun, F. L. Wang, Y. He, L. Liu and C. L. Tian, Angew. Chem., Int. Ed., 2015, 54, 14276–14281 CrossRef CAS PubMed; (d) C. M. B. K. Kourra and N. Cramer, Chem. Sci., 2016, 7, 7007 RSC; (e) S. S. Sun, J. Y. Chen, R. Zhao, D. Bierer, J. Wang, G. M. Fang and Y. M. Li, Tetrahedron Lett., 2019, 60, 1197–1201 CrossRef CAS; (f) Q. Qu, S. Gao, F. Wu, M. Zhang, Y. Li, L. Zhang, D. Bierer, C. Tian, J. Zheng and L. Liu, Angew. Chem., Int. Ed., 2020, 59, 6037 CrossRef CAS; (g) H. Lin, Y. Jiang, Q. Zhang, K. Hu and Z. G. Li, Chem. Commun., 2016, 52, 10389–10391 RSC; (h) K. Hu, H. Geng, Q. Z. Zhang, Q. S. Liu, M. S. Xie, C. J. Sun, W. J. Li, H. C. Lin, F. Jiang, T. Wang, Y. D. Wu and Z. G. Li, Angew. Chem., Int. Ed., 2016, 55, 8013–8017 CrossRef CAS PubMed; (i) Y. Tian, J. X. Li, H. Zhao, X. Z. Zeng, D. Y. Wang, Q. S. Liu, X. G. Niu, X. H. Huang, N. H. Xu and Z. G. Li, Chem. Sci., 2016, 7, 3325–3330 RSC; (j) T. Wang, J. Fan, X. X. Chen, R. Zhao, Y. Xu, D. Bierer, L. Liu, Y. M. Li, J. Shi and G. M. Fang, Org. Lett., 2018, 20(19), 6074–6078 CrossRef CAS; (k) J. Koehbach, M. O'Brien, M. Muttenthaler, M. Miazzo, M. Akcan, A. G. Elliott, N. L. Daly, P. J. Harvey, S. Arrowsmith, S. Gunasekera, T. J. Smith, S. Wray, U. Goransson, P. E. Dawson, D. J. Craik, M. Freissmuth and C. W. Gruber, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 21183–21188 CrossRef CAS PubMed.
- (a) K. Arai, T. Takei, M. Okumura, S. Watanabe, Y. Amagai, Y. Asahina, L. Moroder, H. Hojo, K. Inaba and M. Iwaoka, Angew. Chem., Int. Ed., 2017, 56, 5522–5526 CrossRef CAS; (b) K. Medini, P. W. R. Harris, A. Menorca, K. Hards, G. M. Cook and M. A. Brimble, Chem. Sci., 2016, 7, 2005–2010 RSC; (c) M. Muttenthaler, S. T. Nevin, A. A. Grishin, S. T. Ngo, P. T. Choy, N. L. Daly, S.-H. Hu, C. J. Armishaw, C.-I. Wang, R. J. Lewis, J. L. Martin, P. G. Noakes, D. J. Craik, D. J. Adams and P. F. Alewood, J. Am. Chem. Soc., 2010, 132, 3514–3522 CrossRef CAS PubMed; (d) S. Fiori, S. Pegoraro, S. Rudolph-Bçhner, J. Cramer and L. Moroder, Biopolymers, 2000, 53, 550–564 CrossRef CAS PubMed; (e) S. Pegoraro, S. Fiori, S. Rudolph-Bohner, T. X. Watanabe and L. Moroder, J. Mol. Biol., 1998, 284, 779–792 CrossRef CAS PubMed; (f) Y. Z. Yin, Q. R. Fei, W. D. Liu, Z. R. Li, H. Suga and C. L. Wu, Angew. Chem., Int. Ed., 2019, 58, 4880–4885 CrossRef CAS PubMed; (g) C. Zuo, W.-W. Shi, X.-X. Chen, M. Glatz, B. Riedl, I. Flamme, E. Pook, J. Wang, G.-M. Fang, D. Bierer and L. Liu, Sci. China: Chem., 2019, 62, 1371–1378 CrossRef CAS.
- (a) K. Holland-Nell and M. Meldal, Angew. Chem., Int. Ed., 2011, 50, 5204–5206 CrossRef CAS PubMed; (b) M. Empting, O. Avrutina, R. Meusinger, S. Fabritz, M. Reinwarth, M. Biesalski, S. Voigt, G. Buntkowsky and H. Kolmar, Angew. Chem., Int. Ed., 2011, 50, 5207–5211 CrossRef CAS PubMed; (c) A. White, S. de Veer, G. Wu, P. Harvey, K. Yap, G. J. King and D. Craik, Angew. Chem., Int. Ed., 2020, 59, 11273–11277 CrossRef CAS PubMed.
- (a) M. A. Hossain, K. J. Rosengren, S. Zhang, R. A. D. Bathgate, G. W. Tregear, B. J. van Lierop, A. J. Robinson and J. D. Wade, Org. Biomol. Chem., 2009, 7, 1547–1553 RSC; (b) C. A. Macraild, J. Illesinghe, B. J. Van Lierop, L. Amanda, M. Chebib, B. G. Livett, A. J. Robinson and R. S. Norton, J. Med. Chem., 2009, 52, 755 CrossRef CAS PubMed; (c) T. Wang, Y. F. Kong, Y. Xu, J. Fan, H. J. Xu, D. Bierer, J. Wang, J. Shi and Y. M. Li, Tetrahedron Lett., 2017, 58, 3970 CrossRef CAS; (d) F. L. Feng, Y. Guo, S. J. Li and Y. M. Li, Org. Biomol. Chem., 2015, 13, 6286–6290 RSC; (e) Y. Wu, Y. H. Li, X. Li, Y. Zou, H. L. Liao, L. Liu, Y. G. Chen, D. Bierer and H. G. Hu, Chem. Sci., 2017, 8, 7368 RSC; (f) M. Pan, Q. Y. Zheng, S. Gao, Q. Qu, Y. Y. Yu, M. Wu, H. Lan, Y. L. Li, S. L. Liu, J. B. Li, D. M. Sun, L. N. Lu, T. Wang, W. H. Zhang, J. W. Wang, Y. M. Li, H.-G. Hu, C. L. Tian and L. Liu, CCS Chem., 2019, 1, 476–489 CrossRef CAS.
- (a) G. M. Williams, K. Lee, X. Li, G. J. Cooper and M. A. Brimble, Org. Biomol. Chem., 2015, 13, 4059 RSC; (b) M. A. Hossain, L. M. Haugaard-Kedstrom, K. J. Rosengren, R. A. D. Bathgate and J. D. Wade, Org. Biomol. Chem., 2015, 13, 10895 RSC.
- (a) A. Williamson, Justus Liebigs Ann. Chem., 1851, 77, 37 CrossRef CAS; (b) S. Mandal, S. Mandal, S. K. Ghosh, P. Sar, P. Ghosh, R. Saha and B. Saha, RSC Adv., 2016, 6, 69605 RSC.
- (a) X. E. Hu, Tetrahedron, 2004, 60, 2701–2743 CrossRef CAS; (b) D. Tanner, Angew. Chem., Int. Ed. Engl., 1994, 33, 599–619 CrossRef; (c) B. A. BhanuPrasad, G. Sekar and V. K. Singh, Tetrahedron Lett., 2000, 41, 4677–4679 CrossRef; (d) H. Q. Liu, V. R. Pattabiraman and J. C. Vederas, Org. Lett., 2007, 9, 4211–4214 CrossRef CAS PubMed; (e) C. C. Chen, S. Gao, H. S. Ai, Q. Qu, C. Tian and Y. M. Li, Sci. China: Chem., 2018, 61, 702–707 CrossRef CAS.
- B. McKeever and G. Pattenden, Tetrahedron, 2003, 59, 2071–2712 Search PubMed.
- M. Muttenthaler, A. Andersson, A. D. de Araujo, Z. Dekan, R. J. Lewis and P. F. Alewood, J. Med. Chem., 2010, 53, 8585–8596 CrossRef CAS PubMed.
- (a) T. Nakamura, H. Furunaka, T. Miyata, F. Tokunaga, T. Muta, S. Iwanaga, M. Niwa, T. Takao and Y. Shimonishi, J. Biol. Chem., 1988, 263, 16709 CAS; (b) H. Tamamura, R. Ioma, M. Niwa, S. Funakoshi, T. Murakami and N. Fujii, Chem. Pharm. Bull., 1993, 41, 978 CrossRef CAS PubMed.
- C. Ramalingan, I.-S. Lee and Y.-W. Kwak, Chem. Pharm. Bull., 2009, 57, 591 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02374d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |